

Abstract
Mucosal-associated invariant T (MAIT) cells are a subset of T lymphocytes that respond to microbial metabolites. We defined the populations in different organs and the developmental pathway of mouse and human MAIT cells in the thymus with single-cell RNA sequencing, and phenotypic and metabolic analyses. We showed that the predominant mouse subset, which produced IL-17 (MAIT17), and the subset that produced IFN-γ (MAIT1), not only had greatly different transcriptomes, but also different metabolic states. MAIT17 cells in the different organs had increased lipid uptake, lipid storage, and mitochondrial potential compared to MAIT1 cells. These properties were acquired in the thymus. Human MAIT cells in lung and blood were more homogenous but still differed between tissues. They had increased fatty acid uptake and lipid storage in blood and lung, similar to human CD8 T resident memory cells, but unlike their mouse MAIT17 cells, lacked increased mitochondrial potential. Although mouse and human MAIT cell transcriptomes showed similarities for immature cells in the thymus, in the periphery they diverged more. Analysis of pet store mice demonstrated decreased MAIT17 cells in these so-called “dirty” mice, indicative of an environmental influence on MAIT cell subsets and function.
One Sentence Summary:
Mouse MAIT cell subsets and human MAIT cells show tissue differences, reflected in different gene transcription programs, and they have striking metabolic differences from other T cells, especially for mouse MAIT17 cells, influenced in part by the environment.
Introduction:
Mucosal-associated invariant T (MAIT) cells are found in humans, mice and many other mammals (1). They recognize MR1, a non-polymorphic major histocompatibility complex (MHC)-class I-like protein that binds to 5-(2-oxopropylideneamino)-6-D-ribitylaminouracil (5-OP-RU) and other riboflavin-derived metabolites produced by bacteria and yeast (2–7). MAIT cells are abundant in humans, but relatively rare in laboratory mice (8–10). In humans, MAIT cells have a restricted αβ T cell receptor (TCR), in which the TCR Vα chain comprises a canonical Vα7.2-Jα33 (TRAV1–2-TRAJ33) rearrangement, paired with a limited number of TCR β chains. The mouse MAIT cell TCR is made up predominantly of a homologous Vα19-Jα33 (Trav1-Traj33) TCRα chain associated with a limited set of Vα segments (3, 4, 11). Activated MAIT cells proliferate, rapidly secrete cytokines, and synthesize cytotoxic effector molecules such as perforin and granzymes (12, 13). These properties suggest that MAIT cells function as first responders to microbial infections, while in some cases, contributing to abnormal inflammatory reactions (14, 15), although they also can have other functions, for example in tissue repair (16–18).
MAIT cells originate in the thymus, but their positive selection is mainly dependent on double positive thymocytes (19, 20). A stepwise thymus development process based on the surface expression of CD24 and CD44 is reported for MAIT cells in mouse thymus (9, 21). Similarly, for humans, thymus stages are defined based on expression of CD27 and CD161 (9, 19). Some thymic MAIT cells exhibit effector functions typical of differentiated peripheral T cells, a property found in other innate-like T cells including iNKT cells and γδ T cells (22, 23).
MAIT cell subsets have been validated by several recent reports (21, 24–27). The predominant mouse MAIT cell subset is characterized by the expression of RORγT and other surface markers, as well as IL-17 secretion after activation, and therefore are considered MAIT17 cells, analogous to CD4+ Th17 cells. A T-bet-expressing MAIT1 population that secretes IFNγ also has been characterized (8–10, 28). In humans, most MAIT cells have a mixed MAIT1/MAIT17 transcriptome (29–31), but a significant population of MAIT cells capable of producing IL-17 has been found in tissues (32–34). In contrast to conventional T cells, MAIT cells may encounter their natural cognate antigens during thymic differentiation as metabolites from riboflavin-synthesizing bacteria enter the thymus (25). As a result, even in the thymus MAIT cells appear antigen-experienced, they express memory markers (35) and exhibit effector functions typical of differentiated peripheral T cells, a property found in other innate-like T cells including iNKT cells and γδ T cells (22, 23).
Secretion of particular cytokines are controlled by different metabolic states, as are memory-like vs effector-like states in conventional CD4+ and CD8+ T cells (36–39). The question remains as to which programs MAIT cells adopt at steady-state and the influence of different tissue locations and the environment. Here we have characterized MAIT cell populations in mice and humans from different organs by analyzing transcriptomes, phenotypes and metabolic parameters, and the influence of the laboratory environment.
Results:
Populations of mouse MAIT cells in different tissues
To characterize mouse MAIT cells, we sorted 5-OP-RU loaded mouse MR1 tetramer+ cells (Fig. S1A) from thymus, lung, liver and spleen from C57BL/6J mice and subjected them to single-cell RNA sequencing (scRNA-seq) (Table S1). We used the R package Seurat to perform a dimensional reduction and the Louvain algorithm to cluster cells. scRNA-seq of mouse MAIT cells revealed 11 clusters (Fig. 1A and Table S2). Further details can be obtained in Materials and Methods. Of note, the composition of the clusters was unevenly distributed across the four tissue sources (Fig. 1B and S1B): Cluster 6 was thymus-derived, clusters 3 and 7 were almost entirely from the lung, and clusters 1 and 8 had the highest proportion of cells from the liver, with relatively little representation of lung MAIT cells in those (Fig. 1B and S1B).
Figure 1: Mouse MAIT cell populations in different tissues.
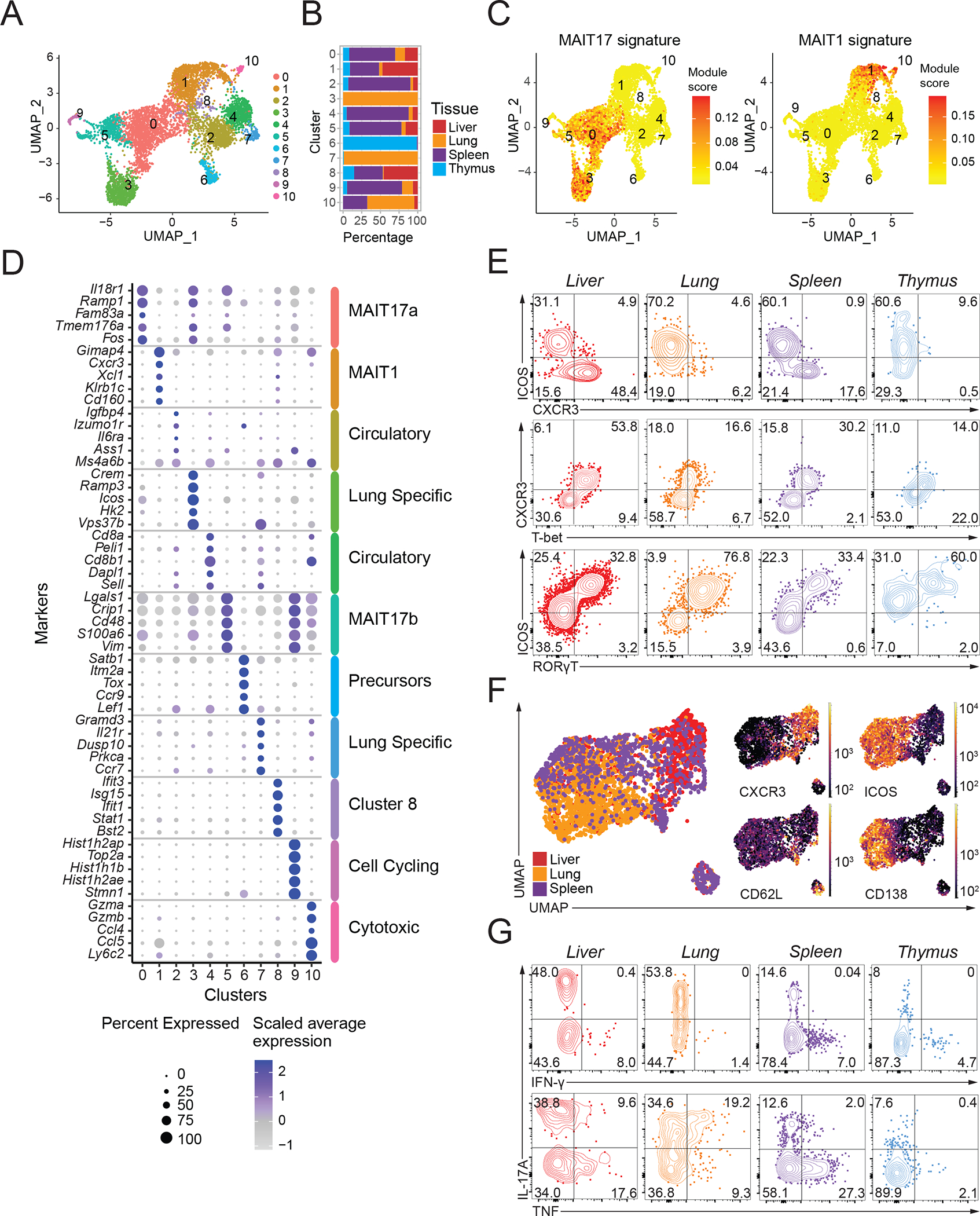
(A) Transcriptomic analysis of 6,080 mouse MAIT cells at steady state was performed using the 10X Genomics platform. Uniform Manifold Approximation and Projection (UMAP) plots were generated by combining scRNA-seq libraries of MAIT cells from thymus, lung, liver and spleen. Clusters were identified by shared nearest neighbor modularity optimization-based clustering algorithm. (B) Bar graph shows for each cluster the tissue origin of the MAIT cells contributing to that cluster. (C) UMAP plot showing the MAIT17 and MAIT1 signature scores for each cell. Signature scores are the difference between the average expression levels of a gene set and control genes for each cell. (D) Dot plot showing top 5 positive marker genes in each cluster. Color gradient and dot size indicate gene expression intensity and the relative proportion of cells within the cluster expressing each gene, respectively. (E) Representative flow cytometry plots showing expression of the indicated surface proteins and transcription factors by MAIT cells from the indicated tissues. (F) Flow cytometry data were acquired using a panel of 17 fluorescent parameters. MAIT cell data from liver, lung and spleen were used to perform UMAP dimensional reduction and unsupervised clustering using the FlowSOM algorithm on the OMIQ software. A total of 3,791 MAIT cells were included in this analysis. (G) Cytokine expression by MAIT cells upon PMA/Ionomycin stimulation in vitro. Intracellular cytokine staining data were representative of 3–4 mice per group, representative of 3 experiments.
Mouse MAIT cell transcriptomes in different clusters could be associated with function as well as preferential tissue location. We used a gene signature for MAIT1 and MAIT17 phenotypes, based on an earlier RNA microarray of MAIT cells (25), to define clusters enriched in these signatures. This analysis showed three MAIT cells clusters (clusters 0, 5 and 3) were enriched for a Th17-like or MAIT17 cell gene expression signature (Fig. 1C, left). For example, the most differentially expressed genes (DEG) in MAIT cells of the largest cluster (cluster 0), including Il18r1, Ramp1, Serpinb1a and Tmem176a (Fig. 1D and Table S2), are also highly expressed in IL-17 producing iNKT cells (NKT17 cells) (24, 40–42). Cluster 1 was enriched for transcripts indicative of a MAIT1 signature (Fig, 1C, right), and also typical of Th1/NKT1 genes (24, 40–42) including Cd160, Klrb1c, Xcl1, Cxcr3 and Gimap4 (Fig. 1D). Additional MAIT cell clusters were distinguished by the expression of cell cycle genes or genes related to cytotoxicity (Fig. 1D).
We used high-parameter flow cytometry and dimensional reduction to validate the tissue differences in MAIT cell subsets. The majority of mouse MAIT cells in different sites could be divided into two populations based on the expression of markers defined from the transcriptomic analysis, ICOS and CXCR3 (Fig. 1E, 1F). While ICOS expression strongly correlated with the expression of RORγT, suggesting ICOS marks MAIT17 cells, CXCR3 correlated with T-bet, suggestive of a MAIT1 population (Fig. 1E, Fig. S1C). It was found earlier that CXCR3 marks MAIT1 cells (21, 26, 43) and ICOS marks MAIT17 cells (10) in the thymus, and in agreement with the data here, recently these proteins were used to identify MAIT1 and MAIT17 cells in the periphery (44). As expected from the transcriptomic analysis, flow cytometry indicated that the ICOS, a surrogate marker for MAIT17 cells, was well-represented in all four organs (Fig. 1E, 1F, Fig. S1D). MAIT17 cells (ICOS+, CXCR3−, CD62L−) could be further divided based on Syndecan-1 (SDC1/CD138) expression into MAIT17a cells (SDC1+), and MAIT17b (SDC1−) populations (Fig. 1F, Fig. S1E). Syndecan-1 has been reported to negatively regulate other innate-like IL-17-producing T cells, including iNKT cells and γδ T cells (45), but in binding to extra cellular matrix proteins, Syndecan-1 expression also could be important for tissue maintenance. MAIT17 subsets had the highest prevalence in lung tissue, while CXCR3+ MAIT1-like cells were most represented in liver (Fig. 1E, 1F, S1D, S1F). This distribution of Th1-like and Th17-like MAIT cells is similar to the iNKT cell functional subsets in different sites (42).
