

Abstract
The effects of mesenchymal stem cells (MSC) on the growth and metastasis of human malignancies including hepatocellular carcinoma (HCC) are controversial, and the underlying mechanisms are not yet understood. The aim of this study was to explore the role of MSC in the progression of HCC. We investigated the effect of MSC on in vitro proliferation and invasion and in vivo tumor growth and pulmonary metastasis of MHCC97‐H HCC cells with a high metastatic potential. The mRNA and protein levels of transforming growth factor‐beta 1 (TGFβ1) and MMP, and their association with the effects of MSC on HCC cells were also evaluated. Co‐culture of MHCC97‐H cells with MSC conditioned medium significantly enhanced in vitro proliferation but inhibited invasiveness. Following MSC treatment of a nude mouse model bearing human HCC, the MSC were predominantly located in the HCC tissues. Compared with controls, MSC‐treated mice exhibited significantly larger tumors (3080.51 ± 1234.78 mm3 vs 2223.75 ± 1000.60 mm3, P = 0.045), but decreased cellular numbers of lung metastases (49.75 ± 18.86 vs 227.22 ± 74.67, P = 0.046). Expression of TGFβ1 and MMP‐2 was significantly downregulated in the MSC‐treated HCC cells. TGFβ siRNA concurrently downregulated expression of TGFβ and MMP‐2 in HCC cells and blocked the MSC‐induced proliferation and invasiveness of MHCC97‐H cells. The MSC enhanced tumor growth but significantly inhibited the invasiveness and metastasis of HCC, possibly through downregulation of TGFβ1. These findings suggest that MSC could be useful in controlling metastatic recurrence of HCC. (Cancer Sci 2010; 101: 2546–2553)
Hepatocellular carcinoma (HCC) is the third leading cause of cancer death in the world, and the second in China. 1 , 2 The extremely poor prognosis of HCC is largely due to the high rate of intra‐hepatic metastases that develop through invasion of the portal vein and spread to other parts of the liver. 3 , 4 Therefore, exploring new therapeutic strategies to control the metastasis of HCC is the key issue for prolonging patient survival. (5)
Recently accumulated evidence indicating that bone marrow stem cells contribute to the development and progression of cancers might be helpful in the diagnosis and treatment of human cancers. 6 , 7 , 8 , 9 , 10 , 11 Bone marrow cells have also been found to be closely associated with liver diseases, and can indirectly influence hepatocarcinogenesis. (12) However, their significance with respect to HCC is far from fully understood.
Mesenchymal stem cells (MSC) from adult bone marrow can be induced to differentiate into a variety of mesenchymal tissues both in vitro and in vivo. 13 , 14 , 15 They can even demonstrate site‐specific differentiation, and have unique immunological characteristics that allow persistence in a xenogeneic environment. (16) Therefore, MSC present an intriguing model in which to investigate the differentiation of stem cells, as well as cell and gene therapy applications. However, little is known about the functional contribution of MSC to tumor growth and progression, and the published results are controversial. 17 , 18 , 19 , 20 The aim of this study was to determine the effects of MSC on the growth and metastasis of human HCC and explore the underlying mechanisms.
Materials and Methods
Cell lines. We used a MSC cell line from Sciencell Research Laboratories (Carlsbad, CA, USA) that was isolated from human bone marrow and characterized by immunofluorescent methods with CD44 and CD90 antibodies. Cells were cultured in alpha‐modified minimum essential medium (α‐MEM, Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco) and 100 U/mL penicillin/streptomycin solution. The fifth to eighth passages of MSC were used in the following experiments.
The human metastatic HCC cell line MHCC97‐H was established at the author’s institute. (21) Cells were cultured at 37°C in high glucose Dulbecco’s modified Eagle’s medium (H‐DMEM) (Gibco) supplemented with 10% FBS.
CyQUANT assay for in vitro cell proliferation of HCC cells after co‐culture with MSC conditioned medium (MSC‐CM). To prepare the MSC‐CM, the MSC cells were seeded (20 000 cells/cm2 in α‐MEM/10% FBS) and incubated overnight. The adhered MSC were washed and further incubated in FBS‐free, high‐glucose DMEM for 24 h, and then the medium was collected and filtered through a 0.2 μm filter.
The MSC‐CM was added to the culture medium of MHCC97‐H cells to a final concentration of 0%, 25% or 50%. After incubation for 2 days, the adhered MHCC97‐H cells were collected and their proliferative activity was evaluated using a CyQUANT Cell Proliferation Assay Kit (Invitrogen, Carlsbad, CA, USA). The optical density (OD) value, which positively correlates with cell number, was determined using a microplate reader at 480/520 nm.
Transwell assay for in vitro invasion of HCC cells. The in vitro invasion assay was performed as described previously. (22) Briefly, 80 μL serum‐free DMEM‐diluted Matrigel (0.8 mg/mL) was added to the Transwell filters (8.0 μm pore size) of a Bodyden chamber (Costar, MA, USA) and incubated at 37°C for 2 h to form a matrix gel. The MHCC97‐H cells (1 × 106 cells) were cultured in FBS‐free DMEM in the presence (50%) or absence of MSC‐CM for 24 h, then 2 × 105 cells were re‐suspended in DMEM and seeded in the upper well of the transwell chamber. As the chemoattractant, 600 μL DMEM containing 10% fetal calf serum was added to the lower chamber. After incubation at 37°C for 48 h, the cells that had invaded across the Matrigel and passed through the transwell filter were stained with Geisma and counted in eight non‐overlapping fields under a light microscope at ×200 magnification.
