

Abstract
Background:
The germline perpetuates genetic information across generations. To maintain the integrity of the germline, transposable elements in the genome must be silenced, as these mobile elements would otherwise engender widespread mutations passed on to subsequent generations. There are several well-established mechanisms that are dedicated to providing defense against transposable elements, including DNA methylation, RNA interference, and the PIWI-interacting RNA pathway.
Objectives:
Recently, several studies have provided evidence that transposon defense is not only provided by factors dedicated to this purpose but also factors with other roles, including in germline development. Many of these are transcription factors. Our objective is to summarize what is known about these “bi-functional” transcriptional regulators.
Materials and methods:
Literature search.
Results and conclusion:
We summarize the evidence that six transcriptional regulators—GLIS3, MYBL1, RB1, RHOX10, SETDB1, and ZBTB16—are both developmental regulators and transposable element-defense factors. These factors act at different stages of germ cell development, including in pro-spermatogonia, spermatogonial stem cells, and spermatocytes. Collectively, the data suggest a model in which specific key transcriptional regulators have acquired multiple functions over evolutionary time to influence developmental decisions and safeguard transgenerational genetic information. It remains to be determined whether their developmental roles were primordial and their transposon defense roles were co-opted, or vice versa.
Keywords: LINE1, germ cell, primordial germ cell, pro-spermatogonia, spermatogonial stem cell, transcription factor, transposon
1 |. INTRODUCTION
The processes of development and transposon defense are distinct functions that have traditionally been studied separately. In this review, we discuss transcriptional regulators that have the surprising ability to function in both processes. We first discuss the known or putative developmental functions of these transcriptional regulators in male germ cells. We then discuss the evidence that these factors silence transposons, what classes of transposons they act on, and how their defense functions might be linked with their developmental functions.
1.1 |. Germ cell development and spermatogenesis
Germ cell development initiates during early embryogenesis when primordial germ cells (PGCs)—the common precursors of spermatozoa and oocytes—are first formed.1,2 These PGCs migrate to the gonadal ridge, whereupon they receive signals that initiate one of two distinct orchestrated programs - oogenesis or spermatogenesis. At ~E12.5 in mice, male PGCs convert into pro-spermatogonia (ProSG; also called gonocytes), which undergo three successive stages – multiplying (M)-ProSG, primary transitional (T1)-ProSG, and secondary transitional (T2)-ProSG. These three ProSG stages are distinguished by whether or not they undergo cell proliferation, as well as their epigenetic reprogramming status.3 At the perinatal stage in mice, T2-ProSG gives rise to spermatogonial stem cells (SSCs), the cell type responsible for supporting spermatogenesis throughout adult life. Like all stem cells, SSCs have the ability to undergo both self-renewal and differentiation.3,4 Their self-renewal is greatly influenced by the niche-factor glial cell line-derived neurotrophic factor (GDNF),5 as well as several non-GDNF-stimulated transcription factors (TFs), including DMRT1, FOXO1, MYC, ZBTB16, and TAF4B.6–10 The differentiation of SSCs leads to the formation of spermatogonial progenitors, which further differentiate to form spermatocytes. The latter undergo meiosis to generate haploid cells called round spermatids, which later differentiate to form elongated spermatids, the immediate precursors of spermatozoa.3 This process of spermatogenesis is a highly orchestrated process that is governed by many TFs, including DMRT1, SOHLH1, SOHLH2, and SOX3.11–13
1.2 |. Transposons
Most eukaryotic genomes are dominated by repetitive DNA, most of which is interspersed between coding regions.14 This repetitive DNA is predominantly derived from the activity of transposable elements (TEs) – many of which are parasitic genetic units that replicate and transpose the new copy to another site by a ‘copy and paste’ mechanism. This process leads—over evolutionary time—to the accumulation of hundreds of thousands of TE copies dispersed through the genome.15 TEs are regarded as a double-edged sword. On the positive side, TEs sometimes acquire functions over evolutionary time that are useful to the host organism. For example, some TEs have been shown to evolve into regulatory sequences and protein-coding domains that have been co-opted to serve the purposes of the host.16 But on the negative side, TEs can be highly deleterious. This follows from the fact that TEs transpose in a relatively random manner in the genome. Thus, they can disrupt gene function or alter gene expression, contributing to genetic disease and cancer.16,17
TEs are broadly characterized as either retrotransposons or DNA transposons. Retrotransposons are transcribed into an RNA intermediate, which can be reverse-transcribed and integrated at a new site in the genome.18 Retrotransposons can be further divided into those that either contain or lack a long terminal repeat (LTR). An example of an LTR-containing retrotransposon is intracisternal A-particles (IAPs), which originally were derived from infectious retroviruses that integrated into the germline.19 Non-LTR retrotransposons include long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs).18 In contrast to retrotransposons, DNA transposons are mobile DNA elements that generate a single-or double-stranded DNA intermediate to move to another genomic location.20 DNA transposons use a ‘cut and paste’ mechanism and thus, unlike retrotransposons, do not accumulate in copy number.
1.3 |. Transposon defense
To protect against the negative effects of TEs, multiple epigenetic and RNA-mediated mechanisms have been postulated to have evolved.21 Such silencing mechanisms are particularly critical for the germline, as they reduce the transmission of TE-induced mutations to subsequent generations.
DNA methylation is a major mechanism that curbs TE transposition.22 The majority of DNA methylation in the mammalian genome occurs in repetitive sequences, which are largely TEs, and encompass the bulk of heterochromatin. The re-methylation of repeat elements following genome-wide demethylation in PGCs occurs progressively in fetal ProSG and is completed by the newborn stage.23 DNA methylation is catalyzed by DNA methyl transferases, including DNMT1, DNMT3A, and DNMT3B.24 DNMT1 is considered to be mainly involved in maintaining methylation marks after DNA replication, and thus it assures faithful propagation of existing DNA methylation patterns. It preferentially methylates hemi-methylated DNA from the parental strand onto the newly synthesized daughter strand during DNA replication. In contrast, DNMT3A and DNMT3B are de novo DNA methyl transferases that generate new DNA methylation marks. DNMT3A and DNMT3B are largely functionally redundant in silencing TEs in germ cells.24
Another DNMT family member—DNMT3L—lacks DNA methyltransferase activity and instead serves to activate other DNMT family members. DNMT3L is particularly important for driving DNA methylation in the germline. Global loss of DNMT3L leads to moderate to severe hypomethylation of repeat elements in ProSG.23 This DNA hypomethylation is associated with reactivation of TEs in male germ cells, meiotic arrest, and male sterility.23,25,26 Thus, DNMT3L is critical for TE defense in germ cells.
An opportunity for TEs to transpose at a high rate is when DNA methylation marks are globally erased in PGCs as a means to epigenetically reprogram the genome.27 This hypomethylated state is maintained when PGCs first become ProSG.28 While genome-wide DNA demethylation provides the benefit of reprogramming the genome for the next generation, it opens the door for TE activation and transposition. Thus, mechanisms have evolved to defend against TEs becoming active when PGCs and ProSG are in this hypo-methylated state.
