

Abstract
(Cancer Sci 2008; 99: 829–835)
In parallel with the completion of a number of whole genome sequences, a number of technologies were developed to allow researchers to investigate the resulting large numbers of genes being characterized. Of these, several DNA‐microarray technologies (essentially miniaturized reverse Northern blots) that allow researchers to quantify the expression of essentially all the genes of a given organism in a single experiment quickly gained predominance due to their relative simplicity, reproducibility, and cost (in the words of Sydney Brenner, ‘the only omics that matters is economics’). The use of DNA microarrays has allowed bench‐side researchers to routinely perform experiments that previously would have been considered mega‐projects. DNA microarray technology has since been followed by various high‐throughput technologies including protein analysis technologies (usually referred to proteomics) and most recently very highly parallelized sequencing techniques. It is striking to us that all these developments have taken place within a short time interval coinciding with the period of the development of our institute.
We have been using DNA microarrays since the technology was introduced to Kyoto University in 2000 in order to compare the gene‐expression profiles of various intermediate stages formed during the differentiation of embryonic stem cells towards defined lineages. At the Center for Developmental Biology (CDB), DNA‐microarray analysis has been provided as a common service to all CDB researchers from the beginning. Through this 7‐year experience of this technology we have become convinced of the impact that omics technologies have had, and will continue to have within our field of research (developmental biology and stem cell biology). However, we have also realized that the true potential of this technology can only be reached through a constant reappraisal of data and methodologies in accordance with the specific demands of individual research fields. In this commentary we will present our opinions on a number of issues that are of relevance to most fields of biological research.
Biologists Don't Do Omics
We have often wondered as to how well those who develop array and associated data analysis methods are aware of the reality of the manner in which the technology is used by basic biologists. Naturally, in the beginning, DNA chips were primarily used by researchers with a background in the development of high‐throughput technologies and statistical analyses. Much of this initial research focused on ways of making some sense of the totality of the data, and either obtaining some novel understanding of the nature in which transcription changes during cellular processes or a means of rapidly identifying large numbers of gene relationships in order to describe the transcriptional networks underlying the processes being examined.( 1 , 2 , 3 , 4 ) At the time, there was a feeling that this kind of analyses would lead to some new understanding of the complexity of the transcriptional networks that underpin so much of cell biology. However, although this initial work (as well as much since then) was both well conceived and executed, we are still waiting for the larger understanding that we were looking forward to, and it is questionable as to whether or not basic biologists have gained much additional wisdom from these studies, though system biologists and bioinformaticians certainly have.
As array analysis has become more mainstream, the makeup of the researchers that have turned to this technology has changed drastically and currently most array experiments are carried out with the aim of identifying some number (preferably not too large) of genes that play important roles in some specific process. For instance, cancer biologists use arrays to identify genes whose expression is gained or lost during the process of oncogenesis, or whose expression is associated with resistance to chemotherapy. Similarly, biologists frequently apply arrays in order to identify genes induced in response to various stimuli. As such, the arrays are used, not to study the overall state of the cells, but rather as a rather powerful substitute for older gene‐fishing strategies (e.g. differential display, subtractive cDNA libraries, brute force EST sequencing, etc.). Although such experiments make use of omics technologies, they do not in our view qualify as ‘doing omics’ since they are not concerned with any holistic issues. Although one might argue that such applications do not fully utilise the capabilities of the available technology, we feel that it is indeed such uses of the technology that have made the greatest impact on biology. We are still convinced of the potential of the various omics technologies to obtain non‐biased and complete views of cellular processes. However, converting these views to some sort of higher understanding is made difficult by a number of issues (some of which we discuss below) that complicate the analysis of the resulting data and such an understanding is likely to elude us for some time.
Hence, in the immediate future we believe that most of the additional biological understanding resulting from the application of omics technologies will result from the identification of important components of the networks that drive biological processes, rather than a true holistic understanding of the networks themselves. The difference between a gene‐fishing study and an omics study can be considered thus: in the former, one sets out to obtain the identities of the components, whereas in the latter, one does not care about the identities of components, merely their interactions.
Biologists in general don't do omics, nor do they tend to do computational analysis or write programs. While we don't think that the former matters much, the latter has meant that most available programs which analyze the large amounts of data generated have been written either by system biologists or bioinformaticians who have traditionally been more concerned with holistic omics questions. The lack of programs fulfulling our somewhat simple requirements has led us to develop our own system of analysis to specifically facilitate gene‐fishing and casual examination of the data. In the development of this system, we were led by the following ideas:
-
1
There is little to be gained by looking at hundreds or thousands of values in one go, as too much detail is easily lost; rather it is better to use specified search criteria and then to inspect what has been selected by the search criteria.