In vivo assays of tumor growth and metastasis in nude mice bearing human HCC. The MHCC97‐H cells (6 × 106 cells) were subcutaneously implanted into 26 nude mice (MHCC97‐H model). Starting on day 3 after implantation, 13 mice were intravenously injected with 5 × 105 MSC three times a week. The other 13 mice were injected with PBS as controls. Mice were examined every 2 days for tumor growth. The volume of the palpable tumor nodule was estimated according to the formula: volume (mm3) = 0.5 a 2 × b, where a is the major tumor diameter and b is the minor diameter perpendicular to the major one. (22) On the 35th day after implantation, the mice were killed and the tumors were removed, weighed and preserved for the following investigations. These experiments were approved by the Shanghai Medical Experimental Animal Care Commission.
Lung metastases were evaluated by hematoxylin and eosin (HE) staining and counted by two independent pathologists in 10 consecutive sections with an interval of 50 μm.
In vivo visualization of MSC in nude mice models. To visualize the distribution of MSC and HCC cells in the nude mice model bearing human HCC, the MHCC97‐H cells were labeled with green fluorescence protein (GFP) by transfection with the plasmid vector pEGFP‐N1 (Clonetech, Mountain View, CA, USA) to obtain the stable cell line GFP‐MHCC97‐H. The MSC were labeled with DAPI (Vector Laboratories, Burlingame, CA, USA) (DAPI‐MSC) according to the manufacturer’s instructions. The GFP‐MHCC97‐H cells (6 × 106 cells) were inoculated subcutaneously into four mice. After tumor formation, each mouse was intravenously injected with 1 × 106 DAPI‐MSC. The mice were killed after 4 days and tumor tissues and livers were collected and cryopreserved in optimum cutting temperature (OCT). The distribution of DAPI‐MSC in 5 μm freshly frozen sections of tumor was determined using a fluorescence microscope. To prolong the observation time, the mice were also treated with 1 × 106 MSC labeled with 5‐bromo‐2‐deoxyuridine (Brdu) (Sigma, Fairfax, VA, USA), and the Brdu‐MSC in tumors were observed after 20 days by immunostaining with Brdu antibody (Sigma).
Determination of MMP‐2 and transformation growth factor‐beta 1 (TGFβ1) mRNA levels by real‐time PCR. Total RNA was extracted from the MHCC97‐H cells and HCC tissues with TRIZOL Reagent (Invitrogen) according to the manufacturer’s instruction. Using SYBR Green Real‐time PCR Master Mix (ToYoBo Co., Osaka, Japan), real‐time RT‐PCR analyses were performed to determine the mRNA levels of MMP2 and TGFβ1. The primers were designed by Premier 5.0 as shown in Table 1. The amplification conditions were 95°C for 9 min, followed by 45 cycles of 95°C for 30 s, 75°C for 30 s and 72°C for 15 s, with a final extension at 72°C for 5 min. β‐actin was used as a control. Curves of increasing reporter dye fluorescence emission were recorded and analyzed with the iCycler iQTM Real‐time PCR Detection System (Bio‐Rad, Hercules, CA, USA) to determine the threshold cycle (Ct) value. The mRNA level was assessed using the formula 2−ΔΔCt, as recommended by the manufacturer (Bio‐Rad).
Table 1.
Primer sequences used for real‐time PCR
| Gene | Primer sequence |
|---|---|
| MMP2 | Sense 5′ ACTGCTGGCTGCCTTAGAACC 3′ |
| Antisense 5′ TCACTATGTGGGCTGAGATGC 3′ | |
| TGFβ1 | Sense 5′ GGCGATACCTCAGCAACCG 3′ |
| Antisense 5′ CTAAGGCGAAAGCCCTCAAT 3′ | |
| β‐actin | Sense 5′ TCGTGCGTGACATTAAGGAG 3′ |
| Antisense 5′ ATGCCAGGGTACATGGTAAT 3′ |
TGFβ1, transforming growth factor‐beta 1.
Western blot analysis and immunostaining of MMP and TGFβ1. Total protein was extracted from the HCC cells cultured with different concentrations of MSC‐CM (0%, 25% or 50%) and the protein concentration was determined using Bradford Reagent. Equal amounts of protein (in 20 μL extract) were heated at 95–100°C for 5 min, separated by 10% SDS‐PAGE, and electrotransferred to the PVDF membrane. The membrane was incubated in blocking buffer for 1 h at room temperature, washed with Tris‐buffered saline/Tween‐20, then incubated with primary antibody against MMP‐2 (M4065, Sigma), MMP‐9 (M9555, Sigma), TGFβ1 (MAB1032, Chemicon, South San Francisco, CA, USA) or β‐actin (MAB1501, Chemicom) in 10 mL dilution buffer (1:1000 dilution) overnight at 4°C. Finally, the membrane was incubated with HRP‐conjugated secondary antibody (1:2000) in 10 mL blocking buffer for 1 h at room temperature, incubated three times with 10 mL LumiGLO (0.5 mL ×20 LumiGLO, 0.5 mL ×20 peroxide) for 1 min at room temperature and exposed to Fuji film.
Five micron paraffin sections of HCC tissues were stained with primary antibodies against MMP‐2, MMP‐9 or TGFβ1. Donkey anti‐rat IgG‐peroxidase (AP189P, Chemicon) and goat anti‐mouse IgG‐peroxidase (A9917, Sigma) were used as the second antibodies.