One means by which TEs are held in check is through histone modifications. Evidence suggests that PGCs and ProSG make use of several histone modifications to suppress TE expression and protect genomic integrity. For example, H2A (H4R3me2s) is a repressive histone mark catalyzed by the arginine methyltransferase, PRMT5, that silences LINE1 and IAP TEs in PGCs.29 Another histone modification that silences TEs, including LINE1 elements, is the dimethylation of lysine (K) 9 in histone 3 (H3) (H3K9me2).30 This histone modification is widely present in spermatogonia (SG) and early spermatocytes (preleptotene to zygotene) but largely disappears at the pachytene stage.31
Another major mechanism that protects against TEs in PGCs and ProSG is the Piwi-interacting (pi) RNA pathway. This pathway is mediated by short (24–31 nt) non-coding RNAs called “piRNAs” that associate with members of the PIWI RNA-binding Argonaute protein family to suppress transposons and thereby maintain germline genome integrity.32 In mice, there are three PIWI family members—PIWIL1, PIWIL2, and PIWIL4 (also known as MIWI, MILI, and MIWI2, respectively)—the latter two of which are expressed in PGCs and/or ProSG. The Piwil2 gene is first detectably expressed in gonadal tissue at E12.5 when PGCs transit to form M-ProSG,33 whereas Piwil4 expression is not detectable until E15.5 in T1-ProSG.34 Both PIWIL2 and PIWIL4 are critical for the suppression of LINE1 elements and other TEs in ProSG, based on analysis of Piwil2- and Piwl4- knockout (KO) mice.34,35 Both KO strains suffer from male sterility.34,35
Mouse germ cells express two distinct populations of piRNAs: “pre-pachytene” and “pachytene.” The pre-pachytene piRNAs are present prenatally in ProSG and postnatally in SG, originate largely from TEs, and primarily target TEs.36 In contrast, pachytene piRNAs are present in more advanced postnatal germ cells (pachytene spermatocytes and round spermatids); while some of this class are derived from repetitive TEs, they mainly originate from unique genomic clusters and are thought to mainly target coding mRNAs.37
PiRNAs suppress gene expression either transcriptionally or post-transcriptionally.38 piRNAs mediate the former by inducing heterochromatin formation. piRNAs mediate the latter by cleaving RNAs complementary with their sequence. One means by which piRNAs repress transcription is by triggering DNA methylation. In ProSG, the PIWIL4-associated protein, SPOCD1, drives piRNA-guided TEs methylation and transcriptional silencing.39
2 |. BI-FUNCTIONAL TRANSCRIPTIONAL REGULATORS
Below, we summarize the evidence that some transcriptional regulators are bi-functional factors: they function in germ cells, including in germ cell development, and were subsequently found to function in TE defense (Figure 1). These transcriptional regulators act at different stages of germ cell development (Figure 1). GLIS3, SETDB1, ZBTB16, RB1, and RHOX10 act in ProSG and/or undifferentiated SG, including SSCs. MYBL1 acts in postnatal germ cells at the pachytene stage.
FIGURE 1.
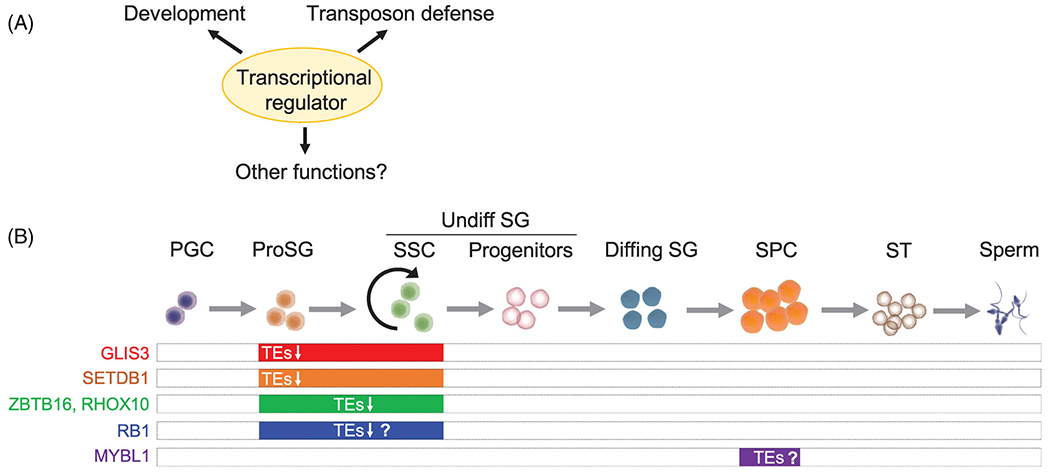
Multi-functional factors acting in the germline. (A) We propose that some individual transcriptional regulators are capable of providing the functions depicted. (B) The 6 germ-cell transcriptional regulators that we discuss in this review. The germ cell stage(s) that they appear to act—including in transposable element (TE) suppression—are indicated. RB1 has functions in ProSG and SSCs, and thus is hypothesized to also suppress TEs in these stages, but its TE suppression function is only based on experiments in somatic cells. In the case of MYBL1, it is known to drive the production of a large subset of piRNAs, raising the possibility that MYBL1 indirectly functions in TE defense, but this has not been directly demonstrated.
2.1 |. RHOX10 and the RHOX cluster
The hallmark of homeobox proteins is they contain a 60 amino-acid domain that binds to specific sequences in DNA or RNA.40 The best-known homeobox protein subfamily is the HOX subfamily. The genes encoding these TFs were originally discovered in flies, where they control key embryonic developmental events that correspond to their position in HOX gene clusters41. Subsequent studies showed that most metazoans contain HOX gene clusters and that these gene clusters encode TFs that drive key embryonic developmental events just as they do in flies.41
More recently, the reproductive homeobox (RHOX) subfamily was discovered.42 The homeobox proteins in this subfamily are all encoded by a single gene cluster on the X chromosome.42–46 Unlike the HOX gene cluster, the RHOX gene cluster is largely confined to mammals, suggesting it is a “mammalian invention” providing functions specifically important for mammals.
All known members of the mouse and human RHOX gene clusters are primarily expressed in cell types in the reproductive tract and placenta,47 raising the possibility that the RHOX proteins are TFs devoted to reproductive functions. In support of the former, RHOX proteins have been shown to function as TFs that bind to specific sequences.48,49 In support of the latter, several studies have shown that RHOX proteins regulate specific genes in Sertoli cells, germ cells, and epididymal cells.48–52
The biological functions of RHOX proteins have primarily been studied in mice. This has been challenging, as there are 33 genes in the mouse Rhox cluster.47 One approach that has been taken is to mutationally inactivate individual Rhox genes. This approach, which has only been applied to a small number of Rhox genes, has revealed that some Rhox genes function in spermatogenesis,50,53,54 while mutational inactivation of Rhox9 has no discernable effects in mice in vivo.55 Another approach to examining the function of Rhox genes has been to knock down their expression in vivo. This approach has successfully demonstrated that the Sertoli cell-expressed Rhox8 gene functions in spermatogenesis,56 but failed to define a function for the germ-cell expressed Rhox3 paralogs because of experimentally validated off-target effects.57
As another approach to defining the biological functions of Rhox genes, Song et al. deleted the entire Rhox cluster in mice.53 They found that global deletion of the Rhox cluster caused strain-specific embryonic lethality. In a mixed genetic background that permitted embryo survival, global Rhox-cluster KO mice were runted and had low viability, but those that survived were found to have progressive spermatogenic defects. Testes weight failed to increase with age; some spermatozoa were produced but the number of spermatozoa failed to increase as these adult Rhox-cluster KO mice aged. Most strikingly, these KO mice exhibited a progressive increase in seminiferous tubules devoid of (or mostly lacking) germ cells, a phenotype consistent with an SSC defect. As further evidence for an SSC defect, tubules that did contain germ cells tended to lack early germ cells. These defects were reproduced in mice conditionally lacking the Rhox cluster specifically in germ cells, demonstrating that these SSC-associated defects are cell autonomous.
