

Abstract
It has been thought that there is a strong relationship between inflammation and carcinogenesis so that the development of cancer occurs with chronic inflammation in many organs. An in‐depth understanding of the mechanism by which inflammation can lead to carcinogenesis may enable the development of drugs targeted at important molecules, providing a powerful tool for preventing cancer development. In this review, we focused on a signal transduction system, the nuclear factor‐kappaB (NF‐κB) pathway, which is thought to play a role in the process leading from inflammation to carcinogenesis, and may thus serve as a candidate for targeted intervention. (Cancer Sci 2008; 99: 836–842)
Abbreviations:
- DSS
dextran sulfate sodium salt
- IKK
IkappaB kinase
- IL
interleukin
- IBD
inflammatory bowel disease
- LPS
lipopolysaccharide
- NF‐κB
nuclear factor‐kappaB
- TNBS
trinitrobenzene sulfonic acid
- TNF
tumor necrosis factor.
The development of cancer in various organs is often associated with chronic inflammation, suggesting a strong relationship between inflammation and carcinogenesis.( 1 ) Recent investigations have shed light on the relationship between inflammation and the carcinogenic process, especially the promotion of carcinogenesis.( 2 ) Abnormal cellular alterations accompanying inflammation, such as oxidative stress, gene mutations, epigenetic changes, and inflammatory cytokine‐induced cell proliferation, have been proposed as carcinogenic factors. The signal transduction pathways for carcinogenesis are also being clarified.( 2 ) An in‐depth understanding of the mechanism by which inflammation can lead to carcinogenesis may enable the development of drugs targeted at important molecules in the process, potentially providing a powerful tool for preventing cancer development.
A summary of the cancers linked to chronic inflammation is presented in Table 1. This association has been described particularly in the digestive system, where the risk for carcinogenesis increases in the presence of chronic conditions such as esophagitis, gastritis, colitis, pancreatitis, and hepatitis. Of particular interest are gastritis and hepatitis, which are chronic inflammations caused, respectively, by Helicobacter pylori and hepatitis virus (HBV, hepatitis B virus; HCV, hepatitis C virus) infections.( 3 , 4 ) The rates of stomach and liver cancer have been shown to decrease after the cause of the infection was eliminated.( 5 , 6 ) A clear definition of the etiology of chronic inflammation should provide clues to the mechanism of carcinogenesis.
Table 1.
Cancers linked to chronic inflammation
| Infection‐related Condition | Tumor | Etiology |
|---|---|---|
| Tuberculosis | Lung cancer | Mycobacterium tuberculosis |
| Chronic gastritis | Gastric cancer | Helicobacter pylori |
| Chronic hepatitis | Hepatocellular carcinoma | Hepatitis B, C virus |
| Mononucleosis | Lymphoma | Epstein‐Barr virus |
| Chronic cervitis | Cervical cancer | Papilloma virus |
| Infection‐unrelated Condition | Tumor | Etiology |
|---|---|---|
| Asbestosis | Lung cancer, mesothelioma | Asbestos |
| Reflux esophagitis | Esophageal cancer | Gastric acid |
| Chronic pancreatitis | Pancreatic cancer | Alcohol |
| Inflammatory bowel disease | Colon cancer | Ulcerative colitis, Crohn's disease |
| Skin inflammation | Melanoma | UV |
In this review, we focused on a signal transduction system, the NF‐κB pathway, which is thought to play a role in the process leading from inflammation to carcinogenesis, and may thus serve as a candidate for targeted intervention.
The NF‐κB signaling pathway
NF‐κB transcription factors are critical regulators of innate immune responses and inflammation.( 7 ) They are assembled by dimerization of two of five subunits: p65 (RelA), c‐Rel, RelB, p50/NF‐κB1, and p52/NF‐κB2.( 8 ) In the absence of stimuli, most NF‐κB dimers are bound to specific inhibitory proteins, IκBs, in the cytoplasm. Many proinflammatory stimuli can activate NF‐κB, mainly through IKK‐dependent phosphorylation and degradation of the IκB inhibitory proteins. Stimulation activates the IKK complex, which consists of two catalytic subunits, IKKα and IKKβ, and a regulatory component, IKKγ/NEMO. For most stimuli, IKK activation occurs primarily through IKKβ,( 9 ) whose absence increases susceptibility to TNF‐α‐mediated apoptosis.( 10 ) The IKK‐dependent phosphorylation of IκBs marks them for ubiquitination and subsequent degradation. NF‐κB, now free of IκB, translocates to the nucleus, where it activates the transcription of target genes, including cytokines, chemokines, and antiapoptotic factors.( 9 ) This pathway is the classical NF‐κB signaling pathway and is one of two major pathways that activate NF‐κB transcription factors (Fig. 1).
Figure 1.
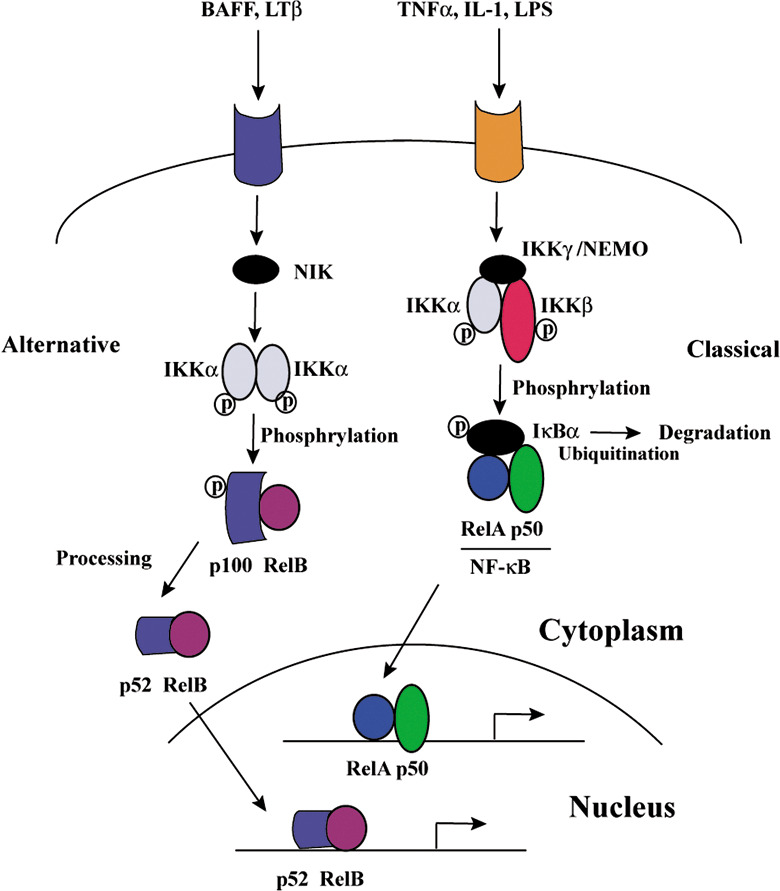
Nuclear factor‐kappaB (NF‐κB) signaling pathways. BAFF, B cell activating factor belonging to the TNF family; IKK, I kappa B kinase; IL, interleukin; LPS, lipopolysaccharide; LT, lymphotoxin; NIK, NF‐κB‐inducing kinase; TNF, tumor necrosis factor.
