

Abstract
We previously reported up‐regulation of T‐LAK cell‐originated protein kinase (TOPK), a novel mitotic kinase, in the great majority of breast cancers. Here we report its critical roles in mitosis, especially in cytokinesis. We found that protein phosphatase 1 alpha (PP1α) inactivation by cyclin‐dependent kinase 1 (CDK1)/cyclin B1 caused enhancement of autophosphorylation of TOPK and resulted in its activation at an early stage of mitosis. Then TOPK interacted with and phosphorylated p97, a member of the AAA+ family of ATPase proteins, through an interaction with p47 protein as an adaptor protein. Interestingly, knockdown of TOPK or p97 in breast cancer cells caused the mitotic failures in the abscission process. This mitotic failure could be rescued by additional exogenous introduction of wild‐type TOPK protein, but not by that of its kinase‐dead form. Our findings suggest that TOPK is indispensable for cancer cell cytokinesis throughout phosphorylation on p97. (Cancer Sci 2009)
Cell mitosis is strictly regulated by activation or inactivation of various protein kinases and phosphatases to control the complicated cell division process, which is essential for continuous cell proliferation.( 1 , 2 ) Cumulative evidence indicates that many evolutionarily conserved serine/threonine protein kinases play significant roles in the regulation of mitotic events. The dysregulation of mitotic kinases might result in two major events related to cancer: unscheduled cell proliferation and aberrant cell division leading to genomic instability. Therefore, numerous mitotic kinases have been investigated and applied to develop targeted therapeutic agents for a wide variety of human cancers.( 3 ) A large number of previous studies have disclosed fundamental roles of mitotic kinases such as CDK1 (cyclin‐dependent kinase 1), NEK2 (NIMA‐related kinase 2), AURKA (Aurora kinase A), and Plk1 (Polo‐like kinase 1) in breast carcinogenesis.( 4 , 5 , 6 , 7 )
Through genome‐wide expression profile analysis of breast cancers,( 8 ) we previously reported up‐regulation of TOPK (T‐LAK cell‐originated protein kinase, also known as PBK; PDZ‐binding kinase) in breast cancers and its potential as a therapeutic molecular target.( 9 ) We also demonstrated that the activated TOPK phosphorylated histone H3 (Ser10) at the M phase and that the TOPK depletion induced by siRNA caused cytokinetic defects of breast cancer cells.( 9 ) Although CDK1/cyclin B1 was reported to phosphorylate TOPK at Thr9 in vitro,( 10 ) how TOPK is activated and contributes to breast cancer cell proliferation remains largely unknown.
Entry into mitosis in mammalian cells has shown to be triggered by activation of the CDK1/cyclin B1 complex that targets many substrates involved in the early mitotic processes such as nuclear lamins, kinesin proteins, and Golgi‐matrix components.( 1 ) However, recent studies have revealed multiple roles of CDK1/cyclin B1 and its involvement in the exit of mitosis through phosphorylations of APC (anaphase‐promoting complex) ubiquitin ligase,( 11 ) as well as in metaphase–anaphase transition and cytokinesis.( 12 , 13 ) In addition, it was reported that the activity of protein phosphatase 1 alpha (PP1α) was inhibited after its phosphorylation (Thr320) by CDK1/cyclin B1 in early to mid‐mitosis.( 14 )
In the present study, we describe a novel mechanism to regulate the activity of TOPK in a cell cycle‐dependent manner by CDK1/cyclin B1 and PP1α. Furthermore, we show that TOPK phosphorylated p97, which is an AAA (ATPase‐associated various cellular activities) ATPase through its interaction with p47 as an adaptor protein, and knockdown of either TOPK or p97 resulted in incomplete cytokinesis. Our findings suggest that mitotic kinase TOPK plays a critical role for cytokinesis of breast cancer cells through the phosphorylation of p97 as its substrate.
Materials and Methods
Cell lines. Human breast cancer cell lines (BT‐549, HCC1937, MCF‐7, MDA‐MB‐231, MDA‐MB‐435S, SKBR3 T47D, and ZR‐75‐1), an immortalized human mammary cell line (HBL100), a human embryonic kidney fibroblast cell line (HEK293T), and a monkey kidney cell line (COS‐7) were purchased from American Type Culture Collection (ATCC, Rockville, MD, USA). A normal human mammary epithelial cell line, HMEC, was purchased from Cambrex BioScience (Walkersville, MD, USA). HBC5, a breast cancer cell line, was a kind gift from Dr Takao Yamori of Molecular Pharmacology, Cancer Chemotherapy Center of the Japanese Foundation for Cancer Research (Tokyo, Japan). All cells were cultured under conditions recommended by their respective depositors.
Recombinant proteins, antibodies, and reagents. The polyclonal antibody to p47 was a kind gift from Dr Akira Kakizuka (Laboratory of Functional Biology, Graduate School of Biostudies, Kyoto University, Kyoto, Japan). Other recombinant proteins, enzymes, antibodies, and chemicals were purchased from commercial sources: active recombinant TOPK protein (Invitrogen, Carlsbad, CA, USA); lambda protein phosphatase and recombinant CDK1/cyclin B1 proteins (New England Biolabs, Ipswich, MA, USA); anti‐TOPK, anti‐CDK1 and anti‐p97 monoclonal antibodies (BD Biosciences, San Jose, CA, USA); anti‐Flag‐tag and anti‐β‐actin monoclonal antibodies (Sigma‐Aldrich, St. Louis, MO, USA); anti‐HA‐tag monoclonal antibody (Roche, Basel, Switzerland); anti‐PP1α and anti‐phospho‐PP1α (T320) polyclonal antibodies (Cell Signaling Technologies, Beverly, MA, USA); anti‐Rb monoclonal antibody (Upstate Biotechnology, Lake Placid, NY, USA); anti‐phospho‐Rb (Ser807/811), anti‐p97, and anti‐PRC1 polyclonal antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA); anti‐cyclin B1, anti‐myc, and anti‐GST‐tag monoclonal antibodies (Santa Cruz Biotechnology); anti‐phosphothreonine or ‐phosphoserine polyclonal antibodies (Invitrogen); okadaic acid (OA), CDK1 inhibitor (CGP74514A) and protease inhibitor cocktail III (Calbiochem, San Diego, CA, USA); and nocodazole (Sigma‐Aldrich).
Cell‐cycle analysis. For fluorescence‐activated cell sorting (FACS) analysis, cultured T47D breast cancer cells were collected and fixed with 70% ethanol, and then kept at 4°C before their use. The cells were incubated with 10 mg/mL of RNaseI in PBS (−) at 37°C for 30 min and stained with 50 μg of propidium iodide (PI) at room temperature for 30 min. Finally, cell suspensions were analyzed with FACscan (Becton Dickinson, Franklin Lakes, NJ, USA). To collect the cells arrested at the G2/M phase, the cells were treated in culture media including 0.3 μg/mL of nocodazole (Sigma‐Aldrich) for at least 16 h. To investigate cell mitosis from prophase or metaphase to the next G1 phase, only mitotic cells were collected by the mitotic shake‐off method, washed with PBS, and released by incubation with fresh culture medium.