-
2
Biological significance trumps statistical significance; meaning that statistically suspect points may be included for further analysis if it is thought that they represent interesting genes, whereas statistically clean points representing boring genes can be discarded.
-
3
Context is vital. Since probe–gene relationships are not always straightforward and gene names themselves are often not very informative, it is important to include additional information pertaining to the probe. This includes both annotation of the probe, the genes it may represent, as well as the signal for that probe in additional samples.
-
4
The reliability of any given expression estimate should be assessable. This can most easily be achieved by allowing the user to view the raw underlying data though there are other ways of achieving this.
-
5
The value of the data is directly related to the number of people that use it. Hence it is important to make the data available to as large a number of people as possible. This is most easily achieved by creating a system that makes casual analysis of the data easy, painless, and, as far as possible, pleasurable. This might seem flippant, but it is perhaps more important than anything else.
To satisfy these requirements, we have built a client‐server system that allows simultaneous access to the expression data by an arbitrary number of researchers (see http://www.cdb.riken.jp/scb/documentation/). This system contains an underlying database, which, in addition to the expression database, contains genomic coordinates for the probes used, along with genomic coordinates and annotation from the Ensembl( 5 ) genome annotation project. Further, it is possible to add additional sequence features such as cDNA libraries and sequences of DNA‐ or RNA‐based probes. This allows probes to be associated with gene loci and allows the incorporation of extensive gene annotation in database lookups.
In contrast to most other programs, this system defaults to display the expression of a single probe‐set across a number of samples encompassing multiple experiments. This provides the user with a kind of dry northern hybridization across all samples available in the database. Since a single probe‐set is shown each time, it is simple to display the underlying raw data for that probe‐set, allowing the user to easily evaluate the reliability of the retrieved data. This is in stark contrast to the commonly used heat map that is used to display the expression of large numbers of genes across a number of classes of samples. The heat map allows much of the information from a set of experiments to be visualized in a single image, but it is not suitable for selecting specific genes since much information from the underlying data is lost (See Fig. 1. for examples of lost information). As such we do not consider it suitable for typical gene‐fishing expeditions; in contrast, although our system displays expression data one at a time, we have built the interface so that it is easy for the user to individually assess the expression of hundreds of genes within a short period of time.
Figure 1.
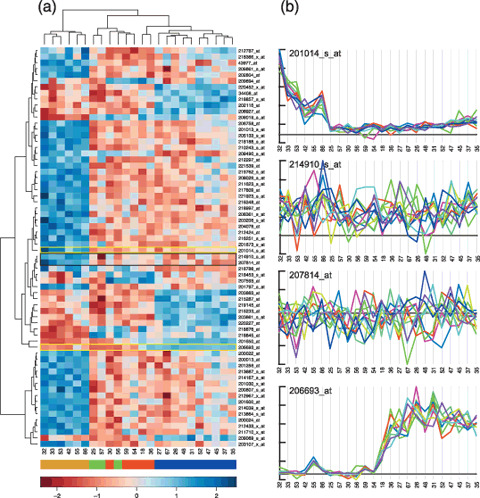
Heatmaps, clustering, and details. We reanalyzed the Neuroblastoma dataset of McArdle et al.( 8 ) using a Bioconductor( 14 ) to estimate expression levels using the RMA( 15 ) algorithm; to select probe‐sets that distinguish individual subtypes (MNA, 11q‐ and HYPD, using McArdle et al.'s inferred classification) using SAM( 16 ); and finally the heatmap_2 function to display and cluster the expression of the selected genes. (a) The resulting heatmap and dendrograms. Although the heatmap demonstrates that the individual subtypes (with some mixture of GNB and HYPD) can be segregated on the basis of the underlying expression, it also clearly indicates a significant heterogeneity within the individual sample classes. With the exception of the MYCN subtypes, there are few probe‐sets that are specific to individual subtypes and there is much variation in the signals within sample subtypes. However, it is difficult to judge the reliability and the likely biological significance of the changes in expression displayed. On Affymetrix arrays, genes are represented by a set of probe‐pairs (referred to as a probe‐set). Expression values can be calculated from these probe‐sets in a number of ways (eg. RMA used for the heatmap), but it is also possible to simply plot the signals from the individual probe‐pairs across a data series. (b) Plots of the underlying probe‐pair data for the probe‐sets indicated by gold boxes (clean data) and the single black box (messy data). The 214910_s_at (APOM) and 207814_at (DEFA6) probe‐sets were chosen as they form the pair with the smallest distance in the dendrogram, implying some special significance. However, the signals from the individual probe‐pairs of these probe‐sets show little or no covariation across the experimental series, suggesting that the inferred expression estimates are unreliable. In stark contrast the raw probe‐pair data from the 201014_s_at (PAICS) and 206693_at (Il‐7) probe‐sets shows a high degree of internal correlation indicating highly reliable data. In addition the plots show that the levels in the MYCN and 11q‐samples respectively deviate significantly from a baseline that lies close to an estimated zero‐signal, indicating a high (to infinity) fold change. None of these details can be inferred from the heatmap display. The numbers beneath the heatmap and plots indicate the sample identifiers used by McArdle et al.; the colour map immediately below indicates the different subtypes as inferred from the expression data( 8 ) as follows: gold MYCN, green GNB, red HYPD, and blue 11q‐. The units of the scale bar at the bottom are in SD (expression values are normalized across the rows of the heatmap to have a mean of 0 and a variance of 1).
