

Abstract
Chemotherapy, radiation, and surgery are the conventional treatment modalities for cancer. The success achieved with these approaches has been limited due to several factors like chemoresistance to drugs, non‐specificity leading to peripheral toxicity, and non‐resectable tumors. To combat these problems, the concept of targeted therapy using immunotoxins was developed. Immunotoxins are chimeric proteins with a cell‐selective ligand chemically linked or genetically fused to a toxin moiety and can target cancer cells overexpressing tumor‐associated antigens, membrane receptors, or carbohydrate antigens. Ligands for these receptors or monoclonal antibodies or single chain variable fragments directed against these antigens are fused with bacterial or plant toxins and are made use of as immunotoxins. Pseudomonas exotoxin, anthrax toxin, and diphtheria toxin are the commonly used bacterial toxins. Ricin, saporin, gelonin, and poke weed antiviral protein are the plant toxins utilized in immunotoxin constructs. Several such fusion proteins are in clinical trials, and denileukin difitox is a FDA‐approved fusion protein. In spite of the promise shown by bacterial‐ and plant toxin‐based chimeric proteins, their clinical application is hampered by several factors like immunogenicity of the toxin moiety and non‐specific toxicity leading to vascular leak syndrome. In order to overcome these problems, a novel generation of immunotoxins in which the cytotoxic moiety is an endogenous protein of human origin like proapoptotic protein or RNase has been developed. This review summarizes the advances in this new class of fusion protein and the future directions to be explored. (Cancer Sci 2009)
Conventional treatment modalities for cancer include chemotherapy, radiation, and surgery. Despite the tremendous strides made in anticancer treatment, mortality rates due to cancers remain alarmingly high due to several reasons. Surgical resection of solid tumors often fails in total elimination of the disease as metastatic cells often remain, leading to relapse. Though most patients initially respond well to chemotherapy, later on chemoresistance is acquired. Besides, chemotherapeutic drugs are non‐specific. Hence, dose escalation and repeated cycles of administration for total elimination of the tumor will lead to peripheral toxicity. Radiotherapy too has deleterious effects on normal cells in the body.
To combat these problems, the concept of targeted therapy was developed. In the early 1900s Paul Ehrlich reasoned that antibodies coupled with toxic compounds could serve as ‘magic bullets’ for killing cancer cells.( 1 ) Immunotoxins are chimeric proteins with a cell‐selective ligand chemically linked or genetically fused to a toxin moiety.( 2 , 3 ) They exploit the phenomenon that cancer cells overexpress several tumor associated antigens, membrane receptors, and carbohydrate antigens. Ligands for these receptors or monoclonal antibodies or single chain variable fragments (scFv) directed against these antigens are fused with bacterial or plant toxins and used as immunotoxins. Pseudomonas exotoxin, anthrax toxin, and diphtheria toxin( 4 ) are the commonly used bacterial toxins. These toxins bring about inactivation of protein synthesis by ADP ribosylation of elongation factor 2, thereby leading to cell death. The normal cell‐binding domains of these proteins are mutated or totally deleted in such fusion proteins. Ricin, saporin, gelonin, and poke weed antiviral protein are plant toxins utilized in immunotoxin constructs. Several of such fusion proteins are in clinical trials and denileukin difitox is a FDA‐approved fusion protein.( 5 )
In spite of the promise shown by bacterial and plant toxin‐based chimeric proteins, they pose several obstacles that limit their clinical application.( 6 ) The toxin part of these fusion proteins elicits a high degree of humoral response in humans. Besides, in developed countries where people are immunized against diphtheria, serum will have circulating antibodies against diphtheria toxin, which will result in neutralization of diphtheria toxin‐based immunotoxins.( 7 , 8 ) Both Pseudomonas exotoxin and diphtheria toxin are large molecules and are difficult to humanize. At sufficiently high concentrations these fusion proteins lead to symptoms like vascular leak syndrome and show some degree of non‐specific toxicity. When the targeted antigen is expressed to some extent on normal cells in vital organs even normal cells will be killed, leading to complications.
In order to overcome these problems, various strategies have been tried. Notable among them are administration of immunosuppressive agents like cyclophosphamide and cyclosporine and monoclonal antibodies like rituximab, polyethylene glycol modification of the toxin, and site‐directed mutagenesis of the toxin to generate less immunogenic variants.( 9 , 10 , 11 , 12 , 13 ) A DNA–protamine based immunotoxin has also been developed.( 14 ) Due to limited success achieved with these strategies in animal models and clinical studies, efforts are being made to generate a new class of immunotoxin in which the cytotoxic moiety is an endogenous protein of human origin like proapoptotic protein or RNase. The targeting moiety in these immunotoxin constructs belongs to one of the two groups mentioned earlier: (i) monoclonal antibody or single chain variable fragment (svFv) specific for the overexpressed tumor‐associated antigen or membrane receptor; or (ii) natural ligand for the membrane receptor like cytokine, growth factor, or peptide hormone. This review summarizes the advances in this new class of fusion protein.
Proapoptotic protein‐based immunotoxins
Apoptosis is an important means for cellular homeostasis and is needed for the correct development and function of multicellular organisms.( 15 ) Cells undergo apoptosis in response to stimuli like irradiation, anticancer drugs, and growth factor deprivation. Caspases that are serine proteases are the major executioners of apoptosis and they get activated by two separate pathways: the death receptor (extrinsic) pathway and mitochondrial (intrinsic) pathway. The former is initiated by the engagement of transmembrane death receptors (Fas, tumor necrosis factor [TNF] receptor, and TNF‐related apoptosis‐inducing ligand [TRAIL] receptor) by their corresponding ligands. Ligand binding leads to receptor trimerization and activation of membrane proximal caspases (caspase 8 or caspase 10), which in turn cleave and activate other effector caspases like caspase 3 and caspase 7. These caspases in turn cleave various cellular substrates, bringing about cellular disassembly. In the intrinsic pathway, caspase 8 cleaves the Bcl‐2 family protein Bid, which then inserts into the mitochondrial membrane evoking release of cytochrome c. Cytochrome c released from the mitochondrial intermembrane space to the cytoplasm complexes with apoptotic protease activating factor‐1 and procaspase‐9 to form the apoptosome, which in return induces activation of caspase 9 and thereby initiates the apoptotic cascade.