Knockdown effects by siRNA oilgonucleotides. We used siRNA oligonucleotides against TOPK and p97 (Sigma Aldrich Japan, Tokyo, Japan) as well as control siRNA for enhanced green fluorescent protein (EGFP) to examine the knockdown effects of those genes on cell morphology or cell‐cycle progression. The sequences targeting each gene were as follows: si‐EGFP (control): 5′‐GCAGCACGACUUCUUCAAG‐3′; si‐TOPK targeting 3′‐UTR of the TOPK gene, 5′‐GUGUGGCUUGCGUAAAUAA‐3′;( 9 ) and si‐p97, 5′‐AAGUAGGGUAUGAUGACAUUG‐3′.( 15 ) T47D or ZR‐75‐1 cells were plated onto 6‐cm dishes (2 × 105 cells/dish) or Col‐1 coated glass slides (1 × 105 cells/slide), and transfected with 100 pmol each of the siRNA duplexes using Lipofectamine RNAiMAX reagent (Invitrogen) according to the manufacturer’s instructions. The cells were collected or immunostained after incubation for 48 h to examine knockdown effects to the TOPK or p97 genes. Cell morphology was examined by phase‐contrast microscopy (Olympus, Tokyo, Japan) at 2 days after the transfection of siRNAs. To examine any arrest on cell cycle, the cells were treated with new culture media containing 0.3 μg/mL of nocodazole and incubated for the following 18 h. For monitoring cell mitosis, T47D cells were seeded at 1 × 105 cells in a 60‐mm dish and transfected with 100 pmol of si‐EGFP or si‐TOPK. Two days after the transfection, the time required for cell mitosis was measured by a Time‐lapse microscopy (Sanyo, Tokyo, Japan). In parallel, the cells were collected and equal amounts (5 μg protein) of whole cell lysates were applied for Western analysis using anti‐TOPK or anti‐p97 monoclonal antibodies to verify knockdown of those proteins. For RNAi‐rescue experiments, T47D cells were transfected with the pCAGGS‐HA‐TOPK construct at 24 h prior to transfection with each siRNA oligonucleotide, in accordance with a previous report.( 16 )
Statistical analysis. To assess TOPK‐knockdown effects on cell morphology, statistical significance was determined by Student’s t‐test using Statview software (SAS institute, Cary, NC, USA) after counting more than 1000 cells. P‐values less than 0.05 were considered to be statistically significant.
Additional methods (constructs; immunocytochemical analysis of TOPK, p47, and p97 proteins; immunoblotting and immunoprecipitation: GST pulldown assay; in vitro phosphorylation; in vivo phosphorylation; and in vitro phosphatase assay) are given in the Supporting Methods.
Results
Autophosphorylation of TOPK protein in mitotic cells. We previously demonstrated that TOPK was up‐regulated and phosphorylated at the M phase in breast cancer cells.( 9 ) To validate this result, we performed immunoblotting analysis using lysates of the T47D and ZR‐75‐1 breast cancer cells at the interphase or at mitosis by the mitotic shake‐off method (see Supporting Methods), and found completely phosphorylated TOPK protein in the mitotic cells (Fig. 1a). Because it has been reported that CDK1/cyclin B1 directly phosphorylate TOPK at Thr9,( 10 ) we firstly confirmed those results, and consequently proved it by observation of their co‐localization and by in vitro kinase assay (Supporting Fig. S1). However, we considered the possibility of autophosphorylation of TOPK during mitosis according to results from our previous in vitro kinase assay.( 9 ) To compare the CDK1‐induced phosphorylation at Thr9 and autophosphorylation of TOPK, T47D breast cancer cells were transfected with wild‐type, T9A, and kinase‐dead (KD) TOPK constructs, and treated with nocodazole. Although the exogenously expressed TOPK proteins were not completely phosphorylated, probably due to abrupt expression with a HA‐tag, immunoblotting with an anti‐HA antibody detected phosphorylated TOPK protein from nocodazole‐treated T47D cells (Fig. 1b). The phosphorylated bands of the cells transfected with the wild‐type or T9A constructs were still observed, while those in the cells with the KD mutant or those in the cell lysates treated with lambda protein phosphatase completely disappeared. These findings suggested that TOPK was predominantly phosphorylated by autophosphorylation rather than phosphorylation (Thr9) by CDK1/cyclin B1.
Figure 1.

Autophosphorylation of T‐LAK cell‐originated protein kinase (TOPK) protein in mitotic cells. (a) TOPK was phosphorylated in mitotic cells. The isolated cells at mitosis (M) or the interphase (I) were analyzed by FACS analysis, and cell lysates were treated with lambda protein phosphatase (λPPase) before immunoblotting with TOPK monoclonal antibody. (b) Autophosphorylation of TOPK in mitotic cells. T47D cells were transfected with wild‐type TOPK (WT), alanine‐substituted mutant at Thr9 (T9A), kinase‐dead (KD), and double mutant (T9A/KD), respectively, and immunoblotting was performed using anti‐HA monoclonal antibody. Both WT and T9A, but neither KD nor T9A/KD, were phosphorylated and showed a slowly migrating band (arrow). (c) Phosphorylation of TOPK was induced by treatment of okadaic acid (OA). Left panels: T47D cells were treated with 50, 100, and 200 nm of OA, and cells were harvested at 3 h after treatment of OA. Right panels: The phosphorylated band, which appeared after treatment with 200 nm of OA for 3 h, was verified by λPPase assay. (d) Interaction of TOPK and protein phosphatase 1 alpha (PP1α). COS‐7 cells were co‐transfected with GST‐fused PP1α (PP1α‐GST), HA‐tagged TOPK (HA‐TOPK), and pulled‐down with equilibrated glutathione sepharose 4B beads or immunoprecipitated with anti‐HA monoclonal antibody, followed by immunoblotting analysis using anti‐GST or HA monoclonal antibodies. (e) The recombinant TOPK (left panel) and endogenous TOPK from mitotic cell lysates (right panel) were dephosphorylated by treatment of recombinant PP1α protein. λPPase served as a positive control for clarification of the band‐shift of TOPK protein.
PP1α regulates the phosphorylation of TOPK. We had previously demonstrated that the phosphorylation of TOPK protein was induced by treatment with OA, which is a potent inhibitor of Ser/Thr protein phosphatases, in T47D breast cancer cells.( 9 ) However, how OA induced phosphorylation of TOPK in breast cancer cells was unclear. Although OA is known to inactivate both PP1α and PP2A (protein phosphatase 2A), the IC50 value of OA in inhibiting PP1α is much higher (70 nm) than that in PP2A (0.4 nm).( 17 ) Moreover, the activity of PP1α was reported to be down‐regulated during cell mitosis.( 14 ) Therefore, we examined phosphorylation levels of TOPK at a high concentration of OA that was expected to inhibit PP1α. We observed that the TOPK phosphorylation was enhanced by addition of 200 nmol/L of OA in the culture medium, but not by low concentrations of OA (less than 200 nm) (Fig. 1c), suggesting the possibility that autophosphorylation of TOPK was enhanced by inactivation of PP1α.
To further examine the interaction of TOPK with PP1α, we co‐transfected two plasmid constructs, which were designed to express GST‐tagged PP1α (PP1α‐GST) and HA‐tagged TOPK (HA‐TOPK) into COS‐7 cells, and then performed co‐immunoprecipitation assays. As shown in Figure 1(d), HA‐TOPK was clearly pulled down with PP1α‐GST (upper panel), and concordantly PP1α‐GST was co‐immunoprecipitated with HA‐TOPK (lower panels). We further examined the in vitro phosphatase activity of PP1α protein on the phosphorylated TOPK protein and found the dephosphorylation of recombinant TOPK protein by the incubation with the recombinant PP1α protein to be similar to that by the lambda phosphatase (λPPase) (Fig. 1e, left panel). In addition, we also found that the endogenous TOPK protein in lysates from T47D cells at the mitotic phase was dephosphorylated in the presence of PP1α protein (Fig. 1e, right panel). Taken together, these findings implied that a protein phosphatase, PP1α, possibly interacts with TOPK and thereby may suppress the activity of TOPK.