Array experiments are carried out for many different purposes, and clearly there is no one method by which the analysis should be carried out. However, for gene‐fishing expeditions we believe that one of the most important criteria is that the expression of selected genes should be individually assessed; our system makes this trivial. In addition, since the data remain in the system after initial analysis, it is easy to re‐analyze the data or to incorporate them in future analyses.
DNA array data and permanence
An important advantage of DNA array data that is often overlooked is that the data is inherently systematically organized which makes it easy to compare data across different experiments and sources. This advantage is most fully appreciated when the data are stored in such a manner that it can easily be accessed from any computer within the user group. Incorporating a large number of samples from different sources into the database allows questions of specificity of expression to be addressed. Although many genes have been described as specific to some cell or tissue type on the basis of an analysis of a small set of samples, any such conclusions are dependent on the range of samples analyzed. As an example, Figure 2ai shows the expression of the Il‐17 gene across 250 samples present in one of our databases. This set of data was not derived from a single experiment, nor a single group, but is a dataset that has grown over time, both as a result of experiments that we and collaborators have performed, and published data that we have incorporated from online databases. As a result of the large number of samples, researchers can easily ask what kind of tissues express a given gene. In the case of Il‐17, a sharp peak of expression can be seen in a small number of samples. A magnified view (Fig. 2b) shows that expression is only observed in a CD3‐CD4‐IL7Rα+α4Bβ7+ population that had been sorted by fluorescence‐activated cell sorting (FACS) analysis from the small intestine of E17.5 embryos. This process is equivalent to doing a large series of Northern blots (or reverse transcription–polymerase chain reactions [RT‐PCRs]) when one becomes interested in a new gene; however, it takes a matter of seconds to perform.
Figure 2.
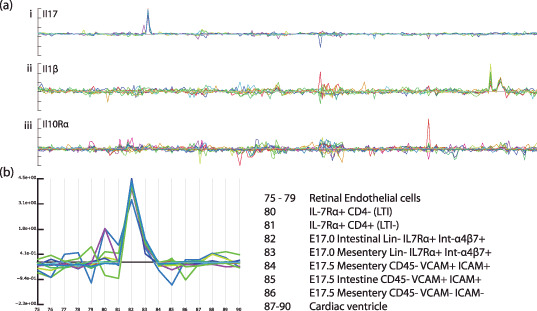
Expression of several interleukins (ILs) and interleukin receptors. (a) Probe‐pair signals for three interleukin‐related genes across a database containing 250 samples: (i) Il‐17, (ii) Il‐1β, and (iii) Il‐10rα. (b) A magnified view of the samples from which expression of Il‐17 can be seen. A clear signal can be seen in both samples 82 and 83, but in none of the other 248 samples in the database. The signal in sample 82 is around six‐times higher than that in 83. The strong deviation from the baseline, in combination with the high extent of correlation of individual probe‐pair signals, provide a high degree of confidence in the specificity of the expression. However, although there is about a six‐fold difference in the signal between 82 and 83, it is difficult to conclude that this represents the relative levels in the sample tissues, since we do not know by how much the signal tends to vary within these samples. In this specific case using replicate samples may not help much, as it is quite likely that the signal in 83 results from a cross‐contamination with tissue from 82, and the only way in which this can clearly be resolved is through in situ measurements.