While, in principal, any of the 33 homeobox genes in the Rhox cluster could be responsible for the SSC-associated defects in Rhox-cluster KO mice, Song et al. considered the best candidate to be Rhox10, as it was the only Rhox gene known to be highly expressed in undifferentiated SG58. To test its role, Song et al. mutationally inactivated Rhox10 and found that this caused the same progressive spermatogenic decline phenotype as Rhox-cluster KO mice.53
To determine the exact nature of the defect, a battery of assays was used. Analysis of SSC markers, including GFRα1, Id4-eGFP, and nuclear FOXO1, demonstrated that Rhox10-null mice had reduced SSCs in newborn mice, indicative of an SSC establishment defect. This was confirmed by germ-cell transplantation analysis, which showed that postnatal day (P) 7–8 Rhox10-null mice had a striking decrease in transplantable colonies. Such an SSC defect could be caused by reduced germ-cell proliferation, reduced germ-cell survival, or impaired germ-cell differentiation. Analysis with proliferation and apoptosis markers indicated that loss of RHOX10 did not have a measurable impact on either the proliferation or apoptosis of newborn germ cells, suggesting instead that RHOX10 is critical for germ cell progression. To directly test this, single-cell RNA-sequencing (scRNAseq) analysis was employed. scRNAseq analysis of FACS-purified Id4-eGFP+ cells at different time points revealed an accumulation of a cell cluster with the molecular characteristics of T1-ProSG in Rhox10-null mice at a time point (P3) when T1-ProSG have already progressed to form later stages of germ cells in wild-type (wt) mice.53 This abnormal accumulation of T1-ProSG in Rhox10-null mice suggests that RHOX10 serves to drive T1-ProSG to form T2-ProSG. Given that T2-ProSG are regarded as the direct precursors of SSCs,3 the T1-to-T2 ProSG progression defect in Rhox10-null mice explains the low frequency of SSCs in newborn Rhox10-null mice.
The notion that RHOX10 drives ProSG differentiation was further supported by Tan et al.49 Using newly identified ProSG markers—DNMT3L and ETV459—they confirmed that Rhox10-null mice accumulate ProSG at P2 and P3 in vivo, a phenotype recapitulated in vitro. Further, using a battery of genome-wide approaches, Tan et al. also identified high-confidence RHOX10 target genes that are candidates to drive ProSG differentiation. Through “rescue” experiments, the authors demonstrated that at least two of these genes act downstream of RHOX10 to promote ProSG differentiation.49
A new wrinkle to this story was provided by Tan et al., who found that RHOX10 suppresses the transposition of LINE1 elements, suggesting this homeobox gene functions to silence parasitic DNA in the germline.60 Using an SN1 single-copy LINE1 transgene reporter mouse line,61 Tan et al. found that SN1 LINE1 copy number was significantly higher in the testes in Rhox10-null mice as compared to littermate wt mice,60 indicative of increased LINE1 transposition as a result of Rhox10 loss. This effect was specific to the testes, not the other adult tissues tested. Mice lacking the entire Rhox-cluster specifically in germ cells also displayed a testes-specific increase in LINE1 copy number. Time-course analysis showed that the increased SN1 LINE1 copy number occurs between E13.5 and E16.5, and was sustained at P0 and adult stages. This indicated that RHOX10 suppresses LINE1 transposition in ProSG. Bisulfite analysis of the LINE1 transgene promoter in testes from E16.5 Rhox10-null mice revealed that this promoter exhibited significantly decreased methylation upon Rhox10 loss. This suggested that one mechanism by which RHOX10 suppresses LINE1 transposition in ProSG is by promoting LINE1 promoter methylation at this stage of germ-cell development.
To identify genes involved in RHOX10-mediated LINE1 suppression, Tan et al. performed RNA sequencing (RNAseq) and ChIP-qPCR analysis of Rhox10-null ProSG in vivo and in vitro, respectively, and found that RHOX10 directly drives the expression of Piwil2, which encodes a key component in the piRNA pathway that protects against TEs.60 Through rescue experiments, they found that RHOX10 inhibits LINE1 transposition through the activation of Piwil2 expression. Thus, the authors identified a Rhox10-Piwil2-LINE1 circuit that serves to provide TE defense in the fetal germline.
In addition to RHOX10, Tan et al. identified several other mouse RHOX family members that also have the ability to suppress LINE1 expression, albeit in vitro.60 Interestingly, many of these other LINE1-suppressing RHOX family members are expressed in different germ-cell developmental stages, for example, the Rhox3 paralogs and Rhox11 are expressed in round spermatids. This raises the possibility that the RHOX gene cluster expanded over evolutionary time as a means to defend the genome against LINE1 elements at different stages of germ cell development. Other Rhox family members are expressed in somatic cells in the reproductive tract, including granulosa cells and Sertoli cells.56,62 Given that LINE1 elements actively transpose in many somatic cells,63,64 this raises the possibility that RHOX TFs expanded over evolutionary time to be expressed in somatic cells in the reproductive tract to defend against TEs.
Tan et al. also found that LINE1 transposition is suppressed by human RHOX family members, indicating that LINE1 suppression is a conserved function of the RHOX protein family. Intriguingly, the ability of human RHOXF2 to suppress LINE1 transposition is lost in mutant forms of RHOXF2 present in infertile human patients. This raises the intriguing possibility that loss of RHOXF2 causes human infertility, in part, because it allows uncontrolled LINE1 transposition in the male germline.
In summary, RHOX10 is a multi-functional homeobox TF that has at least two functions. First, RHOX10 drives the differentiation of ProSG and consequent SSC formation through a molecular mechanism involving at least two downstream genes. Second, RHOX10 defends ProSG against parasitic LINE1 elements in ProSG, and thereby acts as a guardian of the genome. The ability to suppress TE transposition extends to several other RHOX family members, raising the intriguing possibility that the primary impetus for the initial formation and subsequent evolution of this homeobox TF family was to suppress the deleterious effects of TEs.
2.2 |. ZBTB16
ZBTB16 (also known as PLZF) is a member of the POZ and Kruppel zinc finger (POK) protein family. All members of this TF family contain both a Kruppel-like zinc finger and a BTB/POZ domain. The former binds to specific DNA sequences as well as specific proteins, and the latter is an evolutionarily conserved protein-protein-interaction domain that promotes the dimerization of POK family members.
Two studies demonstrated that ZBTB16 has an essential role in SSCs.65,66 Costoya et al. showed that Zbtb16-null mice undergo a progressive loss of SG with age, increased germ cell apoptosis, and the subsequent appearance of “Sertoli cell-only syndrome” seminiferous tubules.65 The appearance of seminiferous tubules lacking all germ cells raised the possibility that Zbtb16-null mice have an SSC defect. This was confirmed by germ-cell transplantation analysis, which showed that germ cells from these mutant mice are unable to repopulate germ cell-depleted recipient testis.65 Buaas et al. came to the same conclusion, based on their analysis of a naturally occurring mouse mutant—luxoid (lu)—that lacks the Zbtb16 gene. These authors obtained several lines of evidence that adult homozygous lu mutant mice have aSSC defect: (i) they largely lack primitive SG in their seminiferous tubules, (ii) some tubules completely lack germ cells, and (iii) germ-cell transplantation analysis showed that lu homozygous mutant germ cells were not able to successfully colonize recipient testes.66 Together, these two studies provide strong evidence that ZBTB16 is essential for SSC maintenance in vivo.
Subsequently, Puszyk et al. discovered that ZBTB16 has a second role in male germ cells – it promotes the silencing of LINE1 retrotransposons.67 These investigators generated a loss-of-function Zbtb16 mutant—which they called “PLZFOFF”—that encodes a form of ZBTB16 that lacks DNA-binding activity. They found that these PLZFOFF mice recapitulate both the testicular phenotype of Zbtb16-null mice65 and the biallelic loss phenotype (PLZF−/−) in humans.68 Analysis of testes and bone marrow from these PLZFOFF mice revealed that many LINE1 elements were upregulated. This was accompanied by hypomethylation of LINE1 promoters, suggesting that ZBTB16 suppresses LINE1 elements by promoting their methylation. MeDIPseq analysis demonstrated that the hypomethylation of TE elements triggered as a result of loss of ZBTB16 was pervasive in both testes and bone marrow. Not only did this methylation de-regulation impact LINE1 elements, but also other interspersed repetitive elements, including SINEs. However, it was not determined whether interspersed repetitive elements besides LINE1 elements were elevated in expression in Zbtb16-null mice. Interestingly, coupled hypomethylation and transcriptional upregulation were not restricted to repetitive elements, as well-established developmental and cell-cycle regulators (such as c-Kit, CrabpI, and Myc) also underwent this kind of regulation in PLZFOFF mice. This dysregulation may have a causal role in the SSC defects caused by ZBTB16 loss.