NF‐κB‐dependent gene expression and apoptosis play crucial roles in numerous cellular processes, and defects in their regulation may contribute to a variety of diseases. In addition to recent tremendous progress in understanding the signaling pathways that lead to NF‐κB activation, signaling mechanisms that negatively regulate these processes are also becoming clear. These molecules include A20, CYLD, and so on. The zinc finger protein A20 has been characterized as a dual inhibitor of NF‐κB activation and apoptosis. Its expression is inducible by a wide variety of stimuli, including cytokines such as TNF‐α and IL‐1. A20‐deficient cells fail to terminate TNF‐induced NF‐κB responses and is critical for limiting inflammation by terminating TNF‐induced NF‐κB responses in vivo.( 11 ) CYLD is a tumor suppressor gene found in cylindromatosis, a disease associated with numerous benign skin tumors. It is reported that CYLD is capable of deubiquitinating the K63 polyubiquitin chain on TRAF2 and IKKγ by direct interaction, leading to the inhibition of NF‐κB activation.( 12 )
Recently, it was reported that the second, or alternative, pathway of NF‐κB activation contributes to the development, survival, and attenuation of apoptosis of B cells. The alternative NF‐κB pathway involves the processing and cleavage of the NF‐κB2/p100 precursor to p52, which are triggered by the phosphorylation of p100 by NF‐κB‐inducing kinase (NIK) and IKKα.( 13 ) This pathway can be activated by stimulation of the lymphotoxin (LT)‐β receptor, the BAFF (B cell activating factor belonging to the TNF family) receptor, or CD40, but not the TNF‐α receptor.( 14 , 15 , 16 ) Then, the p52 molecule forms a heterodimer with another NF‐κB subunit, translocates to the nucleus, and binds to the NF‐κB sites on DNA to induce target gene transcription (Fig. 1). The activation of the alternative NF‐κB pathway and subsequent up‐regulation of target genes is reportedly necessary for the secondary development of lymphoid organs such as the spleen, lymph nodes, and Peyer's patches.( 17 )
NF‐κB activation and carcinogenesis
In the classic chemical carcinogenic model, the carcinogenic process is divided into initiation, promotion, and progression. In reality, however, carcinogenesis does not fit neatly into these three stages, and many mutations, gene amplifications, and epigenetic changes must occur for a normal cell to become a cancer cell. However, in the present report, the carcinogenic process is discussed according to the chemical carcinogenic model (Fig. 2) because it serves as a framework for analyzing the relationship between NF‐κB and carcinogenesis.
Figure 2.
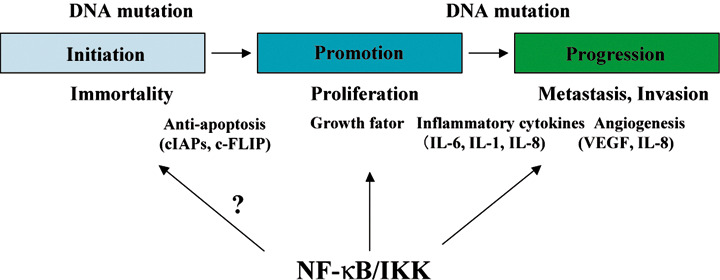
Participation of the nuclear factor‐kappaB (NF‐κB) activation in carcinogenesis. NF‐κB activation participates in many processes in carcinogenesis by the expression of various factors controlling inflammation or growth factors. IKK, I kappa B kinase; IL, interleukin; VEGF, vascular endothelial growth factor.
The initiation of carcinogenesis, which conveys immortality to the cell, is precipitated by DNA mutation; NF‐κB is not thought to have a major role in this step of the process. The first clue linking NF‐κB to cancer was the realization that c‐rel, which is the cellular homolog of the v‐rel oncogene, encodes a NF‐κB subunit and that all of these proteins share the same DNA binding domain, the Rel homology domain.( 18 ) Not surprisingly, overexpression of normal Rel proteins promotes oncogenic transformation.
It is possible that reactive oxygen species (ROS) and nitro compounds produced in chronic inflammation may cause DNA mutations.( 19 ) ROS and nitro compounds are produced by activated neutrophils and macrophages that have infiltrated the inflammatory zone, where these molecules may act on cellular DNA during cell division to induce DNA mutations. Many dividing cells are present in chronic inflammation as a result of repeated cellular organization, destruction, and proliferation under this condition, making inflammatory cells very susceptible to DNA mutations. For example, mutation of the p53 gene has been detected in tissues of chronic rheumatoid arthritis and inflammatory bowel disease, and mutation of the K‐ras gene has been identified in pancreatic tissue during chronic pancreatitis.( 20 , 21 ) These observations indirectly suggest that due to its role as a transcriptional factor for the induction of inflammation, NF‐κB may participate in tumor initiation.
The evidence for the participation of activated NF‐κB in the carcinogenic promotion and progression stages, however, has become clearer in many recent studies. The promotion stage represents the proliferative stage of the immortal cancer cell, and progression is determined by proliferation, antiapoptosis, angiogenesis, invasion, and metastasis.( 22 ) Further mutations can occur during these two stages, and the additional mutations, as well as the initiating mutation, are carried by the proliferating cells. It is thought that several factors controlled by NF‐κB activation participate in these processes (Fig. 2 and Table 2).
Table 2.