It is often felt that DNA array data are not sufficiently reliable and that one should routinely perform many sample replications whenever using DNA arrays. However, this has to be balanced against the cost of performing additional experiments, and the consequent loss in the broadness of the coverage of the data that can be obtained. In the case of gene‐fishing expeditions, it sometimes is an irrelevant requirement; under these circumstances, a small number of genes are selected from the data and subsequently studied in greater detail. This is not done so much to verify the array data, but rather to provide additional information, since usually the array data will only cover a small number of samples and hence does not provide a lot of information. In this case, the number of replications that should be done merely reflects the balance between the cost of identifying false positives/negatives and the cost of the additional replicates. Additionally, the veracity of gene‐expression measurements can often be assessed by inspecting the raw data. In the case of Affymetrix data, genes are represented by sets of short probes (referred to as probe‐sets) providing a number of signal intensities. This allows the reliability of a given measurement to be assessed by considering the behavior of the individual probes across a number of samples. In the case of Il‐17 (Fig. 2ai ), all the individual probes show a clear covariance, indicating that the signal can be considered reliable. In addition, and this is an important point, the signal forms a clear baseline with little variation across all the other samples from which the signal in the indicated sample clearly deviates. Hence, in this case, the context of expression in a large number of unrelated samples directly contributes to the confidence of the measurement in the indicated sample. In the case of the Il‐1β gene (Fig. 2aii ), the data from the individual probe‐pairs display little covariation across most of the sample series, though a small number of samples do have a signal that deviates clearly from the baseline. This is in stark contrast to the signal for the Il‐10Rα gene (Fig. 2aiii ), where little or no covariance is seen throughout the sample series. Although some samples appear to have slightly elevated signals, these are clearly not as reliable as those seen for the Il‐17 and Il‐1β genes. This lack of a clean signal either indicates a lack of expression in the sample series or a failure of the specific probe‐set (some probe‐sets clearly do not work).
However, it should be emphasized that if one needs to identify genes that change their expression by a small amount (e.g. two‐fold), then it is absolutely necessary to perform several replications for individual samples. Of course, considering the error in the individual expression estimates (i.e. looking at the raw data), though seldom done, is still extremely useful in these cases.
Public data repositories
In recent years the numbers of array experiments carried out by biologists has skyrocketed. Public websites have been set up to provide central repositories from which this data can be accessed (e.g. http://www.ncbi.nlm.nih.gov/geo/ and http://www.ebi.ac.uk/arrayexpress/). Since data derived from the same type of array can easily be compared across several experiments (as long as similar protocols were followed), this forms an important data‐mining resource for researchers. As an example, there are many thousands of primary tumor samples available from these repositories, allowing cancer researchers to investigate the expression of genes in more or less any kind of tumor type without having to perform costly experiments.
Although much effort has been expended to provide users with the ability to directly view expression patterns of specified genes directly from these databases, it is still difficult to view the gene‐expression pattern across more than one given experiment, and no databases that we know of provide users with any indication of the errors in the underlying measurements. For these reasons we still consider it useful to download the desired data to a local database where it is possible for the researcher to compile data from a number of different experiments and to compare the expression of genes across the all the samples of interest. This is very much aided by the fact that the data from any given hybridization (i.e. one sample on one chip) provide a wealth of information that allows the quality of that sample and hybridization to be assessed (e.g. the RNA quality of the sample can be addressed). This is rather important, as often one will have very little information about the actual experimental protocols that were followed; this is especially important to consider when dealing with human samples, since these are frequently obtained from autopsies or surgical procedures where there is ample opportunity for the RNA to become degraded.
Recently we were able to use publically available microarray data to bridge discoveries made in developmental biology with clinically relevant issues. As a result of searching for genes that are involved in the differentiation of mesenchymal stem cells, we discovered that Arid3b, which was initially discovered as an RB‐1 binding protein in an AML cell line,( 6 ) is involved in the protection of nascent neural crest‐derived head mesenchymal cells from apoptosis.( 7 ) The knock‐out of this gene results in embryonic lethality due to massive apoptosis of neural‐crest‐derived mesenchymal cells in the branchial arches. This led us to ask whether Arid3b might also be involved in the development of neural‐crest‐derived tumors; in particular, neuroblastoma which is the most frequent infant solid tumor type. In order to determine whether Arid3b is likely to be involved in neuroblastoma development, we initially looked for microarray data from neuroblastoma samples to allow us to investigate the expression of Arid3b in primary tumors. We were able to find appropriate data from ArrayExpres (http://www.ebi.ac.uk/arrayexpress) that, in addition to providing appropriate samples, also provided extensive staging and classification of the samples.( 8 ) From this data we found that Arid3B is expressed in approximately half of the analyzed neuroblastoma samples. An inspection of the detailed description of these particular samples showed that Arid3b is in fact expressed primarily in the more malignant group of samples (in 80% of stage IV samples). It is well known that MYCN is essential for neuroblastoma development. However, MYCN alone is not sufficient to transform mouse embryonic fibroblasts (MEF). Hence, we proceeded to determine whether or not Arid3b can collaborate with MYCN in the transformation of MEF. As expected, while neither MYCN nor Arid3b alone were able to induce transplantable tumors from MEF, all MEF transduced with both genes forming tumors upon transplantation. Thus it is likely that Arid3b is a partner of MYCN in the transformation of neuroblastoma.( 9 ) Currently we are investigating whether neuroblastoma can be induced from neural stem cells by the transduction of these two genes.
