

Abstract
The polyADP‐ribosylation reaction results in a unique post‐translational modification involved in various cellular processes and conditions, including DNA repair, transcriptional control, genomic stability, cell death and transformation. The existence of 17 members of the poly(ADP‐ribose) polymerase (PARP) family has so far been documented, with overlapping functional consequences. PARP‐1 is known to be involved in DNA base excision repair and this explains the susceptibility spectrum of PARP‐1 knockout animals to genotoxic carcinogens. The fact that centrosome amplification is induced by a non‐genotoxic inhibitor of PARP and in PARP‐1 knockout mouse cells, is in line with aneuploidy, which is frequent in cancers. Genetically engineered animal models have revealed that PARP‐1 and VPARP impact carcinogenesis. Furthermore, accumulating experimental evidence supports the utility of PARP and PARG inhibitors in cancer therapy and several clinical trials are now ongoing. Increasing NAD+ levels by pharmacological supplementation with niacin has also been found to exert preventive effects against cancer. In the present review, recent research progress on polyADP‐ribosylation related to neoplasia is summarized and discussed. (Cancer Sci 2007; 98: 1528–1535)
Cancer is a disease that is characterized by various genetic changes, with mutations occurring in many proto‐oncogenes and tumor suppressor genes during a multistep process. These, together with epigenetic alterations, transcriptional deregulation, and aberrations in post‐translational modification, are the forces driving carcinogenesis. Forty years have passed since poly(ADP‐ribose), a biopolymer involved in a unique post‐translational modification, the polyADP‐ribosylation reaction, was first discovered.( 1 , 2 , 3 ) PolyADP‐ribosylation is an NAD+‐dependent enzymatic reaction resulting in covalent modification of acceptor proteins with repeating units of ADP‐ribose residues (Fig. 1). The structure of the biopolymer was characterized some 30 years ago.( 4 ) Originally poly(ADP‐ribose) polymerase (PARP)‐1 was found in the nuclei and shown to be activated by DNA strand breaks. The presence of a salvage pathway of NAD+ synthesis in the nuclei points to the importance of the polyADP‐ribosylation reaction. Subsequent to descriptions of monoADP‐ribosylation of arginine residues of mammalian proteins, cyclic ADP‐ribose formation from NAD+ by ADP‐ribosyl cyclase and NAD+‐dependent histone deacetylases, named sirtuins, is the other enzyme group that uses NAD+ in an important regulatory system for gene transcription. Ironically, NAD+ is also used as the substrate by various microbial toxins for monoADP‐ribosylation reactions.( 5 ) In clear contrast to many other post‐translational modifications, poly(ADP‐ribose) molecules covalently attached to acceptor proteins vary greatly in size, up to several hundred ADP‐ribose residues with branching and large negative charges.( 4 , 6 ) These underlie the unique structural and functional characteristics of polyADP‐ribosylation.
Figure 1.
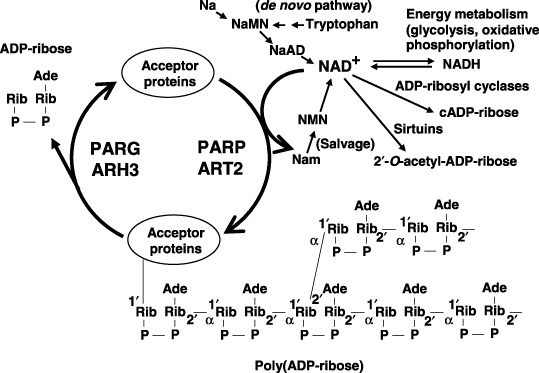
PolyADP‐ribosylation and related reactions. ARH3, ADP‐ribose‐(arginine) protein hydrolase 3; ART2, monoADP‐ribosyl transferase 2; Na, nicotinic acid; NaAD, nicotinic acid adenine dinucleotide; NAD+ (βNAD+), nicotinamide adenine dinucleotide; Nam, nicotinamide; NaMN, nicotinic acid mononucleotide; NMN, nicotinamide mononucleotide; PARG, poly(ADP‐ribose) glycohydrolase; PARP, poly(ADP‐ribose) polymerases.
The present review summarizes recent progress suggesting that polyADP‐ribosylation is dynamic and important for the regulation of critical cell functions, including mechanisms suppressing carcinogenesis. Possible applications in cancer therapy and prevention are also discussed. A recent review of the molecular aspects of polyADP‐ribosylation is helpful.( 7 )
PolyADP‐ribosylation and related reactions
There are now 17 PARP members deduced from genome sequences( 7 ) (Fig. 2). Their differences in the subcellular localizations of PARP and specific expression timing, in part associated with the mitotic apparatus including centrosomes and spindle body, suggest various functions( 3 , 8 ) (Fig. 3, , Table 1). Poly(ADP‐ribose) (PAR) and poly(ADP‐ribose) glycohydrolase (PARG) also localize in the spindle body and centrosomes during mitosis( 9 ) (Fig. 3).
Figure 2.
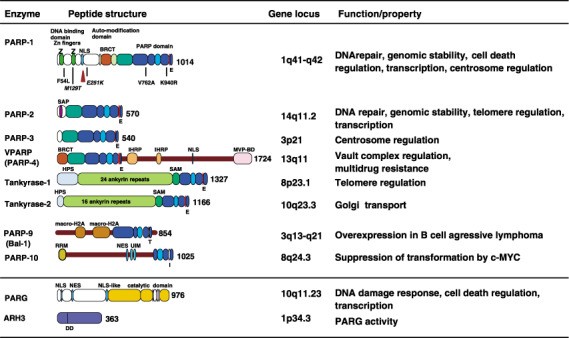
Poly(ADP‐ribose) polymerases (PARP) family proteins relating to carcinogenesis, poly(ADP‐ribose) glycohydrolase (PARG) and ADP‐ribose‐(arginine) protein hydrolase 3 (ARH3). Peptide structures, domains, motifs, gene loci and functions/properties are shown. For PARP‐1, positions of amino acids where single nucleotide polymorphisms (SNP) and mutations (italic) are shown. The caspase cleavage site is shown by the triangle. ARH, ADP‐ribosyl protein hydrolase; BRCT, BRCA1 C‐terminus; HPS, homopolymeric runs of His, Pro, and Ser; IHRP, inter‐α‐trypsin inhibitor family heavy chain‐related protein motif; MVP‐BD, major vault protein binding motif; NES, nuclear export signal; NLS, nuclear localization signal; RRM, RNA recognition motif; SAP, SAF‐A/B, acinus, and PIAS motif; SAM, a sterile α motif; UIM, ubiquitin‐interacting motif. The critical amino acid residue in the PARP domain of PARP‐1 and the residues at the corresponding position for each PARP family protein are shown. The DD residues indicated in ARH3 are critical residues for PARG activity.