Activation of TOPK through inactivation of PP1α by CDK1/cyclin B1. Since PP1α was reported to be inactivated in mitotic cells through phosphorylation at Thr320 by CDK1/cyclin B1 complex,( 14 ) we hypothesized that the inactivation of PP1α during mitosis might be functionally linked to TOPK activation. To investigate this hypothesis, we synchronized T47D cells at the G2/M phase (61–70% were G2/M) by incubation with nocodazole for 16 h, and then subsequently incubated them with a CDK1 inhibitor for up to 4 h (Fig. 2a). Immunoblotting analysis demonstrated that the phosphorylation of TOPK was decreased in a time‐dependent manner (0 to 4 h) after treatment with the CDK1 inhibitor (Fig. 2b, first panel), in concordance with decreased phosphorylation of PP1α at Thr320 (Fig. 2b, third panel). We confirmed the decreased phosphorylation level of the Rb protein (Ser807 and Ser811) that was a hallmark of the activity of the CDK1/cyclin B1 complex (Fig. 2b, fifth panel).
Figure 2.
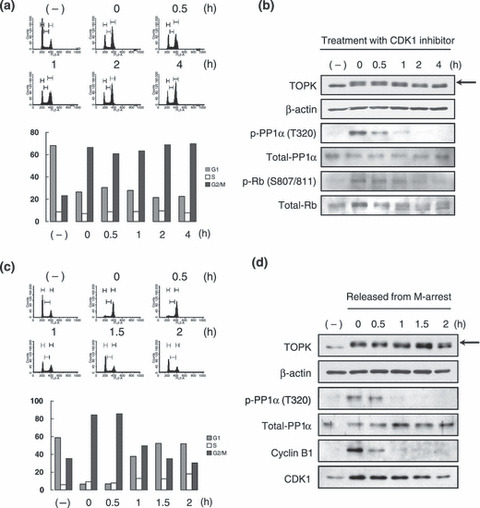
T‐LAK cell‐originated protein kinase (TOPK) was activated by cyclin‐dependent kinase 1 (CDK1) through inactivation of protein phosphatase 1 alpha (PP1α). (a) FACS analysis of M‐phase arrested cells after treatment with CDK1 inhibitor. T47D cells were treated with nocodazole for 16 h, followed by incubation with 25 nmol/L of CDK1 inhibitor (CGP74514A) from 0 to 4 h before FACS analysis. The population (%) of each cell cycle in the indicated time points was graphed. Grey bar, G1 phase; white bar, S phase; black bar, G2/M phase. (b) Expression of TOPK‐related proteins after treatment with CDK1 inhibitor. Equal amounts of total protein were immunoblotted with anti‐TOPK and anti‐total‐Rb monoclonal antibodies and with anti‐phospho‐PP1α (Thr320), anti‐total‐PP1α, and anti‐phospho‐Rb (Ser807/811) polyclonal antibodies. The arrow indicates the phosphorylated TOPK protein. (c) FACS analysis as mitosis progression. Only mitotic T47D cells were isolated and released from mitosis (see the Materials and Methods), followed by FACS analysis. The population (%) of each cell cycle is graphed. Grey bar, G1 phase; white bar, S phase; black bar, G2/M phase. (d) Expression of TOPK‐related proteins during mitosis progression. Equal amounts of total protein were immunoblotted with monoclonal antibodies of anti‐cyclin B1 and anti‐CDK1. The arrow indicates the phosphorylated TOPK protein.
Entry into mitosis is conducted by the activation of the CDK1/cyclin B1 complex, whereas exit of mitosis is driven according to inactivation of the complex through APC‐mediated proteolysis of cyclin B1.( 11 ) Hence, we investigated how TOPK, PP1α, cyclin B1, and CDK1 proteins are regulated from prophase to the next G1 phase by means of FACS and immunoblotting analyses using the isolated mitotic cells (Fig. 2c,d). According to progression of cell mitosis, the proportion of mitotic cells was significantly decreased from 85% to 30% within 2 h after releasing from the synchronization (Fig. 2c). During the M to next G1‐phase transition, we observed dephosphorylation of TOPK (Fig. 2d , first panel) as well as PP1α (Fig. 2d , third panel) in accordance with the reduction of the mitotic‐cell population. Moreover, these decreased phosphorylations of TOPK and PP1α (Thr320) were concordant with the decreased activity of the CDK1/cyclin B1 complex that was reflected by degradation of cyclin B1 protein during the late stages of mitosis (Fig. 2d , fifth panel). Taken together, our findings implied that the phosphorylation of TOPK was regulated by the activities of PP1α and CDK1/cyclin B1 in a mitosis‐dependent manner.
TOPK‐depletion resulted in cytokinetic failure and cell‐cycle arrest. We treated T47D breast cancer cells with TOPK‐specific siRNA (si‐TOPK) or si‐EGFP oligonucleotide (control) for 48 h and confirmed the knockdown of TOPK protein expression in the si‐TOPK‐treated cells (Supporting Fig. S2a). We observed the elongated intercellular bridges (white arrows) in the TOPK‐depleted‐cells by microscopy as well as by immunocytochemical analysis, but not in the control si‐EGFP‐treated cells. The cell population of cytokinetic defects was significantly increased by TOPK‐depletion (P < 0.001), which was accompanied by aberrant formation of the midbodies, indicating an essential role of TOPK protein in completion of cytokinesis (Supporting Fig. S2b). We confirmed the similar results when used another TOPK‐expressing breast cancer cell line, ZR‐75‐1 (Supporting Fig. S2c). To clarify incompletion of cytokinesis in TOPK‐depleted cells in more detail, we examined real‐time imaging of mitotic process of T47D cells by time‐lapse microscopy. Figure 3(a) shows that it took less than 2 h for the cell division from prophase to the next G1 phase in si‐EGFP‐transfected cells (as indicated by white arrows). On the other hand, it took 3.5 h for the cell division from prophase to telophase (white arrows), and a further 6 h for the abscission step (yellow arrows) in TOPK‐depleted cells (Fig. 3b). Additionally, those cytokinetic defects were restored by exogenously expressed wild‐type TOPK, but not by kinase‐dead mutant (Fig. 3c), suggesting that the kinase activity of TOPK is indispensable for cytokinesis.
Figure 3.
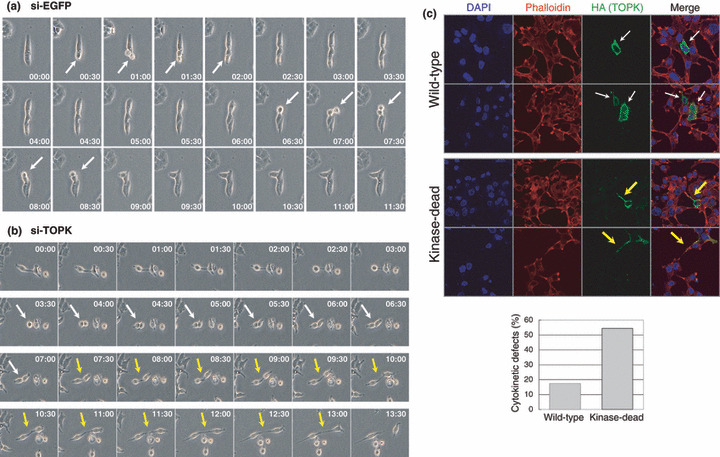
Cytokinetic failure induced by T‐LAK cell‐originated protein kinase (TOPK)‐depletion. (a,b) Two days after transfection with si‐enhanced green fluorescent protein (EGFP) (a) or si‐TOPK (b) in T47D cells, the duration of cell mitosis was measured by time‐lapse microscopy. White and yellow arrows indicate the cells under mitosis and the intercellular bridge, respectively. (c) RNAi rescue experiments. T47D cells were transfected with wild‐type (WT) or kinase‐dead of HA‐tagged TOPK‐expression vectors, and subsequently were transfected with si‐TOPK. Forty‐eight hours after transfection of siRNA, immunocytochemistry was performed. The exogenously expressed TOPK proteins were immunostained with anti‐HA monoclonal antibody (green). The wild‐type of TOPK restored cell morphology of T47D (white arrows), whereas the kinase‐dead did not but still showed intercellular bridges (yellow arrows). The actin structure was stained with Alexa Fluor 594 phalloidin (red), and nuclei were counter‐stained with DAPI (blue). The percentage of cytokinetic defects in the cells expressing the exogenous wild‐type or kinase‐dead of TOPK protein was graphed after counting 50 cells.
