

Abstract
Although unlimited proliferation of cancer cells is supported by multiple signaling pathways involved in the regulation of proliferation, survival, and apoptosis, the molecular mechanisms coordinating these different pathways to promote the proliferation and survival of cancer cells have remained unclear. SAPK and integrin‐ILK signaling pathways play key roles in the promotion of apoptosis and cell proliferation/survival, respectively. Studies of TNFα‐ and H2O2‐induced apoptosis revealed that ASK1, a component of the SAPK system, mediates the TNFα and H2O2 signaling of apoptosis. ASK1 is activated by autophosphorylation of a specific threonine residue (T845) following TNFα stimulation. Our recent studies indicate that PP2Cɛ, a member of the PP2C family, associates with and inactivates ASK1 by dephosphorylating T845. In contrast, PP2Cδ/ILKAP, a second PP2C family member, activates ASK1 by enhancing cellular phosphorylation of T845. PP2Cδ/ILKAP also forms a complex with ILK1 to inhibit the GSK3β‐mediated integrin‐ILK1 signaling in vivo, inhibiting cell cycle progression. These observations raise the possibility that PP2Cδ/ILKAP acts to control the cross‐talk between integrin‐induced and TNFα‐induced signaling pathways, inhibiting the former and stimulating the latter, thereby inhibiting proliferation and survival and promoting the apoptosis of cancer cells. (Cancer Sci 2006; 97: 563–567)
Abbreviations:
- ASK1
apoptosis signal‐regulating kinase 1
- GSK‐3β
glycogen synthase kinase‐3β
- ILK
integrin‐linked kinase
- JNK
c‐Jun N‐terminal kinase
- LNCaP
lymphnode carcinoma of prostate
- MAPK
mitogen‐activated protein kinase
- MEF
mouse embryonic fibroblasts
- MEK
mitogen extracellular signal‐regulated kinase
- MKK
mitogen‐activated protein kinase kinase
- MKKK
mitogen‐activated protein kinase kinase kinase
- PP2C
protein serine/threonine phosphatase 2C
- ROS
reactive oxygen species
- SAPK
stress‐activated protein kinase
- siRNA
short interfering RNA
- TAK1
transforming growth factor‐activated kinase 1
- TNF‐α
tumor necrosis factor‐α.
Unlimited proliferation is typical of cancer cells, a phenotype supported by several distinct biological events, including enhanced cell proliferation, increased cell survival, and decreased cell death. Studies examining the molecular bases of these events have identified a variety of signaling pathways that are involved in their regulation. The mechanisms by which these different signaling pathways are coordinated to regulate proliferation and survival of cancer cells has remained poorly understood.
ROS are normally generated by mitochondria during respiration, but can also be generated by inducible enzyme systems in mammalian cells.( 1 ) Cellular ROS levels increase following exposure to a variety of stress agents, such as anticancer drugs.( 2 ) Upregulation of these highly reactive molecules promotes apoptosis by stimulating the activation of pro‐apoptotic signaling molecules, such as JNK and p38 (Fig. 1).( 3 , 4 ) ROS also function in p53‐induced apoptosis.( 5 )
Figure 1.
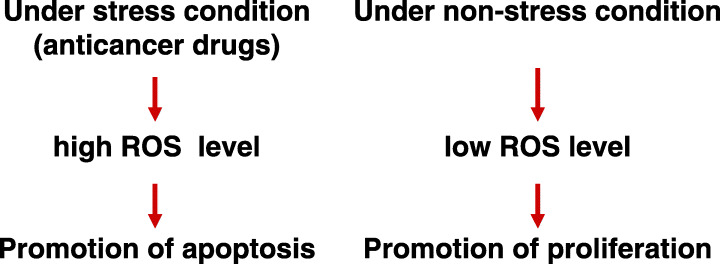
Diverse effects of reactive oxygen species (ROS) on behavior of cancer cells.
In contrast, low levels of ROS stimulate normal cell proliferation under non‐stress conditions; ROS production was reported to be increased in cancer cells( 6 , 7 ) (Fig. 1). Elevated ROS appear to maintain the constitutive activation of transcription factors (NF‐κB and AP‐1) during tumor progression.( 8 ) Overexpression of Mox1, the catalytic subunit of NADPH oxidase, induces superoxide generation, leading to the transformation of NIH 3T3 cells.( 9 ) Thus, ROS might play a critical role in the pathogenesis of cancer. The diverse, and even opposing, effects of ROS on cell behavior reflect the complex nature of the biological functions of these highly reactive molecules.