Nuclear factor‐kappaB (NF‐κB) regulated factors that may be related to carcinogenesis
| Inflammatory cytokine | IL‐1β, IL‐6, TNF‐α |
| Chemokine | IL‐8, MCP‐1 |
| Anti‐apoptosis | cIAPs, c‐FLIP, A20, BclXL |
| Angiogenesis | VEGF, IL‐8 |
| Invasion, metastasis | MMP‐2, MMP‐9 |
IL, interleukin; MMP, matrix metalloproteinase; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor.
Activated inflammatory cells and fibroblasts accumulate in tumor tissue, where they produce growth factors such as epidermal growth factor (EGF) and fibroblast growth factor (FGF), and tumor cells proliferate in response to the binding of growth factors to receptors on tumor cells.( 23 ) Growth factors bind to and activate receptor tyrosine kinases on the cells, and the phosphorylated receptors transmit signals through several different phosphorylation cascades, including the mitogen‐activated protein (MAP) kinases.( 24 ) Although few reports suggest the strong participation of activated NF‐κB in the transcriptional regulation of growth factors for the activation and infiltration of inflammatory cells, the inhibition of NF‐κB activation may reduce the expression of these growth factors. The expression of TNF‐α mainly by macrophages and lymphocytes during inflammation is regulated by NF‐κB, and TNF‐α is a strong regulator of NF‐κB activation. In addition to its central role in inflammation, TNF‐α has been suggested to also concurrently function as an accelerating factor for cell proliferation.( 25 ) In addition, the activation of NF‐κB may affect cell growth through the induction of cyclin expression.( 26 )
The process of antiapoptosis is vital for the maintenance of cancer cells. The inflammatory cytokines TNF‐α and IL‐1, which are produced in chronic inflammation, are strong NF‐κB‐activating factors, and a large number of antiapoptotic factors, including cIAPs, c‐FLIP, A20, and BclXL, are controlled by NF‐κB activation.( 7 ) The production of important antiapoptotic proteins has been demonstrated to occur during carcinogenic promotion.( 27 )
NF‐κB activation inhibits apoptosis as above. However, under certain circumstances NF‐κB activation may promote cell death. For instance, NF‐κB may mediate doxorubicin‐induced cell death in certain type of neuroblastoma cells.( 28 ) Human melanoma cells were protected from UV‐induced apoptosis by NF‐κB down‐regulation.( 29 ) These results suggest that NF‐κB may mediate apoptosis under certain conditions. However, it remains to be demonstrated that NF‐κB has pro‐apoptotic functions in vivo.
The expression of several angiogenic factors is also regulated by NF‐κB. Macrophages as well as tumor cells have been reported to produce vascular endothelial growth factor (VEGF) under the control of NF‐κB activation,( 30 ) and VEGF promotes the proliferation and migration of endothelial cells. The expression of the chemokine IL‐8 by leukocytes during inflammation is regulated by NF‐κB, and IL‐8 functions as a blood vessel growth factor in tumor tissue.( 31 , 32 )
Cancer invasion and metastasis strongly influence a patient's prognosis, and changes in the extracellular matrix as a result of inflammation are critical for cancer cell invasion and metastasis. Matrix metalloproteinases (MMP), which are produced by inflammatory cells and tumor cells, are key players in the degradation of extracellular matrix and basement membranes, and thus are important in tumor invasion. The gelatinases (MMP‐2 and MMP‐9) in particular are prognostic factors in many solid tumors, and their expression is regulated by NF‐κB activation. The clinical application of a MMP inhibitor aimed at preventing metastasis is expected.( 33 ) Recently, IKKα activation was reported to be correlated with a decreased expression of the metastasis suppressor Maspin in prostate cancer metastasis; the alternative NF‐κB signaling pathway was shown to activate IKKα, thus reducing Maspin expression and stimulating metastatic activity.( 34 )
The alternative pathway of NF‐κB activation has been studied in recent years. However, in a few studies the relationship between the alternative pathway and carcinogenesis has been reported. By the analysis of constitutive NF‐κB signaling in multiple myeloma, it was found that mutations causing the inactivation of TRAF3 and NIK result in constitutive activation of the alternative NF‐κB pathway.( 35 ) In addition, the pathway also might be involved in mammary carcinogenesis.( 36 )
The tumor microenvironment and NF‐κB activation
The infiltration of inflammatory cells, primarily macrophages, has been observed in tumors, suggesting that the invading macrophages have a role in the development of neoplasia. Macrophages that infiltrate a tumor locus are named tumor‐associated macrophages (TAMs). TAMs secrete various cytokines and are related to tumor development.( 37 ) With regard to the mechanism of infiltration, cancer cells themselves may bring about inflammatory cell infiltration. Recently, it was reported that NF‐κB is activated by a mutation of the ras gene located downstream of the tyrosine kinase‐type receptor, resulting in increased expression of IL‐8; this may also result in an increased number of tumor vessels.( 38 ) Mutation of the ras gene is believed to occur in 25% of tumors, and if the NF‐κB pathway is actually involved in this way, it may become the target of medical treatment. We have observed the constant expression of IL‐8 in about 25–50% of stomach and colon cancer cell lines (K. Sakamoto and S. Maeda, unpublished observation). In addition, the expression of the chemokine MCP‐1 in neoplastic cells has been reported, and it was proposed that MCP‐1, like IL‐8, participates in inflammatory cell infiltration and increased tumor size.( 39 )
Inflammatory cell infiltration can also be induced by cells in a necrotic or hypoxic state. It has been reported that ROS accumulation, NF‐κB activation, and IL‐8 expression all occur in hypoxic cells and that these contribute to increasing the tumor size.( 40 ) In addition, it has also been suggested that HMGB1, a factor secreted from necrotic cells, activates NF‐κB in neighboring cells at the beginning of liver and colon cancer development, exacerbating inflammation and promoting liver and colon cancer development.( 41 , 42 , 43 ) These observations indicate that necrotizing a tumor without removing the necrotic neoplastic cells may result in the increased survival of the remaining neoplastic cells and actually further the malignancy.