We tested the functional capacity of the MAIT cell subsets by measuring cytokine production following stimulation. There were differences in the degree of activation of cells from different tissues, but MAIT cells capable of producing IL-17A were found in all organs, although least in the thymus (Fig. 1G). IFNγ was not produced by a high percentage of activated MAIT cells by this assay, but IL-17A negative, TNF producing cells, suggestive of MAIT1 cells, were most prevalent in the liver and spleen. In the lung, the TNF producing cells were mostly MAIT17 cells. Overall, the data are consistent with the scRNA-seq and flow cytometry indicating enrichment for MAIT1 cells in liver and spleen. Together, these data indicated a strong correlation between transcriptomic, phenotypic, and functional data in defining MAIT17 states, MAIT1 cells, and a few cell clusters that did not fit into either category. The populations were present, but to varying degrees in all tested tissues, with the important exceptions of the lung-specific and thymic precursor subsets.
Large-scale shifts in gene expression by MAIT thymocytes
The data demonstrated that MAIT cells from mouse thymus were found in most of the clusters with peripheral MAIT cells (Fig. 1B, S1B). These results, along with the cytokine synthesis results (Fig. 1G), confirmed the previously reported presence of mature, functional MAIT cells in the thymus (9, 10, 19, 43). It has been found that immature thymic CD24+CD44− stage 1 precursor cells transition to stage 2 (CD24−CD44−) and give rise to mature CD24−CD44+ MAIT1 and MAIT17 cells (9, 10, 19, 21, 46). To create an unbiased model of MAIT cell thymic differentiation that encompassed the different clusters, we employed the Monocle 3 algorithm (47) to enable pseudo-time ordering based solely on the scRNA-seq of the MAIT cell differentiation stages of thymus cells (Fig. 2A, 2B). The analysis indicated that relatively immature or precursor thymic MAIT cells were divided into two branches: one leading to MAIT1 and another leading to MAIT17 thymus cells (Fig. 2B). Hierarchical clustering of gene expression of the thymic MAIT cell clusters generated 12 modules of genes that tended to be co-expressed in individual cells along the trajectory (Fig. 2C and Table S3).
Figure 2: MAIT cell changes in gene expression during thymus differentiation.
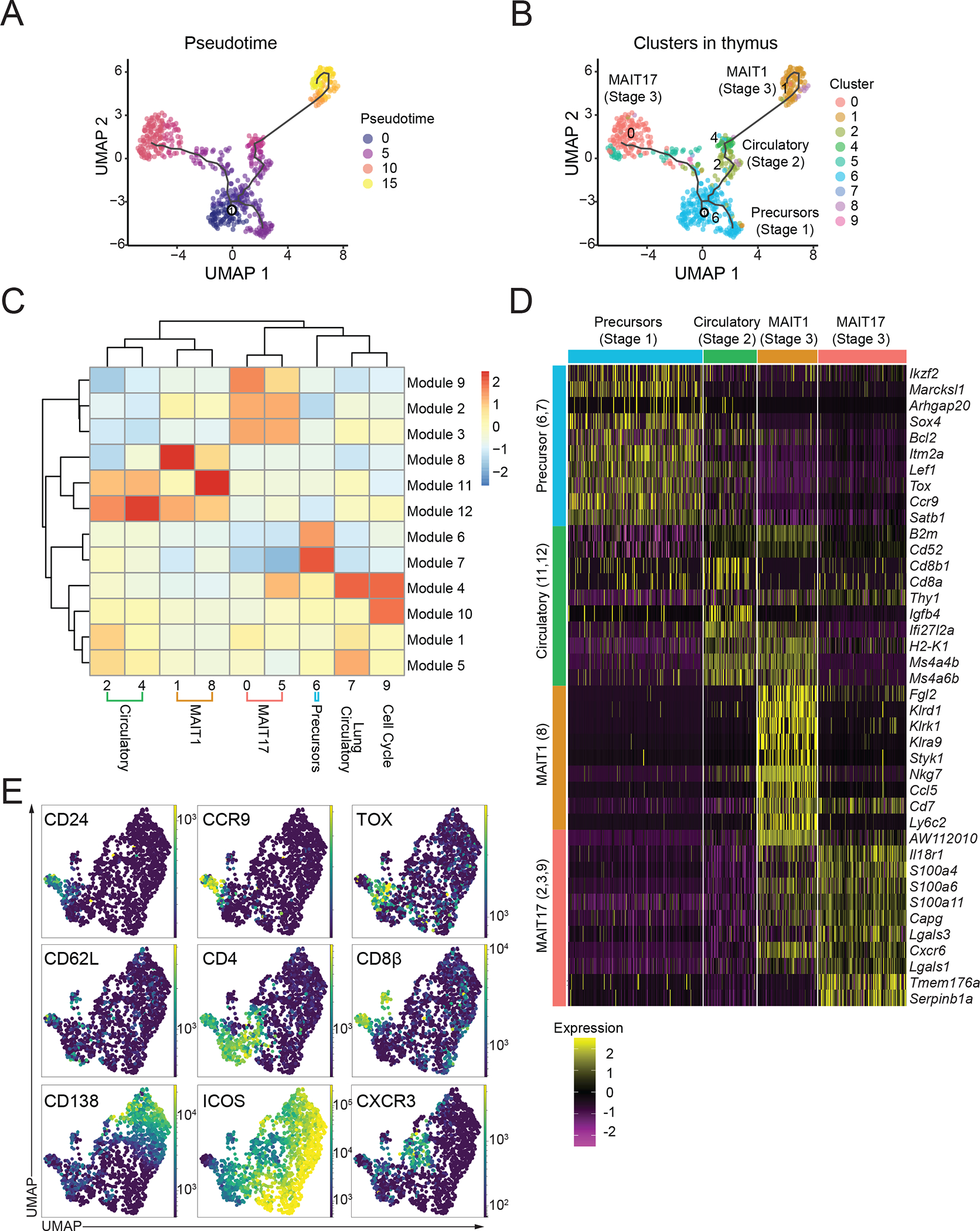
(A) Single-cell trajectories of mouse MAIT thymocytes constructed using Monocle 3. UMAP shows cells colored by pseudotime values along the trajectory. (B) UMAP showing distribution of thymic MAIT cell clusters across branches of single-cell trajectories. Cluster colors and numbers as in Fig 1A. (C) Heatmap showing different stages of thymus MAIT cell differentiation and respective cell clusters on the x-axis and co-regulated gene modules on the y-axis. Modules consist of genes that are differentially expressed along the thymus trajectory path. The legend shows color-coded aggregate scores for each gene module in all the clusters; positive scores indicate higher gene expression. (D) Scaled average expression heatmap of top 10 genes from each thymocyte gene module based on high Morans I value that were expressed in the indicated clusters of MAIT thymocytes. (E) Flow cytometry data were acquired using a panel of 17 different fluorescent parameters. MAIT cell cytometry data from mouse thymus tissue (n=5) were used to perform UMAP dimensional reduction and unsupervised clustering using the FlowSOM algorithm on the OMIQ software. A total of 1,568 MAIT thymocytes from 9-week-old mice were used for the high parameter flow cytometry analysis. All mice were 9-week-old C57BL/6 females.
Based on the expression of key genes, we aligned the gene expression modules with the stages of MAIT cell maturation derived earlier from flow cytometry and functional assays. MAIT cells in thymus-specific cluster 6, labeled as Precursors in Fig. 2C and 2D, had high expression of genes in two modules (6 and 7), which included Sox4, Bcl2, Itm2a, Lef1, Tox, Ccr9, and Satb1 (Fig. 2D, S2A, and Table S3). Flow cytometry confirmed the expression of LEF1 and the higher expression of SATB1 by CD44− MAIT thymocytes (Fig. S2B). Additional analyses confirmed co-expression of TOX protein by cells that coexpressed typical stage 1 genes, such as CD24 and CCR9 (Fig. 2E), further validating the identity of this group as relatively immature or stage 1 thymic MAIT cells. Therefore, this cluster (#6) served as the starting point for the pseudotime analysis. Cells from clusters 2 and 4 lacked expression of both Cd24a, typical of the most immature cells and Cd44, which marks mature or stage 3 cells (Fig. S2C) while the pseudotime trajectory indicated they were close to the precursor population. Furthermore, several genes from the precursor modules, such as Lef1 and Satb1 (Id3 not shown), were expressed but to a lesser extent by these putative stage 2 MAIT cells (Fig. 2D, Table S3). Altogether, this suggested that these cell clusters contained intermediate or stage 2 MAIT cells that shared gene modules 11 and 12 (Stage 2 modules). Characteristic genes included expression of Ms4a4b, Ms4a6b, CD20 family members important for regulating T cell proliferation and survival (48) and Ccr7 (Fig. 2D, Table S3). A recent study indicates that NKT17 cells are generated more rapidly in the thymus than NKT1 cells (49). The in-silico analysis suggested that MAIT17 cells were closer to the precursor stages (Fig. 2B), perhaps reflecting a similar process. Taken together, these data showed that the stages defined on the basis of the expression of CD24 and CD44 in fact encompassed the MAIT cells in the thymus. When an unbiased analysis was undertaken, the stage 3 cells were almost entirely MAIT1 and MAIT17 cells in C57BL/6 mice. The stage 2 classification was likely heterogenous and possibly contained cells with different potentials and/or different degrees of differentiation.
Naïve MAIT cells
MAIT cells are tissue-resident cells in mice (24), but they also are found in circulation, and they are abundant in human peripheral blood (8, 50). A subset of mouse MR1 tetramer-binding cells, especially clusters 2 and 4, exhibited a circulatory gene expression score (Fig. S3A) with increased expression of Sell (encoding CD62L), Ccr7, and Lef1 (Fig. 1D, Fig. S3B). Cells in these clusters were most prevalent in the spleen (Fig. 1B), which we verified by flow cytometry (Fig. S3C). Lymphocytes in these cell clusters had relatively low expression of mRNA encoding Zbtb16b, Rorc, Tbx21, and Cd44 (Fig. 3B), indicating they could be naïve, MR1 tetramer-binding similar to those reported earlier in the thymus and spleen (43). In fact, the CD62L+ tetramer-binding spleen T cells had very little expression of PLZF (Fig. S3D).
Figure 3: CD62L+ MAIT cells are predominantly found in the spleen.
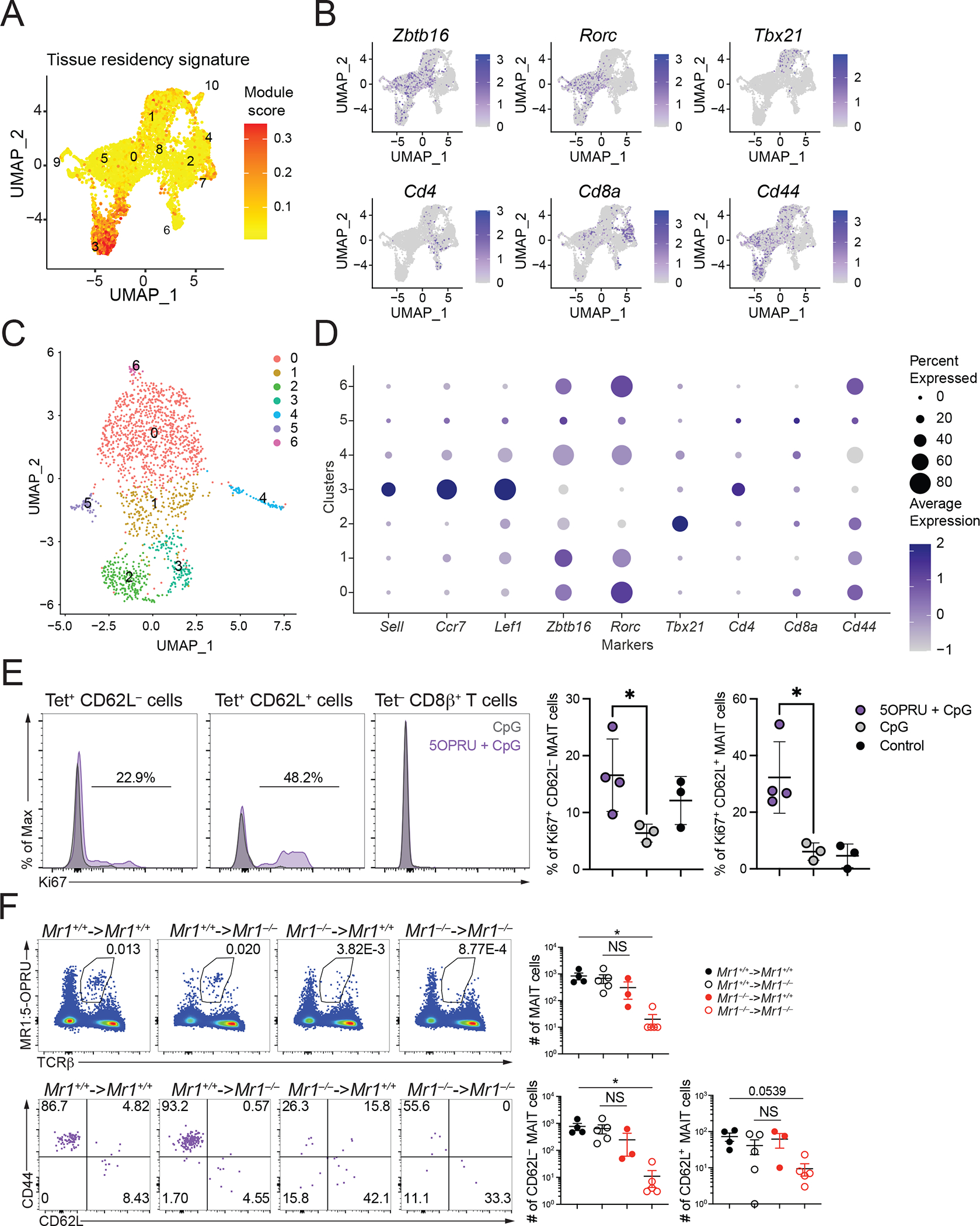
(A) UMAP showing the tissue resident gene expression signature scores from scRNA-seq data from mouse MAIT cells combined from the four liver, lung, spleen and thymus. (B) UMAP feature plots showing expression of key genes in MAIT cells combined from the four organs. (C) UMAP of scRNA-seq dataset of mouse splenic MAIT cells including only cells in which a Trav1 transcript was detected combined from sorted CD62L+ and CD62L− cells. (D) Dot pot of scRNA-seq data from Trav1+ mouse splenic MAIT cells showing expression of key genes in each cluster. (E) C57BL/6 mice were injected at t=0 and t=24h with 5-OP-RU and CpG or CpG alone. Splenic T cells were analyzed for Ki67 expression 48h later. One representative experiment out of two is shown, (F) (top) Number of MAIT cells in the indicated chimeric mice. Donor BM cells were from Mr1+/+ mice (black symbols) or Mr1−/− mice (red symbols); recipients were Mr1+/+ (filled circles) or Mr1−/− mice (open circles). Spleens were harvested after 8 weeks and analyzed by flow cytometry for MR1-tetramer binding cells. (bottom) Expression of CD44 and CD62L on MR1 tetramer-binding spleen cells from chimeric mice. Absolute numbers of CD62L+ and CD62L− tetramer-binding spleen cells are shown. Data analyzed by Kruskal-Wallis with Tukey post-test for multiple comparisons, displayed as mean± SEM, NS: P ≥ 0.05.