Integrins, a large family of adhesion receptors including more than 20 members, mediate highly dynamic cell–cell and cell–extracellular matrix interactions. Integrin‐mediated cell adhesion regulates a wide variety of biological processes, including cell migration, survival, and proliferation. The association and dissociation of integrin and their ligands are achieved by changes in the conformations of integrin molecules. Adoption of the active conformation triggered by intracellular signaling and cytoskeletal assembly, resulting in ligand binding, integrin clustering, and recruitment of cytoplasmic plaque proteins to integrin attachment sites, called focal adhesions.( 10 , 11 ) ILK plays a central role in integrin activation and signaling.( 12 ) ILK is composed of three structurally distinct domains, three ankyrin repeats near the N‐terminus, a short linker sequence, and a C‐terminal kinase domain.
Functional studies of ILK1 revealed that GSK3β is a target of ILK1.( 13 ) GSK3β, an important mediator of developmental signaling through the Wnt/wingless pathway, alters the transcriptional activity of Tcf/Lef factors through phosphorylation of the transcriptional cofactor β‐catenin.( 14 , 15 ) GSK3β‐mediated phosphorylation targets β‐catenin for degradation. Overexpression of ILK1 in epithelial cells induces the phosphorylation of GSK3β at Ser9, an inhibitory modification that results in the stabilization and nuclear translocation of β‐catenin, with concomitant activation of Tcf/Lef transcription factors. Thus, ILK1 is an important intracellular regulator of Wnt/wingless signaling, acting specifically through the modulation of GSK3β activity. Activation of ILK1 leads to cell survival and proliferation.
In this article, we propose a novel mechanism governing the cooperation of ROS induction of apoptosis with the integrin‐induced cellular proliferation.
SAPKs play key roles in ROS‐induced activation of both proliferation and apoptosis
Studies examining ROS‐induced activation of cell growth and apoptosis revealed that SAPKs function in these opposing effects. SAPKs, a subfamily of the MAPK superfamily, are highly conserved from yeast to mammals (Fig. 2).( 16 ) Two distinct classes of SAPKs are present in mammalian cells, the JNK and p38 kinases. Both JNK and p38 are activated by a variety of environmental stresses, regulating diverse cellular functions in response to these challenges. SAPK activation requires phosphorylation by dual‐specificity protein kinases, members of the MKK family, at conserved tyrosine and threonine residues within the catalytic domain. MKK3 and MKK6 specifically phosphorylate p38, whereas MKK7 selectively modifies JNK. MKK4 recognizes both classes of stress‐activated kinases. The MKKs are activated by the phosphorylation of conserved serine and threonine residues. MKKKs, including ASK1, TAK1, the MEK kinase group, and the mixed lineage kinase group of kinases, have been identified, some of which are also activated by phosphorylation (Fig. 2).
Figure 2.
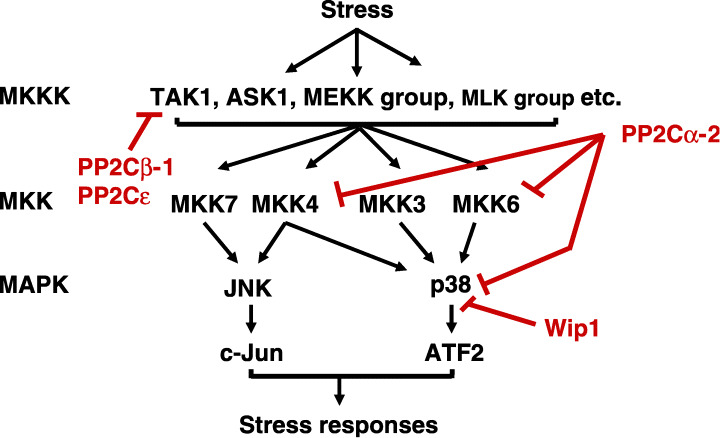
Regulation of stress‐activated protein kinase (SAPK) signal pathways by PP2C family members. The protein kinase cascade of SAPK signaling pathways and the points where the PP2C family members can interfere with the signals are shown. ASK1, apoptosis signal‐regulating kinase 1; ATF2, activating transcription factor; JNK, c‐Jun N‐terminal kinase; MAPK, mitogen‐activated protein kinase; MKK, mitogen‐activated protein kinase kinase; MKKK, mitogen‐activated protein kinase kinase kinase; PP2C, protein serine/threonine phosphatase 2C; TAK1, transforming growth factor‐activated kinase 1.
