

Abstract
Type I interferon (IFN) protein is a cytokine with pleiotropic biological functions that include induction of apoptosis, inhibition of angiogenesis, and immunomodulation. We have demonstrated that intratumoral injection of an IFN‐α‐expressing adenovirus effectively induces cell death of cancer cells and elicits a systemic tumor‐specific immunity in several animal models. On the other hand, reports demonstrated that an elevation of IFN in the serum following an intramuscular delivery of a vector is able to activate antitumor immunity. In this study, we compared the intratumoral and systemic routes of IFN gene transfer with regard to the effect and safety of the treatment. Intratumoral injection of an IFN‐α adenovirus effectively activated tumor‐responsive lymphocytes and caused tumor suppression not only in the gene‐transduced tumors but also in distant tumors, which was more effective than the intravenous administration of the same vector. The expression of co‐stimulatory molecules on CD11c+ cells isolated from regional lymph nodes was enhanced by IFN gene transfer into the tumors. Systemic toxicity such as an elevation of hepatic enzymes was much lower in mice treated by intratumoral gene transfer than in those treated by systemic gene transfer. Our data suggest that the intratumoral route of the IFN vector is superior to intravenous administration, due to the effective induction of antitumor immunity and the lower toxicity. (Cancer Sci 2010)
Interferon‐alpha (IFN‐α) belongs to the group of type I interferons. It is produced by monocytes/macrophages, lymphoblastoid cells, fibroblasts, and plasmacytoid dendritic cells. Interferon‐alpha (IFN‐α) binds to the IFN‐α receptor CD118, and the IFN receptor is coupled to a Janus‐family tyrosine kinase, which phosphorylates signal‐transducing activators of transcription (STATs), and STATs translocate to the nucleus where they activate the transcription of several different genes including the synthesis of host cell proteins that contribute to the inhibition of viral replication.( 1 ) In addition to its antiviral activity, IFN‐α exhibits several other antitumor functions: (i) direct inhibitory effects on tumor cell growth; (ii) radio and chemosensitizing effects; (iii) anti‐angiogenic properties; and (iv) modulation of the immune system.( 2 , 3 , 4 , 5 ) These features might be of special interest for use in cancer treatments.
Among the above antitumor properties, the direct inhibitory effects on tumor cell growth/functions and the interaction of IFN‐α with chemotherapeutic drugs such as 5‐FU have been of particular interest.( 2 , 3 ) The cytokine has been used worldwide for treatment of a variety of cancers including chronic myelogeneous leukemia, melanoma, and renal cancer.( 2 , 5 ) However, clinical experiences with IFN protein therapy for many other solid cancers have generally not been encouraging.( 6 ) In the conventional regimen of IFN clinical trials, the recombinant IFN‐α protein is systemically administered through subcutaneous or intramuscular routes. Since the protein is rapidly degraded in the blood circulation and only a small portion of subcutaneously injected IFN‐α can reach the target sites,( 7 ) the overall limited therapeutic efficacy of treatments based on the IFN‐α protein may reflect the inability to target the cytokine to the right place and at the right dose. Alternative delivery strategies are needed to achieve a safe and effective IFN delivery in a clinical setting. In fact, it was reported that gene‐ and cell‐based delivery of type I IFNs into tumors suppressed growth of various cancers such as breast cancer, prostate cancer, renal cancer, hepatocellular carcinoma, basal cell carcinoma, bladder cancer, and leukemia.( 8 , 9 , 10 , 11 , 12 , 13 ) We have also shown that a single injection of the recombinant IFN‐α protein into a tumor did not result in the suppression of tumor growth, whereas an intratumoral injection of an IFN‐α‐expressing adenovirus effectively induced cell death of cancer cells and did suppress the growth of tumors including pancreatic, colon, and renal cancers.( 14 , 15 , 16 , 17 )
Type I IFNs were long thought to act mainly by suppressing tumor cell proliferation in vivo. However, more recently, it has been established that type I IFNs have important roles in regulating innate and adaptive arms of the immune system: up‐regulation of major histocompatibility complex (MHC) class I gene, promotion of the priming and survival of T cells, enhancement of humoral immunity, increase of the cytotoxic activity of natural killer (NK) cells and CD8+ T cells, and activation of dendritic cells (DCs).( 4 , 5 ) We have also shown that in addition to the direct cytotoxicity in the injected site, intratumoral injection of the IFN‐α adenovirus elicits a significant systemic tumor‐specific immunity in several animal models.( 15 , 16 ) On the other hand, it has been reported that an intramuscular delivery of the IFN‐α gene exhibited inhibition of tumor growth and that CD8+ T cells were required for the antitumor response,( 18 , 19 ) suggesting that an elevation of the IFN‐α level in the serum also activates the antitumor immunity. However, it has not been examined whether the high IFN‐α level in the tumor and blood circulation effectively induces antitumor immunity. It is difficult to inject high amounts of the IFN‐α adenovirus into the muscle of mice without a leakage of the vector into the blood circulation, which may result in a variety of serum IFN‐α levels among mice injected with the same amounts of IFN‐α vector. Therefore, in this study, the adenovirus was injected into the tail vein to express consistent IFN‐α at a high sustained level in the serum, and the antitumor immunity and safety were compared between the intratumoral and systemic routes of IFN gene transfer.