Analysis of NF‐κB activation in an in vivo cancer model
Helicobacter‐related stomach cancer. Helicobacter‐associated stomach cancer is related to the presence of chronic active gastritis. Upon H. pylori infection, gastritis usually develops in the body and pylorus part of the stomach. In a patient with chronic gastritis, atrophic changes of the gastric glands gradually spread over time as a result of inflammation.( 44 ) This change is important for gastric cancer development, and thus eradicating H. pylori or preventing its infection may reduce the incidence of gastric cancer. It was reported that a protein isolated from the bacterial body of H. pylori stimulates macrophages in the epithelium and promotes the secretion of inflammatory cytokines such as TNF‐α or IL‐1.( 45 ) Furthermore, contact between the live bacteria and epithelial cells stimulates the cells to produce abundant IL‐8, which contributes significantly to the inflammatory reaction. The cag pathogenicity island (PAI), which contains more than 30 genes, is believed to be a virulence factor of H. pylori. The presence of the PAI was shown to affect the degree of the inflammatory reaction produced, such that the inflammatory response in gastric mucosa infected with a cag PAI‐negative strain of H. pylori was less than that in gastric mucosa infected with a cag PAI‐positive strain.( 46 ) Some proteins encoded in the PAI are homologs of other bacterial proteins related to the secretion mechanism, suggesting that the PAI proteins participate in secretion. Of particular interest is that a region of the cag PAI is essential for NF‐κB activation and IL‐8 production based on experiments in which mutations were inserted into some cag PAI genes. We have previously reported that the cag PAI of H. pylori encodes a cell type‐specific NF‐κB activator and that IKK, TAK1, TRAF6, and MyD88, which are signaling molecules for IL‐1 or TLRs, are important signal transducers in H. pylori‐infected gastric epithelial cells.( 47 , 48 ) Moreover, Nod1, an intracellular pathogen‐recognition molecule with specificity for Gram‐negative peptidoglycan, has been shown to respond to peptidoglycan delivered by the H. pylori cag PAI.( 49 ) About 50–60% of the H. pylori strains in North American and European countries are cag PAI‐positive, whereas about 95% of the strains in Asian countries are positive, which probably contributes to the high rate of stomach cancer in Asia.( 50 ) In an in vivo experiment in the Mongolian gerbil, infection with bacteria lacking the ability to activate NF‐κB resulted in a milder degree of chronic gastritis than that resulting from wild‐type infection and produced no atrophic gastritis (Fig. 3a).( 51 ) As further support, we recently showed that a NF‐κB inhibitor controlled the degree of gastritis caused by H. pylori infection.( 52 ) These observations suggest that NF‐κB activation is important for the progression from chronic inflammation to carcinogenesis in a Helicobacter infectious disease.
Figure 3.
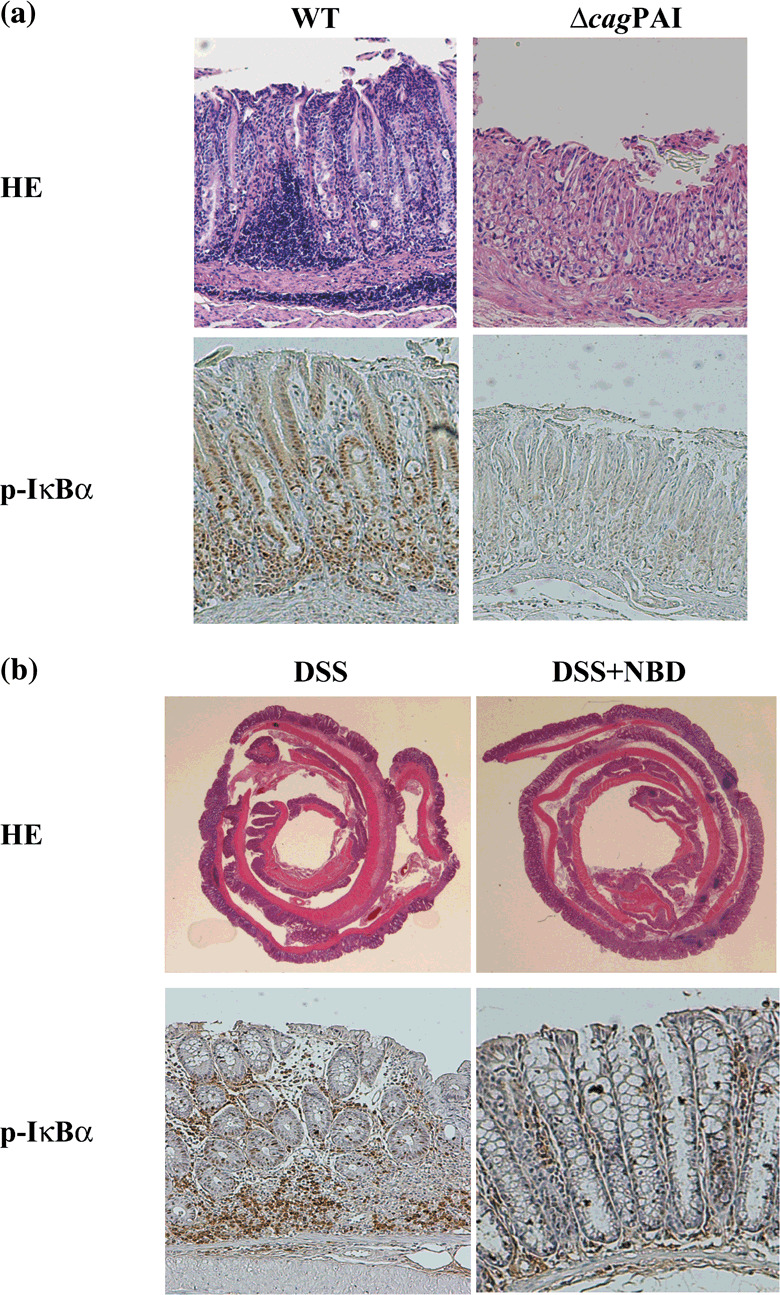
Participation of the nuclear factor‐kappaB (NF‐κB) activation in the Helicobacter pylori (H. pylori) infection and dextran sulfate sodium salt (DSS) colitis models. (a) Hematoxylin–eosin (HE) and phospho‐IκBα stainings of the antrum part 25 weeks after H. pylori infection are shown. By the infection of cag PAI‐negative H. pylori (Δcag) without the activation ability of NF‐κB, degree of the chronic gastritis and phospho‐IκBα staining are extremely slight in comparison with the infection of the wild‐type (WT) (magnification 40 ×). (b) Hematoxylin–eosin (HE) and phospho‐IκBα stainings of the colonic part 10 days after DSS administration are shown. By the treatment of NF‐κB essential modulator (NEMO)‐binding domain (NBD) peptides which inhibit NF‐κB activity, degree of the colitis and phospho‐IκBα staining are extremely slight compared with the absence of NBD peptides (magnification 10 × in HE and 40 × in phospho‐IκBα staining).