SAPKs have been studied primarily in stress responses and apoptosis.( 16 ) Following cellular stress, SAPK activation is important in promoting apoptosis in wide range of cell types, including primary tumor cells and transformed cell lines. Signaling through the JNK pathway is critical in the activation of the mitochondria‐dependent apoptotic pathway, but appears to be dispensable for the apoptotic mechanisms induced by activation of death receptors.( 17 )
Accumulating genetic and biochemical data, however, suggest that SAPKs, especially JNK, contribute to proliferative responses in the absence of stress. JNK1−/– and JNK1−/–JNK2−/–MEFs proliferate more slowly than wild‐type MEFs, achieving lower saturation densities. These results establish that JNK is required for normal MEF proliferation.( 17 )
ASK1 mediates ROS‐induced cell apoptosis
Recent studies have suggested several mechanisms linking ROS induction and SAPK activity. ASK1, an upstream regulator of SAPKs, is inhibited in non‐stressed cells by association with thioredoxin.( 18 ) Increased ROS levels dissociate this complex, enabling the activation of ASK1 and downstream SAPKs.( 19 ) ROS induction is also implicated in TNFα‐induced activation of ASK1.
Studies of the mechanisms controlling TNFα‐ or H2O2‐induced signaling have suggested that early/transient activation of JNK by TNFα or H2O2 correlates with cell survival/proliferation, whereas late/sustained activation leads to apoptosis, raising the possibility that different pathways of JNK activation might regulate cell survival/proliferation and apoptosis.( 20 , 21 ) Using wild‐type and ASK1−/–MEFs, Tobiume et al. demonstrated that ASK1 does not participate in the initial TNFα‐ or H2O2‐induced activation of JNK, but does function in sustained activation of JNK.( 22 ) These results support the idea that ASK1 is specifically responsible for promoting TNFα‐ or H2O2‐induced apoptosis. The induction of apoptosis by TNFα was restored in ASK1−/–MEFs following infection with adenoviruses encoding ASK1.
PP2Cɛ participates in negative regulation of ASK1
Phosphorylation events participate in both the activation and inactivation of SAPK signaling. ASK1 is activated by autophosphorylation at Thr845 following stimulation of upstream signals, but is inactivated by phosphorylation at Ser83 by Akt, suggesting a direct link between Akt and the stress‐activated kinase family.( 23 ) Phosphorylation of both Ser967 and Ser1034 has also been implicated in ASK1 inactivation, although the protein kinases responsible for this phosphorylation event have not been identified.( 24 , 25 ) These observations suggest that protein phosphatases function in both the negative and positive regulation of SAPK signaling.
PP2C is one of the four major protein serine/threonine phosphatase families, PP1, PP2A, PP2B, and PP2C, in eukaryotes. At least 12 distinct PP2C gene products, 2 Cα, 2Cβ, 2 Cγ/FIN13, 2Cδ/ILKAP, 2Cɛ, 2Cζ, 2Cη, Wip1, CaMKP/hFEM2/POPX2, CaMKP‐N/POPX1, NERPP‐2C, and SCOP/PHLPP, have been identified in mammalian cells (Table 1).( 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 , 49 , 50 , 51 ) These 12 PP2C gene products all exhibit Mg2 ± and/or Mn2+‐dependent protein phosphatase activities, sharing six conserved structural motifs. Functional studies of PP2C family members indicated that four of these enzymes, PP2Cα, PP2Cβ, PP2Cɛ, and Wip1, participate in the negative regulation of SAPK signaling (Fig. 2).( 29 , 47 , 52 , 53 , 54 ) PP2Cβ‐1 and PP2Cɛ suppress SAPK signaling by associating with and dephosphorylating TAK1 at different phosphorylation sites( 47 , 54 ) (Li et al., unpublished observations, 2004).
Table 1.