Measurement of MMP‐2 and MMP‐9 activities by gelatin zymography. Equal amounts of protein from HCC tissues were mixed with SDS buffer and incubated for 20 min at 37°C. After incubation, samples (30 μg/lane) were loaded onto a 4.5% (w/v) stacking polyacrylamide gel and separated on a 7.5% (w/v) polyacrylamide gel containing 1 mg/mL gelatin for the detection of MMP‐2 and MMP‐9 activities. After electrophoresis, the gels were soaked in 2.5% Triton X‐100 for 1 h to remove SDS and incubated for 16 h at 37°C in 50 mM Tris–HCl (pH 7.6). Finally, the gels were stained for 1 h in 45% methanol/10% acetic acid containing 0.5% Coomassie brilliant blue G250. Proteolytic activity was detected as clear bands on the blue background of the Coomassie‐blue‐stained gel.
Determination of the TGFβ1 level by immunoassay. The MSC‐CM and MHCC97‐H‐CM were prepared as described above. The TGFβ1 protein levels in the CM and cells were determined using the Quantikine TGFβ1 Immunoassay (R&D, Minneapolis, MN, USA). To investigate the effect of MSC‐CM on TGFβ1 expression in the MHCC97‐H cells, 50% MSC‐CM and TGFβ1 antibody (1 μg in 2 mL) were added to the culture medium of MHCC97‐H cells. After incubation for 24 h, the culture medium was collected to determine the TGFβ1 concentration.
Inhibition of TGFβ using siRNA. siRNA against TGFβ1 (40 pmol TGFβ1 siRNA; Ambion Corp., Austin, TX, USA) was added to the culture medium of MHCC97‐H cells. After 48 h, the cells were harvested to evaluate the protein levels of TGFβ and MMP2 by western blot and investigate the invasive capability by transwell assay. Similarly, 40 pmol TGFβ1 siRNA was added to the MSC culture medium for 48 h, then the adhered MSC were washed and further incubated in FBS‐free high glucose DMEM for 24 h. The MSC‐CM was collected, filtered through a 0.2 μm filter and added to the culture medium of MHCC97‐H to a final concentration of 50%. After 2 days, the adhered MHCC97‐H cells were collected, and their proliferative and invasive activities were evaluated by CyQUANT and Transwell assays, respectively.
Statistical analyses. The data were analyzed using SPSS 11.5 software (SPSS Corp., Chicago, IL, USA). The Student’s t test was used to analyze differences in tumor weight and volume and mRNA expression levels of target genes for independent samples. One‐way anova was used to analyze differences in the CyQuant proliferation assay. Fishers’ exact test was used for the comparison of ratios. All statistical tests were two‐sided and P < 0.05 was considered statistically significant.
Results
Effects of MSC on in vitro proliferation and invasion of HCC cells. After culture with MSC‐CM, the in vitro proliferation of MHCC97‐H cells was significantly increased as determined by the CyQUANT proliferation assay; the OD values of the MHCC97‐H cells cultured with 0%, 25% and 50% of the MSC‐CM were 211.65 ± 54.72, 236.24 ± 57.15 and 283.59 ± 62.16, respectively (F = 3.47, P = 0.02) (Fig. 1A). However, the Transwell invasion assay showed that the number of invaded MHCC97‐H cells was much lower for cells cultured with MSC‐CM than for the controls (11.37 ± 3.54 vs 20.00 ± 5.35, P = 0.002) (Fig. 1B–D). These findings suggest that MSC‐CM significantly enhances in vitro proliferation, but suppresses the invasive ability of HCC cells.
Figure 1.
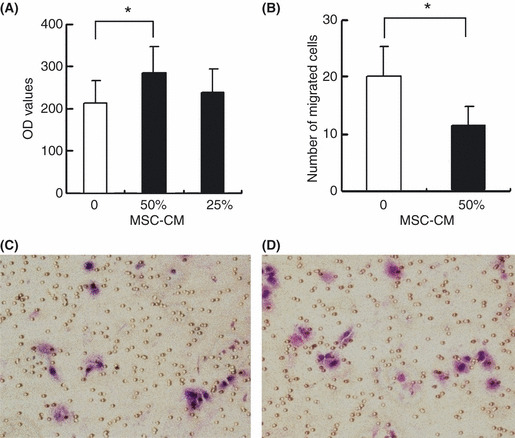
Effects of mesenchymal stem cells (MSC) on in vitro proliferation and invasion of MHCC97‐H cells. (A) The OD value of the MHCC97‐H cells cultured with 50% MSC conditioned medium (MSC‐CM) was significantly increased as determined by CyQUANT assay. (B) Culture with MSC‐CM significantly decreased the number of migrating cells detected by Transwell assay. (C,D) Representative pictures of the Transwell assay observed under light microscope (×20), which indicated that migrating cells cultured with MSC‐CM (C) were significantly decreased compared with the controls (D). Error bar represents the standard error of the mean. *P < 0.05.
Effects of MSC on in vivo tumor growth and metastasis of HCC. Consistent with the above in vitro results, the tumor volume of MHCC97‐H models was much larger in the MSC‐treated group than the controls (3080.51 ± 1234.78 mm3 vs 2223.75 ± 1000.60 mm3, P = 0.045) (Table 2, Fig. 2A), and the average tumor growth rate of the MSC‐treated group was also significantly increased (102.65 ± 63.66 mm3/day vs 72.07 ± 52.13 mm3/day, P = 0.013) (Fig. 2B). These data suggest that MSC enhance in vivo tumor growth. However, the cellular number of lung metastases in the MSC‐treated group was significantly decreased compared with the controls (49.75 ± 18.86 vs 227.22 ± 74.67, P = 0.046) (Table 2, Fig. 2C,D).
