

Abstract
Epithelial–mesenchymal transition (EMT) describes the differentiation switch between polarized epithelial cells and contractile and motile mesenchymal cells, and facilitates cell movements and generation of new tissue types during embryogenesis. Many secreted polypeptides are implicated in the EMT process and their corresponding intracellular transduction pathways form highly interconnected networks. Transforming growth factor‐β, Wnt, Notch and growth factors acting through tyrosine kinase receptors induce EMT and often act in a sequential manner. Such growth factors orchestrate the concerted regulation of an elaborate gene program and a complex protein network, needed for establishment of new mesenchymal phenotypes after disassembly of the main elements of epithelial architecture, such as desmosomes, as well as tight, adherens and gap junctions. EMT of tumor cells occurs during cancer progression and possibly generates cell types of the tumor stroma, such as cancer‐associated myofibroblasts. EMT contributes to new tumor cell properties required for invasiveness and vascular intravasation during metastasis. Here we present some of the current mechanisms that mediate the process of EMT and discuss their relevance to cancer progression. (Cancer Sci 2007; 98: 1512–1520)
Polarized epithelial cell tissues are based on the formation of intercellular tight and adherens junctions. This architectural arrangement of the tissue can be deorganized by epithelial–mesenchymal transition (EMT), whereby epithelial cells disassemble their junctional structures, start expressing mesenchymal cell proteins, remodel their extracellular matrix and become migratory( 1 ) (Fig. 1). EMT is therefore envisioned as a differentiation or morphogenetic process in which new tissue types are generated during embryogenesis, and which contributes to the pathogenesis of disease, such as metastatic cancer and tissue fibrosis.( 2 , 3 , 4 , 5 ) The inverse process of mesenchymal–epithelial transition (MET) describes how transitory mesenchymal cells generate polarized epithelia after migration and homing into new sites of tissue formation (Fig. 1). MET has been described in the context of embryonic development and is also perturbed pathologically in fibrotic disorders.( 5 ) Morphogenetic processes such as EMT or MET are guided by the functional interplay of many signal transduction pathways, usually initiated by secreted polypeptide factors, which aim at regulating a new set of transcriptional and post‐translational events, leading to the generation of new cellular phenotypes. Here, we discuss the role of different pathways in the control of EMT during embryogenesis and in disease.
Figure 1.
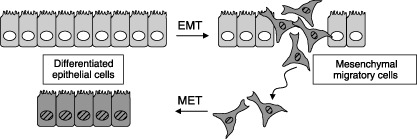
Epithelial–mesenchymal transition (EMT) and mesenchymal–epithelial transition (MET). Cells in an epithelial sheet undergo EMT, generating motile mesenchymal derivatives (darker color/striped nuclei). Mesenchymal cells undergo MET, generating epithelial derivatives.
EMT is important during embryogenesis
During the early stages of embryogenesis, the three germ layers, ectoderm, mesoderm and endoderm, form via an ontogenetic process called gastrulation (which stands for gut formation). While gastrulation in lower chordates involves movements of epithelial cell sheets,( 6 ) in higher vertebrates, the same process evolved a dependency on EMT, which leads to the formation of migratory mesenchyme that progresses along the primitive streak and populates new areas of the embryo that will develop into mesoderm and endoderm.( 1 ) Fibroblast growth factor (FGF) signaling via receptor tyrosine kinases (RTK) promotes mesodermal formation and mesenchymal cell migration through the primitive streak.( 7 ) An important target of FGF signaling during gastrulation is the master regulator of EMT, Snail, which directly represses expression of the epithelial integral component of adherens junctions, E‐cadherin.( 7 , 8 , 9 ) Furthermore, Wnt signaling via β‐catenin and its nuclear partner LEF‐1 is implicated in the EMT process during gastrulation, and stabilization of β‐catenin accelerates the emergence of premature EMT in the ectoderm.( 10 , 11 )
At a later stage of embryogenesis, epithelial cells from the neural tube in the dorsal side of the embryo, the neural crest, undergo EMT, and the mesenchymal cells produced often migrate long distances to new tissue areas, in order to differentiate into several new mesenchymal cell types such as somites, bone and chondrocytes.( 6 ) Neural crest EMT is regulated by dose‐dependent actions of bone morphogenetic proteins (BMP), which are members of the transforming growth factor (TGF)‐β superfamily, and a cohort of transcription factors, including paired‐box, high‐mobility group (HMG), winged‐helix transcription factors and Snail.( 12 ) In addition to neural crest EMT, members of the TGF‐β superfamily cause palatal EMT in the mouse in order to create the connective tissue across the palate.( 13 ) This action is mainly attributed to the TGF‐β member, TGF‐β3, because mice lacking the TGF‐β3 gene exhibit a cleft palate phenotype. In chicken and mice, the heart valve and septa form after a prominent EMT process of intracardial cells that respond in a sequential manner to Notch signaling, which induces expression of TGF‐β2 in order to induce Snail expression and repress E‐cadherin.( 14 , 15 ) Interestingly, the inverse scenario is also possible, as TGF‐β signaling can induce expression of Notch ligands, such as Jagged‐1, which activate Notch signaling, leading to EMT and epithelial cell cycle arrest in cell models in vitro.( 16 , 17 )
A surrogate model system for morphogenetic processes that normally occur during embryonic development, is the in vitro culture in 3‐D gels composed of extracellular matrix components.( 18 ) Such model systems have confirmed the critical role of the above signaling pathways in regulation of branching morphogenesis and the creation of organotypic epithelial tubes.( 18 ) Interestingly, recent approaches that have tried to model the factors that determine the position and frequency of branching tubes have revealed that the local activity of autocrine TGF‐β signaling acts as an inhibitor of branching site selection.( 19 )
It is therefore apparent that major developmental signaling pathways mediated by, for example, RTK, Notch, Wnt and TGF‐β provide primary inputs that change the fate of embryonic epithelial cells to mesenchymal derivatives with enhanced migratory and differentiation capacity, critical for the morphogenesis of many vital organs and tissues.
Different signaling pathways provide the necessary stimuli that trigger EMT
We will now discuss, in more detail, signal transduction pathways and critical mediators of EMT in the context of in vitro cell models and in vivo mouse tumor models (Fig. 2). The hepatocyte growth factor (HGF) that signals via the RTK c‐Met and the Erk mitogen‐activated protein kinase (MAPK) cascade, was among the first to be observed to play roles in reshaping epithelial differentiation towards a scattering phenotype that was characterized by robust down‐regulation of E‐cadherin and had critical links to tumor metastasis.( 20 ) The HGF pathway has recently been linked to the regulation of the transcription factor Snail, a major inducer of EMT.( 21 ) In a similar fashion, FGF signaling via its RTK receptor system mediates EMT.( 22 ) Recent work has established, at least in frog embryos, that FGF signaling promotes mesodermal differentiation by enhancing embryonic TGF‐β/nodal signaling.( 23 ) The mechanism depends on p53 phosphorylation in response to FGF‐induced activation of the Erk MAPK that enables the interaction of phosphorylated p53 with the TGF‐β/nodal signal transducers, Smads, in the nucleus. Phosphatidylinositol‐3′‐kinase (PI3K), which is a critical intracellular signaling mediator of RTK but also of many other transduction pathways, plays critical roles in the establishment of EMT, and provides cross‐talk between growth factor signaling, integrin receptors and small GTPases of the Rho family that control cytoskeletal organization.( 24 , 25 ) A similar nodal role has been recently established for the p38 MAPK, which signals down‐regulation of E‐cadherin during gastrulation.( 26 )
Figure 2.
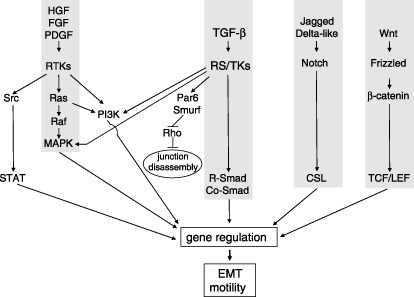
Major signal transduction pathways that induce epithelial–mesenchymal transition (EMT). Hepatocyte growth factor (HGF), fibroblast growth factor (FGF) and platelet‐derived growth factor (PDGF) signal via receptor tyrosine kinases (RTK) towards the central Ras‐Raf‐MAPK pathway or towards the PI3K pathway and the Src‐STAT pathway. Transforming growth factor (TGF)‐β signals via receptor serine/threonine kinases (RS/TK) towards the central R‐Smad/Co‐Smad pathway or towards the PI3K and MAPK pathways. Alternatively, the TGF‐β receptor signals towards the polarity protein Par6, thus recruiting the ubiquitin ligase Smurf, which degrades Rho and leads to disassembly of tight junctions in epithelial cells. Jagged and Delta‐like ligands signal via Notch receptors towards the transcription factor CSL. Wnt ligands signal via Frizzled receptors towards β‐catenin and the transcription factors LEF‐1/TCF. All these pathways modulate gene expression and lead to EMT and cell motility.