Aberrations in the apoptotic mechanisms lead to diseases like cancer.( 16 , 17 ) In cancer the proteins involved in induction of apoptosis are mutated or non‐functional leading to a reduced sensitivity toward apoptosis. In some cases, the antiapoptotic proteins are upregulated. Hence, it is of therapeutic interest to design chimeric proteins that selectively target cancer cells and tip the balance of cellular fate toward apoptosis. Endogenous proteins that have distinct roles in apoptotic pathways are fused with cytokines, peptide hormones, or antibody fragments to produce chimeric proteins with anticancer activity. Following are the major proapoptotic proteins utilized for this purpose.
Bcl‐2 family proteins. The Bcl‐2 family, the best‐characterized protein family involved in the regulation of apoptosis, is divided into two groups: the antiapoptotic proteins such as Bcl‐2 and Bcl‐xL; and the proapoptotic proteins, which include Bax and Bak.( 18 , 19 ) The former prevents apoptosis by sequestering proforms of the caspases or by inhibiting the release of mitochondrial apoptotic factors into the cytoplasm. The latter after oligomerization acts on mitochondrial permeability transition pores and induces the release of mitochondrial apoptotic factors into the cytoplasm. Among the prodeath members, Bak and Bax have been categorized as the key regulators of cytochrome c release.( 20 , 21 ) Other prodeath members, mainly BH3‐only proteins, are thought to directly induce Bak–Bax oligomerization or to antagonize the antideath Bcl‐2 members, and are regulated by caspases. The antiapoptotic Bcl‐2 members prevent mitochondrial protein release by interacting with and inhibiting both Bak–Bax and BH3‐only proteins. The apoptotic function of Bik, Bax, and Bak was exploited to form Interleukin‐2‐Bax (IL‐2–Bax)( 22 ) and Gonadotropin‐releasing Hormone‐Bik‐Bax‐Bak (GnRHBik–Bax–Bak)( 23 ) chimeras, which were found to be specifically cytotoxic to cancers expressing the corresponding receptor.
DNA fragmentation factor 40. DNA fragmentation factor (DFF) 40 or caspase‐activated DNase is a nuclease that acts downstream in the apoptotic cascade. In vivo it is found attached to its inhibitor DFF45 or inhibitor of CAD.( 24 ) Upon activation by caspase 3 or granzyme B, DFF40 is translocated to the nucleus where it brings about cleavage of internucleosomal DNA, leading to apoptosis.( 25 ) GnRH was genetically fused to DFF40 and the partially purified chimeric protein was found to be cytotoxic to adenocarcinoma cell lines, which are GnRHR positive.( 26 )
Granzyme B. Granzymes are serine proteases present along with perforins in the cytoplasmic granules of natural killer cells and cytotoxic T lymphocytes. Binding of the cytotoxic T lymphocytes to a virus‐infected cell or transformed cell results in transmembrane pore formation in the target cell by perforins and granzyme entry into the cell by the granule exocytose pathway. Granzyme release into the cell is triggered by perforin when it perforates the granzyme endosomal membrane. Five granzymes (A, B, H, M, and tryptase) have been identified in humans, of which granzyme B is the most potent. It cleaves substrates at key aspartic acid residues. It cleaves several procaspases and also caspase substrates like inhibitor of CAD. It also cleaves the protein Bid, involved in altering the mitochondrial membrane permeability, and thus plays a major role in inducing apoptosis.( 27 , 28 , 29 ) This apoptosis‐inducing activity of granzyme B was redirected to eliminate cancer cells by fusing it with ligands like vascular endothelial growth factor,( 30 ) transforming growth factor α, and scFv specific for the membrane antigens ErbB2( 31 ) and gp240.( 32 ) The chimeric proteins were found to be cytotoxic and gave rise to characteristic apoptotic features like DNA laddering and cytochrome c release from mitochondria in the target cells.
Tumor necrosis factor‐related apoptosis‐inducing ligand. TRAIL is a member of the TNF super family and is involved in the elimination of cancer cells and virus‐infected cells by T cells and natural killer cells.( 33 , 34 ) TRAIL is expressed as a homotrimeric type 2 transmembrane protein or as its proteolytic product, which is soluble TRAIL (sTRAIL). TRAIL has been found to bind to five receptors: death receptor 4 (DR4/TRAIL‐R1), death receptor 5 (DR5/TRAIL‐R2), decoy receptors DcR1 and DcR2 and also the soluble receptor osteoprotegrin.( 35 ) The binding of TRAIL to DR4 or DR5 recruits caspase 8 via Fas‐Associated protein with Death Domain (FADD), leading to the formation of death‐inducing signaling complex and the subsequent caspase‐mediated apoptosis.( 36 , 37 ) TRAIL activity is modulated mainly by low‐affinity binding to the decoy receptors, which lack the death domain.( 38 ) TRAIL has been found to show selective toxicity toward tumor cells.( 39 ) It is presumed that it is due to the overexpression of TRAIL receptors in tumor cells or the high rate of expression of decoy receptors in normal cells. These attractive properties led to the development of several therapeutics targeting TRAIL receptors. These include recombinant sTRAIL, agonistic antibodies and gene therapy agents that express TRAIL.
The efficacy of antibody‐based therapies is often hampered as the tumor cells adopt tactics such as target antigen shedding and masking to escape immune surveillance. Target antigen expression may be downregulated due to therapy. In this context, therapeutic strategies that bring about bystander effects are important. A promising strategy that has been developed uses TRAIL‐based immunotoxins that target cancer cells overexpressing growth factor receptors and bring about the bystander effect. The bystander effect is based on the principle that targeted tumor cells are not only eliminated, but are also exploited to convey a therapeutic effect toward neighboring tumor cells that lack expression of the target antigen. Three chimeric proteins, scFv54–sTRAIL, scFv425–sTRAIL, and scFvCD7–sTRAIL, which target epithelial glycoprotein‐2, epidermal growth factor receptor, and CD7‐positive cancer cells, respectively, have been developed.( 40 , 41 , 42 , 43 ) All three of the proteins after binding to their target cells could effectively get rid of nearby target cells lacking the target antigen by crosslinking the agonistic TRAIL receptors on them.
