

Abstract
Cancer epigenetics is rapidly moving into a translational phase, and knowledge on how aberrant DNA methylation is induced is becoming important. Aging, chronic inflammation, and viral infections are known to promote methylation of non‐core regions of promoter CpG islands (CGI). The non‐core methylation and ‘seeds of methylation’, scattered methylation in a CGI, are considered to serve as triggers for dense methylation of a promoter CGI, which permanently represses expression of its downstream gene. Decreased gene transcription is an important factor that promotes induction of dense methylation. The presence of the CGI methylator phenotype (CIMP), in which methylation of multiple CGI was observed, is under dispute. Some gastric cancer cell lines have increased rates of de novo methylation, and neuroblastoma cases with CIMP show qualitatively different prognosis from those without. This strongly supports the presence of CIMP, but it seems to contain multiple entities. Limited knowledge is available for epimutagens, the chemicals that induce DNA demethylation or methylation. We have developed an assay system to detect demethylating agents, and an assay system for methylating agents is necessary. Efforts in the field on how aberrant methylation is induced will lead to new cancer prevention, diagnostics, and therapeutics. (Cancer Sci 2005; 96: 206 –211)
DNA methylation of a CpG island (CGI) in the promoter region of a tumor‐suppressor gene represses its transcription. (1) The first example was identified for the RB gene in sporadic retinoblastomas in 1993 2 , 3 followed by VHL, (4) CDKN2A (p16), 5 , 6 CDH1 (E‐cadherin), 7 , 8 and hMLH1. (9) Now, many tumor‐suppressor genes are known to be inactivated by methylation of their promoter CGI in a wide variety of cancers. (1) Methylation of a promoter CGI excludes some methylation‐sensitive transcription factors, such as CTCF, and recruits methyl‐CpG binding proteins, such as MeCP2 and MBD1‐MBD3. (10) These methyl‐CpG binding proteins further recruit histone deacetylases, histone methyltransferases, and heterochromatin proteins. (11) It is believed that changes in chromatin structure will block the access of transcription complex to DNA, and repress transcription.
In parallel with the mechanistic studies on how DNA methylation leads to gene silencing, the search for genomic regions aberrantly methylated in cancers has also made a lot of progress. (12) In the late 1990s, before the human genome sequence was available, several genome‐wide screening methods were developed, such as restriction landmark genomic scanning‐methylation, methylation‐sensitive‐representational difference analysis (MS‐RDA), methylation‐sensitive‐arbitrarily primed PCR, and methylated CpG island amplification‐RDA. 13 , 14 , 15 , 16 , 17 These methods revealed that cancers harbor many aberrantly methylated genomic regions. Now, owing to completion of the sequencing of the human genome, it has become evident that, even if limited to CGI in promoter regions or putative promoter regions (5′ regions) of genes, most cancers have multiple aberrant methylations. 18 , 19 , 20 , 21 These aberrant methylations are considered to provide a good source of tumor markers, (22) and targets for chemotherapeutics. 23 , 24
In contrast with these rapidly progressing areas of epigenetics, the etiology of aberrant DNA methylation needs more attention. What induces DNA methylation and how? As there are many excellent reviews on epigenetics and cancer, 1 , 3 , 10 , 22 here, we would like to focus on recent advancements on how aberrant DNA methylation is induced, based on our recent findings. 19 , 20 , 21 , 25 , 26 , 27 , 28 , 29
Factors known to be associated with DNA methylation. Factors that are known to be associated with methylation of CGI include aging, chronic inflammation, and viral infections. 30 , 31 , 32 , 33 , 34 , 35
It was first reported that a NotI restriction site in exon 1 of estrogen receptor (ESR) was methylated in normal colon mucosa in association with aging. (30) Further, the N33 transcription start site and MYOD exon 1 were methylated in normal colon mucosa in association with aging, while p16, THBS1, HIC‐1 and CALCA were not. (31) These findings led to the establishment of a concept that some regions of CGI are methylated in association with aging in normal tissues, which has been confirmed by many subsequent studies using human samples and also in rats. 36 , 37 However, it is noteworthy that age‐related methylation of tumor‐suppressor genes applies mostly to exonic or far upstream regions within a promoter CGI, and that, even within the same promoter CGI, a small region covering the transcription start site is kept unmethylated. 33 , 36 , 38
Chronic inflammation is also known to be associated with increased methylation. Normal‐appearing colon mucosa of cases with ulcerative colitis are associated with increased methylation of p16 exon 1, MYOD far upstream region, and CSPG2 exon 1.( 32 , 33 ) As in the case of age‐related methylation, a region covering the p16 transcription start site was spared from methylation. (33) Tobacco smoke, which contains various carcinogens and also induces inflammation, is also known to be associated with methylation of p16 exon 1. (39) Along with the inflammation induced by viral infections as described below, chronic inflammation is considered as one of the factors that induce DNA methylation.
It is well established that viral DNA is methylated upon infection into mammalian cells. (40) It is becoming recognized that not only the viral DNA but also cellular DNA can be methylated as a consequence of viral infection. (41) Stomach cancers positive for Epstein‐Barn (EB) virus are known to have more methylated CGI than those without. 34 , 35 As with age‐related and inflammation‐induced methylation, methylation of p16 was present in its exon 1. In hepatocellular carcinomas, for which a precise comparison between virus‐positive cancers and negative ones is very difficult, aberrant methylation was detected even in non‐cancerous liver tissues showing chronic hepatitis or liver cirrhosis. (42) Detailed analysis of MGMT and hMLH1 promoter CGI showed that weak methylation was present in far upstream regions. (43) Also, in the disease course of adult T‐cell leukemia, endogenous genes, including p16, were shown to be methylated. (44) These points indicate that infection by EB virus, hepatitis viruses, and possibly other viruses triggers cellular machineries to methylate these viruses, which erroneously methylate endogenous genes.