Materials and Methods
Tumor cell line and recombinant adenovirus vectors. CT26 and Renca are weakly immunogenic BALB/c‐derived colon and renal cancer cell lines, respectively, which were obtained from the American Type Culture Collection (Rockville, MD, USA). CT26 and Renca cells were confirmed to express MHC class I molecules (H‐2Kd and H‐2Dd) by flow cytometry; however, the heterogeneous MHC expression was observed in the subcutaneous tumors (data not shown). Cells were maintained in RPMI containing 10% FBS, 2 mM L‐glutamine, and 0.15% sodium bicarbonate (complete RPMI). The recombinant adenovirus vectors expressing mouse interferon‐α (Ad‐mIFN) or alkaline phosphatase (Ad‐AP) were prepared as described.( 15 , 20 ) The recombinant adenoviruses are based on the serotype 5 with a deletion of the entire E1 region and a part of the E3 region, and have the CAG promoter, which is a hybrid of the cytomegalovirus immediate early enhancer sequence and the chicken β‐actin/rabbit β‐globin promoter, in the deleted E1 region.( 21 ) A cesium chloride‐purified virus was desalted using a sterile Bio‐Gel P‐6 DG chromatography column (Econopac DG 10; BioRad, Hercules, CA, USA) and diluted for storage in a 13% glycerol/PBS solution. All viral preparations were free of the E1+ adenovirus by PCR assay.( 22 )
In vivo tumor inoculation and IFN‐α gene transfer. Tumor cells were prepared in a total volume of 100 μL PBS (5 × 106 of Renca and 1 × 106 of CT26 cells) and injected subcutaneously into the leg of BALB/c mice (Charles River Japan, Kanagawa, Japan). When the subcutaneous tumor was established (∼0.5 cm in diameter), 50 μL of Ad‐mIFN or control vector (Ad‐AP) was injected once into the tumors or into the tail vein. The injection of 1 × 108 PFU (plaque forming unit) of Ad‐AP showed that 70–80% of the cells were stained in the entire CT26 tumors. The shortest (r) and longest (l) tumor diameters were measured and the tumor volume was determined as r 2 × l/2.
To deplete NK cells before and during the treatment with IFN‐α gene transfer, BALB/c nude mice (Charles River Japan) received intraperitoneal injections of 0.5 mg of anti‐asialo GM1 antibody targeting NK cells (Wako Pure Chemical Industries, Tokyo, Japan). Administration of antibody started 2 days after the inoculation of CT26 cells, and the injection was repeated every 5–6 days throughout the entire experimental period. Flow cytometry showed that more than 80% of NK cells were depleted in the antibody‐treated mice.
Reverse transcription–polymerase chain reaction (RT‐PCR) and ELISA analyses of IFN‐α expression. Subcutaneous tumors were collected 4 days after the intratumoral or intravenous injection of Ad‐mIFN or Ad‐AP in the CT26 tumor‐bearing BALB/c mice. To examine the expression of the IFN‐α gene in the tumors, RT‐PCR amplification was carried out using total RNA in a 50 μL of the PCR mixture with the following primer sets: IFN‐α upstream (5′‐GATGGTTCTGGCTGTGATGAG‐3′) and downstream (5′‐GATGTTCAGGATCTGCTGGGT‐3′) primers; β‐actin upstream (5′‐CCTCTATGCCAACACAGTGC‐3′) and downstream (5′‐ATACTCCTGCTTGCTGATCC‐3′) primers. In total, 33 cycles (β‐actin, 25 cycles) of the PCR were carried out at 94°C for 1 min, 60°C for 1 min, and 72°C for 2 min. The PCR products were electrophoresed on a 2% agarose gel. Interferon‐alpha (IFN‐α) concentration in the serum, tumor and organs was measured by enzyme‐linked immunosorbent assay (ELISA; Immunotech, Marseille Cedex, France).
ELISpot assays. Interferon‐gamma (IFN‐γ) ELISpot kits (BD Bioscience, San Jose, CA, USA) were used according to the manufacturer’s instructions. CT26 cells were inoculated into the BALB/c mice, and 7 days later 1 × 108 PFU of Ad‐mIFN or Ad‐AP was administered into the tumors or into the tail vein. Twenty days later, splenocytes were harvested from the mice, and the splenocytes (1 × 105) and mitomycin C (MMC)‐treated CT26 cells (1 × 104) were co‐cultured for 20 h at 37°C in complete RPMI medium in 96‐well plates pre‐coated with antimouse IFN‐γ antibody (BD Bioscience) in triplicate. After aspirating the cells away and washing the wells with de‐ionized water, a biotinylated antimouse IFN‐γ antibody (2 μg/mL) was added and incubated for 2 h at room temperature. After extensive washing, a streptavidin–horseradish peroxidase solution was added and incubated for 1 h at room temperature. Then, an aminoethyl carbazole substrate solution was added, and the plate was incubated for 15 min. Spots were counted under a stereomicroscope.
Flow cytometry of co‐stimulatory molecules. The regional lymph nodes were collected from the CT26 tumor‐bearing mice 10 days after the intratumoral or intravenous injection of Ad‐mIFN or Ad‐AP (1 × 108 PFU). The 1 × 106 of cells from the lymph nodes were incubated at 4°C for 30 min with phycoerythrin‐conjugated hamster antimouse CD11c monoclonal antibody (HL3; IgG; BD Biosciences), FITC‐conjugated rat antimouse CD40 monoclonal antibody (3/23; IgG; BD Biosciences), FITC‐conjugated hamster antimouse CD80 monoclonal antibody (16‐10A1; IgG; BD Biosciences), FITC‐conjugated rat antimouse CD86 monoclonal antibody (GL1; IgG; BD Biosciences), or isotype control antibody (IgG), and then washed twice with PBS containing 2% BSA. Flow cytometry was carried out using a FACScan system (BD Bioscience).
Immunohistochemistry. Immunostaining was performed using the streptavidin–biotin–peroxidase complex techniques (Nichirei, Tokyo, Japan). Consecutive cryostat tissue sections (5 μm) were mounted on glass slides and fixed in 99.5% ethanol for 20 min. After blocking with normal rat serum, the sections were stained with rat antimouse CD4 and CD8 antibodies (BD Biosciences). The cryostat sections were also processed for the terminal deoxynucleotidyltransferase‐mediated dUTP‐digoxigenin nick‐end‐labeling (TUNEL) assay (Intergen Company, Purchase, NY, USA). Negative controls without primary antibodies were examined in all cases. The sections were counterstained with methylgreen.
Statistical analysis. Two‐sided t‐tests were used to validate the significance of the observed differences, which were considered statistically significant when P < 0.05.