In addition to RTK signaling, polypeptide factors that signal via G‐protein coupled receptors, such as endothelin‐1, also mobilize signaling cascades that ultimately target Snail and the regulation of E‐cadherin expression during the establishment of EMT.( 27 ) Despite the established role of RTK signaling in EMT, recent studies on the regulation of epithelial cell polarity clearly demonstrate that oncogenic pathways whose signaling involves RTK may not be sufficient to elicit EMT; while they are capable of destroying polarity and tight junction assembly, they fail to induce a mesenchymal, migratory phenotype.( 28 ) The latter observation is compatible with the overwhelming evidence that EMT in various carcinomas involves an intricate interplay of multiple signaling pathways, including the tumor suppressor TGF‐β, as well as platelet‐derived growth factor (PDGF), Wnt and Notch, which cause activation of downstream Erk MAPK, β‐catenin and nuclear factor‐κB (NF‐κB) pathways( 2 ) (Fig. 3). In addition, many of these pathways activate intracellular reactive oxygen species (ROS), which inhibit the activity of phosphatases, thus affecting MAPK signaling and effectors of the EMT response.( 29 ) We will therefore attempt to highlight here this complex signaling network by emphasizing the roles of TGF‐β, while citing new examples of alternative pathways that contribute to EMT and tumor metastasis.
Figure 3.
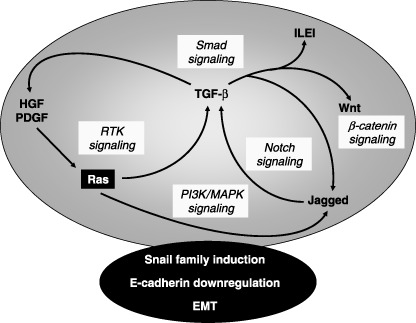
Autocrine/paracrine growth factor crosstalk during epithelial–mesenchymal transition (EMT). Autocrine or paracrine loops of growth factors induce expression and secretion of one another. Ras in the black box represents its oncogenic form that often is required in order to stimulate a robust autocrine crosstalk. The various signaling pathways involved are highlighted in light gray. The integrated signaling network (large gray ellipse) coordinates the action of transcription factors (such as Snail family members), which down‐regulate E‐cadherin among other gene responses in order to establish EMT.
TGF‐β is a major inducer of EMT
TGF‐β signaling does not only contribute to EMT during embryonic development, but also induces EMT during cancer progression in mouse models in vivo. TGF‐β signals via two distinct receptor serine/threonine kinases, the type I and type II receptors; after ligand binding, the type II receptor trans‐phosphorylates the type I receptor, which then phosphorylates cytoplasmic Smad proteins (Smad2 and Smad3). Activated Smad2/3 then form complexes with Smad4, which, upon entry to the nucleus, bind to chromatin and regulate expression of genes that play critical roles in the control of cell proliferation, differentiation (including EMT), apoptosis and cell migration.( 30 ) Furthermore, the TGF‐β receptors activate alternative signaling effectors, such as MAPK, PI3K and small GTPases of the Rho family that contribute to both gene regulation and cytoplasmic signaling involved in cell motility, apoptosis and EMT.( 31 )
Transgenic mice that misexpress TGF‐β1 in keratinocytes and are exposed to a chemical carcinogenesis protocol activate secretion of TGF‐β3, which further enhances EMT progression and development of spindle carcinomas that become invasive in an accelerated fashion.( 32 , 33 ) The fact that human skin carcinomas oversecrete TGF‐β1 and deregulate their TGF‐β type II receptor suggests a similar scenario in human skin malignancies.( 34 ) EMT induced by oncogenic stimuli depends on TGF‐β signaling because in skin carcinogenesis, in mouse mammary carcinomas that express the ras or raf oncogenes, as well as in liver and colon carcinomas, inhibitors of the TGF‐β pathway block EMT; interestingly, the TGF‐β inhibitors also block carcinoma invasiveness and metastasis.( 33 , 35 , 36 , 37 , 38 ) According to these mouse models of cancer progression, carcinoma cells secrete abnormally high doses of bioactive TGF‐β, which sensitizes both carcinoma and surrounding stromal cells, leading to escape from the primary growth suppressive and pro‐apoptotic response to TGF‐β, but permitting the establishment of EMT. These models of cancer progression seem to be different from in vitro EMT models of normal epithelial cells, which undergo EMT at the same time as their proliferation is arrested.( 39 ) The control of the cell cycle is therefore linked to the process of EMT, as it has been recently demonstrated in epithelial cells that undergo EMT only when their cycle is arrested at the G1/S border. In contrast, the same cells undergo apoptosis when their cycle is stalled at the G2/M boundary.( 40 ) This is relevant to the current model of the role of TGF‐β during cancer progression, in which TGF‐β suppresses normal epithelial and benign adenoma cell growth but also promotes aggressive carcinoma EMT, invasiveness and metastasis.( 41 )
In all in vitro cell models of EMT analyzed so far, TGF‐β down‐regulates various epithelial proteins, including E‐cadherin, the tight junction protein ZO‐1 and specific keratins, and up‐regulates certain mesenchymal proteins such as fibronectin, fibroblast‐specific protein 1, α‐smooth muscle actin and vimentin (reviewed in( 42 )). We have reported that signaling pathways of the TGF‐β/activin branch that activate Smad2 and Smad3, can induce EMT.( 39 , 43 ) In contrast, pathways of the BMP branch that activate Smad1, Smad5 and Smad8, do not induce robust EMT,( 39 ) and can even inhibit EMT promoted by TGF‐β in normal mammary and lens epithelial cells.( 44 , 45 ) Such in vitro studies, but also tumor analyses in mouse models have additionally established that Smad signaling mediates the EMT response to TGF‐β family members, because mutant Smad proteins that block endogenous Smad signaling, Smad‐specific RNA interference (RNAi), tissue‐specific Smad knockouts, and a mutant type I receptor for TGF‐β that cannot activate Smad2 or Smad3, all block this specific epithelial cell response (reviewed in( 42 )). Interestingly, comparative analysis of Smad2 versus Smad3 liver‐specific knockout mice has recently confirmed that TGF‐β‐driven EMT of hepatocytes depends on Smad3 and not Smad2; in contrast, Smad2 seemed to counteract the EMT response thus acting as a suppressor of hepatocyte dedifferentiation.( 46 )
Smad signaling normally depends on the common mediator Smad4. It was therefore surprising that a recent study showed that the EMT response was not perturbed at all in human immortalized keratinocytes and colon carcinoma cells in which Smad4 was depleted using RNAi.( 47 ) It is possible that low activity of the Smad signaling pathway, provided by a remaining low level of Smad4, is sufficient to induce EMT. This is a plausible explanation, as a parallel RNAi experiment targeting Smad4 in cultured cell models and tissue‐specific knockout of Smad4 in the mammary gland and the pancreas, have all confirmed an important role of Smad4 in EMT of these epithelial tissue types.( 48 , 49 , 50 ) Furthermore, Smad4 has been recently shown to be indispensable for the transcriptional mechanism that down‐regulates E‐cadherin expression in response to TGF‐β, a hallmark of EMT.( 51 )
The mechanism by which Smad signaling elicits the EMT response involves regulation of several genes, whose products either are necessary or need to be eliminated for EMT to take place. Large‐scale gene expression studies of cell models undergoing EMT in response to TGF‐β, or in vivo models of carcinoma invasiveness and metastasis, have revealed many potential regulators of EMT.( 39 , 52 , 53 , 54 , 55 , 56 ) Some of these genes that have already been functionally dissected will be discussed later.