Figure 3.
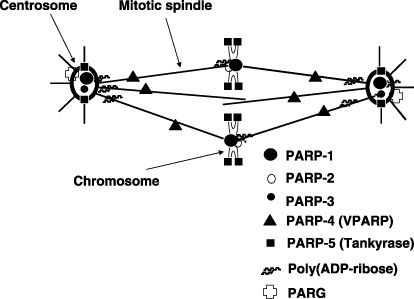
Localization of poly(ADP‐ribose) polymerase (PARP) family members, poly(ADP‐ribose) glycohydrolase (PARG) and poly(ADP‐ribose) at the mitotic apparatus.
Table 1.
Phenotypic outcome of dysfunction in polyADP‐ribosylation
| Enzyme | Subject | Outcome | Method |
|---|---|---|---|
| PARP‐1 | Carcinogenesis | Susceptibility (induced by alkylating agents)↑ | KO mice |
| Susceptibility (in aged mice)↑ | KO mice | ||
| Genomic instability | SCE↑ | KO mice | |
| Gene amplification↑ | KO mice | ||
| Micronuclei↑, chromosomal aberration↑ | Antisense | ||
| Centrosome amplification↑, Ploidy↑ | KO mice | ||
| Deletion mutation (after BHP treatment)↑ | KO mice | ||
| Transcriptional dysregulation↑ | KO mice and Drosophila | ||
| DNA damage response | DNA repair↓ | KO mice, antisense | |
| Lethality of alkylating agents and γ‐irradiation↑ | KO mice | ||
| Cell death induced by oxidative stress↓ | KO mice | ||
| Differentiation | Differentiation to trophoblast lineage↑ | KO mice | |
| PARP‐2 | Genomic instability | Chromosomal aberration↑ | KO mice |
| Ploidy↑ | KO mice | ||
| Aberration of spermatogenesis↑ | KO mice | ||
| DNA damage response | DNA repair↓ | KO mice | |
| Lethality of alkylating agents and γ‐irradiation↑ | KO mice | ||
| Differentiation | Adipocyte differentiation↓ | KO mice | |
| Tankyrase 1 | Mitosis control | Aberration in chromosomal segregation↑ | siRNA |
| PARG | DNA damage response | Lethality of alkylating agents and γ‐irradiation↑ | KO mice |
| Neuronal dysregulation | Neuronal degeneration↑ | KO Drosophila | |
| VPARP | Carcinogenesis | Carcinogenesis induced by urethane↑ | KO mice |
KO, knock‐out; PARG, poly(ADP‐ribose) glycohydrolase; PARP, poly(ADP‐ribose) polymerase; SCE, sister chromatid exchanges; VPARP, vault‐associated PARP.
MonoADP‐ribosylation reactions. Post‐translational modification by single ADP‐ribose residues is termed monoADP‐ribosylation and is catalyzed by various viral and bacterial toxins,( 5 ) and eukaryotic enzymes using NAD+ as the substrate. Mammalian monoADP‐ribosyl transferases (ART) 1–7 have already been reported and ART2 is located on cell surfaces and is able to catalyze autopolyADP‐ribosylation.( 10 ) Of note, pierisin, isolated from Pierisis rapae, is demonstrated to monoADP‐ribosylate guanine residues of DNA and induce apoptosis of various cancer cells.( 11 )
DNA Repair
Single‐strand breaks and base excision repair. PARP‐1 is activated by single‐ and double‐strand breaks (SSB and DSB, respectively) and binds to such DNA strand breaks with zinc finger motifs (Fig. 2). After hydrogen peroxide treatment or SSB induction, foci of poly(ADP‐ribose) appear in nuclei within several minutes, followed by foci of X‐ray repair cross‐complementing I (XRCCI). In PARP‐1−/– cells, these are not detected. XRCCI has a high affinity for poly(ADP‐ribose), and polyADP‐ribosylated PARP‐1 is suggested to bind and recruit XRCCI to SSB.( 12 )
PARP‐1 knockout cells show increased sensitivity to alkylating agents, topoisomerase (topo) I inhibitors and γ‐irradiation.( 13 ) Increased levels of SSB and DSB and a delay in DNA repair are observed in PARP‐1−/– mouse embryonic fibroblast (MEF) after alkylating damage. When mutation frequency was measured in the red/gam gene using the gpt‐Δ transgenic mouse system, the frequency of deletions, particularly those accompanying rearrangements, is increased in the livers of PARP‐1−/– mice after treatment with N‐bis(2‐hydroxypropyl)nitrosamine (BHP).( 14 ) After alkylation of bases in DNA, glycosylases active in base excision repair (BER) first remove alkylated DNA bases. During the BER process, strand breaks and gaps are produced, and PARP‐1 is activated and recruits XRCC1 and Ligase III‐α complex in certain conditions, as illustrated in Fig. 4. PARP‐1 may possibly protect the introduced DNA gaps (Fig. 4). In the absence of PARP‐1, stalled BER may cause unligated SSB and may further induce DSB, and DNA fill‐in reactions in short‐patch or long‐patch repair processes may be disturbed. The condensin I complex also supports BER through interactions with PARP‐1 and XRCC1.( 15 ) Furthermore, PARP‐1 or PARP‐2 can induce reactivation of stalled DNA topo I in covalent complexes and stimulate DNA strand break sealing.( 16 )
Figure 4.
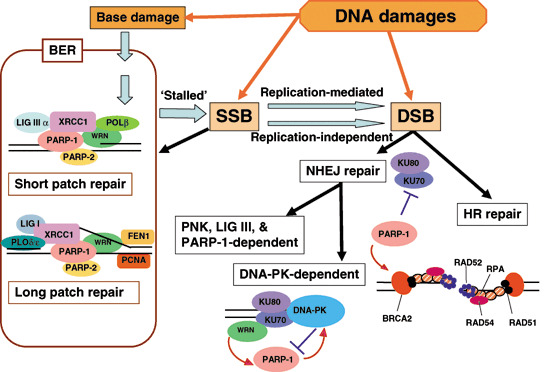
Model for involvement of poly(ADP‐ribose) polymerase (PARP) in DNA repair. DNA damage directly produces base damage, single‐strand breaks (SSB) and double‐strand breaks (DSB). SSB may be left unligated accidentally in the process of base excision repair (BER). SSB are possibly converted to DSB in a replication‐mediated manner or replication‐independently. Involvement of PARP in short‐patch and long‐patch repairs of BER, non‐homologous end‐joining (NHEJ) repair, and homologous recombination (HR) repair is shown. FEN1, flap endonuclease I; LIG, DNA ligase; PCNA, proliferating cell nuclear antigen; PNK, polynucleotide kinase; POL, DNA polymerase.