Our experience is a clear example of the value of array data available from public repositories; this is especially true for clinically derived samples, which are often very difficult for basic researcher to obtain. In the future we hope that such data will help to bridge the chasm that separates clinical from basic researchers.
Despite the obvious attractions of using publically available data, there are at least two issues that should be considered. The first of these is the quality and the reliability of the actual expression measurements. However, this can quite easily be overcome since individual hybridizations provide sufficient information to assess the quality of the samples and the procedure followed. In addition, the veracity of individual measurements can be assessed by an inspection of the raw data. A more pressing issue is the quality of sample annotation that is frequently encountered whilst perusing these databases; this is presumably a result of rather over‐complicated standards having been developed that create a great deal of confusion and trouble for those submitting data to these repositories. Additionally the sample annotation data have not always been presented in a very clear manner, and we have resorted to writing scripts to automate the downloading of sample annotation from these databases. This, however, may not be too much of an issue when obtaining a limited number of samples, and in any case, we are optimists and believe that the situation will improve in the future.
We should also mention that in order to be able to compare expression measurements between different experiments obtained from different sources, it is sometimes necessary and usually prudent to obtain the raw underlying data and to use them to derive expression measurements or view directly if appropriate programs are used. This is necessary since there are a number of different means of calculating expression estimators from array data and these will give different incomparable values depending on the parameters chosen. Whereas this is not a problem for researchers who are familiar with DNA array data, it would be more convenient if the data were accessible from an integrated database system that could be easily browsed. Although there are a number of difficulties in achieving this, there are many groups that are currently involved in such endeavours and we are hopeful that this will be possible in the foreseeable future.
The stemness that never was: the trouble with complexity
The strength of DNA array analysis is its ability to deal globally with gene expression. However, dealing with such data requires an understanding of some simple statistical realities. This remains true both for simple gene‐fishing expeditions and for attempts to find some kind of overall patterns in the data. However, it is far more important for the latter, since in this scenario the data obtained from the array experiments are used not only as a lead for further research but as a means to make biological conclusions. In either scenario the pertinent issues stem from the simple fact that if you create a large random dataset and then proceed to look for specific patterns within that set you are quite likely to find those patterns. Hence it is necessary to have some way of deciding whether or not patterns that are found have some biological meaning. The two analyses described below both failed to take any such precautions and as a result were able to make spectacularly unsupported conclusions.
In October 2002 two papers were published in Science ( 10 , 11 ) in which a comparison of the transcriptomes of hematopoietic, neural, and embryonic stem cells was used to identify transcripts specifically and commonly expressed in the three stem cell populations. Although both groups identified more than 200 transcripts each, a comparison of the identified transcripts found only six of those to be in common to the two groups.( 12 ) The discrepancy between these analyses was attributed to a number of factors though the data analysis methods used were not questioned. Since we have an interest in stem cell biology, we obtained the two datasets and performed some rudimentary analyses. Contrary to the published findings, our analysis suggested that in fact neither dataset supported the presence of a set of genes that are commonly and specifically expressed in all the three stem cell signatures. Rather our analysis indicated that both groups had in fact looked rather too hard to find some set of genes that they could define as being associated with general stemness, leading both groups to identify a number of irrelevant genes. Given this, it is not surprising that there was very little overlap between the findings of the two groups. In fact, this was alluded to in a technical comment published shortly afterwards,( 12 ) but was never spelt out very clearly, and confusion around the perceived discrepancies between the two publications persisted for some time afterwards.
Both groups based their conclusions on the intersection of lists of transcripts determined to be selectively expressed in the three individual stem‐cell populations. These lists were generated by simple binary comparisons between the stem‐cell data and individual control populations. In the case of Ramalho Santos et al.,( 11 ) hematopoietic stem cells (HSC) were compared to bone marrow cells (BM), neural stem cells (NSC) to cells of the lateral ventricles (LVB), and embryonic stem cells (ES) to both non‐stem populations. This mode of analysis is reasonable if one wants to identify genes that are expressed in stem cells but are down‐regulated upon differentiation. However, in this case the control populations used in the comparisons must be both immediate descendants of the stem cells and form reasonably homogenous populations. Neither of these criteria was met by the control populations used by Ramalho‐Santos et al. The homogeneity of the non‐stem samples is a serious issue, as if a gene is active in the stem‐cell population, but is turned off in 50% of the descendants, then this would falsely be identified as stem‐enriched.