This attractive feature makes sTRAIL‐based immunotoxins worthy of consideration as a substitute for existing targeted therapies.
FASL. Fas‐ligand (FASL) is an effector molecule involved in the death pathway of apoptosis induction. To exploit this property, several FASL agonists like anti‐FAS antibodies and multimeric soluble forms of FASL were developed. In spite of their tumoricidal activity toward cell lines and primary tumors, their clinical application in humans was not possible as they were found to be lethal to mouse models.( 44 , 45 ) It was discovered that oligomeric, multimeric, and aggregated forms of FASL in recombinant souble FASL (sFASL) preparations lead to systemic toxicity whereas a homotrimeric form showed toxicity only upon selective targeting to a tumor cell.( 46 ) Hence, fusion proteins using this form of sFASL were tried for. sFASL was genetically fused to anti‐CD7 scFv( 47 ) and was found to be highly toxic to CD7+ T‐cell acute lymphoblastic leukemia (T‐ALL) and acute myeloblastic leukemia (AML) cell lines (Table 1; Fig. 1).
Table 1.
Proapoptotic protein‐based immunotoxins, their effector and targeting moieties, and respective cellular targets
| Immunotoxin | Cytotoxic moiety | Targeting moiety | Target | Reference |
|---|---|---|---|---|
| IL‐2–Bax | Bax | IL‐2 | CTCL, renal cell cancer, melanoma | 22 |
| GnRH–Bik–Bax–Bak | Bik–Bax–Bak | GnRH | Adenocarcinomas | 23 |
| GnRH–DFF40 | DFF40 | GnRH | Adenocarcinomas | 26 |
| GrB–VEGF121 | Granzyme B | VEGF | Solid tumors | 30 |
| GrB5 | Granzyme B | Anti‐ErbB2 scFv | Epithelial cell tumors | 31 |
| GrB–scFVMEL | Granzyme B | Anti‐gp240 scFV | Melanoma | 32 |
| scFv54–sTRAIL | sTRAIL | Anti‐EGP2 scFv | Colorectal, breast carcinomas | 40 |
| scFv425–sTRAIL | sTRAIL | Anti‐EGFR scFv | Squamous cell carcinomas | 41,42 |
| ScFvCD7–sTRAIL | sTRAIL | Anti‐CD7scFV | Leukemias | 43 |
| ScFvCD7–FASL | FASL | Anti‐CD7scFV | Leukemias | 47 |
TCL, Cutaneous T cell lymphoma; DFF, DNA fragmentation factor; EGFR, Epidermal growth factor receptor; EGP, Epithelial glyco protein; FASL, Fas ligand; GnRH, Gonadotropin‐releasing hormone; GrB, granzyme B; IL, Interleukin; scFVMEL, Single chain fragment variable targeting the melanoma gp240 antigen; sTRAIL, soluble TRAIL; TRAIL, tumor necrosis factor‐related apoptosis‐inducing ligand; VEGF, Vascular endothelial growth factor.
Figure 1.
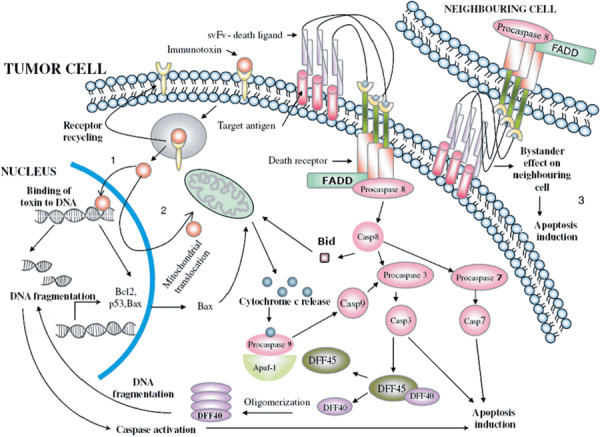
Mechanism of action of proapoptotic protein‐based immunotoxins. The immunotoxin binds to the cancer cell by means of its targeting domain and undergoes internalization. It is released into the cytoplasm from the endosome and the receptor is degraded or recycled back to the surface. From the cytosol, it embarks upon specific pathways depending on the constituent cytoxic moiety. (1) Conjugates with DNA fragmentation factor (DFF) 40 moves to the nucleus by means of its nuclear localization signal, binds to DNA, and cleaves it. Damaged DNA serves as an apoptotic signal resulting in cytochrome c release from the mitochondria and caspase activation culminating in cell death. In addition, the activated caspases release endogenous DFF40 from its inhibitor DFF45, thus resulting in an amplification loop. (2) Certain other conjugates move to the nucleus where they exert their effect on the transcription of specific genes and then translocate back to the cytoplasm for action on mitochondria, subsequent cytochrome c release, caspase activation, and ultimately apoptosis. (3) Conjugates with death factors like sFASL or soluble tumor necrosis factor‐related apoptosis‐inducing ligand as effector molecules, after binding to the target antigen, crosslink with death receptors on autologous as well as neighboring tumor cells, activating the death pathway of apoptosis induction. The bystander effect is produced on tumor cells that lack the target antigen. Casp, caspase; sFASL, souble FASL.
The results obtained with all these apoptotic protein‐based immunotoxins are encouraging. As all of the above‐mentioned chimeric proteins are of human origin, they are expected to display reduced immunogenicity in human recipients. Besides, as target cells are killed via induction of apoptosis, systemic damage is unlikely. The apoptotic cells shrink and condense, whereas the organelles and plasma membrane maintain their integrity. The dead cells are rapidly engulfed by macrophages and are eliminated before any of their contents can leak outside to cause a systemic response. Thus, targeted apoptosis inducing chimeric proteins should open new avenues in the battle against cancer.
RNase‐based immunotoxins. Standard anticancer drugs like doxorubicin and cisplatin exert their cytotoxic effects by damaging DNA. The damaged DNA serves as a death signal for the cells to undergo apoptosis. But limited success has been achieved in the clinical management of many cancers due to chemoresistance exhibited by them. Numerous genetic and epigenetic changes in the cancer cell lead to chemoresistance. Intrinsic DNA repair mechanisms as well as non‐functional tumor suppressor protein p53 lead to cancer cells that can resist these drugs and survive and metastasize. Hence, chimeric proteins that bring about RNA damage and induce apoptosis without the involvement of p53 are attractive alternatives to standard DNA‐damaging anticancer drugs.