Table 2.
Effects of MSC on tumor growth and metastasis of HCC
| MSC injection | No. | Mean ± SD | P | |
|---|---|---|---|---|
| Tumor volume (mm3) | + | 13 | 3080.51 ± 1234.78 | 0.045 |
| − | 13 | 2223.75 ± 1000.60 | ||
| Tumor/bodyweight | + | 13 | 0.11 ± 0.04 | 0.055* |
| − | 13 | 0.08 ± 0.04 | ||
| Lung metastasis (%) | + | 13 | 67% | 0.610 |
| − | 13 | 64% | ||
| No. lung metastases | + | 13 | 2.88 ±2.48 | 0.087 |
| − | 13 | 7.22 ± 6.40 | ||
| Cellular numbers of metastases | + | 13 | 49.75 ± 18.86 | 0.046 |
| − | 13 | 227.22 ± 74.67 |
HCC, hepatocellular carcinoma; MSC, mesenchymal stem cells; SD, standard deviation. The Student’s t‐test was used to assess the statistical difference in tumor volume between the MSC injection group and the No‐MSC injection group. *Using the mean of the ratio of tumor/bodyweight as the cut‐off point, mice with the higher ratio in each group were analyzed by Fishers’ exact test.
Figure 2.
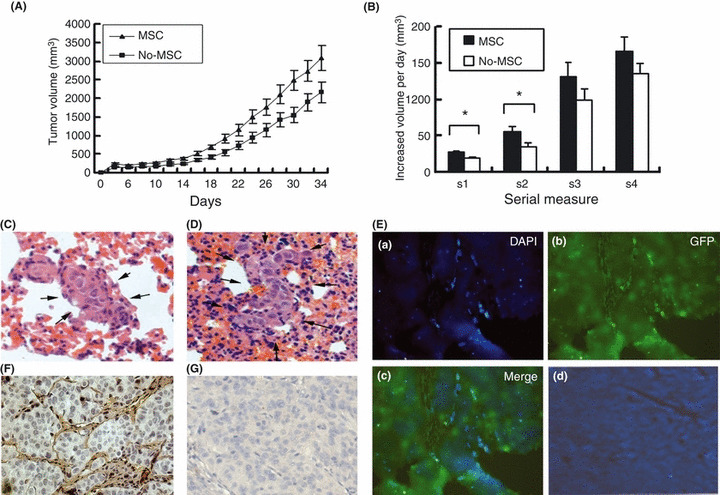
In vivo distribution of mesenchymal stem cells (MSC) and effects of MSC on tumor growth and metastasis in nude mice bearing human hepatocellular carcinoma (HCC). (A) Tumor growth of the MSC‐treated group (MSC) was significantly increased compared with the controls (No‐MSC). (B) The rate of tumor volume increase in MHCC97‐H models was also significantly larger in the MSC group than in the No‐MSC group. Lung metastasis in the MSC‐treated models (C) was much lower than in the controls (D) (HE staining, ×20). (E) More DAPI‐MSC (stained blue) were found in HCC tissues (green) (a–c) than in normal liver tissues (d) (×10). (F) Brdu‐MSC were mainly found in tumor stroma. (G) HCC tissues without injection of Brdu‐MSC are shown as a negative control (×20).
In vivo distribution of MSC in nude mice models. More MSC were found in the tumor site than in normal liver tissues on the fourth day after intravenous injection of MSC into the mice (Fig. 2E). Moreover, even at the 20th day after injection, the MSC were still mainly present in the tumor stroma (Fig. 2F,G).
Effect of MSC on TGFβ1 expression in HCC cells. After 48 h of in vitro co‐culture with MSC‐CM, the expression level of TGFβ1 in MHCC97‐H cells was significantly decreased (Fig. 3A). Similarly, in the MHCC97‐H in vivo model, the TGFβ1 mRNA level in the HCC tissues was significantly decreased in the MSC‐treated mice compared with the controls (1.34 ± 1.01 vs 2.42 ± 2.03, P = 0.047). When the tumors were grouped according to the median level of TGFβ1, a lower level of TGFβ1 correlated with a larger tumor size (3076.97 ± 1416.52 mm3 for low TGFβ1 vs 2272.87 ± 783.36 mm3 for high TGFβ1, P = 0.044). Furthermore, the TGFβ1 expression level was much lower in the non‐metastatic group than the metastatic group (1.19 ± 0.8 vs 2.31 ± 1.7, P = 0.05). These findings were further confirmed by immunohistochemical staining (Fig. 3B,C).
Figure 3.