Despite the importance of Smad signaling as an inducer of EMT, most in vitro cell studies implicate alternative, non‐Smad signaling effectors as major regulators of EMT. As already discussed above, transformed carcinoma cells expressing the ras oncogene, but also immortalized keratinocytes and normal mammary epithelial cells, respond to TGF‐β and exhibit highly active Erk, p38 and Jun‐N‐terminal kinase (JNK) MAPK, PI3K, Rho GTPase and NF‐κB signaling during induction of EMT.( 35 , 36 , 52 , 57 , 58 , 59 , 60 , 61 , 62 , 63 , 64 , 65 ) At least in the case of NF‐κB activation, which cooperates functionally with Smad signaling, the MAPK kinase kinase, TGF‐β‐activated kinase (TAK) 1, may link the TGF‐β receptors to the activation of IκB kinase 2 (IKK‐2), which phosphorylates and induces degradation of IκBα, thus releasing active NF‐κB.( 66 ) Additionally, integrin receptors can signal together with TGF‐β for the activation of the p38 MAPK, which can also contribute to EMT.( 67 , 68 ) Integrin‐linked kinase (ILK) gene expression can be induced by Smad signaling,( 69 ) and ILK contributes to TGF‐β‐induced EMT,( 70 ) or to BMP‐7‐induced ureteric bud formation, a morphogenetic process of renal epithelial cells.( 71 ) An alternative signaling mechanism activated by TGF‐β involves protein kinase A (PKA) and the signal transducer and activator of transcription 3 (STAT3), which mediate both EMT and apoptotic responses, via as yet uncharacterized downstream effector mechanisms.( 72 )
A new signaling mechanism during EMT induced by TGF‐β makes a link between the TGF‐β receptors and the polarity complex that regulates epithelial polarization, the activity of which needs to be reversed during EMT.( 73 ) According to the proposed mechanism, TGF‐β type I receptors are located in the tight junctions of polarized epithelial cells and interact with the integral membrane protein occludin and the polarity protein Par6.( 73 ) When TGF‐β signaling starts, the type II receptor is recruited to tight junctions and phosphorylates not only the type I receptor, but also the type I receptor‐associated Par6, which leads to degradation of the small GTPase RhoA and tight junction disorganization (Fig. 2).
In addition to tight junction dissolution during EMT, adherens junction disassembly is equally important, and a novel cellular mechanism proposes that E‐cadherin levels are down‐regulated after clathrin‐mediated endocytosis and lysosomal degradation in mammary epithelial cells undergoing EMT in response to the cooperating signals of TGF‐β and oncogenic Raf.( 74 ) Because TGF‐β induces expression of the adaptor protein disabled‐2 (Dab2) that participates in clathrin‐mediated endocytosis and protects mammary cells from apoptosis, while permitting EMT to occur,( 75 ) it is tempting to speculate that the pathway of lysosomal degradation of E‐cadherin might be induced by the TGF‐β/Dab2 signal. Yet another new signaling mechanism of TGF‐β‐induced EMT suggests that Ras and PI3K probably activate Src‐family tyrosine kinases, which phosphorylate α‐ and β‐catenin, thus leading to destabilization of E‐cadherin/catenin complexes and destruction of the adherens junctions in pancreatic carcinoma cells.( 76 ) These novel signaling mechanisms downstream of the TGF‐β receptors promise additional regulatory pathways that govern EMT and tumor cell invasiveness, and the possible involvement of Smad protein function in such membrane‐proximal mechanisms warrants future investigations.
In conclusion, a complex signaling mechanism downstream of TGF‐β induces EMT responses. Non‐Smad and Smad signals mediate the direct disorganization of epithelial cell adherens junctions, while a genomic program must be critical for the differentiation change and the generation of the mesenchymal cell phenotype, as will be discussed later. The latter underscores the importance of gene targets of the TGF‐β pathway and their role in EMT.
Gene targets of TGF‐β signaling establish cross‐talk with many other pathways during EMT
Because the targets of TGF‐β signaling are many (a few hundred measured by genome‐wide expression analyses in cell models in vitro), we divide their discussion into three parts: (i) gene targets that mediate activation of another signaling pathway, thus establishing cross‐talk with TGF‐β, which is necessary for EMT (Fig. 3); (ii) gene targets that encode transcription factors and that establish a hierarchical gene network which controls the EMT differentiation switch (Fig. 4); and (iii) gene targets that define the mesenchymal phenotype (Fig. 4).
Figure 4.
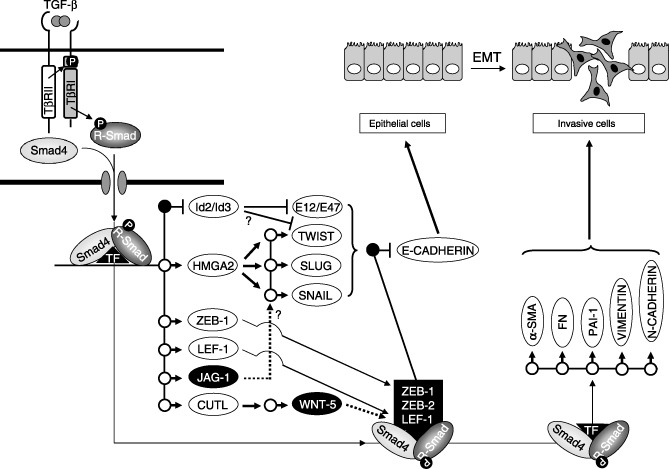
The transcriptional program of transforming growth factor (TGF)‐β that elicits epithelial–mesenchymal transition (EMT). TGF‐β activates Smad complexes that act together with other transcription factors (TF) and induce expression of several other transcription factors. Gene targets are shown as circles. Black circles represent gene repression, while white circles represent gene induction. Id2 and Id3 inhibit E‐box‐binding proteins such as E12/E47 and possibly Twist (?). HMGA2 induces further expression of Twist, Slug or Snail. All these transcriptional repressors down‐regulate E‐cadherin expression. ZEB‐1 and LEF‐1 are transcriptionally induced and together with ZEB‐2 participate in transcriptional complexes with Smads, and repress the E‐cadherin gene. Jagged‐1 (JAG‐1) activates the Notch pathway (dotted arrow), which possibly induces expression of Snail (?). CUTL induces expression of Wnt‐5, which activates the β‐catenin pathway (dotted arrow), thus mobilizing transcription factor LEF‐1. Finally, Smad complexes induce expression of α‐smooth muscle actin (α‐SMA), fibronectin (FN), PAI‐1, vimentin and N‐cadherin, which contribute to the establishment of the motile, mesenchymal cell.
TGF‐β regulates the status of other signaling pathways
A major example in this signaling scenario that leads to EMT has emerged during the past year and involves transcriptional induction of ligands of the PDGF family by the concerted action of the TGF‐β and oncogenic Ras pathways. The first clear evidence was reported from genome‐wide transcriptomic studies in hepatocellular carcinomas that undergo EMT, become invasive and show enhanced in vivo metastasis in response to TGF‐β and oncogenic Ras stimuli.( 35 , 77 ) In this cell model, the PDGF‐A isoform and the two forms of the PDGF receptor (α and β) were all up‐regulated in cells undergoing EMT, causing autocrine stimulation of cells. Critically, blocking PDGF signaling in the hepatocytes alleviated the ability of TGF‐β to elicit EMT and invasive cell behavior. The downstream effectors of PDGF signaling were identified as PI3K and nuclear β‐catenin, the former being important for a survival signal that protects hepatocarcinoma cells from anoikis and promotes their invasive spread during metastasis.( 78 ) An independent study has established the mechanism by which PDGF regulates β‐catenin function.( 79 ) Accordingly, the PDGF receptor directly phosphorylates the RNA helicase p68, which disrupts the cytoplasmic complex of β‐catenin with axin and glycogen synthase 3′ kinase β (GSK3β), translocates to the nucleus together with β‐catenin in a Wnt‐independent manner, and mediates formation of nuclear complexes with the transcription factor LEF‐1, thus leading to EMT. It appears therefore, that under the primary instruction of TGF‐β, PDGF signaling engages major components of the Wnt pathway, thus taking over the critical transcriptional control of the EMT transcriptome, in a Wnt‐independent manner. In a parallel model of breast cancer metastasis, TGF‐β induces EMT, which up‐regulates PDGF ligands and receptors, leading to PI3K activation and pro‐survival signals.( 80 ) This study provided new evidence that PDGF receptor expression can serve as a marker for metastatic breast cancer, and suggests that inhibitors of the PDGF receptor kinase, such as imatinib, could be used therapeutically as an anti‐metastatic agent.