TOPK interacted with p97 via p47 during mitosis. To investigate the signaling pathway phosphorylated by the TOPK kinase, we attempted to identify substrate(s) of TOPK by in vitro protein pulldown assay using GST‐tagged TOPK (GST‐TOPK) and GST protein as a control. Through a comparison of proteins pulled‐down together with GST‐TOPK and GST proteins, we identified a protein of approximately 45‐kDa in a lane corresponding to the proteins co‐immunoprecipitated with the GST‐TOPK protein, but not in that with the control GST (Supporting Fig. S3). MALDI‐TOF analysis defined this protein to be a p47 protein, an adaptor of p97 that belongs to AAA+ ATPase.( 18 ) It has been reported that p47 forms a complex with p97 during mitosis.( 19 ) Therefore, to validate the interaction between TOPK and the p47/p97 complex, the protein pools co‐precipitated with GST‐TOPK were immunoblotted with anti‐p47 and anti‐p97 polyclonal antibodies, respectively. As shown in Figure 4(a) , both of the endogenous p47 and p97 proteins were included in co‐precipitates with GST‐TOPK in nocodazole‐treated T47D cells, but not in those from non‐treated cells, indicating a possibility of their interaction in a mitosis‐phase‐specific manner. To further validate their in vivo interaction, we performed co‐transfection of HA‐tagged TOPK (HA‐TOPK) with GST‐tagged p47 (GST‐p47) or myc‐tagged p97 (p97‐myc) constructs into COS‐7 cells, in which p47 expression was undetectable (data not shown), and then proteins were precipitated. Immunoblotting analysis to the precipitates using anti‐HA antibodies revealed that HA‐TOPK was co‐precipitated with GST‐p47 but not with p97‐myc (Supporting Fig. S4a). On the other hand, when all of the p97‐myc, GST‐p47, and HA‐TOPK constructs were co‐transfected, followed by immunoprecipitation with myc‐tag, we detected that HA‐TOPK was co‐precipitated with p97‐myc and the interaction was dependent on the expression of GST‐p47 (Supporting Fig. S4b). Furthermore, we confirmed the high expression of p47 and p97 proteins in the majority of the breast cancer cell lines examined, but not in the normal cells, HMEC (Supporting Fig. S4c). In addition, we observed the subcellular localization of endogenous p47 in the cytoplasm of interphase cells and diffusion in mitotic cells after breakdown of nuclear membrane (white arrows), similar to that of TOPK and p97 (Supporting Fig. S4d). It is notable that that TOPK and p97 clearly localized in the midbody during cytokinesis (Supporting Fig. S4e). Taken together, these findings suggest that p47 mediates protein interaction between TOPK and p97, and that their interaction plays a key role during the mitosis of breast cancer cells.
Figure 4.
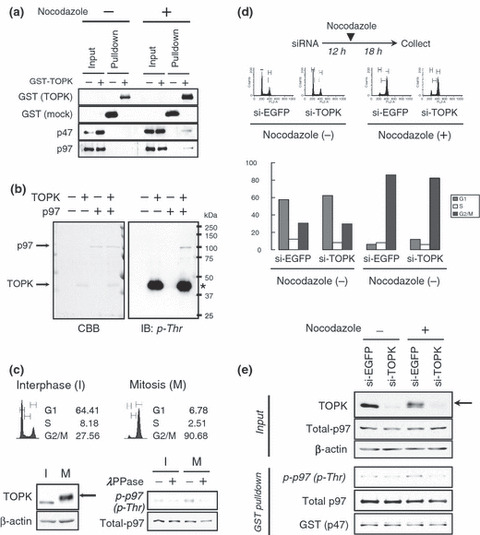
T‐LAK cell‐originated protein kinase (TOPK) phosphorylates p97 at the M‐phase. (a) p47/p97 complex interacts with TOPK in the mitotic cells. GST‐pulldown assay was performed with the cell lysates from T47D treated with/without nocodazole. The protein pools co‐precipitated with GST or GST‐TOPK were immunoblotted with anti‐GST and anti‐TOPK monoclonal antibodies, and with anti‐p47 and anti‐p97 polyclonal antibodies. (b) In vitro kinase assay of recombinant TOPK and p97 proteins using non‐labeled ATP. Each of 5 μg of recombinant p97 protein was incubated with 1 μg of active TOPK for 2 h at 30°C. After reaction, the protein samples were loaded on SDS‐PAGE gel followed by Coomassie Brilliant Blue (CBB) staining and immunoblotting with anti phospho‐Thr polyclonal antibody. The asterisk indicates autophosphorylation of TOPK. (c) Mitotic phosphorylation of p97. The isolated T47D cells at interphase (I) or mitosis (M) were analyzed by FACS analysis, and cell lysates were immunoblotted with anti‐TOPK monoclonal antibody (left) or phospho‐Thr specific polyclonal antibody (right) after treatment with lambda protein phosphatase (λPPase). β‐actin and total p97 served as loading controls. (d) TOPK‐depletion attenuates mitotic phosphorylation of p97. After transfection with si‐EGFP or si‐TOPK, T47D cells were incubated for 12 h and then treated with/without 0.3 μg/mL of nocodazole for additional 18 h before cell collection. The population (%) of each cell cycle is graphed. Grey bar, G1 phase; white bar, S phase; black bar, G2/M phase. (e) Equal amounts of total protein were immunoblotted with anti‐TOPK monoclonal antibody to show knockdown and phosphorylation (arrow) of TOPK protein induced by si‐TOPK and nocodazole, respectively. After in vitro binding and pulldown assays with GST‐tagged p47 protein, equal amounts of endogenous p97 proteins were co‐precipitated with sepharose 4B beads and immunoblotted with phospho‐Thr specific polyclonal antibody. Total p97 and GST‐p47 served as loading controls for the GST‐pulldown assay.
TOPK phosphorylates p97 during mitosis. To examine the possibility of whether the p47/p97 complex is a substrate for TOPK kinase, we first performed in vitro kinase assay using Flag‐tagged p47 protein (Flag‐p47) that was exogenously expressed and immunoprecipitated from COS‐7 cells in which p97 protein was highly expressed. Our results revealed that endogenous p97 protein was co‐immunoprecipitated with Flag‐p47 (Supporting Fig. S5a), and that the co‐immunoprecipitated p97 protein was phosphorylated by recombinant TOPK protein in vitro (Supporting Fig. S5b). We further confirmed the phosphorylation of purified myc‐tagged p97 protein by TOPK in vitro using an anti‐phospho‐Thr specific polyclonal antibody (Fig. 4b), but not by an anti‐phospho‐Ser antibody (Supporting Fig. S5c).
To further investigate phosphorylation of the endogenous p97 protein by TOPK during mitosis, we collected T47D breast cancer cells at the M phase by the mitotic shake‐off method, and then performed immunoblotting using an anti‐phospho‐Thr antibody. We observed the phosphorylation of endogenous TOPK protein (Fig. 4c, left panels) and p97 protein (Fig. 4c, right panels) in mitotic cell lysates (M), whereas the phosphorylation was hardly detectable in the cells in the interphase (I). We also investigated the phosphorylation status of p97 during mitosis in TOPK‐depleted cells. Our previous studies found that at least 24 h were required to significantly knockdown TOPK expression at the protein level. Hence, T47D cells were incubated for 12 h after transfection with si‐TOPK or si‐EGFP (control), and further incubated in the culture media with or without nocodazole for 18 h (see the Materials and Methods). We then collected the mitotic cells from each of the control (si‐EGFP)‐treated and si‐TOPK‐treated cells. As a result, when the cells were collected (30 h from the beginning of si‐TOPK transfection), TOPK protein was significantly depleted and the cells were arrested at pro‐ or meta‐phases (Fig. 4d). Moreover, by immunoblotting with anti‐TOPK monoclonal antibody, we validated that TOPK protein was significantly decreased in si‐TOPK‐treated cells although it was phosphorylated by treatment with nocodazole (Fig. 4e). Then, each of cell lysates was incubated with GST‐p47 recombinant protein to pull down the endogenous p97 protein. The total amount of p97 was not affected by treatment with si‐RNA or nocodazole, but the phosphorylation level of the p97 protein was significantly reduced in the TOPK‐depleted cells. Conversely, the phosphorylation of p97 was induced by exogenous introduction of wild‐type TOPK at the mitotic phase of HEK293T cells (Supporting Fig. S6). Therefore, we speculate that p97 is a novel mitotic substrate for the TOPK kinase.