Colitis and colitis‐associated cancer. As described above, the mutation of a bacterial factor was used to analyze the role of NF‐κB in the relationship between inflammation and carcinogenesis in a model of stomach cancer. However, a similar approach has not been possible for studying carcinogenesis in other internal organs. The knockout of a major NF‐κB‐related factor has only recently been achieved in a mouse because knocking out RelA or IKKβ, which are critical molecules for NF‐κB activation, is fatal in mice. Thus, a conditional‐knockout mouse was produced by crossbreeding a transgenic mouse specifically expressing Cre recombinase with a mouse carrying a Cre‐loxP site in the ikkβ gene; an analysis was performed recently using the conditional‐knockout mouse. Colitis‐associated cancer (CAC) occurs in patients suffering from inflammatory bowel diseases such as ulcerative colitis and Crohn's disease. In mice, CAC can be induced by the injection of the carcinogen azoxymethane (AOM). After AOM injection, DSS is added to the drinking water of the mouse to produce inflammation at the tumor site, providing a model of inflammation and carcinogenesis. The conditional inactivation of the ikkβ gene within the enterocytes resulted in a marked decrease in the incidence of CAC but not tumor size. An analysis of the enterocytes in IKKβ‐deleted mice shortly after exposure to AOM plus DSS revealed increased apoptosis of the enterocytes. When IKKβ was deleted in myeloid cells, including mature macrophages, dendritic cells, and neutrophils, the tumor load was reduced by 50%. Thus, the deletion of IKKβ in myeloid cells, but not in enterocytes, diminished the proliferation of AOM‐exposed enterocytes. These results suggest that IKKβ‐driven NF‐κB contributes to the development of CAC through distinct cell type‐specific mechanisms: in enterocytes, NF‐κB activates antiapoptotic genes and thereby suppresses the apoptotic elimination of preneoplastic cells; in myeloid cells, NF‐κB promotes the production of cytokines that act as growth factors for transformed enterocytes.( 53 ) One of these growth factors was subsequently identified as IL‐6, which is encoded by a NF‐κB target gene. The inhibition of IL‐6 signaling by antagonistic anti‐IL‐6 receptor antibodies inhibited tumor growth, similar to IKKβ ablation in myeloid cells.( 54 )
Recently, to test for its potential clinical applicability, we investigated whether the NF‐κB essential modulator (NEMO)‐binding domain (NBD) peptide, which is an amino‐terminal α‐helical region of NEMO associated with a carboxyl‐terminal segment of IKKα and IKKβ and has been shown to block the association of NEMO with IKKβ and to inhibit NF‐κB activity,( 55 ) reduces inflammatory injury in mice with colitis. In two colitis models, DSS in the drinking water of mice and a trinitrobenzene sulfonic acid enema marked NF‐κB activation, and expression of proinflammatory cytokines were observed in colonic tissues. The NBD peptide ameliorated colonic inflammatory injury through the down‐regulation of proinflammatory cytokines mediated by NF‐κB inhibition in both models (Fig. 3b).( 56 ) These results indicate that an IKKβ‐targeted NF‐κB blockade using the NBD peptide might be an promising therapeutic approach for inflammatory bowel disease.
Liver cancer. Although IKKβ‐deficient mice die at mid‐gestation from uncontrolled liver apoptosis,( 10 ) the deficiency of IKKβ in hepatocytes did not influence their function or growth.( 57 ) However, with the injection of concanavalin A (ConA) to produce a mouse acute hepatitis model, liver damage became worse in the hepatocyte‐specific IKKβ knockout mouse, and TNF‐α, which was expressed in T cells in response to ConA, exhibited antiapoptosis activity. This result suggests that IKKβ/NF‐κB activation in hepatocytes is important for surviving TNF‐α‐mediated acute hepatitis and that the inactivation of NF‐κB leaves the hepatocytes vulnerable to injury.( 57 )
The participation of IKKβ in carcinogenesis was examined by using diethylnitrosamine (DEN) as a chemical carcinogen. Unlike the carcinogens used in CAC carcinogenesis, DEN does not require assistance from inflammation‐inducing tumor promoters when given to 2‐week‐old mice. Compared to intestinal epithelial cells, hepatocytes in the knockout group showed about three times the number of tumors and larger tumor diameters. In knockout mice, hepatocyte death increased with the increase in ROS during the acute reaction to carcinogen administration. Furthermore, cell death was accompanied by an inflammation reaction, and the elevated hepatocyte death rate enhanced compensatory proliferation due to the strong regenerative capacity of the liver.( 42 )
Decreased NF‐κB activity and elevated JNK activity have been shown to promote TNF‐α‐induced cell death.( 58 ) Accordingly, the ablation of hepatocyte IKKβ results in higher DEN‐induced JNK activity and greater cell death. Prolonged JNK activation in the absence of NF‐κB depends on the accumulation of ROS. Interestingly, the reduction of ROS production after the administration of an antioxidant (BHA) was shown to control liver cell death and proliferation, and to reduce carcinogenesis.( 42 ) The hepatocyte‐specific deletion of IKKβ augmented DEN‐induced hepatocyte death, cytokine‐driven compensatory proliferation, and increased tumorigenesis, whereas the disruption of JNK1 abrogated these responses.( 59 )
Much of the observed compensatory proliferation depends on the production by non‐parenchymal cells of factors such as TNF‐α, IL‐6, and hepatocyte growth factor (HGF),( 60 ) and IKKβ/NF‐κB activation is important for the production of these cytokines. Correspondingly, the incidence of hepatocellular carcinoma decreased in mice with IKKβ knockout in myeloid cells, including Kupffer cells, which are specialized macrophages in the liver, as well as hepatocytes. These results suggest that IKKβ/NF‐κB activation in myeloid cells is important for the production of liver growth factors, presenting the possibility of preventing liver cancer by inhibiting IKKβ/NF‐κB activation.( 42 ) Indeed, when we compared DEN‐induced hepatocarcinogenesis between IL‐6 knockout mice and wild‐type controls, a marked reduction in cancer incidence was seen in IL‐6 knockout mice. Interestingly, a gender disparity was observed in the reaction of mice to DEN, but the ablation of IL‐6 abolished the gender difference in hepatocarcinogenesis in mice.( 61 )
The participation of the IKKβ/NF‐κB signaling pathway in carcinogenesis differs among organs, cells, and models. The relationship between an inflammation cell and a neoplastic cell, which is instigated by NF‐κB activation, is critical for tumor organization (Fig. 4).
