

Abstract
Infection with cagA‐positive Helicobacter pylori is associated with the development of gastric adenocarcinoma. The cagA gene product CagA is injected directly from the bacterium into the bacterium‐attached gastric epithelial cells via the type‐IV secretion system. Upon membrane localization and subsequent tyrosine phosphorylation by Src family kinases, CagA functions as a scaffolding adaptor and interacts with a number of host proteins that regulate cell growth, cell motility and cell polarity in both CagA phosphorylation‐dependent and phosphorylation‐independent manners. Of special interest is the interaction of CagA with the SHP‐2 tyrosine phosphatase, gain‐of‐function mutations that of which have recently been found in a variety of human malignancies. The CagA–SHP‐2 interaction is entirely dependent on CagA tyrosine phosphorylation and, through the complex formation, SHP‐2 is catalytically activated and induces morphological transformation with elevated cell motility. Intriguingly, structural diversity of the tyrosine phosphorylation sites of CagA accounts for the differential activity of individual CagA to bind and activate SHP‐2. Deregulation of SHP‐2 and other intracellular signaling molecules by H. pylori CagA may predispose cells to accumulate multiple genetic and epigenetic changes involved in gastric carcinogenesis. Furthermore, the differential potential of individual CagA to disturb cellular functions indicates that H. pylori strains carrying biologically more active CagA are more virulent than those with less active CagA and are more closely associated with gastric carcinoma. (Cancer Sci 2005; 96: 835–843)
Helicobacter pylori, a spiral‐shaped bacterium that colonizes the human gastric mucosa, is estimated to inhabit at least half of the world's human population. Since its first report in 1984 by Marshall and Warren, H. pylori has been recognized as the etiological agent of gastric diseases such as chronic atrophic gastritis and peptic ulcers. H. pylori infection is primarily acquired in childhood, transmission occurring through a oral–oral or fecal–oral mode primarily within families, and infection is lifelong in the majority of cases. Furthermore, the plasticity of the H. pylori genome generates a huge diversity of strains, with each strain showing differences in their genome sequence by more than 20%.( 1 ) Thus, even a single strain may generate multiple variants and select those that adapt to an individual host environment during long‐term colonization.( 2 ) Despite the genomic diversity of H. pylori, epidemiological studies have revealed the importance of several genetic elements, such as cag pathogenicity islands (PAI), in the development of gastroduodenal disorders.
Gastric adenocarcinoma is the second most common cause of cancer‐related deaths worldwide, with 876 000 estimated new cases and 405 000 estimated deaths in the year 2000.( 3 ) Gastric adenocarcinoma is histopathologically subdivided into intestinal type and diffuse type. Intestinal‐type gastric adenocarcinoma, which occurs in older people, is more common than diffuse type, which affects younger people with poor prognosis. A small subset of diffuse‐type gastric adenocarcinoma is of familial origin, caused by mutations in the E‐cadherin gene.( 4 ) Recent epidemiological studies as well as infection studies using animal models have indicated that H. pylori plays a critical role in the development of both intestinal and diffuse types of gastric adenocarcinoma.( 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 ) However, the molecular mechanisms by which H. pylori stimulates the development of gastric lesions leading to gastric carcinoma remain largely unknown.
Helicobacter pylori CagA, a bacterial intruder
Helicobacter pylori strains can be divided into two major subpopulations based on their ability to produce a 120–145‐kDa immunodominant protein called cytotoxin‐associated gene A (CagA) antigen.( 13 , 14 ) The cagA gene that encodes CagA is localized at one end of the cag PAI, a 40‐kb DNA segment that was most likely incorporated into the H. pylori genome by a process of horizontal transfer. Approximately 60% of H. pylori strains isolated in Western countries carry cag PAI, whereas almost all of the East Asian isolates are cag PAI‐positive. The cag PAI DNA segment contains 31 putative genes (open reading frames), including cagA and those encoding components of a molecular ‘syringe’ termed the type IV secretion system,( 15 ) through which macromolecules are delivered from the inside to the outside of the bacterium. Clinically, infection with the cagA‐positive H. pylori strain has been associated with higher grades of gastric mucosal inflammation as well as severe atrophic gastritis and has been suggested to play an important role in the development of gastric carcinoma.( 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 )
Upon attachment of cagA‐positive H. pylori to the gastric epithelial cell, the CagA protein is injected directly into the cell via the cag PAI‐encoded type IV secretion system (Fig. 1).( 24 , 25 , 26 , 27 , 28 ) Translocated CagA then localizes to the inner surface of the plasma membrane, where it undergoes tyrosine phosphorylation by several members of the Src family kinases (SFK) such as c‐Src, Fyn, Lyn and Yes.( 29 , 30 ) Phosphorylation of CagA by SFK occurs in the absence of any stimuli, indicating that SFK are constitutively activated in gastric epithelial cells. In general, tyrosine phosphorylation plays a crucial role in transmitting intracellular signaling for growth, movement or differentiation in mammalian cells. Accordingly, the finding raised the intriguing possibility that, upon tyrosine phosphorylation, the bacterial protein disturbs signal transduction and thereby provokes cellular dysfunction that eventually leads to cell transformation.
Figure 1.