As for p16 and hMLH1 genes, the above described factors do not induce methylation in critical regions for gene transcription (core regions), small regions covering their transcription start sites (Figs 1d, e), 12 , 45 , 46 , 47 but in other regions (non‐core regions), such as exon 1 and far upstream regions (Fig. 1b). This shows that the core regions in promoter CGI are protected from de novo methylation. This leads to a hypothesis that the influence of the above factors can be more readily analyzed using non‐core regions within promoter CGI or CGI outside promoter regions.
Figure 1.
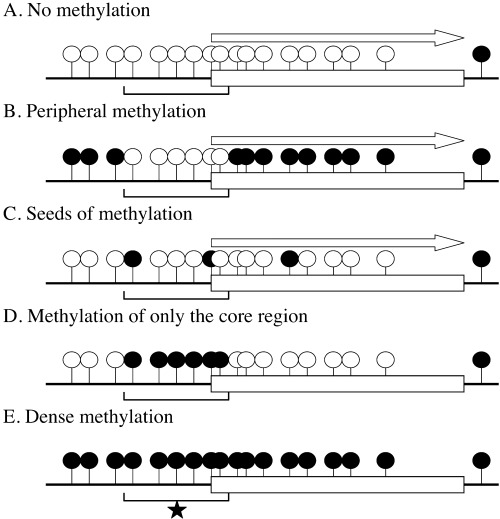
Various types of methylation in a promoter CGI. When the core region (shown by brackets)* is methylated, transcription of the downstream gene is blocked (d, e). However, even if non‐core regions are methylated, transcription is not blocked (b). ‘Seeds of methylation’ (c) is drawing attention as a precursor for dense methylation (e). Open lollipops show unmethylated CpG sites, and closed lollipops show methylated CpG sites.
Dietary factors, such as folate deficiency and choline deficiency, are known to induce genomic hypomethylation, through induction of deficiency of methyl donors, such as S‐adenosylmethionine. (48)
Molecular mechanisms for methylation induction. When a gene is silenced by methylation of its promoter CGI, the CGI is usually densely methylated (Fig. 1e), as is known for CDH1, VHL, p16, hMLH1, and many other genes. 19 , 25 , 38 , 45 , 46 , 47 Before this dense methylation is formed, two types of precursor methylation are considered to be present. One is methylation of non‐core regions within a CGI (Fig. 1b), and the other is ‘seeds of methylation’, scattered methylation (Fig. 1c). The above‐mentioned factors, aging, chronic inflammation, and viral infection, are involved in induction of non‐core methylation and possibly ‘seeds of methylation’.
Non‐core methylation is frequently observed for various genes, including p16, and hMLH1, and does not block gene transcription. 45 , 46 , 47 Although a direct demonstration that non‐core methylation leads to dense methylation with a significant frequency is not yet available, this model is widely believed. This is because methylation of core regions is almost always associated with methylation of non‐core regions in cancer and normal cells, and because dense methylation in a cancer is often associated with methylation of non‐core regions in its surrounding tissues. However, a quantitative correlation between the degree of non‐core methylation and the chance of dense methylation needs to be carefully examined.
‘Seeds of methylation’ is emerging as an important precursor. 28 , 49 , 50 Stirzaker et al. introduced GSTP1 with diminished promoter activity into LNCaP prostate cancer cells. When the construct was not methylated initially, little methylation was induced after 22 days. In contrast, when the construct was initially sparsely methylated with HpaII methylase, a high degree of methylation all over the CGI was induced after 22 days. Also, we found that some gastric cancer cell lines tend to have scattered methylation, and that the scattered methylation leads to dense methylation with a low frequency. (28) These observations demonstrated that ‘seeds of methylation’ is an important precursor to dense methylation of a CGI.
Diminished transcription is also considered as a factor that promotes induction of dense methylation. Clark and colleagues demonstrated that even if ‘seeds of methylation’ are present, introduced GSTP1 with active promoter was not methylated. 49 , 50 De Smet et al. demonstrated that impaired promoter activity or lack of cellular capacity for transcription promote re‐methylation of demethylated MAGE‐A1. (51) As circumstantial evidence, when we made genome‐wide screenings for genes methylated in pancreatic and breast cancers, most of the genes whose 5′‐CGI were methylated had low or no expression in normal counterpart cells. 20 , 21
Antisense RNA to HBA2 was shown to induce methylation of its promoter CGI. (52) Short interfering RNA targeted to CDH1 and EF1A promoter CGI was shown to induce methylation of these CGI. 53 , 54 These mechanisms are very interesting since even transient expression of these RNA could lead to permanent inactivation of respective genes, and these CGI‐specific mechanisms for methylation induction could be involved in physiological processes to induce tissue‐specific methylation patterns. Also, a leukemia protein, PML‐RAR fusion protein, has been shown to induce methylation of its target sequence by recruiting DNA methyltransferases. (55) A great deal of research is necessary on how sequence‐specific methylation is induced in physiological processes, such as embryonic development.