Another transcriptional target of TGF‐β that links to tumor cell invasiveness and to the Wnt signaling pathway is the homeobox transcription factor CUTL1.( 81 ) CUTL1 induces expression of many genes that regulate cell motility, tumor cell invasiveness and extracellular matrix deposition, and CUTL1 has been proposed as a poor prognosis marker for metastatic breast carcinoma. The secreted factor Wnt5A is one of the major transcriptional targets of CUTL1.( 82 ) Wnt5A plays critical roles in the induction of EMT and invasiveness of pancreatic tumor cells, and serves as a prognostic marker of invasive pancreatic adenocarcinoma. Wnt signaling mobilizes Axin2, which blocks the activity of GSK3β and thus leads to Snail protein stabilization and enhanced transcriptional activity, thus facilitating the onset of EMT.( 83 )
Additional signaling pathways that are mobilized by the primary TGF‐β stimulus include the Notch pathway that was discussed above in the embryogenesis section, but also novel cytokines, such as the secreted interleukin‐like EMT‐inducer (ILEI), whose expression is stimulated by TGF‐β at the translational level, and which independently causes EMT, invasive growth of carcinomas and metastasis of breast cancer models.( 84 ) The signaling pathway elicited by ILEI remains unknown; however, it has been reported that ILEI might lead to sequential waves of chemokine secretion by carcinoma cells. This interesting possibility is in agreement with recent and parallel evidence that the chemokine CXCL12/stromal cell‐derived factor‐1 (SDF‐1), which signals via the receptor CXCR4 and downstream PI3K/Akt effectors, leads to EMT of oral squamous cell carcinomas, and promotes lymph node metastasis in mouse models.( 85 )
In conclusion, TGF‐β orchestrates the activity of many other signaling pathways contributing to EMT (Fig. 3). It should be noted that many of these pathways (e.g. Ras/MAPK, Notch) themselves induce TGF‐β secretion and activity, thus causing an amplification of EMT.
A transcription factor network downstream of TGF‐β
TGF‐β signals via Smads the transcriptional repression of Id genes, while BMP Smad signaling induces and stabilizes expression of the same Ids in epithelial cells( 44 , 86 ) (Fig. 4). The Id transcriptional regulators are known inhibitors of differentiation and they also inhibit EMT.( 44 , 87 ) Repression of Id gene expression by TGF‐β is required for subsequent down‐regulation of E‐cadherin and ZO‐1 and establishment of EMT.( 44 ) For example, Id2 repression by TGF‐β permits binding of the basic helix‐loop‐helix (bHLH) factors E12/E47 to the E‐cadherin promoter, which leads to its repression.( 87 ) In contrast, BMP signaling leads to high Id levels, which supposedly interfere with proper bHLH protein function, and via an unknown mechanism preserve epithelial differentiation. We have proposed that regulation of Id gene expression explains the established competition between TGF‐β and BMP signaling, because BMP induces MET in a dominant fashion relative to TGF‐β, which mediates EMT.( 5 , 44 , 45 , 88 ) We therefore propose that in early stage carcinomas, few cells undergo EMT and reduce their Id levels. These cells could possibly represent cancer stem cells. Upon metastasis, the transitory cells in the new site of tumor growth could again increase their Id levels in order to support their proliferation and survival, and in this sense Id protein regulation serves the purpose of metastatic spread. Experimental evidence shows that high Id2 levels can be measured in bone and lung metastases (reviewed in( 89 )); however, evidence for a decrease in Id levels during transient carcinoma invasiveness or intravasation is still lacking.
In addition to Id, transcriptional repressors of the E‐cadherin gene, such as members of the Snail family of zinc finger proteins (Snail, Slug), two‐handed zinc finger/homeodomain proteins (ZEB1, ZEB2), bHLH proteins (E12/E47, Twist) and high‐mobility group box‐containing proteins (LEF‐1), are involved in the EMT response to TGF‐β( 9 ) (Fig. 4). These repressors recognize E‐box DNA sequences located near the transcriptional initiation site of the E‐cadherin gene, and recruit transcriptional co‐repressors and histone deacetylases. As already discussed throughout the present review, regulation of E‐cadherin is a central event during EMT. TGF‐β transcriptionally induces Snail gene expression via Smad3 or via activation of the Erk and PI3K pathways.( 90 , 91 ) Smad proteins interact with ZEB1 or ZEB2, thus forming repressor complexes on the E‐box region of the E‐cadherin gene but also on other gene targets.( 9 , 92 , 93 ) TGF‐β can also activate LEF‐1 activity directly via Smad signaling during normal palate development or in mammary epithelial cells that are transformed by a synthetic fos‐estrogen receptor oncogene,( 13 , 56 , 94 , 95 ) or indirectly via PDGF or Wnt signaling pathways, as discussed before. In contrast, in chickens, TGF‐β induces expression of Slug during EMT that promotes normal development of the heart valves.( 96 ) It is not currently understood whether the many transcriptional mechanisms that aim at repressing E‐cadherin expression act independently from each other or in concert downstream of TGF‐β, or even whether they represent tissue‐specific scenarios. Our recent work provides new insight to this problem, as the high mobility group factor, HMGA2, has been identified as a new regulator of EMT, the expression of which is transcriptionally induced by TGF‐β/Smad signaling, and which represents a known regulator of mesenchymal differentiation during embryogenesis.( 97 ) Interestingly, HMGA2 induces expression of the transcriptional regulators Snail, Slug and Twist, while depletion of HMGA2 in mammary epithelial cells prevents EMT. This finding suggests that TGF‐β may be capable of eliciting a hierarchical network of transcriptional changes that involves many, if not all, of the above regulators of E‐cadherin.
Establishing the mesenchymal and migratory phenotype
Although regulation of E‐cadherin is a central event during EMT, this differentiation response is polygenic as already described, and thus many additional gene targets should be regulated by TGF‐β or any of the other signaling pathways involved. In most cases, it is accepted that the previously described transcription factors, that is, Snail, Slug, Twist or bHLH proteins, are responsible for the down‐regulation of all necessary epithelial gene expression, particularly those genes that contribute to the assembly of junctional complexes, such as claudins, connexins, occludin, ZO‐family genes, and more.( 98 ) Interestingly, the same transcription factors seem to be required for the induction of the mesenchymal phenotype, as, for example, Snail induces expression of fibronectin or vitronectin,( 98 ) and Twist induces expression of the serine/threonine kinase Akt2, which is an effector of PI3K and a critical signaling regulator of cell survival during EMT( 99 ) (Fig. 4). However, the pathways and genes that define mesenchymal differentiation emerging from epithelial precursors remain relatively underexplored. This is emphasized by the recent finding that transcriptional induction of a major marker of fibroblastic cells produced via EMT, fibroblast‐specific protein 1 (FSP1) is mediated by a transcriptional complex between CArG box‐binding factor A (CBF‐A) and KRAB‐associated protein 1 (KAP‐1).( 100 )
Cancer cell invasiveness is thought to be directly linked to the process of EMT. Motility, and an ability to degrade or remodel the extracellular matrix, is a common feature of invasive cells. The unifying model of EMT currently suggests that migratory and matrix remodeling (fibrotic) features of tumor cells may not only depend on the action of fibroblasts in the tumor environment, but rather characterize different stages of differentiation of the original carcinoma cell.( 3 )
Focusing again on TGF‐β and its role during carcinoma motility, a plethora of cellular mechanisms can be listed that involve both Smad‐dependent gene regulation and activation of alternative signaling effectors in the carcinoma cell. For example, microarray screens have identified regulators of actin dynamics downstream of TGF‐β, such as the guanine exchange factor NET1, which leads to sustained activation of Rho GTPases, thus supporting actin reorganization.( 101 ) TGF‐β also induces expression of α3β1‐integrin and promotes motility and invasiveness of hepatocellular carcinoma cells.( 102 ) In metastatic breast carcinoma cells, autocrine TGF‐β1 signals via the PI3K pathway to induce in vitro motility.( 103 ) Such examples bring about common signaling mechanisms that are involved in the establishment of EMT and also contribute to the process of carcinoma motility.
How relevant is EMT to cancer progression and metastasis?