Results
Distribution of interferon‐α in various organs following intratumoral or intravenous administration of Ad‐mIFN. To confirm the in vivo expression of the IFN‐α gene, total RNA of CT26 subcutaneous tumors was subjected to an RT‐PCR analysis of the gene 4 days after the intratumoral or intravenous injection of Ad‐mIFN or control Ad‐AP (1 × 108 PFU). Intratumoral IFN‐α gene transfer yielded a significant level of IFN‐α expression in the tumor, whereas the intravenous route did not show the IFN‐α band there (Fig. 1a). The IFN‐α expression was not detected in the tumor injected with Ad‐AP (Fig. 1a).
Figure 1.
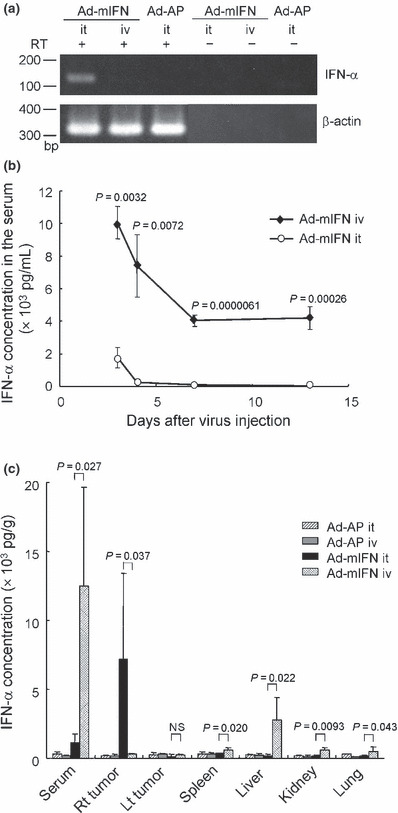
Distribution of interferon‐alpha (IFN‐α) in the sera, subcutaneous tumors, and various organs. (a) Expression of IFN‐α gene in subcutaneous tumors. The 1 × 108 PFU (plaque forming unit) of recombinant adenovirus vectors expressing mouse interferon‐α (Ad‐mIFN) or alkaline phosphatase (Ad‐AP) was injected into the CT26 subcutaneous tumor or into the tail vein, and 4 days later the subcutaneous tumor was isolated, and IFN‐α expression was examined by RT‐PCR analysis. (b) Time course of IFN‐α expression in the serum. The 1 × 108 PFU of Ad‐mIFN was injected into the subcutaneous tumor or into the tail vein, and IFN‐α concentration in the serum was measured by ELISA 3, 4, 7, and 13 days after the injection (n = 4). iv, intravenous injection; it, intratumoral injection. (c) Interferon‐alpha (IFN‐α) concentration in the sera, tumors, and organs. The 1 × 108 PFU of adenoviruses was injected into the subcutaneous tumor or into the tail vein, and 3 days later IFN‐α concentration was measured (n = 4). Rt, right; Lt, left.
To determine the kinetics of IFN‐α expression in the serum, the IFN‐α levels were measured 3, 4, 7, and 13 days after the intratumoral or intravenous injection of Ad‐mIFN. In the mice treated by intratumoral IFN‐α gene transfer, the cytokine was slightly elevated at Day 3 and returned to a base line by Day 4, whereas in the mice with the intravenous injection of Ad‐mIFN the peak levels in the serum were observed at Day 3 and the expression continued for more than 13 days, demonstrating that the intravenous gene transfer leads to the sustained high IFN‐α expression in the blood circulation (Fig. 1b).
Then, to examine the in vivo distribution of IFN‐α expression, we inoculated CT26 cells into both legs of BALB/c mice, followed by adenovirus (1 × 108 PFU) injection either into the right tumors or into the tail veins. Three days later the IFN‐α levels were measured in the serum, subcutaneous tumors, and various organs such as the spleen, liver, kidney, and lung. In the mice treated by intratumoral IFN‐α gene transfer, the serum showed a slight elevation of IFN‐α, whereas its concentration was highest in the right virus‐injected tumor (Fig. 1c), which was approximately 10‐fold higher than the serum as previously described.( 14 , 15 , 17 , 23 ) In the mice treated by intravenous IFN‐α gene transfer, the IFN‐α level was significantly elevated in the serum and various organs including the liver but not in the subcutaneous tumors compared with the intratumoral IFN‐α gene transfer (Fig. 1c). The IFN‐α was not elevated in the serum, tumors, or organs in the mice that received the administration of Ad‐AP.
Antitumor effects of intratumoral and intravenous adminis‐tration of Ad‐mIFN. To examine the in vivo antitumor effect of the IFN‐α gene transduction, various amounts (5 × 106, 5 × 107, 1 × 108, and 5 × 108 PFU) of adenoviruses were injected into right tumors or into the tail veins in mice with CT26 tumors on both legs. The intravenous injection of Ad‐mIFN did not show a definite suppressive effect except at the highest dose examined (5 × 108 PFU), which resulted in tumor suppression comparable with that of the intratumoral injection (Fig. 2a). The intratumoral injection of Ad‐mIFN showed remarkable tumor suppressive effects in not only the vector‐injected right tumors but also in the vector‐uninjected left tumors at all doses examined (Fig. 2a,b). Tumor volumes were not changed in the mice treated by intratumoral and intravenous injections of Ad‐AP at any doses (Fig. 2a), which were similar to those in the no treatment group (data not shown).
Figure 2.