If a gene is specifically expressed in all stem‐cell types, then by definition, it should be expressed at low or non‐detectable levels in all or most non‐stem‐cell populations. Furthermore, if we consider the level of expression to be related to functionality, then we would expect that it should be expressed at similar levels within all stem‐cell types. However, neither Ramalho‐Santos et al.( 11 ) or Ivanova et al.( 10 ) appear to have made any efforts to consider the expression levels of the genes they identified across the entire set of samples; thus relying completely on the appropriateness of their criteria for selecting genes as being enriched in individual stem‐cell populations to select biologically meaningful genes. The fallacy in this is that it allows genes to be identified as being associated with a specific stem‐cell population, whilst being expressed at a much higher level in other non‐stem‐cell populations. Thus Asf‐1 was identified as being enriched in the NSC sample, whilst actually being expressed at a much higher level in the bone marrow non‐stem‐cell sample (Fig. 3a). Similarly neurochondrin was classified as HSC‐enriched although its expression is much higher in the LVB samples (Fig. 3b). It can be argued that it is inappropriate to compare the level of expression between different cell types such as HSC and LVB cells; however, it is difficult to make this argument, whilst at the same time arguing that it is appropriate to compare whole unsorted bone marrow with highly purified HSCs.
Figure 3.
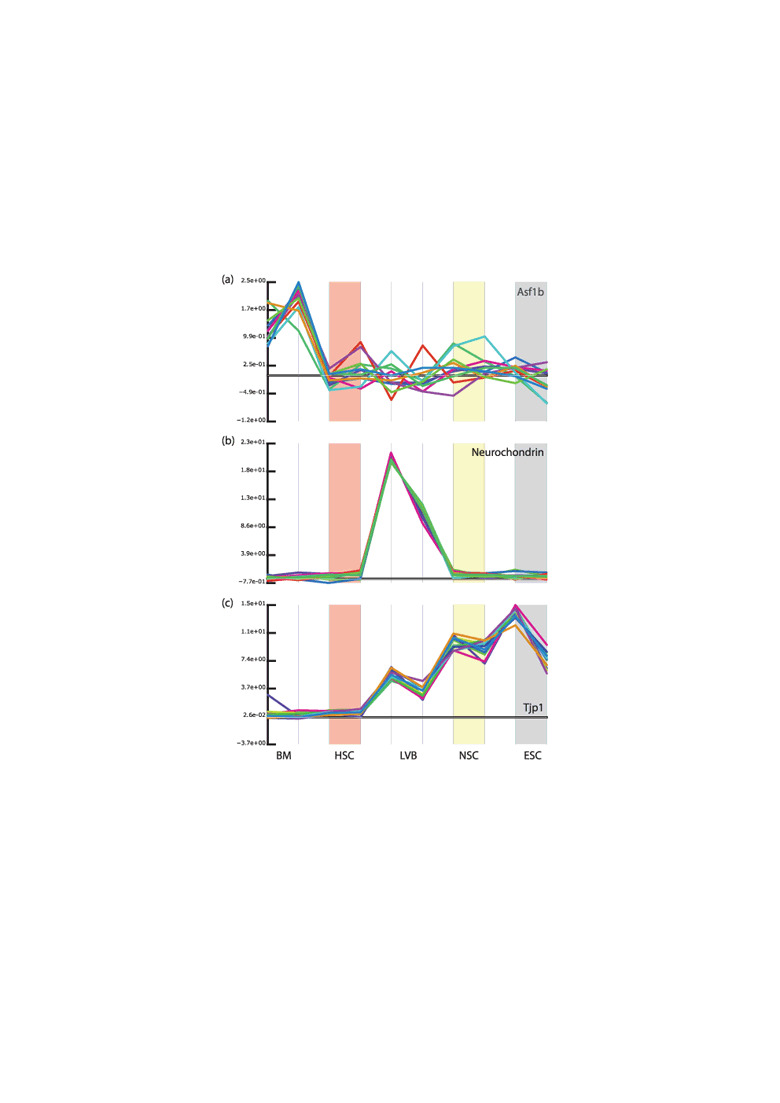
Examples of genes identified as enriched in specific stem cell types. We obtained the raw data from Ramalho‐Santos et al.( 11 ) and used our system to inspect the expression patterns of a a number of genes identified as being enriched in the different stem‐cell populations. (a) Asf1b (97364_at). This probe‐set was identified as being enriched in the neural stem cell (NSC) population (indicated by light yellow background). Its expression is indeed higher in the NSC samples than in the lateral ventral brain (LVB; immediately to the left of the NSC), but this level of expression is a small fraction of that seen in the bone marrow sample (BM; to the far left of the plot). (b) Neurochondrin (99633_at). This gene was identified as being specifically expressed in hematopoietic stem cells (HSC; indicated by light red background) as its expression in the BM fraction is somewhat lower. However, it is far more highly expressed in the LVB sample. (c) Tjp1 (99935_at). This is one of the genes identified as being specifically and commonly expressed by all three stem cell populations. Indeed, it is expressed in the HSC, NSC, and embryonic stem cell (ESC; indicated by a light grey background). However, its expression in the HSC fraction is extremely low (much less than that seen in the LVB sample) and it seems unlikely that this represents a functional level of expression. (The values plotted above were not produced in an identical manner to those by Ramalho‐Santos et al.; however, identical results are seen when the values they reported are used).