Ribonucleases can be considered as toxins due to their inherent ability to degrade RNA and thus causing cell death via inhibition of protein synthesis. Bacterial RNases like binase from Bacillus intermedius, the fungal RNase α‐sarcin from Aspergillus giganteus, the amphibian RNase ranpirnase or onconase from Rana pipiens and bovine semen RNase were found to be cytotoxic.( 48 ) It is safe to assume that the ripples of degradation of RNA are felt beyond the protein synthesis machinery as RNA is also present in other cellular components such as signal recognition particles involved in directing proteins to the endoplasmic reticulum. Due to this and the regulatory role played by small non‐coding RNA, any cleavage of RNA might result in altered gene expression. The success achieved in the ongoing clinical trials with onconase( 49 , 50 ) in the treatment of human mesotheliomas sparked off interest in the possible use of endogenous RNases in immunotoxin conjugates.( 51 )
Ribonucleases constitute a large superfamily spread across several species. Human RNases are encoded by at least eight different genes out of which five (RNases 1–5) have been identified at the protein level. In addition to being ribonucleolytic, these are involved in a plethora of physiological activities like antitumor, antibacterial, antiviral, antihelminthic, and angiogenic activities. RNases of human origin utilized in immunotoxin constructs are described below.
Human pancreatic RNase1. This enzyme is present in low levels in the pancreas and in other organs and body fluids. In contrast, its bovine homolog is secreted in large amounts by the pancreas.( 52 ) This hints that human pancreatic RNase does not play much of a role in digestion. The mature protein comprises 128 amino acids preceded by a 28‐amino acid signal peptide and exhibits differential post‐translational processing based on tissue of origin.( 53 ) It is devoid of any inherent toxicity and has not been attributed with any of the physiological activities characteristic of other RNases. On the other hand, it undergoes internalization and displays cell‐killing ability when fused with ligands or antibody fragments specific for overexpressed membrane receptors. These attractive features led to the development of several immunoRNases with human pancreatic RNase1 as the cell‐killing moiety.( 54 , 55 , 56 , 57 , 58 )
RNase2 or eosinophil‐derived neurotoxin. This enzyme is present predominantly in eosinophils and in organs like the spleen and liver.( 59 ) The mature protein is composed of 134 residues and is synthesized as a preprotein with a 27‐amino acid leader peptide. Its primary structure bears 35% identity with that of RNase1 and 67% identity with RNase3. Eosinophil‐derived neurotoxin (EDN) is reported to be several‐fold catalytically more active than RNase3. This enzyme exhibits neurotoxic properties and is capable of inducing the Gordon phenomenon in rabbits.( 60 ) EDN also has antihelminthic and antiviral properties that are linked to its ribonucleolytic activity. EDN has been used as a ribonucleolytic partner in an immunoRNase, targeting leukemic cell lines with overexpression of transferrin receptor.( 61 ) Although the results obtained are encouraging it is not known whether the neurotoxic activity of EDN will be a setback for its therapeutic use.
RNase3 or eosinophil cationic protein. Eosinophil cationic protein (ECP) is present in the large granules of eosinophils and the mature protein is made up of 133 amino acids. It is antihelminthic, neurotoxic, and has been attributed with in vitro antibacterial activity. ECP is the most stable among all human RNases( 62 ) and is markedly different from other RNases in its ability to inhibit growth of a variety of cell lines.( 63 ) These desirable features make ECP a promising partner in RNase chimeras for therapeutic use in cancers.( 61 )
RNAse5 or angiogenin. Angiogenin is a 14‐kDa plasma protein with both angiogenic and ribonucleolytic activities and has 65% homology with RNase A.( 64 , 65 ) Angiogenin levels in serum are often used as indicators of the progression of several cancers.( 66 , 67 , 68 ) Though not cytotoxic by itself, it exhibits cytotoxicity when fused with a ligand and internalized upon binding. It can abolish cellular protein synthesis by hydrolyzing cellular tRNA. Though the principal role of angiogenin is angiogenesis, due to its tRNase activity in serum, it is speculated that it forms part of the host defense system. Several immunoRNases that make use of the ribonucleolytic property of angiogenin have been made for selective destruction of cancer cells (Table 2; Fig. 2).( 69 , 70 , 71 )
Table 2.
RNase‐based immunotoxins, their effector and targeting moieties and respective cellular targets
| Immunotoxin | Cytotoxic moiety | Targeting moiety | Target | Reference |
|---|---|---|---|---|
| RNase–EGF | Pancreatic RNase | Epidermal growth factor | Squamous cell carcinoma | 55 |
| hpRNase–FGF | Pancreatic RNase | Fibroblast growth factor | Melanomas, astronoma | 56 |
| hpRNase–IL‐2 | Pancreatic RNase | Interleukin‐2 | Leukemias, lymphoma | 57 |
| ERB–HPR | Pancreatic RNase | Anti‐ErbB2 scFv | Ovary, lung, breast carcinoma | 58 |
| EDN–scFv | Eosinophil derived neurotoxin | Anti‐TFRC scFv | Carcinomas, sarcoma | 61 |
| ECP–scFv | Eosinophil cationic protein | Anti‐TFRC scFv | Carcinomas, sarcoma | 61 |
| Ang–EGF | Angiogenin | Epidermal growth factor | Squamous cell carcinoma | 69 |
| CH2–Ang | Angiogenin | Anti‐TFRC scFv | Carcinomas, sarcoma | 70 |
| Ang–CD30 | Angiogenin | CD30 ligand | Hodgkin's lymphoma | 71 |
Ang, Angiogenin; ECP, Eosinophil cationic protein; EDN, Eosinophil derived neurotoxin; EGF, Epidermal growth factor; EGP, Epithelial glycoprotein; FGF, Fibroblast growth factor; IL, Interleukin; TFRC, Transferrin receptor.
Figure 2.