Intratumoral interferon‐alpha (IFN‐α) gene transfer induces a systemic antitumor effect. CT26 cells were inoculated in both legs in BALB/c mice, and 7 days later recombinant adenovirus vectors expressing mouse interferon‐α (Ad‐mIFN) or alkaline phosphatase (Ad‐AP) was injected into the subcutaneous tumor on the right leg or into the tail vein. (a) Growth suppression of the CT26 subcutaneous tumors 20 days after the tumor inoculation. Various amounts (5 × 106, 5 × 107, 1 × 108, and 5 × 108 PFU [plaque forming unit]) of adenoviruses were injected, and 13 days after the virus injection tumor volume was measured (n = 6). In the mice treated by intravenous injection of adenoviruses, tumor volumes on both legs were combined to calculate the average. (b) Time course of CT26 tumor growth following intratumoral IFN‐α gene transfer. The 5 × 107 PFU of adenoviruses was injected, and tumor volume was measured at indicated days (n = 6). (c) Interferon‐alpha (IFN‐α) levels after the injection of various amounts of Ad‐mIFN. Various amounts of Ad‐mIFN were injected into the subcutaneous tumor or into the tail vein, and 3 days later IFN‐α concentration was measured in the serum and tumor (n = 3). The IFN‐α concentration in the tumors injected with 5 × 108 PFU of Ad‐mIFN was 13895.3 ± 5916.1 pg/g. (d) Time course of Renca tumor growth. The 1 × 108 PFU of adenoviruses was injected, and tumor volume was measured at indicated days (n = 4). In the mice treated by intravenous injection of adenoviruses, tumor volumes for both legs were combined to calculate the average. (e) Survival of mice treated by intratumoral or intravenous injection of adenoviruses. The 1 × 108 PFU of adenoviruses was injected in the Renca tumor‐bearing mice, and a time to death after tumor inoculation was assessed (n = 6). (f) Growth suppression of CT26 and Renca tumors. CT26 cells were inoculated on the right legs and on the backs and Renca cells were inoculated on the left legs in BALB/c mice, and then 1 × 108 PFU of Ad‐mIFN was injected into the CT26 tumor on the right leg (n = 6).
To compare the IFN‐α levels in the tumors with the antitumor effects, an IFN‐α concentration was examined 3 days after the gene transfer. The IFN‐α concentration in the tumors following the intratumoral gene transfer was elevated in a dose‐dependent manner, and the levels were much higher than those for the intravenous injections at all doses (Fig. 2c). An intravenous injection of less than 1 × 108 PFU of Ad‐mIFN did neither elevate the IFN‐α concentration in the tumors nor suppress the tumor growth. By contrast, the intravenous injection of the high dose (5 × 108 PFU) resulted in 1522.2 pg/g of IFN‐α in the tumors, which was close to that (2054.7 pg/g) after an intratumoral injection of low dose (5 × 106 PFU) of Ad‐mIFN (Fig. 2c), and significantly suppressed tumor growth (Fig. 2a). The increase of the IFN‐α level in the tumor might be related to induction of an effective antitumor immunity.
To confirm the antitumor activity of intratumoral IFN‐α gene transfer in a different tumor cell line, 1 × 108 PFU of Ad‐mIFN was injected into Renca tumor‐bearing mice. The antitumor activity of an intratumoral injection was also evident against the renal cancer cells (Fig. 2d), and resulted in a statistically significant improvement in the survival of the treated mice as compared with the Ad‐AP‐injected animals (Fig. 2e).
Then, we inoculated CT26 cells on the right legs and on the backs and Renca cells on the left legs of BALB/c mice, and 1 × 108 PFU of Ad‐mIFN was injected into CT26 tumors on the right legs. Interferon‐alpha (IFN‐α) gene transfer suppressed the growth of not only the right leg CT26 tumors but also the back CT26 tumors; and the growth of Renca tumors, which were not transduced with IFN‐α gene, on the left legs was also significantly suppressed compared with the Ad‐AP injection (Fig. 2f). Certain tumor‐associated antigens (TAAs) might be shared by CT26 and Renca tumors. Alternatively, the activation of NK cells by the IFN‐α expression may be responsible for this phenomenon as later described.
Cell‐death induction and infiltration of CD4+ and CD8+ cells in the tumors. To examine whether the IFN‐α expression induces cell death in the tumors, TUNEL assay was performed using tumors of mice treated by IFN‐α gene transfer. The assay revealed massive cell death of cancer cells in Ad‐mIFN‐injected tumors as shown in our previous reports,( 17 ) but TUNEL‐positive cells were also recognized albeit relatively sparsely in the vector‐uninjected tumors in the mice treated by intratumoral IFN‐α gene transfer (Fig. 3a). The difference in numbers of TUNEL‐positive cells is probably due to presence of the direct cell‐death induction by the IFN‐α expression in the injected but not uninjected tumors.
Figure 3.
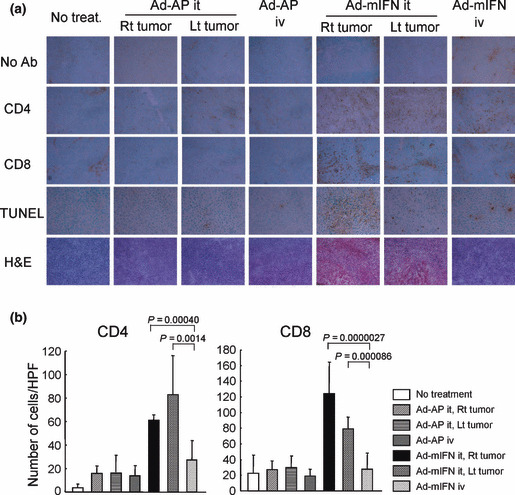
Infiltration of CD4+ and CD8+ T cells into the tumors. (a) Immunohistochemical staining of CT26 tumors. Ten days after the administration of adenoviruses, the fresh frozen sections of subcutaneous tumors were processed for immunohistochemistry with anti‐CD4 and anti‐CD8 antibodies, and also processed for TUNEL staining (×400). Ab, antibody; H&E, hematoxylin–eosin. (b) Number of CD4+ and CD8+ T cells in CT26 tumors. Positive cells were counted in 10 representative high power view fields (HPF, ×400) under microscope.
To determine whether the different therapeutic efficacies between the intratumoral and intravenous IFN‐α gene transfer are associated with different T‐cell responses, we analyzed the infiltration of CD4+ and CD8+ cells into the subcutaneous tumors by immunostaining 10 days after the vector injection. The intratumoral IFN‐α expression significantly increased the infiltration of CD4+ and CD8+ T cells in the tumors on both legs, compared to the intravenous administration of Ad‐mIFN (Fig. 3a,b). No increases of those cells were observed in the Ad‐AP‐treated mice. The infiltrated immune cells could induce an antitumor effect against tumors, in particular at distant sites.