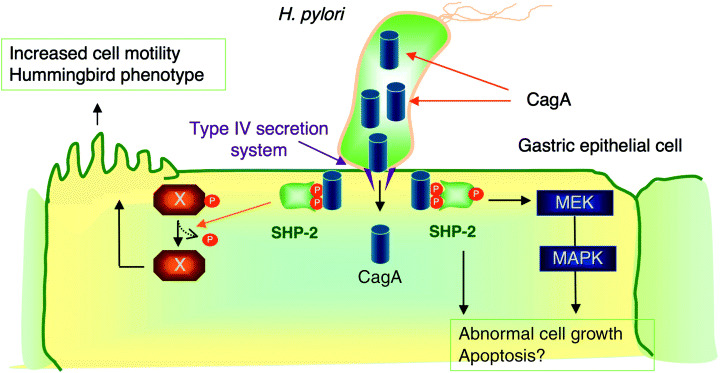
Physical and functional interaction between H. pylori CagA and SHP‐2. CagA is translocated from H. pylori into H. pylori‐attached gastric epithelial cells via the bacterial type IV secretion system. The translocated CagA protein localizes to the inner surface of the plasma membrane and undergoes tyrosine phosphorylation by Src family kinases. Upon tyrosine phosphorylation, CagA binds specifically to the SH2 domain‐containing tyrosine phosphatase SHP‐2 and activates the phosphatase activity. The CagA‐activated SHP‐2 potentiates Erk MAP kinase activity, which regulates both cell growth and cell morphology. SHP‐2 also dephosphorylates a cellular substrate (shown as X in the figure), which is involved in induction of the hummingbird phenotype that is associated with elevated cell motility.
EPIYA motif, the site of CagA tyrosine phosphorylation
The tyrosine phosphorylation site of CagA is characterized by the presence of a unique Glu‐Pro‐Ile‐Tyr‐Ala (EPIYA) motif, which is present in multiple numbers in the carboxy‐terminal region of the protein (Fig. 2a).( 31 , 32 ) From the sequences flanking these EPIYA motifs, four distinct EPIYA segments, EPIYA‐A, ‐B ‐C and ‐D, each of which contains a single EPIYA motif, have been identified in the CagA protein.( 32 , 33 , 34 ) The representative CagA protein of Western H. pylori isolates, such as those from Europe, North America and Australia, possesses a 32‐amino acid EPIYA‐A and a 40‐amino acid EPIYA‐B segment followed by a 34‐amino acid EPIYA‐C segment (‘A‐B‐C’‐type CagA) (Fig. 2a). Intriguingly, the 34‐amino acid EPIYA‐C segment multiplies variably, mostly from one time to three times, among different Western CagA species as a result of homologous recombination or misaligned replication of a 102‐bp cagA gene segment, which codes for the 34‐amino acid EPIYA‐C segment. The tyrosine residue that constitutes the EPIYA‐C site is the major site of tyrosine phosphorylation in Western CagA by SFK in gastric epithelial cells, whereas those present within the EPIYA‐A and EPIYA‐B segments are only weakly phosphorylated in the cells.( 31 , 32 ) Most CagA proteins of H. pylori isolated in East Asian countries such as Japan, Korea and China (East Asian CagA) also possess the EPIYA‐A and EPIYA‐B segments but not the repeatable EPIYA‐C segment. Instead, they have a distinct EPIYA‐containing sequence, termed the EPIYA‐D segment, which is unique to East Asian CagA species (Fig. 2b).( 32 ) East Asian CagA is therefore regarded as ‘A‐B‐D’‐type CagA. The EPIYA‐D site within the EPIYA‐D segment represents the major tyrosine phosphorylation site of East Asian CagA. In addition to its role in tyrosine phosphorylation, the EPIYA motif acts as a membrane‐targeting signal of CagA in gastric epithelial cells, although CagA–membrane interaction does not require EPIYA tyrosine phosphorylation.( 34 ) Accordingly, the EPIYA motif has a dual function in membrane association and tyrosine phosphorylation in CagA.
Figure 2.
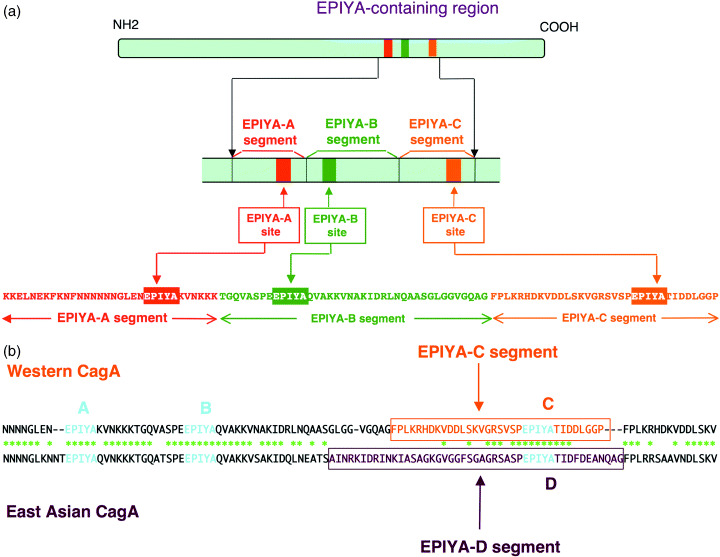
Molecular anatomy of the EPIYA‐containing region of CagA. (a) Subdivision of the EPIYA‐containing region. Prevalent CagA proteins of Western H. pylori isolates contain three EPIYA motifs, which are designated EPIYA‐A, EPIYA‐B and EPIYA‐C sites, based on the sequence surrounding each of the EPIYA motifs. Accordingly, the EPIYA‐containing region of CagA can be subdivided into the EPIYA‐A segment, the EPIYA‐B segment and the EPIYA C segment. The border of each segment was determined by comparing sequences of the EPIYA‐containing regions, which were made by extensive genetic recombination among various Western CagA isolates.( 34 ) (b) Comparison of the EPIYA‐containing region between Western and East Asian CagA species. Both CagA species have the conserved EPIYA‐A and EPIYA‐B segments. Following the EPIYA‐A and EPIYA‐B segments, Western CagA possesses the EPIYA‐C segment whereas East Asian CagA possesses the EPIYA‐D segment, whose sequence is unique to East Asian CagA.