The processes of embryonic development that rely on EMT for the generation of new tissue types are co‐opted by tumors, reflecting their inherent uncontrolled proliferation, which leads to spatial expansion.( 1 , 2 , 4 ) Cancer stem cells may be the critical contributors of EMT in the context of the growing tumor.( 104 ) If this is true, EMT in the tumor context essentially represents mesenchymal differentiation from tumor epithelial stem cells. The latter idea is compatible with studies of embryonic stem cells that are capable of undergoing EMT under in vitro culture conditions.( 105 ) In fact, a recent report on a mouse model of hepatocellular carcinoma progression and metastasis, suggests that sequential signal transduction by TGF‐β, which then induces PDGF secretion and PDGF receptor activation, cooperates with β‐catenin signaling to produce a small population of carcinoma cells that seem to act as cancer stem cells.( 78 )
The hypothesis that EMT during cancer progression may primarily affect the rare cancer stem cells is compatible with the low‐frequency observation of transitory mesenchymal cells within or near the mass of a growing tumor that becomes invasive and metastatic. Based on the difficulty of observing such rare cell types using classical histochemical techniques, many oncologists and tumor pathologists have disputed the relevance of EMT in cancer.( 106 ) A more objective view of the role of EMT during advanced tumor progression and metastasis has considered the fact that EMT can be transient and reversible, and that it represents only one of the steps required by carcinomas to establish productive expansion via invasiveness and intravasation to the neighboring vasculature.( 107 ) This is also compatible with the ability of epithelial cell sheets to migrate without the need of disseminating single migratory cells, possibly via the action of proteins such as podoplanin, a regulator of actin dynamics, as has recently been demonstrated.( 108 ) However, mechanisms such as epithelial sheet migration do not exclude the presence or significance of EMT as discussed here. Despite the apparent difficulties in studying tumor‐related EMT in vivo, recent advances in imaging technology and transgenic mouse models promise possibilities that transitory mesenchymal cells derived from carcinomas might become easier to follow.( 109 )
Because tumors are 3‐D tissues with a complex architecture and a plethora of interconnected cell types and surrounding extracellular matrix, it is obvious that EMT represents only part of the processes of tumor cell invasiveness and metastasis. Starting with a primary carcinoma, EMT of the transformed carcinoma cells can produce migratory mesenchymal derivatives. Such migratory cells use extracellular matrix structures (e.g. collagen fibers) to reach the pericyte/endothelial wall of nearby blood vessels in order to start the process of intravasation.( 109 ) In this context, EMT can be distinguished from events that initiate disruption of epithelial cell polarity, such as apical membrane differentiation and tight junction organization, but fail to generate true migratory cell derivatives.( 1 ) Finally, EMT of primary carcinomas may give rise to myoepithelial cells that are associated with the tumor, and which become major regulators of the extracellular matrix and providers of several growth factors, cytokines and chemokines that reshape the landscape of the tumor microenvironment as malignancy progresses.( 3 ) The latter feature of EMT in association with the hypothesis that EMT affects cancer stem cells may in the end be proven as the major biological roles of EMT in cancer progression.
Contribution of the tumor stroma on EMT and tumor metastasis
In addition to reshaping the differentiation fate of the carcinoma cells, EMT may also affect cells in the tumor microenvironment, such as cancer‐associated fibroblasts or myofibroblasts, immune cells and microvessels. The developmental origin of tumor‐associated fibroblasts is unclear, and EMT of carcinoma cells has been suggested to be a possible source of such cell types.( 3 ) This has been documented in human breast cancer cells that undergo EMT but retain certain epithelial markers, such as specific keratins, and function as direct ‘feeders’ of carcinoma cells that regulate their proliferation.( 110 ) The ‘feeding’ process naturally involves chemokines, growth and angiogenic factors and among the many, TGF‐β plays a primary role in promoting secretion of many other cytokines by the fibroblasts.( 111 ) In addition to TGF‐β, PDGF and basic FGF are also produced by cancer‐associated fibroblasts. Such growth factors that signal via RTK explain the mitogenic effects of TGF‐β as explained above in the discussion of EMT. Another TGF‐β‐inducible factor secreted by fibroblasts is connective tissue growth factor (CTGF), which induces mitogenesis of fibroblasts or neighboring cells.( 112 ) Interestingly, CTGF has recently been established as a prognostic marker for breast carcinoma metastasis to bone.( 54 )
The tumor stroma also contains the so‐called ‘activated’ myofibroblasts, which are characterized by the expression of α‐smooth muscle actin, and which have been proposed to provide migratory cues for metastatic carcinoma cells, one of them being TGF‐β.( 113 ) Expression of N‐cadherin seems to characterize the invasive properties of the myofibroblasts. In the invasive front of squamous cell carcinomas that secrete TGF‐β, stromal myofibroblasts are derived via EMT.( 114 ) TGF‐β signaling in the stromal myofibroblasts further induces expression of HGF, which promotes enhanced carcinoma proliferation and invasion. This tumor model has been confirmed independently after fibroblast‐specific knockout of the TGF‐β type II receptor.( 115 ) Fibroblasts that cannot receive TGF‐β signals promote prostate neoplasms and invasive squamous cell carcinomas in the forestomach of the knockout mice. These tumors had an excessive stroma with its fibroblasts secreting high HGF amounts that stimulated proliferation of the adjacent epithelial cells. When the TGF‐β type II receptor was deleted from the mammary gland fibroblasts, normal mammary ductal development was inhibited significantly, because epithelial cell numbers decreased, while the knockout fibroblasts increased in numbers.( 116 ) A mixture of receptor knockout mammary fibroblasts with mammary carcinoma cells, when xenografted into mice, led to high tumor growth and invasion, which correlated with over‐production of HGF, macrophage‐stimulating protein (MSP) and other mitogenic factors by the knockout fibroblasts.
Such studies concentrating on cancer‐associated fibroblasts and their regulatory roles promise new designs for anti‐tumor therapy.
EMT and MET are established in vivo Processes in fibrotic disease
In addition to cancer, EMT has been documented as a prominent cellular process that contributes to tissue fibrosis. The best studied cases are the development of fibrosis in the kidneys of patients with chronic renal failure (reviewed in( 5 )). A major role has been ascribed to TGF‐β in the context of such fibrotic disorders of the kidney. Accordingly, renal tubular epithelial cells undergo EMT and deposit high levels of extracellular matrix in response to the abnormally high levels of bioactive TGF‐β detected in these kidney lesions in patients suffering from diabetic nephropathy or in experimental animals where a protocol of ureteral obstruction is applied.( 69 , 91 ) The end result is tubulointerstitial fibrosis, which obstructs the proper filtering function of the kidney glomeruli.
A number of inhibitors that target signaling proteins of various pathways have proven efficacious for treatment of kidney fibrosis at least in the context of experimental mouse models. Some of these inhibitor studies could potentially be translated to therapeutic application in humans. For example, TGF‐β seems to activate the intracellular kinase and proto‐oncogene, c‐Abl, during signaling in fibroblasts of injured kidneys. Based on this observation, progression of lung and kidney fibrosis in experimental mouse and rat models could be blocked by inhibitors of the c‐Abl kinase, such as imatinib.( 117 ) Another attractive target for intervention is ILK, which contributes to EMT in cell models in vitro; in mouse models of kidney fibrosis in vivo, dominant‐negative mutants of ILK effectively block progression of fibrosis.( 69 ) These findings suggest that combinations of drugs that attack multiple signaling enzymes might provide efficient pharmacological regimens that would eliminate EMT‐based processes during tissue fibrosis.
In addition to TGF‐β and the intracellular kinases that impact on EMT in fibrotic disorders, BMP induce the inverse process of MET, and in this way antagonize the actions of TGF‐β on fibrotic kidney epithelial cells in vitro.( 88 ) A similar mode of action of the BMP acting as TGF‐β antagonists has been observed in additional in vitro cell models, such as normal mammary and lens epithelial cells.( 44 ) More important is the in vivo relevance of this process that has been demonstrated during kidney fibrosis( 5 ). In this case, BMP‐7 acts on adult renal fibroblasts and promotes MET, which has therapeutic effects because the injured kidney regenerates and heals. We therefore suggest that additional members of the BMP family might be potent inducers of MET in various organs, and thus, in addition to kidney fibrosis, such processes may have clinical importance even in cases of cancer progression.
Perspectives
The process of EMT downstream of various signaling factors becomes gradually explored in greater depth. A future challenge is the understanding of complex signaling networks operating during EMT in vivo. Additional important regulators of EMT are also expected to be discovered in the years to come. A focus on EMT also promises the generation of new drugs against cancer cell invasiveness and metastasis, but also against tissue fibrosis. Based on the role of multiple signaling pathways that establish EMT, combinations of drugs that affect TGF‐β, Notch, Wnt and RTK signaling might become important in anticancer therapy.
Acknowledgments
Due to space limitations, only selected literature is cited. Funding of the authors’ work is provided by the Ludwig Institute for Cancer Research, the Swedish Cancer Society, the Swedish Research Council and the Marie Curie Research Training Network (RTN) ‘EpiPlastCarcinoma’ under the European Union FP6 program. The authors thank all past and present members of the TGF‐β signaling group for their contributions to the scientific work emanating from their laboratory.