Given that ZBTB16 is a TF, one might expect that it would repress LINE1 transcription by binding to LINE1 promoters. However, ChIPseq analysis by Puszyk et al. demonstrated that rather than occupying LINE1 promoters, ZBTB16 occupies a subregion of the LINE1 open reading frame (ORF) 2.67 This occupancy was associated with increased DNA methylation and decreased histone H3 acetylation level at the 5’ UTR regulatory region of LINE1 elements. This suggests (but does not prove) that ZBTB16 occupancy of the LINE1 coding region serves to suppress LINE1 expression by a mechanism that somehow methylates LINE1 promoter elements. As evidence of functionality, the PLZF-binding sites in the LINE1 ORF2 region were found to be conserved in mice and humans.67 In the future, it will be intriguing to dissect how the binding of ZBTB16 in LINE1 coding regions drives downstream events in LINE1 promoters to ultimately suppress LINE1 transcription.
Cellular stress has previously been implicated in unleashing TEs to undergo transposition.69 This led Puszyk et al. to wonder if ZBTB16 could have a role in stress-induced LINE1 activation. In support of this hypothesis, a previous study showed that cellular stress (in the form of the cytokine IL-3) elicits the re-localization of ZBTB16 from the nucleus to the cytoplasm of human CD34+ CD71− myeloid progenitors.70 Puszyk et al. replicated this finding in the human hematopoietic cell line, KG1a, exposed to heat stress. Given that ZBTB16 can only directly regulate transcription in the nucleus, such a nuclear-to-cytoplasm localization shift would be predicted to unleash LINE1 elements and allow their transposition, which Puszyk et al. found to be the case. This was further validated using a LINE1 reporter in HEK293T cells.67 Together, these data support a model in which nuclear ZBTB16 is recruited to LINE1 elements to promote their methylation, transcriptional repression, and impaired transposition.
In summary, ZBTB16 is an intriguing multi-functional TF that has critical roles in SSC maintenance and TE defense. In the future, it will be critical to dissect its exact role(s) in SSC maintenance (e.g., SSC self-renewal vs. inhibition of SSC differentiation). It will also be important to determine whether it drives related processes, such as the initial formation of SSCs perinatally. In support, ZBTB16 was recently shown to act downstream of RHOX10 to drive ProSG differentiation and SSC establishment in vitro.49 With regard to ZBTB16’s LINE1 defense role, it will be interesting to determine what stages of germ cell development are impacted and whether its LINE1 defense role is conserved in humans. Finally, it will be important to determine the molecular mechanisms employed by ZBTB16 to suppress LINE1 elements. In the Perspective, below, we discuss the possibility that ZBTB16 acts downstream of RHOX10 to drive LINE1 defense.
2.3 |. RB1
RB1 (also known as simply “RB”) is a transcriptional co-receptor that serves to inhibit the cell proliferation of a wide array of cell types.71 It performs this inhibitory role through its interaction with members of the E2F family of transcription factors.72
To determine whether RB1 has a role in the proliferative block that ProSG undergoes when they transition between the M- and T1-ProSG stage (see Introduction), Spiller etal. made use of global Rb1-null mice.73 Given that these mice suffer from embryonic lethality at ~E14.5, these authors addressed the role of RB1 using ex vivo organ cultures of testicular cells from Rb1-null mouse fetuses.73 They elected to generate these organ cultures from E14.5 embryos, as this is the time point when proliferative quiescence normally initiates in ProSG; that is, the M-ProG/T1-ProSG transition. Using the proliferation markers MKI67 and pHH3, as well as bromodeoxyuridine incorporation, they observed that wt E14.5 ProSG exhibited little proliferation in these organ cultures, as expected. In contrast, E14.5 Rb1-null ProSG were actively proliferating, indicative of loss of the G1/G0 arrest that normally occurs in T1-ProSG. In an attempt to simulate ProSG reaching the fully quiescent state (which normally occurs at ~E16.5 in vivo), the authors also cultured these organoids for 2 days. They found that this two-day culture caused the proliferative quiescence phenotype of the mutant ProSG to be lost. One interpretation of this finding is that RB1 only functions to promote the initial period of mitotic arrest in T1-ProSG. Another possibility is that this in vitro study failed to fully capture RB1’s functions in vivo.
To determine the role of RB1 in germ cells in vivo, a subsequent study—Du et al.—conditionally knocked out Rb1 at the stage proceeding to the ProSG stage – in PGCs (using Blimp1-Cre mice). They found that these Rb1-cKOBlimp1 mice have normal germ cell number and germ cell proliferation at the M-ProSG stage (at E13.5).74 However, at the beginning of the T1-ProSG stage (E14.5), when proliferation normally begins to taper, only 14% of Rb1-cKOBlimp1 ProSG were non-proliferative (as compared to 41% of wt control germ cells, based on the proliferation marker MKI67). The proliferative quiescence defect was even more striking in T1-ProSG at E16.5: only 35% of Rb1-cKOBlimp1 ProSG were MKI67− as compared with 98% MKI67− ProSG in control mice. Du et al. also found that a 6-fold higher proportion of Rb1-cKOBlimp1 ProSG undergoes apoptosis than control ProSG (at E16.5, but not at E14.5). Thus, RB1 not only promotes mitotic arrest of ProSG but also promotes their survival.
ProSG gives rise to two types of SG in rodents. One type is SSCs, the testicular stem cells that permit long-term spermatogenesis. The other type is differentiating SG which gives rise to the first wave of spermatogenesis. To investigate the role of RB1 in this bifurcation of the spermatogenic pathway, Yang et al. conditionally knocked out Rb1 in mouse ProSG (by mating Ddx4-Cre mice with Rb1-floxed mice).75 Yang et al. found these cKO mice are able to undergo the first wave of spermatogenesis, but not subsequent SSC-dependent spermatogenesis, as indicated by a progressive germ cell-loss phenotype.
It remains to be determined why RB1 is required for SSC-dependent spermatogenesis. One possibility is ProSG must undergo a proliferative quiescence “priming stage” before they can form SSCs. Another possibility is that RB1 drives SSC-dependent spermatogenesis by a mechanism independent of its mitotic arrest-promoting role. For example, it might regulate the transcription of specific genes (through its known role as a transcriptional co-receptor) in ProSG to permit SSC generation, or alternatively, in SSCs, to promote SSC maintenance. Thus, a fundamental question is: does RB1 only function in ProSG or does it also have functions after the ProSG stage?
To directly investigate whether RB1 functions after the ProSG stage, Yang et al.75 conditionally knocked out Rb1 at the spermatogonial progenitor stage (by mating with Neurog3- and Stra8-Cre mice). They found that Rb1-cKONeurog3 and Rb1-cKOStra8 male mice are fertile but have modest but significantly reduced testes weight (by 9% and 21% compared to controls, respectively). The seminiferous tubules in these mutant mice had heterogenous morphology; some with disrupted spermatogenesis, others with normal spermatogenesis. Evidence suggests that RB1 functions in SSCs. For example, Yang et al. found that RB1 knockdown in undifferentiated SG cell cultures reduced SSC frequency, as assessed by the germ-cell transplantation analysis. In further support of a role in SSCs, Hu et al.76 conditionally knocked out Rb1 in ProSG (using Ddx4-Cre mice) and found that these Rb1-cKO male mice become sterile by ~2 months of age. Histological analysis showed that these Rb1-cKO mice displayed a striking depletion of early spermatogenic cell types by P28, consistent with an SSC defect. This interpretation was verified by analysis of Rb1-cKO mice between P5 and P28. GFRα1+ Asingle SG was present at a normal density at P5 but thereafter declined dramatically in Rb1-cKO testes. These authors found the loss of GFRα1+ Asingle SG in Rb1-cKO testes is not due to their inhibited proliferation or increased apoptosis. Instead, these cells lacked the capacity for self-renewal, based on BrdU-tracing analysis of the mitotic progeny of Rb1-mutant Asingle SG.