SHP‐2, a cellular target of tyrosine‐phosphorylated CagA
Upon tyrosine phosphorylation by SFK, CagA acquires the ability to bind specifically to SHP‐2, a cytoplasmic protein tyrosine phosphatase having two tandem‐repeated Src homology‐2 (SH2) domains, termed N‐SH2 and C‐SH2, on the N‐terminal half and a protein tyrosine phosphatase (PTP) domain on the C‐terminal half (Fig. 1).( 31 , 32 , 33 ) The physical complex formed between CagA and SHP‐2 is detectable not only in cells infected in vitro or transfected with CagA but also in gastric mucosa of patients infected with cagA‐positive H. pylori.( 35 ) An SH2 domain functions as a protein module that specifically recognizes and interacts with a phosphotyrosyl peptide. Indeed, the CagA–SHP‐2 interaction requires both of the functional N‐SH2 and C‐SH2 domains of SHP‐2, indicating that two of the tyrosine‐phosphorylated EPIYA sites, either in cis or trans, are actively involved in the stable complex formation between CagA and SHP‐2.( 32 ) The crystal structure of SHP‐2 indicates that the N‐SH2 domain occludes the catalytic cleft of the PTP domain, blocking substrate access.( 36 ) This closed (inactive) structure keeps the basal phosphatase activity level of SHP‐2 low. Binding of tyrosine‐phosphorylated CagA to the SH2 domains induces a conformational change in SHP‐2 that relieves inhibition of the PTP domain by the N‐SH2 domain, resulting in the activation of SHP‐2 phosphatase activity.
Pathophysiological consequence of the CagA–SHP‐2 interaction
Numerous studies have indicated that SHP‐2 participates in signal transduction downstream of growth factor and cytokine receptors to regulate cellular responses, including proliferation, morphogenesis and cell motility.( 37 ) Consistent with this finding, gastric epithelial cells expressing either infected CagA or transfected CagA elicit cell‐morphological transformation, termed the hummingbird phenotype, which is characterized by elongated cell‐shape with dramatic cytoskeletal rearrangements.( 24 , 31 ) Time‐lapse video microscopic analysis has revealed that the hummingbird phenotype represents highly elevated motility of cells.( 38 ) Inhibition of CagA tyrosine phosphorylation or disruption of the CagA–SHP‐2 complex inhibits induction of the hummingbird phenotype. Furthermore, constitutive or conditional knockdown of SHP‐2 abolishes induction of hummingbird cells by CagA.( 39 ) Thus, CagA‐deregulated SHP‐2 plays a crucial role in induction of the hummingbird phenotype by CagA in gastric epithelial cells.
Physiologically, SHP‐2 is known to activate Erk MAP kinase by both Ras‐dependent and ‐independent mechanisms.( 37 ) Consistent with this, expression of CagA in gastric epithelial cells results in the sustained activation of Erk MAP kinase activity.( 38 ) As prolonged Erk activity has been suggested to play an important role in the G1 to S phase progression,( 40 ) CagA may predispose gastric epithelial cells to deregulated growth by inducing sustained Erk activation. Interestingly, Erk MAP kinase activity is also required for induction of the hummingbird phenotype by CagA, suggesting the role of sustained Erk activation in cell morphology and movement as well.( 40 )
Differences in SHP‐2 binding activity between East Asian and Western CagA proteins
East Asian CagA and Western CagA proteins possess distinctly structured tyrosine phosphorylation and SHP‐2‐binding sites, EPIYA‐D and EPIYA‐C, respectively (Fig. 2b). The sequence flanking EPIYA‐D of East Asian CagA perfectly matches the consensus high‐affinity binding sequence for the SH2 domains of SHP‐2 (pY‐[V/T/A/I/S]‐X‐[L/I/V]‐X‐[F/W]) (Fig. 3a).( 32 , 33 , 41 ) In contrast, the sequences flanking EPIYA‐C of Western CagA differ from the consensus SHP‐2 binding sequence by a single amino acid at the pY + 5 position. As a result, the EPIYA‐D site in East Asian CagA exhibits stronger SHP‐2 binding and greater morphogenetic activity than EPIYA‐C of Western CagA (Fig. 3b).
Figure 3.
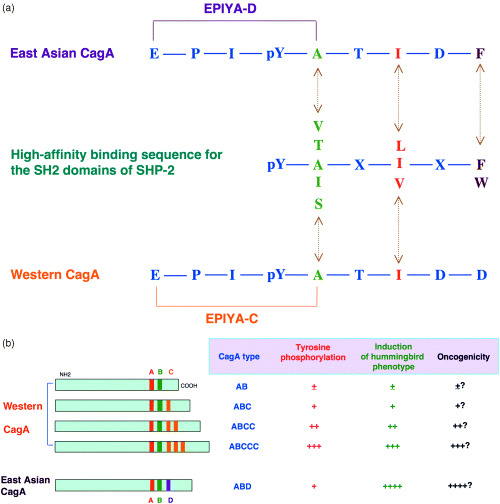
(a) The high‐affinity binding sequence for the SH2 domains of SHP‐2. The consensus SHP‐2‐binding sequence is aligned with the EPIYA‐C site of Western CagA and the EPIYA‐D site of East Asian CagA. (b) The influence of EPIYA‐repeat polymorphism on the pathophysiological activities of CagA. EPIYA‐C and EPIYA‐D sites are major tyrosine phosphorylation sites of Western CagA and East Asian CagA, respectively. EPIYA‐D site of East Asian CagA binds SHP‐2 more strongly than does EPIYA‐C site of Western CagA. As a result, East Asian CagA exhibits greater activity to induce hummingbird cells than Western CagA. Among Western CagA species, those having larger numbers of EPIYA‐C exhibit stronger SHP‐2 binding activity and greater activity to induce the hummingbird cells than those having less numbers of EPIYA‐C do.
Among Western CagA species, the number of EPIYA‐C sites is correlated directly with the levels of CagA tyrosine phosphorylation, SHP‐2 binding activity and the morphogenetic activity of CagA.( 32 ) This finding suggests that Western CagA proteins possessing a greater number of EPIYA‐C are biologically more active than those having a smaller number of EPIYA‐C (Fig. 3b).