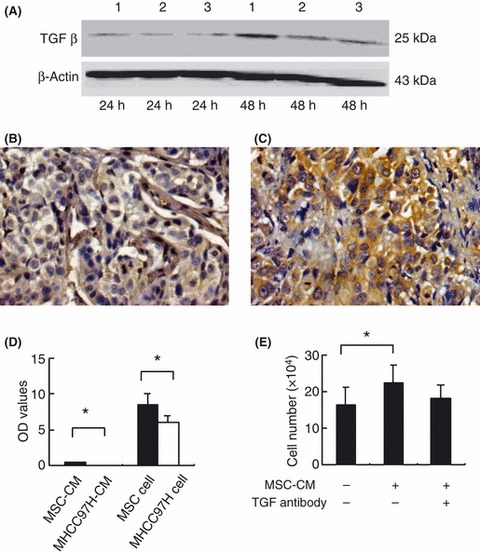
Mesenchymal stem cells (MSC) induced downregulation of transforming growth factor‐beta 1 (TGFβ1), which correlated with tumor growth of hepatocellular carcinoma (HCC). (A) Western blot analysis of protein expression levels of TGFβ1 (25 kD, under non‐reducing conditions) in MHCC97‐H cells cultured with MSC conditioned medium (MSC‐CM) for 24 and 48 h; lanes 1–3 show MHCC97‐H cells cultured with 0%, 50% and 25% of MSC‐CM, respectively. (B,C) Immunohistochemical staining showed that the expression level of TGFβ1 in MSC‐treated HCC tissues (B) was significantly lower than that of controls (C) (×20). (D) Immunoassays showed that the TGFβ1 level in MSC‐CM was significantly higher than in MHCC97‐H‐CM and that the MSC themselves expressed a higher level of TGFβ1 than MHCC97‐H cells. OD, optical density. (E) The TGFβ1 antibody blocked the induction of proliferation in MHCC97‐H cells cultured with MSC‐CM. Error bars denote the standard error of the mean. *P < 0.05.
Immunoassays showed that MSC secreted a higher level of TGFβ1 than MHCC97‐H cells: the TGFβ1 level in MSC‐CM (0.41 ± 0.05) was significantly higher than that of MHCC97‐H‐CM (0.045 ± 0.01, P < 0.001), and a consistent difference was also observed between MSC and MHCC97‐H cells themselves (8.58 ± 1.38 vs 5.89 ± 1.00, P = 0.011) (Fig. 3D). The proliferative ability of MHCC97‐H was significantly increased in cells cultured with MSC‐CM compared with no CM (16.45 ± 4.73 vs 22.45 ± 4.84, P = 0.045); however, when TGFβ1 antibody was added, no significant difference was observed between culture with or without MSC‐CM (16.45 ± 4.73 vs 18.02 ± 3.68, P = 0.586) (Fig. 3E).
Downregulation of MMP‐2 in MSC‐treated MHCC97‐H cells. Compared with the MHCC97‐H cells cultured alone, cells cultured with MSC‐CM for 48 h showed significant downregulation of both mRNA and protein levels of MMP‐2 as detected by real‐time PCR (1.70 ± 0.59 vs 0.39 ± 0.18, P = 0.034) (Fig. 4A) and western blot (Fig. 4B). In addition, both the expression level and activity of MMP‐2 were significantly inhibited in the HCC tissues of MSC‐treated mice models compared with the controls (1.13 ± 0.55 vs 1.61 ± 1.52, P = 0.050) (Fig. 4C). Moreover, downregulation of MMP‐2 correlated with inhibition of HCC metastasis, since the MSC‐treated mice exhibited lower levels of metastasis than the controls (0.86 ± 0.30 vs 1.46 ± 0.84, P = 0.032). Reduced expression of MMP‐2 was also observed in the HCC tissues of the MSC‐treated mice by immunohistochemical staining (Fig. 4D). However, there was no significant change in MMP‐9 expression in the MHCC97‐H cell line co‐cultured with MSC‐CM (Fig. 4B), or in the HCC tissues of MSC‐treated mice (Fig. 4D). These findings suggest that the inhibitory effect of MSC on HCC metastasis might be related to the downregulation of MMP‐2.
Figure 4.
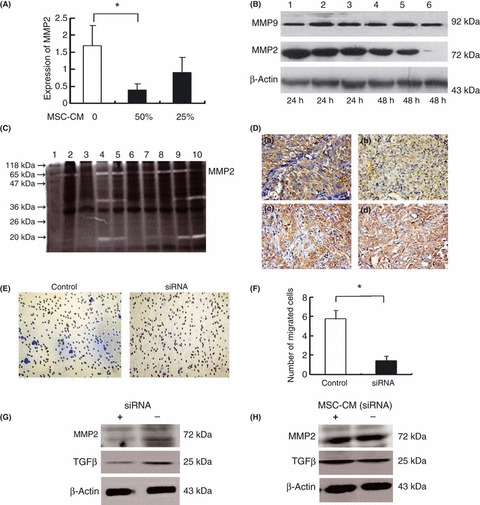
Effect of mesenchymal stem cells (MSC) on MMP‐2 and MMP‐9 expression in MHCC97‐H cells. Real‐time PCR (A) and western blot analysis (B) showed that both mRNA and protein levels of MMP‐2 in MHCC97‐H were decreased by culture with MSC conditioned medium (MSC‐CM), whereas there was no significant change in the expression of MMP‐9. Lanes 1–3, or 4–6 correspond to 0%, 25% and 50% MSC‐CM, respectively. (C) In zymography analysis, the amount of pro‐MMP‐2 (72 kD) was significantly lower in the MSC‐treated mice (lanes 2, 3, 6, 7) than in the controls (lanes 4, 5, 8–10). (D) Immunohistochemical staining showed lower expression of MMP‐2 in hepatocellular carcinoma (HCC) tissues following MSC treatment (a) compared with the controls (b), whereas no significant alteration in MMP‐9 expression was observed between MSC‐treated animals (c) and controls (d) (×20). (E,F) Transformation growth factor‐beta 1 (TGFβ1) siRNA significantly inhibited the invasive capability of MHCC97‐H cells compared with the controls, as evaluated by Transwell assays. *P < 0.05. (G) Western blot analysis showed that TGFβ1 siRNA concurrently downregulated expression of TGFβ1 and MMP‐2 in MHCC97‐H cells under non‐reducing conditions. (H) The TGFβ1 siRNA significantly blocked the inhibitory effect of MSC on the expression of TGFβ and MMP‐2 in MHCC97‐H cells under non‐reducing conditions.