The MAIT cell CD62L+ population is small, and therefore we tested for expression of rearranged Trav1 genes in sorted, CD62L+ and CD62L− 5-OP-RU-MR1-tetramer-binding spleen cells by scRNA-seq (Table S4, S5). Indeed, only a minority of the spleen, CD62L+ MR1 tetramer binding cells expressed a Trav1 transcript (Fig. S3E). Some of the MR1 tetramer binding could be due to interactions of CD8 molecules with MR1 (51). A combined analysis of the CD62L+ and CD62L− spleen MR1 tetramer-binding cells that expressed Trav1+ revealed different clusters MR1 tetramer binding in uniform manifold approximation and projection (UMAP) space, including MAIT17 cells with more Rorc expression and MAIT1 cells with Tbx21 expression (Fig. S3F). Importantly, cluster 3 Trav1+ tetramer-binding cells, derived from the CD62L+ population, showed increased expression of Sell, Ccr7 and Lef1 (Fig. S3F). To test if CD62L+ tetramer+ binding cells were capable of specifically reacting to the canonical MAIT cell antigen, we injected wild-type mice with 5-OP-RU combined with CpG, which previously had been shown to increase the MAIT cell population (52), and analyzed the spleens. Both CD62L+ and CD62L− tetramer+ populations increased Ki67 staining after stimulation with 5-OPRU + CpG, but not in controls with only CpG injection (Fig. 3E). In contrast, CD8β+ splenic T cells did not show increased Ki67. These data show that a subset of the CD62L+ MAIT cells were responsive to antigen in vivo.
We analyzed bone marrow chimeric mice to determine if the CD62L+ MR1 tetramer binding cells were selected by bone marrow-derived cells, as are mature MAIT cells, or by radiation resistant cells. We generated reciprocal bone marrow chimeric mice and evaluated the tetramer-binding populations in the spleen and thymus. Tetramer binding cells were decreased approximately 100-fold in spleen and thymus when MR1 was not expressed by either population. The data suggest that CD62L+ MAIT cells in spleen (Fig. 3F) and thymus (Fig. S3F) could be selected either by bone marrow-derived or by irradiation-resistant cells, consistent with a previous report on thymus, CD62L+, naïve MAIT cells (25). There was a trend toward decreased total MAIT cells, and CD62L− MAIT cells when MR1 was not expressed by bone marrow derived cells, as well as decreased expression of CD44 with Mr1−/− donor bone marrow into MR1 expressing hosts, although this did not reach statistical significance. Overall, although the circulatory population contained cells that are not MAIT cells, it also contained antigen responsive cells with less mature or naive phenotype.
Mouse MAIT17 cells are metabolically active
Following activation and differentiation, CD4+ and CD8+ T cells profoundly change their cellular metabolism. Effector-like as opposed to memory-like and tissue-resident states are controlled by divergent metabolic programs, relying on glycolytic versus mitochondrial or fatty acid oxidation metabolism, respectively (37, 38). Analysis of the scRNA-seq signature scores for oxidative phosphorylation, mitochondrial genes, fatty acid metabolism and glycolysis showed that MAIT17 cell clusters had the most enrichment for oxidative phosphorylation genes (Fig. S4A).
We measured MAIT cell metabolic parameters by flow cytometry to validate these findings. We quantified the uptake of fatty acids and glucose, cytoplasmic lipid droplet content, and the activity of mitochondria with membrane-potential sensitive MitoTracker Deep Red FM. Large differences were evident for all measures during the differentiation of total thymic MAIT cells, stages gated as in Fig. S4B, with stages 1 and 2 significantly less active metabolically compared to stage 3. The predominant stage 3 or mature MAIT cells in the thymus were MAIT17 cells, and compared to mature MAIT1 thymus cells, MAIT17 thymocytes showed higher levels for all the metabolic parameters, except glucose uptake (Fig 4A and 4B). These data indicated that thymic MAIT17 cells adopted a distinctly active metabolic phenotype during their functional differentiation.
Figure 4: Mouse MAIT cell subsets have distinct metabolic features.
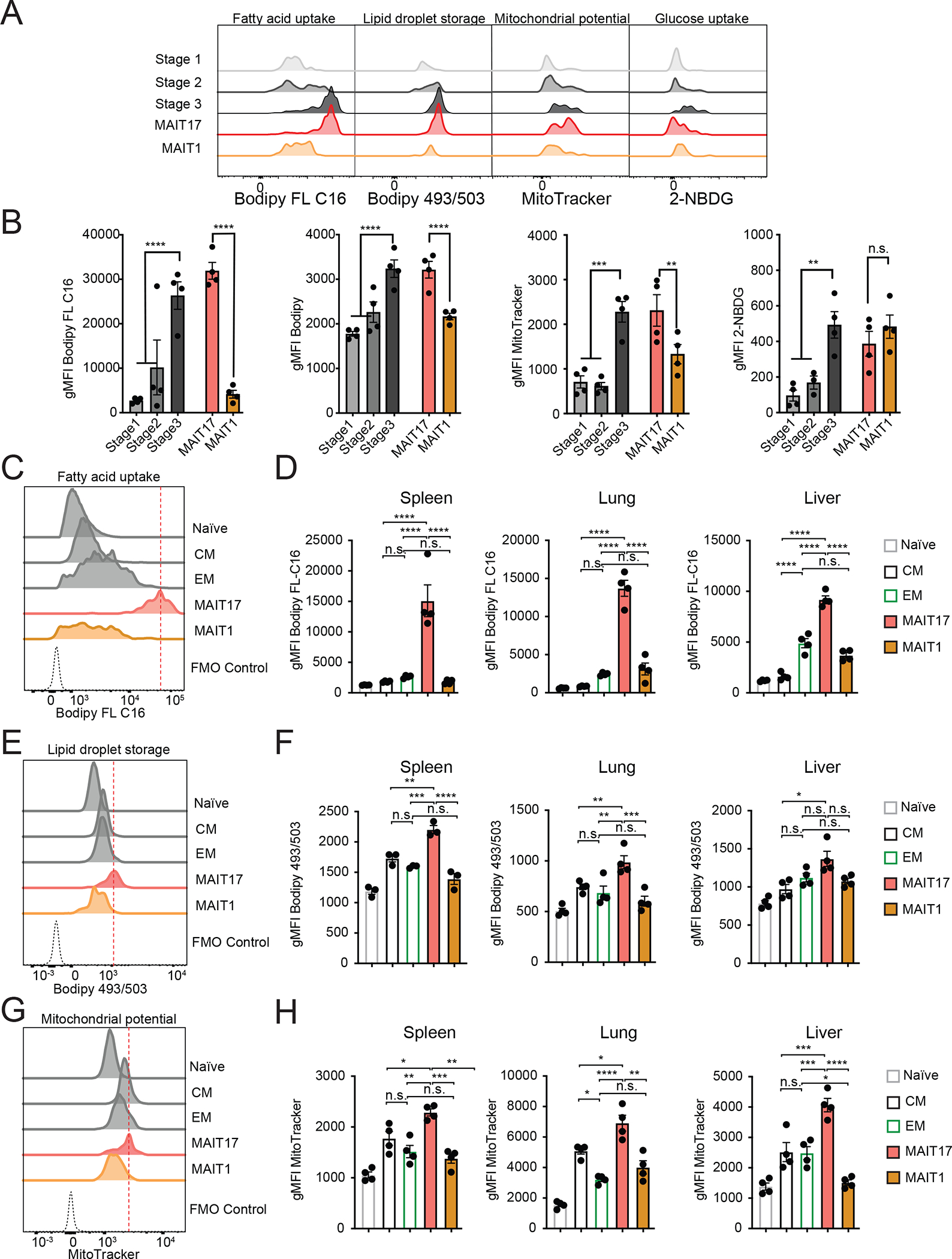
(A-B) Metabolic parameters of MAIT thymocytes were quantified for the thymus MAIT cell differentiation stages 1–3 and mature MAIT1 and MAIT17 thymocytes. Representative histograms (A) and quantification (B) are depicted as geometric mean of fluorescence intensity (gMFI). Neutral lipid droplets were quantified by Bodipy 493/503 fluorescence (left), fatty acid uptake was quantified as intensity of Bodipy FL C16 fluorescence (center left), mitochondrial content was quantified as MitoTracker Deep Red FM fluorescence (center right) and glucose consumption by uptake of 2-NBDG (right). (C-H) Cells were isolated from spleen, lung and liver and metabolic parameters were quantified in CD8+ naïve, central memory (CM) and effector memory (EM) TCRβ+ CD8+ T cells and MAIT cell subsets. Representative histograms (C) and quantification (D) of fatty acid uptake in the indicated cell types. Representative histograms (E) and quantification (F) of neutral lipid droplet content. Representative histograms (G) and quantification (H) of mitochondrial content. Data from 3–4 mice per group, representative of ≥3 experiments. Data analyzed by one-way ANOVA with Dunnett’s post-test for multiple comparisons, displayed as mean± SEM, *P <0.05, **P <0.01 ***P <0.001 and ****P <0.0001.
We compared peripheral mouse MAIT cells to CD8+ naïve, central memory (CM) and effector memory (EM) T cells, gated as Fig S4B. As in the thymus, the subsets differed: MAIT17 cells in all sites had significantly elevated uptake of fatty acids, lipid content, and mitochondrial potential compared either to MAIT1 cells, or the CD8+ memory T cell subsets (Fig. 4C–H), except for the lipid droplet content of liver MAIT17 cells. In contrast, a time course analysis showed that MAIT1 cells had a higher glucose uptake compared to MAIT17 cells in liver and spleen (Fig. S4C). Therefore, MAIT1 cells may have partially supported the generation of ATP through consumption of glucose, while MAIT17 cells preferentially metabolized fatty acids. Together, the findings indicated that adoption of a differential metabolic program by MAIT17 cells occurred in the thymus and correlated with the functional differentiation they underwent rather than their ultimate tissue localization.
Human MAIT cells in different tissues
In order characterize human MAIT cell populations in different sites and to assess the homology of human and mouse MAIT cell subsets, we carried out scRNA-seq of sorted human MAIT cells from thymus, peripheral blood and lung (Table S6). Lung and peripheral blood were obtained from the same adult donors, including uninvolved sections of the lung in patients undergoing surgery for early-stage lung cancer (53). MAIT cells were identified as Vα7.2+, human MR1 5-OP-RU tetramer+ cells as shown in Fig. S5A. Human MAIT cells could also be divided into multiples clusters in UMAP space (Fig. 5A and Table S7) and several clusters were tissue-specific (Fig. 5B and S5B). Importantly, demultiplexing analysis indicated that the clusters contained cells from multiple donors (Fig. S5C). There was one nearly completely lung specific subset (cluster 4) and several thymus-specific clusters (Fig. 5B and S5B). A caveat is that the thymus tissue was from young children and the other tissues from adults. The largest MAIT cell group (cluster 0) consisted mostly of cells from PBMCs and they expressed the highest expression of genes for NK receptors, NCR3, KLRB1, and GZMK (Fig. 5C). We also performed a separate scRNA-seq of MAIT cells from PBMCs of healthy donors (Table S8). Although differences in the methodology did not permit combination of the data in a UMAP plot (see Materials and Methods), the results indicated that similar to MAIT cells from PMBCs from cancer patients, there were few major clusters of MAIT cells from PBMCs (Fig. S5D, Table S9) and these cells had expression of some MAIT1 signature genes, including NK receptors and molecules involved in cell killing such as KLRD1 and NKG7, as well as ZEB2, and GIMAP4 (Fig. S5D, S5E). Cluster 2 from the thymus also showed an enrichment of a MAIT1 cell signature score (Fig. 5D), like MAIT cells from peripheral blood, and cluster 2 thymocytes also showed the strong expression of Zbtb16 transcripts along with MAIT cells from PMBCs (Fig. S5F). Regardless of these features, the separation of MAIT1 cells from MAIT17 cells was much less evident than in the mouse. Many of the human MAIT cells had a more mixed gene expression pattern, including those from the lung (clusters 3 and 4) (Fig. 5D), and notably, Rorc transcripts were not highly concentrated in one cluster (Fig. S5F). Similar to the mouse, human lung MAIT cell clusters were enriched for a tissue-residency signature genes, whereas a circulatory gene expression signature was in clusters of MAIT cells from PBMCs (Fig. 5D).