Inhibition of SFK by CagA
In addition to SHP‐2, tyrosine phosphorylated CagA has been shown to bind to the SH2 domain of the C‐terminal Src kinase (Csk).( 42 ) Through the interaction, CagA stimulates the kinase activity of Csk, which in turn phosphorylates SFK at the C‐terminal inhibitory tyrosine residue. As a result, CagA inhibits SFK activity through Csk activation. Because SFK are kinases that phosphorylate CagA, this finding indicates the presence of a feedback mechanism that attenuates phosphorylation‐dependent activities of CagA.( 42 ) Most notably, the CagA–Csk interaction may be important in limiting SHP‐2 deregulation by CagA, which otherwise elicits acute and serious toxicity toward gastric epithelial cells. Thus, the negative feedback loop ensures a long‐term equilibrium between cagA‐positive H. pylori and the human host for decades without excess mucosal damage to the host.
In addition to Csk‐mediated inhibition, CagA is capable of directly inhibiting SFK activity in a phosphorylation‐dependent manner.( 43 ) CagA‐mediated inhibition of SFK activity is associated with a decrease in the level of tyrosine‐phosphorylated cortactin. Because cortactin plays a critical role in the actin cytoskeletal rearrangement, reduced tyrosine phosphorylation of cortactin by CagA may also play a role in induction of the hummingbird phenotype by CagA.
Phosphorylation‐independent biological activities of CagA
Having shown that the membrane‐bound and tyrosine‐phosphorylated CagA specifically recruits SH2 domain‐containing proteins such as SHP‐2 and Csk, it can be concluded that the bacterial protein functionally mimics mammalian scaffolding adaptors such as Gab and insulin receptor substrate family proteins.( 44 ) Notably, however, there is no sequence similarity between CagA and any of the known mammalian proteins.
In addition to the functions of CagA as a phosphorylation‐dependent scaffolding adaptor, recent studies have revealed phosphorylation‐independent activities of CagA, which are also involved in cell growth and cell motility. CagA binds Grb2 and activates Ras in a manner independent of CagA tyrosine phosphorylation.( 45 ) The CagA–Grb2 interaction is also involved in elevated cell motility known as the scattering phenotype. Upon complex formation, CagA promotes proliferation of gastric epithelial cells through activation of the Ras‐MAP kinase pathway. CagA also interacts with the c‐Met hepatocyte growth factor (HGF) receptor.( 46 ) Although the complex formation requires infection of cells with H. pylori which causes tyrosine phosphorylation of c‐Met, it is again independent of CagA tyrosine phosphorylation. The interaction between CagA and c‐Met indicates that CagA potentiates HGF‐dependent intracellular signaling that elicits cell morphological changes. Interaction of CagA with phospholipase C‐gamma (PLCγ) has also been reported, although the functional consequence of the interaction remains to be elucidated.( 46 )
Recent studies have revealed that translocated CagA associates with tight‐junctional proteins such as ZO‐1, causing an ectopic assembly of tight‐junction components at sites of bacterial attachment and altering the function of the apical‐junctional complex.( 47 ) Hence, long‐term CagA delivery to polarized epithelia causes a disruption of the epithelial barrier function and dysplastic changes in epithelial cell morphology, alterations that have been shown to play a role in carcinogenesis. Importantly, the CagA activity on apical junctions does not require CagA tyrosine phosphorylation. More recently, Franco et al. reported that β‐catenin, a major component of adherens junctions, is deregulated in gastric epithelial cells expressing CagA.( 48 ) Although the mechanism underlying β‐catenin activation by CagA remains to be elucidated, results indicate that Wnt/β‐catenin signaling, which plays a crucial role in colon carcinogenesis, is also involved in the development of gastric carcinoma.
Deregulation of transcription factors by CagA. CagA is capable of indirectly deregulating transcription factors through multiple distinct mechanisms. Hirata et al. showed that CagA activates serum responsive element‐dependent transcription in a manner independent of CagA phosphorylation.( 49 ) Brandt et al. recently showed that CagA is capable of activating NF‐κB, which in turn induces interleukin‐8 expression.( 50 ) As injection of the H. pylori‐derived proteoglycan via the type IV injection system of H. pylori also stimulates NF‐κB in gastric epithelial cells,( 51 ) these results indicate that H. pylori activates NF‐κB through multiple distinct mechanisms.
More recently, CagA was found to activate the nuclear factor of activated T cells (NFAT) in gastric epithelial cells.( 52 ) Expression of CagA in gastric epithelial cells activates the calcium‐dependent serine/threonine phosphatase calcineurin and induces translocation of cytoplasmic NFAT to the nucleus, where it transactivates NFAT‐dependent genes. The CagA activity toward NFAT is again independent of CagA phosphorylation. Although the mechanism by which CagA activates the calcineurin‐NFAT system needs further investigation, the reported CagA–PLCγ interaction might trigger Ca2+ mobilization and subsequent activation of calcineurin.( 46 ) Surprisingly, one of the NFAT‐dependent genes activated by CagA in gastric epithelial cells is p21Cip1 cyclin‐dependent kinase inhibitor.( 52 ) Thus, whereas CagA activates a growth‐promoting signal via SHP‐2 deregulation, it simultaneously inhibits progression of the cell cycle through NFAT activation. As a result, CagA may cause proliferation, apoptosis or differentiation, depending on the cellular setting. Intriguingly, another H. pylori virulence factor, vacuolating toxin VacA, counteracts the activity of CagA to activate NFAT.( 52 ) This finding indicates that VacA also plays a role in determining the fate of gastric epithelial cells expressing CagA (Fig. 4).
Figure 4.