Figure 4.
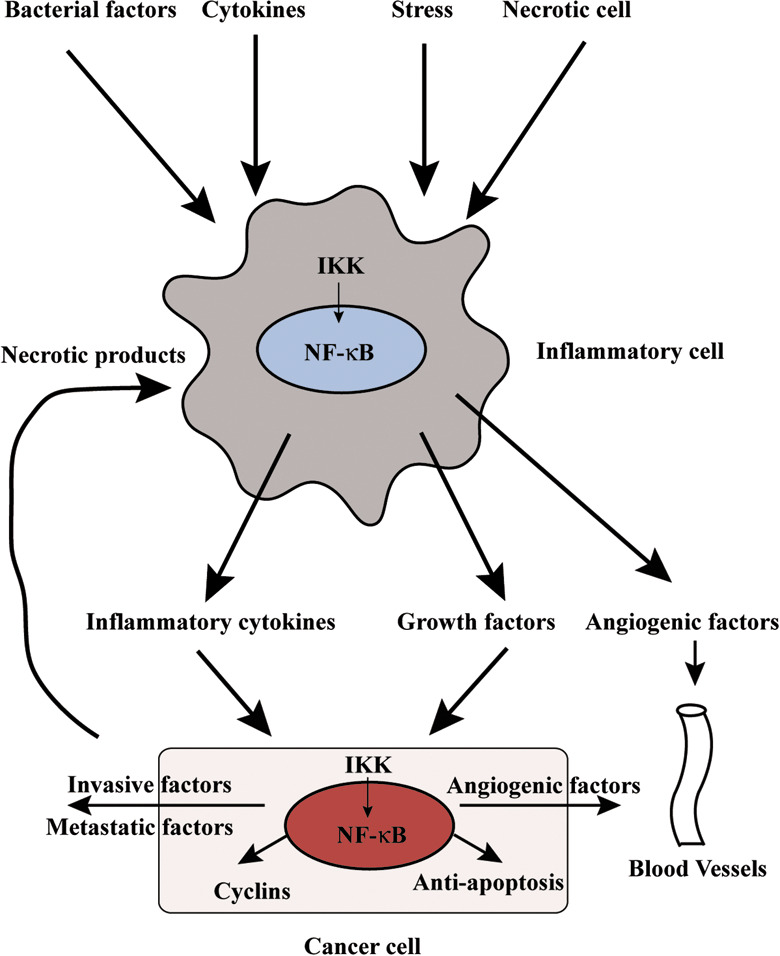
Relationship of nuclear factor‐kappaB (NF‐κB) activation to an inflammation cell and a neoplastic cell. NF‐κB‐regulated factors, which are growth factors, inflammatory cytokines, and angiogenic factors are produced in an inflammation cell around the neoplastic cell by bacteria factor, various cytokines, or stress, leading to the proliferation and progression of the neoplastic cell. Furthermore, NF‐κB activation in the neoplastic cell controls expression of antiapoptotic, cell‐cycle associated genes and participates in further malignancy. IKK, I kappa B kinase.
NF‐κB inhibition as a treatment target for cancer
As NF‐κB in inflammatory cells serves an important immune function, and its absence can result in severe immunodeficiency, prolonged and strong inhibition of NF‐κB might not be practical for cancer prevention. The application of a NF‐κB inhibitor may prove useful as an anticancer therapy. Given the antiapoptosis effect of NF‐κB, its inhibition should result in apoptosis. Furthermore, constitutive NF‐κB activation is observed in many cancer cells and is assumed to be involved in malignancy of the cancer.( 62 ) Research aimed at developing IKKβ/NF‐κB inhibitors is currently flourishing. In addition to NBD peptide as above, several NF‐κB inhibitors, such as proteasome inhibitors, BAY11, soy isoflavone genistein, parthenolide, and dehydroxymethylepoxyquinomicin (DHMEQ), have been developed. It has also been demonstrated that steroids and non‐steroidal anti‐inflammatory drugs block NF‐κB.( 63 ) However, few cell types will experience apoptosis solely via NF‐κB inhibition, and it is likely that a NF‐κB inhibitor will be effective only in combination with other anticancer agents or radiotherapy. As activation of NF‐κB by some agents or stimuli may mediate apoptosis under certain conditions,( 62 ) as described above, it will be necessary to investigate the function of NF‐κB, pro‐apoptotic or antiapoptotic, before the usage of inhibitors against such tumors.
Conclusions
The mechanism of NF‐κB activation and its connection with carcinogenesis are gradually being elucidated. The function of NF‐κB in inflammation‐associated carcinogenesis appears to be critical, and the development a NF‐κB inhibitor for clinical application is expected in the future. Nevertheless, the use of a NF‐κB inhibitor may have unintended effects on various divergent signaling pathways, and it will be necessary to carefully and completely investigate the basic science of these pathways so that any potential side‐effects can be considered.
