

Abstract
It has been estimated that >20% of all malignancies are initiated or exacerbated by inflammation. Until recently, the molecular basis of this process has not been clarified. However, recent studies have uncovered the molecular mechanism of intracellular signaling pathways of inflammatory cytokines such as tumor necrosis factor (TNF)‐α, interferon (IFN)‐γ and interleukin (IL)‐6. Three major transcription factors including NF‐κB, STAT1 and STAT3 have been shown to play major roles in transmitting inflammatory cytokine signals to the nucleus. One function of NF‐κB and STAT3 in tumor cells is the promotion of cell growth and cell survival through the induction of target genes, whose products promote cell division and inhibit apoptosis. In addition, NF‐κB and STAT1 are important transcription factors that induce inflammatory mediators from inflammatory cells, especially macrophages, while STAT3 often antagonizes this process. STAT1 is generally believed to be an anti‐oncogene because it promotes apoptosis through p53, but it could promote inflammation‐mediated tumor development by enhancing tissue injury, remodeling, fibrosis and inflammation. Hence, the inhibition of NF‐κB and STATs offers a strategy for treatment of a variety of malignancies and can convert inflammation‐induced tumor growth into inflammation‐induced tumor regression. (Cancer Sci 2006; 97)
Inflammation and cancer
The immune system has been called a ‘double‐edged sword’ because of its ability to fight infectious pathogens on the one hand and to produce autoimmunity on the other. The immune system also serves as a tumor suppressor as immune surveillance on the one hand and as an initiator and/or promoter of tumors on the other( 1 ) (Fig. 1). Clinical observations support the idea of a strong association of chronic inflammation and cancer.( 1 , 2 ) For example, chronic inflammations induced by the hepatitis C virus (HCV), Helicobacter pylori and ulcerative colitis (UC) are important risk factors for malignancies such as hepatocellular carcinoma (HCC), gastric cancer and colon cancer, respectively.( 3 , 4 ) The finding that the long‐term use of non‐steroidal anti‐inflammatory drugs (NSAIDs) decreases cancer risk (a 40–50% reduction in the case of colon cancer) strengthens the proposed link between inflammation and cancer.( 5 ) Furthermore, several non‐infectious causes of chronic inflammation (such as cigarette smoke, asbestos and silica) also increase the risk of developing cancer. Pleural malignant mesothelioma, for example, is primarily due to asbestos, and the number of deaths in Japan is estimated to increase in the future.( 6 )
Figure 1.
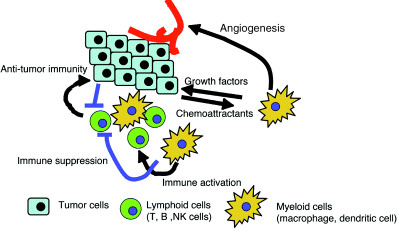
Role of inflammatory cells on tumor development.
In addition to epidemiological data that link inflammation with cancer, polymorphisms in the inflammatory genes including tumor‐necrosis factor (TNF), interleukin‐1 (IL‐1) and Toll‐like receptors (TLRs) are reported to be associated with non‐Hodgkin's lymphoma, stomach cancer and prostate cancer, respectively.( 7 ) Recent progress in our understanding of signal transduction mechanisms of innate immune systems also provides additional evidence for the link between inflammation and cancer. The nucleotide‐binding oligomerization domain (NOD) proteins, NOD1 and NOD2, have important roles in innate immunity as sensors of microbial components derived from bacterial peptidoglycan. Mutations in the gene that encodes NOD2 occur in a subpopulation of patients with Crohn's disease, and abnormalities in NOD1 function might lead to the development of chronic infection with H. pylori and to the subsequent development of peptic ulcer disease and gastric carcinoma.( 8 )
An over‐simplistic interpretation of two‐step carcinogenesis is: (i) initiation by activating or disabling mutations in molecules that regulate the cellular circuits and capacitors controlling cell division, survival and senescence; and (ii) promotion by cellular and extracellular signals leading to immortalized cells that are resistant to growth‐inhibitory signals, apoptosis and antitumor immunity. Inflammation could contribute to both. Persistent inflammation leads to tissue damage, resulting in increased cellular turnover. Nitric oxide (NO) and reactive oxygen species (ROS) from inflammatory cells may induce DNA damage, which increases the possibility of the emergence of cells possessing a high risk of malignant transformation. In addition, various factors from inflammatory cells could promote tumor development, as shown in Table 1.
Table 1.
Macrophage products with potential to influence tumorigenesis
| Biological effect | Factor s and molecules |
|---|---|
| Growth and survival | bFGF, EGF, HGF, PDGF, IL‐6, TNF, polyamine, PGE2, WNT7b |
| Angiogenesis | VEGF, MMP‐9, IL‐1, IL‐8, uPA, CXCL‐1,CXCL‐8,HIF‐1α, HIF‐2α, PGE2 |
| Tissue invasion | Chemokines, PGE2, MMP‐9, uPA, TIMP, plasmin |
| DNA damage | ROS, NO |
| Suppressors of immune responses | IL‐10, TGF‐β, IDO, arginase, PGE2, ROS, NO |
| Activation of immune responses | IL‐12, TNF, CD80, CD86, MHC, CD40, chemokines, BAFF |
IL, interleukin; MMP, matrix metalloproteinases; NO, nitric oxide; ROS, reactive oxygen species; TIMP, tissue inhibitor of metalloproteinases; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor.
Roles of myeloid cells in cancer development and progression
Tumors arise in the context of stroma, which includes lymphocytes, myeloid cells, fibroblasts and connective tissue. Myeloid cells (also known as innate immune cells) including macrophages, dendritic cells, granulocytes, eosinophils and mast cells, have a remarkable ability to produce a variety of factors and small chemicals that can influence tumorigenesis.( 9 ) Figure 1 and Table 1 show factors from activated macrophages that promote the growth and survival of tumors, angiogenesis, tissue invasion and metastases.