DSB and homologous recombination repair. DSB repair is mainly carried out by error‐prone non‐homologous end‐joining (NHEJ) and error‐free homologous recombination (HR) pathways (Fig. 4). Interactions of the DNA‐dependent protein kinase (DNA‐PK) complex, PARP‐1 and Werner syndrome protein (WRN) seem to be involved in balancing these pathways.( 17 ) Ku70/80 in the DNA‐PK complex has a high affinity for DSB, and this affinity is reduced by polyADP‐ribosylation.( 17 ) In PARP‐1−/– chicken DT 40 cells, the HR pathway is substantially inhibited by the Ku protein, indicating that PARP‐1 functions in suppressing Ku protein blockage of HR repair.( 18 ) WRN is recruited by interaction with Ku70/80 to DSB and is necessary for full activation of PARP‐1.( 17 ) The presence of an alternative DSB pathway involving PARP‐1, PNK and DNA ligase III has also been suggested.
In HR repair, after bridging DSB by Rad50, Mre11 and the NBS‐1 complex, the DSB terminus with a 3′‐overhang structure is protected by Rad51 and BRCA2 (Fig. 4). In the absence of BRCA2, PARP‐1 may possibly function to protect DSB ends from nuclease attack because BRCA2‐deficient cancer cells are highly sensitive to PARP inhibitors and exhibit an increased frequency of DSB.( 19 , 20 ) Some DSB‐repair deficient cells, including examples that are ATM‐deficient, also show hypersensitivity to PARP inhibitors.( 21 )
PARP‐2 and DNA repair. PARP‐2 is mainly present in centromeres during interphase and recognizes DNA loop structures. It is also activated by DNA damage.( 7 ) After γ‐irradiation, an increased level of DNA strand breaks was observed in the centromere regions of PARP‐2 knockout cells, these also demonstrating increased sensitivity to alkylating agents.( 22 ) PARP‐2 also supports BER and interacts with PARP‐1, XRCC1, DNA polymerase‐β and DNA ligase III. The functions of PARP‐1 and PARP‐2 in BER may be complementary only in a part and it remains to be determined whether their roles differ depending on the local chromatin structures.
PARG and DNA repair responses. PARP‐1 and PARG are suggested to form a complex that breaks down poly(ADP‐ribose) to ADP‐ribose, resulting in regeneration of ATP molecules, which are required for DNA repair.( 23 ) PARG has been found to re‐localize at sites of DNA breaks induced by UV‐A laser microirradiation in HeLa cells,( 24 ) but further evidence of involvement in DNA repair needs to be obtained.
Transcriptional control
PARP‐1 acts as a co‐activator and co‐repressor of transcription. In PARP‐1−/– mice, NF‐κB‐dependent inducible nitric oxide synthase (iNOS) gene expression is substantially reduced.( 25 ) Acetylation of lysine residues near the BRCT motif of PARP‐1 is required for activation of transcription.( 25 ) PARP‐1 also acts as a co‐activator of retinoic acid‐inducible retinoic acid receptor (RAR)‐dependent transcription of the RARβ gene, by binding to an inactive mediator that is then activated by RAR.( 26 ) Subsequently the co‐repressor complex is released, and recruited histone acetyltransferase complex activates transcription. PARP‐1 also functions as a co‐activator in β‐catenin/TCF4‐dependent transcription.( 27 ) In estrogen receptor (ER)‐dependent transcription of the ER gene, auto‐polyADP‐ribosylation stimulates formation of transcriptional complexes.( 28 ) During transcriptional activation of the ER gene, PARP‐1 interacts with topo II‐β and transient DSB are induced, which is necessary for transcription.( 29 ) The polyADP‐ribosylation reaction is required for transcriptional activation of wide regions of chromatin through ‘puff formation’.( 30 )
DNA methylation, imprinting and PARP. Hypomethylation of the global genome and local DNA hypermethylation frequently occur from the early stages of carcinogenesis. In this context the finding that PARP inhibitors enhance DNA methylation of the HTF9 gene promoter is of interest.( 31 ) DNA methyltransferase (DNMT) 1 possesses two poly(ADP‐ribose) binding motifs and DNMT activity is repressed after its binding to poly(ADP‐ribose).( 32 )
In cancer cells, loss of imprinting is also observed. CTCF (CCCTC binding factor), which binds to the non‐methylated maternal allele of the insulator domain in the H19 imprinting control region (ICR), is preferentially polyADP‐ribosylated.( 33 ) More than 140 CTCF target sites have been found to be polyADP‐ribosylated and chromatin insulator functions are sensitive to PARP inhibition. It remains to be clarified which members of the PARP family are involved in the regulation of imprinting.
Macrodomain and PARP‐1. The release of histone from chromatin by polyADP‐ribosylation, in the so‐called ‘histone shuttle model’, may enable dynamic conversion of local chromatin structures. Besides histones, various proteins bind poly(ADP‐ribose) in cellular extracts. PARP‐9 (Fig. 2), PARP‐14 and PARP‐15 all contain macrodomains that consist of hydrophobic amino acids and a helix structure. Recently, the macrodomains of macroH2A and PARP‐9 were demonstrated to bind monoADP‐ribose and poly(ADP‐ribose).( 34 ) There is thus a possibility that the local or cellular ADP‐ribose metabolic state is translated into transcriptional regulation through macrodomains.
Differentiation control
In early studies, PARP inhibitors were shown to modulate differentiation processes. In human promyelocytic leukemia, HL‐60 cells, stimulation of differentiation into granulocytes was accompanied by loss of c‐myc gene amplification.( 35 ) In H‐ras‐transformed NIH3T3 cells, PARP inhibitors also caused loss of amplified H‐ras and c‐myc oncogenes and reversal of the transformed phenotype.( 35 )
During teratocarcinoma‐like tumor formation from mouse embryonic stem (ES) cells, induction of the trophoblast lineage, including trophoblast giant cells (TGC), was observed in tumors derived from PARP‐1−/– ES cells.( 36 ) The properties of TGC are similar to those of the syncytiotrophoblastic giant cells (STGC) observed in human germ cell tumors. PARP‐1 deficiency may be related to induction of STGC during human germ cell tumor development.