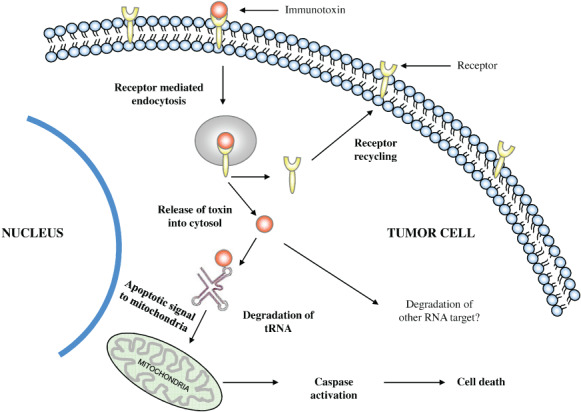
Mechanism of action of RNase‐based immunotoxins. Upon binding to the target antigen or receptor, the immunotoxin undergoes receptor‐mediated endocytosis. From the acidic compartment of the endosome, immunotoxin is released into the cytoplasm whereas the receptor is either degraded in the endosome or recycled back to the cell surface. The released conjugate degrades tRNA (as in the case of angiogenin‐based proteins) or other RNA targets. Degraded RNA leads to protein synthesis inhibition and cell death and also serves as an apoptotic signal to mitochondria, triggering apoptosis.
Human RNase‐based immunotoxins could be used to circumvent the problems of non‐specific toxicity and immunogenicity associated with bacteria‐ or plant‐based toxins. Although the majority of them are less potent in vitro in comparison to their counterparts using plant‐ or bacteria‐based toxins, they seem to be less toxic to animals and may selectively kill target cells in vivo at safer doses. The rate of protein synthesis inhibition by ricin‐ and diphtheria toxin‐based conjugates increases proportional to the square root of the toxin concentration. This might lead to a reduced log kill of target cells. In RNase conjugates there is a linear relationship between the rate of protein synthesis inhibition and toxin concentration, which is advantageous for attaining an increased log cell kill.
Concluding remarks
Though in vitro and in vivo efficacy studies done using humanized immunotoxins appear promising, we should exercise caution in avoiding an overstated interpretation of the results. It is a well‐known fact that drugs that kill cancer cells in tissue culture often fail to act on the same cells when grown as a tumor in animal models. Cancer cell lines are not true representatives of the parent tumors from which they are derived. They are made up of rapidly dividing identical cells whereas tumors are made up of heterogeneous populations of cells whose characteristics are decided by the tumor microenvironment. Cell lines might show altered properties due to prolonged culture and hence a heightened response to anticancer drugs.
Animal models with human xenografts are also intrinsically flawed due to various reasons and hence drugs proven to be effective in preclinical in vivo models may give disappointing results in clinical trials. In spite of the great deal of homology between the mouse and human genomes, there remains the fact that mice are not humans. A human tumor xenograft transplant is located in a murine environment, which will influence its sensitivity to chemotherapeutic drugs. The blood supply and neovascularization is provided by the host; the stroma is murine. Ideally the tumor should be located in an orthotopic position as growth properties of a tumor may differ in different locations. As orthotopic transplantation is technically difficult, researchers often go for subcutaneous sites, which have reduced metastatic potential. Certain biological mechanisms like replicative senescence and apoptosis are not perfectly conserved in mice and humans. These differences might contribute to the heightened sensitivity to anticancer drugs exhibited by animal models.
A critical factor that determines the success of an immunotoxin in terms of its specificity and cytotoxicity is the antigen that it targets. Ideally, the target antigen should be exclusive for tumor cells and should be capable of mediating internalization after immunotoxin binding. But tumor‐specific antigens are rare or non‐existent. Hence, the only alternative is to target antigens that are significantly overexpressed on tumor cells so that peripheral toxicity is limited. If the tumor cells express some antigenic variants that are absent on normal cells, monoclonal antibodies specific for the variants can be designed and used in immunotoxin constructs. With the advent of technologies like laser capture microscopy and DNA microarray an enormous chunk of data regarding gene expression in cancer cells is being generated. Hence we can remain hopeful that novel antigens that can be targeted for anticancer therapies will be identified. The cytotoxicity of proapoptotic protein‐based immunotoxins largely depends on the sensitivity of cells to apoptosis. Many cancer cells develop resistance to apoptosis by mechanisms like inactivation of p53, and upregulation of inhibitors of apoptosis like BCL‐2 and BCL‐xL. Various small molecule inhibitors of these antiapoptotic proteins like ABT‐737 have been found to be successful in sensitizing cancer cells to apoptosis. Combinatorial therapy using these small molecule inhibitors and immunotoxins should be the strategy adopted to eliminate cancer cells.
Endosomal sequestration of drugs is a factor that significantly curtails the availability of anticancer drugs for their cellular target and thereby their cytotoxicity. Tannock and Lee( 72 ) have demonstrated that this can be brought down by coadministration of basic compounds like chloroquine and omeprazole with anticancer drugs like doxorubicin. It is worth trying the same strategy with humanized immunotoxins. Yu et al. have developed a chimeric protein with truncated apoptosis‐inducing factor as the cell‐killing moiety for targeting HER2‐overexpressing tumor cells.( 73 ) This fusion protein had the translocation domain of Pseudomonas exotoxin for facilitating cytosolic release. This protein could kill HER2‐positive cells in culture as well as mouse models bearing human xenografts. In order to reduce the bacterial segments and thus the immunogenicity, they went on to develop granzyme B–caspase 3‐containing immunoproapoptotic conjugates with synthetic or bacteria‐derived furin‐sensitive sequences.( 74 ) These proteins effectively induced cell death and reduced tumor size in animal models. Thus we can conclude that incorporation of furin‐sensitive sequences in humanized immunotoxins is a good strategy for yielding efficacious proteins with sparse immunogenicity.
In vitro studies have proven that proper intracellular routing to the cytosol and resistance to inhibition by ubiquitous RNase inhibitors are the critical factors that determine the efficacy of human RNase‐based conjugates. RNase conjugates with RNase residues involved in binding to RNase inhibitors mutated, showed greater cytotoxicity, generating more potent RNase‐based immunotoxins. It has been proven that normally non‐toxic RNases like human RNase A and angiogenin become cytotoxic when artificially routed through the Golgi apparatus in the presence of retinoic acid. Following receptor binding, the mode of internalization of the bound RNase conjugate decides its efficient release into the cytosol and therefore its cytotoxicity. Hence we can speculate that fusion of RNase inhibitor‐resistant RNase variants with cell‐selective ligands that are trafficked efficiently into the cytosol may be an attractive strategy for developing potent immunotoxins.