Natural killer (NK) cells also play a role in antitumor effect of intratumoral Ad‐mIFN administration. It has been reported that IFN‐α enhances the activity of NK cells( 4 ) in addition to the activation of cytotoxic T lymphocyte. To determine whether the absence of T cells abrogates IFN‐α‐mediated therapeutic efficacy, we inoculated CT26 cells in both legs of BALB/c nude mice, and then injected 5 × 107 PFU of Ad‐mIFN into the tumor on the right leg or into the tail vein. The intravenous IFN‐α gene transfer showed modest tumor growth inhibition compared with the Ad‐AP injection, whereas a marked suppression of both vector‐injected and vector‐uninjected tumors was observed after intratumoral IFN‐α gene transfer (Fig. 4a). The depletion of NK cells with anti‐asialo GM1 antibody cancelled the antitumor effect of vector‐uninjected tumors on the left legs almost completely, whereas significant growth suppression was still recognized in the Ad‐mIFN‐injected tumors on the right legs (Fig. 4b), possibly due to the direct cell‐death induction by IFN‐α expression. The results suggest that the NK cells in the regional tumor are activated by the IFN‐α expression and contribute to systemic antitumor immunity, which is consistent with our previous report.( 15 ) Interestingly, the intravenous injection of 5 × 107 PFU of Ad‐mIFN suppressed the tumor growth in immune‐deficient nude mice (Fig. 4a) but not in immune‐competent mice (Fig. 2b). The difference may be explained by the fact that activity of NK cells is more elevated in nude mice compared with immune‐competent mice, and by the possibility that an increase of the serum IFN‐α concentration effectively enhances the cytotoxicity of the NK cells more than that of T cells. Although it could be due to augmented IFN‐α production since nude mice have less ability to exclude adenovirus, the serum IFN‐α concentration of nude mice 4 days after the intravenous injection of Ad‐mIFN (1 × 108 PFU) was 5444.1 ± 244.7 pg/mL, which was not significantly different from the value (7402.9 ± 1912.1 pg/mL) of BALB/c mice (Fig. 1b).
Figure 4.

Suppression of CT26 tumors in nude mice. (a) The CT26 cells were inoculated on both legs of BALB/c nude mice, and 7 days later 5 × 107 PFU (plaque forming unit) of recombinant adenovirus vectors expressing mouse interferon‐α (Ad‐mIFN) or alkaline phosphatase (Ad‐AP) was injected into the right tumor or into the tail vein. (b) BALB/c nude mice were treated with anti‐asialo GM1 antibody, and the CT26 tumors were injected with 5 × 107 PFU of Ad‐mIFN (n = 7). Tumor volumes 10 days after interferon‐alpha (IFN‐α) gene transfer are presented.
Expansion of tumor‐responsive lymphocytes after intratumoral IFN‐α gene transfer. To examine the expansion of tumor‐responsive lymphocytes after intratumoral or intravenous IFN‐α gene transfer in immune‐competent BALB/c mice, the splenocytes were harvested 20 days after the vector administration, and stimulated with MMC‐treated CT26 cells or syngeneic lymphocytes. ELISpot assay showed that the intravenous IFN‐α gene transfer slightly increased the number of IFN‐γ‐secreting cells in response to CT26 cells compared with the control Ad‐AP injection, and that the intratumoral IFN‐α gene transfer resulted in a significantly higher number of IFN‐γ‐positive spots than the intavenous injection (Fig. 5a, left), suggesting that the IFN‐α expression in the tumor effectively expanded tumor‐responsive immune cells including cytotoxic T cells and NK cells. Spot numbers of lymphocytes from CT26 tumor‐bearing mice with no treatment were similar to those with the Ad‐AP injection (data not shown). Intratumoral IFN‐α gene transfer did not significantly change the number of IFN‐γ‐positive spots for syngeneic lymphocytes, whereas the intravenous route of the same vector increased the number of spots for lymphocytes (Fig. 5a, right). The elevation of the IFN‐α concentration in the serum may enhance the lymphocyte reactivity nonspecifically.
Figure 5.

Activation of antigen‐presenting cells isolated from the mice treated by interferon‐alpha (IFN‐α) gene transfer. (a) Expansion of tumor‐responsive T cells after intratumoral injection of recombinant adenovirus vector expressing mouse interferon‐α (Ad‐mIFN). ELISpot assay of interferon‐gamma (IFN‐γ)‐producing cells was performed in response to stimulation of CT26 cells. Ad‐mIFN or recombinant adenovirus vector expressing alkaline phosphatase (Ad‐AP) was injected into the CT26 tumor or into the tail vein and 20 days later splenocytes were co‐cultured with CT26 cells or lymphocytes, and stained with biotinylated antimouse IFN‐γ antibody to detect captured IFN‐γ (n = 3). Lymphocytes were isolated from the spleen of a naïve BALB/c mouse. (b) Expression of co‐stimulatory molecules on CD11c+ cells. Flow cytometry of CD40, CD80, and CD86 expressions was performed on CD11c+ cells isolated from regional lymph nodes of the mice treated by intratumoral or intravenous injection of Ad‐mIFN or Ad‐AP (n = 3–4). The frequency of CD11c+ cells (upper left) per lymphocytes, and CD40+ (upper right), CD80+ (lower left), and CD86+ (lower right) cells per CD11c+ cells are presented. (c) Number of IFN‐γ‐producing cells by the stimulation of CD11c+ cells. CD11c+ cells were isolated from the tumors of treated mice. CD4+ T cells were isolated from the spleens of CT26 tumor‐bearing mice, and co‐cultured with CT26 cells and the isolated CD11c+ cells for 3 days, and then the number of IFN‐γ‐producing CD4+ T cells was counted. AP‐CD11c+, CD11c+ cells isolated from tumors in mice treated by Ad‐AP. mIFN‐CD11c+, CD11c+ cells isolated from tumors in mice treated by Ad‐mIFN.