Cell‐cycle controls
PARP‐1 is also involved in cell‐cycle check‐point control after DNA damage. After γ‐irradiation, p53‐dependent induction of the p21 and mdm2 genes is attenuated by PARP inhibitors and suppression of G1 arrest and enhancement of G2 arrest are observed.( 2 , 37 ) After treatment with neocarzinostatin, an increased level of γ‐H2AX, a marker of DSB, was observed, accompanied by augmented p53 phosphorylation at the ser 18 residue in PARP‐1−/– MEF. This accompanied enhancement of kinase activity of the ATM protein.( 38 ) In addition, S‐phase entry from G0 phase was found to be delayed in several cell types by PARP‐1 deficiency.( 2 )
Role of PARP‐1 in cell death regulation
During the course of carcinogenesis, various types of cell death stress, including oxidative stress induced by inflammation and energy depletion, may be operating. Survival may be associated with mutations or epigenetic alteration of genes responsible for cell death pathways. PARP‐1 dependent cell death occurs after treatment with N‐methyl‐N′‐nitro‐N‐nitrosoguanidine (MNNG) or massive oxidative stress. After MNNG treatment, NAD+ depletion and translocation of apoptosis‐inducing factor‐1 (AIF‐1) from mitochondria to nuclei is normally observed, but this type of cell death is lacking in PARP‐1 knockout cells resistant to oxidative stress,( 39 ) and streptozotocin‐induced pancreatic β‐cell death.( 40 ) PARP inhibitors enhanced development of streptozotocin‐induced pancreatic insulinomas in rats, suggesting that PARP‐1 dependent cell death is involved in the prevention of carcinogenesis.( 41 ) Because more than half of cancers feature mutation in the p53‐dependent apoptosis pathway, PARP‐1 dependent cell death could be a good target for cancer therapy.
PARG−/– mice lacking the 110 kDa isoform are hypersensitive to alkylating agents and γ‐irradiation, with reduced automodification activity of PARP‐1.( 42 ) PARG−/– ES cells also exhibit increased sensitivity to MMS treatment and γ‐irradiation, apoptosis occurring within a much shorter period than in PARG+/+ ES cells.( 43 ) This was linked to a marked increase of polyADP‐ribosylated proteins in nuclei and a reduction in NAD+ levels. The cytotoxicity of MNNG and menadione is increased in PARG−/– cells.( 44 ) These results suggest that PARG activity is involved in survival after DNA damage. The poly(ADP‐ribose) polymer itself induces cell‐death through induction of AIF release from mitochondria.( 45 ) This is consistent with a large accumulation of polyADP‐ribosylated compounds and neuronal cell death in PARG knockout Drosophila. ( 46 ) Thus, there is a possibility that Parg inhibitors might enhance chemo‐ or radiation therapy of cancers.
Chromosomal stability
It is well known that cancer cells are generally characterized by extensive genomic instability. Centrosome amplification is frequently observed and could be a cause of chromosomal missegregation between daughter cells.
A century ago, a hypothesis was proposed that malignant tumors arise through defects of centrosome functions that lead to improper cell divisions resulting in aneuploidy.( 3 ) Many reports have appeared documenting that certain post‐translational modifications, including phosphorylation and ubiquitylation, occur in centrosomes and regulate their function.( 3 ) Because PARP‐1, PARP‐3, PARP‐4 and PARP‐5, as well as polyADP‐ribosylated proteins and PARG, are found in the mitotic apparatus (Fig. 3), polyADP‐ribosylation might be involved in the regulation of fidelity of correct separation of chromosomes during mitosis.( 3 , 47 ) It is interesting to note that 3‐aminobenzamide (3‐AB), an inhibitor of PARP, seems to be not mutagenic (non‐genotoxic) to Salmonella, but does cause centrosome amplification in mammalian cells.( 47 ) They also may induce sister chromatid exchanges.( 5 ) These data suggest that non‐genotoxic compounds like PARP inhibitors might induce chromosomal instability, which could promote carcinogenesis.
Cellular transformation
PARP inhibitors like benzamide decrease in vitro transformation of human fibroblasts induced by various types of carcinogens, such as benzo[a]pyrene and MNNG.( 48 ) In contrast, transformation of mouse C3H10T1/2 cells by ethylnitrosourea (ENU) or ethylmethanesulfonate was elevated by PARP inhibitors.( 5 ) These effects are possibly related to transcriptional control by PARP family members. PARP‐10 was recently identified as a c‐myc interacting protein, suppressing cellular transformation induced by c‐MYC and E1A protein.( 49 ) PARP‐10 polyADP‐ribosylates itself, as well as core histones, and may be indirectly involved in the regulation of c‐myc.
Animal models of tumorigenesis
PARP‐1 −/– mice show increased susceptibility to carcinogenesis induced by alkylating agents, including BHP, and azoxymethane.( 2 , 50 ) In contrast, there is no such difference regarding carcinogenesis induced by a heterocyclic amine, IQ (2‐amino‐3‐methylimidazo [4,5‐f]quinoline) and 4‐nitroqinoline 1‐oxide (4NQO), both of which give rise to bulky DNA adducts.( 51 , 52 ) Therefore, there are carcinogen specific effects concerning the involvement of PARP‐1 in carcinogenesis. Alkylation damage to DNA bases may be repaired mainly by BER, while bulky DNA adducts induced by IQ and 4NQO may be targeted by nucleotide excision repair (NER). Susceptibility to carcinogenesis might thus be explained by the involvement of PARP‐1 in the repair pathway for BER, but not for NER.