In Neurospora, mutation of histone methyltransferase abolished DNA methylation, showing that histone methylation is indispensable for DNA methylation. (56) In mammalian cells, DNA methylation is known to induce histone modification such as histone deacetylation. (10) At the same time, recruitment of histone H3‐Lys9 methyltransferase, SETDB1, along with heterochromatin protein 1 (HP1) to a euchromatic silenced gene‐induced DNA methylation. (57) Also, temporal observation of DNA‐demethylated cancer cells revealed that histone H3‐Lys9 methylation preceded DNA re‐methylation. (58) Although no reports have shown that histone modification is indispensable for DNA methylation in mammalian cells, histone methylation at H3‐Lys9 seems to promote DNA methylation also in mammalian cells. The relationship among ‘seeds of DNA methylation’, induction of histone H3‐Lys9 methylation, and dense DNA methylation needs to be clarified.
CpG island methylator phenotype (CIMP). Multiple CGI are methylated in some cancers. In 1999, Toyota et al. found that a subset of colon cancers had methylation of multiple CGI using ‘Methylated in tumors (MINT)’ clones they originally isolated. (59) They found a biological meaning in this subset, that is, this subset had a significantly higher incidence of hMLH1 methylation than the others, and designated this phenotype as CIMP. CIMP was also observed in stomach and pancreatic cancers. 60 , 61 However, after a detailed analysis using 35 non‐biased loci, most of which are derived from CGI, in 207 colorectal cancer samples, Yamashita et al. concluded that the number of cancers with specific numbers of methylated loci obeyed a normal distribution of random events, and qualitative distinction between CIMP(+) and CIMP(–) is impossible. (62)
We would like to note that several factors should be considered to solve this distortion. First, to honor the connotation of the word ‘methylator’, the rate of occurrence of methylation events in a defined number of cell divisions should be analyzed (Fig. 2a). If a cancer had originated from a precursor cell with many aberrantly methylated loci and high fidelity in its replicating methylated status, this is still observed as ‘CIMP’ (Fig. 2b). Second, the loci used are different. Different CGI show different susceptibility to methylation, and, even within a CGI, different regions show different susceptibility. 12 , 26 , 63 Third, CGI methylation that can affect cellular growth should be avoided, which was carefully done by Yamashita et al. (62)
Figure 2.
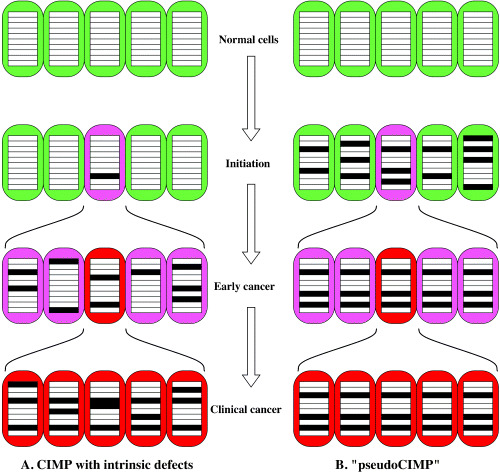
CGI methylator phenotype (CIMP) and ‘pseudoCIMP.’ Open rectangles show unmethylated CGI, and closed rectangles show methylated CGI. In cancer cells (shown in pink or red) with intrinsic defects (a), aberrant methylation keeps occurring. After several clonal selections during multistep carcinogenesis, all cancer cells come to have methylation of multiple CGI. In contrast, if a cancer cell is derived from a precursor cell with methylation of multiple CGI (b), cancer cells derived from it will also display methylation of multiple CGI.
To address the first point, whether or not some cancer cells have increased rates of occurrence of methylation events, we analyzed occurrence of de novo methylation in a defined generation of culture in four gastric cancer cell lines, two of which had multiple CGI methylated and the other two had few. (28) The former two cell lines showed increased rates of de novo methylation, which was measured as decreased fidelity in replicating CpG methylation patterns, and the increased rates resulted in the rare appearance of fully methylated molecules (Fig. 3). The latter two cell lines showed limited numbers of de novo methylation, and did not show appearance of fully methylated molecules at all. Interestingly, the increase of de novo methylation was observed in only limited CGI. This finding clearly demonstrated that at least some gastric cancer cell lines have intrinsic defects that manifest as increased de novo methylation that can lead to appearance of densely methylated CGI. The molecular mechanism for the increased rate of de novo methylation needs to be clarified.
Figure 3.
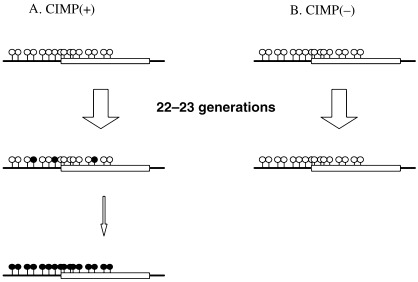
Decreased fidelity in replicating methylation patterns and induction of dense methylation. (28) Two gastric cancer cell lines positive for CGI methylator phenotype (CIMP) displayed scattered methylation after 22–23 generations (a). This led to appearance of densely methylated DNA molecules, although the frequency was rare. In contrast, two different gastric cancer cell lines without CIMP did not display scattered methylation (b).