PP2C family members
| Size (kDa) | Isoforms | Tissue distribution | References | |
|---|---|---|---|---|
| PP2Cα | 42, 36 | α‐1, α‐2 | ubiquitous | 27, 29, 33 |
| PP2Cβ | 42–55 | β‐1(βs)‐β‐5, | β‐1, βx: ubiquitous | 31, 32, 34–36 |
| βx (βl) | β‐2: brain, heart β‐3–5: testis, liver, intestine | |||
| PP2Cγ (FIN13) | 59 | ubiquitous | 37, 38 | |
| PP2Cδ (ILKAP) | 43 | ubiquitous | 39, 40 | |
| PP2Cɛ | 34 | rich in brain, heart, testis | 47 | |
| PP2Cζ | 56 | testis | 48 | |
| PP2Cη | 45 | ubiquitous | 49 | |
| Wip1 | 66 | 41 | ||
| CaMKP (hFEM2, POPX2) | 49 | ubiquitous | 42, 43, 45 | |
| NERPP‐2C | 55 | NERPP‐55 | brain | 46 |
| 80 | NERPP‐80 | |||
| CaMKP‐N (POPX1) | 83 | rich in brain, testis | 44, 45 | |
| SCOP (PHLPP) | 140 | 50, 51 |
Recent evidence suggested that PP2Cɛ participates in the regulation of both TAK1 and ASK1 signaling (Fig. 3) (Saito et al., unpublished observations, 2006). In coexpression and coimmunoprecipitation studies, ASK1 was able to associate with PP2Cɛ, as seen with TAK1. The in vivo binding of endogenous PP2Cɛ to endogenous ASK1 was also observed in mouse brain. PP2Cɛ inhibited ASK1‐induced AP‐1 activation in a dose‐dependent manner. PP2Cɛ also suppressed the H2O2‐enhanced phosphorylation of ASK1 Thr845, suggesting that PP2Cɛ is involved in the negative regulation of ASK1 (Fig. 3).
Figure 3.
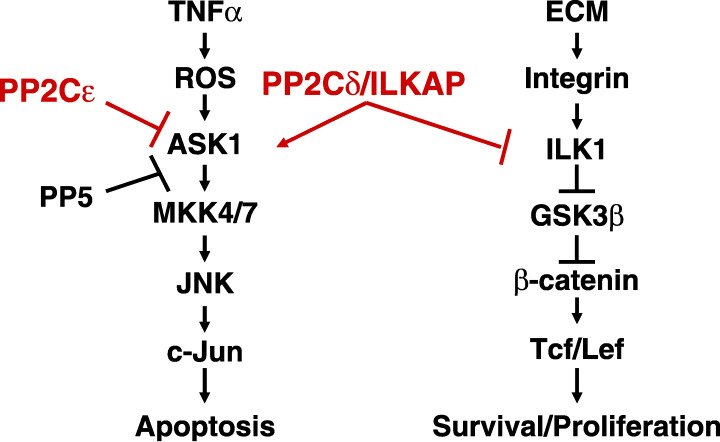
Regulation of TNF‐α‐ASK1 and integrin‐ILK1 signal pathways by PP2Cɛ, PP5 and PP2Cδ/ILKAP. ASK1, apoptosis signal‐regulating kinase 1; GSK‐3β, glycogen synthase kinase‐3β; ILK, integrin‐linked kinase; JNK, c‐Jun N‐terminal kinase; MKK, mitogen‐activated protein kinase kinase; PP2C, protein serine/threonine phosphatase 2C; ROS, reactive oxygen species; TNF‐α, tumor necrosis factor‐α.
Morita et al. previously reported that ASK1 was negatively regulated by PP5, a protein Ser/Thr phosphatase that is distinct from the PP2C family (Fig. 3).( 55 ) Interestingly, PP5 was also able to dephosphorylate Thr845 of ASK1. Furthermore, H2O2 treatment of cells induced the association of PP5 with ASK1, suggesting that PP5 participates in the negative feedback regulation of the ASK1 signaling pathway. In contrast, PP2Cɛ bound to ASK1 under non‐stress conditions and dissociated from ASK1 on H2O2 treatment of cells, suggesting that PP2Cɛ contributes to keeping the ASK1 signaling pathway in an inactive state by associating with and dephosphorylating ASK1 (Saito et al., unpublished observations, 2006). Thus, PP2Cɛ and PP5 appear to play different roles in regulating the ASK1 pathway, although they both dephosphorylate the same site of ASK1.