In PARP‐1−/–p53−/– mice, an increased frequency of spontaneous development of carcinomas and lymphomas has been observed. Medulloblastomas also develop at the age of 16 weeks, accompanied by activation of the hedgehog pathway through overexpression of the GLI (Greig cephalopolysyndactyly syndrome) gene.( 53 ) It is notable that medulloblastomas are also observed in Ligase IV−/–p53−/– mice, which are defective in NHEJ repair.( 2 ) The combination of DNA‐PKc mutations (SCID mutations) with PARP‐1 deficiency has resulted in increased incidence of T‐cell lymphoma and partial recovery of V[D]J recombination in T cells.( 54 ) Ku80 heterozygous mutations with PARP‐1 homozygous mutations have caused enhanced frequency of hepatocellular carcinoma development in aged mice.( 55 ) In addition, WRNΔhe1 /Δhe1 PARP‐1 null mice show an increased incidence of spontaneous tumors.( 56 ) Furthermore, in aged PARP‐1−/– mice, the incidence of spontaneous development of hepatocellular tumors is increased.( 55 ) Recently, it was shown that vault‐associated PARP (VPARP) knockout mice exhibit elevated susceptibility to carcinogenesis induced by urethane in the lungs and by dimethylhydrazine in the colon.( 57 )
Human cancer
In human cancers, increased expression of the PARP‐1 gene has been reported in Ewing's sarcomas,( 2 ) and in malignant lymphomas.( 2 ) In contrast, decreased expression has been observed in several gastric and colon cancer cell lines,( 2 ) as well as in some breast cancers.( 58 )
Relations of genetic alterations in PARP‐1 gene with carcinogenesis have been reported by several authors. The heterologous Met129Thr mutation in the PARP‐1 gene has been reported in the germ cell tumor.( 59 ) The Val762Ala single nucleotide polymorphism (SNP) was found to impact prostate cancer in Caucasians, the Ala/Ala allele being associated with a two‐fold increase in susceptibility.( 60 ) The 762Ala variant showed decreased PARP activity and reduced interaction with XRCC1 compared with the 762Val variant. With esophageal and lung cancers, a two‐fold increase in risk with the Ala/Ala allele was observed in Chinese smokers.( 61 , 62 ) It is also noteworthy that a combination effect of the 762Ala allele of the PARP‐1 gene and the 399Gln allele of the XRCC‐1 gene has been reported. It should be noted that the 762Ala allele frequency is much higher in Asian compared to Caucasian populations. The relation of Val762Ala as well as Lys940Arg to the risk of lung cancer was investigated in Japan, but no associations were detected.( 63 ) Genetic differences in the population or variation in the profile of environmental exposure to carcinogens may have exerted an influence.
PARP‐9 contains two macroH2A domains that could repress transcription when localized sufficiently close to a promoter. This PARP was originally found as BAL1 (B‐Aggressive Lymphoma 1) and is expressed at significantly higher levels in fatal high‐risk diffuse large B‐cell lymphomas (DLBCL) than in curable low‐risk tumors.( 64 ) Increased PARP‐9 expression in DLBCL is associated with an activated peripheral B‐cell phenotype and high rates of tumor cell migration.
Inhibition of PARP and PARG for potentiation of anticancer drugs
Earlier work in the 1980s was focused on the effects of PARP inhibitors, benzamide and 3‐AB. 3‐AB was demonstrated to enhance the cytotoxic effect of dimethylsulfate,( 65 ) when used in combination with bleomycin as an anticancer drug in Ehrlich ascites mammary cancer cells.( 66 ) Recently a more potent inhibitor, AG14361 and the AG014669 derivative (Fig. 5), were developed in England and are now undergoing Phase II clinical trials with the DNA methylating anticancer drug, temozolomide, for malignant melanomas.( 67 ) The BRCA2 gene required for HR repair is mutated in some cancer cells. Inhibitors of PARP that stall DNA replication forks and cause DSB might thus be expected to kill such mutated cells.( 19 , 20 ) However, because the BRCA2 deficient human cell line, CAPAN‐1, was not found to be sensitive to PARP inhibition,( 68 ) further studies are still necessary to understand what factors affect the consequences of PARP inhibition. A specific inhibitor for PARP5 (tankyrase 1) affecting telomerase function is also suggested to be a potential telomere‐directed anticancer target.( 69 )
Figure 5.
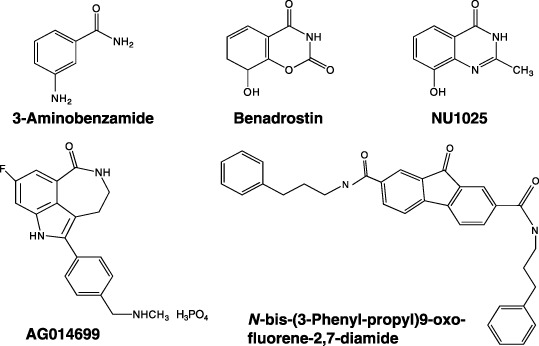
Developed poly(ADP‐ribose) polymerase (PARP) and poly(ADP‐ribose) glycohydrolase (PARG) inhibitors. Structures of PARP inhibitors, 3‐aminobenzamide,( 65 ) benadrostin,( 2 ) NU1025, AG014699,( 67 ) and PARG inhibitor N‐bis(3‐phenyl‐propyl)9‐oxo‐fluorene‐2,7‐diamide( 70 ) are shown.
So far, PARG inhibitors have not been intensively studied as anticancer agents. Nobotanin B, adenosine diphosphate (hydroxymethyl)‐pyrrolidinediol (ADP‐HPD), as well as its cell‐permeable derivative, 8‐octylamino‐ADP‐HPD, have been reported. Pargamicin was also recently reported.( 2 ) An approach to using a PARG inhibitor, N‐bis‐(3‐phenyl‐propyl)9‐oxo‐fluorene‐2,7‐diamide (Fig. 5), in combination with temozolomide to treat temozolomide‐resistant cancers, has been published.( 70 ) Therefore, PARG could be a new molecular target for cancer chemotherapy.
Chemoprevention
The substrate of PARP, NAD+, is synthesized using nicotinic acid mononucleotide by the kynurenic pathway starting with L‐tryptophan or using niacin (nicotinic acid, vitamin B3) supplied in the diet (Fig. 1). It has been reported that in rats maintained under a niacin‐deficient diet, the level of NAD+ in bone marrow is decreased, with even more extensive reduction in poly(ADP‐ribose).( 71 ) The animals were found to show greatly elevated susceptibility to ENU, particularly regarding induction of leukemias. In contrast, rats supplemented with niacin or nicotinamide in the diet had increased NAD+ and basal and ENU‐treated poly(ADP‐ribose) levels in bone marrow. ENU‐induced carcinogenesis was furthermore slowed.( 71 ) Niacin deficiency was also shown to enhance skin cancer development with preventive effects of supplementation. Tashtoush et al. showed that myristyl nicotinate, a derivative of nicotinic acid, can be used as a topical agent increasing NAD+ content by 40%.( 72 ) Because NAD+ is used in various physiological processes, the preventive effects of niacin could be due to a combinatory influence on ADP‐ribosylation enzymes and other NAD‐requiring reactions.
Concluding remarks
Carcinogenesis is multistage and may involve not only gene mutations but also abnormal dynamics of chromosomal organization, possibly also caused by non‐genotoxic factors. For a better understanding of the entirety of neoplasia, more needs to be learned from basic biological as well as clinical features. Research on polyADP‐ribosylation has progressed rapidly, as evidenced by the multitude of details available on the website, PARP link (http://parplink.u‐strasbg.fr/index.html). Considering the fact that many post‐translational modifications are actively involved in regulation of key reactions, interplay among processes like polyADP‐ribosylation, monoADP‐ribosylation, phosphorylation, acetylation, methylation, and ubiquitination of key proteins deserves greater attention. This research field might best be termed ‘Proteomodificomics (PMM)’. Progress of PMM in today's post‐genome era, with collaboration from various scientists and clinicians, should help establish new concepts for understanding the characteristics of clinical cancer that should lead to better diagnosis, treatment and prevention.