Tumor‐associated macrophages (TAMs) can also interfere with T cell responses to cancer (Fig. 1). Activated macrophages produce immunosuppressive cytokines, such as IL‐10 and transforming growth factor (TGF)‐β, which inhibit antigen presentation by dendritic cells and induce regulatory T cells (a subset of immunosuppressive T cells).( 10 ) TGF‐β also suppresses the transcription of genes encoding multiple key proteins of the ‘cytotoxic program’ of CD8+ cytotoxic T cells (CTL), such as perforin and granzymes.( 11 ) Macrophages produce indoleamine‐2,3‐dioxygenase (IDO), which converts tryptophan to kynurenine and suppresses T cell proliferation.( 12 ) NO produced by inducible nitric oxide synthase (iNOS) inhibits IL‐2 receptor signaling and induces T cell apoptosis.( 13 ) Recently, tumor‐associated myeloid cells, which include Gr‐1+ immature myeloid cells and F4/80+ macrophages, but not conventional activated macrophages, have been shown to play an important role in the suppression of antitumor immunity depending on arginase and NO.( 14 ) Thus, ongoing inflammatory responses within the inflammatory stroma of tumors have the capacity to not only produce tumor‐promoting factors but to also profoundly suppress the ability of the host antitumor immune responses. Therefore, simultaneous suppression of tumor‐promoting inflammation and activation of antitumor immunity is an ideal strategy for cancer treatment, however, this is very hard because both use similar intracellular mechanisms.
Transcription factors activated by inflammatory cytokines and pathogen‐associated molecular patterns
Induction of inflammation by invading microorganisms is dependent on activation of macrophages and dendritic cells, which is initiated by recognizing conserved motifs of microbial origin, also known as pathogen‐associated molecular patterns (PAMPs) through TLRs and NODs.( 15 ) Recently, a number of endogenous molecules such as heat shock proteins (HSPs) and the high mobility group box 1 (HMGB1) protein have also been shown to be capable of inducing the release of proinflammatory cytokines from the monocytes/macrophages via the TLRs.( 16 ) Then, chronic inflammation develops through the action of various inflammatory mediators including proinflammatory cytokines, chemokines and small chemical mediators. Among them, proinflammatory cytokines such as TNF‐α, IL‐1, IL‐6, IL‐12 and IFN play essential roles. These proinflammatory cytokines are mostly produced from activated myeloid and lymphoid cells and further activate these inflammatory cells. Such proinflammatory cytokines have been implicated in inflammation‐associated tumors. Those proinflammatory cytokines, as well as TLRs and NODs, ultimately activate NF‐κB, AP‐1, STAT1 and STAT3 transcription factors (Fig. 2). In myeloid cells, these transcription factors are used to produce inflammatory mediators, cytokines and growth factors. In target tumors or pretumorous cells, these transcription factors regulate cell proliferation, antiapoptosis, immune regulation and invasion. Therefore, these transcription factors play multiple roles in inflammation‐linked tumor development depending on the cell types. The general schema of this situation is shown in Fig. 3a.
Figure 2.
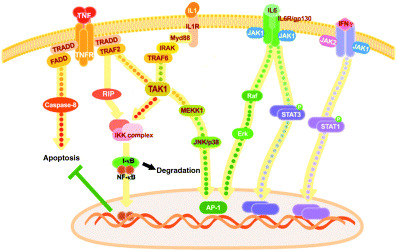
Signal transduction pathways of proinflammatory cytokines.
Figure 3.
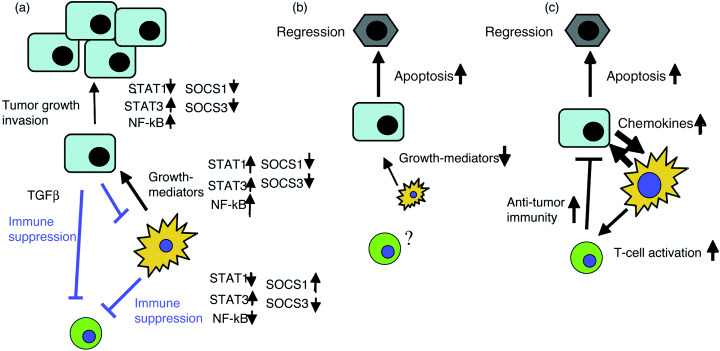
Role of inflammatory transcription factors on tumor development and the mechanism of tumor regression by blocking of NF‐kB or STAT3. (a) Tumor progression state with tumor‐associated inflammation and suppression of antitumor immunity. In tumor cells, STAT3 and NF‐kB are activated via cytokines from myeloid cells and support proliferation and antiapoptosis. This is associated with reduced expression of suppressors of cytokine signaling (SOCS)1 and SOCS3. Reduced STAT1 activation is often associated with highly malignant tumors. In myeloid cells, for growth promoting factor production, NF‐kB, STAT1 and STAT3 usually work positively, while SOCS1 and SOCS3 work negatively. However, in the status of the suppression of antitumor immunity, STAT3 is activated and suppresses NF‐kB and STAT1. Enhanced SOCS1 and reduced SOCS3 expression is associated with this situation. (b) When the NF‐kB pathway is blocked, cytokine production from myeloid cells is blocked and tumor cell apoptosis is accelerated. However, NF‐kB blockage may weaken antitumor immunity. (c) When STAT3 is blocked, tumor cell apoptosis is enhanced and antitumor immunity is reinforced by increasing chemokine production from tumor cells and by enhancing dendritic activation and cytotoxic T cells (CTL) activation. However, this may enhance inflammation which supplies tumor‐promoting factors.
NF‐κB pathway
NF‐κB can be activated by a number of stimuli and more than 100 genes are induced on its activation.( 17 ) So far, two important NF‐κB signaling pathways have been described.( 18 ) The classical pathway is activated by a variety of inflammatory signals including TNF‐α, IL‐1, viruses, TLRs, NODs and antigen receptors, resulting in the coordinated expression of multiple inflammatory and innate immune genes. Signal transduction pathways from receptors to NF‐κB have been extensively studied. A number of adaptor molecules are involved in this process. A simple view of this pathway is illustrated in Figure 2. The alternative pathway is strictly dependent on IKK‐α homodimers and is activated by lymphotoxin β, B cell‐activating factor belonging to the TNF family (BAFF), and CD40 ligand (CD40L). The alternative pathway is important for survival of premature B cells and development of secondary lymphoid organs, but may have little importance in inflammation‐associated tumors.