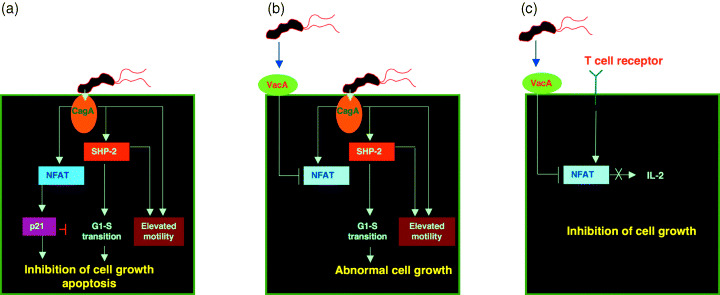
A proposed model for functional interaction between CagA and VacA. (a) H. pylori‐injected CagA deregulates SHP‐2 and other cellular target molecules that promote cell proliferation. Simultaneously, CagA activates NFAT and thereby induces NFAT‐dependent genes such as p21 Cip.1 . Elevated p21Cip1 then arrests gastric epithelial cells in G1 phase. Such G1‐arrested cells subsequently undergo senescence, apoptosis or intestinal trans‐differentiation known as intestinal metaplasia. (b) When gastric epithelial cells simultaneously encounter VacA and CagA, VacA counteracts nuclear translocation of NFAT by CagA and thus abolishes induction of p21Cip1 in CagA‐expressing cells. Accordingly, in the presence of adequate levels of VacA, CagA stimulates deregulated cell growth. (c) VacA inhibits production of T‐cell growth factor, interleukin‐2 (IL‐2), by suppressing NFAT activity.
Role of CagA in gastric carcinogenesis
Development of gastric adenocarcinoma is a multistep process that requires qualitative as well as quantitative alterations in the expression of oncogenes and tumor suppressor genes, lasting for several decades. During cagA‐positive H. pylori infection, gastric epithelial cells are continuously exposed to the injection of CagA from the bacteria. The injected CagA binds and deregulates SHP‐2 and other intracellular signaling molecules in both tyrosine phosphorylation‐dependent and ‐independent manners, generating deregulated signals for cell growth and cell movement. CagA also disrupts cell–cell junctions, destroying normal epithelial architecture.
Among the various CagA activities that disturb cellular functions, deregulation of SHP‐2 by CagA is of potential importance in gastric carcinogenesis because mutations in PTPN11, the gene encoding human SHP‐2, have been identified in human malignancies.( 53 , 54 ) Most of the reported cases carry missense mutations in exons 3 and 8, which encode segments of the N‐SH2 domain and the PTPase domain, respectively. Somatic SHP‐2 mutations are found in 35% of cases of sporadic juvenile myelomonocytic leukemia, 5–10% of cases of childhood myelodysplastic syndrome, 7% of cases of B‐cell acute lymphoblastic leukemia, 5% of cases of acute myelocytic leukemia and in some solid tumors such as neuroblastoma. Molecular modeling of SHP‐2 indicates that such mutations weaken the autoinhibitory interaction and hence constitutively activate SHP‐2 phosphatase activity. Accordingly, deregulation of SHP‐2 by CagA functionally mimics the gain‐of‐function mutation of SHP‐2 that is associated with human malignancies (Fig. 5). A potential role of SHP‐2 in the development of gastric cancer has also been suggested by results of a recent study demonstrating that genetically engineered mice lacking the SHP‐2 binding site on the interleukin‐6 family coreceptor gp130 develop intestinal‐type gastric adenocarcinoma with extremely high frequency.( 55 )
Figure 5.
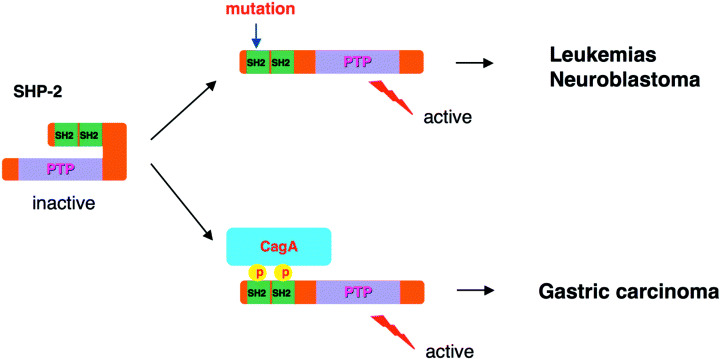
Involvement of SHP‐2 in human malignancies. Point mutations in PTPN11, a human gene that encodes SHP‐2, have been associated with childhood leukemias and some solid tumors. The mutations abolish the inhibitory interaction between the N‐SH2 domain and the phosphatase domain of SHP‐2 and therefore produce gain‐of‐function mutations. Upon complex formation with CagA, SHP‐2 is fixed to its active state, mimicking the gain‐of‐function mutation of SHP‐2.
cagA‐positive H. pylori infection induces progressive inflammatory changes in the gastric mucosa that leads to gastric cancer: superficial gastritis, atrophic gastritis, intestinal metaplasia, dysplasia, carcinoma.( 56 ) Because CagA–SHP‐2 complexes are detectable primarily in atrophic mucosa, the complex may be involved in the development of atrophic gastritis and the transition from atrophy to intestinal metaplasia.( 35 ) Possibly, CagA‐triggered abnormal signals that deregulate cell growth, cell–cell contact and cell migration may enhance epithelial cell turnover as a result of increased cell proliferation and subsequent apoptosis. Such an elevated cell turnover obviously increases the risk of damaged cells acquiring precancerous genetic changes. In this regard, results of recent studies using a Helicobacter‐infected mouse model have led to the surprising conclusion that gastric adenocarcinoma originates from circulating bone marrow‐derived cells (BMDC), not from resident gastric cells.( 57 ) If this is also the case in humans, chronic mucosal inflammation caused by CagA‐positive H. pylori may exhaust gastric stem cells and eventually lead to depletion of the resident stem cell pool, resulting in recruitment and settlement of BMDC into gastric mucosa. It has also been suggested that BMDC do not differentiate properly, resulting in progression to metaplasia, dysplasia and gastric cancer. This caveat obviously warrants further investigation.