Association between TGFβ and MMP‐2. To assess the possible relationship between TGFβ and MMP‐2 expression, HCC tissues were divided into two groups according to the median level of TGFβ1. The MMP‐2 expression level in HCC tissues of the low TGFβ1 group was significantly lower than that of the high TGFβ1 group (1.25 ± 0.31 vs 1.82 ± 0.50, P = 0.047), indicating a significant correlation between expression of TGFβ and MMP‐2.
To further prove the association between TGFβ1 and MMP‐2 expression, siRNA against TGFβ was used to block TGFβ expression in both the MHCC97‐H cells and MSC. Treatment with TGFβ siRNA significantly inhibited the invasive ability of MHCC97‐H cells (1.43 ± 0.50 vs 5.75 ± 0.88, P = 0.002) (Fig. 4E,F) and resulted in concurrent downregulation of TGFβ1 and MMP‐2 expression (Fig. 4G). Treatment of MSC with TGFβ siRNA blocked the inhibitory effect of MSC‐CM on TGFβ and MMP‐2 expression in MHCC97‐H cells (Fig. 4H). Moreover, there was no significant change in the proliferation of MHCC97H cells (OD values, 153.02 ± 92.45 vs 163.49 ± 94.96, P = 0.886), or in the numbers of migrated cells detected by Transwell assay (12.50 ± 5.18 vs 15.37 ± 4.56, P = 0.259) before and after culture with CM from TGFβ siRNA‐treated MSC. These data suggest that TGFβ1 expression is closely correlated with expression of MMP‐2, and plays an important role in the effect of MSC on HCC progression.
Discussion
Mesenchymal stem cells have long been known to contribute to the maintenance and regeneration of connective tissues, the formation of fibroblasts and wound repair through engraftment. (23) In vivo engraftment is not merely an intrinsic function of MSC but also depends on appropriate external signals from the microenvironment. A tumor can be considered a special wound that never heals, since the formation of tumor stroma closely resembles wound healing and scar formation. (24) In this context, the tumor microenvironment might provide a special niche for engraftment of MSC. 25 , 26 Indeed, in this study, we consistently observed a larger number of MSC distributed within the tumor site compared with the non‐tumor liver tissues.
The differentiation of stem cells is determined not only by inherent cellular properties but also by the cellular niche, which is composed of extracellular matrix and other non‐cellular constituents. (27) Although the differentiation of MSC in tumor tissues is still controversial, many studies have suggested that the MSC is a potential precursor for tumor stroma. (17) Our observation that MSC were mainly distributed in the tumor stroma supports this view. It is known that, during tumor progression, cancer and stromal cellular responses co‐evolve and communicate with each other through the secretion of growth factors, chemokines and cytokines. (25) Therefore, if the MSC is one of the origins of cancer stroma, the engrafted MSC will change the microenvironment of the tumor in complicated ways to influence the growth and metastasis of the tumor. Experiments using the MSC‐CM gave similar results to the co‐culture with the MSC themselves, providing evidence that the effects of MSC on cancer cells are mainly due to the parasecretion of MSC, which induces changes in the microenvironment of HCC.
The most interesting, albeit perplexing, results of this study involve the dual roles of MSC in tumor growth and metastasis of HCC. Previous gene expression profiling indicated that TGFβ signaling was one of the most important pathways in the roles of MSC. (28) We believed the diverse effects of MSC on tumor growth and metastasis of HCC might, at least in part, be due to the modulation of TGFβ. Clinical and experimental results show that TGFβ has diverse and often conflicting roles in tumor progression even within the same tumor types. On one hand, components of the TGFβ pathway are important tumor suppressor genes; absence or malfunction of one or more signaling proteins in this pathway is believed to result in loss of growth regulation. On the other hand, abnormal expression of TGFβ by tumor cells is associated with increased invasiveness in various carcinomas including HCC. 29 , 30 , 31 , 32 , 33
The function of TGF‐β1 might be time‐bound. In normal epithelial cells and early tumors, TGFβ acts as a tumor suppressor, inhibiting proliferation and inducing apoptosis of tumor cells. In contrast, during tumor progression, TGFβ becomes an oncogenic factor inducing proliferation, angiogenesis, invasion and metastasis, as well as suppressing the anti‐tumoral immune response. 33 , 34 Transformation growth factor‐beta has been found to enhance the cell proliferation of colon cancer; (35) inhibition of TGFβ signaling reduces pancreatic adenocarcinoma growth and invasiveness. (36) Similarly, the serum concentration of TGFβ is elevated as tumors progress in HCC patients, and tumor‐derived TGFβ could activate Smads and constantly suppress p151NK4B expression to accelerate the proliferation and malignant progression of HCC. (37) In the present study, MSC exhibited higher expression levels of TGFβ1 than HCC cells, and co‐culture of HCC cells with MSC‐CM induced downregulation of TGFβ1 expression by a feedback mechanism. Thus, it appears that TGFβ1 expression levels of HCC cells are influenced by MSC and moreover. And the effects of TGFβ1 downregulation induced by MSC on HCC growth and metastasis are consistent with a previous report of breast cancer, in which TGFβ signaling was shown to suppress de novo mammary cancer formation but promote metastasis of tumors in genetic mouse models, and treatment with TGFβ neutralizing antibodies or receptor kinase inhibitors strongly inhibited development of lung and bone metastases, but did not significantly affect tumor cell proliferation or apoptosis. (38)
Matrix metalloproteinases are known to play important roles in the progression and metastasis of several cancers including HCC. Previous studies (42) have reported interplay between TGFβ and MMP. MMP‐9 and MMP‐2 proteolytically cleave latent TGFβ, providing a novel and potentially important mechanism for TGFβ activation. (39) Conversely, TGFβ can induce expression of MMP and modulates the net balance of MMP/TIMP through the TGFβ/smad pathway. 40 , 41 These findings indicate a positive feedback loop between activation of TGFβ1 and MMP‐2. In the present study, we have shown that MSC significantly downregulated expression of both TGFβ1 and MMP‐2. The correlation between MMP‐2 and the TGFβ1 pathway observed in our study suggests that MSC can break down this loop and suppress the upregulation of MMP‐2 expression otherwise induced by the TGFβ1 pathway.