In addition to its developmental roles in germ cells, Montoya et al. obtained evidence that RB1 functions in TE defense. First, Montoya et al. found that molecular complexes composed of RB1 and E2F transcription factors interact with LINE1 elements.77 Second, Montoya et al. found that triple-KO fibroblasts harboring mutations in Rb1 and its orthologs, p107 and p130, have reduced heterochromatic marks (e.g., H3K9me3) at LINE1 elements, accompanied by increased LINE1 expression.77 Third, RB1 is known to cooperatively regulate several genes in conjunction with ZBTB16,78 which, as described in the “ZBTB16” section above, is known to transcriptionally silence LINE1 elements and inhibit their transposition in germ cells.67 This raises the intriguing possibility that RB1 and ZBTB16 cooperate to defend against LINE1 elements, but this remains untested (see Perspective). Also, untested is whether RB1 provides transposon defense in germ cells. At present, the only evidence that RB1 functions in transposon defense come from experiments in somatic cells.77
In summary, RB1 is a transcriptional co-receptor that drives one of the hallmark features of ProSG—their mitotic silencing—and is also critical for long-term spermatogenesis. RB1 is also essential at a later stage of germ-cell development—in undifferentiated SG—where it functions to allow for normal spermatogenesis, but its precise role in undifferentiated SG is not yet clear. There is tantalizing evidence that RB1 is a bi-functional factor that not only has developmental roles but also functions in LINE1 defense, but whether it performs this role in germ cells remains to be tested.
2.4 |. GLIS3
GLIS Family Zinc Finger 3 (GLIS3) is a nuclear protein harboring 5 C2H2-type zinc finger domains that serve as both a transcriptional activator and repressor.79,80 In the male reproductive tract, GLIS3 is transiently expressed in PGCs and then re-expressed in ProSG. GLIS3 expression persists until the undifferentiated SG stage and then is downregulated before the differentiating SG stage postnatally.81,82 Two studies have examined the function of GLIS3 in germ cells.
Kang et al. conducted their studies on Glis3-hypomorphic male mice.81 They observed that these mutant mice have reduced numbers of germ cells (TRA98+ cells) relative to control wt mice at P1; a defect that persists at P4 and then becomes more severe by P7. While an obvious possible mechanism for this reduction in germ cell number would be either increased germ cell apoptosis or impaired germ cell proliferation, Kang et al. found no evidence for either of these mechanisms, implying instead that germ cells in Glis3-hypomorphic mice have a germ-cell progression defect. In support, P7 Glis3-hypomorphic mice were found to have an excess of germ cells expressing cytoplasmic FOXO1, a ProSG marker,83 and fewer germ cells expressing nuclear FOXO1, an SSC marker.83 This raised the possibility that Glis3-hypomorphic mice have an SSC establishment defect, which was supported by the finding that Glis3-hypomorphic mice exhibit low expression of many undifferentiated SG-enriched genes at P7. This defect persists even at P28, indicative of a developmental block, not a developmental delay. Together, these data suggest that GLIS3 promotes the ProSG-to-SSC conversion step, but more work needs to be done to confirm this.
A more recent study—Ungewitter et al.—examined a strain of Glis3-mutant mice with a null mutation in Glis382. Their analysis revealed that these Glis3-null mice have a ~40% reduction in ProSG at E15.5, implying that GLIS3 not only functions in late-stage (T2) ProSG (above) but also in early-stage (T1) ProSG. These authors did not observe increased ProSG apoptosis in Glis3-null mice, suggesting that the ProSG defect in these KO mice is instead due to decreased ProSG proliferation or impaired ProSG progression. In support of the latter, they found that critical differentiation genes, including Nanos2, Nanos3, Stra8, and Rec8, are dis-regulated in E14.5 Glis3-null mice testes. Furthermore, GLIS3 has been implicated in the differentiation of other cell lineages.84,85
Intriguingly, Ungewitter et al. also found that Glis3-null mice exhibit upregulation of a large battery of TE defense genes. RNAseq analysis revealed that all three major retrotransposon classes—LINE, SINE, and LTR (ERV)—exhibited upregulated expression in Glis3-null E14.5 testes. This suggests that GLIS3 is involved in broad TE silencing in ProSG. With regard to mechanism, these authors found that several genes involved in piRNA biosynthesis, catabolism, and metabolism are downregulated in Glis3-null mice, raising the possibility that GLIS3 suppresses TEs by positively regulating the piRNA pathway. Downregulated piRNA pathway genes include Ddx4, Piwil1, Piwil2, Piwil4, Mael, Tdrd1, Tdrd5, and Tdrd9. Other downregulated genes include Dnmt3l and Morc1, the former of which promote TE methylation and subsequent transcriptional repression; and the latter of which encodes a retrotransposon silencer.86 As additional evidence that GLIS3 serves as a TE defense factor, the authors found that increasing GLIS3 expression at E12.5 (using a Rosa26-driven Glis3 mouse line crossed with Dppa3-creER) increases the expression of several TE defense genes, including Dnm3tl, Piwil1, and Piwil4. Together, these data support the possibility that GLIS3 is a key TF that suppresses TEs by broadly transcriptionally activates several TE defense pathways in ProSG and perhaps later stages.
In summary, GLIS3 appears to be a multi-functional TF involved in both germ cell development and TE defense. GLIS3 is essential for normal T1-ProSG accumulation. Some evidence suggests it promotes the T2-ProSG-to-SSC conversion step. Its loss causes widespread activation of TEs in ProSG, accompanied by repressed piRNA pathway functions, strongly suggesting that GLIS3 serves to defend the male germline against TEs.
2.5 |. SETDB1
SETDB1 (also known as KMT1E or ESET) is a histone lysine N-methyltransferase responsible for the di- and tri-methylation of H3K9. Consistent with the well-established role of methylated H3K9 in transcriptional repression, SETDB1 has been found to promote gene silencing through heterochromatin formation in a wide variety of cell lineages.87
Given that SETDB1 catalyzes the formation of a key repressive histone mark, Mochizuki et al. hypothesized that this methyltransferase has functions in the early embryo.88 To test this hypothesis, these investigators conditionally knocked out Setdb1 in the epiblast (by breeding Setdb1-floxed mice with Sox2-Cre mice). They found that these conditional (c) KO mice exhibited a drastic reduction in nascent PGCs, indicating that SETDB1 is critical for PGC formation.
To test whether SETDB1 has roles in germ cells after the PGC formation stage, Liu et al. conditionally knocked out Setdb1 in migrating PGCs (by breeding Setdb1-floxed mice with Tnap-Cre mice, which express CRE as early as E9.5).89 They found that these cKO mice had reduced M-ProSG number (at E13.5), reduced postnatal gonad size (at P10), and reduced testis size (examined at 5 weeks and in adults) compared to wt littermate mice. Histological analysis of P10 and P35 testes showed that many seminiferous tubules in Setdb1-cKO mice are devoid of germ cells. While it is uncertain why not all seminiferous tubules in these cKO mice have this phenotype, it is likely to be due to incomplete CRE-mediated recombination, as the authors did not observe transmission of the deleted allele in offspring. They found that neither proliferation nor apoptosis genes were broadly dysregulated, suggesting that SETDB1 does not act in ProSG by driving their proliferation or survival. This implied that, instead, SETDB1 acts by promoting ProSG progression, but this requires further experimentation verification.