PP2Cδ/ILKAP participates in positive regulation of ASK1
In contrast to PP2Cɛ, PP2Cδ/ILKAP acts as an activator of TNFα‐induced SAPK signaling (Fig. 3). The expression of PP2Cδ/ILKAP, originally identified by Tong et al., is induced by cellular stresses, including ethanol treatment or ultraviolet irradiation.( 39 ) Recently, we determined that expression of PP2Cδ/ILKAP in 293 cells enhanced both the TNFα‐induced activation of AP1 and the phosphorylation of JNK and p38. PP2Cδ expression in HEK293 cells, however, had no effect on TNFα‐induced, early/transient activation of JNK or p38, but further enhanced the sustained activation of these MAPKs (Toriumi et al., unpublished observations, 2006). In these cells, PP2Cδ/ILKAP associated with ASK1, activating this kinase by enhancing the phosphorylation of T845. These observations suggest that PP2Cδ might act as a positive regulator of TNFα‐induced apoptosis (Fig. 3). It remains unknown, however, whether PP2Cδ mediates this activation by dephosphorylating any of the inhibitory phosphorylation sites (Ser83, Ser967, and Ser1034) of ASK1.
PP2Cδ/ILKAP suppresses integrin signaling by inhibiting ILK1
Leung‐Hagesteijin et al. isolated PP2Cδ/ILKAP in a yeast two‐hybrid screen designed to identify proteins interacting with ILK1.( 40 ) PP2Cδ/ILKAP and ILK1 were coprecipitated from HEK293 cell lysates, independent of ILK1 and PP2Cδ/ILKAP catalytic activities. Exogenous expression of recombinant, catalytically active PP2Cδ/ILKAP in HEK293 cells inhibited endogenous ILK1 protein kinase activity (Fig. 3). PP2Cδ/ILKAP did not affect Raf‐1 kinase activity. Inactive PP2Cδ/ILKAP did not affect ILK1 activity. PP2Cδ/ILKAP expression inhibited integrin‐stimulated phosphorylation of GSK3β at Ser9, leading to inactivation of Tcf/Lef. Conversely, silencing of endogenous PP2Cδ/ILKAP with siRNA stimulated GSK3β phosphorylation at Ser9. These results suggested that PP2Cδ/ILKAP selectively inhibits GSK3β‐mediated ILK1 signaling in vivo by forming a complex with ILK1 (Fig. 3).( 40 )
PP2Cδ/ILKAP suppresses oncogenic transformation by inhibition of ILK1
The function of PP2Cδ/ILKAP in the regulation of oncogenic transformation was investigated by Kumar et al.( 56 ) In LNCaP prostate carcinoma cells, both transient and stable expression of PP2Cδ/ILKAP suppressed ILK kinase activity; PP2Cδ/ILKAP‐mediated inhibition of ILK downregulated GSK3β Ser9 phosphorylation, which could be reversed by overexpression of wild‐type, but not dominant‐negative, ILK. The expression levels of cyclin D1, a target of ILK‐GSK3β signaling, were enhanced by siRNA‐mediated downregulation of PP2Cδ/ILKAP, suggesting that ILK1 antagonism modulates cell cycle progression. Exogenous expression of PP2Cδ/ILKAP increased the proportion of LNCaP cells in G1 phase in comparison to that seen in cells transfected with vector alone; siRNA suppression of PP2Cδ/ILKAP expression increased the entry of cells into S phase, consistent with ILK antagonism. Anchorage‐independent growth of LNCaP cells was inhibited by PP2Cδ/ILKAP overexpression, suggesting a critical role for this protein in the suppression of cellular transformation.( 56 ) Taken together, these results indicate that endogenous PP2Cδ/ILKAP activity inhibits ILK‐GSK3β signaling, suggesting that PP2Cδ/ILKAP activity inhibits oncogenic transformation (Fig. 3).
Conclusions
In this article, we discussed the regulation by PP2C family members of the TNFα‐ASK1‐JNK and integrin‐ILK‐GSK3β‐β‐catenin signaling pathways, which participate in the stimulation of apoptosis and cell proliferation/survival, respectively. The TNFα‐ASK1‐JNK pathway is inactivated by PP2Cɛ and activated by PP2Cδ/ILKAP. Both of these PP2C family members regulate ASK1 activity, although they exert opposite effects (Fig. 3). These observations suggest that PP2Cδ/ILKAP functions as an apoptosis‐promoting factor. PP2Cδ/ILKAP, however, is also able to suppress the integrin‐ILK‐GSK3β‐β‐catenin pathway by inhibition of ILK1. Accumulating evidence indicates that PP2Cδ/ILKAP inhibits cell proliferation and oncogenic transformation through its effects on this pathway. These observations raise the possibility that PP2Cδ might control the cross‐talk between the integrin‐induced and TNFα‐induced signaling pathways, inhibiting the former and stimulating the latter to promote the apoptosis of cancer cells (Fig. 3).