In summary, the above findings indicate that MSC can change the stromal microenvironment and suppress metastasis of HCC. Downregulation of TGFβ plays a crucial role in this process. Therefore, MSC could represent a novel strategy to control the metastasis of HCC through modulation of the host microenvironment. However, further attention should be given to balancing the disparate effects of MSC on tumor growth and metastasis.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This work was supported in part by China National Key Projects for Infectious Disease (2008ZX10002‐021), China National Natural Science Foundation for Distinguished Young Scholars (30325041), the China National High‐tech Research and Development Program (863 Program) (2006AA02Z473), and Program of Shanghai Subject Chief Scientist (08XD14008). The authors would like to thank Mrs Qiong Xue, Dong‐Mei Gao, Rui‐Xia Sun and Jie Chen, and Drs Hai‐Ying Zeng, Teng‐Fang Zhu and Jun Chen for their help with the animal experiments and cell culture.
References
- 1. Parkin D, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin 2005; 55: 74–108. [DOI] [PubMed] [Google Scholar]
- 2. He J, Gu D, Wu X et al. Major causes of death among men and women in China. N Engl J Med 2005; 353: 1124–34. [DOI] [PubMed] [Google Scholar]
- 3. Portolani N, Coniglio A, Ghidoni S et al. Early and late recurrence after liver resection for hepatocellular carcinoma: prognostic and therapeutic implications. Ann Surg 2006; 243: 229–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ye Q, Qin L, Forgues M et al. Predicting hepatitis B virus‐positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat Med 2003; 9: 416–23. [DOI] [PubMed] [Google Scholar]
- 5. Qin L, Tang Z. Recent progress in predictive biomarkers for metastatic recurrence of human hepatocellular carcinoma: a review of the literature. J Cancer Res Clin Oncol 2004; 130: 497–513. [DOI] [PubMed] [Google Scholar]
- 6. Coussens L, Tinkle C, Hanahan D, Werb Z. MMP‐9 supplied by bone marrow‐derived cells contributes to skin carcinogenesis. Cell 2000; 103: 481–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lyden D, Hattori K, Dias S et al. Impaired recruitment of bone‐marrow‐derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med 2001; 7: 1194–201. [DOI] [PubMed] [Google Scholar]
- 8. Chanda D, Isayeva T, Kumar S et al. Therapeutic potential of adult bone marrow‐derived mesenchymal stem cells in prostate cancer bone metastasis. Clin Cancer Res 2009; 15: 7175–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sasportas L, Kasmieh R, Wakimoto H et al. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc Natl Acad Sci U S A 2009; 106: 4822–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fritz V, Jorgensen C. Mesenchymal stem cells: an emerging tool for cancer targeting and therapy. Curr Stem Cell Res Ther 2008; 3: 32–42. [DOI] [PubMed] [Google Scholar]
- 11. Karnoub A, Dash A, Vo A et al. Mesenchymal stem cells within tumor stroma promote breast cancer metastasis. Nature 2007; 449: 557–63. [DOI] [PubMed] [Google Scholar]
- 12. Alison M, Lovell M. Liver cancer: the role of stem cells. Cell Prolif 2005; 38: 407–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pittenger M, Mackay A, Beck S et al. Multilineage potential of adult human mesenchymal stem cells. Science 1999; 284: 143–7. [DOI] [PubMed] [Google Scholar]
- 14. Minguell J, Erices A, Conget P. Mesenchymal stem cells. Exp Biol Med (Maywood) 2001; 226: 507–20. [DOI] [PubMed] [Google Scholar]
- 15. Jiang Y, Jahagirdar B, Reinhardt R et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 2002; 418: 41–9. [DOI] [PubMed] [Google Scholar]
- 16. Liechty K, MacKenzie T, Shaaban A et al. Human mesenchymal stem cells engraft and demonstrate site‐specific differentiation after in utero transplantation in sheep. Nat Med 2000; 6: 1282–6. [DOI] [PubMed] [Google Scholar]
- 17. Studeny M, Marini F, Dembinski J et al. Mesenchymal stem cells: potential precursors for tumor stroma and targeted‐delivery vehicles for anticancer agents. J Natl Cancer Inst 2004; 96: 1593–603. [DOI] [PubMed] [Google Scholar]
- 18. Fierro F, Sierralta W, Epuñan M, Minguell J. Marrow‐derived mesenchymal stem cells: role in epithelial tumor cell determination. Clin Exp Metastasis 2004; 21: 313–9. [DOI] [PubMed] [Google Scholar]
- 19. Ohlsson L, Varas L, Kjellman C, Edvardsen K, Lindvall M. Mesenchymal progenitor cell‐mediated inhibition of tumor growth in vivo and in vitro in gelatin matrix. Exp Mol Pathol 2003; 75: 248–55. [DOI] [PubMed] [Google Scholar]