References
- 1. Hay ED. The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev Dyn 2005; 233: 706–20. [DOI] [PubMed] [Google Scholar]
- 2. Huber MA, Kraut N, Beug H. Molecular requirements for epithelial‐mesenchymal transition during tumor progression. Curr Opin Cell Biol 2005; 17: 548–58. [DOI] [PubMed] [Google Scholar]
- 3. Radisky DC, Kenny PA, Bissell MJ. Fibrosis and cancer: Do myofibroblasts come also from epithelial cells via EMT? J Cell Biochem; 101: 830–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thiery J‐P, Sleeman JP. Complex networks orchestrate epithelial‐mesenchymal transitions. Nat Rev Mol Cell Biol 2006; 7: 131–42. [DOI] [PubMed] [Google Scholar]
- 5. Zeisberg M, Kalluri R. The role of epithelial‐to‐mesenchymal transition in renal fibrosis. J Mol Med 2004; 82: 175–81. [DOI] [PubMed] [Google Scholar]
- 6. Trainor PA, Melton KR, Manzanares M. Origins and plasticity of neural crest cells and their roles in jaw and craniofacial evolution. Int J Dev Biol 2003; 47: 541–53. [PubMed] [Google Scholar]
- 7. Ciruna B, Rossant J. FGF signaling regulates mesoderm cell fate specification and morphogenetic movement at the primitive streak. Dev Cell 2001; 1: 37–49. [DOI] [PubMed] [Google Scholar]
- 8. Carver EA, Jiang R, Lan Y, Oram KF, Gridley T. The mouse snail gene encodes a key regulator of the epithelial‐mesenchymal transition. Mol Cell Biol 2001; 21: 8184–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Peinado H, Portillo F, Cano A. Transcriptional regulation of cadherins during development and carcinogenesis. Int J Dev Biol 2004; 48: 365–75. [DOI] [PubMed] [Google Scholar]
- 10. Kemler R, Hierholzer A, Kanzler B et al . Stabilization of β‐catenin in the mouse zygote leads to premature epithelial‐mesenchymal transition in the epiblast. Development 2004; 131: 5817–24. [DOI] [PubMed] [Google Scholar]
- 11. Mohamed OA, Clarke HJ, Dufort D. Β‐catenin signaling marks the prospective site of primitive streak formation in the mouse embryo. Dev Dyn 2004; 231: 416–24. [DOI] [PubMed] [Google Scholar]
- 12. Meulemans D, Bronner‐Fraser M. Gene–regulatory interactions in neural crest evolution and development. Dev Cell 2004; 7: 291–9. [DOI] [PubMed] [Google Scholar]
- 13. Nawshad A, LaGamba D, Hay ED. Transforming growth factor β (TGFβ) signalling in palatal growth, apoptosis and epithelial mesenchymal transformation (EMT). Arch Oral Biol 2004; 49: 675–89. [DOI] [PubMed] [Google Scholar]
- 14. Timmerman LA, Grego‐Bessa J, Raya A et al . Notch promotes epithelial‐mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev 2004; 18: 99–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Person AD, Klewer SE, Runyan RB. Cell biology of cardiac cushion development. Int Rev Cytol 2005; 243: 287–335. [DOI] [PubMed] [Google Scholar]
- 16. Zavadil J, Cermak L, Soto‐Nieves N, Böttinger EP. Integration of TGF‐β/Smad and Jagged1/Notch signalling in epithelial‐to‐mesenchymal transition. EMBO J 2004; 23: 1155–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Niimi H, Pardali K, Vanlandewijck M, Heldin CH, Moustakas A. Notch signaling is necessary for epithelial growth arrest by TGF‐β. J Cell Biol 2007; 176: 695–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. O’Brien LE, Zegers MM, Mostov KE. Opinion: Building epithelial architecture: insights from three‐dimensional culture models. Nat Rev Mol Cell Biol 2002; 3: 531–7. [DOI] [PubMed] [Google Scholar]
- 19. Nelson CM, Vanduijn MM, Inman JL, Fletcher DA, Bissell MJ. Tissue geometry determines sites of mammary branching morphogenesis in organotypic cultures. Science 2006; 314: 298–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol 2003; 4: 915–25. [DOI] [PubMed] [Google Scholar]
- 21. Grotegut S, Von Schweinitz D, Christofori G, Lehembre F. Hepatocyte growth factor induces cell scattering through MAPK/Egr‐1‐mediated upregulation of Snail. EMBO J 2006; 25: 3534–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Savagner P, Yamada KM, Thiery JP. The zinc‐finger protein slug causes desmosome dissociation, an initial and necessary step for growth factor‐induced epithelial‐mesenchymal transition. J Cell Biol 1997; 137: 1403–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cordenonsi M, Montagner M, Adorno M et al . Integration of TGF‐β and Ras/MAPK signaling through p53 phosphorylation. Science 2007; 315: 840–3. [DOI] [PubMed] [Google Scholar]
- 24. Irie HY, Pearline RV, Grueneberg D et al . Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial‐mesenchymal transition. J Cell Biol 2005; 171: 1023–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Larue L, Bellacosa A. Epithelial‐mesenchymal transition in development and cancer: role of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene 2005; 24: 7443–54. [DOI] [PubMed] [Google Scholar]
- 26. Zohn IE, Li Y, Skolnik EY, Anderson KV, Han J, Niswander L. p38 and a p38‐interacting protein are critical for downregulation of E‐cadherin during mouse gastrulation. Cell 2006; 125: 957–69. [DOI] [PubMed] [Google Scholar]
- 27. Rosano L, Spinella F, Di Castro V et al . Endothelin‐1 promotes epithelial‐to‐mesenchymal transition in human ovarian cancer cells. Cancer Res 2005; 65: 11 649–57. [DOI] [PubMed] [Google Scholar]
- 28. Aranda V, Haire T, Nolan ME et al . Par6‐aPKC uncouples ErbB2 induced disruption of polarized epithelial organization from proliferation control. Nat Cell Biol 2006; 8: 1235–45. [DOI] [PubMed] [Google Scholar]
- 29. Wu WS. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev 2006; 25: 695–705. [DOI] [PubMed] [Google Scholar]
- 30. Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev 2005; 19: 2783–810. [DOI] [PubMed] [Google Scholar]
- 31. Moustakas A, Heldin C‐H. Non‐Smad TGF‐β signals. J Cell Sci 2005; 118: 3573–84. [DOI] [PubMed] [Google Scholar]
- 32. Cui W, Fowlis DJ, Bryson S et al . TGFβ1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell 1996; 86: 531–42. [DOI] [PubMed] [Google Scholar]
- 33. Portella G, Cumming SA, Liddell J et al . Transforming growth factor β is essential for spindle cell conversion of mouse skin carcinoma in vivo: implications for tumor invasion. Cell Growth Differ 1998; 9: 393–404. [PubMed] [Google Scholar]
- 34. Han G, Lu SL, Li AG et al . Distinct mechanisms of TGF‐β1‐mediated epithelial‐to‐mesenchymal transition and metastasis during skin carcinogenesis. J Clin Invest 2005; 115: 1714–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gotzmann J, Huber H, Thallinger C et al . Hepatocytes convert to a fibroblastoid phenotype through the cooperation of TGF‐β1 and Ha‐Ras: steps towards invasiveness. J Cell Sci 2002; 115: 1189–202. [DOI] [PubMed] [Google Scholar]
- 36. Janda E, Lehmann K, Killisch I et al . Ras and TGFβ cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol 2002; 156: 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lehmann K, Janda E, Pierreux CE et al . Raf induces TGFβ production while blocking its apoptotic but not invasive responses: a mechanism leading to increased malignancy in epithelial cells. Genes Dev 2000; 14: 2610–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Oft M, Heider KH, Beug H. TGFβ signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol 1998; 8: 1243–52. [DOI] [PubMed] [Google Scholar]
- 39. Valcourt U, Kowanetz M, Niimi H, Heldin C‐H, Moustakas A. TGF‐β and the Smad signaling pathway support transcriptomic reprogramming during epithelial‐mesenchymal cell transition. Mol Biol Cell 2005; 16: 1987–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang Y, Pan X, Lei W, Wang J, Song J. Transforming growth factor‐β1 induces epithelial‐to‐mesenchymal transition and apoptosis via a cell cycle‐dependent mechanism. Oncogene 2006; 25: 7235–44. [DOI] [PubMed] [Google Scholar]
- 41. Akhurst RJ, Derynck R. TGF‐β signaling in cancer – a double‐edged sword. Trends Cell Biol 2001; 11: S44–51. [DOI] [PubMed] [Google Scholar]
- 42. Pardali K, Moustakas A. Actions of TGF‐β as tumor suppressor and prometastatic factor in human cancer. Biochim Biophys Acta 2007; 1775: 21–62. [DOI] [PubMed] [Google Scholar]