Another piece of support for the presence of CIMP came from identification of a prognostic marker for neuroblastomas, one of the most common pediatric solid tumors. We made a genome‐wide screening for differences in DNA methylation between neuroblastomas with a good prognosis and those with a poor prognosis by MS‐RDA, and found that multiple CGI were methylated in the latter. (29) The multiple methylation was a very strong prognostic factor, surpassing the currently most reliable prognostic marker, N‐myc amplification. This showed that methylation of multiple CGI, CIMP, was underlined by an important biological mechanism(s). Importantly, specific CGI were useful to sensitively detect CIMP in neuroblastomas; and MINT clones, which are good markers for CIMP in colon cancers, were not methylated in neuroblastomas with CIMP.
In contrast with the findings that support the presence of CIMP, we could not classify breast cancers into CIMP(+) and (–) groups using 13 CGI (Miyamoto et al. manuscript in preparation). Many different entities seem to be included in the current concept of ‘CIMP’, and careful description on how ‘CIMP’ was analyzed is requisite in future studies.
Chemicals that cause epigenetic alterations: Epimutagens. Some chemicals are reported to induce methylation or demethylation of CpG sites and CGI, in some cases, and are designated as epimutagens (Table 1). (64) Only a limited number of chemicals are known to induce methylation, including nickel, butyrate, and arsenic. 65 , 66 , 67 However, the meaning of these methylation changes, especially whether or not they can induce permanent changes in gene expression, needs to be carefully interpreted. It is possible that exposure to chemicals first induce gene expression changes, and then the altered gene expression induce DNA methylation (or demethylation) at limited number of CpG sites.
Table 1.
List of chemicals reported to alter methylation statuses, epimutagens
| Chemical | Characteristics | Reference |
|---|---|---|
| Hypermethylation | ||
| Butyrate | short‐chain fatty acid | (66) |
| Nickel | metal | (65) |
| Arsenic | metal | (67) |
| Phenobarbital | tumor promoting agent | (70) |
| NNK | tobacco‐specific carcinogen | (71) |
| Vinyl carbamate | urethane‐derived carcinogen | (71) |
| Methylene chloride | occupational carcinogen | (71) |
| Hypomethylation | ||
| 5‐aza‐C, 5‐aza‐dC | cytidine analog | (24) |
| Zebularine | cytidine analog | (68) |
| Ethionine | methionine analog | (72) |
| Arsenic | metal | (73, 74) |
| Procaine | anesthetic agent | (75) |
| Procainamide | antiarrhythmic agent | (76, 77) |
| Hydralazine | antihypertensive agent | (77) |
| Valproic acid | antiepileptic agent | (78) |
| EGCG | major polyphenol from green tee | (69) |
| Trichloroacetic acid | corrosive agent | (79) |
5‐aza‐C, 5‐azacytidine; 5‐aza‐dC. 5‐aza‐2′‐deoxycitidine.
In contrast, the action of a group of demethylating agents, 5‐aza‐2′‐deoxycytidine (5‐aza‐dC) and its derivatives, 24 , 68 is well documented. These chemicals are incorporated into DNA strands, and will trap DNA methyltransferase 1 (DNMT1). The trapped DNMT1 is degraded, and demethylation of cellular DNA is induced. (–)‐Epigallocatechin‐3‐gallate (EGCG), a polyphenol in green tea, was recently shown to inhibit DNMT1 and induce demethylation of multiple CGI, and demethylation is proposed as one of its cancer‐preventive mechanisms. (69) However, for other chemicals with ‘demethylating activities’, again, their significance needs careful interpretation.
In spite of the potential influence on human health, the number of known epimutagens is very small, possibly because there are no efficient assay systems for them. Taking this into account, we developed a detection system for demethylating agents using an endogenous promoter CGI (Fig. 4a). (27) We first searched for a CGI that is silenced in a cancer cell line, but can drive ample gene expression. Then, we inserted a Hyg R ‐EGFP marker gene downstream of the CGI by homologous recombination. When the CGI was demethylated by 5‐aza‐dC, expression of the Hyg R ‐EGFP marker gene was detected. To detect methylating agents (Fig. 4b), we need a promoter CGI that can be readily methylated, and a repression system of gene expression. The construction is also under way.
Figure 4.

Strategy to detect demethylating agents (a), (27) and methylating agents (b) using an endogenous promoter CGI. (a) In a human colon cancer cell line HCT116, the Hyg R ‐EGFP marker gene was introduced into a downstream exon of the silenced FLJ32130 gene by homologous recombination. As long as the promoter CGI was kept methylated, the recombinant Hyg R ‐EGFP fusion gene was not expressed. However, by treatment with a known demethylating agent, 5‐aza‐dC, the promoter CGI was demethylated and the Hyg R ‐EGFP expression was detected by its mRNA and fluorescence. (b) A repressor for transcription of a marker gene should be introduced into a downstream exon of a promoter CGI that can be readily methylated. The marker gene also must be introduced. As long as the CGI is kept unmethylated, the repressor is transcribed and the marker is not transcribed. However, if methylation is induced in the promoter CGI, the repressor is not expressed, and the marker will be transcribed.
Future directions. No single mechanism will be able to explain how methylation of CGI is induced. It is mostly unknown which proteins regulate the frequency of de novo methylation and where it takes place. A particularly interesting issue is how ‘seeds of methylation’ lead to dense methylation of a CGI. A mechanism that unifies the methylation status of multiple CpG sites within a CGI is likely to exist.
Distinction between aberrant methylation and physiological methylation is very difficult, considering the presence of age‐related methylation. In addition to the essentially confusing nature of methylation, inadequate description or analysis of methylation makes the situation worse. Does a study analyze the methylation status collectively of CGI or of a CpG site? Is the CGI located in the promoter region or not? Is methylation of the analyzed region critical for transcription repression? Precise analysis will open up new paths to cancer prevention, diagnostics, and treatment.