The examples described above also highlight an additional issue. It is often believed that stem cells of various types express many of the genes associated with their derivatives at a low level, and this belief seems to be reinforced by microarray experiments. However, the two above examples are of genes associated with differentiated cells of unrelated lineages being expressed in the stem cells (HSCs expressing neurochondrin, and NSCs expressing Asf‐1) at low levels. This is to be expected, since we looked for genes with such characteristics from the the set of genes selected by Ramalho‐Santos et al., in order to demonstrate the problems with the selection criteria they used. In doing so, we wish to make two points:( 1 ) first, if you look for something, you’re quite likely to find it, and( 2 ) second, the fact that you can find low‐level expression of progeny‐associated genes in stem cells does not in itself mean very much. It should be emphasized that low‐level expression of large numbers of genes does not appear to be a stem‐cell‐specific property; the number of genes that we, and others, have detected as expressed in stem cells does not markedly differ from non‐stem populations. We do not wish to suggest that the above ideas are incorrect per se; only that data from microarray experiments do not in themselves argue for this.
Similarly most of the genes selected by these groups as being enriched in all three stem cell populations showed markedly different expression between the different stem‐cell types. One of these, Tjp‐1 is indeed expressed at a much lower level in the HSC population than in the NSC and ES cells. It is also expressed at a much higher level in the LVB sample (Fig. 3c) than in the HSC cells; whereas the difference between the NSC and LVB samples is rather marginal (approximately two‐fold). Although a two‐fold difference is often used as an arbitrary threshold, it is questionable whether or not this is reasonable in this case where one sample (LVB) is expected to be made up of a much more heterogeneous population than the other sample (NSC). It would also be surprising if the very marginal expression of the Tjp‐1 in the HSC population really indicates an expression level that is functional.
In order to determine whether or not the data of Ramalho‐Santos et al. argue for or against the presence of a common subset of genes expressed specifically in stem cells, we selected a number of genes by comparing their maximum mean (of the two replicates) stem signal versus the maximal mean non‐stem signal, and then plotted the expression levels of those genes in the three in a triangular area (Fig. 4).( 13 ) This allows us to visualize the expression levels of the genes in the three samples in one plot, and lets us see if the transcripts can be classified into distinct groups by their expression levels in stem‐cell samples. Transcripts expressed at a similar level in all three samples will be plotted in the central part of the triangle; since the transcripts were selected by a comparison against the maximal non‐stem‐cell expression level (arbitrary three‐fold level), any that are expressed at a similar level in the stem‐cell populations must be expressed at lower levels in all of the non‐stem samples. Most of the transcripts are either specific to one of the three stem‐cell populations or are shared between the NSC and ESC populations. Strikingly, very few transcripts map to the center of the plot indicating that there is no large set of genes that can be considered as a stem‐cell signature.
Figure 4.
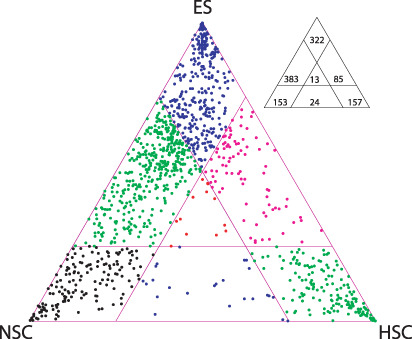
Relative expression of transcripts identified as expressed preferentially in at least one of the three stem cell populations. Relative expression in the three stem cell populations of 1147 probe‐sets whose maximal stem signal was at least three times the maximum non‐stem signal and the maximal stem cell signal was at least 10. Many genes are expressed at similar levels in the embryonic stem (ES) and neural stem cell (NSC) samples. However, there are only a very small number of genes whose expression is similar in all three stem cell populations (as indicated by a lack of points in the central area of the triangle). HSC, hematopoietic stem cells.
Our re‐analysis of the datasets from the above papers indicates that conclusions regarding a distinct genetic program for stem cells were reached rather prematurely. In fact, this can be seen very simply by looking at the expression patterns of the selected genes. It can also be seen by inspecting the enrichment scores that were used to select the transcripts. Fortunel et al. showed that those genes that were selected as commonly enriched in the stem‐cell populations were also those that had the lowest enrichment scores.( 12 ) In other words, common transcripts were found by ‘scraping the bottom of the barrel’ of enrichment, or as we like to think of it, ‘by harvesting the statistical noise’. At this point we should emphasize that further analysis of similar datasets may find some such common genetic program, but if so, we do not expect that it will be one specific to stem cells.