References
- 1. Shacter E, Weitzman SA. Chronic inflammation and cancer. Oncology (Williston Park) 2002; 16 (217–26): 29; discussion 30–2. [PubMed] [Google Scholar]
- 2. Coussens LM, Werb Z. Inflammation and cancer. Nature 2002; 420: 860–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nomura A, Stemmermann GN, Chyou PH, Kato I, Perez‐Perez GI, Blaser MJ. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N Engl J Med 1991; 325: 1132–6. [DOI] [PubMed] [Google Scholar]
- 4. Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology 2004; 127: S35–50. [DOI] [PubMed] [Google Scholar]
- 5. Uemura N, Okamoto S, Yamamoto S et al . Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 2001; 345: 784–9. [DOI] [PubMed] [Google Scholar]
- 6. Yoshida H, Shiratori Y, Moriyama M et al . Interferon therapy reduces the risk for hepatocellular carcinoma: national surveillance program of cirrhotic and noncirrhotic patients with chronic hepatitis C in Japan. IHIT Study Group. Inhibition of hepatocarcinogenesis by interferon therapy. Ann Intern Med 1999; 131: 174–81. [DOI] [PubMed] [Google Scholar]
- 7. Karin M, Lin A. NF‐kappaB at the crossroads of life and death. Nat Immunol 2002; 3: 221–7. [DOI] [PubMed] [Google Scholar]
- 8. Hoffmann A, Baltimore D. Circuitry of nuclear factor kappaB signaling. Immunol Rev 2006; 210: 171–86. [DOI] [PubMed] [Google Scholar]
- 9. Ghosh S, Karin M. Missing pieces in the NF‐kappaB puzzle. Cell 2002; 109 (Suppl): S81–96. [DOI] [PubMed] [Google Scholar]
- 10. Li Z‐W, Chu W, Hu Y et al . The IKKβ subunit of IκB kinase (IKK) is essential for NF‐κB activation and prevention of apoptosis. J Exp Med 1999; 189: 1839–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee EG, Boone DL, Chai S et al . Failure to regulate TNF‐induced NF‐kappaB and cell death responses in A20‐deficient mice. Science 2000; 289: 2350–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kovalenko A, Chable‐Bessia C, Cantarella G, Israel A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF‐kappaB signalling by deubiquitination. Nature 2003; 424: 801–5. [DOI] [PubMed] [Google Scholar]
- 13. Pomerantz JL, Baltimore D. Two pathways to NF‐kappaB. Mol Cell 2002; 10: 693–5. [DOI] [PubMed] [Google Scholar]
- 14. Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF‐induced NEMO‐independent processing of NF‐kappa B2 in maturing B cells. Nat Immunol 2002; 3: 958–65. [DOI] [PubMed] [Google Scholar]
- 15. Coope HJ, Atkinson PG, Huhse B et al . CD40 regulates the processing of NF‐kappaB2 p100 to p52. EMBO J 2002; 21: 5375–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dejardin E, Droin NM, Delhase M et al . The lymphotoxin‐beta receptor induces different patterns of gene expression via two NF‐kappaB pathways. Immunity 2002; 17: 525–35. [DOI] [PubMed] [Google Scholar]
- 17. Bonizzi G, Karin M. The two NF‐kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol 2004; 25: 280–8. [DOI] [PubMed] [Google Scholar]
- 18. Kabrun N, Enrietto PJ. The Rel family of proteins in oncogenesis and differentiation. Semin Cancer Biol 1994; 5: 103–12. [PubMed] [Google Scholar]
- 19. Bartsch H, Nair J. Oxidative stress and lipid peroxidation‐derived DNA‐lesions in inflammation driven carcinogenesis. Cancer Detect Prev 2004; 28: 385–91. [DOI] [PubMed] [Google Scholar]
- 20. Yamanishi Y, Boyle DL, Rosengren S, Green DR, Zvaifler NJ, Firestein GS. Regional analysis of p53 mutations in rheumatoid arthritis synovium. Proc Natl Acad Sci USA 2002; 99: 10 025–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tada M, Komatsu Y, Kawabe T et al . Quantitative analysis of K‐ras gene mutation in pancreatic tissue obtained by endoscopic ultrasonography‐guided fine needle aspiration: clinical utility for diagnosis of pancreatic tumor. Am J Gastroenterol 2002; 97: 2263–70. [DOI] [PubMed] [Google Scholar]
- 22. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70. [DOI] [PubMed] [Google Scholar]
- 23. Zumkeller W, Schofield PN. Growth factors, cytokines and soluble forms of receptor molecules in cancer patients. Anticancer Res 1995; 15: 343–8. [PubMed] [Google Scholar]
- 24. Sebolt‐Leopold JS, Herrera R. Targeting the mitogen‐activated protein kinase cascade to treat cancer. Nat Rev Cancer 2004; 4: 937–47. [DOI] [PubMed] [Google Scholar]
- 25. Szlosarek PW, Balkwill FR. Tumour necrosis factor alpha. a potential target for the therapy of solid tumours. Lancet Oncol 2003; 4: 565–73. [DOI] [PubMed] [Google Scholar]
- 26. Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS Jr. NF‐kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol 1999; 19: 5785–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Karin M. Nuclear factor‐kappaB in cancer development and progression. Nature 2006; 441: 431–6. [DOI] [PubMed] [Google Scholar]
- 28. Bian X, McAllister‐Lucas LM, Shao F et al . NF‐kappa B activation mediates doxorubicin‐induced cell death in N‐type neuroblastoma cells. J Biol Chem 2001; 276: 48 921–9. [DOI] [PubMed] [Google Scholar]
- 29. Ivanov VN, Ronai Z. p38 protects human melanoma cells from UV‐induced apoptosis through down‐regulation of NF‐kappaB activity and Fas expression. Oncogene 2000; 19: 3003–12. [DOI] [PubMed] [Google Scholar]
- 30. Kiriakidis S, Andreakos E, Monaco C, Foxwell B, Feldmann M, Paleolog E. VEGF expression in human macrophages is NF‐kappaB‐dependent. studies using adenoviruses expressing the endogenous NF‐kappaB inhibitor IkappaBalpha and a kinase‐defective form of the IkappaB kinase 2. J Cell Sci 2003; 116: 665–74. [DOI] [PubMed] [Google Scholar]
- 31. Strieter RM, Polverini PJ, Arenberg DA et al . Role of C‐X‐C chemokines as regulators of angiogenesis in lung cancer. J Leukoc Biol 1995; 57: 752–62. [DOI] [PubMed] [Google Scholar]
- 32. Yoshida S, Ono M, Shono T et al . Involvement of interleukin‐8, vascular endothelial growth factor, and basic fibroblast growth factor in tumor necrosis factor alpha‐dependent angiogenesis. Mol Cell Biol 1997; 17: 4015–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer 2002; 2: 161–74. [DOI] [PubMed] [Google Scholar]