Acknowledgments
The authors are grateful to Drs K. Moriguchi, K. Miyamoto, and S. Yamashita for critical reading of the manuscript.
References
- 1. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3: 415–28. [DOI] [PubMed] [Google Scholar]
- 2. Ohtani‐Fujita N, Fujita T, Aoike A, Osifchin NE, Robbins PD, Sakai T. CpG methylation inactivates the promoter activity of the human retinoblastoma tumor‐suppressor gene. Oncogene 1993; 8: 1063–7. [PubMed] [Google Scholar]
- 3. Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer 2004; 4: 143–53. [DOI] [PubMed] [Google Scholar]
- 4. Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, Samid D, Duan DS, Gnarra JR, Linehan WM, Baylin SB. Silencing of the VHL tumor‐suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci USA 1994; 91: 9700–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gonzalez‐Zulueta M, Bender CM, Yang AS, Nguyen T, Beart RW, Van Tornout JM, Jones PA. Methylation of the 5′‐CpG island of the p16/CDKN2 tumor suppressor gene in normal and transformed human tissues correlates with gene silencing. Cancer Res 1995; 55: 4531–5. [PubMed] [Google Scholar]
- 6. Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Baylin SB, Sidransky D. 5′‐CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med 1995; 1: 686–92. [DOI] [PubMed] [Google Scholar]
- 7. Yoshiura K, Kanai Y, Ochiai A, Shimoyama Y, Sugimura T, Hirohashi S. Silencing of the E‐cadherin invasion‐suppressor gene by CpG methylation in human carcinomas. Proc Natl Acad Sci USA 1995; 92: 7416–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Graff JR, Herman JG, Lapidus RG, Chopra H, Xu R, Jarrard DF, Isaacs WB, Pitha PM, Davidson NE, Baylin SB. E‐cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res 1995; 55: 5195–9. [PubMed] [Google Scholar]
- 9. Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair‐defective human tumor cell lines. Cancer Res 1997; 57: 808–11. [PubMed] [Google Scholar]
- 10. Jaenisch R, Bird A. Epigenetic regulation of gene expression. how the genome integrates intrinsic and environmental signals. Nat Genet 2003; 33 (Suppl.): 245–54. [DOI] [PubMed] [Google Scholar]
- 11. Fujita N, Watanabe S, Ichimura T, Tsuruzoe S, Shinkai Y, Tachibana M, Chiba T, Nakao M. Methyl‐CpG binding domain 1 (MBD1) interacts with the Suv39h1‐HP1 heterochromatic complex for DNA methylation‐based transcriptional repression. J Mol Biol 2003; 278: 24132–8. [DOI] [PubMed] [Google Scholar]
- 12. Ushijima T. Detection and interpretation of altered methylation in cancer cells. Nature Rev Cancer 2005; 5: 223–31. [DOI] [PubMed] [Google Scholar]
- 13. Kawai J, Hirotsune S, Hirose K, Fushiki S, Watanabe S, Hayashizaki Y. Methylation profiles of genomic DNA of mouse developmental brain detected by restriction landmark genomic scanning (RLGS) method. Nucl Acids Res 1993; 21: 5604–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ushijima T, Morimura K, Hosoya Y, Okonogi H, Tatematsu M, Sugimura T, Nagao M. Establishment of methylation‐sensitive‐representational difference analysis and isolation of hypo‐ and hypermethylated genomic fragments in mouse liver tumors. Proc Natl Acad Sci USA 1997; 94: 2284–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gonzalgo ML, Liang G, Spruck CH 3rd, Zingg JM, Rideout WM 3rd, Jones PA. Identification and characterization of differentially methylated regions of genomic DNA by methylation‐sensitive arbitrarily primed PCR. Cancer Res 1997; 57: 594–9. [PubMed] [Google Scholar]
- 16. Huang TH, Laux DE, Hamlin BC, Tran P, Tran H, Lubahn DB. Identification of DNA methylation markers for human breast carcinomas using the methylation‐sensitive restriction fingerprinting technique. Cancer Res 1997; 57: 1030–4. [PubMed] [Google Scholar]
- 17. Toyota M, Ho C, Ahuja N, Jair KW, Li Q, Ohe‐Toyota M, Baylin SB, Issa JP. Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification. Cancer Res 1999; 59: 2307–12. [PubMed] [Google Scholar]
- 18. Suzuki H, Gabrielson E, Chen W, Anbazhagan R, Van Engeland M, Weijenberg MP, Herman JG, Baylin SB. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat Genet 2002; 31: 141–9. [DOI] [PubMed] [Google Scholar]
- 19. Kaneda A, Kaminishi M, Yanagihara K, Sugimura T, Ushijima T. Identification of silencing of nine genes in human gastric cancers. Cancer Res 2002; 62: 6645–50. [PubMed] [Google Scholar]
- 20. Hagihara A, Miyamoto K, Furuta J, Hiraoka N, Wakazono K, Seki S, Fukushima S, Tsao MS, Sugimura T, Ushijima T. Identification of 27 5′‐CpG islands aberrantly methylated and 13 genes silenced in human pancreatic cancers. Oncogene 2004; 23: 8705–10. [DOI] [PubMed] [Google Scholar]