Summary
Microarray data have much to offer basic biologists. Today, there is a vast amount of data available from centralized repositories that allow gene expression to be assessed from almost any kind of tissue, and there is much that can be gained from this data whether one is performing microarray experiments or not. We tend to think that the value of this resource will increase as it grows, in the same manner as the value of centralized sequence repositories have gone from being of marginal utility to an everyday necessity for basic researchers. However, expression data are more complex than sequence data and we are still some way off having systems that make it sufficiently easy for these vast resources to be fully utilized.
We should also emphasize that there are a number of pitfalls when dealing with large datasets of great complexity. Many of these pitfalls can be simply avoided by closely looking at the data, whether that be the raw data from which expression levels have been calculated, the expression levels themselves, or statistics derived from them. In essence this isn't actually very different from how research should normally be carried out, and for us there is little difference between looking carefully at cells under a microscope, or looking carefully at large sets of numbers.
Acknowledgment
This work was supported by a grant for Regenerative Medicine Realization Projects from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
References
- 1. Jakt LM, Cao L, Cheah KS, Smith DK. Assessing clusters and motifs from gene expression data. Genome Res 2001; 11: 112–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kuznetsov VA, Knott GD, Bonner RF. General statistics of stochastic process of gene expression in eukaryotic cells. Genetics 2002; 161: 1321–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Spellman PT, Sherlock G, Zhang MQ et al . Comprehensive identification of cell cycle‐regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell 1998; 9: 3273–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tavazoie S, Hughes JD, Campbell MJ, Cho RJ, Church GM. Systematic determination of genetic network architecture. Nature Genet 1999; 22: 281–5. [DOI] [PubMed] [Google Scholar]
- 5. Flicek P, Aken BL, Beal K et al . Ensembl 2008. Nucleic Acids Res 2008; 36 (Database issue): D707–D714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Numata S, Claudio PP, Dean C, Giordano A, Croce CM. Bdp, a new member of a family of DNA‐binding proteins, associates with the retinoblastoma gene product. Cancer Res 1999; 59: 3741–7. [PubMed] [Google Scholar]
- 7. Takebe A, Era T, Okada M, Martin Jakt L, Kuroda Y, Nishikawa S. Microarray analysis of PDGFR alpha+ populations in ES cell differentiation culture identifies genes involved in differentiation of mesoderm and mesenchyme including ARID3b that is essential for development of embryonic mesenchymal cells. Dev Biol 2006; 293: 25–37. [DOI] [PubMed] [Google Scholar]
- 8. McArdle L, McDermott M, Purcell R et al . Oligonucleotide microarray analysis of gene expression in neuroblastoma displaying loss of chromosome 11q. Carcinogenesis 2004; 25: 1599–609. [DOI] [PubMed] [Google Scholar]
- 9. Kobayashi K, Era T, Takebe A, Jakt LM, Nishikawa S. ARID3B induces malignant transformation of mouse embryonic fibroblasts and is strongly associated with malignant neuroblastoma. Cancer Res 2006; 66: 8331–6. [DOI] [PubMed] [Google Scholar]
- 10. Ivanova NB, Dimos JT, Schaniel C, Hackney JA, Moore KA, Lemischka IR. A stem cell molecular signature. Science (New York, NY) 2002; 298: 601–4. [DOI] [PubMed] [Google Scholar]
- 11. Ramalho‐Santos M, Yoon S, Matsuzaki Y, Mulligan RC, Melton DA. ‘ Stemness’: transcriptional profiling of embryonic and adult stem cells. Science (New York, NY) 2002; 298: 597–600. [DOI] [PubMed] [Google Scholar]
- 12. Fortunel NO, Otu HH, Ng HH et al . Comment on ‘Stemness’: transcriptional profiling of embryonic and adult stem cells’ and ‘a stem cell molecular signature. Science (New York, NY) 2003; 302: 393; author reply. [DOI] [PubMed] [Google Scholar]
- 13. Sakurai H, Era T, Jakt LM et al . In vitro modeling of paraxial and lateral mesoderm differentiation reveals early reversibility. Stem Cells (Dayton, Ohio) 2006; 24: 575–86. [DOI] [PubMed] [Google Scholar]
- 14. Gentleman RC, Carey VJ, Bates DM et al . Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004; 5: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Irizarry RA, Hobbs B, Collin F et al . Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics (Oxford, England) 2003; 4: 249–64. [DOI] [PubMed] [Google Scholar]
- 16. Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 2001; 98: 5116–21. [DOI] [PMC free article] [PubMed] [Google Scholar]