References
- 1. Kamata H, Hirata H. Redox regulation of cellular signalling. Cell Signal 1999; 11: 1–14. [DOI] [PubMed] [Google Scholar]
- 2. Jabs T. Reactive oxygen intermediates as mediators of programmed cell death in plants and animals. Biochem Pharmacol 1999; 57: 231–45. [DOI] [PubMed] [Google Scholar]
- 3. Benhar M, Dalyot I, Engelberg D, Levitzki A. Enhanced ROS production in oncogenically transformed cells potentiates c‐jun N‐terminal kinase and p38 mitogen‐activated protein kinase activation and sensitization to genotoxic stress. Mol Cell Biol 2001; 21: 6913–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Davis WJ, Ronai Z, Tew KD. Cellular thiols and reactive oxygen species in drug‐induced apoptosis. J Pharmacol Exp Ther 2001; 296: 1–6. [PubMed] [Google Scholar]
- 5. Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53‐induced apoptosis. Nature 1997; 389: 300–5. [DOI] [PubMed] [Google Scholar]
- 6. Burdon RH. Superoxide and hydrogen peroxide in relation to mammalian cell proliferation. Free Radic Biol Med 1995; 18: 775–94. [DOI] [PubMed] [Google Scholar]
- 7. Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res 1991; 51: 794–8. [PubMed] [Google Scholar]
- 8. Gupta A, Rosenberger SF, Bowden GT. Increased ROS levels contribute to elevated transcription factor and MAP kinase activities in malignantly progressed mouse keratinocyte cell lines. Carcinogenesis 1999; 20: 2063–73. [DOI] [PubMed] [Google Scholar]
- 9. Suh YA, Arnold RS, Lassegue B et al. Cell transformation by the superoxide‐generating oxidase Mox1. Nature 1999; 401: 79–82. [DOI] [PubMed] [Google Scholar]
- 10. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell 2002; 110: 673–87. [DOI] [PubMed] [Google Scholar]
- 11. Brakebusch C, Fässler R. The integrin–actin connection, an eternal love affair. EMBO J 2003; 22: 2324–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hannigan GE, Leung‐Hagesteijn C, Fitz‐Gibbon L et al. Regulation of cell adhesion and anchorage‐dependent growth by a new β1‐integrin‐linked protein kinase. Nature 1996; 379: 91–6. [DOI] [PubMed] [Google Scholar]
- 13. Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Phosphoinositide‐3‐OH kinase‐dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin‐linked kinase. Proc Natl Acad Sci USA 1998; 95: 11211–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Novak A, Hsu SC, Leung‐Hagesteijn C et al. Cell adhesion and the integrin‐linked kinase regulate the LEF‐1 and β‐catenin signaling pathways. Proc Natl Acad Sci USA 1998; 95: 4374–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Troussard AA, Tan C, Yoganathan TN, Dedhar S. Cell–extracellular matrix interactions stimulate the AP‐1 transcription factor in an integrin‐linked kinase‐ and glycogen synthase kinase 3‐dependent manner. Mol Cell Biol 1999; 19: 7420–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell 2000; 103: 239–52. [DOI] [PubMed] [Google Scholar]
- 17. Tournier C, Hess P, Yang DD et al. Requirement of JNK for stress‐induced activation of the cytochrome c‐mediated death pathway. Science 2000; 288: 870–4. [DOI] [PubMed] [Google Scholar]
- 18. Saitoh M, Nishitoh H, Fujii M et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal‐regulating kinase (ASK) 1. EMBO J 1998; 17: 2596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu H, Nishitoh H, Ichijo H, Kyriakis JM. Activation of apoptosis signal‐regulating kinase 1 (ASK1) by tumor necrosis factor receptor‐associated factor 2 requires prior dissociation of the ASK1 inhibitor thioredoxin. Mol Cell Biol 2000; 20: 2198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guo YL, Baysal K, Kang B, Yang LJ, Williamson JR. Correlation between sustained c‐Jun N‐terminal protein kinase activation and apoptosis induced by tumor necrosis factor‐α in rat mesangial cells. J Biol Chem 1998; 273: 4027–34. [DOI] [PubMed] [Google Scholar]