- 43. Piek E, Moustakas A, Kurisaki A, Heldin C‐H, Ten Dijke P. TGF‐β type I receptor/ALK‐5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J Cell Sci 1999; 112: 4557–68. [DOI] [PubMed] [Google Scholar]
- 44. Kowanetz M, Valcourt U, Bergström R, Heldin C‐H, Moustakas A. Id2 and Id3 define the potency of cell proliferation and differentiation responses to tranforming growth factor β and bone morphogenetic protein. Mol Cell Biol 2004; 24: 4241–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Saika S, Ikeda K, Yamanaka O et al . Adenoviral gene transfer of BMP‐7, Id2, or Id3 suppresses injury‐induced epithelial‐to‐mesenchymal transition of lens epithelium in mice. Am J Physiol Cell Physiol 2006; 290: C282–9. [DOI] [PubMed] [Google Scholar]
- 46. Ju W, Ogawa A, Heyer J et al . Deletion of Smad2 in mouse liver reveals novel functions in hepatocyte growth and differentiation. Mol Cell Biol 2006; 26: 654–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Levy L, Hill CS. Smad4 dependency defines two classes of transforming growth factor β (TGF‐β) target genes and distinguishes TGF‐β‐induced epithelial‐mesenchymal transition from its antiproliferative and migratory responses. Mol Cell Biol 2005; 25: 8108–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li W, Qiao W, Chen L et al . Squamous cell carcinoma and mammary abscess formation through squamous metaplasia in Smad4/Dpc4 conditional knockout mice. Development 2003; 130: 6143–53. [DOI] [PubMed] [Google Scholar]
- 49. Bardeesy N, Cheng KH, Berger JH et al . Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev 2006; 20: 3130–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Deckers M, Van Dinther M, Buijs J et al . The tumor suppressor Smad4 is required for transforming growth factor β‐induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res 2006; 66: 2202–9. [DOI] [PubMed] [Google Scholar]
- 51. Takano S, Kanai F, Jazag A et al . Smad4 is essential for down‐regulation of E‐cadherin induced by TGF‐β in pancreatic cancer cell line PANC‐1. J Biochem (Tokyo) 2007; 141: 345–51. [DOI] [PubMed] [Google Scholar]
- 52. Zavadil J, Bitzer M, Liang D et al . Genetic programs of epithelial cell plasticity directed by transforming growth factor‐β. Proc Natl Acad Sci U S A 2001; 98: 6686–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jechlinger M, Grunert S, Tamir IH et al . Expression profiling of epithelial plasticity in tumor progression. Oncogene 2003; 22: 7155–69. [DOI] [PubMed] [Google Scholar]
- 54. Kang Y, Siegel PM, Shu W et al . A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003; 3: 537–49. [DOI] [PubMed] [Google Scholar]
- 55. Xie L, Law BK, Aakre ME et al . Transforming growth factor β‐regulated gene expression in a mouse mammary gland epithelial cell line. Breast Cancer Res 2003; 5: R187–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. LaGamba D, Nawshad A, Hay ED. Microarray analysis of gene expression during epithelial‐mesenchymal transformation. Dev Dyn 2005; 234: 132–42. [DOI] [PubMed] [Google Scholar]
- 57. Bhowmick NA, Ghiassi M, Bakin A et al . Transforming growth factor‐β1 mediates epithelial to mesenchymal transdifferentiation through a RhoA‐dependent mechanism. Mol Biol Cell 2001; 12: 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ellenrieder V, Hendler SF, Boeck W et al . Transforming growth factor β1 treatment leads to an epithelial‐mesenchymal transdifferentiation of pancreatic cancer cells requiring extracellular signal‐regulated kinase 2 activation. Cancer Res 2001; 61: 4222–8. [PubMed] [Google Scholar]
- 59. Bakin AV, Rinehart C, Tomlinson AK, Arteaga CL. p38 mitogen‐activated protein kinase is required for TGFβ‐mediated fibroblastic transdifferentiation and cell migration. J Cell Sci 2002; 115: 3193–206. [DOI] [PubMed] [Google Scholar]
- 60. Grande M, Franzén A, Karlsson JO, Ericson LE, Heldin NE, Nilsson M. Transforming growth factor‐β and epidermal growth factor synergistically stimulate epithelial to mesenchymal transition (EMT) through a MEK‐dependent mechanism in primary cultured pig thyrocytes. J Cell Sci 2002; 115: 4227–36. [DOI] [PubMed] [Google Scholar]
- 61. Huber MA, Azoitei N, Baumann B et al . NF‐κB is essential for epithelial‐mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest 2004; 114: 569–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xie L, Law BK, Chytil AM, Brown KA, Aakre ME, Moses HL. Activation of the Erk pathway is required for TGF‐β1‐induced EMT in vitro. Neoplasia 2004; 6: 603–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Davies M, Robinson M, Smith E, Huntley S, Prime S, Paterson I. Induction of an epithelial to mesenchymal transition in human immortal and malignant keratinocytes by TGF‐β1 involves MAPK, Smad and AP‐1 signalling pathways. J Cell Biochem 2005; 95: 918–31. [DOI] [PubMed] [Google Scholar]
- 64. Ao M, Williams K, Bhowmick NA, Hayward SW. Transforming growth factor‐β promotes invasion in tumorigenic but not in nontumorigenic human prostatic epithelial cells. Cancer Res 2006; 66: 8007–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Santibanez JF. JNK mediates TGF‐β1‐induced epithelial mesenchymal transdifferentiation of mouse transformed keratinocytes. FEBS Lett 2006; 580: 5385–91. [DOI] [PubMed] [Google Scholar]
- 66. Shim JH, Xiao C, Paschal AE et al . TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev 2005; 19: 2668–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bhowmick NA, Zent R, Ghiassi M, McDonnell M, Moses HL. Integrin β1 signaling is necessary for transforming growth factor‐β activation of p38 MAPK and epithelial plasticity. J Biol Chem 2001; 276: 46 707–13. [DOI] [PubMed] [Google Scholar]
- 68. Bates RC, Bellovin DI, Brown C et al . Transcriptional activation of integrin β6 during the epithelial‐mesenchymal transition defines a novel prognostic indicator of aggressive colon carcinoma. J Clin Invest 2005; 115: 339–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Li Y, Yang J, Dai C, Wu C, Liu Y. Role for integrin‐linked kinase in mediating tubular epithelial to mesenchymal transition and renal interstitial fibrogenesis. J Clin Invest 2003; 112: 503–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lee YI, Kwon YJ, Joo CK. Integrin‐linked kinase function is required for transforming growth factor β‐mediated epithelial to mesenchymal transition. Biochem Biophys Res Commun 2004; 316: 997–1001. [DOI] [PubMed] [Google Scholar]
- 71. Leung‐Hagesteijn C, Hu MC, Mahendra AS et al . Integrin‐linked kinase mediates bone morphogenetic protein 7‐dependent renal epithelial cell morphogenesis. Mol Cell Biol 2005; 25: 3648–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yang Y, Pan X, Lei W et al . Regulation of transforming growth factor‐β1‐induced apoptosis and epithelial‐to‐mesenchymal transition by protein kinase a and signal transducers and activators of transcription 3. Cancer Res 2006; 66: 8617–24. [DOI] [PubMed] [Google Scholar]
- 73. Ozdamar B, Bose R, Barrios‐Rodiles M, Wang HR, Zhang Y, Wrana JL. Regulation of the polarity protein Par6 by TGFβ receptors controls epithelial cell plasticity. Science 2005; 307: 1603–9. [DOI] [PubMed] [Google Scholar]
- 74. Janda E, Nevolo M, Lehmann K, Downward J, Beug H, Grieco M. Raf plus TGFβ‐dependent EMT is initiated by endocytosis and lysosomal degradation of E‐cadherin. Oncogene 2006; 25: 7117–30. [DOI] [PubMed] [Google Scholar]
- 75. Prunier C, Howe PH. Disabled‐2 (Dab2) is required for transforming growth factor β‐induced epithelial to mesenchymal transition (EMT). J Biol Chem 2005; 280: 17 540–8. [DOI] [PubMed] [Google Scholar]
- 76. Vogelmann R, Nguyen‐Tat MD, Giehl K, Adler G, Wedlich D, Menke A. TGFβ‐induced downregulation of E‐cadherin‐based cell‐cell adhesion depends on PI3‐kinase and PTEN. J Cell Sci 2005; 118: 4901–12. [DOI] [PubMed] [Google Scholar]
- 77. Gotzmann J, Fischer AN, Zojer M et al . A crucial function of PDGF in TGF‐β‐mediated cancer progression of hepatocytes. Oncogene 2006; 25: 3170–85. [DOI] [PubMed] [Google Scholar]
- 78. Fischer AN, Fuchs E, Mikula M, Huber H, Beug H, Mikulits W. PDGF essentially links TGF‐β signaling to nuclear β‐catenin accumulation in hepatocellular carcinoma progression. Oncogene 2007; 26: 3395–405. [DOI] [PubMed] [Google Scholar]