Acknowledgments
We thank Dr T. Sugimura, President Emeritus of the National Cancer Center, Tokyo, for continuous encouragement, and Dr O. Hayaishi, Professor Emeritus of Kyoto University, as well as Dr T. Takamura for their suggestions. Our appreciation is also extended to Drs S. Hanai, M. Kanai, K. Uchida, H. Ogino, A. Gunji, and A. Shibata, and many other collaborators, for their contributions to PARP research. Because of limitation in page length, we apologize that many of the references could not be directly cited. This work was supported in part by Grant‐in‐Aid for Cancer Research from the Ministry of Health, Labour and Welfare, and Grant‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan and Hishi‐no‐mi Grant‐in‐Aid for Cancer Research.
References
- 1. Sugimura T. Poly (adenosine diphosphate ribose). Prog Nucl Acid Res Mol Biol 1973; 13: 127–51. [DOI] [PubMed] [Google Scholar]
- 2. Masutani M, Nakagama H, Sugimura T. Poly(ADP‐ribosyl)ation in relation to cancer and autoimmune disease. Cell Mol Life Sci 2005; 62: 769–83. [DOI] [PubMed] [Google Scholar]
- 3. Miwa M, Kanai M, Uchida M, Uchida K, Hanai S. Roles of poly(ADP‐ribose) metabolism in the regulation of centrosome duplication and in the maintenance of neuronal integrity. In: Buerkle A, ed. Poly(ADP‐Ribosyl)ation. Georgetown, Texas: Landes Bioscience, 2006: 51–60 [Google Scholar]
- 4. Miwa M, Saikawa N, Yamaizumi Z, Nishimura S, Sugimura T. Structure of poly(adenosine diphosphate ribose): identification of 2′‐[1″‐ribosyl‐2″‐(or 3″‐) (1″′‐ribosyl) ]adenosine‐5′,5″,5″′‐tris (phosphate) as a branch linkage. Proc Natl Acad Sci USA 1979; 76: 595–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Althaus FR, Richter C. ADP‐ribosylation of proteins. Enzymology and biological significance. Mol Biol Biochem Biophys 1987; 37: 1–237. [PubMed] [Google Scholar]
- 6. Hayashi K, Tanaka M, Shimada T, Miwa M, Sugimura T. Size and shape of poly(ADP‐ribose): examination by gel filtration, gel electrophoresis and electron microscopy. Biochem Biophys Res Commun 1983; 112: 102–7. [DOI] [PubMed] [Google Scholar]
- 7. Schreiber V, Dantzer F, Ame JC, De Murcia G. Poly(ADP‐ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 2006; 7: 517–28. [DOI] [PubMed] [Google Scholar]
- 8. Kanai M, Uchida M, Hanai S, Uematsu N, Uchida K, Miwa M. Poly(ADP‐ribose) polymerase localizes to the centrosomes and chromosomes. Biochem Biophys Res Commun 2000; 278: 385–9. [DOI] [PubMed] [Google Scholar]
- 9. Ohashi S, Kanai M, Hanai S et al . Subcellular localization of poly(ADP‐ribose) glycohydrolase in mammalian cells. Biochem Biophys Res Commun 2003; 307: 915–21. [DOI] [PubMed] [Google Scholar]
- 10. Morrison AR, Moss J, Stevens LA et al . ART2, A T cell surface mono‐ADP‐ribosyltransferase, generates extracellular poly(ADP‐ribose). J Biol Chem 2006; 81: 33 363–72. [DOI] [PubMed] [Google Scholar]
- 11. Takamura‐Enya T, Watanabe M, Totsuka Y et al . Mono (ADP‐ribosyl)ation of 2′‐deoxyguanosine residue in DNA by an apoptosis‐inducing protein, pierisin‐1, from cabbage butterfly. Proc Natl Acad Sci USA 2001; 98: 12 414–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. El‐Khamisy SF, Masutani M, Suzuki H, Caldecott KW. A requirement for PARP‐1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucl Acids Res 2003; 31: 5526–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. De Murcia JM, Niedergang C, Trucco C et al . Requirement of poly(ADP‐ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci USA 1997; 94: 7303–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shibata A, Kamada N, Masumura K et al . Parp‐1 deficiency causes an increase of deletion mutations and insertions/rearrangements in vivo after treatment with an alkylating agent. Oncogene 2005; 24: 1328–37. [DOI] [PubMed] [Google Scholar]
- 15. Heale JT, Ball AR Jr, Schmiesing JA et al . Condensin I interacts with the PARP‐1‐XRCC1 complex and functions in DNA single‐strand break repair. Mol Cell 2006; 21: 837–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Malanga M, Althaus FR. Poly(ADP‐ribose) reactivates stalled DNA topoisomerase I and induces DNA strand break resealing. J Biol Chem 2004; 279: 5244–8. [DOI] [PubMed] [Google Scholar]
- 17. Von Kobbe C, Harrigan JA, May A et al . Central role for the Werner syndrome protein/poly(ADP‐ribose) polymerase 1 complex in the poly (ADP‐ribosyl) ation pathway after DNA damage. Mol Cell Biol 2003; 23: 8601–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hochegger H, Dejsuphong D, Fukushima T et al . Parp‐1 protects homologous recombination from interference by Ku and ligase IV in vertebrate cells. Embo J 2006; 25: 1305–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bryant HE, Schultz N, Thomas HD et al . Specific killing of BRCA2‐deficient tumours with inhibitors of poly(ADP‐ribose) polymerase. Nature 2005; 434: 913–17. [DOI] [PubMed] [Google Scholar]
- 20. Farmer H, McCabe N, Lord CJ et al . Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434: 917–21. [DOI] [PubMed] [Google Scholar]
- 21. Bryant HE, Helleday T. Inhibition of poly(ADP‐ribose) polymerase activates ATM which is required for subsequent homologous recombination repair. Nucl Acids Res 2006; 34: 1685–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Menissier de Murcia J, Ricoul M, Tartier L et al . Functional interaction between PARP‐1 and PARP‐2 in chromosome stability and embryonic development in mouse. Embo J 2003; 22: 2255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oei SL, Ziegler M. ATP for the DNA ligation step in base excision repair is generated from poly(ADP‐ribose). J Biol Chem 2000; 275: 23 234–9. [DOI] [PubMed] [Google Scholar]