Acknowledgments
We acknowledge the help rendered by Mr Dhandapani in preparing the illustrations.
References
- 1. Ehrlich P. The relationship existing between chemical constitution, distribution, and pharmacological action. In: Himmelweite F, Marquardt M, Dale H, eds. The Collected Papers of Paul Ehrlich. London: Pergamon Press, 1956; 596–618. [Google Scholar]
- 2. Pastan I. Targeted therapy of cancer with recombinant immunotoxins. Biochim Biophys Acta 1997; 1333: C1–C6. [DOI] [PubMed] [Google Scholar]
- 3. Krietman RJ. Immunotoxins for targeted cancer therapy. AAPS J 2006; 8: 532–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Potala S, Sahoo SK, Verma RS. Targeted therapy of cancer using diphtheria toxin‐derived immunotoxins. Drug Discov Today 2008; 13: 809–15. [DOI] [PubMed] [Google Scholar]
- 5. Piascik P. FDA approves fusion protein for treatment of lymphoma. J Am Pharm Assoc 1999; 39: 571–2. [DOI] [PubMed] [Google Scholar]
- 6. Frankel AE. Reducing the Immune Response to Immunotoxin Commentary re R. Hassan et al. Pretreatment with rituximab does not inhibit the human immune response against the immunogenic protein LMB‐1. Clin Cancer Res 2004; 10: 16–8. [DOI] [PubMed] [Google Scholar]
- 7. Hall PD, Virella G, Willoughby T, Atchley DH, Kreitman RJ, Frankel AE. Antibody response to DT‐GM, a novel fusion toxin consisting of truncated diphtheria toxin (DT) linked to human granulocyte‐macrophage colony‐stimulating factor (GM), during a phase I trial of patients with relapsed or refractory acute myeloid leukemia. Clin Immunol 2001; 100: 191–7. [DOI] [PubMed] [Google Scholar]
- 8. Hertler AA, Spitler LE, Frankel AE. Humoral immune response to a ricin A chain immunotoxin in patients with metastatic melanoma. Cancer Drug Deliv 1987; 4: 245–53. [DOI] [PubMed] [Google Scholar]
- 9. Oratz R, Speyer JL, Wernz JC, et al . Antimelanoma monoclonal antibody ricin A chain immunoconjugate (XMMME‐001‐RTA) plus cyclophosphamide in the treatment of metastatic malignant melanoma: results of a phase II trial. J Biol Response Mod 1990; 9: 345–54. [PubMed] [Google Scholar]
- 10. Selvaggi K, Saria EA, Schwartz R, et al . Phase I/II study of murine monoclonal antibody–ricin A chain (XOMAZYMEMel) immunoconjugate plus cyclosporine A in patients with metastatic melanoma. J Immunother 1993; 13: 201–7. [DOI] [PubMed] [Google Scholar]
- 11. Siegall CB, Haggerty WG, Warner GL, et al . Prevention of immunotoxin‐induced immunogenicity by coadministration with CTLA4Ig enhances antitumor efficacy. J Immunol 1997; 159: 5168–73. [PubMed] [Google Scholar]
- 12. Jin FS, Youle RJ, Johnson VG, et al . Suppression of the immune response to immunotoxins with anti‐CD4 monoclonal antibodies. J Immunol 1991; 146: 1806–11. [PubMed] [Google Scholar]
- 13. Tsutsumi Y, Onda M, Nagata S, Lee B, Kreitman RJ, Pastan I. Site‐specific chemical modification with polyethylene glycol of recombinant immunotoxin anti‐Tac (Fv)‐PE38 (LMB‐2) improves antitumor activity and reduces animal toxicity and immunogenicity. Proc Natl Acad Sci USA 2000; 97: 8548–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen SY, Zani C, Khouri Y, Marasco WA. Design of a genetic immunotoxin to eliminate toxin immunogenicity. Gene Ther 1995; 2: 116–23. [PubMed] [Google Scholar]
- 15. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 2007; 35: 495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jia L, Srinivasula SM, Liu FT, et al . Apaf‐1 protein deficiency confers resistance to cytochrome c‐dependent apoptosis in human leukemic cells. Blood 2001; 98: 414–21. [DOI] [PubMed] [Google Scholar]
- 17. Soengas MS, Capodieci P, Polsky D, et al . Inactivation of the apoptosis effector Apaf‐1 in malignant melanoma. Nature 2001; 409: 207–11. [DOI] [PubMed] [Google Scholar]
- 18. Adams JM, Cory S. Life‐or‐death decisions of the Bcl‐2 protein family. Trends Biochem Sci 2001; 26: 61–6. [DOI] [PubMed] [Google Scholar]
- 19. Cory S, Adams JM. The Bcl2 family: regulators of the cellular life‐or‐death switch. Nature Rev Cancer 2002; 2: 647–56. [DOI] [PubMed] [Google Scholar]
- 20. Xiang J, Chao DT, Korsmeyer SJ. BAX‐induced cell death may not require interleukin 1β‐converting enzyme‐like proteases. Proceedings of the Natl Acad Sci USA 1993, 14 559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jurgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci USA 1998; 95: 4997–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aqeilan R, Yarkoni S, Galski HL. Interleukin 2–Bax: a novel prototype of human chimeric proteins for targeted therapy. FEBS Lett 1999; 457: 271–6. [DOI] [PubMed] [Google Scholar]
- 23. Azar Y, Galski HL. GnRH‐Bik/Bax/Bak chimeric proteins target and kill adenocarcinoma cells: the general use of pro‐apoptotic proteins of the Bcl‐2 family as novel killing components of targeting chimeric proteins. Apoptosis 2000; 5: 531–42. [DOI] [PubMed] [Google Scholar]
- 24. Halenbeck R, MacDonald H, Roulston A, Chen TT, Conroy L, Williams LT. CPAN, a human nuclease regulated by the caspase‐sensitive inhibitor DFF45. Curr Biol 1998; 8: 537–40. [DOI] [PubMed] [Google Scholar]
- 25. Woo EJ, Kim YG, Kim MS, et al . Structural mechanism for inactivation and activation of CAD/DDFF40 in the apoptotic pathway. Mol Cell 2004; 14: 531–9. [DOI] [PubMed] [Google Scholar]