Figure 5: Human MAIT cell populations in different organs.
(A) UMAP plots of transcriptomic analysis of human MAIT cells generated by combining three individual scRNA-seq libraries from human thymus (n=5), lung (n=4) and PBMCs matched from the lung donors (n=4) (Supplementary Table S7). (B) Bar graph shows the contribution of MAIT cells from different tissues to individual clusters. (C) Dot plot showing top 5 positive marker genes for each cluster. (D) UMAP showing the MAIT1, MAIT17, tissue residency and circulating signature scores for each cell. (E) UMAP (left) of human MAIT cells from thymus with cells ordered in pseudotime and UMAP showing distribution of thymic MAIT cells (right) across branches of single-cell trajectories. Cells are colored and numbered by clusters as in Fig. 5A. (F) Heatmap showing different stages of development and respective cell clusters on the x-axis and co-regulated gene modules on the y-axis. Modules were generated with genes that are differentially expressed along the trajectory path. The legend shows color-coded aggregate module scores for gene modules for cells in each cluster; positive scores indicated higher gene expression. (G) Scaled average expression heatmap of top 10 genes from modules that were expressed in indicated stages of MAIT cell development based on high Morans I value as shown in Fig 5F. These genes were selected based on their expression changes as the cells progressed along the MAIT cell developmental trajectory.
To investigate the gene expression dynamics underlying the human MAIT cell differentiation program, we created a pseudo-time ordering for human thymus MAIT cell transcriptomes (Fig. 5E), and we identified modules of genes that were co-expressed (Fig. 5F and Table S10). The analysis revealed a precursor gene module (module 2) found in immature cell clusters 6 and 9 (Fig. 5G and Fig. S6A) and gene modules (4 and 5) enriched for genes associated with cytotoxicity that might be considered MAIT1-like (Fig. 5G). Pseudotime analysis suggested that cluster 9 stage 1 MAIT cells differentiated into stage 2-like cells (cluster 12) with CD27 expression (Fig. S6B). These cells then branched into stage 3-like cells (cluster 2) that expressed KLRB1 (Fig. S6B). A separate branch gave rise to cluster 1, enriched in gene modules 3 and 7. This MAIT cell cluster had higher expression of circulatory or precursor transcripts, such as LEF1 and CCR7 (Fig. 5C), and also LRRN3, which was co-expressed with LEF1 and CCR7 and marks naïve human T cells (54). These data are consistent with the notion that this group of human MAIT cells were not fully differentiated and may be similar to mouse stage 2 thymocytes and other less mature cells, although their differentiation potential is unknown.
We validated markers from the human MAIT single cell RNA-seq dataset by flow cytometry. Similar to the transcriptomic dataset, human MAIT cells from each tissue clustered separately when performing dimensional reduction, in this case with the Opt-SNE algorithm (Fig. S6C). Of note, three transcription factors and three surface markers (CD44, CD27 and CD8) generated tissue-exclusive regions in the Opt-SNE. Accordingly, human thymus MAIT had lower expression of PLZF, CD161 and CD44, as predicted by the single cell transcriptomic dataset. Similar to mouse MAIT cells, progenitor populations from the thymus expressed high levels of SATB1 transcript, which was confirmed by flow cytometry (Fig. S6D).
Human MAIT cells have increased fatty acid uptake and storage
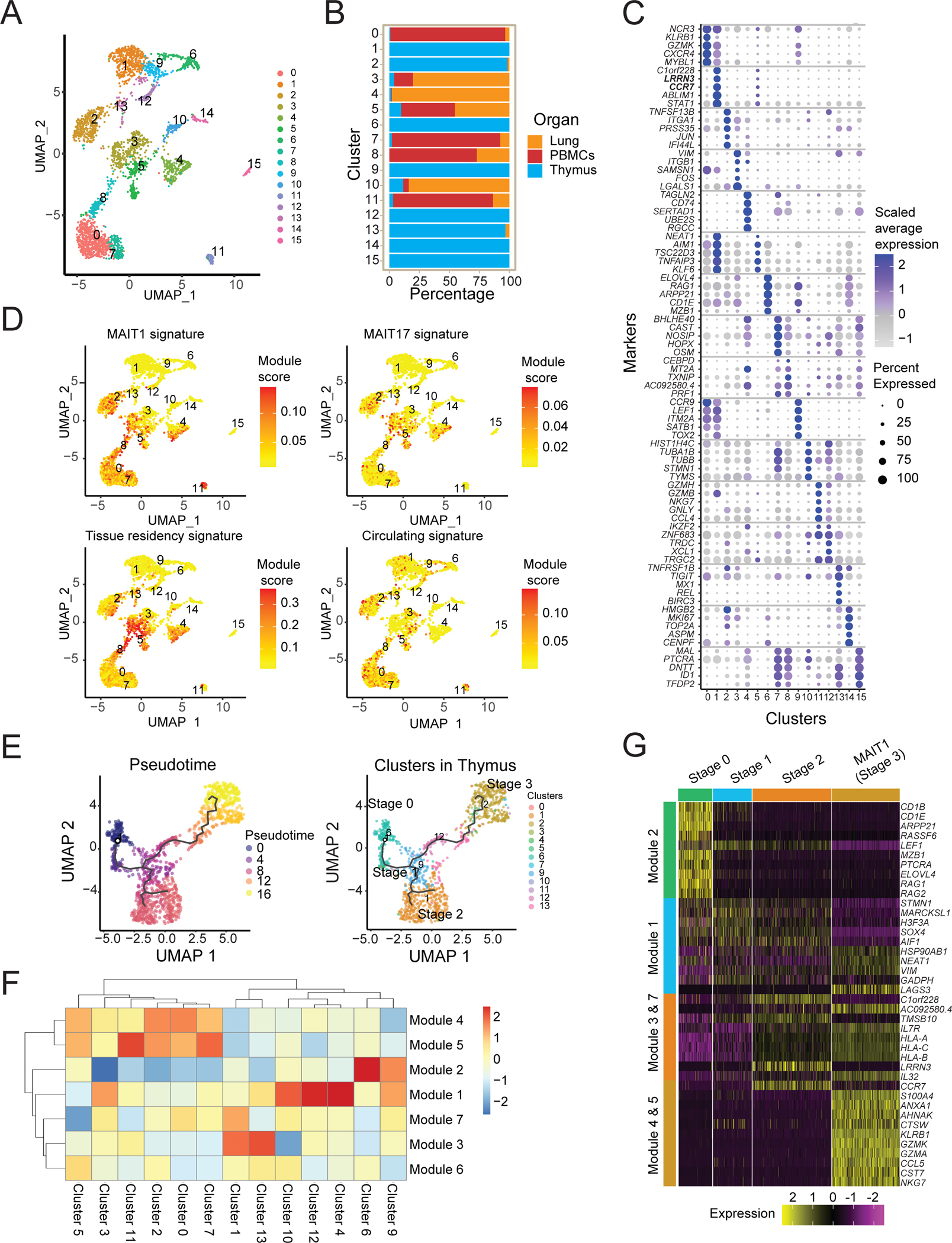
We calculated metabolic signature scores for the transcriptomes of human MAIT cell clusters (Fig. S7A). We also functionally tested the metabolic activity of human MAIT cells from blood and lung tissue and compared them to naïve, effector and memory CD8+ T cell populations (gated as in Fig. S7B). The data indicated that the lung and blood CD8+ memory T cell subsets were higher for their metabolic readouts compared to naïve T lymphocytes (Fig. 6A–6D). As reported previously (53, 55), the CD103+ subset of CD8 T lymphocytes, a phenotype correlated with tissue-resident memory T cells (TRM), had the highest fatty acid uptake considering CD8 T cells. We analyzed MAIT cell phenotypic subsets, including the largest population of MAIT cells (CD103−, CD161+), a smaller population of CD103−, CD161− MAIT cells, and in human lung, a population of CD103+, CD161+ MAIT cells (Fig. S7B). The human MAIT cell subsets had a larger reservoir of stored lipids and also actively took up higher amounts of fatty acids, compared to naïve T cells and similar to TRM (Fig. 6A–6D). In contrast, the mitochondrial potential in all human MAIT cell subsets was not significantly different from naïve or effector CD8+ T cells. Therefore, the human MAIT cell metabolic parameters were not restricted to a phenotypic subset, as defined here, or to lung as opposed to PMBCs (Fig. 6A–6D). They resembled mouse MAIT17 cells with high lipid stores and uptake of fatty acids, but unlike their mouse counterparts, did not have a higher mitochondrial potential.
Figure 6: Human MAIT cell metabolic parameters differ from naïve CD8+ T cells.
Cells were isolated from paired samples of human lung biopsies (A and B) or blood (C and D) and metabolic parameters were quantified in CD8+ T cell and MAIT cell subsets. TCRβ+ CD8+ T cells excluding MAITs were subdivided into naïve, central memory (CM), effector memory (EM) and resident memory (RM) subsets based on expression of CD45RA, CCR7 and CD103. Representative histograms (A, C) and quantification (B, D) of fatty acid uptake (left) was measured as gMFI of Bodipy FL C16. Neutral lipid droplet content (middle) was measured as gMFI of Bodipy 493/503 fluorescence. Mitochondrial potential is indicated as gMFI of MitoTracker Deep Red FM signal (right). Data combined from 2 experiments and 3 donors (A-B) or from 3 experiments and 3 donors (C-D). Data were analyzed by one-way ANOVA with Dunnett’s post-test for multiple comparisons, displayed as mean ± SEM, *P <0.05, **P <0.01 ***P <0.001 and ****P <0.0001.
Homology of human and mouse MAIT cell populations
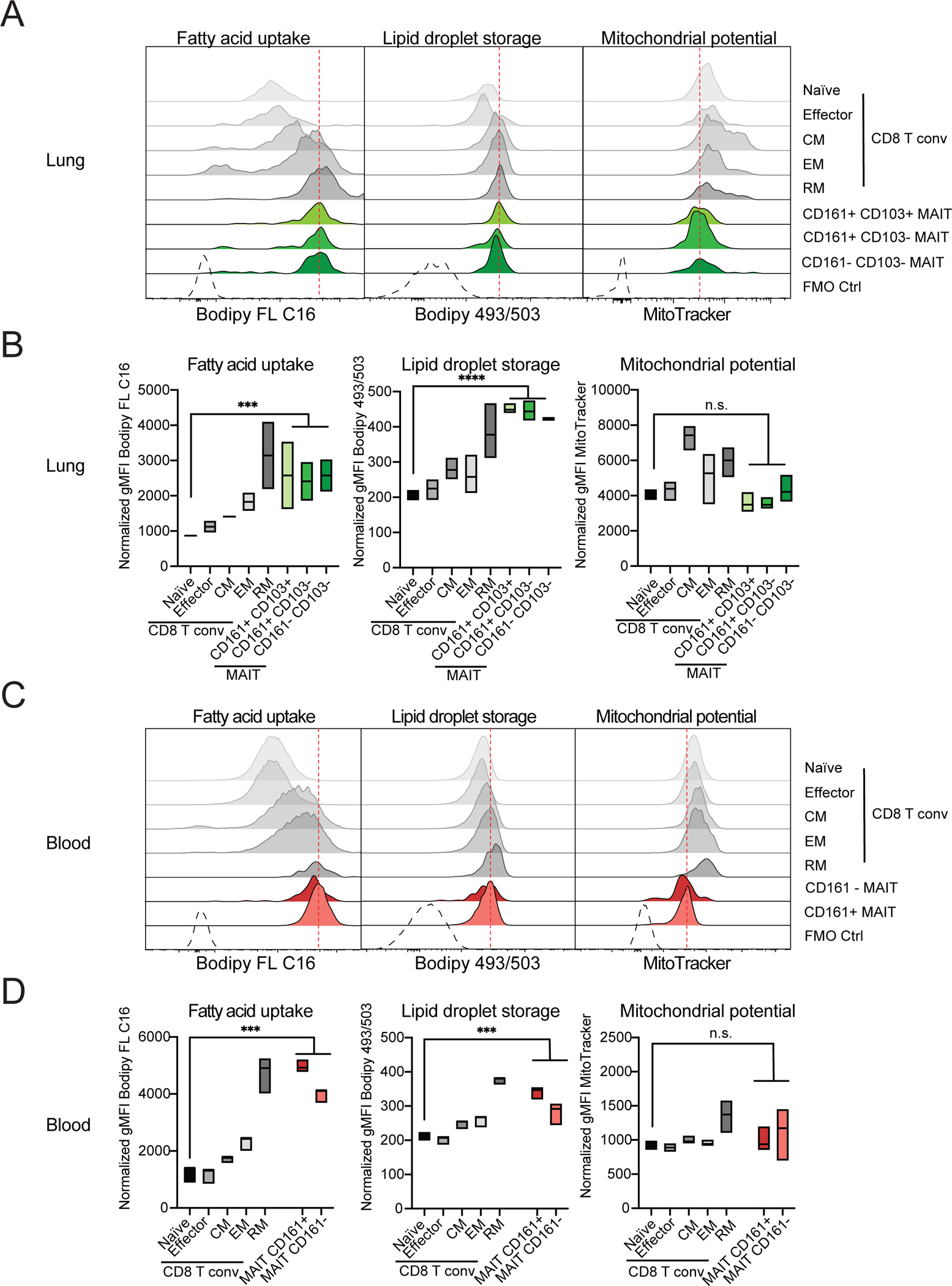
To evaluate the similarities in the transcriptional signatures of human and mouse MAIT cell subsets, we performed integration of the human and mouse datasets (56). Fourteen integrated MAIT cell clusters were identified (Fig. 7A), including some that were tissue specific (Fig. 7B). Several clusters contained MAIT cells from both species (Fig. 7B and Fig. S8A), particularly for precursors in the thymus. MAIT cells in integrated cluster (i-cluster) 5 consisted of precursor cells from mouse (cluster 6) and humans (cluster 9) (Fig. 7B, S8A) with expression of ITM2A, CCR9 and TOX and other genes characteristic of the thymus differentiation of these cells (Fig. 7C). Cells in i-clusters 0 and 3 also contained mouse and human MAIT thymocytes, similar to thymus stage 2 MAIT cells transcriptomes (Fig. 7C and Fig. S8B). Thymic MAIT cell subsets, however, did not completely overlap, as stage 0 human MAIT thymocytes expressing RAG1 and RAG2 (cluster 6) were not found in mice (Fig. S8A).