- 24. Ame JC, De Murcia G. Regulation of PARG recruitment at DNA damage sites. Med Sci 2005; 11 (Suppl 1): 41. [Google Scholar]
- 25. Hassa PO, Haenni SS, Buerki C et al . Acetylation of poly(ADP‐ribose) polymerase‐1 by p300/CREB‐binding protein regulates coactivation of NF‐kappaB‐dependent transcription. J Biol Chem 2005; 280: 40 450–64. [DOI] [PubMed] [Google Scholar]
- 26. Pavri R, Lewis B, Kim TK et al . PARP‐1 determines specificity in a retinoid signaling pathway via direct modulation of mediator. Mol Cell 2005; 18: 83–96. [DOI] [PubMed] [Google Scholar]
- 27. Idogawa M, Masutani M, Shitashige M et al . Ku70 and poly(ADP‐Ribose) Polymerase‐1 competitively regulate {beta}‐catenin and T‐cell factor‐4‐mediated gene transactivation: possible linkage of DNA damage recognition and Wnt signaling. Cancer Res 2007; 67: 911–18. [DOI] [PubMed] [Google Scholar]
- 28. Kim MY, Mauro S, Gevry N, Lis JT, Kraus WL. NAD+‐dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP‐1. Cell 2004; 119: 803–14. [DOI] [PubMed] [Google Scholar]
- 29. Ju BG, Lunyak VV, Perissi V et al . A topoisomerase IIbeta‐mediated dsDNA break required for regulated transcription. Science 2006; 312: 1798–802. [DOI] [PubMed] [Google Scholar]
- 30. Tulin A, Spradling A. Chromatin loosening by poly(ADP)‐ribose polymerase (PARP) at Drosophila puff loci. Science 2003; 299: 560–2. [DOI] [PubMed] [Google Scholar]
- 31. Zardo G, Caiafa P. The unmethylated state of CpG islands in mouse fibroblasts depends on the poly(ADP‐ribosyl)ation process. J Biol Chem 1998; 273: 16 517–20. [DOI] [PubMed] [Google Scholar]
- 32. Pleschke JM, Kleczkowska HE, Strohm M, Althaus FR. Poly(ADP‐ribose) binds to specific domains in DNA damage checkpoint proteins. J Biol Chem 2000; 275: 40 974–80. [DOI] [PubMed] [Google Scholar]
- 33. Yu W, Ginjala V, Pant V et al . Poly(ADP‐ribosyl)ation regulates CTCF‐dependent chromatin insulation. Nat Genet 2004; 36: 1105–10. [DOI] [PubMed] [Google Scholar]
- 34. Karras GI, Kustatscher G, Buhecha HR et al . The macro domain is an ADP‐ribose binding module. Embo J 2005; 24: 1911–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shima H, Nakayasu M, Aonuma S, Sugimura T, Nagao M. Loss of the MYC gene amplified in human HL‐60 cells after treatment with inhibitors of poly (ADP‐ribose) polymerase or with dimethyl sulfoxide. Proc Natl Acad Sci USA 1989; 86: 7442–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nozaki T, Masutani M, Watanabe M et al . Syncytiotrophoblastic giant cells in teratocarcinoma‐like tumors derived from Parp‐disrupted mouse embryonic stem cells. Proc Natl Acad Sci USA 1999; 96: 13 345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nozaki T, Masutani M, Akagawa T, Sugimura T, Esumi H. Suppression of G1 arrest and enhancement of G2 arrest by inhibitors of poly(ADP‐ribose) polymerase: possible involvement of poly(ADP‐ribosyl)ation in cell cycle arrest following gamma‐irradiation. Jpn J Cancer Res 1994; 85: 1094–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Watanabe F, Fukazawa H, Masutani M et al . Poly(ADP‐ribose) polymerase‐1 inhibits ATM kinase activity in DNA damage response. Biochem Biophys Res Commun 2004; 319: 596–602. [DOI] [PubMed] [Google Scholar]
- 39. Yu SW, Wang H, Poitras MF et al . Mediation of poly(ADP‐ribose) polymerase‐1‐dependent cell death by apoptosis‐inducing factor. Science 2002; 297: 259–63. [DOI] [PubMed] [Google Scholar]
- 40. Masutani M, Suzuki H, Kamada N et al . Poly(ADP‐ribose) polymerase gene disruption conferred mice resistant to streptozotocin‐induced diabetes. Proc Natl Acad Sci USA 1999; 96: 2301–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yamagami T, Miwa A, Takasawa S, Yamamoto H, Okamoto H. Induction of rat pancreatic B‐cell tumors by the combined administration of streptozotocin or alloxan and poly(adenosine diphosphate ribose) synthetase inhibitors. Cancer Res 1985; 45: 1845–9. [PubMed] [Google Scholar]
- 42. Cortes U, Tong WM, Coyle DL et al . Depletion of the 110‐kilodalton isoform of poly(ADP‐ribose) glycohydrolase increases sensitivity to genotoxic and endotoxic stress in mice. Mol Cell Biol 2004; 24: 7163–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Masutani M, Gunji A, Ogino H et al . Functional analysis of poly (ADP‐ribose) glycohydrolase: Hypersensitivity to DNA damaging agents and spontaneous development of renal lesions under Parg‐deficiency. Med Sci 2005; 11 (Suppl 1): 22. [Google Scholar]
- 44. Koh DW, Dawson TM, Dawson VL. Mediation of cell death by poly (ADP‐ribose) polymerase‐1. Pharmacol Res 2005; 52: 5–14. [DOI] [PubMed] [Google Scholar]
- 45. Andrabi SA, Kim NS, Yu SW et al . Poly(ADP‐ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci USA 2006; 103: 18 308–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hanai S, Kanai M, Ohashi S et al . Loss of poly(ADP‐ribose) glycohydrolase causes progressive neurodegeneration in Drosophila melanogaster . Proc Natl Acad Sci USA 2004; 101: 82–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kanai M, Tong WM, Sugihara E, Wang ZQ, Fukasawa K, Miwa M. Involvement of poly(ADP‐Ribose) polymerase 1 and poly (ADP‐Ribosyl)ation in regulation of centrosome function. Mol Cell Biol 2003; 23: 2451–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kun E, Kirsten E, Milo GE, Kurian P, Kumari HL. Cell cycle‐dependent intervention by benzamide of carcinogen‐induced neoplastic transformation and in vitro poly(ADP‐ribosyl)ation of nuclear proteins in human fibroblasts. Proc Natl Acad Sci USA 1983; 80: 7219–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yu M, Schreek S, Cerni C et al . PARP‐10, a novel Myc‐interacting protein with poly(ADP‐ribose) polymerase activity, inhibits transformation. Oncogene 2005; 24: 1982–93. [DOI] [PubMed] [Google Scholar]