Antigen presentation by DCs isolated from vector‐injected tumors. After antigen capture, and in response to inflammatory stimuli, DCs mature and migrate to lymph nodes to initiate immunity. To verify whether the expression of IFN‐α in the tumors augments the maturation of the antigen presenting cells, we isolated CD11c+ cells from the regional lymph nodes of tumor‐bearing mice treated with adenoviruses, and examined the expression of co‐stimulatory molecules in the cells. Flow cytometry showed that the frequency of CD11c+ cell per lymphocyte increased and the expressions of CD40, CD80, and CD86 were up‐regulated in CD11c+ cells isolated from the mice treated by intratumoral IFN‐α gene transfer. The expression of CD86 in CD11c+ cells after intravenous injections of Ad‐mIFN was slightly increased compared with the Ad‐AP‐injected mice (Fig. 5b). The results indicate that IFN‐α expression in the tumor effectively enhances the maturation of CD11c+ cells in the regional lymph nodes more than the high systemic level of IFN‐α does.
Then, to examine whether the enhanced expression of co‐stimulatory molecules on DCs is associated with antigen presentation, CD4+ T cells derived from the spleen of CT26 tumor‐bearing control mice were co‐cultured for 3 days with the CD11c+ cells isolated from the tumors in the vector‐treated mice and MMC‐treated CT26 cells. The production of IFN‐γ protein from the isolated CD11c+ cells per se was minimal in the in vitro culture (data not shown). ELISpot analysis showed that a primary culture with CD11c+ cells isolated from the tumors treated by Ad‐mIFN resulted in a higher number of IFN‐γ‐secreting CD4+ T cells than cultures with CD11c+ cells isolated from tumors in the intravenously Ad‐mIFN‐, intratumorally Ad‐AP‐, and intravenously Ad‐AP‐injected mice (Fig. 5c), demonstrating that the antigen presentation capacity of DCs was significantly enhanced by IFN‐α expression in the tumor.
Lower systemic toxicity after intratumoral injection of Ad‐mIFN. To compare the toxicity between the administration routes of the IFN‐α vector, different doses (1 × 108, 5 × 108, 1 × 109, and 3 × 109 PFU) of Ad‐mIFN or Ad‐AP were injected into the subcutaneous tumors or into the tail veins of mice. All three mice treated by intravenous injection of 3 × 109 PFU of Ad‐mIFN died within 4 days after the vector administration, whereas all the mice treated by the intratumoral injection of the same amount of Ad‐mIFN survived and appeared healthy up to 5 days, when the mice were sacrificed to evaluate serum enzyme values. In the mice that received the intravenous IFN‐α gene transfer, the injection of 1 × 109 PFU significantly elevated the aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels (more than 100‐fold higher than the average value in normal mice) in the serum, and the injection of 5 × 108 PFU slightly elevated the ALT level (5.7‐fold higher than the average value) (Fig. 6). Although the intratumoral injection of Ad‐mIFN slightly increased the AST level in a dose‐dependent manner, the values were similar to the intratumoral injection of Ad‐AP, confirming the lower systemic toxicity after the intratumoral IFN‐α gene transfer. Regarding albumin and creatinine, there was no significant difference among the treated mice (Fig. 6).
Figure 6.

Elevation of hepatic enzymes after intravenous injection of recombinant adenovirus vector expressing mouse interferon‐α (Ad‐mIFN). Different doses (1 × 108, 5 × 108, 1 × 109, and 3 × 109 PFU) of Ad‐mIFN or recombinant adenovirus vector expressing alkaline phosphatase (Ad‐AP) were injected into the subcutaneous tumor or into the tail vein. Five days later the selected blood chemistries (albumin, creatinine, aspartate aminotransferase (AST), and alanine aminotrans‐ferase (ALT)) were examined (n = 3).
Discussion
It has been reported that the modes of antitumor responses of IFN‐α gene therapy manifested three aspects: direct antiproliferative effect,( 1 ) stimulation of antitumor immunity,( 4 ) and anti‐angiogenesis activity.( 24 ) Our previous reports also showed that an intratumoral IFN‐α gene transfer induces a marked regional antitumor effect and an effective systemic immunity.( 15 , 16 ) This immunity was not attributable to the elevated level of the IFN‐α in the systemic circulation, because we found a little leakage of the cytokine following the intratumoral injection of the IFN‐expressing adenovirus.( 15 , 17 ) On the other hand, the elevation of the systemic IFN‐α level after intramuscular delivery of the IFN‐α gene was reported to exhibit an inhibition of tumor growth by the infiltration of CD8+ T cells.( 18 , 19 ) Therefore, in this study, we compared the induction of systemic antitumor immunity by a high IFN‐α level in the tumor and blood circulation. The antitumor effects and general toxicities in the mice treated by intratumoral or intravenous injection of Ad‐mIFN at various doses are summarized in Table 1. The tumor growth suppression was recognized at the lowest dose (5 × 106 PFU) and the toxicity was not observed at levels of less than 1 × 109 PFU in the mice treated by the intratumoral IFN‐α gene transfer, whereas the antitumor effect only appeared at 5 × 108 PFU, and hepatic toxicity was observed at the 5 × 108 PFU level, in the mice treated by intravenous gene transfer. The results suggested that the intratumoral route of IFN vector is superior to intravenous administration, due to the effective induction of an antitumor immunity and the lower toxicity.
Table 1.
Summary of antitumor effects and toxicities by intratumoral and intravenous injection of Ad‐mIFN at various doses
| Route | Dose | Antitumor effect | Toxicity |
|---|---|---|---|
| Intratumoral injection | 5 × 106 | ++ | NE |
| 5 × 107 | ++ | NE | |
| 1 × 108 | ++ | – | |
| 5 × 108 | ++ | NE | |
| 1 × 109 | NE | – | |
| 3 × 109 | NE | + | |
| Intravenous injection | 5 × 106 | – | NE |
| 5 × 107 | – | NE | |
| 1 × 108 | – | – | |
| 5 × 108 | ++ | + | |
| 1 × 109 | NE | +++ | |
| 3 × 109 | NE | +++ |
Antitumor effect was evaluated on the basis of tumor growth compared with recombinant adenovirus vector expressing alkaline phosphatase (Ad‐AP)‐injected tumors: +, >70%; ++, 30–70%; +++, <30%. Toxicity was evaluated on the basis of the elevation of aspartate aminotransferase (AST) or alanine aminotransferase (ALT) values compared with average in normal mice: +, >5‐fold elevation; ++, 5–100‐fold elevation; +++, >100‐fold elevation or death. Ad‐mIFN, recombinant adenovirus vector expressing mouse interferon‐α; NE, not evaluated.