The classic NF‐κB pathway activates the tripartite IKK complex, leading to phosphorylation‐induced I‐κB degradation.( 18 ) The best characterized components of the classic IKK complex are I‐κB‐kinase‐1 (IKK‐1 or IKK‐α), IKK‐2 (IKK‐β) and the NF‐κB essential modulator (NEMO) (also called IKK‐γ). IKK‐2, the essential kinase for the classic NF‐κB signaling pathway, phosphorylates I‐κB, followed by ubiquitination and eventual proteasomal degradation. Following I‐κB degradation, NF‐κB translocates to the nucleus, where it binds and activates κB motif‐containing promoters. The most predominant NF‐κB dimer activated by the classical pathway is p65:p50. Translocation of p65:p50 to the nucleus results in the transcription of a myriad of proinflammatory genes, such as cytokines (IL‐1, IL‐6, TNF‐α, GM‐CSF, IL‐4), chemokines (IL‐8, RANTES, macrophage inflammatory protein‐1α, monocyte chemotactic protein‐1), pro‐angiogenic factors (vascular endothelial growth factor [VEGF]), adhesion molecules (vascular cell adhesion molecule‐1, intercellular adhesion molecule‐1, E‐selectin), antiapoptotic proteins (Bcl2, Bcl‐XL, cIAP, FLIP) and inducible enzymes (iNOS, COX‐2, MMP‐9). Most of them overlap with the target genes of STAT1 and STAT3 activated by inflammatory stimuli. This is because the promoter region of many of those genes contains GAS or ISRE sites (STAT‐binding elements or IRF binding elements) in addition to κB (NF‐κB binding element) sites. Therefore, those target genes are often additively or synergistically activated in combination with NF‐κB activating stimulation and STAT‐activating cytokines.
An interesting feature of the TNF signaling network is the existence of extensive cross‐talk between apoptosis, NF‐κB and JNK signaling.( 19 ) In the absence of NF‐κB activity, cellular susceptibility to TNF‐induced apoptosis increases, whereas the enforced activation of NF‐κB protects against apoptosis (Fig. 2). Moreover, TNF‐induced JNK activation is stronger and more prolonged in cells lacking NF‐κB, resulting in the promotion of apoptosis.
STAT
Cytokines including IFN‐γ, IL‐6 and IL‐12 use the so called the JAK/STAT pathway (Fig. 2). These receptors lack intrinsic kinase activity but are associated with Janus kinases (JAKs), a family of protein tyrosine kinases.( 20 ) Upon ligand binding to cytokine receptors and subsequent receptor dimerization, JAKs are activated to phosphorylate tyrosine residues in the intracellular domain of cytokine receptors. These phosphorylated tyrosines then become docking sites for a number of intracellular proteins, most representatively a family of signal transducers and activators of transcription (STATs). STATs, after tyrosine phosphorylation at the C‐terminal region by JAKs, dimerize and translocate to the nucleus, where they induce the expression of their target genes by binding to GAS (γ‐activated sequence) or other specific motifs in the promoter region.
Various combinations of JAKs (JAK1, JAK2, JAK3 and TYK2) and STATs (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b and STAT6) are activated to transduce cytokine signals, but recent gene targeting studies have clarified the non‐redundant and specific roles of each JAK and STAT member in different cytokines. Among them, STAT1 is relatively specific to IFNs, STAT3 is activated by IL‐6 and other gp130‐related cytokines, STAT4 is activated by IL‐12, and STAT6 is specifically activated by IL‐4 and IL‐13. STAT5 is activated by various cytokines including IL‐2, IL‐3, erythropoietin and growth hormone.
Among STATs, STAT1 and STAT3 are strongly associated with tumors. Constitutive STAT3 activation and STAT1 repression are often observed in many types of tumors.( 21 , 22 ) This is because STAT3 functions as an oncogene, which promotes cell proliferation and survival. In contrast, STAT1 induces apoptosis. STAT1 is essential for IFN‐γ signaling and promotes tissue damage and apoptosis induced by NK cells and CTLs. Thus, STAT1 probably plays a positive role in the early stage of inflammation‐associated tumors by accelerating inflammation but inhibits tumorigenesis in the late stage by inducing apoptosis. Conversely, STAT3 is activated by anti‐inflammatory cytokine IL‐10 and suppresses the NF‐κB pathway.( 23 ) The molecular mechanism of this cross‐talk has not been completely understood. Thus, STAT3 functions as a tumor promoter in tumor cells but suppresses inflammation by infiltrated myeloid and lymphoid cells. In contrast, STAT1 inhibits tumor cell growth, but enhances inflammation in myeloid cells. Therefore, STAT activation in tumors plays a dual role in cancer development. The concept of this schema is described in Figure 3a.
AP‐1 and interferon regulatory factors
Inflammatory cytokines also activate the MAP kinase cascade.( 24 ) IL‐1 and TNF‐α activate MAP kinase kinase kinases (MAPKKKs) including TAK1 and apoptosis‐signal regulated kinase 1 (ASK1), resulting in the activation of c‐Jun NH2‐terminal kinase (JNK) and p38. Conversely, IL‐6 activates Erk, which induces c‐fos, forming AP‐1 transcription factors (Fig. 2). It has been shown that p38 is essential for proinflammatory cytokine production.
Inflammatory cytokine production is regulated by another family of transcription factors called interferon regulatory factors (IRFs). IRF‐3 is an essential factor for IFN‐β production, which is initiated by the TRIF (Toll/IL‐1 receptor domain‐containing adaptor inducing IFN‐β)‐mediated pathway and cytosolic RNA helicases RIG‐I (retinoic acid inducible gene I).( 25 ) HCV serine protease NS3‐4A inactivates the RIG‐I pathway to block IFN‐β production.( 26 ) IRF‐5 is shown to be an important component for proinflammatory cytokine production through TLRs.( 25 ) IRF‐7 has been shown to be critical for induction of IFN‐α.( 27 ) Because of space limitations, we will not discuss these transcription factors in the present review.
Suppressors of cytokine signaling
A number of mechanisms have been reported to modulate cytokine signaling to prevent this overreaction of cytokines. Recent accumulating evidence indicates that a family of proteins, suppressors of cytokine signaling (SOCS) and cytokine‐inducible SH2 protein (CIS), are important negative regulators for cytokine signaling.( 28 ) Gene‐disrupted mice and human cancer studies have unexpectedly revealed significant roles of SOCS proteins in tumor development.
There are eight CIS/SOCS family proteins; each has a central SH2 domain, an amino‐terminal domain of variable length and sequence, and a carboxyl‐terminal 40‐amino‐acid module known as the SOCS box. The SOCS family members best characterized to date are CIS, SOCS1, SOCS2 and SOCS3. CIS and SOCS2 bind to phosphorylated tyrosine residues on activated (phosphorylated) cytokine receptors and inhibit the recruitment and activation of STATs (especially STAT5). Both SOCS1 and SOCS3 can inhibit JAK tyrosine kinase activity because they have the kinase inhibitory region (KIR) in their N‐terminal domain, which is proposed to function as a pseudo substrate. While SOCS1 directly binds to the activation loop of JAKs through its SH2 domain, the SOCS3 SH2 domain binds to the cytokine receptor (e.g. Y757 [mouse] or Y759 [human] of gp130). Because binding of the SH2 domain of SOCS3 is relatively specific to receptors for STAT3‐activating cytokines, such as IL‐6, G‐CSF and leptin, the effect of SOCS3 seems to be relatively restricted to STAT3. In contrast, SOCS1 is strongly induced by a small number of cytokines such as IFN‐γ, and the dominant effect of SOCS1 is relatively specific to STAT1.