Figure 7: Divergent mouse and human peripheral MAIT cell subsets.
(A) Aggregated UMAP representation of scRNA-seq data from mouse and human MAIT cells. (B) Mouse and human cells shown in separate UMAPs by tissue and species with the same coordinates as in Fig 7A. (C) Dot plot showing top 5 genes in each integrated cluster across both mouse and human cells. Color gradient and size of dots indicate gene expression intensity and the relative proportion of cells (within the cluster) that expressed each gene respectively.
Other MAIT cell clusters contained a highly unbalanced representation of cells from the two species. For example, cells in i-cluster 3 expressed genes typical of MAIT1 cells, such as KLRC1 and NKG7 (Fig. S8B), and were prevalent in mouse liver and spleen, but represented to a much lesser extent in human PBMCs (Fig. 7C and Fig. S8A). Also, i-cluster 1 had a MAIT17 signature, including expression of TMEM176A and IL18R1, with mouse cells from different organs but only a few human cells, consistent with the prevalence of MAIT17 cells in mice (Fig. 7C and Fig. S8B). Furthermore, human and mouse lung cells did not cluster together. Integrated i-cluster 2 consisted of lung mouse MAIT cells, but only a few human lung MAIT cells were present, demonstrating that the lung gene expression signature varied between the two species.
Environmental influences on MAIT cells
The divergent transcriptional signatures in peripheral MAIT cells might reflect genetic and/or environmental differences between the two species. To further understand how the environment can affect the properties of MAIT cells, we have studied outbred mice from pet stores, so-called dirty mice, SPF controls and neonatal SPF mice cross-fostered with pet store mothers. The lung and liver were analyzed, and the effect was greatest in the lung. The number of lung MAIT cells was not greatly increased by exposure to the non-SPF flora (Fig. S9A, S9B). We found alterations in MAIT cell phenotype that were environment-dependent, however, and likely related to the microflora. Pet store mice had fewer SCD1+ MAIT cells in the lung (Fig. S9A), a marker of MAIT17 cells. The decrease in SCD1+ MAIT cells also was observed in cross-fostered C57BL/6 mice compared to their SPF counterparts, which eliminated a role for genetic differences (Fig. S9A). These phenotypic changes were accompanied by a decrease in lung MAIT cells capable of secreting IL-17A (Fig. 8A and 8B). TNF secretion was also decreased, but this was associated with lung MAIT17 cells more than MAIT1 cells. Genetic heterogeneity may have obscured other differences, but cross-fostered mice also had increased percentages of T-bet+ lung MAIT cells, and a small but significant reduction in RORγt expressing MAIT cells (Fig. 8B). KLRG1+ lung MAIT cells were increased (Fig. S9B). KLRG1+ lung MAIT cells were virtually undetectable at steady-state in C57BL/6 mice, but this was found to be a marker for memory-like MAIT1 cells (57). These data suggested that removal from SPF conditions significantly decreased MAIT17 cells with perhaps an increase in MAIT1 cells.
Figure 8: Pet shop mice MAIT cells have an altered phenotype.
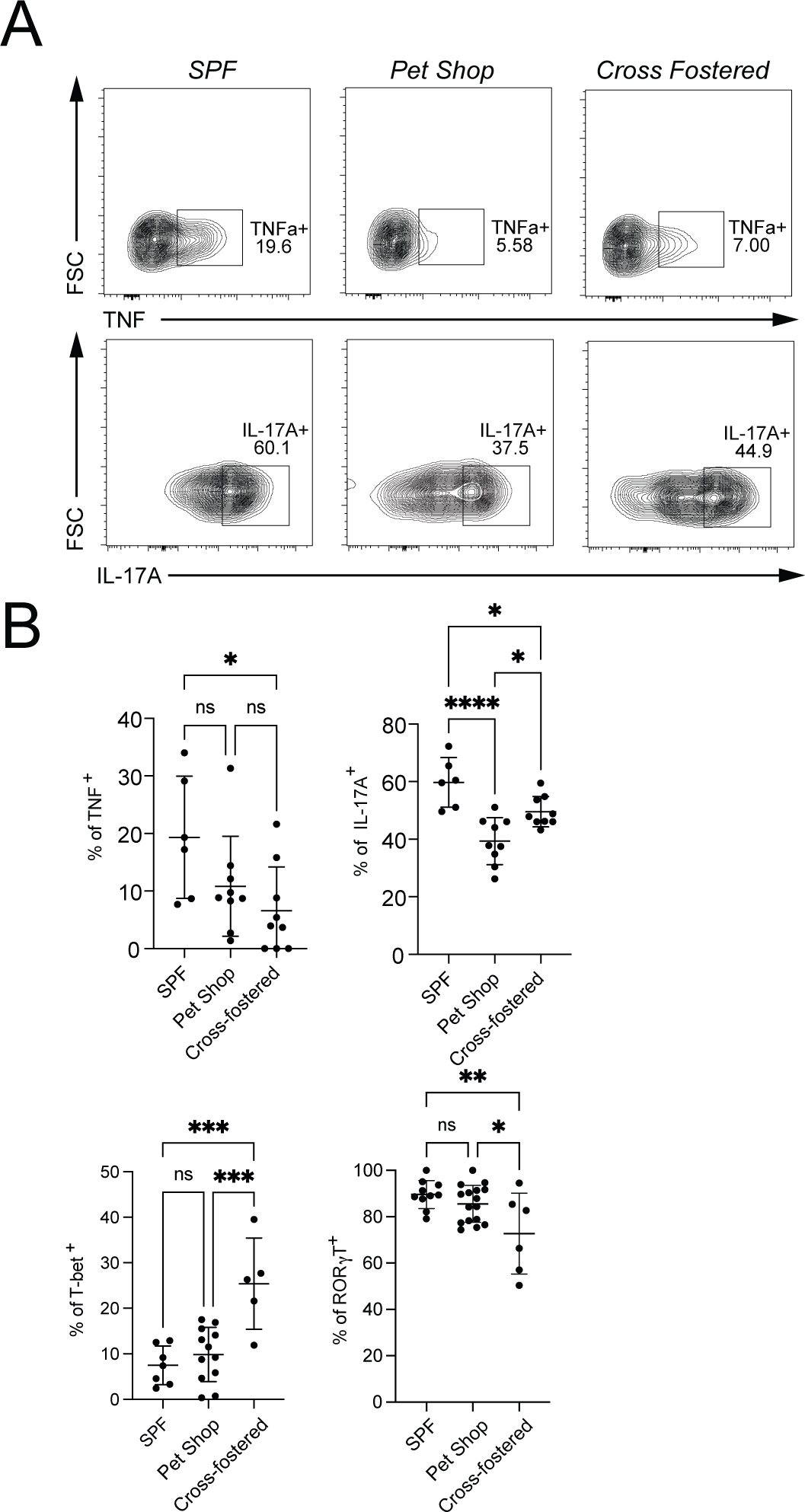
(A) Representative flow cytometry showing the percentage of MAIT cells expressing TNF and IL-17A from lungs of the indicated mice. (B) Cumulative data of expression of TNF, IL-17A, T-bet and RORγT analyzed by one-way ANOVA with Tukey test displayed as mean ± S.D. SPF mice n = 10, Pet shop mice n=16 and Cross-fostered mice n=6, *P <0.05, **P <0.01 ***P <0.001 and ****P <0.0001.
Discussion
Here we have used a combination of scRNA-seq, phenotypic and metabolic analyses to characterize mouse MAIT cell subsets, tissue-specific differences, and the conservation of MAIT cells between mouse and human. Measurement of metabolic parameters indicated that the majority of mature MAIT cells had a different metabolic state from most CD8+ T cells, characterized in both species by increased fatty acid uptake. Additionally, the metabolic states of mouse MAIT1 and MAIT17 cells at steady-state were strikingly different. In contrast, human MAIT cells were more homogenous. While some clusters or groups of mouse and human MAIT cells defined by scRNA-seq were well-represented in both species, particularly those in the thymus, other peripheral MAIT cell subpopulations had major quantitative and qualitative differences between mouse and human, which could partially reflect environmental as well as genetic influences.
In the thymus, we identified multiple clusters of mouse and human MAIT cells, including precursors unique to the thymus and clusters of mature MAIT thymocytes that also were present in the periphery. When analyzed by single-cell trajectory analysis, we generated a model in which mouse thymus MAIT cell precursors matured into stage 2-like MAIT cells and then diverged into MAIT1 and MAIT17 subsets. The finding of mature thymic MAIT1 and MAIT17 subsets agrees with several others (9, 10, 19, 25), but here we also showed that maturation from stages 1–3 is accompanied by corresponding changes in metabolic parameters, with stage 3 cells, predominantly MAIT17, similar to peripheral MAIT17 cells. Unlike another report (26), we did not find evidence for MAIT2 cells, which may reflect the difference between C57BL/6 and BALB/c strain mice.
Our data detected a predominantly spleen population of MR1 tetramer binding cells with a stage 2-like phenotype, which included Trav1+ MAIT cells that were CD62L+. We found that in chimeric mice these cells could be selected by MR1 expression on bone marrow derived or irradiation resistant cells, as was reported previously for this CD62L+ population in the thymus (25), but full maturation including high CD44 expression required MR1 expression by hematopoietic cells. It remains uncertain why these naïve-like cells were exported from the thymus and if they have an important function.
The evidence indicated human thymus MAIT cells differentiated from precursors mostly into a mixed population with mixed but somewhat more MAIT1 than MAIT17 cell characteristics. In agreement with an earlier study (10), an early MAIT cell subset (human cluster 6) with cells expressing RAG1 and RAG2 found in humans was absent in the mouse thymus. This might reflect differences in the kinetics of differentiation, for example, the transition from the most immature stage (human cluster 6) might be slower in the human compared to the mouse thymus, however, the very young age of the thymus donors could have been an issue. Analysis of human thymus from older individuals would be essential to confirm if there is a difference in the kinetics. In addition, the pseudotime analysis suggested some human MAIT thymus precursors differentiated to cells that expressed CCR7 and have other features similar to mouse stage 2 MAIT thymocytes (Fig. 5).
Overall, our data showed that populations of mouse and human MAIT cells have metabolic features that distinguished them from naïve CD8+ and some memory CD8+ T lymphocytes. The predominant mouse MAIT17 cell subset had a metabolism characterized by fatty acid uptake and storage and mitochondrial activity, while mouse MAIT1 cells were more active in glucose uptake. This metabolic difference was observed across tissues, including in the thymus. Similar findings were recently reported when γδ T cells that produce IFNγ were compared to those that produce IL-17, highlighting similarities between populations of innate-like T lymphocytes (58). Recently, we reported that weeks after bacterial infection, populations of mouse lung, memory-like MAIT1 and MAIT17 cells have metabolisms that are different from one another, and also from steady-state lung MAIT17 cells. Furthermore, these antigen-adapted MAIT1 and MAIT17 cells also greatly alter their metabolism after activation (57). Therefore, MAIT cell metabolic features are altered both short-term and long-term after antigenic stimulation.
Using CD161 and CD103 staining, subsets of human MAIT cells from lung and blood could not be divided into metabolic subgroups by surface phenotype. Instead, human MAIT cells in blood and lung shared similar metabolic features, most comparable to tissue-resident CD8+ memory T cells for uptake and storage of fatty acids, but unlike mouse MAIT17 cells, without an increased mitochondrial potential. There may be differences in other tissues, because transcriptomic analysis shows human liver MAIT cells have higher expression of genes related to fatty acid metabolism and oxidative phosphorylation compared to those in blood (59). The metabolic phenotype we measured resembles the controlled activation state of epithelial-resident T cells (60). A previous study also showed generally low mitochondrial activity in bulk human MAIT cells from peripheral blood, with an ability to rapidly reactivate metabolic and effector pathways upon stimulation (61) dependent on increased MYC protein expression (62). Therefore, as in mouse, these metabolic features alter rapidly after short-term activation. Increased mitochondrial potential and activity was functionally linked to increased IL-17 production by human MAIT cells (63) consistent with a connection between mitochondrial activity, IL-17 production by MAIT cells and the activated metabolic state of steady-state mouse MAIT17 cells.