- 26. Yehudah BA, Aqeilan R, Robashkevich D, Galski HL. Using apoptosis for targeted cancer therapy by a new gonadotropin releasing hormone–DNA fragmentation factor 40 chimeric protein. Clin Cancer Res 2003; 9: 1179–90. [PubMed] [Google Scholar]
- 27. Shi L, Kam CM, Powers JC, Aebersold R, Greenberg AH. Purification of three cytotoxic lymphocyte granule serine proteases that induce apoptosis through distinct substrate and target cell interactions. J Exp Med 1992; 176: 1521–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Heusel JW, Wesselschmidt RL, Shresta S, Russell JH, Ley TJ. Cytotoxic lymphocytes require granzyme B for the rapid induction of DNA fragmentation and apoptosis in allogeneic target cells. Cell 1994; 76: 977–87. [DOI] [PubMed] [Google Scholar]
- 29. Talanian RV, Yang XH, Turbow J, et al . Granule‐mediated killing: pathways for granzyme B‐initiated apoptosis. J Exp Med 1997; 186: 1323–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu Y, Cheung LH, Thorpe P, Rosenblum MG. Mechanistic studies of a novel, human fusion toxin composed of vascular endothelial growth factor (VEGF) 121 and the serine protease granzyme B: directed apoptotic events in vascular endothelial cells. Mol Cancer Ther 2003; 2: 949–59. [PubMed] [Google Scholar]
- 31. Dälken B, Giesübel U, Knauer SK, Wels WS. Targeted induction of apoptosis by chimeric granzyme B fusion proteins carrying antibody and growth factor domains for cell recognition. Cell Death Differ 2006; 13: 576–85. [DOI] [PubMed] [Google Scholar]
- 32. Liu Y, Zhang W, Niuy T, et al . Targeted apoptosis activation with GrB/scFvMEL modulates melanoma growth, metastatic spread, chemosensitivity, and radiosensitivity. Neoplasia 2006; 8: 125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cleveland JL, Ihle JN. Contenders in FasL/TNF death signaling. Cell 1995; 81: 479–82. [DOI] [PubMed] [Google Scholar]
- 34. Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A. Induction of apoptosis by Apo‐2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem 1996; 271: 12 687–90. [DOI] [PubMed] [Google Scholar]
- 35. LeBlanc HN, Ashkenazi A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ 2003; 10: 66–75. [DOI] [PubMed] [Google Scholar]
- 36. Kischkel FC, Lawrence DA, Chuntharapai A, et al . Apo2L/TRAIL‐dependent recruitment of endogenous FADD and caspase‐8 to death receptors 4 and 5. Immunity 2000; 12: 611–20. [DOI] [PubMed] [Google Scholar]
- 37. Sprick MR, Weigand MA, Rieser E, et al . FADD/MORT1 and caspase‐8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity 2000; 12: 599–609. [DOI] [PubMed] [Google Scholar]
- 38. Pan G, Ni J, Wei YF, Yu G, Gentz R, Dixit VM. An antagonist decoy receptor and a death domain‐containing receptor for TRAIL. Science 1997; 277: 815–18. [DOI] [PubMed] [Google Scholar]
- 39. Carlo‐Stella C, Lavazza C, Locatelli A, Vigano L, Gianni AM, Gianni L. Targeting TRAIL agonistic receptors for cancer therapy. Clin Cancer Res 2007; 13: 2313–17. [DOI] [PubMed] [Google Scholar]
- 40. Bremer E, Kuijlen J, Samplonius D, Walczak H, De Leij L, Helfrich W. Target cell‐restricted and ‐enhanced apoptosis induction by an scFv:sTRAIL fusion protein with specificity for the pancarcinoma associated antigen EGP2. Intl J Cancer 2004; 109: 281–90. [DOI] [PubMed] [Google Scholar]
- 41. Bremer E, Samplonius D, Kroesen BJ, Genne L, De Leij L, Helfrich W. Exceptionally potent anti‐tumor bystander activity of an scFv:sTRAIL fusion protein with specificity for EGP2 towards target antigen negative tumor cells. Neoplasia 2004; 6: 636–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bremer E, Samplonius D, Genne L, et al . Simultaneous inhibition of EGFR signaling and enhanced activation of TRAIL‐R‐mediated apoptosis induction by an scFv:sTRAIL fusion protein with specificity for human EGFR. J Biol Chem 2005; 280: 10 025–33. [DOI] [PubMed] [Google Scholar]
- 43. Bremer E, Samplonius DF, Peipp M, et al . Target cell‐restricted apoptosis induction of acute leukemic T‐cells by a recombinant TRAIL fusion protein with specificity for human CD7. Cancer Res 2005; 65: 3380–8. [DOI] [PubMed] [Google Scholar]
- 44. Ogasawara J, Watanabe‐Fukunaga R, Adachi M, et al . Lethal effect of the anti‐FAS antibody in mice. Nature 1993; 364: 806–9. [DOI] [PubMed] [Google Scholar]
- 45. Rensing‐Ehl A, Frei K, Flury R, et al . Local FAS/APO‐1 (CD95) ligand‐mediated tumor cell killing in vivo . Eur J Immunol 1995; 25: 2253–8. [DOI] [PubMed] [Google Scholar]
- 46. Schneider P, Holler N, Bodmer JL, et al . Conversion of membrane‐bound FAS (CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J Exp Med 1998; 187: 1205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bremer E, Cate BT, Samplonius DF, De Leij LFMH, Helfrich W. CD7‐restricted activation of Fas‐mediated apoptosis: a novel therapeutic approach for acute T‐cell leukemia. Blood 2006; 107: 2863–70. [DOI] [PubMed] [Google Scholar]
- 48. Makarov AM, Ilinskaya NO. Cytotoxic ribonucleases: molecular weapons and their targets. FEBS Lett 2003; 540: 15–20. [DOI] [PubMed] [Google Scholar]
- 49. Mikulski SM, Grossman AM, Carter PW, Shogen K, Costanzi JJ. Phase 1 human clinical trial of ONCONASE (P‐30 protein) administered intravenously on a weekly schedule in cancer patients with solid tumors. Int J Oncol 1993; 3: 57–64. [DOI] [PubMed] [Google Scholar]
- 50. Mikulski SM. Phase II trial of onconase in patients with advanced malignant mesothelioma: analysis of survival. Fifth International Meeting on Ribonucleases May 12–16, 1999; Warrenton, VA.