TOPK and p97 are indispensable for cytokinesis. To elucidate the biological roles of p97 in breast cancer cells, we knocked down p97 expression in T47D (Fig. 5a,b) and ZR‐75‐1 (Fig. 5c,d) breast cancer cells by siRNA treatment. Subsequently, we observed by microscopy (Fig. 5a,c) and immunocytochemistry (Fig. 5b,d) that the p97‐depletion resulted in incomplete cytokinesis of these cells, remaining connected by intercellular bridges (yellow arrows), and thus significantly increased the cell population of cytokinetic defects (P < 0.0001). These results indicated that the absence of p97 caused the failure of cytokinesis, similar to morphologic changes observed by the knocking down of TOPK as shown in Figure S2, and therefore allowed us to conclude that the TOPK–p97 pathway could be indispensable for cytokinesis.
Figure 5.
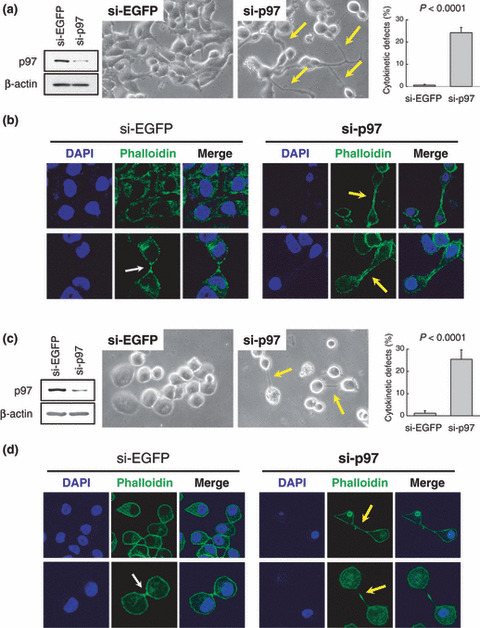
p97 is indispensable for cytokinesis. (a) Knockdown of p97 resulted in cytokinetic defects in T47D cells. Forty‐eight hours after transfection, the p97 protein was significantly depleted in si‐p97‐treated T47D cells compared with si‐enhanced green fluorescent protein (EGFP)‐treated cells. β‐actin served as a loading control for immunoblotting analysis. By observation of cell morphology using a phase contrast microscopy, the p97‐depleted cells showed intercellular bridges as indicated by yellow arrows. The percentage of cells with cytokinetic defects were graphed after counting more than 1000 cells (P < 0.0001, Student’s t‐test). (b) Immunocytochemical staining of the T47D cells. To clarify a shape of cell, the actin structure was stained with Alexa Fluor 488 phalloidin (green), and nuclei were counter‐stained with DAPI (blue). The p97‐depleted cells failed in the cytokinesis and showed elongated intercellular bridges (yellow arrows). The white arrow indicates midbody formation of si‐EGFP‐treated cells at the cytokinesis. (c,d) Knockdown of p97 resulted in cytokinetic defects in ZR‐75‐1 cells. The depletion of p97 protein, cell shape, and percentages of the cells with cytokinetic defects were validated as mentioned above.
Discussion
We previously suggested that TOPK is a novel mitotic kinase indispensable for breast cancer cell growth and highly phosphorylated in the mitotic cells.( 9 ) Although it has been reported that CDK1/cyclin B1 complex directly phosphorylated TOPK at Thr9 by in vitro experiments,( 10 ) our results implied that autophosphorylation of TOPK was likely to be more critical for its activation during mitosis than the phosphorylation at Thr9 by CDK1 (Fig. 1b). Indeed, our results were consistent with previous findings that CDK1/cyclin B1 phosphorylation was not sufficient to activate TOPK.( 20 ) We further demonstrated that the activated CDK1/cyclin B1 complex enhanced autophosphorylation of TOPK indirectly through suppression of PP1α activity at the entry of mitosis (Fig. 2a,b); inactivation of PP1α resulted in elevation of the autophosphorylation level of TOPK. Furthermore, we demonstrated that TOPK was gradually dephosphorylated at the exit of mitosis due to restoration of PP1α, that was induced by CDK1 inactivation (Fig. 2c,d). It is notable that two proteins of opposite biological roles, protein kinase CDK1 and protein phosphatase PP1α, comprise an axis to regulate the cell cycle‐dependent activation of TOPK. This switching mechanism is similar to other protein kinases such as Aurora kinase A, ATM, and Nek2, which are activated by autophosphorylation but negatively regulated by protein phosphatases.( 21 , 22 , 23 )
Although several studies have reported that knockdown of TOPK resulted in cytokineic defects, ( 9 , 10 , 24 ) its biological role in cytokinesis has not been understood. In this study, we observed that the TOPK‐depleted cells had an aberrant midbody structure and were connected with elongated intercellular bridges (Supporting Fig. S2). The real‐time imaging of mitotic cells demonstrated that the TOPK‐depleted cells resulted in delayed cytokinesis without cell cleavage, indicating an indispensable role for the abscission process, which is the final step of cytokinesis, the separation of two daughter cells (Fig. 3). These findings strongly suggest that TOPK is a mitotic kinase required to terminate cancer cell cytokinesis through phosphorylation of essential substrates involved in the cytokinesis. Consequently, we identified the p47/p97 complex proteins as candidate substrates of TOPK and ascertained that TOPK interacted directly with p47 but indirectly with p97 (Fig. 4a and Supporting Fig. S4). Furthermore, we discovered that TOPK is responsible for the mitotic phosphorylation of p97 (Fig. 4b–e).
The p97 protein, one of the AAA+ ATPase members, has been reported to be involved in various cellular events: ERAD (ER‐associated protein degradation), ubiquitin‐dependent proteolysis, spindle disassembly, and membrane fusion.( 18 ) These distinct molecular functions of p97 are known to be regulated by several adaptors and interacting proteins.( 25 ) Consistent with these reports, we have shown that adaptor protein p47 contributes to the interaction between TOPK and p97. The critical roles of p97 in mitosis have been suggested in various species, from yeast to mammalian cells.( 26 ) For example, the yeast homolog of p97, cdc48 protein, was reported to regulate spindle disassembly at the end of mitosis and to coordinate cellular morphologies during the M–G1 transition.( 27 , 28 ) We in this study demonstrated by siRNA experiments that knockdown of p97 in breast cancer cells caused cell‐cycle arrest at the boundary of the M and next G1 phase without cell abscission (Fig. 5). These results of cytokinetic defects might be induced from aberrant formation of the midbodies, where the abscission initiates, as observed in TOPK‐depleted cells (Supporting Fig. S2b). Considering phosphorylation of p97 by TOPK at the mitotic phase, our findings imply for the first time that TOPK plays a critical role in cell cytokinesis through phosphorylation of p97 as its substrate. Although further analysis of the biological roles of TOPK‐induced phosphorylation in p97 will be necessary, the phosphorylation may regulate the cell cycle‐dependent abilities of p97.