Increased production of IL-17 by human MAIT cells has been observed, prominently in children with severe asthma (34) and in the bronchiolar lavage from children with community-acquired pneumonia (33). IL-17A producing lung MAIT cells were the majority and/or even more frequent than those producing IFNγ, but decreasing with age and only in young children. Furthermore, in the pneumonia study, a MAIT17 gene expression signature was identified, but these cells also expressed IFNG and TBET. Therefore, it is uncertain if some of the IL-17A producing cells also secreted IFNγ or if they maintain IL-17A secretion as a stable MAIT17 function similar to mice (64). We conclude that an intrinsic, highly specialized MAIT17 subset either is very infrequent or absent in the young thymus or steady-state adult tissues, although it may be present in specialized circumstances.
There is evidence that obesity and metabolic alterations can alter MAIT cell function at steady-state. This has not only been observed in mice (65), but there was increased MAIT cell production of IL-17 by MAIT cells from obese individuals (63, 66), although IFNγ-producing cells remained more numerous. Furthermore, supplementation with the TCA metabolite alpha-ketoglutarate augmented human MAIT cell effector capacity (66) providing a further connection between metabolism and MAIT cell function. These data and other studies (57) suggest there is a causal link between metabolism and MAIT cell function.
MAIT cell specificity is highly conserved, and the data revealed the most similarity between differentiating human and mouse MAIT cells in the thymus. Even in peripheral MAIT cells, some homologous genes were regulated similarly in the two species. Furthermore, strong tissue differences were observed. For example, lung MAIT cells in both species were different from their counterparts in other tissues. Mouse and human lung MAIT cells did not align well, however, in the integration analysis, nor did the transcriptome of mouse spleen MAIT1 cells align with human MAIT1 cells from PBMC. Undoubtedly, genetic differences between mice and humans influence the frequency and function of MAIT cell populations, but it is also possible that the highly controlled, standard SPF conditions of laboratory mouse housing have an influence as well. Exposure to the intestinal microbiome is not only necessary for MAIT cell thymic development (46, 67), but also differences in the microbiome can influence the number and function of skin MAIT cells (67). Our data indicated that exposure to a less controlled environment in the lung decreased cells with a MAIT17 phenotype and function, with an increase in T-bet+ and KLRG1+ MAIT cells in cross-fostered mice. Further study is required to determine the extent to which mouse and human MAIT cell transcriptomes and function can be attributed to microbial exposures or other environmental factors.
MATERIALS AND METHODS
Animals
Inbred mice were bred and housed under specific pathogen-free conditions in the vivarium of the La Jolla Institute for Immunology (La Jolla, CA). C57BL/6J mice were purchased from Jackson laboratories. Pet shop mice were analyzed immediately after purchase or housed in a vivarium maintained by the University of California, San Diego. We used SPF (specific pathogen free) C57BL/6 mice of similar weight as controls because the precise age of pet store mice was unknown. For cross-fostering, breeding pairs of SPF B6 mice and pet shop mice were simultaneously set up when individual mice reached approximately 6 weeks of age. SPF B6 pups born within 48 h were used for cross-fostering. After the birth of both SPF B6 and pet shot litters, the pet shop litters were removed and replaced with similar numbers of pups from the SPF B6 litters. Litters from SPF B6 breeders were then nursed by pet shop mothers until weaning. Cross-fostered male and female mice were analyzed when they were approximately 8 weeks of age. All procedures were approved by the La Jolla Institute for Immunology or University of California San Diego Animal Care and Use Committee for pet shop and cross-fostered mice and are compliant with the ARRIVE standards.
Antibodies and tetramers
Mouse and human MR1 tetramers loaded with either 5-OP-RU or 6-FP were obtained from the NIH Tetramer Core Facility. Fluorochrome-conjugated monoclonal antibodies were purchased from eBioscience, BD Bioscience, or BioLegend. Antibodies with clone indicated in parentheses: anti-mouse CD45 (30-F11); anti-mouse IgD (clone 11–26c.2a); anti-mouse γδ TCR (clone GL3); anti-mouse CD4 (clone GK1.5 or RM4–5); anti-mouse CD8α (clone 53–6.7); anti-mouse CD8β (clone H35–17.2); anti-mouse CD138 (clone 281–2); anti-mouse TOX (clone TXRX10); anti-mouse CD19 (clone 1D3); anti-mouse CCR9 (clone CW-1.2); anti-mouse CD24 (clone M1/69); rabbit polyclonal anti-mouse LEF1 (C12A5); anti-mouse SATB1 (clone 14/SATB1); anti-mouse IFN-γ (clone XMG1.2); anti-mouse TNF (clone MP6-XT22); anti-mouse IL-17A (clone TC11–18H10); anti-mouse CD69 (clone H1.2F3); anti-mouse T-bet (clone O4–46); anti-mouse RORγT (clone Q31–378 or B2D); anti-mouse IL-18R1 (clone BG/IL18Ra); anti-mouse CD11b (clone. M1/70); anti-mouse CD62L (clone MEL-14); anti-mouse CD45R/B220 (clone RA3–6B2); anti-mouse CD11c (clone N418); anti-mouse ICOS (clone C398.4A); anti-mouse CXCR3 (clone CXCR3–173); anti-mouse TCRβ (clone H57–597), anti-mouse CD44 (clone MI7); anti-mouse KLRG1 (clone 2F1); anti-human CD3 (clone OKT3); anti-human CD8α (clone RPA-T8); anti-human CD161 (clone HP-3G10); anti-human Vα7.2 (clone 3C10); anti-human CD19 (clone HIB19); anti-human CCR7 (clone 150503); anti-human CD45RA (clone HI100) and anti-human CD103 (clone Ber-ACT8).
Isolation of mouse cells
Splenocytes and thymocytes were harvested by mechanical disruption on 70 μm cell strainers followed by red blood cell (RBC) lysis and washed with Hank’s Balanced Salt Solution (HBSS) (Gibco) supplemented with 10% FBS. Lung tissue was digested with STEMCELL spleen dissociation medium, and mechanically dissociated using GentleMACS Dissociator (Miltenyi). Cells were strained though a 70 μm filter and washed with HBSS supplemented with 10% FBS followed by RBC lysis. Liver cells were harvested by mechanical disruption on 70 μm cell strainer followed by 34% Percoll gradient before RBC lysis and washing.
Flow cytometry
For staining of cell surface molecules, cells were suspended in staining buffer (PBS, 1% bovine serum albumin (BSA), and 0.01% NaN3) and first reacted with using PE- or APC-conjugated MR1 tetramers at a dilution of 1:300 in staining buffer for 45 minutes at room temperature followed by surface staining with fluorochrome-conjugated antibody at 0.1–1 μg/107 cells. Cells were stained with Live/Dead Yellow (ThermoFisher) at 1:500 and Fc receptors were blocked with 2.4G2 antibody at 1:500 and Free Streptavidin at 1:1000 for 15 min at 4°C. After washing, cells were stained with cell surface-specific antibodies for 30 minutes on ice. For cytokine staining, cells were previously stimulated with 100 ng/ml of PMA and 1 μg/ml of Ionomycin for 1h at 37°C and then incubated in GolgiStop and GolgiPlug (both from BD PharMingen) for 2 h at 37°C. For intracellular staining, cells were fixed with CytoFix (BD) for 20 min, and permeabilized with Perm 1X solution (ThermoFisher) with intracellular antibodies overnight. For high-parameter flow cytometry experiments, data were acquired on Fortessa or Symphony S6 (BD Biosciences), data were processed with DIVA (BD Bioscience) and analyzed with FlowJo v10.7 (BD). Opt-tSNE and UMAP dimensional reduction as well as FlowSOM algorithm clustering of flow cytometry data was performed in OMIQ software (OMIQ Inc.).
Cell enrichment and cell sorting
For MAIT cell enrichment before sorting, negative selection of cells was carried out using biotinylated antibodies against CD11b (clone M1/70), CD11c (clone M418), F4/80 (clone BM8.1), CD19 (clone 1D3), and TER-119 (clone TER-119). These were used together with Rapidspheres (STEMCELL Technologies) and either the Big Easy (STEMCELL Technologies) or Easy eight magnets (STEMCELL Technologies) and protocols from Stem Cell Technologies. MAIT cells were sorted using a FACSAria III (BD Biosciences). For some sorting and sequencing experiments, a mouse MR1 tetramer loaded with 6-FP was used as a control to discard non-MAIT cells that could still bind non-specifically to the MR1:5-OP-RU tetramer.
Human tissue and cell preparation
Postnatal human thymus was tissue obtained from children with congenital heart disease undergoing cardiac surgery at Rady Children’s Hospital, San Diego, CA. Only patients who meet the inclusion criteria and sign informed consent, are included in the study. Five thymus samples were obtained, from a 2-year-old male, 2-year-old female, two 13-month-old males and a 4-year-old female. Thymus tissue was processed by mechanical dissociation into a single cell suspension, strained and viable lymphocytes were purified by Lymphoprep (STEMCELL) density gradient centrifugation before cryopreservation. For lung and peripheral blood samples, written, informed consent was obtained from all subjects from the Institutional Review Board of La Jolla Institute for Immunology and the Southampton and South West Hampshire Research Ethics Board. Newly diagnosed, untreated patients with non-small cell lung cancer were prospectively recruited once referred. Freshly resected tumor tissue, and where available, matched adjacent non-tumor lung tissue, was obtained from patients with lung cancer following surgical resection. Four lung tissue samples were used that provided non-tumor lung tissue, obtained from a 76-year-old female, 63-year-old female, 66-year-old male and 48-year-old male. Tissues were macroscopically dissected and slowly frozen in 90% FBS (Thermo Fisher Scientific) and 10% DMSO (Sigma) for storage, until samples could be prepared. Cryopreserved non-tumor lung tissue was mechanically dissociated and enzymatically digested at 37°C as previously described (53). Briefly, lung tissue was minced with a scalpel and digested enzymatically with 0.15 IU/mL of D-Liberase (Roche) and 800 U/mL of DNase I (Sigma-Aldrich) for 15 min at 37°C. Then it was disaggregated into a single-cell suspension by passing it through a 70 μm strainer and rinsed with cold buffer (1× phosphate-buffered saline (PBS), 2 mM EDTA, 0.5% BSA). PBMCs were either obtained from the same patients that provided lung tissue or from six healthy donors (all male donors, ages between 19–35). PBMCs were isolated using density gradient before cryopreservation.
Metabolic assays
Cytometry-based metabolic assays have been described previously (68). Briefly, cells were stained with MitoTracker Deep-Red FM (Life Technologies) at 100 nM concentration, 37 °C, 5 % CO2 for 30–45 minutes in RPMI1640 (Gibco) containing 5 % FBS. For glucose uptake measurements, cells were incubated in glucose-free media containing 5 μg/ml 2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2-Deoxyglucose (2-NBDG, ThermoFisher) and 2.5% FBS at 37 °C, 5 % CO2 for 30 minutes, unless indicated otherwise. For lipid droplet quantification, cells were incubated in media containing 1 μg/ml Bodipy 493/503 (ThermoFisher) for 30 min. Uptake of fatty acids was quantified after incubation with 1uM 4,4-Difluoro-5,7-Dimethyl-4-Bora-3a,4a-Diaza-s-Indacene-3-Hexadecanoic acid (Bodipy-FL C16, ThermoFisher) at 37 °C, 5 % CO2 for 30 minutes. Optimal incubation periods for metabolic dye and metabolite uptake depended on the tissue and required fluorescence intensity, but only exceeded 45 minutes where indicated. Data were acquired using Fortessa or LSR II flow cytometers (BD Biosciences) and analyzed with FlowJo v10.7 software (BD Life Sciences). Metabolic marker fluorescence intensity depended on the instrument type and laser intensity, and therefore did not permit inter-experiment comparisons.
MAIT cell in vitro culture
Mouse CD62L+ and CD62L− MAIT cells subpopulations were isolated by cell sorting MR1:5-OP-RU+ TCRβ+ cells from the spleen of C57BL/6 mice. Cells were either cultured at a 1:25 ratio with anti-CD3/CD8 microbeads (Gibco, ThermoFisher) or co-cultured 1:1 ratio with peritoneal macrophages pulsed with 5-OP-RU (100 μg/ml). Cells were cultured for 10 days in RPMI 10% FBS supplemented with IL-2 (10 ng/ml) and IL-7 (20 ng/ml), and then evaluated for transcription factor expression or cytokine production after re-stimulations with PMA and Ionomycin, as indicated above. All cytometric analysis were made on MR1:5-OP-RU+ TCRβ+ cells.
In vivo MAIT cell stimulation
C57BL/6 mice were administered injected i.v. with a combination of 20 ng/ml of TLR9 agonist CpG (ODN 1585, InvivoGen) and 5 nmol of 5-OP-RU, two times, 24 h between doses. Antigen was prepared from 5-A-RU HCl as described (69). After 48h, splenocytes were analyzed for Ki-67 expression by flow cytometry.
MR1 KO bone marrow chimeras
Mr1+/+ C57BL/6 or Mr1−/− mice on the C57BL/6 background were irradiated with 900 rads and reconstituted i.v. with 10 × 106 bone marrow cells from either Mr1+/+ or Mr1−/− mice depleted with anti-CD4 and anti-CD8 antibodies. Thymus and spleen were harvested after 8 weeks and analyzed by flow cytometry for MR1 tetramer-binding T cells, long with CD44 and CD62L antibodies.