The function of the SOCS‐box is the recruitment of the ubiquitin‐transferase system. The SOCS box interacts with Elongins B and C, Cullin‐5 or Cullin‐2, Rbx‐1 and E2.( 29 ) Thus, CIS/SOCS family proteins, as well as other SOCS box‐containing molecules, probably function as E3 ubiquitin ligases and mediate the degradation of proteins associated through their N‐terminal regions. Therefore, SOCS1 and SOCS3 seem to combine specific inhibition (i.e. kinase inhibition by KIR) and a generic mechanism of targeting interacting proteins for proteasomal degradation.
IKK/NF‐kB links inflammation to cancer
Based on many functions of NF‐κB target genes, a close relationship between NF‐κB and cancer has already been extensively reviewed.( 7 , 30 , 31 ) Therefore, in this review, only several recent important reports are reviewed. Tanaka's group established an inflammation‐dependent colon tumor‐model in mice, in which tumors developed after a single dose of genotoxic colonic carcinogen, azoxymethane (AOM), followed by dextran sodium sulfate (DSS) in drinking water.( 32 ) In this model, deletion of IKK‐β in intestinal epithelial cells did not decrease inflammation, but rather, led to a dramatic decrease in tumor incidence without affecting tumor size.( 33 ) In contrast, deletion of IKK‐β in myeloid cells resulted in a significant decrease in tumor size. IKK‐β deletion diminishes the expression of proinflammatory cytokines that may serve as tumor growth factors. One of these cytokines, IL‐6, has been shown to play an important role in tumor growth, while TGF‐β has been shown to antagonize IL‐6 function.( 34 ) Thus, the specific inactivation of the IKK/NF‐κB pathway in both epithelial cells and inflammatory cells can attenuate the formation of inflammation‐associated tumors (Fig. 3b).
The positive role of NF‐κB in inflammation‐associated cancer was also demonstrated in Mdr2‐deficient mice, which develop cholestatic hepatitis followed by HCC.( 35 ) In this model, the inflammatory process triggered chronic activation of NF‐κB in hepatocytes, most likely through the enhanced production of TNF‐α by adjacent inflammatory cells. Suppression of NF‐κB activity in Mdr2 −/− mice from birth to 7 months of age had no effect on the early phases of tumorigenesis. By contrast, NF‐κB inhibition in later stages of tumor development resulted in apoptosis of transformed hepatocytes and failure to progress to HCC. Therefore, the suppression of NF‐κB in tumors, which was associated with high levels of TNF‐α, could induce apoptosis of tumor cells. This is probably because the caspase and JNK pathways downstream of TNF‐R1 become predominant in the absence of NF‐κB activation (2, 3).
NF‐κB activation plays a critical role in inflammation‐driven tumor progression, as demonstrated in a syngeneic colon and mammary cancer xenograft mouse model.( 36 ) After metastasized tumors were established, the mice were given a sublethal dose of lipopolysaccharide (LPS) to elicit systemic inflammation, which stimulated tumor growth. LPS‐induced metastatic growth response in this model depends on both TNF‐α from host hematopoietic cells and NF‐κB activation in tumor cells. Remarkably, the inhibition of NF‐κB in carcinoma cells converts the LPS‐induced growth response to LPS‐induced tumor regression. This conversion is not dependent on TNF‐α but on another member of the TNF superfamily, TRAIL, whose receptor is induced in NF‐κB‐deficient cancer cells by IFNs from host inflammatory cells. Thus, NF‐κB inhibition in tumor cells in combination with IFN administration may promote the activity of TRAIL to achieve enhanced tumor apoptosis.
From the examples provided by these extensive studies, the inhibition of NF‐κB in both tumor cells and inflammatory cells seems to be an effective therapeutic way to prevent cancer progression (Fig. 3b).
STAT3 functions as an oncogene
Accumulating evidence suggests that STAT3 is an oncogenic transcription factor( 21 , 22 ) (Fig. 3a,c). STAT3 is persistently phosphorylated in many types of human cancer cell lines or primary tumors, including HCC, breast cancer, prostate cancer, and head and neck cancer, and several hematological malignancies. Furthermore, STAT3 is necessary for v‐src‐induced transformation of fibroblasts, and a constitutively active mutant of STAT3 can transform fibroblasts in cooperation with other transcription factors.( 37 ) Several target genes of STAT3 have been identified using human tumor cell lines and rodent fibroblasts. Proteins that regulate cell survival, including Bcl‐2, Bcl‐xL, mcl‐1 and Fas, are direct targets of STAT3. Cell cycle regulators, cyclin D1, cyclin E1 and p21, can be activated by STAT3 in certain contexts. In addition, other transcription factors, including c‐myc, c‐jun and c‐fos, are themselves STAT3 targets.( 38 ) VEGF and TGF‐β have been shown to be targets of STAT3 and to contribute to tumor angiogenesis and fibrosis, respectively.( 39 ) Another important target of STAT3 is the tissue inhibitor of metalloproteinases‐1 (TIMP‐1), which inhibits matrix metalloproteinases (MMPs) and could be involved in tissue remodeling.( 40 )
A link between STAT3 and p53 has also been found.( 41 ) In this case, STAT3 functions as a transcriptional repressor of p53 expression. Blocking STAT3 in cancer cells upregulates the expression of p53, leading to p53‐mediated tumor cell apoptosis.