The underlying mechanism of the antitumor immunity induced by IFN‐α gene transfer is not fully understood. In this study, we noticed the role of IFN‐α in the maturation and function of DCs. Dendritic cells (DCs) are antigen‐presenting cells specialized to induce T‐cell responses against cells exposing foreign peptides including TAAs, and DCs show an effective cross‐priming with antigens from apoptotic tumor cells.( 25 , 26 ) As shown in Figure 3a and our previous report,( 17 ) a high IFN‐α level after intratumoral injection of an adenovirus vector induced apoptotic cell death in CT26 subcutaneous tumors, and abundant apoptosis induced by intratumoral IFN‐α expression may expose TAAs in large quantity and enhance the uptake of released TAAs by DCs. Moreover, recent data have shown that type I IFN itself can act as an important signal for differentiation and maturation of DC: DCs generated after a few days of treatment with IFN‐α showed the mature phonotype.( 4 , 27 ) It is known that maturation of DCs is associated with up‐regulation of the co‐stimulatory molecules, and enhancement of their antigen‐presenting function.( 28 ) In this study also, the CD11+ cells isolated from regional sites of mice treated by intratumoral IFN gene transfer showed up‐regulations of CD40, CD80, and CD86 (Fig. 5b) and the enhancement of antigen‐presentation capacity (Fig. 5c). Finally, TAA‐specific T cells activated by DCs could efficiently infiltrate into the CT26 tumors, leading to antitumor effect. Therefore, the effective apoptosis induction and direct immunomodulatory effect of IFN‐α on DCs may contribute to the observed augmentation of the tumor specific immunity.
Intravenous administration of an adenovirus results in gene delivery primarily to the liver.( 29 , 30 ) We previously reported a significant production of IFN‐α in the liver after an intravenous injection of 1 × 107 of an IFN‐α‐expressing adenovirus vector in the dimethylnitrosamine‐induced cirrhotic rat, but no IFN‐α was detected in the serum.( 31 ) The transgene expression was mainly observed in the fibrous septa but not in hepatocytes in a cirrhotic liver, whereas it was detected in septa as well as in hepatocytes in the normal rats.( 31 ) In this study, we intravenously injected 1 × 108 PFU of Ad‐mIFN to examine the biodistribution of IFN‐α expression in various mice organs (Fig. 1c). The difference in the amount of the injected virus and the histopathology of the liver may be the reason that high IFN‐α levels were sustained in the serum in this study.
It is well known that a systemic administration of IFN‐α protein often induces severe adverse effects such as flu‐like symptoms, leucopenia, liver dysfunction, and mental depression.( 32 ) With respect to the safety of a local IFN‐α gene therapy, it is noteworthy that the intratumoral IFN‐α gene transfer showed very limited general toxicity such as an elevation of hepatic enzymes, since the IFN‐α concentration in the serum was approximately 6–68‐fold lower in the intratumorally Ad‐mIFN‐injected mice compared with the intravenously Ad‐mIFN‐injected mice (Fig. 1b). The apparent difference in the IFN‐α concentration between the tumor and serum is an important finding especially from the viewpoint of safety of local IFN‐α gene therapy. As one of the reasons for a little leakage of the IFN‐α protein from the tumors into blood circulation, we recently reported that extracellular matrix proteins such as fibronectin in tumors directly interact with IFN‐α and retain the cytokine.( 23 )
In summary, our preclinical study suggests that a regional adenovirus‐mediated gene transfer of IFN‐α is one of the promising new approaches to cancer. The strategy may deserve an evaluation in a future clinical trial for intractable cancer.
Acknowledgments
This work was supported in part by a Grant‐in‐Aid for the 3rd‐Term Comprehensive 10‐Year Strategy for Cancer Control from the Ministry of Health, Labour and Welfare of Japan, by Grants‐in‐Aid for Cancer Research from the Ministry of Health, Labour and Welfare of Japan, and by the Program for Promotion of Foundation Studies in Health Science of the National Institute of Biomedical Innovation (NiBio). H. Hara and T. Udagawa are awardees of a Research Resident Fellowship from the Foundation for Promotion of Cancer Research.