On the basis of these findings we propose the following model to describe the mechanism accounting for activation of TOPK and for the TOPK‐p47/p97 pathway during mitosis (Fig. 6). At the entry into mitosis, CDK1 is dephosphorylated by cdc25s, and rapidly translocates from the cytoplasm to the nucleus after nuclear envelope breakdown.( 29 ) The active CDK1/cyclin B1 complex phosphorylates PP1α at Thr320, leading to inactivation of PP1α, resulting in enhancement of TOPK autophosphorylation at an early stage of mitosis. Subsequently, the activated TOPK phosphorylates p97 through their interaction with p47 and the complex formation of TOPK‐p47‐p97. On the other hand, CDK1/cyclin B1 is gradually inactivated due to APC‐dependent degradation of cyclin B1, resulting in reactivation of PP1α when the cells exit mitosis.( 11 ) The restored PP1α dephosphorylates and inactivates TOPK to the steady level at the exit of mitosis. Because TOPK and p47/p97 are sequentially regulated as mitosis progresses, any interruption of these regulations is supposed to lead to cell‐cycle arrest and the failure of cell division as we observed by depletion of either TOPK or p97 (3, 5).
Figure 6.

Schema of the roles of T‐LAK cell‐originated protein kinase (TOPK) in cancer cell mitosis. At an early stage of mitosis, TOPK autophosphorylation is induced though the inactivation of protein phosphatase 1 alpha (PP1α) by cyclin‐dependent kinase (CDK1)/cyclin B1 complex. Then, the activated TOPK phosphorylates p97 through its interaction with p47. Finally, the restored PP1α dephosphorylates and inactivates TOPK to the steady level at the exit of mitosis.
In summary, we here report that TOPK is a novel mitotic kinase strictly regulated by CDK1 and PP1α, and plays an indispensable role in terminating cell cytokinesis. In addition, we suggest that the interaction of TOPK‐p47‐p97 plays a crucial role in cell abscission.
Supporting information
Fig. S1. Cyclin‐dependent (CDK1) phosphorylates T‐LAK cell‐originated protein kinase (TOPK) at Thr9 in vitro. (a) Co‐localization of TOPK, CDK1, and cyclin B1 at the M‐phase. T47D cells were treated with nocodazole (0.3 μg/mL for 18 h) and immunostained individually with monoclonal antibodies of TOPK (red), CDK1 (green), and cyclin B1 (green). Nuclei were counterstained with DAPI (blue) and arrows indicate the cells at mitosis. (b) In vitro kinase assay using recombinant proteins of CDK1/cyclin B1 and TOPK (Escherichia coli). Each of 5 μg of inactive TOPK‐recombinant protein, wild‐type (WT), and an alanine‐substituted mutant (T9A) generated by E. coli was incubated with five units of CDK1/cyclin B1 complex (New England Biolabs, Ipswitch, MA, USA). After reactions for 1 h at 30°C, the mixtures were loaded on SDS‐PAGE and autoradiographed (upper panel). The phosphorylated band of TOPK was diminished by competitive incubation with a peptide against the N‐terminus of TOPK protein (TOPK1‐18 peptide: MEGISNFKTPSKLSEKKK).
Fig. S2. (a) Cytokinetic failure in T47D cells after T‐LAK cell‐originated protein kinase (TOPK) ‐depletion. TOPK expression was drastically suppressed in si‐TOPK‐treated T47D cells compared with si‐EGFP‐treated cells. β‐Actin served as a control for immunoblotting analysis. Cellular morphology was observed by phase‐contrast microscopy at 2 days after transfection with si‐EGFP or si‐TOPK (upper panels), and the percentage of cells with cytokinetic defects was graphed after counting more than 1000 cells (middle panels). To clarify a shape of cell, the actin structure was stained with Alexa Fluor 594 phalloidin (red), and nuclei were counterstained with DAPI (blue) by immunocytochemistry (lower panels). The arrows indicate intercellular bridges. (b) Immunocytochemical staining with a protein marker for the midbody structure. The T47D cells transfected with si‐EGFP or si‐TOPK were immunostained with anti‐PRC1 polyclonal antibody (green) to validate formation of the midbody structure during cytokinesis. (c) Cytokinetic failure in ZR‐75‐1 cells after TOPK‐depletion. Similar to the results of T47D cells (a), many more cells of cytokinetic defects with the intercellular bridges (arrows) were observed in the TOPK‐depleted ZR‐75‐1 cells.
Fig. S3. Identification of a T‐LAK cell‐originated protein kinase (TOPK)‐interacting protein, p47. (a) T47D cells were transfected with plasmids encoding GST‐only, GST‐tagged TOPK protein of full‐length (1‐322), and GST‐tagged TOPK protein depleted with the C‐terminal ETDV motif (1‐318). After nocodazole‐treatment (0.3 μg/mL for 18 h) and GST pulldown, the co‐precipitates with GST‐tagged proteins were loaded on SDS‐PAGE followed by immunoblotting and silver staining. A differentially appeared protein band in the lanes of GST‐TOPKs (red arrow) was collected and analyzed by mass spectrometry. (b) Results from mass spectrometry. Mascot analysis (http://www.matrixscience.com) of peptide mass fingerprinting predicted the isolated sample as human p47 protein.
Fig. S4. T‐LAK cell‐originated protein kinase (TOPK) interacts with p47/p97. (a) TOPK directly interacts with p47, but not with p97. COS‐7 cells were co‐transfected with HA‐TOPK, GST‐p47, and p97‐myc. Subsequently, co‐precipitants with GST‐p47 (left panels) or p97‐myc (right panels) proteins were collected by GST‐pulldown assay or immunoprecipitation, respectively (see the Materials and Methods). The arrow indicates HA‐TOPK co‐precipitated with GST‐p47. The asterisk indicates a heavy chain of immunoglobulin from anti‐myc monoclonal antibody used for immunoprecipitation. (b) TOPK binds to p97 via p47 as an adaptor. COS‐7 cells were tri‐transfected with GST‐p47, myc‐p97, or HA‐TOPK constructs. The complex among those proteins were immunoprecipitated using anti‐myc monoclonal antibody, and immunoblotted with anti‐myc, ‐GST, ‐HA, and ‐TOPK monoclonal antibodies, respectively. The asterisk indicates a heavy chain of immunoglobulin from anti‐myc monoclonal antibody. (c) Equal amounts of total protein were prepared from breast cancer cell lines (BT‐549, HBC5, HCC1937, MCF‐7, MDA‐MB‐231, MDA‐MB‐435S, SKBR3, T47D, and ZR‐75‐1), HBL100, and a human mammalian epithelial cell line (HMEC). After SDS‐PAGE and membrane transfer, the proteins were immunoblotted with an anti‐TOPK monoclonal antibody and anti‐p47 or ‐p97 polyclonal antibodies. β‐Actin served as a control for immunoblotting analysis. (d,e) Subcellular localizations of p47, p97, and TOPK in T47D were observed by immunocytochemistry using an anti‐p47 polyclonal antibody or Alexa Fluor 488 phalloidin (green) and anti‐p97 or ‐TOPK monoclonal antibodies (red). Nuclei were counter‐stained with DAPI (blue). White and yellow arrows indicate mitotic cells (d) and the midbody structure of the cells at cytokinesis (e), respectively.