The molecular basis of hyper STAT3 activation in tumor cells has been poorly understood. One mechanism is autocrine production of IL‐6 from tumor cells and paracrine activation by IL‐6 and its related cytokines from stroma and infiltrated inflammatory cells. Indeed, the IL‐6 levels are usually high in tumor patients.( 42 ) Many oncogenic tyrosine kinases including the EGF receptor could phosphorylate STAT3. In addition, STAT3 is activated by various viral factors, such as HCV core protein,( 43 ) the Kaposi's sarcoma‐associated herpes virus (KSHV)‐encoded latency‐associated nuclear antigen (LANA),( 44 ) and viral IL‐6 and viral IL‐10 encoded in human herpes viruses. Reduced expression of SOCS3 could be a mechanism of constitutive activation of STAT3.( 45 , 46 )
STAT3 and inflammation‐associated tumor
STAT3 activation is recognized as an important link between inflammation and cancer. Ernst et al. generated a unique mouse model of gastric cancer that resembles human intestinal‐type adenocarcinoma.( 47 ) Their mice (gp130(757F/F) mice) carry a point mutation (Y757F) that disrupts SOCS3 and SHP2‐binding on the IL‐6 family receptor gp130. As a result, these mice show hyperactivation of STAT3, resulting in chronic gastric inflammation and distal stomach tumors. These phenotypes were dependent on the hyperactivation of STAT3, because gp130 (757F/F) mice in the STAT3(+/–) background prevented systemic inflammation and suppressed the growth of spontaneously arising gastric adenomas.( 48 ) More recently, this gp130 (757F/F) mutant mouse was crossed with IL‐6 deficient mice and mice lacking mature T and B cells.( 49 ) Surprisingly, the lack of IL‐6 or T and B cells did not have an impact on tumor growth. Rather, gp130(757F/F)/IL‐6(−/–) mice showed approximately 10–20‐fold more submucosal tumor invasion. This is associated with greater IL‐11 (which also uses gp130 as a receptor component) levels and MMP‐13 and MMP‐9 production. MMP‐13 was largely restricted to tumor‐associated stroma, and MMP‐9 was also expressed in polymorphonuclear cells and a subset of epithelial cells. Therefore, increased submucosal invasion in gp130(757F/F)/IL‐6(−/–) mice was most likely facilitated by augmented metalloproteinase activity driven by elevated IL‐11 levels. These studies found an important role of the STAT3 system in tumor invasion. However, the cytokine that activates gp130 and induces gastric cell tumor (that is not IL‐6) has not yet been identified. Identification of this cytokine could provide an important clue to prevent inflammation‐associated gastric tumor in humans.
In these gastric tumor models, one important target gene of STAT3 has been shown to be Smad7.( 50 ) The hyperactivation of STAT3 conferred resistance to the cytostatic effect of TGF‐β on cells through transcriptional induction of inhibitory Smad7. In addition, we have shown that STAT3 enhances TGF‐β production from hepatocytes (Ogata et al., unpublished data, 2006). Thus, STAT3 probably contributes to immunosuppression by producing TGF‐β from tumor cells, while STAT3 protects cells from the antiproliferative effect of TGF‐β by inducing Smad7. These studies provide a novel link for the cross‐talk between STAT and Smad signaling in gastric homeostasis.
STAT3 and antitumor immunity
As discussed, STAT3 is shown to upregulate the expression of numerous genes that are important for tumor progression. However, STAT3 also functions as an important transcriptional repressor. Especially, STAT3 mediates the anti‐inflammatory effect of IL‐10, which represses NF‐κB‐ and STAT1‐mediated inflammatory gene expression.( 40 ) We proposed a molecular basis for the reason why IL‐6 is proinflammatory while IL‐10 is anti‐inflammatory.( 50 ) In macrophages, STAT3 activation by IL‐6 is transient, while STAT3 activation by IL‐10 lasts a long time. We demonstrated that SOCS3 is a key regulator of this divergent action of these two cytokines. In macrophages lacking the SOCS3 gene or carrying a mutation of the SOCS3‐binding site (Y759F; equivalent to Y757F in previous gastric tumor mouse model) in gp130, not only IL‐10, but also IL‐6, suppressed LPS‐induced TNF‐α production. SOCS3 protein was strongly induced by both IL‐6 and IL‐10 in the presence of LPS but selectively inhibited IL‐6‐, but not IL‐10‐, mediated STAT3 activation because of selective binding of SOCS3 to gp130 but not to the IL‐10 receptor. Therefore, the constitutive activation of STAT3 by IL‐10 could suppress the NF‐κB and STAT1 pathways, while the transient activation of STAT3 by IL‐6 probably functions as proinflammatory.
Thus, constitutive STAT3 activation in tumor cells probably plays a preventive role in the initiation of inflammatory responses during cancer development. Indeed, active STAT3 in tumor cells has been shown to suppress the expression of proinflammatory mediators such as IL‐6, IP‐10 and RANTES, while blocking of STAT3 results in enhanced production of inflammatory mediators.( 51 ) In addition, we have shown that active STAT3 upregulates anti‐inflammatory mediators (immune suppressors) TGF‐β and IL‐10 (our unpublished observation). Therefore, blocking STAT3 in tumor cells promotes innate immunity, leading to tumor‐specific T‐cell responses (Fig. 3c). Constitutive activation of STAT3 has been shown to suppress dendritic cell maturation (inhibition of induction of maturation factors, such as TNF‐α, IL‐12, CD‐40, MHC molecules and T cell costimulators).( 52 ) Therefore, blocking of STAT3 in hematopoietic cells could enhance antitumor immunity. This was confirmed by using hematopoietic cell‐specific STAT3‐deficient mice. Kortylewski et al. observed a markedly enhanced function of dendritic cells, T cells, natural killer (NK) cells and neutrophils in tumor‐bearing mice with STAT3‐deficient hematopoietic cells, resulting in tumor regression.( 53 ) In addition, targeting STAT3 with a small‐molecule drug (CPA‐7) induces T cell‐ and NK cell‐dependent growth inhibition of established tumors.( 53 )
These studies elucidated the importance of STAT3 in both tumor cells and immune cells for tumor cell proliferation as well as suppression of antitumor immunity. Thus, suppression of STAT3 in vivo could be an effective way to prevent cancer development (Fig. 3c).
STAT1: Anti‐oncogene and inflammation promoter
In contrast to STAT3, STAT1 phosphorylation levels are usually low in tumor cells.( 21 ) While STAT3 promotes cell proliferation and antiapoptosis, STAT1 generally induces growth inhibition and apoptosis (Fig. 3a). STAT1 promotes tissue damage induced by NK and CTL, while STAT3 protects cells from the killing effect of these cells (Fig. 3a).
STAT1 is essential for the signaling of IFNs.( 20 ) Thus, STAT1 could be essential for protection from virus‐induced tumors. However, STAT1 is important for Th‐1 development of helper T cells, induction of proinflammatory mediators, such as iNOS and COX‐2 in myeloid cells, and activation of macrophages. STAT1 also induces various IFN‐inducible genes, many of which promote inflammation. Thus STAT1 functions as a promoter of inflammation‐mediated tumor progression.