References
- 1. Caraglia M, Marra M, Pelaia G et al. Alpha‐interferon and its effects on signal transduction pathways. J Cell Physiol 2005; 202: 323–35. [DOI] [PubMed] [Google Scholar]
- 2. Pfeffer LM, Dinarello CA, Herberman RB et al. Biological properties of recombinant alpha‐interferons: 40th anniversary of the discovery of interferons. Cancer Res 1998; 58: 2489–99. [PubMed] [Google Scholar]
- 3. Gutterman JU. Cytokine therapeutics: lessons from interferon alpha. Proc Natl Acad Sci USA 1994; 91: 1198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Santini SM, Lapenta C, Santodonato L, D’Agostino G, Belardelli F, Ferrantini M. IFN‐alpha in the generation of dendritic cells for cancer immunotherapy. Handb Exp Pharmacol 2009; 188: 295–317. [DOI] [PubMed] [Google Scholar]
- 5. Ferrantini M, Capone I, Belardelli F. Dendritic cells and cytokines in immune rejection of cancer. Cytokine Growth Factor Rev 2008; 19: 93–107. [DOI] [PubMed] [Google Scholar]
- 6. Einhorn S, Grander D. Why do so many cancer patients fail to respond to interferon therapy? J Interferon Cytokine Res 1996; 16: 275–81. [DOI] [PubMed] [Google Scholar]
- 7. Salmon P, Le Cotonnec JY, Galazka A, Abdul‐Ahad A, Darragh A. Pharmacokinetics and pharmacodynamics of recombinant human interferon‐beta in healthy male volunteers. J Interferon Cytokine Res 1996; 16: 759–64. [DOI] [PubMed] [Google Scholar]
- 8. Zhang JF, Hu C, Geng Y et al. Treatment of a human breast cancer xenograft with an adenovirus vector containing an interferon gene results in rapid regression due to viral oncolysis and gene therapy. Proc Natl Acad Sci USA 1996; 93: 4513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hottiger MO, Dam TN, Nickoloff BJ, Johnson TM, Nabel GJ. Liposome‐mediated gene transfer into human basal cell carcinoma. Gene Ther 1999; 6: 1929–35. [DOI] [PubMed] [Google Scholar]
- 10. Ahmed CM, Wills KN, Sugarman BJ et al. Selective expression of nonsecreted interferon by an adenoviral vector confers antiproliferative and antiviral properties and causes reduction of tumor growth in nude mice. J Interferon Cytokine Res 2001; 21: 399–408. [DOI] [PubMed] [Google Scholar]
- 11. Benedict WF, Tao Z, Kim CS et al. Intravesical Ad‐IFNalpha causes marked regression of human bladder cancer growing orthotopically in nude mice and overcomes resistance to IFN‐alpha protein. Mol Ther 2004; 10: 525–32. [DOI] [PubMed] [Google Scholar]
- 12. Studeny M, Marini FC, Dembinski JL et al. Mesenchymal stem cells: potential precursors for tumor stroma and targeted‐delivery vehicles for anticancer agents. J Natl Cancer Inst 2004; 96: 1593–603. [DOI] [PubMed] [Google Scholar]
- 13. De Palma M, Mazzieri R, Politi LS et al. Tumor‐targeted interferon‐alpha delivery by Tie2‐expressing monocytes inhibits tumor growth and metastasis. Cancer Cell 2008; 14: 299–311. [DOI] [PubMed] [Google Scholar]
- 14. Hatanaka K, Suzuki K, Miura Y et al. Interferon‐alpha and antisense K‐ras RNA combination gene therapy against pancreatic cancer. J Gene Med 2004; 6: 1139. [DOI] [PubMed] [Google Scholar]
- 15. Ohashi M, Yoshida K, Kushida M et al. Adenovirus‐mediated interferon alpha gene transfer induces regional direct cytotoxicity and possible systemic immunity against pancreatic cancer. Br J Cancer 2005; 93: 441–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hara H, Kobayashi A, Yoshida K et al. Local interferon‐alpha gene therapy elicits systemic immunity in a syngeneic pancreatic cancer model in hamster. Cancer Sci 2007; 98: 455–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hara H, Kobayashi A, Narumi K et al. Intratumoral interferon‐alpha gene transfer enhances tumor immunity after allogeneic hematopoietic stem cell transplantation. Cancer Immunol Immunother 2009; 58: 1007–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Horton HM, Anderson D, Hernandez P, Barnhart KM, Norman JA, Parker SE. A gene therapy for cancer using intramuscular injection of plasmid DNA encoding interferon alpha. Proc Natl Acad Sci USA 1999; 96: 1553–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li S, Zhang X, Xia X et al. Intramuscular electroporation delivery of IFN‐alpha gene therapy for inhibition of tumor growth located at a distant site. Gene Ther 2001; 8: 400–7. [DOI] [PubMed] [Google Scholar]
- 20. Aoki K, Barker C, Danthinne X, Imperiale MJ, Nabel GJ. Efficient generation of recombinant adenoviral vectors by Cre‐lox recombination in vitro . Mol Med 1999; 5: 224–31. [PMC free article] [PubMed] [Google Scholar]
- 21. Niwa H, Yamamura K, Miyazaki J. Efficient selection for high‐expression transfectants with a novel eukaryotic vector. Gene 1991; 108: 193–9. [DOI] [PubMed] [Google Scholar]
- 22. Zhang WW, Koch PE, Roth JA. Detection of wild‐type contamination in a recombinant adenoviral preparation by PCR. BioTechniques 1995; 18: 444–7. [PubMed] [Google Scholar]
- 23. Yoshida K, Kondoh A, Narumi K, Yoshida T, Aoki K. Extracellular matrix interacts with interferon alpha protein: retention and display of cytotoxicity. Biochem Biophys Res Commun 2008; 376: 299–304. [DOI] [PubMed] [Google Scholar]
- 24. Indraccolo S, Gola E, Rosato A et al. Differential effects of angiostatin, endostatin and interferon‐alpha(1) gene transfer on in vivo growth of human breast cancer cells. Gene Ther 2002; 9: 867–78. [DOI] [PubMed] [Google Scholar]
- 25. Hart DN. Dendritic cells: unique leukocyte populations which control the primary immune response. Blood 1997; 90: 3245–87. [PubMed] [Google Scholar]
- 26. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature 1998; 392: 245–52. [DOI] [PubMed] [Google Scholar]
- 27. Santini SM, Lapenta C, Logozzi M et al. Type I interferon as a powerful adjuvant for monocyte‐derived dendritic cell development and activity in vitro and in Hu‐PBL‐SCID mice. J Exp Med 2000; 191: 1777–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cella M, Sallusto F, Lanzavecchia A. Origin, maturation and antigen presenting function of dendritic cells. Curr Opin Immunol 1997; 9: 10–6. [DOI] [PubMed] [Google Scholar]
- 29. Jaffe HA, Danel C, Longenecker G et al. Adenovirus‐mediated in vivo gene transfer and expression in normal rat liver. Nat Genet 1992; 1: 372–8. [DOI] [PubMed] [Google Scholar]
- 30. Cristiano RJ, Smith LC, Kay MA, Brinkley BR, Woo SL. Hepatic gene therapy: efficient gene delivery and expression in primary hepatocytes utilizing a conjugated adenovirus‐DNA complex. Proc Natl Acad Sci USA 1993; 90: 11548–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Suzuki K, Aoki K, Ohnami S et al. Adenovirus‐mediated gene transfer of interferon alpha improves dimethylnitrosamine‐induced liver cirrhosis in rat model. Gene Ther 2003; 10: 765–73. [DOI] [PubMed] [Google Scholar]
- 32. Taguchi T. Clinical studies of recombinant interferon alfa‐2a (Roferon‐A) in cancer patients. Cancer 1986; 57: 1705–8. [DOI] [PubMed] [Google Scholar]