CagA polymorphism and gastric cancer
Whereas some human populations with high incidences of H. pylori infection, such as those in East Asian countries (Japan, Korea and China), have high incidences of gastric carcinoma, other highly infected populations, such as populations in central Africa, do not. This enigma might be explained at least in part by the structural polymorphism of CagA proteins among H. pylori strains circulating in different geographic areas. As noted previously, East Asian H. pylori and Western H. pylori possess CagA proteins with distinctly structured tyrosine phosphorylation/SHP‐2‐binding sites, EPIYA‐D and EPIYA‐C. The EPIYA‐D site exhibits stronger SHP‐2 binding and greater morphogenetic activity than does the EPIYA‐C site (Fig. 3). Furthermore, the degrees of inflammation, activity of gastritis, and atrophy are significantly higher in patients infected with East Asian cagA‐positive strains than in patients infected with CagA‐negative or Western cagA‐positive strains.( 58 , 59 ) Thus, populations infected with East Asian cagA‐positive H. pylori may be at greater risk for gastric cancer than those infected with Western cagA‐positive or cagA‐negative strains (Fig. 6). Furthermore, among the Western CagA species, the number of EPIYA‐C sites is directly correlated with the levels of tyrosine phosphorylation, SHP‐2 binding activity and morphogenetic activity of CagA. Thus, Western CagA proteins with a greater number of EPIYA‐C sites are pathophysiologically more virulent and thus more carcinogenic. This notion has been supported by the results of recent work by Argent et al. demonstrating that five of six Western H. pylori strains harvested from gastric carcinoma patients possessed multiple EPIYA‐C sites, whereas 18 of 19 Western H. pylori strains isolated from non‐cancer patients possessed a single EPIYA‐C site.( 60 )
Figure 6.
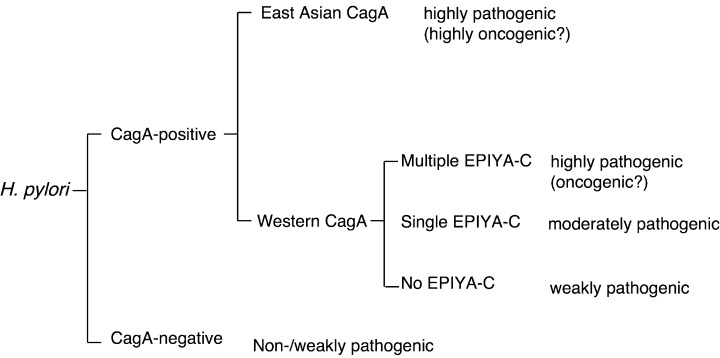
Classification of H. pylori based on the EPIYA‐repeat polymorphism of CagA. H. pylori is divided into CagA‐positive and CagA‐negative strains. CagA‐negative H. pylori is non‐pathogenic or only weakly pathogenic and is not involved in gastric carcinogenesis. CagA‐positive H. pylori is subdivided into those carrying East Asian CagA and those carrying Western CagA. Virulence of H. pylori carrying Western CagA is determined at least partly by the number of EPIYA‐C. Those having CagA with greater number of EPIYA‐C are more virulent and more closely associated with severe atrophic gastritis and gastric carcinoma than those having CagA with less EPIYA‐C. H. pylori strains carrying East Asian CagA are biologically more active than most if not all Western‐type H. pylori and individuals infected with this type of H. pylori are at the highest risk of developing gastric carcinoma.
Conclusions
Helicobacter pylori infection is declining as the standard of living rises. However, huge human populations have already been infected with cagA‐positive H. pylori and are at a higher risk of developing gastric carcinoma in the short or medium term. Interaction of CagA with SHP‐2, the first phosphatase that acts as a bona fide oncoprotein in human malignancy, is one of the key determinants for the development of gastric carcinoma associated with cagA‐positive H. pylori infection.
CagA is divided into two major types: East Asian CagA and Western CagA. Further studies should make it possible to identify additional CagA polymorphisms that account for the differential degree of virulence. From the clinical standpoint, elucidation of cagA‐positive H. pylori strains with the highest potential of developing gastric carcinoma will be extremely important. Recent studies have shown that eradication of H. pylori in humans appears to lower the risk of developing gastric carcinoma.( 61 ) Thus, systemic eradication of oncogenic H. pylori from a high‐risk population would dramatically reduce the worldwide incidence of gastric cancer. Furthermore, understanding molecular mechanisms underlying the H. pylori–gastric cell interaction is not only important in developing more effective therapies for gastric cancer but also crucial in understanding the genesis of inflammation‐associated cancers.
Acknowledgments
We thank Dr Takeshi Azuma for valuable discussions. We also thank the members of the Division of Molecular Oncology, Institute for Genetic Medicine, Hokkaido University, for help.