- 79. Yang L, Lin C, Liu ZR. P68 RNA helicase mediates PDGF‐induced epithelial mesenchymal transition by displacing Axin from beta‐catenin. Cell 2006; 127: 139–55. [DOI] [PubMed] [Google Scholar]
- 80. Jechlinger M, Sommer A, Moriggl R et al . Autocrine PDGFR signaling promotes mammary cancer metastasis. J Clin Invest 2006; 116: 1561–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Michl P, Ramjaun AR, Pardo OE et al . CUTL1 is a target of TGFβ signaling that enhances cancer cell motility and invasiveness. Cancer Cell 2005; 7: 521–32. [DOI] [PubMed] [Google Scholar]
- 82. Ripka S, Konig A, Buchholz M et al . WNT5A – target of CUTL1 and potent modulator of tumor cell migration and invasion in pancreatic cancer. Carcinogenesis 2007; 8: 1178–87. [DOI] [PubMed] [Google Scholar]
- 83. Yook JI, Li XY, Ota I et al . A Wnt‐Axin2‐GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol 2006; 8: 1398–406. [DOI] [PubMed] [Google Scholar]
- 84. Waerner T, Alacakaptan M, Tamir I et al . ILEI: a cytokine essential for EMT, tumor formation, and late events in metastasis in epithelial cells. Cancer Cell 2006; 10: 227–39. [DOI] [PubMed] [Google Scholar]
- 85. Onoue T, Uchida D, Begum NM, Tomizuka Y, Yoshida H, Sato M. Epithelial‐mesenchymal transition induced by the stromal cell‐derived factor‐1/CXCR4 system in oral squamous cell carcinoma cells. Int J Oncol 2006; 29: 1133–8. [PubMed] [Google Scholar]
- 86. Kang Y, Chen CR, Massagué J. A self‐enabling TGFβ response coupled to stress signaling. Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell 2003; 11: 915–26. [DOI] [PubMed] [Google Scholar]
- 87. Kondo M, Cubillo E, Tobiume K et al . A role for Id in the regulation of TGF‐β‐induced epithelial‐mesenchymal transdifferentiation. Cell Death Differ 2004; 11: 1092–101. [DOI] [PubMed] [Google Scholar]
- 88. Zeisberg M, Shah AA, Kalluri R. Bone morphogenic protein‐7 induces mesenchymal to epithelial transition in adult renal fibroblasts and facilitates regeneration of injured kidney. J Biol Chem 2005; 280: 8094–100. [DOI] [PubMed] [Google Scholar]
- 89. Perk J, Iavarone A, Benezra R. Id family of helix‐loop‐helix proteins in cancer. Nat Rev Cancer 2005; 5: 603–14. [DOI] [PubMed] [Google Scholar]
- 90. Peinado H, Quintanilla M, Cano A. Transforming growth factor β‐1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J Biol Chem 2003; 278: 21 113–23. [DOI] [PubMed] [Google Scholar]
- 91. Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF‐β1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest 2003; 112: 1486–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Comijn J, Berx G, Vermassen P et al . The two‐handed E box binding zinc finger protein SIP1 downregulates E‐cadherin and induces invasion. Mol Cell 2001; 7: 1267–78. [DOI] [PubMed] [Google Scholar]
- 93. Vandewalle C, Comijn J, De Craene B et al . SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell‐cell junctions. Nucl Acids Res 2005; 33: 6566–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Eger A, Stockinger A, Park J et al . β‐Catenin and TGFβ signalling cooperate to maintain a mesenchymal phenotype after FosER‐induced epithelial to mesenchymal transition. Oncogene 2004; 23: 2672–80. [DOI] [PubMed] [Google Scholar]
- 95. Martinez‐Alvarez C, Blanco MJ, Perez R et al . Snail family members and cell survival in physiological and pathological cleft palates. Dev Biol 2004; 265: 207–18. [DOI] [PubMed] [Google Scholar]
- 96. Romano LA, Runyan RB. Slug is an essential target of TGFβ2 signaling in the developing chicken heart. Dev Biol 2000; 223: 91–102. [DOI] [PubMed] [Google Scholar]
- 97. Thuault S, Valcourt U, Petersen M, Manfioletti G, Heldin C‐H, Moustakas A. Transforming growth factor‐β employs HMGA2 to elicit epithelial‐mesenchymal transition. J Cell Biol 2006; 174: 175–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Barrallo‐Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival implications in development and cancer. Development 2005; 132: 3151–61. [DOI] [PubMed] [Google Scholar]
- 99. Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD, Wang LH. Twist transcriptionally up‐regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res 2007; 67: 1979–87. [DOI] [PubMed] [Google Scholar]
- 100. Venkov CD, Link AJ, Jennings JL et al . A proximal activator of transcription in epithelial‐mesenchymal transition. J Clin Invest 2007; 117: 482–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Shen X, Li J, Hu PP, Waddell D, Zhang J, Wang X‐F. The activity of guanine exchange factor NET1 is essential for transforming growth factor‐β‐mediated stress fiber formation. J Biol Chem 2001; 276: 15 362–8. [DOI] [PubMed] [Google Scholar]
- 102. Giannelli G, Fransvea E, Marinosci F et al . Transforming growth factor‐β1 triggers hepatocellular carcinoma invasiveness via α3β1 integrin. Am J Pathol 2002; 161: 183–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Dumont N, Bakin AV, Arteaga CL. Autocrine transforming growth factor‐β signaling mediates Smad‐independent motility in human cancer cells. J Biol Chem 2003; 278: 3275–85. [DOI] [PubMed] [Google Scholar]
- 104. Prindull G. Hypothesis: cell plasticity, linking embryonal stem cells to adult stem cell reservoirs and metastatic cancer cells? Exp Hematol 2005; 33: 738–46. [DOI] [PubMed] [Google Scholar]
- 105. Ullmann U, In't Veld P, Gilles C et al . Epithelial‐mesenchymal transition process in human embryonic stem cells cultured in feeder‐free conditions. Mol Hum Reprod 2007; 13: 21–32. [DOI] [PubMed] [Google Scholar]
- 106. Tarin D, Thompson EW, Newgreen DF. The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res 2005; 65: 5996–6000; discussion–1. [DOI] [PubMed] [Google Scholar]
- 107. Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res 2006; 66: 8319–26. [DOI] [PubMed] [Google Scholar]
- 108. Wicki A, Lehembre F, Wick N, Hantusch B, Kerjaschki D, Christofori G. Tumor invasion in the absence of epithelial‐mesenchymal transition: podoplanin‐mediated remodeling of the actin cytoskeleton. Cancer Cell 2006; 9: 261–72. [DOI] [PubMed] [Google Scholar]
- 109. Wang W, Goswami S, Sahai E, Wyckoff JB, Segall JE, Condeelis JS. Tumor cells caught in the act of invading: their strategy for enhanced cell motility. Trends Cell Biol 2005; 15: 138–45. [DOI] [PubMed] [Google Scholar]
- 110. Petersen OW, Nielsen HL, Gudjonsson T et al . Epithelial to mesenchymal transition in human breast cancer can provide a nonmalignant stroma. Am J Pathol 2003; 162: 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Micke P, Östman A. Exploring the tumour environment: cancer‐associated fibroblasts as targets in cancer therapy. Expert Opin Ther Targets 2005; 9: 1217–33. [DOI] [PubMed] [Google Scholar]
- 112. Igarashi A, Okochi H, Bradham DM, Grotendorst GR. Regulation of connective tissue growth factor gene expression in human skin fibroblasts and during wound repair. Mol Biol Cell 1993; 4: 637–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. De Wever O, Westbroek W, Verloes A et al . Critical role of N‐cadherin in myofibroblast invasion and migration in vitro stimulated by colon‐cancer‐cell‐derived TGF‐β or wounding. J Cell Sci 2004; 117: 4691–703. [DOI] [PubMed] [Google Scholar]
- 114. Lewis MP, Lygoe KA, Nystrom ML et al . Tumour‐derived TGF‐β1 modulates myofibroblast differentiation and promotes HGF/SF‐dependent invasion of squamous carcinoma cells. Br J Cancer 2004; 90: 822–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Bhowmick NA, Chytil A, Plieth D et al . TGF‐β signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004; 303: 848–51. [DOI] [PubMed] [Google Scholar]
- 116. Cheng N, Bhowmick NA, Chytil A et al . Loss of TGF‐β type II receptor in fibroblasts promotes mammary carcinoma growth and invasion through upregulation of TGF‐α‐, MSP‐ and HGF‐mediated signaling networks. Oncogene 2005; 24: 5053–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Wang S, Wilkes MC, Leof EB, Hirschberg R. Imatinib mesylate blocks a non‐Smad TGF‐β pathway and reduces renal fibrogenesis in vivo. FASEB J 2005; 19: 1–11. [DOI] [PubMed] [Google Scholar]