Although STAT1‐deficient mice develop no spontaneous tumors, they are highly susceptible to chemical carcinogen‐induced tumorigenesis. An anti‐oncogenic feature of STAT1 is partly dependent on p53. Crossing the STAT1 knockout mice into a p53‐deficient background yields animals that develop tumors more rapidly, and with a broader spectrum of tumor types than is seen with p53 single mutants. Moreover, the levels of p53 are regulated by IFN/STAT1.( 54 )
STAT1 has been directly implicated in modulating apoptosis. For example, cells lacking STAT1 are less susceptible to TNF‐ or hypoxia‐induced cell death than wild type cells.( 55 )
As mentioned, tumor‐associated macrophages are able to inhibit a T cell‐mediated immune response in vitro via the induction of T cell apoptosis through arginase and NO. Analysis of STAT knockout mice indicates that STAT1, but not STAT3 or STAT6, is responsible for the activity of tumor‐associated macrophages.( 14 ) IFN‐γ from CTL is thought to be an important factor in tumor regression. These data suggest that blocking of STAT1 in inflammation‐associated cancer could reduce inflammation but simultaneously enhance tumor cell survival and reduce antitumor immunity. Thus the activation of STAT1 by cytokines including IFNs might be a reasonable choice for cancer treatment.
SOCS and human cancer
The antitumor activity of SOCS1 has been reported by several groups.( 26 ) SOCS1 may inhibit the development and/or progression of HCC, because SOCS1 expression is significantly reduced in HCC cells, which can be explained by the inactivation of the SOCS1 promoter due to the hypermethylation of CpG islands.( 56 , 57 ) SOCS1 was most frequently methylated (65%) among known methylated genes in HCC. The methylation of SOCS1, APC and p15 was more frequently seen in HCV‐positive HCC than in HCV/HBV‐negative HCC. These data suggest that promotion of the hypermethylation SOCS1 gene is an important event in HCC development and that viral infection may play a role in gene methylation.
SOCS1 deletion in tumor cells may enhance IL‐6‐mediated cell proliferation. In addition, we found that SOCS1‐silencing frequently occurred in the non‐tumor region in HCV‐infected patients.( 57 ) We also observed the hyperactivation of STAT1 and reduced activation of STAT3 in the liver of chemical carcinogen‐treated SOCS1+/– mice. SOCS1‐heterozygous mice are hypersensitive to dimethylnitrosamine‐induced hepatocarcinogenesis,( 57 ) and SOCS1‐deficient mice develop very severe colitis accompanied by spontaneous colon tumor.( 58 ) Interestingly, colon tumor development in SOCS1‐deficient mice is dependent on IFN‐γ but not on TNF‐α. Thus, the reduced expression of SOCS1 probably enhances tissue injury and inflammation, which promotes turnover of epithelial cells. SOCS1 could be a novel anti‐oncogene that suppresses inflammation‐induced carcinogenesis.
Like SOCS1, SOCS3 may also be involved in the development and progression of malignancies. Reduced expression of SOCS3 has been observed in human cancers and is associated with constitutive STAT3 activation. The methylation of the SOCS3 gene has been reported in lung cancer, HCC and squamous cell carcinoma (SCC).( 45 , 46 ) We also observed reduction of SOCS3 expression in the HCC region in HCV‐infected patients (Ogata et al., unpublished data, 2006) SOCS3 levels were inversely correlated with STAT3 activation in the non‐HCC regions. Using liver‐specific SOCS3‐deficient mice, we demonstrated that SOCS3 deletion in the liver resulted in the hyperactivation of STAT3 (Ogata et al., unpublished data, 2006) and enhanced carcinogen‐induced hepatic tumor development (Ogata et al., unpublished data). Thus, silencing of the SOCS3 gene promotes tumorigenesis. These data suggest that upregulation of SOCS1 and SOCS3 expression or administration of SOCS‐mimicking compounds may be effective for cancer treatment.
Therapeutic implications
As discussed, the inhibition of IKK‐β/NF‐κB and JAK/STAT3 appears to have multi‐inhibitory effects on tumor promotion by affecting not only tumor cell growth/survival but also the activity of infiltrating mononuclear cells. Because conventional chemotherapeutic drugs target malignant cells only, targeting not only tumor cells but also the inflammatory component of tumors is an attractive strategy. Anti‐inflammatory cytokine signal therapy combined with established cytocidal drugs, death cytokines, or therapeutic radiation could be more effective than alone.( 59 )
NF‐κB inhibitors could simultaneously induce apoptosis of tumor cells and reduce tumorigenic mediator production from inflammatory cells.( 30 ) Conversely, STAT3 inhibitors could also induce tumor cell apoptosis or growth arrest( 38 , 39 ) but enhance antitumor immunity.( 53 ) To extend anti‐inflammatory cytokine therapy to humans, the turning point of inflammation from antitumor immunity to a tumor promoting event must be identified. It is notable that 5,6‐dimethylxanthenone‐4‐acetic acid has been recently shown to induce an effective antitumor immune response by enhancing proinflammatory cytokine levels and modulating tumor‐associated macrophages.( 60 )
In addition, the impact of inflammatory cytokines on tumor development differs from one experimental model to another. For example, IL‐6 antagonist gp130‐Fc prevented colon tumor development induced by AOM/DSS,( 33 ) while IL‐6 depletion by gene targeting enhanced the invasion of gastric tumors of gp130(757F/F) mice.( 48 ) Thus, the most effective inhibitors must be chosen for each tumor type.
At present, numerous small compounds are developed as IKK inhibitors, NF‐κB suppressors, JAK inhibitors and STAT3 suppressors. Some of them are already approved by the US Food and Drug Administration (FDA), including thalidomide (an IKK inhibitor) and bortezomib (an inhibitor of I‐κB degradation).( 31 )
A number of small peptides that inhibit protein–protein interactions during signal transduction or nuclear translocation of NF‐κB or STAT3 have been developed. Other experimental reagents that suppress NF‐κB and STAT activation have been generated; for example, cell‐permeable SOCS3, SOCS1‐mimic peptide (Tkip), antisense oligodeoxynucleotides (ODNs) ribozymes, small interfering RNAs (siRNAs) and decoy ODNs for NF‐κB, STAT3, SOCS1 and SOCS3.( 30 , 31 , 60 ) These reagents were successfully used for preventing inflammation or related diseases, but they may also be useful to inflammation‐associated tumor. For example, SOCS1‐suppressing siRNA has been shown to strongly promote antitumor immunity of dendritic cells.( 61 )
Because the interaction between epithelial cell and commensal bacteria through TLRs plays an important role in intestinal inflammation, depletion of bacteria or inhibition of TLRs may be valuable for treating colon tumors. In connection with this, a number of reports have suggested that probiotics can reduce colon cancer risk.( 62 )
Conclusion
In summary, great strides have been made toward understanding the molecular machinery that links inflammation and cancer. The remaining challenges, however, include identifying the mechanisms that lead to and maintain the activation of NF‐κB and STAT3 in both the malignant and the inflammatory components of the tumor. In addition, we do not clearly understand the mechanism of conversion of antitumor immunity to tumor‐supporting inflammation. Nevertheless, the translation of basic findings described here into clinical practice is already underway.