- 51. Rybak SM, Newton DL. Natural and engineered cytotoxic ribonucleases: therapeutic potential. Exp Cell Res 1999; 253: 325–35. [DOI] [PubMed] [Google Scholar]
- 52. Weickmann JL, Elson M, Glitz DG. Purification and characterization of human pancreatic ribonuclease. Biochemistry 1981; 20: 1272–8. [DOI] [PubMed] [Google Scholar]
- 53. Beintema JJ, Weitzes P, Weicksmann J, Glitz JJ. The amino acid sequence of human pancreatic ribonuclease. Anal Biochem 1984; 136: 48–64. [DOI] [PubMed] [Google Scholar]
- 54. Zewe M, Rybak SM, Dubel S, et al . Cloning and cytotoxicity of a human pancreatic RNase immunofusion. Immunotechnology 1997; 3: 127–13. [DOI] [PubMed] [Google Scholar]
- 55. Psarras K, Ueda M, Yamamura T. Human pancreatic RNase1‐epidermal growth factor fusion; an entirely human ‘immunotoxin analog’ with cytotoxic properties against squamous cell carcinomas. Protein Eng Des Sel 1998; 11: 1285–92. [DOI] [PubMed] [Google Scholar]
- 56. Futami J, Seno M, Ueda M, Tada H, Yamada H. Inhibition of cell growth by a fused protein of human ribonuclease 1 and human basic fibroblast growth factor. Protein Eng Des Sel 1999; 12: 1013–19. [DOI] [PubMed] [Google Scholar]
- 57. Psarras K, Ueda M, Tanabe M, et al . Targeting activated lymphocytes with an entirely human immunotoxin analogue: human pancreatic RNase1–human IL‐2 fusion. Cytokine 2000; 12: 786–90. [DOI] [PubMed] [Google Scholar]
- 58. De Lorenzo C, Nigro A, Piccoli R, et al . A new RNase‐based immunoconjugate selectively cytotoxic for ErbB2‐overexpressing cells. FEBS Lett 2002; 516: 208–12. [DOI] [PubMed] [Google Scholar]
- 59. Rosenberg HF. The eosinophil ribonucleases. Cell Mol Life Sci 1998; 54: 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gordon MH. Remarks on Hodgkin's disease: a pathologic agent in the glands and its application in diagnosis. BMJ 1933; 1: 641–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Newton DL, Nicholls PJ, Rybak SM, Youle RJ. Expression and characterization of recombinant human eosinophil‐derived neurotoxin and eosinophil‐derived neurotoxin‐anti‐transferrin receptor sFv. J Biol Chem 1994; 269: 26 739–45. [PubMed] [Google Scholar]
- 62. Maeda T, Mahara K, Kitazoe M, et al . RNase 3 (ECP) is an extraordinarily stable protein among human pancreatic‐type RNases. J Biochem 2002; 132: 737–42. [DOI] [PubMed] [Google Scholar]
- 63. Maeda T, Kitazoe M, Tada H, et al . Growth inhibition of mammalian cells by eosinophil cationic protein. FEBS J 2002; 269: 306–16. [DOI] [PubMed] [Google Scholar]
- 64. Shapiro R, Riordan JF, Vallee BL. The characteristic ribonuclease activity of angiogenin. Biochem Insert 1986; 25: 3527–32. [DOI] [PubMed] [Google Scholar]
- 65. Saxena SK, Rybak SM, Davey RT Jr, Youle RJ, Ackerman EJ. Angiogenin is a cytotoxic, tRNA specific ribonuclease in the RNase A superfamily. J Biol Chem 1992; 267: 21 982–6. [PubMed] [Google Scholar]
- 66. Li D, Bell J, Brown A, Berry CL. The observation of angiogenin and basic fibroblast growth factor gene expression in human colonic adenocarcinomas gastric adenocarcinomas, and hepatocellular carcinomas. J Pathol 1994; 172: 171–5. [DOI] [PubMed] [Google Scholar]
- 67. Shimoyama S, Gansauge F, Gansauge S, Negri G, Oohara T, Beger HG. Increased angiogenin expression in pancreatic cancer is related to cancer aggressiveness. Cancer Res 1996; 56: 2703–6. [PubMed] [Google Scholar]
- 68. Chopra V, Dinh TV, Hannigan EV. Serum levels of interleukins, growth factors and angiogenin in patients with endometrial cancer. J Cancer Res Clin Oncol 1997; 123: 167–72. [DOI] [PubMed] [Google Scholar]
- 69. Yoon JM, Han SH, Kown OB, Kim SH, Park MH, Kim BK. Cloning and cytotoxicity of fusion proteins of EGF and angiogenin. Life Sci 1999; 64: 1435–45. [DOI] [PubMed] [Google Scholar]
- 70. Newton DL, Pollock D, DiTullio P, et al . Antitransferrin receptor antibody–RNase fusion protein expressed in the mammary gland of transgenic mice. J Immunol Meth 1999; 231: 159–67. [DOI] [PubMed] [Google Scholar]
- 71. Huhn M, Sasse S, Tur MK, et al . Human angiogenin fused to human CD30 ligand (Ang–CD30L) exhibits specific cytotoxicity against CD30‐positive lymphoma. Cancer Res 2001; 61: 8737–42. [PubMed] [Google Scholar]
- 72. Lee CM, Tannock IF. Inhibition of endosomal sequestration of basic anticancer drugs: influence on cytotoxicity and tissue penetration. Br J Cancer 2006; 94: 863–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yu JC, Jia TL, Meng LY, et al . Selective proapoptotic activity of a secreted recombinant antibody/AIF fusion protein in carcinomas over expressing HER2. Gene Ther 2006; 13: 313–20. [DOI] [PubMed] [Google Scholar]
- 74. Wang T, Zhao J, Ren LJ, et al . Recombinant immunoproapoptotic proteins with furin site can translocate and kill HER2‐positive cancer cells. Cancer Res 2007; 67: 11 830–9. [DOI] [PubMed] [Google Scholar]