- 21. Roulston A, Reinhard C, Amiri P, Williams LT. Early activation of c‐Jun N‐terminal kinase and p38 kinase regulate cell survival in response to tumor necrosis factor α. J Biol Chem 1998; 273: 10232–9. [DOI] [PubMed] [Google Scholar]
- 22. Tobiume K, Matsuzawa A, Takahashi T et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep 2001; 2: 222–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. Akt phosphorylates and negatively regulates apoptosis signal‐regulating kinase 1. Mol Cell Biol 2001; 21: 893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goldman EH, Chen L, Fu H. Activation of apoptosis signal‐regulating kinase 1 by reactive oxygen species through dephosphorylation at serine 967 and 14‐3‐3 dissociation. J Biol Chem 2004; 279: 10442–9. [DOI] [PubMed] [Google Scholar]
- 25. Fujii K, Goldman EH, Park HR, Zhang L, Chen J, Fu H. Negative control of apoptosis signal‐regulating kinase 1 through phosphorylation of Ser‐1034. Oncogene 2004; 23: 5099–104. [DOI] [PubMed] [Google Scholar]
- 26. Tamura S, Li MG, Komaki K, Sasaki M, Kobayashi T. Roles of mammalian protein phosphatase 2C family members in the regulation of cellular functions. In: Arino J, Alexander DR eds. Topics in Current Genetics: Protein Phosphatases. Vol. 5. Heidelberg: Springer‐Verlag, 2004; 91–105. [Google Scholar]
- 27. Tamura S, Lynch KR, Larner J et al. Molecular cloning of rat type 2C (IA) protein phosphatase mRNA. Proc Nat Aca Sci USA 1989; 86: 1796–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mann DJ, Campbell DG, McGowan CH, Cohen PT. Mammalian protein serine/threonine phosphatase 2C: cDNA cloning and comparative analysis of amino acid sequences. Biochim Biophys Acta 1992; 1130: 100–4. [DOI] [PubMed] [Google Scholar]
- 29. Takekawa M, Maeda T, Saito H. Protein phosphatase 2Calpha inhibits the human stress‐responsive p38 and JNK MAPK pathways. EMBO J 1998; 17: 4744–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miyamoto T, Fiorenza MT, Zhao Y, Hasuike S, Westphal H. Molecular cloning and expression analysis of MPPα‐2, a novel mouse transcript detected in a differential screen of pituitary libraries. Biochim Biophys Acta 2002; 1577: 109–12. [DOI] [PubMed] [Google Scholar]
- 31. Wenk J, Trompeter HI, Pettrich KG, Cohen PT, Campbell DG, Mieskes G. Molecular cloning and primary structure of a protein phosphatase 2C isoform. FEBS Lett 1992; 297: 135–8. [DOI] [PubMed] [Google Scholar]
- 32. Terasawa T, Kobayashi T, Murakami T et al. Molecular cloning of a novel isotype of Mg (2+)‐dependent protein phosphatase beta (type 2Cβ) enriched in brain and heart. Arch Biochem Biophys 1993; 307: 342–9. [DOI] [PubMed] [Google Scholar]
- 33. Kato S, Kobayashi T, Terasawa T et al. The cDNA sequence encoding mouse Mg (2+)‐dependent protein phosphatase α. Gene 1994; 145: 311–12. [DOI] [PubMed] [Google Scholar]
- 34. Kato S, Terasawa T, Kobayashi T et al. Molecular cloning and expression of mouse Mg(2+)‐dependent protein phosphatase β‐4 (type 2Cβ‐4). Arch Biochem Biophys 1995; 318: 387–93. [DOI] [PubMed] [Google Scholar]
- 35. Marley AE, Kline A, Crabtree G, Sullivan JE, Beri RK. The cloning expression and tissue distribution of human PP2Cβ. FEBS Lett 1998; 431: 121–4. [DOI] [PubMed] [Google Scholar]
- 36. Seroussi E, Shani N, Ben‐Meir D et al. Uniquely conserved non‐translated regions are involved in generation of the two major transcripts of protein phosphatase 2Cβ. J Mol Biol 2001; 312: 439–51. [DOI] [PubMed] [Google Scholar]
- 37. Guthridge MA, Bellosta P, Tavoloni N, Basilico C. FIN13, a novel growth factor‐inducible serine‐threonine phosphatase which can inhibit cell cycle progression. Mol Cell Biol 1997; 17: 5485–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Travis SM, Welsh MJ. PP2Cγ: a human protein phosphatase with a unique acidic domain. FEBS Lett 1997; 412: 415–19. [DOI] [PubMed] [Google Scholar]