- 50. Tsutsumi M, Masutani M, Nozaki T et al . Increased susceptibility of poly (ADP‐ribose) polymerase‐1 knockout mice to nitrosamine carcinogenicity. Carcinogenesis 2001; 22: 1–3. [DOI] [PubMed] [Google Scholar]
- 51. Ogawa K, Masutani M, Kato K et al . Parp‐1 deficiency does not enhance liver carcinogenesis induced by 2‐amino‐3‐methylimidazo[4,5‐f]quinoline in mice. Cancer Lett 2006; 236: 32–8. [DOI] [PubMed] [Google Scholar]
- 52. Gunji A, Uemura A, Tsutsumi M et al . Parp‐1 deficiency does not increase the frequency of tumors in the oral cavity and esophagus of ICR/129Sv mice by 4‐nitroquinoline 1‐oxide, a carcinogen producing bulky adducts. Cancer Lett 2006; 241: 87–92. [DOI] [PubMed] [Google Scholar]
- 53. Tong WM, Ohgaki H, Huang H, Granier C, Kleihues P, Wang ZQ. Null mutation of DNA strand break‐binding molecule poly(ADP‐ribose) polymerase causes medulloblastomas in p53(‐/‐) mice. Am J Pathol 2003; 162: 343–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Morrison C, Smith GC, Stingl L, Jackson SP, Wagner EF, Wang ZQ. Genetic interaction between PARP and DNA‐PK in V (D) J recombination and tumorigenesis. Nat Genet 1997; 17: 479–82. [DOI] [PubMed] [Google Scholar]
- 55. Tong WM, Cortes U, Hande MP et al . Synergistic role of Ku80 and poly (ADP‐ribose) polymerase in suppressing chromosomal aberrations and liver cancer formation. Cancer Res 2002; 62: 6990–6. [PubMed] [Google Scholar]
- 56. Lebel M, Lavoie J, Gaudreault I, Bronsard M, Drouin R. Genetic cooperation between the Werner syndrome protein and poly(ADP‐ribose) polymerase‐1 in preventing chromatid breaks, complex chromosomal rearrangements, and cancer in mice. Am J Pathol 2003; 162: 1559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Raval‐Fernandes S, Kickhoefer VA, Kitchen C, Rome LH. Increased susceptibility of vault poly(ADP‐ribose) polymerase‐deficient mice to carcinogen‐induced tumorigenesis. Cancer Res 2005; 65: 8846–52. [DOI] [PubMed] [Google Scholar]
- 58. Bieche I, De Murcia G, Lidereau R. Poly(ADP‐ribose) polymerase gene expression status and genomic instability in human breast cancer. Clin Cancer Res 1996; 2: 1163–7. [PubMed] [Google Scholar]
- 59. Shiokawa M, Masutani M, Fujihara H et al . Genetic alteration of poly(ADP‐ribose) polymerase‐1 in human germ cell tumors. Jpn J Clin Oncol 2005; 35: 97–102. [DOI] [PubMed] [Google Scholar]
- 60. Lockett KL, Hall MC, Xu J et al . The ADPRT V762A genetic variant contributes to prostate cancer susceptibility and deficient enzyme function. Cancer Res 2004; 64: 6344–8. [DOI] [PubMed] [Google Scholar]
- 61. Hao B, Wang H, Zhou K et al . Identification of genetic variants in base excision repair pathway and their associations with risk of esophageal squamous cell carcinoma. Cancer Res 2004; 64: 4378–84. [DOI] [PubMed] [Google Scholar]
- 62. Zhang X, Miao X, Liang G et al . Polymorphisms in DNA base excision repair genes ADPRT and XRCC1 and risk of lung cancer. Cancer Res 2005; 65: 722–6. [PubMed] [Google Scholar]
- 63. Sakiyama T, Kohno T, Mimaki S et al . Association of amino acid substitution polymorphisms in DNA repair genes TP53, POLI, REV1 and LIG4 with lung cancer risk. Int J Cancer 2005; 114: 730–7. [DOI] [PubMed] [Google Scholar]
- 64. Aguiar RC, Yakushijin Y, Kharbanda S, Salgia R, Fletcher JA, Shipp MA. BAL is a novel risk‐related gene in diffuse large B‐cell lymphomas that enhances cellular migration. Blood 2000; 96: 4328–34. [PubMed] [Google Scholar]
- 65. Durkacz BW, Omidiji O, Gray DA, Shall S. (ADP‐ribose)n participates in DNA excision repair. Nature 1980; 283: 593–6. [DOI] [PubMed] [Google Scholar]
- 66. Kawamitsu H, Miwa M, Tanaka Y et al . Inhibitors of poly(adenosine diphosphate ribose) polymerase potentiate the antitumor activity of bleomycin against Ehrlich ascites carcinoma. J Pharmacobiodyn 1982; 5: 900–4. [DOI] [PubMed] [Google Scholar]
- 67. Calabrese CR, Almassy R, Barton S et al . Anticancer chemosensitization and radiosensitization by the novel poly(ADP‐ribose) polymerase‐1 inhibitor AG14361. J Natl Cancer Inst 2004; 96: 56–67. [DOI] [PubMed] [Google Scholar]
- 68. Gallmeier E, Kern SE. Absence of specific cell killing of the BRCA2‐deficient human cancer cell line CAPAN1 by poly(ADP‐ribose) polymerase inhibition. Cancer Biol Ther 2005; 4: 703–6. [DOI] [PubMed] [Google Scholar]
- 69. Seimiya H, Muramatsu Y, Ohishi T, Tsuruo T. Tankyrase 1 as a target for telomere‐directed molecular cancer therapeutics. Cancer Cell 2005; 7: 25–37. [DOI] [PubMed] [Google Scholar]
- 70. Tentori L, Leonetti C, Scarsella M et al . Poly(ADP‐ribose) glycohydrolase inhibitor as chemosensitiser of malignant melanoma for temozolomide. Eur J Cancer 2005; 41: 2948–57. [DOI] [PubMed] [Google Scholar]
- 71. Kirkland JB. Niacin and carcinogenesis. Nutr Cancer 2003; 46: 110–18. [DOI] [PubMed] [Google Scholar]
- 72. Tashtoush BM, Qasem J, Williams JD, Dewald TP, Jacobson EL, Jacobson MK. Analysis and stability study of myristyl nicotinate in dermatological preparations by high‐performance liquid chromatography. J Pharm Biomed Anal 2007; 43: 893–9. [DOI] [PubMed] [Google Scholar]