Fig. S5. In vitro kinase assay with T‐LAK cell‐originated protein kinase (TOPK) and immunoprecipitated proteins. (a) The candidate proteins to interact with TOPK were expressed in COS‐7 cells and immunoprecipitated using an anti‐Flag antibody. The endogenous p97 protein was co‐precipitated with Flag‐p47 and appeared in the SDS‐PAGE gel of CBB staining (right panel). (b) In vitro kinase assay of TOPK and the precipitated proteins using radioisotope‐labelled ATP. Each protein pool was reacted with 0.5 μg of active TOPK for 30 min at 30°C and autoradiographed. P1 is another candidate protein that may interact with TOPK, but not phosphorylated by TOPK. The asterisk indicates autophosphorylated TOPK. (c) In vitro kinase assay with recombinant proteins of TOPK, p47 and p97. The myc‐tagged p97 recombinant protein (1 μg) was incubated with the active TOPK (0.5 μg) with/without GST‐tagged p47 (0.5 μg). After reactions for 2 h at 30°C, the proteins were loaded on SDS‐PAGE and analyzed by CBB staining and immunoblotting with anti‐myc or ‐GST monoclonal antibodies and anti‐phospho‐Thr or ‐Ser specific polyclonal antibodies.
Fig. S6. In vivo phosphorylation of p97 by exogenously expressed T‐LAK cell‐originated protein kinase (TOPK). HEK293T cells were cotransfected with p97‐myc and HA‐tagged wild‐type (WT) or kinase‐dead (KD) of TOPK. Before cell collection, the cells were treated with 0.1 μg/mL of nocodazole for 18 h. After immunoprecipitation with myc‐tag antibody, equal amounts of p97‐myc protein were precipitated and then immunoblotted with anti‐phospho‐Thr polyclonal antibody. The arrow indicates the phosphorylated TOPK protein.
Supporting Methods.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
We thank Dr Akira Kakizuka for p47 polyclonal antibody; Kyoko Kijima, Kie Naito, Yoshiko Fujisawa, Masahiko Ajiro, and Jung‐Won Kim for excellent technical support; and Drs Chikako Fukukawa, Tomomi Ueki, and Arata Shimo for helpful discussions.
References
- 1. Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol 2001; 2: 21–32. [DOI] [PubMed] [Google Scholar]
- 2. Trinkle‐Mulcahy L, Lamond AI. Mitotic phosphatases: no longer silent partners. Curr Opin Cell Biol 2006; 18: 623–31. [DOI] [PubMed] [Google Scholar]
- 3. Li J, Li S. Mitotic kinases: the key to duplication, segregation, and cytokinesis errors, chromosomal instability, and oncogenesis. Pharmacol Ther 2006; 111: 974–84. [DOI] [PubMed] [Google Scholar]
- 4. Yuan J, Yan R, Kramer A et al. Cyclin B1 depletion inhibits proliferation and induces apoptosis in human tumor cells. Oncogene 2004; 23: 5843–52. [DOI] [PubMed] [Google Scholar]
- 5. Hayward DG, Clarke RB, Faragher AJ, Pillai MR, Hagan IM, Fry AM. The centrosomal kinase Nek2 displays elevated levels of protein expression in human breast cancer. Cancer Res 2004; 64: 7370–6. [DOI] [PubMed] [Google Scholar]
- 6. Wang X, Zhou YX, Qiao W et al. Overexpression of aurora kinase A in mouse mammary epithelium induces genetic instability preceding mammary tumor formation. Oncogene 2006; 25: 7148–58. [DOI] [PubMed] [Google Scholar]
- 7. Spankuch B, Kurunci‐Csacsko E, Kaufmann M, Strebhardt K. Rational combinations of siRNAs targeting Plk1 with breast cancer drugs. Oncogene 2007; 26: 5793–807. [DOI] [PubMed] [Google Scholar]
- 8. Nishidate T, Katagiri T, Lin ML et al. Genome‐wide gene‐expression profiles of breast‐cancer cells purified with laser microbeam microdissection: identification of genes associated with progression and metastasis. Int J Oncol 2004; 25: 797–819. [PubMed] [Google Scholar]
- 9. Park JH, Lin ML, Nishidate T, Nakamura Y, Katagiri T. PDZ‐binding kinase/T‐LAK cell‐originated protein kinase, a putative cancer/testis antigen with an oncogenic activity in breast cancer. Cancer Res 2006; 66: 9186–95. [DOI] [PubMed] [Google Scholar]
- 10. Matsumoto S, Abe Y, Fujibuchi T et al. Characterization of a MAPKK‐like protein kinase TOPK. Biochem Biophys Res Commun 2004; 325: 997–1004. [DOI] [PubMed] [Google Scholar]
- 11. Kraft C, Herzog F, Gieffers C et al. Mitotic regulation of the human anaphase‐promoting complex by phosphorylation. EMBO J 2003; 22: 6598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goto H, Kiyono T, Tomono Y et al. Complex formation of Plk1 and INCENP required for metaphase–anaphase transition. Nat Cell Biol 2006; 8: 180–7. [DOI] [PubMed] [Google Scholar]
- 13. Neef R, Gruneberg U, Kopajtich R et al. Choice of Plk1 docking partners during mitosis and cytokinesis is controlled by the activation state of Cdk1. Nat Cell Biol 2007; 9: 436–44. [DOI] [PubMed] [Google Scholar]
- 14. Kwon YG, Lee SY, Choi Y, Greengard P, Nairn AC. Cell cycle‐dependent phosphorylation of mammalian protein phosphatase 1 by cdc2 kinase. Proc Natl Acad Sci U S A 1997; 94: 2168–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wojcik C, Yano M, DeMartino GN. RNA interference of valosin‐containing protein (VCP/p97) reveals multiple cellular roles linked to ubiquitin/proteasome‐dependent proteolysis. J Cell Sci 2004; 117: 281–92. [DOI] [PubMed] [Google Scholar]
- 16. Zhu C, Lau E, Schwarzenbacher R, Bossy‐Wetzel E, Jiang W. Spatiotemporal control of spindle midzone formation by PRC1 in human cells. Proc Natl Acad Sci U S A 2006; 103: 6196–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ammosova T, Washington K, Debebe Z, Brady J, Nekhai S. Dephosphorylation of CDK9 by protein phosphatase 2A and protein phosphatase‐1 in Tat‐activated HIV‐1 transcription. Retrovirology 2005; 2: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Uchiyama K, Kondo H. p97/p47‐Mediated biogenesis of Golgi and ER. J Biochem 2005; 137: 115–9. [DOI] [PubMed] [Google Scholar]
- 19. Uchiyama K, Jokitalo E, Kano F et al. The localization and phosphorylation of p47 are important for Golgi disassembly‐assembly during the cell cycle. J Cell Biol 2003; 161: 1067–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gaudet S, Branton D, Lue RA. Characterization of PDZ‐binding kinase, a mitotic kinase. Proc Natl Acad Sci U S A 2000; 97: 5167–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Katayama H, Zhou H, Li Q, Tatsuka M, Sen S. Interaction and feedback regulation between STK15/BTAK/Aurora‐A kinase and protein phosphatase 1 through mitotic cell division cycle. J Biol Chem 2001; 276: 46219–24. [DOI] [PubMed] [Google Scholar]
- 22. Goodarzi AA, Jonnalagadda JC, Douglas P et al. Autophosphorylation of ataxia‐telangiectasia mutated is regulated by protein phosphatase 2A. EMBO J 2004; 23: 4451–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mi J, Guo C, Brautigan DL, Larner JM. Protein phosphatase‐1alpha regulates centrosome splitting through Nek2. Cancer Res 2007; 67: 1082–9. [DOI] [PubMed] [Google Scholar]
- 24. Fujibuchi T, Abe Y, Takeuchi T et al. Expression and phosphorylation of TOPK during spermatogenesis. Dev Growth Differ 2005; 47: 637–44. [DOI] [PubMed] [Google Scholar]
- 25. Schuberth C, Buchberger A. UBX domain proteins: major regulators of the AAA ATPase Cdc48/p97. Cell Mol Life Sci 2008; 65: 2360–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Meyer H, Popp O. Role(s) of Cdc48/p97 in mitosis. Biochem Soc Trans 2008; 36: 126–30. [DOI] [PubMed] [Google Scholar]
- 27. Cao K, Nakajima R, Meyer HH, Zheng Y. The AAA‐ATPase Cdc48/p97 regulates spindle disassembly at the end of mitosis. Cell 2003; 115: 355–67. [DOI] [PubMed] [Google Scholar]
- 28. Cao K, Zheng Y. The Cdc48/p97‐Ufd1‐Npl4 complex: its potential role in coordinating cellular morphogenesis during the M‐G1 transition. Cell Cycle 2004; 3: 422–4. [PubMed] [Google Scholar]
- 29. Takizawa CG, Morgan DO. Control of mitosis by changes in the subcellular location of cyclin‐B1‐Cdk1 and Cdc25C. Curr Opin Cell Biol 2000; 12: 658–65. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Cyclin‐dependent (CDK1) phosphorylates T‐LAK cell‐originated protein kinase (TOPK) at Thr9 in vitro. (a) Co‐localization of TOPK, CDK1, and cyclin B1 at the M‐phase. T47D cells were treated with nocodazole (0.3 μg/mL for 18 h) and immunostained individually with monoclonal antibodies of TOPK (red), CDK1 (green), and cyclin B1 (green). Nuclei were counterstained with DAPI (blue) and arrows indicate the cells at mitosis. (b) In vitro kinase assay using recombinant proteins of CDK1/cyclin B1 and TOPK (Escherichia coli). Each of 5 μg of inactive TOPK‐recombinant protein, wild‐type (WT), and an alanine‐substituted mutant (T9A) generated by E. coli was incubated with five units of CDK1/cyclin B1 complex (New England Biolabs, Ipswitch, MA, USA). After reactions for 1 h at 30°C, the mixtures were loaded on SDS‐PAGE and autoradiographed (upper panel). The phosphorylated band of TOPK was diminished by competitive incubation with a peptide against the N‐terminus of TOPK protein (TOPK1‐18 peptide: MEGISNFKTPSKLSEKKK).