References
- 1. Salama N, Guillemin K, McDaniel TK, Sherlock G, Tompkins L, Falkow S. A whole‐genome microarray reveals genetic diversity among Helicobacter pylori strains. Proc Natl Acad Sci USA 2000; 97: 14 668–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest 2004; 113: 321–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Parkin DM, Bray FI, Devesa SS. Cancer burden in the year 2000. The global picture. Eur J Cancer 2001; 37: S4–S66 . [DOI] [PubMed] [Google Scholar]
- 4. Guilford P, Hopkins J, Harraway J et al. E‐cadherin germline mutations in familial gastric cancer. Nature 1998; 392: 402–5. [DOI] [PubMed] [Google Scholar]
- 5. Forman D, Newell DG, Fullerton F et al. Association between infection with Helicobacter pylori and risk of gastric cancer: evidence form a prospective investigation. Br Med J 1991; 302: 1302–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Parsonnet J, Friedman GD, Vandersteen DP et al. Helicobacter pylori and the risk of gastric carcinoma. N Engl J Med 1991; 325: 1127–31. [DOI] [PubMed] [Google Scholar]
- 7. Nomura A, Stemmermann GN, Chyou PH, Kato I, Perez GI, Blaser MJ. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N Engl J Med 1991; 325: 1132–6. [DOI] [PubMed] [Google Scholar]
- 8. Watanabe T, Tada M, Nagai H, Sasaki S, Nakao M. Helicobacter pylori infection induces gastric cancer in Mongolian gerbils. Gastroenterology 1998; 115: 642–8. [DOI] [PubMed] [Google Scholar]
- 9. Honda S, Fujioka T, Tokieda M, Satoh R, Nishizono A, Nasu M. Development of Helicobacter pylori‐induced gastric carcinoma in Mongolian gerbils. Cancer Res 1998; 58: 4255–9. [PubMed] [Google Scholar]
- 10. Sugiyama A, Maruta F, Ikeno T et al. Helicobacter pylori infection enhances N‐methyl‐N‐nitrosourea‐induced stomach carcinogenesis in the Mongolian gerbil. Cancer Res 1998; 58: 2067–9. [PubMed] [Google Scholar]
- 11. Shimizu N, Inada K, Nakanishi H et al. Helicobacter pylori infection enhances glandular stomach carcinogenesis in Mongolian gerbils treated with chemical carcinogenesis. Carcinogenesis 1999; 20: 669–76. [DOI] [PubMed] [Google Scholar]
- 12. Uemura N, Okamoto S, Yamamoto S et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 2001; 345: 784–9. [DOI] [PubMed] [Google Scholar]
- 13. Covacci A, Censini S, Bugnoli M et al. Molecular characterization of the 128‐kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc Natl Acad Sci USA 1993; 90: 5791–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tummuru MK, Cover TL, Blaser MJ. Cloning and expression of a high‐molecular‐mass major antigen of Helicobacter pylori: evidence of linkage to cytotoxin production. Infect Immun 1993; 61: 1799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Akopyants NS, Clifton SW, Kersulyte D et al. Analyses of the cag pathogenicity island of Helicobacter pylori . Mol Microbiol 1998; 28: 37–53. [DOI] [PubMed] [Google Scholar]
- 16. Kuipers EJ, Perez‐Perez GI, Meuwissen SG, Blaser MJ. Helicobacter pylori and atrophic gastritis: importance of the cagA status. J Natl Cancer Inst 1995; 87: 1777–80. [DOI] [PubMed] [Google Scholar]
- 17. Crabtree JE, Taylor JD, Wyatt JI et al. Mucosal IgA recognition of Helicobacter pylori 120 kDa protein, peptic ulceration, and gastric pathology. Lancet 1991; 338: 332–5. [DOI] [PubMed] [Google Scholar]
- 18. Blaser MJ, Perez‐Perez GI, Kleanthous H et al. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res 1995; 55: 2111–15. [PubMed] [Google Scholar]
- 19. Parsonnet J, Friedman GD, Orentreich N, Vogelman H. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut 1997; 40: 297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rugge M, Busatto G, Cassaro M et al. Patients younger than 40 years with gastric carcinoma –Helicobacter pylori genotype and associated gastritis phenotype. Cancer 1999; 85: 2506–11. [PubMed] [Google Scholar]
- 21. Shimoyama T, Fukuda S, Tanaka M, Mikami T, Munakata A, Crabtree JE. CagA seropositivity associated with development of gastric cancer in a Japanese population. J Clin Pathol 1998; 51: 225–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nomura M, Lee J, Stemmermann GN, Nomura RY, Perez‐Perez GI, Blaser MJ. Helicobacter pylori CagA seropositivity and gastric carcinoma risk in a Japanese American population. J Infect Dis 2002; 186: 1138–44. [DOI] [PubMed] [Google Scholar]
- 23. Kikuchi S, Crabtree JE, Forman D, Kurosawa M. Association between infections with CagA‐positive or ‐negative strains of Helicobacter pylori and risk for gastric cancer in young adults. Am J Gastroenterol 1999; 94: 3455–9. [DOI] [PubMed] [Google Scholar]
- 24. Segal ED, Cha J, Lo J, Falkow S, Tompkins LS. Altered states: Involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori . Proc Natl Acad Sci USA 1999; 96: 14 559–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Asahi M, Azuma T, Ito S et al. Helicobacter pylori CagA protein can be tyrosine phosphorylated in gastric epithelial cells. J Exp Med 2000; 191: 593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stein M, Rappuoli R, Covacci A. Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag‐driven host cell translocation. Proc Natl Acad Sci USA 2000; 97: 1263–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 2000; 287: 1497–500. [DOI] [PubMed] [Google Scholar]
- 28. Backert S, Ziska E, Brinkmann V et al. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell Microbiol 2000; 2: 155–64. [DOI] [PubMed] [Google Scholar]
- 29. Selbach M, Moese S, Hauck CR, Meyer TF, Backert S. Src is the kinase of the Helicobacter pylori CagA protein in vitro and in vivo . J Biol Chem 2002; 277: 6775–8. [DOI] [PubMed] [Google Scholar]
- 30. Stein M, Bagnoli F, Halenbeck R, Rappuoli R, Fantl WJ, Covacci A. c‐Src/Lyn kinases activate Helicobacter pylori CagA through tyrosine phosphorylation of the EPIYA motifs. Mol Microbiol 2002; 43: 971–80. [DOI] [PubMed] [Google Scholar]
- 31. Higashi H, Tsutsumi R, Muto S et al. SHP‐2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 2002; 295: 683–6. [DOI] [PubMed] [Google Scholar]