Single-cell RNA sequencing
Cells were sorted into a low retention 1.5 mL collection tubes containing 500 μL of a solution of PBS: FBS (1:1) supplemented with RNase inhibitor (1:100). After sorting, ice-cold PBS was added to make up to a volume of 1.4 mL. Cells were then centrifuged (5 min, 600 g, 4°C) and the supernatant was carefully aspirated, leaving 5 to 10 μL. The cell pellet was gently resuspended in 25 μL of resuspension buffer (0.22 μm filtered ice-cold PBS supplemented with ultra-pure BSA; 0.04%, Sigma-Aldrich). Following that, 33 μL of the cell suspension was transferred to a PCR-tube and single-cell libraries prepared as per the manufacturer’s instructions (10x Genomics). Samples were processed using 10X v2 chemistry for the mouse dataset and 10X v3 chemistry for the human dataset, as per the manufacturer’s recommendations; 11 and 12 cycles were used for cDNA amplification and library preparation, respectively. Libraries were quantified and pooled according to equivalent molar concentrations and sequenced on Illumina HiSeq 2500 or NovaSeq sequencing platform with the following read lengths: read 1 – 101 cycles; read 2 – 101 cycles; and i7 index - 8 cycles. For additional human PBMC MAIT cells and mouse Spleen MAIT cells, 5’ chemistry to include TCR V(D)J sequencing and Feature Barcoding (Hashtag or CITE-seq) to these datasets. Due to the disparity in chemistry, these datasets were analyzed separately to avoid batch effects. For these same experiments, CITE-seq method used TotalSeq-C (BioLegend) Hashtag antibodies C0302, C0303, C0305, C0307 and an anti-mouse barcoded CD62L (Clone MEL-14, BioLegend) C0112. All antibodies were added to staining cocktails with other fluorescent antibodies before cell sorting.
Single-cell transcriptome analysis
Single-cell libraries from mouse cells were mapped with Cell Ranger’s count pipeline using mm10 genome reference. To normalize for sequencing depth among the different gene expression libraries, multiple libraries were aggregated in Cell Ranger with Cell Ranger’s aggr command. The aggregated data were then imported into the R environment where Seurat (2.1.0) was used to filter cells and find clusters. No further aggregation or integration of libraries from mouse MAIT cells was performed in Seurat. Cells with less than 200 genes and more than 2,500 genes were discarded. Furthermore, cells with more than 5% UMIs coming from mitochondrial genes were filtered out. Genes expressed in less than 3 cells were also excluded from further analysis. This filtering step to eliminate low quality cells and doublets resulted in 6,080 cells with 13,503 genes for downstream analyses. The gene expression matrix was then normalized and scaled for downstream analysis. The FindVariableFeatures() function was used to detect the 2000 most variables genes. Principal Component Analysis was performed on the scaled data and based on the elbow plot, 20 principal components (PCs) were selected for clustering, default resolution (0.6) was used and a perplexity of 100 was chosen for the t-SNE dimensionality reduction. This dataset was further split up into 3 tissue types – spleen (3,145 cells), lung (1,313 cells), thymus (535 cells) which were then analyzed individually using the same steps. The first 15, 8, and 21 principal components, respectively, were used for clustering. We next assessed if there were any batch effects. Briefly, by comparing different batch-effect correction methods using the scIB pipeline (70), we identified that scANVI (71) displayed the best overall performance score and importantly conserved biological differences between batches (Fig. S10A). By running this benchmark tool (scANVI), we found minimal batch effects in our dataset, as shown by minimal changes in the values of two classical label conservation metrics (72, 73) obtained without (PCA-based aggregation shown in Fig. 1A) and with scANVI batch-effect correction method (Fig. S10B). Importantly, even after applying batch-effect correction, we did not find major differences the cluster annotations of the cells when compared to their clustering pattern without batch effect correction, i.e., scANVI-based batch-effect correction does not alter the cluster annotations that were obtained without batch-effect correction (Fig. S10C). Therefore, to preserve biological changes, we did not include additional batch effect correction in our analysis. Furthermore, differences in count and gene content distributions across batches were not significant and further supported the presence of negligible batch effects (Fig. S10D).
Single-cell libraries from human cells were mapped with Cell Ranger’s count pipeline using the GRCh38–1.2.0 genome reference. Multiple libraries were aggregated in Cell Ranger with Cell Ranger’s aggr command. Aggregated data was then imported into the R environment where Seurat (v3.9.9.9008) was used to filter cells, normalize and find clusters. Cells with less than 200 genes and more than 20,000 UMIs were excluded from further analysis. Furthermore, cells with more than 15% UMIs coming from mitochondrial genes were filtered out. Genes expressed in less than 3 cells were ignored. This resulted in a total of 3,020 cells from all tissues with 17,626 genes for downstream analyses. The gene expression matrix was then normalized and scaled using log normalization. The FindVariableFeatures() function was used to detect the 2000 most variables genes. Principal Component Analysis was performed on the scaled data, and based on the elbow plot, 18 PCs were selected for clustering and a resolution of 0.9 was used. To determine the clusters’ enriched genes (markers), Seurat’s FindAllMarkers function was used with test.use = “MAST” (Adjusted p-value < 0.05 and |log fold change| > 0.25). For analyzing human thymus, cells (1,316) from thymus tissue type were selected and analyzed using the same steps as listed above. 25 PCs were used for clustering of the thymus cells. For additional spleen CD62L+ and CD62L− MAIT cells subpopulations, cellranger count libraries were aggregated with cellranger aggr pipeline. Aggregated data was imported into Seurat (v3) to filter cells, normalize counts and find clusters. Cells with less than 200 genes and more than 20,000 UMIs were discarded, as well as cells with more than 5% of genes coming from mitochondria. Additionally, cells were filtered for TCR contigs containing the Trav1 gene. A total of 1,703 cells with 13,670 expressed genes were used for downstream analyses. Most variable genes, normalization and scaling was done identically to other datasets. Based on elbow plot, 20 PCs and a resolution of 0.9 were used to find clusters. Enriched markers were determined by using the MAST algorithm, as above. For additional human PBMC MAIT cell dataset, after cellranger count and aggregation, data was imported into Seurat (v3) to filter cells, normalize counts and find clusters. Cells with less than 200 genes and more than 20,000 UMIs were discarded, as well as cells with more than 5% of genes coming from mitochondria. A total of 12,594 cells and 17,574 genes were detected. Most variable genes, normalization and scaling was done identically to other datasets. Based on elbow plot, 21 PCs and a resolution of 0.3 was used to find clusters. Enriched markers were determined by using the MAST algorithm, as above.
Human-mouse data integration
Single-cell sequencing data from human and mouse MAIT cells was integrated using Seurat’s (3.0.2) alignment method (56). Briefly, we identified cross-dataset pairs of cells matching biological states (anchors). These pairs are used to correct technical differences between conditions. Default parameters were used, and the following was tailored: the first 15 principal components based on the elbow plot; and resolution 0.5 was used to identify the clusters in the integrated data. Gene names between species were matched by converting mouse gene names to uppercase.
Signature plots
Signature module scores were calculated with Seurat’s AddModuleScore function using default parameters. This function calculates the average expression levels of a gene set of interest, subtracted by the aggregated expression of control gene sets, randomly selected from genes binned by average expression. Gene lists used for MAIT1 and MAIT17 analysis were obtained from Legoux et al, 2019 (25). Gene lists for tissue resident memory and circulating signatures were obtained from Milner et al., 2017 (74). Gene lists for glycolysis were obtained from MSigDB geneset KEGG GLYCOLYSIS_GLUCONEOGENESIS. A gene list for oxidative phosphorylation was obtained from MSigDB geneset HALLMARK_OXIDATIVE_PHOSPHORYLATION. A gene list for fatty acid metabolism was obtained from MSigDB geneset KEGG_FATTY_ACID_METABOLISM. A gene list for mitochondrial gene was obtained from MSigDB geneset MITOCHONDRIAL_GENE_EXPRESSION.
Single-cell trajectory analysis
A wrapper script from R package SeuratWrappers v0.3.0 was used for calculating trajectories with Monocle3 (75) v0.2.3.0. Seurat’s object with Seurat clustering and UMAP coordinates was converted into a Monocle3 object. A single monocle partition was used for all cells. Monocle3 function learn_graph was used to fit a principal graph for the partition used. The cells were then ordered using the function order_cells, which calculates where each cell falls in pseudotime. Monocle3 helper function get_earliest_principal_node was used to specify the root node of the trajectory. Trajectory UMAPs were plotted using the function plot_cells with Monocle3 object as input. The function graph_test was used to find genes that are differentially expressed on different paths through the trajectory with the option neighbor_graph=“principal_graph”. To avoid baising of the trajectory by either cell death or cycling cells, ribosomal genes and TCR genes were excluded from this analysis. The trajectory-variable genes were then collected into co-regulated modules using the function find_gene_modules. Monocle3’s function aggregate_gene_expression was used to calculate aggregate expression of genes in each module for all the clusters. These module scores were then plotted in a heatmap using R package pheatmap v1.0.12 with options cluster_rows=TRUE, cluster_cols=TRUE, scale=“column”, clustering_method=“ward.D2”. The modules were further combined into stages based on their functionality/annotation. Top 10 genes were selected for each stage with high morans_I value calculated by the function graph_test earlier. The Module heatmap was generated using Seurat’s function DoHeatmap. It shows scaled average expression for the top 10 genes grouped by cells in each stage.
Demultiplexing
PLINK (v2.1.4) from Illumina Genome Studio plugins was used to convert and export Illumina genotype data into PLINK data format. PLINK is again used with the “--recode vcf” option to convert PLINK data format to VCF. snpQC package (76) was used to detect low quality SNPs. SNPs failing in >5% of the samples and SNPs with Illumina’s gene call scores <0.2 in <90% of the samples were excluded for downstream analysis. The alignment files generated by CellRanger count program (v3.1.0) were split by cell barcode using samtools (v1.9) (76, 77). Each individual cell-specific BAM file was run through Freebayes (v0.9.21) (77, 78) with the SNP array variants as input to catalog matching SNPs for each cell.
The score for each sample-cell barcode pair were calculated as shown in the below formula:
where Gn is the number of genotype calls for this barcode that match sample n and GANY is the total number of genotype calls made for this barcode. Scores for each sample were then ranked from highest to lowest and the score for the highest-ranking sample was compared to that of the second highest. If the ratio between these two was 1.3 or greater and at least 300 genotype calls were made for the cell, the sample with the highest score was assigned.
Statistical Methods
All graphs and statistical analysis were generated using Prism 9 software (GraphPad Software, San Diego, CA) or R version 3.5.1. Data are plotted as mean ± standard deviation or mean ± standard error of the mean (SEM), and statistical significance was determined by using unpaired t test. Significance for multiparameter comparisons was determined by one-way ANOVA with Dunnett’s post-test, paired t test or one-way ANOVA with post hoc Tukey test.
Data visualization tool
Visualization web-based platform was constructed using R package Shiny v1.7.1, customizing it with CSS theme and htmlwidgets. The app was fed with mouse and human Seurat and Monocle objects, subsetting only the elementary data in order to optimize memory resources. Plots are constructed in real time using Seurat and Monocle V3 functions using the single cell data object selected by the user. Data visualization tool is available at https://mait.lji.org.
Supplementary Material
Table S1: Quality of mouse single cell RNA sequencing libraries.
Table S3: Coregulated genes identified in different modules as defined by Monocle analysis of mouse thymus MAIT cells.
Table S2: Transcripts enriched in each of the clusters defined by single cells sequencing analysis of mouse MAIT cells combined from four tissues.
Table S4: Quality of single cell RNA sequencing libraries from spleen CD62L+ and CD62L− mouse spleen MAIT cells.
Table S6: Quality of human single cell RNA sequencing libraries.
Table S5: Transcripts enriched in each of the clusters defined by single cells sequencing analysis of spleen CD62L+ and CD62L− mouse spleen MAIT cells.
Table S7: Transcripts enriched in each of the clusters defined by single cells sequencing analysis of human MAIT cells.
Table S8: Quality of human PBMCS single cell RNA sequencing library.
Table S9: Transcripts enriched in each of the clusters defined by single cells sequencing analysis of human PBMCs.
Table S10: Coregulated genes identified in different modules as defined by Monocle analysis of human thymus MAIT cells.
Acknowledgement
We would like to thank Dr. Anusha Preethi Ganesan for help with human lung dissociation protocol and Ms. Gina Levi for coordinating receipt of human thymus samples. We thank the La Jolla Institute (LJI) Flow Cytometry Core for assisting with cell sorting and sequencing Core for performing scRNA sequencing. We thank Mr. Woo and Mr. Alzetani for access to surgical material, Dr. Serena Chee, Ben Johnson, Alice Appleford and Sophie Matthew for collection, processing, and storage of the tissues. We thank Dr. David Lewinsohn for reagents to generate 5-OP-RU.
Funding
Supported by the following grants-
National Institutes of Health grant AI105215 (MK)
National Institutes of Health grant AI71922 (MK)
National Institutes of Health grant AI137230 (MK)
Wellcome Trust grant 210842_Z_18_Z to (TR)
National Institutes of Health grant AI108651 (LF L)
National Institutes of Health grant AI163813 (LFL)
UCSD Program in Immunology seed grant (LFL and SMH)
Lung tissue collection in the UK was supported by funding from the Whittaker fund and iCURE, by the Wessex Clinical Research Network and the National Institute of Health Research, UK.