- 34. Luo JL, Tan W, Ricono JM et al . Nuclear cytokine‐activated IKKalpha controls prostate cancer metastasis by repressing Maspin. Nature 2007; 446: 690–4. [DOI] [PubMed] [Google Scholar]
- 35. Keats JJ, Fonseca R, Chesi M et al . Promiscuous mutations activate the noncanonical NF‐kappaB pathway in multiple myeloma. Cancer Cell 2007; 12: 131–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Demicco EG, Kavanagh KT, Romieu‐Mourez R et al . RelB/p52 NF‐kappaB complexes rescue an early delay in mammary gland development in transgenic mice with targeted superrepressor IkappaB‐alpha expression and promote carcinogenesis of the mammary gland. Mol Cell Biol 2005; 25: 10 136–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res 2006; 66: 605–12. [DOI] [PubMed] [Google Scholar]
- 38. Sparmann A, Bar‐Sagi D. Ras‐induced interleukin‐8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 2004; 6: 447–58. [DOI] [PubMed] [Google Scholar]
- 39. Varney ML, Johansson SL, Singh RK. Tumour‐associated macrophage infiltration, neovascularization and aggressiveness in malignant melanoma: role of monocyte chemotactic protein‐1 and vascular endothelial growth factor‐A. Melanoma Res 2005; 15: 417–25. [DOI] [PubMed] [Google Scholar]
- 40. Mizukami Y, Jo WS, Duerr EM et al . Induction of interleukin‐8 preserves the angiogenic response in HIF‐1alpha‐deficient colon cancer cells. Nat Med 2005; 11: 992–7. [DOI] [PubMed] [Google Scholar]
- 41. Ulloa L, Messmer D. High‐mobility group box 1 (HMGB1) protein: friend and foe. Cytokine Growth Factor Rev 2006; 17: 189–201. [DOI] [PubMed] [Google Scholar]
- 42. Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine‐driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005; 121: 977–90. [DOI] [PubMed] [Google Scholar]
- 43. Maeda S, Hikiba Y, Shibata W et al . Essential roles of high‐mobility group box 1 in the development of murine colitis and colitis‐associated cancer. Biochem Biophys Res Commun 2007; 360: 394–400. [DOI] [PubMed] [Google Scholar]
- 44. Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest 2007; 117: 60–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bodger K, Crabtree JE. Helicobacter pylori and gastric inflammation. Br Med Bull 1998; 54: 139–50. [DOI] [PubMed] [Google Scholar]
- 46. Censini S, Lange C, Xiang Z et al . cag, a pathogenicity island of Helicobacter pylori, encodes type I‐specific and disease‐associated virulence factors. Proc Natl Acad Sci USA 1996; 93: 14 648–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hirata Y, Ohmae T, Shibata W et al . MyD88 and TNF receptor‐associated factor 6 are critical signal transducers in Helicobacter pylori‐infected human epithelial cells. J Immunol 2006; 176: 3796–803. [DOI] [PubMed] [Google Scholar]
- 48. Maeda S, Yoshida H, Ogura K et al . H. pylori activates NF‐kappaB through a signaling pathway involving IkappaB kinases, NF‐kappaB‐inducing kinase, TRAF2, and TRAF6 in gastric cancer cells. Gastroenterology 2000; 119: 97–108. [DOI] [PubMed] [Google Scholar]
- 49. Viala J, Chaput C, Boneca IG et al . Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol 2004; 5: 1166–74. [DOI] [PubMed] [Google Scholar]
- 50. Maeda S, Yoshida H, Ikenoue T et al . Structure of cag pathogenicity island in Japanese Helicobacter pylori isolates. Gut 1999; 44: 336–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ogura K, Maeda S, Nakao M et al . Virulence factors of Helicobacter pylori responsible for gastric diseases in Mongolian gerbil. J Exp Med 2000; 192: 1601–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yanai A, Maeda S, Shibata W et al . Activation of IkappaB kinase and NF‐kappaB is essential for Helicobacter pylori‐induced chronic gastritis in Mongolian gerbils. Infect Immun 2008; 76: 781–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Greten FR, Eckmann L, Greten TF et al . IKKbeta links inflammation and tumorigenesis in a mouse model of colitis‐associated cancer. Cell 2004; 118: 285–96. [DOI] [PubMed] [Google Scholar]
- 54. Becker C, Fantini MC, Schramm C et al . TGF‐beta suppresses tumor progression in colon cancer by inhibition of IL‐6 trans‐signaling. Immunity 2004; 21: 491–501. [DOI] [PubMed] [Google Scholar]
- 55. May MJ, D’Acquisto F, Madge LA, Glockner J, Pober JS, Ghosh S. Selective inhibition of NF‐kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science 2000; 289: 1550–4. [DOI] [PubMed] [Google Scholar]
- 56. Shibata W, Maeda S, Hikiba Y et al . Cutting edge: the IkappaB kinase (IKK) inhibitor, NEMO‐binding domain peptide, blocks inflammatory injury in murine colitis. J Immunol 2007; 179: 2681–5. [DOI] [PubMed] [Google Scholar]
- 57. Maeda S, Chang L, Li ZW, Luo JL, Leffert H, Karin M. IKKbeta is required for prevention of apoptosis mediated by cell‐bound but not by circulating TNFalpha. Immunity 2003; 19: 725–37. [DOI] [PubMed] [Google Scholar]
- 58. Tang G, Minemoto Y, Dibling B et al . Inhibition of JNK activation through NF‐kappaB target genes. Nature 2001; 414: 313–17. [DOI] [PubMed] [Google Scholar]
- 59. Sakurai T, Maeda S, Chang L, Karin M. Loss of hepatic NF‐kappa B activity enhances chemical hepatocarcinogenesis through sustained c‐Jun N‐terminal kinase 1 activation. Proc Natl Acad Sci USA 2006; 103: 10 544–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fausto N. Liver regeneration. J Hepatol 2000; 32: 19–31. [DOI] [PubMed] [Google Scholar]
- 61. Naugler WE, Sakurai T, Kim S et al . Gender disparity in liver cancer due to sex differences in MyD88‐dependent IL‐6 production. Science 2007; 317: 121–4. [DOI] [PubMed] [Google Scholar]
- 62. Luo JL, Kamata H, Karin M. IKK/NF‐kappaB signaling. balancing life and death – a new approach to cancer therapy. J Clin Invest 2005; 115: 2625–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nakanishi C, Toi M. Nuclear factor‐kappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer 2005; 5: 297–309. [DOI] [PubMed] [Google Scholar]