References
- 1. Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 2005; 7: 211–7. [DOI] [PubMed] [Google Scholar]
- 2. Coussens LM, Werb Z. Inflammation and cancer. Nature 2002; 420: 860–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Koike K. Molecular basis of hepatitis C virus‐associated hepatocarcinogenesis: lessons from animal model studies. Clin Gastroenterol Hepatol 2005; 10 (Suppl. 2): 132–5. [DOI] [PubMed] [Google Scholar]
- 4. Roder DM. The epidemiology of gastric cancer. Gastric Cancer 2002; 5 (Suppl. 1): 5–11. [DOI] [PubMed] [Google Scholar]
- 5. Thun MJ et al. Nonsteroidal anti‐inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst 2002; 94: 252–66. [DOI] [PubMed] [Google Scholar]
- 6. Manning CB, Vallyathan V, Mossman BT. Diseases caused by asbestos: mechanisms of injury and disease development. Int Immunopharmacol 2002; 2: 191–200. [DOI] [PubMed] [Google Scholar]
- 7. Karin M, Greten FR. NF‐kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol 2005; 5: 749–59. [DOI] [PubMed] [Google Scholar]
- 8. Strober W, Murray PJ, Kitani A, Watanabe T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nature Rev Immunol 2006; 6: 9–20. [DOI] [PubMed] [Google Scholar]
- 9. Nathan C, Sporn M. Cytokines in context. J Cell Biol 1991; 113: 981–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Horwitz DA, Zheng SG, Gray JD. The role of the combination of IL‐2 and TGF‐beta or IL‐10 in the generation and function of CD4+ CD25+ and CD8+ regulatory T cell subsets. J Leukoc Biol 2003; 74: 471–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Trapani JA. The dual adverse effects of TGF‐beta secretion on tumor progression. Cancer Cell 2005; 8: 349–50. [DOI] [PubMed] [Google Scholar]
- 12. Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med 1999; 189: 1363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bobe P, Benihoud K, Grandjon D, Opolon P, Pritchard LL, Huchet R. Nitric oxide mediation of active immunosuppression associated with graft‐versus‐host reaction. Blood 1999; 94: 1028–37. [PubMed] [Google Scholar]
- 14. Kusmartsev S, Gabrilovich DI. STAT1 signaling regulates tumor‐associated macrophage‐mediated T cell deletion. J Immunol 2005; 174: 4880–91. [DOI] [PubMed] [Google Scholar]
- 15. Takeda K, Akira S. Toll‐like receptors in innate immunity. Int Immunol 2005; 17: 1–14. [DOI] [PubMed] [Google Scholar]
- 16. Tsan MF. Toll‐like receptors, inflammation and cancer. Semin Cancer Biol 2006; 16: 32–7. [DOI] [PubMed] [Google Scholar]
- 17. Pahl HL. Activators and target genes of Rel/NF‐kB transcription factors. Oncogene 1999; 18: 6853–66. [DOI] [PubMed] [Google Scholar]
- 18. Bonizzi G, Karin M. The two NF‐kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol 2004; 25: 280–8. [DOI] [PubMed] [Google Scholar]
- 19. Chen G, Goeddel DV. TNF‐R1 signaling: a beautiful pathway. Science 2002; 296: 1634–5. [DOI] [PubMed] [Google Scholar]
- 20. O'Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell 2002; 109: 121–31. [DOI] [PubMed] [Google Scholar]
- 21. Haura EB, Turkson J, Jove R. Mechanisms of disease: Insights into the emerging role of signal transducers and activators of transcription in cancer. Nat Clin Pract Oncol 2005; 2: 315–24. [DOI] [PubMed] [Google Scholar]
- 22. Hodge DR, Hurt EM, Farrar WL. The role of IL‐6 and STAT3 in inflammation and cancer. Eur J Cancer 2005; 41: 2502–12. [DOI] [PubMed] [Google Scholar]
- 23. Lang R. Tuning of macrophage responses by Stat3‐inducing cytokines: molecular mechanisms and consequences in infection. Immunobiology 2005; 210: 63–76. Review. [DOI] [PubMed] [Google Scholar]
- 24. Kaminska B. MAPK signalling pathways as molecular targets for anti‐inflammatory therapy‐from molecular mechanisms to therapeutic benefits. Biochim Biophys Acta 2005; 1754: 253–62. [DOI] [PubMed] [Google Scholar]
- 25. Honda K, Yanai H, Takaoka A, Taniguchi T. Regulation of the type I IFN induction: a current view. Int Immunol 2005; 17: 1367–78. [DOI] [PubMed] [Google Scholar]
- 26. Meylan E, Curran J, Hofmann K et al. Cardif is an adaptor protein in the RIG‐I antiviral pathway and is targeted by hepatitis C virus. Nature 2005; 437: 1167–72. [DOI] [PubMed] [Google Scholar]
- 27. Honda K, Ohba Y, Yanai H et al. Spatiotemporal regulation of MyD88‐IRF‐7 signalling for robust type‐I interferon induction. Nature 2005; 434: 1035–40. [DOI] [PubMed] [Google Scholar]
- 28. Kubo M, Hanada T, Yoshimura A. Suppressors of cytokine signaling and immunity. Nat Immunol 2003; 4: 1169–76. [DOI] [PubMed] [Google Scholar]
- 29. Kamura T, Maenaka K, Kotoshiba S et al. VHL‐box and SOCS‐box domains determine binding specificity for Cul2‐Rbx1 and Cul5‐Rbx2 modules of ubiquitin ligases. Genes Dev 2004; 18: 3055–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Luo JL, Kamata H, Karin M. IKK/NF‐kappaB signaling: balancing life and death – a new approach to cancer therapy. J Clin Invest 2005; 115: 2625–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li Q, Withoff S, Verma IM. Inflammation‐associated cancer: NF‐kappaB is the lynchpin. Trends Immunol 2005; 26: 318–25. [DOI] [PubMed] [Google Scholar]
- 32. Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H. A novel inflammation‐related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci 2003; 94: 965–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Greten FR et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis‐associated cancer. Cell 2004; 118: 285–96. [DOI] [PubMed] [Google Scholar]
- 34. Becker C et al. TGF‐beta suppresses tumor progression in colon cancer by inhibition of IL‐6 trans‐signaling. Immunity 2004; 21: 491–501. [DOI] [PubMed] [Google Scholar]