- 39. Tong Y, Quirion R, Shen SH. Cloning and characterization of a novel mammalian PP2C isozyme. J Biol Chem 1998; 273: 35282–90. [DOI] [PubMed] [Google Scholar]
- 40. Leung‐Hagesteijn C, Mahendra A, Naruszewicz I, Hannigan GE. Modulation of integrin signal transduction by ILKAP, a protein phosphatase 2C associating with the integrin‐linked kinase, ILK1. EMBO J 2001; 20: 2160–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fiscella M, Zhang H, Fan S et al. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53‐dependent manner. Proc Natl Acad Sci USA 1997; 94: 6048–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kitani T, Ishida A, Okuno S, Takeuchi M, Kameshita I, Fujisawa H. Molecular cloning of Ca(2+)/calmodulin‐dependent protein kinase phosphatase. J Biochem 1999; 125: 1022–8. [DOI] [PubMed] [Google Scholar]
- 43. Tan KM, Chan SL, Tan KO, Yu VC. The Caenorhabditis elegans sex‐determining protein FEM‐2 and its human homologue, hFEM‐2, are Ca(2+)/calmodulin‐dependent protein kinase phosphatases that promote apoptosis. J Biol Chem 2001; 276: 44193–202. [DOI] [PubMed] [Google Scholar]
- 44. Takeuchi M, Ishida A, Kameshita I, Kitani T, Okuno S, Fujisawa H. Identification and characterization of CaMKP‐N, nuclear calmodulin‐dependent protein kinase phosphatase. J Biochem 2001; 130: 833–40. [DOI] [PubMed] [Google Scholar]
- 45. Koh CG, Tan EJ, Manser E, Lim L. The p21‐activated kinase PAK is negatively regulated by POPX1 and POPX2, a pair of serine/threonine phosphatases of the PP2C family. Curr Biol 2002; 12: 317–21. [DOI] [PubMed] [Google Scholar]
- 46. Labes M, Roder J, Roach A. A novel phosphatase regulating neurite extension on CNS inhibitors. Mol Cell Neurosci 1998; 12: 29–47. [DOI] [PubMed] [Google Scholar]
- 47. Li MG, Katsura K, Nomiyama H et al. Regulation of the interleukin‐1‐induced signaling pathways by a novel member of the protein phosphatase 2C family (PP2Cɛ). J Biol Chem 2003; 278: 12013–21. [DOI] [PubMed] [Google Scholar]
- 48. Kashiwaba M, Katsura K, Ohnishi M et al. A novel protein phosphatase 2C family member (PP2Cζ) is able to associate with ubiquitin conjugating enzyme 9. FEBS Lett 2003; 538: 197–202. [DOI] [PubMed] [Google Scholar]
- 49. Komaki K, Katsura K, Li MG et al. Molecular cloning of PP2Cη, a novel member of the protein phosphatase 2C family. Biochim Biophys Acta 2003; 1630: 130–7. [DOI] [PubMed] [Google Scholar]
- 50. Shimizu K, Okada M, Takano A, Nagai K. SCOP, a novel gene product expressed in a circadian manner in rat suprachiasmatic nucleus. FEBS Lett 1999; 458: 363–9. [DOI] [PubMed] [Google Scholar]
- 51. Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell 2005; 18: 13–24. [DOI] [PubMed] [Google Scholar]
- 52. Hanada M, Kobayashi T, Ohnishi M et al. Selective suppression of stress‐activated protein kinase pathway by protein phosphatase 2C in mammalian cells. FEBS Lett 1998; 437: 172–6. [DOI] [PubMed] [Google Scholar]
- 53. Takekawa M, Adachi M, Nakahata A et al. p53‐inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK‐p53 signaling in response to UV radiation. EMBO J 2000; 19: 6517–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hanada M, Ninomiya‐Tsuji J, Komaki K et al. Regulation of the TAK1 signaling pathway by protein phosphatase 2C. J Biol Chem 2001; 276: 5753–9. [DOI] [PubMed] [Google Scholar]
- 55. Morita K, Saitoh M, Tobiume K et al. Negative feedback regulation of ASK1 by protein phosphatase 5 (PP5) in response to oxidative stress. EMBO J 2001; 20: 6028–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kumar AS, Naruszewicz I, Wang P, Leung‐Hagesteijn C, Hannigan GE. ILKAP regulates ILK signaling and inhibits anchorage‐independent growth. Oncogene 2004; 23: 3454–61. [DOI] [PubMed] [Google Scholar]