Utilized equipment was supported by-
National Institutes of Health grant S10RR027366 (BD FACSAria II)
National Institutes of Health grant S10OD025052 (NovaSeq 6000)
National Institutes of Health grant S10-OD016262 (Illumina HiSeq 2500).
Footnotes
This manuscript has been accepted for publication in Science Immunology. This version has not undergone final editing. Please refer to the complete version of record at www.scienceimmunology.org. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS.
Competing interests
Authors declare that they have no competing interests.
Code availability
The code developed for the analyses performed in this study is available upon request.
Data and Software Availability
Single-cell RNA sequencing data generated for this study are deposited at the Gene Expression Omnibus under accession number GSE189483, GSE189484 and GSE189485. Data visualization tool is available at https://mait.lji.org.
References
- 1.Treiner E et al. , Mucosal-associated invariant T (MAIT) cells: an evolutionarily conserved T cell subset. Microbes Infect 7, 552–559 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Kjer-Nielsen L et al. , MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 491, 717–723 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Tilloy F et al. , An invariant T cell receptor alpha chain defines a novel TAP-independent major histocompatibility complex class Ib-restricted alpha/beta T cell subpopulation in mammals. J Exp Med 189, 1907–1921 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Treiner E et al. , Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature 422, 164–169 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Gold MC et al. , Human mucosal associated invariant T cells detect bacterially infected cells. PLoS Biol 8, e1000407 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le Bourhis L et al. , Antimicrobial activity of mucosal-associated invariant T cells. Nat Immunol 11, 701–708 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Corbett AJ et al. , T-cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature 509, 361–365 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Rahimpour A et al. , Identification of phenotypically and functionally heterogeneous mouse mucosal-associated invariant T cells using MR1 tetramers. J Exp Med 212, 1095–1108 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koay HF et al. , A three-stage intrathymic development pathway for the mucosal-associated invariant T cell lineage. Nat Immunol 17, 1300–1311 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Koay H-F et al. , A divergent transcriptional landscape underpins the development and functional branching of MAIT cells. Science immunology 4, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gapin L, Check MAIT. J Immunol 192, 4475–4480 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chandra S, Kronenberg M, Activation and Function of iNKT and MAIT Cells. Adv Immunol 127, 145–201 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Legoux F, Salou M, Lantz O, MAIT Cell Development and Functions: the Microbial Connection. Immunity 53, 710–723 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Xiao X, Cai J, Mucosal-Associated Invariant T Cells: New Insights into Antigen Recognition and Activation. Front Immunol 8, 1540 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hinks TSC, Zhang XW, MAIT Cell Activation and Functions. Front Immunol 11, 1014 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hinks TSC et al. , Activation and In Vivo Evolution of the MAIT Cell Transcriptome in Mice and Humans Reveals Tissue Repair Functionality. Cell Rep 28, 3249–3262 e3245 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamichhane R et al. , TCR- or Cytokine-Activated CD8(+) Mucosal-Associated Invariant T Cells Are Rapid Polyfunctional Effectors That Can Coordinate Immune Responses. Cell Rep 28, 3061–3076 e3065 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Leng T et al. , TCR and Inflammatory Signals Tune Human MAIT Cells to Exert Specific Tissue Repair and Effector Functions. Cell Reports 28, 3077–3091.e3075 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koay HF, Godfrey DI, Pellicci DG, Development of mucosal-associated invariant T cells. Immunol Cell Biol 96, 598–606 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seach N et al. , Double-positive thymocytes select mucosal-associated invariant T cells. J Immunol 191, 6002–6009 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Koay HF et al. , Diverse MR1-restricted T cells in mice and humans. Nat Commun 10, 2243 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pellicci DG, Koay HF, Berzins SP, Thymic development of unconventional T cells: how NKT cells, MAIT cells and γδ T cells emerge. Nature Reviews Immunology, (2020). [DOI] [PubMed] [Google Scholar]
- 23.Garner LC, Klenerman P, Provine NM, Insights Into Mucosal-Associated Invariant T Cell Biology From Studies of Invariant Natural Killer T Cells. Front Immunol 9, 1478 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salou M et al. , A common transcriptomic program acquired in the thymus defines tissue residency of MAIT and NKT subsets. J Exp Med 216, 133–151 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Legoux F et al. , Molecular mechanisms of lineage decisions in metabolite-specific T cells. Nat Immunol 20, 1244–1255 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Lee M et al. , Single-cell RNA sequencing identifies shared differentiation paths of mouse thymic innate T cells. Nature Communications 11, 4367 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drashansky TT et al. , BCL11B is positioned upstream of PLZF and RORγt to control thymic development of mucosal-associated invariant T cells and MAIT17 program. iScience 24, 102307 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Z et al. , Characterization and Purification of Mouse Mucosal-Associated Invariant T (MAIT) Cells. Current Protocols in Immunology 127, 1–30 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Leeansyah E et al. , Arming of MAIT Cell Cytolytic Antimicrobial Activity Is Induced by IL-7 and Defective in HIV-1 Infection. PLOS Pathogens 11, e1005072 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dias J et al. , The CD4−CD8− MAIT cell subpopulation is a functionally distinct subset developmentally related to the main CD8+ MAIT cell pool. Proceedings of the National Academy of Sciences 115, E11513–E11522 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dias J, Leeansyah E, Sandberg JK, Multiple layers of heterogeneity and subset diversity in human MAIT cell responses to distinct microorganisms and to innate cytokines. Proceedings of the National Academy of Sciences 114, E5434–E5443 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dusseaux M et al. , Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood 117, 1250–1259 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Lu B et al. , IL-17 production by tissue-resident MAIT cells is locally induced in children with pneumonia. Mucosal Immunol 13, 824–835 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lezmi G et al. , Evidence for a MAIT-17-high phenotype in children with severe asthma. J Allergy Clin Immunol 144, 1714–1716 e1716 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Napier RJ, Adams EJ, Gold MC, Lewinsohn DM, The Role of Mucosal Associated Invariant T Cells in Antimicrobial Immunity. Front Immunol 6, 344 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van der Windt GJ, Pearce EL, Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol Rev 249, 27–42 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van der Windt GJ et al. , Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 36, 68–78 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Sullivan D, The metabolic spectrum of memory T cells. Immunol Cell Biol 97, 636–646 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Geltink RIK, Kyle RL, Pearce EL, Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu Rev Immunol 36, 461–488 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee YJ et al. , Lineage-Specific Effector Signatures of Invariant NKT Cells Are Shared amongst γδ T, Innate Lymphoid, and Th Cells. (2016), vol. 197, pp. 1460–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Georgiev H, Ravens I, Benarafa C, Forster R, Bernhardt G, Distinct gene expression patterns correlate with developmental and functional traits of iNKT subsets. Nat Commun 7, 13116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Engel I et al. , Innate-like functions of natural killer T cell subsets result from highly divergent gene programs. Nat Immunol 17, 728–739 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Legoux F et al. , Microbial metabolites control the thymic development of mucosal-associated invariant T cells. Science 366, 494–499 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Tao H et al. , Differential controls of MAIT cell effector polarization by mTORC1/mTORC2 via integrating cytokine and costimulatory signals. Nat Commun 12, 2029 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jaiswal AK et al. , Syndecan-1 Regulates Psoriasiform Dermatitis by Controlling Homeostasis of IL-17–Producing γδ T Cells. The Journal of Immunology 201, 1651–1661 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Martin E et al. , Stepwise development of MAIT cells in mouse and human. PLoS Biol 7, e54 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trapnell C et al. , The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu H et al. , MS4a4B, a CD20 homologue in T cells, inhibits T cell propagation by modulation of cell cycle. PLoS One 5, e13780 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bortoluzzi S et al. , Brief homogeneous TCR signals instruct common iNKT progenitors whose effector diversification is characterized by subsequent cytokine signaling. Immunity 54, 2497–2513 e2499 (2021). [DOI] [PubMed] [Google Scholar]
- 50.Gherardin NA et al. , Human blood MAIT cell subsets defined using MR1 tetramers. Immunol Cell Biol 96, 507–525 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Souter MNT et al. , CD8 coreceptor engagement of MR1 enhances antigen responsiveness by human MAIT and other MR1-reactive T cells. Journal of Experimental Medicine 219, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Z et al. , Mucosal-associated invariant T-cell activation and accumulation after in vivo infection depends on microbial riboflavin synthesis and co-stimulatory signals. Mucosal Immunology 10, 58–68 (2017). [DOI] [PubMed] [Google Scholar]
- 53.Ganesan AP et al. , Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat Immunol 18, 940–950 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cano-Gamez E et al. , Single-cell transcriptomics identifies an effectorness gradient shaping the response of CD4(+) T cells to cytokines. Nat Commun 11, 1801 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Konjar S, Veldhoen M, Dynamic Metabolic State of Tissue Resident CD8 T Cells. Front Immunol 10, 1683 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stuart T et al. , Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Riffelmacher T et al. , Divergent metabolic programmes control two populations of MAIT cells that protect the lung. Nat Cell Biol 25, 877–891 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lopes N et al. , Distinct metabolic programs established in the thymus control effector functions of gammadelta T cell subsets in tumor microenvironments. Nat Immunol 22, 179–192 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lamichhane R et al. , Human liver-derived MAIT cells differ from blood MAIT cells in their metabolism and response to TCR-independent activation. European Journal of Immunology, (2021). [DOI] [PubMed] [Google Scholar]
- 60.Konjar S et al. , Mitochondria maintain controlled activation state of epithelial-resident T lymphocytes. Sci Immunol 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zinser ME et al. , Human MAIT cells show metabolic quiescence with rapid glucose-dependent upregulation of granzyme B upon stimulation. Immunol Cell Biol 96, 666–674 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kedia-Mehta N et al. , The proliferation of human mucosal-associated invariant T cells requires a MYC-SLC7A5-glycolysis metabolic axis. Sci Signal 16, eabo2709 (2023). [DOI] [PubMed] [Google Scholar]
- 63.Carolan E et al. , Altered distribution and increased IL-17 production by mucosal-associated invariant T cells in adult and childhood obesity. J Immunol 194, 5775–5780 (2015). [DOI] [PubMed] [Google Scholar]
- 64.Chandra S et al. , Development of Asthma in Inner-City Children: Possible Roles of MAIT Cells and Variation in the Home Environment. The Journal of Immunology 200, 1995–2003 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Toubal A et al. , Mucosal-associated invariant T cells promote inflammation and intestinal dysbiosis leading to metabolic dysfunction during obesity. Nat Commun 11, 3755 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brien AO et al. , Targeting mitochondrial dysfunction in MAIT cells limits IL-17 production in obesity. Cell Mol Immunol 17, 1193–1195 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Constantinides MG et al. , MAIT cells are imprinted by the microbiota in early life and promote tissue repair. Science (New York, N.Y.) 366, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Riffelmacher T et al. , Autophagy-Dependent Generation of Free Fatty Acids Is Critical for Normal Neutrophil Differentiation. Immunity 47, 466–480 e465 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li K et al. , Synthesis, stabilization, and characterization of the MR1 ligand precursor 5-amino-6-D-ribitylaminouracil (5-A-RU). PLoS One 13, e0191837 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Luecken MD et al. , Benchmarking atlas-level data integration in single-cell genomics. Nature Methods 19, 41–50 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu C et al. , Probabilistic harmonization and annotation of single-cell transcriptomics data with deep generative models. Mol Syst Biol 17, e9620 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lütge A et al. , CellMixS: quantifying and visualizing batch effects in single-cell RNA-seq data. Life Sci Alliance 4, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Korsunsky I et al. , Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Milner JJ et al. , Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature 552, 253–257 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cao J et al. , The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gondro C, Porto-Neto LR, Lee SH, SNPQC--an R pipeline for quality control of Illumina SNP genotyping array data. Anim Genet 45, 758–761 (2014). [DOI] [PubMed] [Google Scholar]
- 77.Li H et al. , The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Garrison E, Marth G, Haplotype-based variant detection from short-read sequencing.arXiv preprint arXiv:1207.3907 [q-bio.GN] (2012). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Quality of mouse single cell RNA sequencing libraries.
Table S3: Coregulated genes identified in different modules as defined by Monocle analysis of mouse thymus MAIT cells.
Table S2: Transcripts enriched in each of the clusters defined by single cells sequencing analysis of mouse MAIT cells combined from four tissues.
Table S4: Quality of single cell RNA sequencing libraries from spleen CD62L+ and CD62L− mouse spleen MAIT cells.
Table S6: Quality of human single cell RNA sequencing libraries.
Table S5: Transcripts enriched in each of the clusters defined by single cells sequencing analysis of spleen CD62L+ and CD62L− mouse spleen MAIT cells.
Table S7: Transcripts enriched in each of the clusters defined by single cells sequencing analysis of human MAIT cells.
Table S8: Quality of human PBMCS single cell RNA sequencing library.
Table S9: Transcripts enriched in each of the clusters defined by single cells sequencing analysis of human PBMCs.
Table S10: Coregulated genes identified in different modules as defined by Monocle analysis of human thymus MAIT cells.
Data Availability Statement
Single-cell RNA sequencing data generated for this study are deposited at the Gene Expression Omnibus under accession number GSE189483, GSE189484 and GSE189485. Data visualization tool is available at https://mait.lji.org.