- 35. Pikarsky E et al. NF‐kappaB functions as a tumour promoter in inflammation‐associated cancer. Nature 2004; 431: 461–6. [DOI] [PubMed] [Google Scholar]
- 36. Luo JL, Maeda S, Hsu LC, Yagita H, Karin M. Inhibition of NF‐kappaB in cancer cells converts inflammation‐induced tumor growth mediated by TNFalpha to TRAIL‐mediated tumor regression. Cancer Cell 2004; 6: 297–305. [DOI] [PubMed] [Google Scholar]
- 37. Joo A, Aburatani H, Morii E, Iba H, Yoshimura A. STAT3 and MITF cooperatively induce cellular transformation through upregulation of c‐fos expression. Oncogene 2004; 23: 726–34. [DOI] [PubMed] [Google Scholar]
- 38. Hirano T, Ishihara K, Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL‐6 family of cytokine receptors. Oncogene 2000; 19: 2548–5. [DOI] [PubMed] [Google Scholar]
- 39. Xu Q, Briggs J, Park S et al. Targeting Stat3 blocks both HIF‐1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 2005; 24: 5552–60. [DOI] [PubMed] [Google Scholar]
- 40. Williams L, Bradley L, Smith A, Foxwell B. Signal transducer and activator of transcription 3 is the dominant mediator of the anti‐inflammatory effects of IL‐10 in human macrophages. J Immunol 2004; 172: 567–76. [DOI] [PubMed] [Google Scholar]
- 41. Niu G, Wright KL, Ma Y et al. Role of Stat3 in regulating p53 expression and function. Mol Cell Biol 2005; 25: 7432–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Giannitrapani L, Cervello M, Soresi M et al. Circulating IL‐6 and sIL‐6R in patients with hepatocellular carcinoma. Ann N Y Acad Sci 2002; 963: 46–52. [DOI] [PubMed] [Google Scholar]
- 43. Yoshida T, Hanada T, Tokuhisa T et al. Activation of STAT3 by the hepatitis C virus core protein leads to cellular transformation. J Exp Med 2002; 196: 641–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Muromoto R, Okabe K, Fujimuro M et al. Physical and functional interactions between STAT3 and Kaposi's sarcoma‐associated herpes virus‐encoded LANA. FEBS Lett 2005. [DOI] [PubMed]
- 45. He B, You L, Uematsu K et al. SOCS‐3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc Natl Acad Sci USA 2003; 100: 14133–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Niwa Y, Kanda H, Shikauchi Y et al. Methylation silencing of SOCS‐3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene 2005; 24: 6406–17. [DOI] [PubMed] [Google Scholar]
- 47. Tebbutt NC, Giraud AS, Inglese M et al. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT‐mediated trefoil gene activation in gp130 mutant mice. Nat Med 2002; 8: 1089–97. [DOI] [PubMed] [Google Scholar]
- 48. Jenkins BJ, Grail D, Nheu T et al. Hyperactivation of Stat3 in gp130 mutant mice promotes gastric hyperproliferation and desensitizes TGF‐beta signaling. Nat Med 2005; 11: 845–52. [DOI] [PubMed] [Google Scholar]
- 49. Howlett M, Judd LM, Jenkins B et al. Differential regulation of gastric tumor growth by cytokines that signal exclusively through the coreceptor gp130. Gastroenterology 2005; 129: 1005–18. [DOI] [PubMed] [Google Scholar]
- 50. Yasukawa H, Ohishi M, Mori H et al. IL‐6 induces an anti‐inflammatory response in the absence of SOCS3 in macrophages. Nature Immunol 2003; 4: 551–6. [DOI] [PubMed] [Google Scholar]
- 51. Wang T, Niu G, Kortylewski M et al. Regulation of the innate and adaptive immune responses by Stat‐3 signaling in tumor cells. Nat Med 2004; 10: 48–54. [DOI] [PubMed] [Google Scholar]
- 52. Park SJ, Nakagawa T, Kitamura H et al. IL‐6 regulates in vivo dendritic cell differentiation through STAT3 activation. J Immunol 2004; 173: 3844–54. [DOI] [PubMed] [Google Scholar]
- 53. Kortylewski M, Kujawski M, Wang T et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med 2005; 11: 1314–21. [DOI] [PubMed] [Google Scholar]
- 54. Takaoka A, Hayakawa S, Yanai H et al. Integration of interferon‐alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 2003; 424: 516–23. [DOI] [PubMed] [Google Scholar]
- 55. Kumar A, Commane M, Flickinger T, Horvath CM, Stark GR. Defective TNF‐alpha‐induced apoptosis in STAT1‐null cells due to low constitutive levels of caspases. Science 1997; 278: 1630–2. [DOI] [PubMed] [Google Scholar]
- 56. Yoshikawa H, Matsubara K, Qian GS et al. SOCS‐1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth‐suppression activity. Nat Genet 2001; 28: 29–35. [DOI] [PubMed] [Google Scholar]
- 57. Yoshida T, Ogata H, Kamio M et al. SOCS1 is a suppressor of liver fibrosis and hepatitis‐induced carcinogenesis. J Exp Med 2004; 199: 1701–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chinen T, Kobayashi T, Ogata H et al. Suppressor of cytokine signaling‐1 regulates inflammatory bowel disease in which both interferon‐gamma and interleukin‐4 are involved. Gastroenterology 2006; 130: 373–88. [DOI] [PubMed] [Google Scholar]
- 59. Jassar AS, Suzuki E, Kapoor V et al. Activation of tumor‐associated macrophages by the vascular disrupting agent 5,6‐dimethylxanthenone‐4‐acetic acid induces an effective CD8+ T‐cell‐mediated antitumor immune response in murine models of lung cancer and mesothelioma. Cancer Res 2005; 65: 11752–61. [DOI] [PubMed] [Google Scholar]
- 60. Karin M, Yamamoto Y, Wang QM. The IKK NF‐kB system: a treasure trove for drug development. Nat Rev Drug Discov 2004; 3: 17–26. [DOI] [PubMed] [Google Scholar]
- 61. Shen L, Evel‐Kabler K, Strube R, Chen SY. Silencing of SOCS1 enhances antigen presentation by dendritic cells and antigen‐specific anti‐tumor immunity. Nat Biotechnol 2004; 22: 1546–53. [DOI] [PubMed] [Google Scholar]
- 62. Guarner F, Malagelada JR. Gut flora in health and disease. Lancet 2003; 361: 512–9. [DOI] [PubMed] [Google Scholar]