By what molecular mechanism does SETDB1 act in ProSG? Past studies have shown that SETDB1 catalyzes the formation of the repressive histone marks, H3K9me2 and H3K9me3, in other cell lineages.90 Thus, a likely mechanism is SETDB1 does the same in the germline lineage. In support, Mochizuki et al. and Liu et al. found that cKO mice conditionally lacking functional Setdb1 in developing PGCs and ProSG, respectively, have decreased H3K9me3 occupancy and an overall increase in gene expression.89,88 SETDB1 was found to also increase the level of another repressive histone mark—H3K37me3—in the germline,89 probably through an indirect mechanism, as it is known that SETDB1 modulates PRC2, a regulator of H3K27me3 level.91
Mochizuki et al. pinpointed three SETDB1-regulated genes as key regulators of PGC formation.88 These three genes—Dppa2, Otx2, and Utf1—were found to be involved in the BMP signaling events required to drive PGC formation. Through gain- and loss-of-function experiments in cell aggregates containing PGC-like cells, Mochizuki et al. found that SETDB1 negatively regulates and occupies the flanking regions of these three genes. They then showed that these three genes serve to negatively regulate BMP signaling. Together, the data support a model in which SETDB1 promotes PGC formation by repressing the transcription of three BMP signaling regulators, which leads to positive BMP signaling, a key event in PGC generation.
Not only does SETDB1 serve as a developmental regulator of PGCs, but it may regulate self-renewal vs. differentiation decisions in undifferentiated SG. An et al. found that the knockdown of SETDB1 in undifferentiated SG cultures reduces the frequency of SSCs in these cultures, as determined by germ-cell transplantation analysis.92 This strongly suggests that SETDB1 promotes SSC maintenance. While the molecular mechanism for this function is not known, a tantalizing possibility is that SETDB1 silences SG differentiation genes to allow for SSC self-renewal.
Together, the data from Mochizuki et al.,88 Liu et al.,89 and An et al.92 support the possibility that SETDB1 has many developmental roles in germ cells, including promoting the initial formation of PGCs, their progression in the embryo, as well as SSC maintenance after birth.
Intriguingly, Liu et al. found that SETDB1 not only has developmental roles, but it silences retrotransposons89. These authors found that ablation of Setdb1 decreases H3K9me3 occupancy at widespread ERV loci (including the ERVK and ERV1 subfamilies), accompanied by upregulation of their expression in ProSG. The repressive histone mark, H3K27me3, was also present at increased levels at ERV loci, presumably by an indirect mechanism (see above). LINE1 elements also had decreased H3K9me3 and H3K27me3 occupancy, but most LINE1 elements exhibited little or no increased expression. A possible explanation for the lack of LINE1 activation is epigenetic compensation. In support, most LINE1 elements in Setdb1-cKO mice were found to exhibit increased DNA methylation. This hypermethylation response was widespread, as Setdb1-cKO ProSG exhibited a ~2.5-fold increase in DNA methylation globally. In contrast, ERV family members typically avoided this putative counter-regulatory mechanism. Indeed, Liu et al. found that the ERV family members exhibiting the highest level of H3K9me3 in wt mice and strongest transcriptional induction in Setdb1-cKO mice (e.g., IAPLTR1 and IAPLTR1a), exhibited decreased DNA methylation.
In summary, SETDB1 is a chromatin regulator involved in both germ cell development and TE defense. Evidence suggests it has roles in PGC formation, ProSG progression, and SSC maintenance. In ProSG, SETDB1 serves to defend the genome specifically against ERV TEs.
2.6 |. MYBL1
MYBL1 (also called A-MYB) is a TF that binds to DNA through a helix-loop-helix domain. This TF is part of a small protein family that includes two other family members - MYB (also called C-MYB) and MYBL2 (also called B-MYB).
MYBL1 is unique among these three family members in being highly expressed in male germ cells, raising the possibility it has roles in spermatogenesis. Indeed, Toscani et al. showed that Mybl1-null mice have meiotic arrest at the early pachytene stage, and generate no spermatozoa.93 The morphology of the seminiferous tubules in Mybl1-null mice is abnormal, including vacuolization of the Sertoli cell cytoplasm. While SG and pre-leptotene spermatocytes are normal, most pachytene primary spermatocytes exhibit degeneration, thereby pinpointing the defect between the pre-leptotene to the pachytene stage. This was confirmed by the lack of detectable expression of the pachytene markers PGK-2 and HSP70-2, both of which begin expression at the leptotene stage. Post-meiotic germ-cell stages (i.e., spermatids and spermatozoa) were completely absent, indicative of a complete block.
As another approach to discern the function of MYBL1, Bolcun-Filas et al. examined mice harboring a missense mutation in Mybl1 (derived from a large-scale mutagenesis genetic screen to identify infertile mice). These Mybl1repro9 mutant mice were found to have subtle defects in autosomal synapsis at the pachytene stage, a high incidence of un-synapsed sex chromosomes, incomplete double-strand break repair on synapsed pachytene chromosomes, and little DNA crossing over.94 As further evidence of a pachytene defect, microarray analysis of whole testes from P14 and P17 Mybl1repro9 mutant and wt mice revealed that meiotic genes were preferentially misregulated in KO testes. Using ChIP-chip analysis, putative direct targets of MYBL1 in pachytene spermatocytes were identified, including genes encoding proteins known to be involved in the processes found to be a defect in these Mybl1repro9 mutant mice, including double-stranded DNA repair, synapsis, crossing over, and pachytene cell-cycle progression. These genes encode proteins that are candidates to act downstream of MYBL1 in meiosis.
Together, the results from Bolcun-Filas et al.94 and Toscani et al.93 suggested that MYBL1 is a master transcriptional regulator that controls several different events necessary for the progression of germ cells through the pachytene stage of meiosis.
A subsequent study—Li et al.—found that MYBL1 is critical for another function during meiosis - pachytene piRNA production.95 This study found that MYBL1 drives the production of two classes of RNAs critical for piRNA biosynthesis: (i) piRNA precursors and (ii) mRNAs encoding core piRNA biogenesis factors, including PIWIL1. MYBL1 was found to also promote the expression of several other RNA-silencing-pathway genes, including Tdrd1, Tdrd5, Tdrd6, Tdrd12, Pld6, and Mael. MYBL1 is likely to directly regulate all these genes, as it was found to occupy their promoter regions.
The finding that MYBL1 promotes the transcription of both piRNA genes and genes encoding piRNA biogenesis proteins led Li et al. to propose that MYBL1 operates in a coherent feedforward loop that ensures robust accumulation of pachytene piRNAs.95 Such feedforward loops are known to amplify initiating signals to increase target gene expression. They also serve to act as switches that respond to sustained signals but ignore transient signals. The notion that MYBL1 is regulated by a feedforward loop is also supported by the finding that MYBL1 positively autoregulates its own expression.95
Recently, another study—Maezawa et al.96—obtained evidence that MYBL1 drives piRNA biogenesis through the action of super-enhancers (SEs) – enhancer clusters that drive unusually high levels of transcription97. Maezawa et al. defined a SE as any extensive chromatin region enriched for acetylated [ac] H3K27, which is generally regarded as an enhancer mark. Among the SEs this study identified were “meiotic SEs,” based on their being present in pachytene spermatocytes, not earlier or later stages. The TF most enriched within H3K27ac peaks at meiotic SEs was MYBL1. As evidence that MYBL1 is functionally important at these meiotic SEs, Mybl1-null pachytene spermatocyte chromosome spreads had dramatically reduced H3K27ac signal as compared to wt pachytene spermatocyte chromosome spreads. Intriguingly, many of the 518 MYBL1-bound meiotic SEs overlapped with pachytene piRNA gene clusters and piRNA biogenesis genes, suggesting that the expression of these piRNA-related genes is driven by SEs through the action of MYBL1. Together, the data from Maezawa et al.96 suggests that MYBL1 drives the formation of meiotic SEs that, in turn, drive the high transcription of pachytene piRNA pre-mRNA genes and piRNA biogenesis factor genes. It remains to be determined what proportion of piRNAs depend on this MYBL1/SE-dependent mechanism for their expression during meiosis.