Fig. S2. (a) Cytokinetic failure in T47D cells after T‐LAK cell‐originated protein kinase (TOPK) ‐depletion. TOPK expression was drastically suppressed in si‐TOPK‐treated T47D cells compared with si‐EGFP‐treated cells. β‐Actin served as a control for immunoblotting analysis. Cellular morphology was observed by phase‐contrast microscopy at 2 days after transfection with si‐EGFP or si‐TOPK (upper panels), and the percentage of cells with cytokinetic defects was graphed after counting more than 1000 cells (middle panels). To clarify a shape of cell, the actin structure was stained with Alexa Fluor 594 phalloidin (red), and nuclei were counterstained with DAPI (blue) by immunocytochemistry (lower panels). The arrows indicate intercellular bridges. (b) Immunocytochemical staining with a protein marker for the midbody structure. The T47D cells transfected with si‐EGFP or si‐TOPK were immunostained with anti‐PRC1 polyclonal antibody (green) to validate formation of the midbody structure during cytokinesis. (c) Cytokinetic failure in ZR‐75‐1 cells after TOPK‐depletion. Similar to the results of T47D cells (a), many more cells of cytokinetic defects with the intercellular bridges (arrows) were observed in the TOPK‐depleted ZR‐75‐1 cells.
Fig. S3. Identification of a T‐LAK cell‐originated protein kinase (TOPK)‐interacting protein, p47. (a) T47D cells were transfected with plasmids encoding GST‐only, GST‐tagged TOPK protein of full‐length (1‐322), and GST‐tagged TOPK protein depleted with the C‐terminal ETDV motif (1‐318). After nocodazole‐treatment (0.3 μg/mL for 18 h) and GST pulldown, the co‐precipitates with GST‐tagged proteins were loaded on SDS‐PAGE followed by immunoblotting and silver staining. A differentially appeared protein band in the lanes of GST‐TOPKs (red arrow) was collected and analyzed by mass spectrometry. (b) Results from mass spectrometry. Mascot analysis (http://www.matrixscience.com) of peptide mass fingerprinting predicted the isolated sample as human p47 protein.
Fig. S4. T‐LAK cell‐originated protein kinase (TOPK) interacts with p47/p97. (a) TOPK directly interacts with p47, but not with p97. COS‐7 cells were co‐transfected with HA‐TOPK, GST‐p47, and p97‐myc. Subsequently, co‐precipitants with GST‐p47 (left panels) or p97‐myc (right panels) proteins were collected by GST‐pulldown assay or immunoprecipitation, respectively (see the Materials and Methods). The arrow indicates HA‐TOPK co‐precipitated with GST‐p47. The asterisk indicates a heavy chain of immunoglobulin from anti‐myc monoclonal antibody used for immunoprecipitation. (b) TOPK binds to p97 via p47 as an adaptor. COS‐7 cells were tri‐transfected with GST‐p47, myc‐p97, or HA‐TOPK constructs. The complex among those proteins were immunoprecipitated using anti‐myc monoclonal antibody, and immunoblotted with anti‐myc, ‐GST, ‐HA, and ‐TOPK monoclonal antibodies, respectively. The asterisk indicates a heavy chain of immunoglobulin from anti‐myc monoclonal antibody. (c) Equal amounts of total protein were prepared from breast cancer cell lines (BT‐549, HBC5, HCC1937, MCF‐7, MDA‐MB‐231, MDA‐MB‐435S, SKBR3, T47D, and ZR‐75‐1), HBL100, and a human mammalian epithelial cell line (HMEC). After SDS‐PAGE and membrane transfer, the proteins were immunoblotted with an anti‐TOPK monoclonal antibody and anti‐p47 or ‐p97 polyclonal antibodies. β‐Actin served as a control for immunoblotting analysis. (d,e) Subcellular localizations of p47, p97, and TOPK in T47D were observed by immunocytochemistry using an anti‐p47 polyclonal antibody or Alexa Fluor 488 phalloidin (green) and anti‐p97 or ‐TOPK monoclonal antibodies (red). Nuclei were counter‐stained with DAPI (blue). White and yellow arrows indicate mitotic cells (d) and the midbody structure of the cells at cytokinesis (e), respectively.
Fig. S5. In vitro kinase assay with T‐LAK cell‐originated protein kinase (TOPK) and immunoprecipitated proteins. (a) The candidate proteins to interact with TOPK were expressed in COS‐7 cells and immunoprecipitated using an anti‐Flag antibody. The endogenous p97 protein was co‐precipitated with Flag‐p47 and appeared in the SDS‐PAGE gel of CBB staining (right panel). (b) In vitro kinase assay of TOPK and the precipitated proteins using radioisotope‐labelled ATP. Each protein pool was reacted with 0.5 μg of active TOPK for 30 min at 30°C and autoradiographed. P1 is another candidate protein that may interact with TOPK, but not phosphorylated by TOPK. The asterisk indicates autophosphorylated TOPK. (c) In vitro kinase assay with recombinant proteins of TOPK, p47 and p97. The myc‐tagged p97 recombinant protein (1 μg) was incubated with the active TOPK (0.5 μg) with/without GST‐tagged p47 (0.5 μg). After reactions for 2 h at 30°C, the proteins were loaded on SDS‐PAGE and analyzed by CBB staining and immunoblotting with anti‐myc or ‐GST monoclonal antibodies and anti‐phospho‐Thr or ‐Ser specific polyclonal antibodies.
Fig. S6. In vivo phosphorylation of p97 by exogenously expressed T‐LAK cell‐originated protein kinase (TOPK). HEK293T cells were cotransfected with p97‐myc and HA‐tagged wild‐type (WT) or kinase‐dead (KD) of TOPK. Before cell collection, the cells were treated with 0.1 μg/mL of nocodazole for 18 h. After immunoprecipitation with myc‐tag antibody, equal amounts of p97‐myc protein were precipitated and then immunoblotted with anti‐phospho‐Thr polyclonal antibody. The arrow indicates the phosphorylated TOPK protein.
Supporting Methods.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item