- 32. Higashi H, Tsutsumi R, Fujita A et al. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc Natl Acad Sci USA 2002; 99: 14 428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hatakeyama M. Oncogenic mechanisms of Helicobacter pylori CagA protein. Nature Rev Cancer 2004; 4: 688–94. [DOI] [PubMed] [Google Scholar]
- 34. Higashi H, Yokoyama K, Fujii Y et al. EPIYA motif is a membrane targeting signal of Helicobacter pylori virulence factor CagA in mammalian cells. J Biol Chem 2005; 280: 23 130–7. [DOI] [PubMed] [Google Scholar]
- 35. Yamazaki S, Yamakawa A, Ito Y et al. The CagA protein of Helicobacter pylori is translocated into epithelial cells and binds to SHP‐2 in human gastric mucosa. J Infect Dis 2003; 187: 334–7. [DOI] [PubMed] [Google Scholar]
- 36. Hof P, Pluskey S, Dhe‐Paganon S, Ech MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP‐2. Cell 1998; 92: 441–50. [DOI] [PubMed] [Google Scholar]
- 37. Neel BG, Gu H, Pao L. The ‘Shp’ing news: SH2 domain‐containing tyrosine phosphatases in cell signaling. Trends Biochem Sci 2003; 28: 284–93. [DOI] [PubMed] [Google Scholar]
- 38. Higashi H, Nakaya A, Tsutsumi R et al. Helicobacter pylori CagA induces Ras‐independent morphogenetic response through SHP‐2 recruitment and activation. J Biol Chem 2004; 279: 17205–16. [DOI] [PubMed] [Google Scholar]
- 39. Higuchi M, Tsutsumi R, Higashi H, Hatakeyama M. Conditional gene silencing utilizing the lac repressor reveals a role of SHP‐2 in cagA ‐positive Helicobacter pylori pathogenicity. Cancer Sci 2004; 95: 442–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roovers K, Assoian RK. Integrating the MAP kinase signal into the G1 phase cell cycle machinery. Bioessays 2000; 22: 818–26. [DOI] [PubMed] [Google Scholar]
- 41. De Souza D, Fabri LJ, Nash A et al. SH2 domains from suppressor of cytokine signaling‐3 and protein tyrosine phosphatases SHP‐2 have similar binding specificities. Biochemistry 2002; 41: 9229–36. [DOI] [PubMed] [Google Scholar]
- 42. Tsutsumi R, Higashi H, Higuchi M, Okada M, Hatakeyama M. Attenuation of Helicobacter pylori CagA–SHP‐2 signaling by interaction between CagA and C‐terminal Src kinase. J Biol Chem 2003; 278: 3664–70. [DOI] [PubMed] [Google Scholar]
- 43. Selbach M, Moese S, Hurwitz R, Hauck CR, Meyer TF, Backert S. Helicobacter pylori CagA protein induces cortactin dephosphorylation and actin rearrangement by c‐Src inactivation. EMBO J 2003; 22: 515–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hatakeyama M. Helicobacter pylori CagA – a potential bacterial oncoprotein that functionally mimics the mammalian Gab family of adaptor proteins. Microbes Infect 2003; 5: 143–50. [DOI] [PubMed] [Google Scholar]
- 45. Mimuro H, Suzuki T, Tanaka J, Asahi M, Haas R, Sasakawa C. Grb2 is a key mediator of Helicobacter pylori CagA protein activities. Mol Cell 2002; 10: 745–55. [DOI] [PubMed] [Google Scholar]
- 46. Churin Y, Al‐Ghoul L, Kepp O, Meyer TF, Birchmeier W, Naumann M. Helicobacter pylori CagA protein targets the c‐Met receptor and enhances the motogenic response. J Cell Biol 2003; 161: 249–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical‐junctional complex by Helicobacter pylori CagA. Science 2003; 300: 1430–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Franco AT, Israel DA, Washington MK et al. Activation of β‐catenin by carcinogenic Helicobacter pylori . Proc Natl Acad Sci USA 2005; 102: 10646–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hirata Y, Maeda S, Mitsuno Y et al. Helicobacter pylori CagA protein activates serum response element‐driven transcription independently of tyrosine phosphorylation. Gastroenterology 2002; 123: 1962–71. [DOI] [PubMed] [Google Scholar]
- 50. Brandt S, Kwok T, Hartig R, Konig W, Backert S. NF‐κB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci USA 2005; 102: 9300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Viala J, Chaput C, Boneca IG et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol 2004; 5: 1166–74. [DOI] [PubMed] [Google Scholar]
- 52. Yokoyama K, Higashi H, Ishikawa S et al. Functional antagonism between Helicobacter pylori CagA and vacuolating toxin VacA in control of the NFAT signaling pathway in gastric epithelial cells. Proc Natl Acad Sci USA 2005; 102: 9661–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tartaglia M, Niemeyer CM, Fragale A et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet 2003; 34: 148–50. [DOI] [PubMed] [Google Scholar]
- 54. Bentires‐Alj M, Paez JG, David FS et al. Activating mutations of the Noonan syndrome‐associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res 2004; 64: 8816–20. [DOI] [PubMed] [Google Scholar]
- 55. Judd LM, Alderman BM, Howlett M et al. Gastric cancer development in mice lacking the SHP2 binding site on the IL‐6 family co‐receptor gp130. Gastroenterology 2004; 126: 196–207. [DOI] [PubMed] [Google Scholar]
- 56. Correa P, Haenszel W, Cuello C, Tannenbaum S, Archer M. A model for gastric cancer epidemiology. Lancet 1975; 2: 58–60. [DOI] [PubMed] [Google Scholar]
- 57. Houghton J, Stoicov C, Nomura S et al. Gastric cancer originating from bone marrow‐derived cells. Science 2004; 306: 1568–71. [DOI] [PubMed] [Google Scholar]
- 58. Azuma T, Yamazaki S, Yamakawa A et al. Association between diversity in the Src homology 2 domain‐containing tyrosine phosphatase binding site of Helicobacter pylori CagA protein and gastric atrophy and cancer. J Infect Dis 2004; 189: 820–7. [DOI] [PubMed] [Google Scholar]
- 59. Azuma T, Ohtani M, Yamazaki Y, Higashi H, Hatakeyama M. Meta‐analysis of the relationship between CagA seropositivity and gastric cancer. Gastroenterology 2004; 126: 1926–7. [DOI] [PubMed] [Google Scholar]
- 60. Argent RH, Kidd M, Owen RJ, Thomas RJ, Limb MC, Atherton JC. Determinants and consequences of different levels of CagA phosphorylation for clinical isolates of Helicobacter pylori . Gastroenterology 2004; 127: 514–23. [DOI] [PubMed] [Google Scholar]
- 61. Wong BC, Lam SK, Wong WM et al. Helicobacter pylori eradication to prevent gastric cancer in a high‐risk region of China: a randomized controlled trial. JAMA 2004; 291: 187–94. [DOI] [PubMed] [Google Scholar]