In summary, MYBL1 appears to be a master TF critical for several events during the pachytene stage of meiosis, including (i) sex-chromosome synapsis, (ii) double-strand DNA break repair on synapsed pachytene chromosomes, (iii) DNA crossing over, (iv) transcriptional induction of piRNA precursor genes during meiosis, and (v) transcriptional induction of genes encoding key components of the pachytene piRNA pathway as well as other RNA-silencing pathways. Given that a proportion of pachytene piRNAs are derived from TEs,98,99 the latter two functions raise the possibility that MYBL1 provides defense against TEs during the process of meiosis. However, there is, as of yet, no direct evidence for this.
3 |. PERSPECTIVE
In this review, we make the case that some transcriptional regulators have the unique ability to both regulate developmental events in germ cells and protect these cells from deleterious TEs. Below are some questions about these “bi-functional” factors to consider for the future.
First, might some of the putative bi-functional factors described in this review work together?
One likely example of this is the TFs RHOX10 and ZBTB16, as RHOX10 was recently shown-through rescue experiments-to act upstream of ZBTB16 to drive ProSG differentiation and SSC establishment in vitro.49 While it remains to be determined whether this molecular circuit operates in vivo, it is known that ZBTB16 is expressed in ProSG in vivo65 and RHOX10 drives ProSG differentiation and the initial formation of SSCs in vivo.49,53 It will also be intriguing to determine whether ZBTB16 also acts downstream of RHOX10 to provide LINE1 defense. A third factor that may also be involved is the mitotic silencing factor RB1. Evidence suggests that RB1 silences LINE1 elements,77 and RB1 is known to cooperatively regulate several genes in conjunction with ZBTB16.78 Thus, it is conceivable that RB1, ZBTB16, and RHOX10 all function together in a molecular circuit that confers defense against TEs.
Second, did the bi-functional factors described in this review originally have only one activity and then acquired a second activity over time?
For example, were they originally developmental regulators and then acquired a TE defense role later? Or were these factors originally TE defense factors that later acquired other roles, such as the ability to influence germ cell development and differentiation? Functional analysis of these factors in different species has the potential to provide an answer to this question.
Third, do the developmental defects in organisms lacking these factors result from TE-induced defects?
We think this is unlikely to be true for RHOX10 for at least two reasons. First, the molecular mechanisms by which RHOX10 acts in development and TE defense have been partially defined, and they are different. RHOX10 drives ProSG diff through the TFs DMRT1 and ZBTB16,49 while it suppresses LINE1 transposition through the RNA-silencing factor PIWIL2.60 Second, RHOX10 acts on these two events at different developmental time points: its LINE1 defense role is exerted at fetal stages, while its ProSG differentiation role is exerted perinatally.49,60 In the case of the other factors we have discussed, it is possible, in some cases, that loss of their TE defense role leads to the defects observed. For example, the reduction in ProSG number in Glis3-null mice and Setdb1-null mice could be due to the activation of LINE1 elements or other TEs in these mutant mice. In support, it is known that elevated expression of LINE1 ORFs can cause deleterious endonuclease and reverse transcriptase activities.100–102 In particular, overly active LINE1s are known to cause DNA damage accumulation, checkpoint activation, and cell death.101,102
Fourth, might some of these factors regulate developmental events by acting on repetitive elements that have been integrated into developmental genes over evolutionary time?
This possibility is supported by the finding that one-quarter of expressed genes contain a LINE1 retrotransposon in their 3’UTR or in their introns.103 Thus, perhaps some of the factors we have described in this review regulate developmental genes by binding to former TEs that have been incorporated into the regulatory circuitry of these developmental genes. If this was the case, such factors would regulate development and promote TE defense by a common mechanism – by binding to repetitive elements.
Fifth, what is the breadth of TE defense provided by the bi-functional factors described in this review?
Might the factors we described here function in cell stages or types beyond those demonstrated in the studies to date? For example, since evidence suggests that ZBTB16, GLIS3, and RB1 have developmental roles in SSCs,65,66,75,81 these factors might also have TE defense roles in SSCs. A related question is – what is the breadth of TEs impacted by the factors discussed here? For example, ZBTB16 was found to suppress LINE1 element expression,67 but other TEs and repetitive elements were not examined. Given that Zbtb16-mutant mice not only exhibit hypomethylation of LINE1 elements but also SINEs, this raises the possibility that SINE activity is also suppressed by ZBTB16. In some cases, the co-regulation could be causal. As a case in point, LINE1 elements are known to facilitate the transposition of at least two other subtypes of non-LTR retrotransposons: SINES and SVAs.104–106
The field has just begun to uncover how developmental decisions and “selfish DNA” defense might be linked. It is only natural that evolutionary forces might select proteins that function in both processes.
ACKNOWLEDGMENTS
We apologize to those researchers whose work could not be discussed, as well as to all the authors whose original work could not be cited due to space limitations. This work was supported by the National Institutes of Health [grant number R01 GM119128 to Kun Tan and Miles F. Wilkinson].
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
REFERENCES
- 1.Saffman EE, Lasko P. Germline development in vertebrates and invertebrates. Cell Mol Life Sci. 1999;55(8-9):1141–1163. 10.1007/s000180050363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gomperts M, Garcia-Castro M, Wylie C, Heasman J. Interactions between primordial germ cells play a role in their migration in mouse embryos. Development. 1994;120(1):135–141. 10.1242/dev.120.1.135 [DOI] [PubMed] [Google Scholar]
- 3.Tan K, Wilkinson MF. A single-cell view of spermatogonial stem cells. Curr Opin Cell Biol. 2020;67:71–78. 10.1016/j.ceb.2020.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tan K, Wilkinson MF. Human spermatogonial stem cells scrutinized under the single-cell magnifying glass. Cell Stem Cell. 2019;24(2):201–203. 10.1016/j.stem.2019.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meng X, Lindahl M, Hyvonen ME, et al. Regulation of cell fate decision of undifferentiated spermatogonia by GDNF. Science. 2000;287(5457):1489–93. 10.1126/science.287.5457.1489 [DOI] [PubMed] [Google Scholar]