- 20. Zhu Y, Sun Z, Han Q et al. Human mesenchymal stem cells inhibit cancer cell proliferation by secreting DKK‐1. Leukemia 2009; 23: 925–33. [DOI] [PubMed] [Google Scholar]
- 21. Li Y, Tang Y, Ye L et al. Establishment of a hepatocellular carcinoma cell line with unique metastatic characteristics through in vivo selection and screening for metastasis‐related genes through cDNA microarray. J Cancer Res Clin Oncol 2003; 129: 43–51. [DOI] [PubMed] [Google Scholar]
- 22. Giannelli G, Fransvea E, Marinosci F et al. Transforming growth factor‐beta1 triggers hepatocellular carcinoma invasiveness via alpha3beta1 integrin. Am J Pathol 2002; 161: 183–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Prockop D. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science 1997; 276: 71–4. [DOI] [PubMed] [Google Scholar]
- 24. Dvorak H. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 1986; 315: 1650–9. [DOI] [PubMed] [Google Scholar]
- 25. Littlepage L, Egeblad M, Werb Z. Coevolution of cancer and stromal cellular responses. Cancer Cell 2005; 7: 499–500. [DOI] [PubMed] [Google Scholar]
- 26. Son B, Marquez‐Curtis L, Kucia M et al. Migration of bone marrow and cord blood mesenchymal stem cells in vitro is regulated by stromal‐derived factor‐1‐CXCR4 and hepatocyte growth factor‐c‐met axes and involves matrix metalloproteinases. Stem Cells 2006; 24: 1254–64. [DOI] [PubMed] [Google Scholar]
- 27. Scadden D. The stem‐cell niche as an entity of action. Nature 2006; 441: 1075–9. [DOI] [PubMed] [Google Scholar]
- 28. Kulterer B, Friedl G, Jandrositz A et al. Gene expression profiling of human mesenchymal stem cells derived from bone marrow during expansion and osteoblast differentiation. BMC Genomics 2007; 8: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Woodward J, Rennie I, Burn J, Sisley K. A potential role for TGFbeta in the regulation of uveal melanoma adhesive interactions with the hepatic endothelium. Invest Ophthalmol Vis Sci 2005; 46: 3473–7. [DOI] [PubMed] [Google Scholar]
- 30. Katabami K, Mizuno H, Sano R et al. Transforming growth factor‐beta1 upregulates transcription of alpha3 integrin gene in hepatocellular carcinoma cells via Ets‐transcription factor‐binding motif in the promoter region. Clin Exp Metastasis 2005; 22: 539–48. [DOI] [PubMed] [Google Scholar]
- 31. Blobe G, Schiemann W, Lodish H. Role of transforming growth factor beta in human disease. N Engl J Med 2000; 342: 1350–8. [DOI] [PubMed] [Google Scholar]
- 32. Seoane J. The TGFBeta pathway as a therapeutic target in cancer. Clin Transl Oncol 2008; 10: 14–9. [DOI] [PubMed] [Google Scholar]
- 33. Padua D, Massagué J. Roles of TGFbeta in metastasis. Cell Res 2009; 19: 89–102. [DOI] [PubMed] [Google Scholar]
- 34. Welm AL. TGFbeta primes breast tumor cells for metastasis. Cell 2008; 133: 27–8. [DOI] [PubMed] [Google Scholar]
- 35. Chow JY, Cabral JA, Chang J, Carethers JM. TGFbeta modulates PTEN expression independently of SMAD signaling for growth proliferation in colon cancer cells. Cancer Biol Ther 2008; 7: 1694–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gaspar NJ, Li L, Kapoun AM et al. Inhibition of transforming growth factor beta signaling reduces pancreatic adenocarcinoma growth and invasiveness. Mol Pharmacol 2007; 72: 152–61. [DOI] [PubMed] [Google Scholar]
- 37. Matsuzaki K, Date M, Furukawa F et al. Autocrine stimulatory mechanism by transforming growth factor beta in human hepatocellular carcinoma. Cancer Res 2000; 60: 1394–402. [PubMed] [Google Scholar]
- 38. Tan AR, Alexe G, Reiss M. Transforming growth factor‐beta signaling: emerging stem cell target in metastatic breast cancer? Breast Cancer Res Treat 2009; 115: 453–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yu Q, Stamenkovic I. Cell surface‐localized matrix metalloproteinase‐9 proteolytically activates TGF‐beta and promotes tumor invasion and angiogenesis. Genes Dev 2000; 14: 163–76. [PMC free article] [PubMed] [Google Scholar]
- 40. Kwak H, Park M, Cho H et al. Transforming growth factor‐beta1 induces tissue inhibitor of metalloproteinase‐1 expression via activation of extracellular signal‐regulated kinase and Sp1 in human fibrosarcoma cells. Mol Cancer Res 2006; 4: 209–20. [DOI] [PubMed] [Google Scholar]
- 41. Zavadil J, Bitzer M, Liang D et al. Genetic programs of epithelial cell plasticity directed by transforming growth factor‐beta. Proc Natl Acad Sci U S A 2001; 98: 6686–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cox G, O’Byrne KJ. Matrix metalloproteinase and cancer. Anticancer Res 2001; 21: 4207–20. [PubMed] [Google Scholar]