- 21. Miyamoto K, Fukutomi T, Akashi‐Tanaka S, Hasegawa T, Asahara T, Sugimura T, Ushijima T. Identification of 20 genes aberrantly methylated in human breast cancers. Int J Cancer in press. [DOI] [PubMed]
- 22. Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer 2003; 3: 253–66. [DOI] [PubMed] [Google Scholar]
- 23. Issa JP, Garcia‐Manero G, Giles FJ, Mannari R, Thomas D, Faderl S, Bayar E, Lyons J, Rosenfeld CS, Cortes J, Kantarjian HM. Phase 1 study of low‐dose prolonged exposure schedules of the hypomethylating agent 5‐aza‐2′‐deoxycytidine (decitabine) in hematopoietic malignancies. Blood 2004; 103: 1635–40. [DOI] [PubMed] [Google Scholar]
- 24. Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004; 429: 457–63. [DOI] [PubMed] [Google Scholar]
- 25. Miyamoto K, Asada K, Fukutomi T, Okochi E, Yagi Y, Hasegawa T, Asahara T, Sugimura T, Ushijima T. Methylation‐associated silencing of heparan sulfate d‐glucosaminyl 3‐O‐sulfotransferase‐2 (3‐OST‐2) in human breast, colon, lung and pancreatic cancers. Oncogene 2003; 22: 274–80. [DOI] [PubMed] [Google Scholar]
- 26. Ushijima T, Watanabe N, Okochi E, Kaneda A, Sugimura T, Miyamoto K. Fidelity of the methylation pattern and its variation in the genome. Genome Res 2003; 13: 868–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Okochi‐Takada E, Ichimura S, Kaneda A, Sugimura T, Ushijima T. Establishment of a detection system for demethylating agents using an endogenous promoter CpG island. Mutat Res 2004; 568: 187–94. [DOI] [PubMed] [Google Scholar]
- 28. Ushijima T, Watanabe N, Shimizu K, Miyamoto K, Sugimura T, Kaneda A. Decreased fidelity in replicating CpG methylation patterns in cancer cells. Cancer Res 2005; 65: 11–7. [PubMed] [Google Scholar]
- 29. Abe M, Ohira M, Kaneda A, Yagi Y, Yamamoto S, Kitano Y, Takato T, Nakagawara A, Ushijima T. CpG island methylator phenotype is a strong determinant of poor prognosis in neuroblastomas. Cancer Res 2005; 65: 828–34. [PubMed] [Google Scholar]
- 30. Issa JP, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet 1994; 7: 536–40. [DOI] [PubMed] [Google Scholar]
- 31. Ahuja N, Li Q, Mohan AL, Baylin SB, Issa JP. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res 1998; 58: 5489–94. [PubMed] [Google Scholar]
- 32. Hsieh CJ, Klump B, Holzmann K, Borchard F, Gregor M, Porschen R. Hypermethylation of the p16INK4a promoter in colectomy specimens of patients with long‐standing and extensive ulcerative colitis. Cancer Res 1998; 58: 3942–5. [PubMed] [Google Scholar]
- 33. Issa JP, Ahuja N, Toyota M, Bronner MP, Brentnall TA. Accelerated age‐related CpG island methylation in ulcerative colitis. Cancer Res 2001; 61: 3573–7. [PubMed] [Google Scholar]
- 34. Osawa T, Chong JM, Sudo M, Sakuma K, Uozaki H, Shibahara J, Nagai H, Funata N, Fukayama M. Reduced expression and promoter methylation of p16 gene in Epstein‐Barr virus‐associated gastric carcinoma. Jpn J Cancer Res 2002; 93: 1195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kang GH, Lee S, Kim WH, Lee HW, Kim JC, Rhyu MG, Ro JY. Epstein‐barr virus‐positive gastric carcinoma demonstrates frequent aberrant methylation of multiple genes and constitutes CpG island methylator phenotype‐positive gastric carcinoma. Am J Pathol 2002; 160: 787–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Abe M, Okochi E, Kuramoto T, Kaneda A, Takato T, Sugimura T, Ushijima T. Cloning of the 5′ upstream region of the rat p16 gene and its role in silencing. Jpn J Cancer Res 2002; 93: 1100–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Waki T, Tamura G, Sato M, Motoyama T. Age‐related methylation of tumor suppressor and tumor‐related genes: an analysis of autopsy samples. Oncogene 2003; 22: 4128–33. [DOI] [PubMed] [Google Scholar]
- 38. Graff JR, Herman JG, Myohanen S, Baylin SB, Vertino PM. Mapping patterns of CpG island methylation in normal and neoplastic cells implicates both upstream and downstream regions in de novo methylation. J Mol Biol 1997; 272: 22322–9. [DOI] [PubMed] [Google Scholar]
- 39. Kim DH, Nelson HH, Wiencke JK, Zheng S, Christiani DC, Wain JC, Mark EJ, Kelsey KT. p16 (INK4a) and histology‐specific methylation of CpG islands by exposure to tobacco smoke in non‐small cell lung cancer. Cancer Res 2001; 61: 3419–24. [PubMed] [Google Scholar]
- 40. Doerfler W. Uptake of foreign DNA by mammalian cells via the gastrointestinal tract in mice: methylation of foreign DNA – a cellular defense mechanism. Curr Top Microbiol Immunol 1995; 197: 209–24. [DOI] [PubMed] [Google Scholar]
- 41. Muller K, Heller H, Doerfler W. Foreign dna integration. genome‐wide perturbations of methylation and transcription in the recipient genomes. J Mol Biol 2001; 276: 14271–8. [DOI] [PubMed] [Google Scholar]
- 42. Kondo Y, Kanai Y, Sakamoto M, Mizokami M, Ueda R, Hirohashi S. Genetic instability and aberrant DNA methylation in chronic hepatitis and cirrhosis – A comprehensive study of loss of heterozygosity and microsatellite instability at 39 loci and DNA hypermethylation on 8 CpG islands in microdissected specimens from patients with hepatocellular carcinoma. Hepatology 2000; 32: 970–9. [DOI] [PubMed] [Google Scholar]
- 43. Matsukura S, Soejima H, Nakagawachi T, Yakushiji H, Ogawa A, Fukuhara M, Miyazaki K, Nakabeppu Y, Sekiguchi M, Mukai T. CpG methylation of MGMT and hMLH1 promoter in hepatocellular carcinoma associated with hepatitis viral infection. Br J Cancer 2003; 88: 521–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yasunaga J, Taniguchi Y, Nosaka K, Yoshida M, Satou Y, Sakai T, Mitsuya H, Matsuoka M. Identification of aberrantly methylated genes in association with adult T‐cell leukemia. Cancer Res 2004; 64: 6002–9. [DOI] [PubMed] [Google Scholar]
- 45. Gonzalgo ML, Hayashida T, Bender CM, Pao MM, Tsai YC, Gonzales FA, Nguyen HD, Nguyen TT, Jones PA. The role of DNA methylation in expression of the p19/p16 locus in human bladder cancer cell lines. Cancer Res 1998; 58: 1245–52. [PubMed] [Google Scholar]
- 46. Miyakura Y, Sugano K, Konishi F, Ichikawa A, Maekawa M, Shitoh K, Igarashi S, Kotake K, Koyama Y, Nagai H. Extensive methylation of hMLH1 promoter region predominates in proximal colon cancer with microsatellite instability. Gastroenterol 2001; 121: 1300–9. [DOI] [PubMed] [Google Scholar]
- 47. Deng G, Chen A, Hong J, Chae HS, Kim YS. Methylation of CpG in a small region of the hMLH1 promoter invariably correlates with the absence of gene expression. Cancer Res 1999; 59: 2029–33. [PubMed] [Google Scholar]
- 48. Poirier LA. The effects of diet, genetics and chemicals on toxicity and aberrant DNA methylation: an introduction. J Nutr 2002; 132: 2336S–9S. [DOI] [PubMed] [Google Scholar]
- 49. Song JZ, Stirzaker C, Harrison J, Melki JR, Clark SJ. Hypermethylation trigger of the glutathione‐S‐transferase gene (GSTP1) in prostate cancer cells. Oncogene 2002; 21: 1048–61. [DOI] [PubMed] [Google Scholar]
- 50. Stirzaker C, Song JZ, Davidson B, Clark SJ. Transcriptional gene silencing promotes DNA hypermethylation through a sequential change in chromatin modifications in cancer cells. Cancer Res 2004; 64: 3871–7. [DOI] [PubMed] [Google Scholar]
- 51. De Smet C, Loriot A, Boon T. Promoter‐dependent mechanism leading to selective hypomethylation within the 5′ region of gene MAGE‐A1 in tumor cells. Mol Cell Biol 2004; 24: 4781–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tufarelli C, Stanley JA, Garrick D, Sharpe JA, Ayyub H, Wood WG, Higgs DR. Transcription of antisense RNA leading to gene silencing and methylation as a novel cause of human genetic disease. Nat Genet 2003; 34: 157–65. [DOI] [PubMed] [Google Scholar]
- 53. Kawasaki H, Taira K. Induction of DNA methylation and gene silencing by short interfering RNAs in human cells. Nature 2004; 431: 211–7. [DOI] [PubMed] [Google Scholar]
- 54. Morris KV, Chan SW, Jacobsen SE, Looney DJ. Small interfering RNA‐induced transcriptional gene silencing in human cells. Science 2004; 305: 1289–92. [DOI] [PubMed] [Google Scholar]
- 55. Di Croce L, Raker VA, Corsaro M, Fazi F, Fanelli M, Faretta M, Fuks F, Lo Coco F, Kouzarides T, Nervi C, Minucci S, Pelicci PG. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 2002; 295: 1079–82. [DOI] [PubMed] [Google Scholar]
- 56. Tamaru H, Selker EU. A histone H3 methyltransferase controls DNA methylation in Neurospora crassa. Nature 2001; 414: 277–83. [DOI] [PubMed] [Google Scholar]
- 57. Ayyanathan K, Lechner MS, Bell P, Maul GG, Schultz DC, Yamada Y, Tanaka K, Torigoe K, Rauscher FJ. 3rd. Regulated recruitment of HP1 to a euchromatic gene induces mitotically heritable, epigenetic gene silencing: a mammalian cell culture model of gene variegation. Genes Dev 2003; 17: 1855–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bachman KE, Park BH, Rhee I, Rajagopalan H, Herman JG, Baylin SB, Kinzler KW, Vogelstein B. Histone modifications and silencing prior to DNA methylation of a tumor suppressor gene. Cancer Cell 2003; 3: 89–95. [DOI] [PubMed] [Google Scholar]
- 59. Toyota M, Ahuja N, Ohe‐Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA 1999; 96: 8681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Toyota M, Ahuja N, Suzuki H, Itoh F, Ohe‐Toyota M, Imai K, Baylin SB, Issa JP. Aberrant methylation in gastric cancer associated with the CpG island methylator phenotype. Cancer Res 1999; 59: 5438–42. [PubMed] [Google Scholar]
- 61. Ueki T, Toyota M, Sohn T, Yeo CJ, Issa JP, Hruban RH, Goggins M. Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res 2000; 60: 1835–9. [PubMed] [Google Scholar]
- 62. Yamashita K, Dai T, Dai Y, Yamamoto F, Perucho M. Genetics supersedes epigenetics in colon cancer phenotype. Cancer Cell 2003; 4: 121–31. [DOI] [PubMed] [Google Scholar]
- 63. Feltus FA, Lee EK, Costello JF, Plass C, Vertino PM. Predicting aberrant CpG island methylation. Proc Natl Acad Sci USA 2003; 100: 12253–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Holliday R, Ho T. DNA methylation and epigenetic inheritance. Methods 2002; 27: 179–83. [DOI] [PubMed] [Google Scholar]
- 65. Lee YW, Klein CB, Kargacin B, Salnikow K, Kitahara J, Dowjat K, Zhitkovich A, Christie NT, Costa M. Carcinogenic nickel silences gene expression by chromatin condensation and DNA methylation: a new model for epigenetic carcinogens. Mol Cell Biol 1995; 15: 2547–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Boffa LC, Mariani MR, Parker MI. Selective hypermethylation of transcribed nucleosomal DNA by sodium butyrate. Exp Cell Res 1994; 211: 420–3. [DOI] [PubMed] [Google Scholar]
- 67. Mass MJ, Wang L. Arsenic alters cytosine methylation patterns of the promoter of the tumor suppressor gene p53 in human lung cells: a model for a mechanism of carcinogenesis. Mutat Res 1997; 386: 263–77. [DOI] [PubMed] [Google Scholar]
- 68. Cheng JC, Weisenberger DJ, Gonzales FA, Liang G, Xu GL, Hu YG, Marquez VE, Jones PA. Continuous zebularine treatment effectively sustains demethylation in human bladder cancer cells. Mol Cell Biol 2004; 24: 1270–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, Welsh W, Yang CS. Tea polyphenol (‐) ‐epigallocatechin‐3‐gallate inhibits DNA methyltransferase and reactivates methylation‐silenced genes in cancer cell lines. Cancer Res 2003; 63: 7563–70. [PubMed] [Google Scholar]
- 70. Watson RE, Goodman JI. Effects of phenobarbital on DNA methylation in GC‐rich regions of hepatic DNA from mice that exhibit different levels of susceptibility to liver tumorigenesis. Toxicol Sci 2002; 68: 51–8. [DOI] [PubMed] [Google Scholar]
- 71. Pulling LC, Vuillemenot BR, Hutt JA, Devereux TR, Belinsky SA. Aberrant promoter hypermethylation of the death‐associated protein kinase gene is early and frequent in murine lung tumors induced by cigarette smoke and tobacco carcinogens. Cancer Res 2004; 64: 3844–8. [DOI] [PubMed] [Google Scholar]
- 72. Shivapurkar N, Wilson MJ, Poirier LA. Hypomethylation of DNA in ethionine‐fed rats. Carcinogenesis 1984; 5: 989–92. [DOI] [PubMed] [Google Scholar]
- 73. Zhao CQ, Young MR, Diwan BA, Coogan TP, Waalkes MP. Association of arsenic‐induced malignant transformation with DNA hypomethylation and aberrant gene expression. Proc Natl Acad Sci U S A 1997; 94: 10907–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chen H, Li S, Liu J, Diwan BA, Barrett JC, Waalkes MP. Chronic inorganic arsenic exposure induces hepatic global and individual gene hypomethylation: implications for arsenic hepatocarcinogenesis. Carcinogenesis 2004; 25: 1779–86. [DOI] [PubMed] [Google Scholar]
- 75. Villar‐Garea A, Fraga MF, Espada J, Esteller M. Procaine is a DNA‐demethylating agent with growth‐inhibitory effects in human cancer cells. Cancer Res 2003; 63: 4984–9. [PubMed] [Google Scholar]
- 76. Lin X, Asgari K, Putzi MJ, Gage WR, Yu, X , Cornblatt BS, Kumar A, Piantadosi S, DeWeese TL, De Marzo AM, Nelson WG. Reversal of GSTP1 CpG island hypermethylation and reactivation of pi‐class glutathione S‐transferase (GSTP1) expression in human prostate cancer cells by treatment with procainamide. Cancer Res 2001; 61: 8611–6. [PubMed] [Google Scholar]
- 77. Segura‐Pacheco B, Trejo‐Becerril C, Perez‐Cardenas E, Taja‐Chayeb L, Mariscal I, Chavez A, Acuna C, Salazar AM, Lizano M, Duenas‐Gonzalez A. Reactivation of tumor suppressor genes by the cardiovascular drugs hydralazine and procainamide and their potential use in cancer therapy. Clin Cancer Res 2003; 9: 1596–603. [PubMed] [Google Scholar]
- 78. Detich N, Bovenzi V, Szyf M. Valproate induces replication‐independent active DNA demethylation. J Mol Biol 2003; 278: 27586–92. [DOI] [PubMed] [Google Scholar]
- 79. Tao L, Li Y, Kramer PM, Wang W, Pereira MA. Hypomethylation of DNA and the insulin‐like growth factor‐II gene in dichloroacetic and trichloroacetic acid‐promoted mouse liver tumors. Toxicology 2004; 196: 127–36. [DOI] [PubMed] [Google Scholar]