- 6.Zhang T, Oatley J, Bardwell VJ, Zarkower D. DMRT1 Is required for mouse spermatogonial stem cell maintenance and replenishment. PLoS Genet. 2016;12(9):e1006293. 10.1371/journal.pgen.1006293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hobbs RM, Seandel M, Falciatori I, Rafii S, Pandolfi PP. Plzf regulates germline progenitor self-renewal by opposing mTORC1. Cell. 2010;142(3):468–479. 10.1016/j.cell.2010.06.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lovasco LA, Gustafson EA, Seymour KA, de Rooij DG, Freiman RN. TAF4b is required for mouse spermatogonial stem cell development. Stem Cells. 2015;33(4):1267–1276. 10.1002/stem.1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goertz MJ, Wu Z, Gallardo TD, Hamra FK, Castrillon DH. Foxo1 is required in mouse spermatogonial stem cells for their maintenance and the initiation of spermatogenesis. J Clin Invest. 2011;121(9):3456–3466. 10.1172/JCI57984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanatsu-Shinohara M, Tanaka T, Ogonuki N, et al. Myc/Mycn-mediated glycolysis enhances mouse spermatogonial stem cell self-renewal. Genes Dev. 2016;30(23):2637–2648. 10.1101/gad.287045.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suzuki H, Ahn HW, Chu T, et al. SOHLH1 and SOHLH2 coordinate spermatogonial differentiation. Dev Biol. 2012;361(2):301–312. 10.1016/j.ydbio.2011.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matson CK, Murphy MW, Griswold MD, Yoshida S, Bardwell VJ, Zarkower D. The mammalian doublesex homolog DMRT1 is a transcriptional gatekeeper that controls the mitosis versus meiosis decision in male germ cells. Dev Cell. 2010;19(4):612–624. 10.1016/j.devcel.2010.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laronda MM, Jameson JL. Sox3 functions in a cell-autonomous manner to regulate spermatogonial differentiation in mice. Endocrinology. 2011;152(4):1606–15. 10.1210/en.2010-1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Britten RJ, Kohne DE. Repeated sequences in DNA. Hundreds of thousands of copies of DNA sequences have been incorporated into the genomes of higher organisms. Science. 1968;161(3841):529–40. 10.1126/science.161.3841.529 [DOI] [PubMed] [Google Scholar]
- 15.Platt RN 2nd, Vandewege MW, Ray DA. Mammalian transposable elements and their impacts on genome evolution. Chromosome Res. 2018;26(1-2):25–43. 10.1007/s10577-017-9570-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Payer LM, Burns KH. Transposable elements in human genetic disease. Nat Rev Genet. 2019;20(12):760–772. 10.1038/s41576-019-0165-8 [DOI] [PubMed] [Google Scholar]
- 17.Hancks DC, Kazazian HH Jr. Roles for retrotransposon insertions in human disease. Mob DNA. 2016;7:9. 10.1186/s13100-016-0065-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wells JN, Feschotte C. A field guide to eukaryotic transposable elements. Annu Rev Genet. 2020;54:539–561. 10.1146/annurev-genet-040620-022145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stocking C, Kozak CA. Murine endogenous retroviruses. Cell Mol Life Sci. 2008;65(21):3383–98. 10.1007/s00018-008-8497-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007;41:331–68. 10.1146/annurev.genet.40.110405.090448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deniz O, Frost JM, Branco MR. Regulation of transposable elements by DNA modifications. Nat Rev Genet. 2019;20(7):417–431. 10.1038/s41576-019-0106-6 [DOI] [PubMed] [Google Scholar]
- 22.Greenberg MVC, Bourc’his D. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 2019;20(10):590–607. 10.1038/s41580-019-0159-6 [DOI] [PubMed] [Google Scholar]
- 23.Kato Y, Kaneda M, Hata K, et al. Role of the Dnmt3 family in de novo methylation of imprinted and repetitive sequences during male germ cell development in the mouse. Hum Mol Genet. 2007;16(19):2272–80. 10.1093/hmg/ddm179 [DOI] [PubMed] [Google Scholar]
- 24.Chen T, Li E. Establishment and maintenance of DNA methylation patterns in mammals. Curr Top Microbiol Immunol. 2006;301:179–201. 10.1007/3-540-31390-7_6 [DOI] [PubMed] [Google Scholar]
- 25.Bourc’his D, Bestor TH. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature. 2004;431(7004):96–9. 10.1038/nature02886 [DOI] [PubMed] [Google Scholar]
- 26.Hata K, Kusumi M, Yokomine T, Li E, Sasaki H. Meiotic and epigenetic aberrations in Dnmt3L-deficient male germ cells. Mol Reprod Dev. 2006;73(1):116–22. 10.1002/mrd.20387 [DOI] [PubMed] [Google Scholar]
- 27.Smallwood SA, Kelsey G. De novo DNA methylation: a germ cell perspective. Trends Genet. 2012;28(1):33–42. 10.1016/j.tig.2011.09.004 [DOI] [PubMed] [Google Scholar]
- 28.McCarrey JR. Toward a more precise and informative nomenclature describing fetal and neonatal male germ cells in rodents. Biol Reprod. 2013;89(2):47. 10.1095/biolreprod.113.110502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim S, Gunesdogan U, Zylicz JJ, et al. PRMT5 protects genomic integrity during global DNA demethylation in primordial germ cells and preimplantation embryos. Mol Cell. 2014;56(4):564–79. 10.1016/j.molcel.2014.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Di Giacomo M, Comazzetto S, Sampath SC, Sampath SC, O’Carroll D. G9a co-suppresses LINE1 elements in spermatogonia. Epigenetics Chromatin. 2014;7:24. 10.1186/1756-8935-7-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tachibana M, Nozaki M, Takeda N, Shinkai Y. Functional dynamics of H3K9 methylation during meiotic prophase progression. EMBO J. 2007;26(14):3346–59. 10.1038/sj.emboj.7601767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iwasaki YW,Siomi MC,Siomi H. PIWI-Interacting RNA: Its Biogenesis and Functions. Annu Rev Biochem. 2015;84:405–33. 10.1146/annurev-biochem-060614-034258 [DOI] [PubMed] [Google Scholar]
- 33.Kuramochi-Miyagawa S, Kimura T, Yomogida K, et al. Two mouse piwi-related genes: miwi and mili. Mech Dev. 2001;108(1-2):121–33. 10.1016/s0925-4773(01)00499-3 [DOI] [PubMed] [Google Scholar]
- 34.Kuramochi-Miyagawa S, Watanabe T, Gotoh K, et al. DNA methylation of retrotransposon genes is regulated by Piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev. 2008;22(7):908–17. 10.1101/gad.1640708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kojima-Kita K, Kuramochi-Miyagawa S, Nagamori I, et al. MIWI2 as an Effector of DNA Methylation and Gene Silencing in Embryonic Male Germ Cells. Cell Rep. 2016;16(11):2819–2828. 10.1016/j.celrep.2016.08.027 [DOI] [PubMed] [Google Scholar]
- 36.Fu Q, Wang PJ. Mammalian piRNAs: Biogenesis, function, and mysteries. Spermatogenesis. 2014;4:e27889. 10.4161/spmg.27889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gan H, Lin X, Zhang Z, et al. piRNA profiling during specific stages of mouse spermatogenesis. RNA. 2011;17(7):1191–203. 10.1261/rna.2648411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watanabe T, Lin H. Posttranscriptional regulation of gene expression by Piwi proteins and piRNAs. Mol Cell. 2014;56(1):18–27. 10.1016/j.molcel.2014.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zoch A, Auchynnikava T, Berrens RV, et al. SPOCD1 is an essential executor of piRNA-directed de novo DNA methylation. Nature. 2020;584(7822):635–639. 10.1038/s41586-020-2557-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garcia-Fernandez J. The genesis and evolution of homeobox gene clusters. Nat Rev Genet. 2005;6(12):881–92. 10.1038/nrg1723 [DOI] [PubMed] [Google Scholar]
- 41.Krumlauf R. Hox genes in vertebrate development. Cell. 1994;78(2):191–201. 10.1016/0092-8674(94)90290-9 [DOI] [PubMed] [Google Scholar]
- 42.Maclean JA 2nd, Chen MA, Wayne CM, et al. Rhox: a new homeobox gene cluster. Cell. 2005;120(3):369–82. 10.1016/j.cell.2004.12.022 [DOI] [PubMed] [Google Scholar]
- 43.Jackson M, Watt AJ, Gautier P, et al. A murine specific expansion of the Rhox cluster involved in embryonic stem cell biology is under natural selection. BMC Genomics. 2006;7:212. 10.1186/1471-2164-7-212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morris L, Gordon J, Blackburn CC. Identification of a tandem duplicated array in the Rhox alpha locus on mouse chromosome X. Mamm Genome. 2006;17(2):178–87. 10.1007/s00335-005-0138-4 [DOI] [PubMed] [Google Scholar]
- 45.Wang X, Zhang J. Remarkable expansions of an X-linked reproductive homeobox gene cluster in rodent evolution. Genomics. 2006;88(1):34–43. 10.1016/j.ygeno.2006.02.007 [DOI] [PubMed] [Google Scholar]
- 46.Geyer CB, Eddy EM. Identification and characterization of Rhox13, a novel X-linked mouse homeobox gene. Gene. 2008;423(2):194–200. 10.1016/j.gene.2008.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.MacLean JA 2nd, Wilkinson MF. The Rhox genes. Reproduction. 2010;140(2):195–213. 10.1530/REP-10-0100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.MacLean JA 2nd, Hu Z, Welborn JP, et al. The RHOX homeodomain proteins regulate the expression of insulin and other metabolic regulators in the testis. J Biol Chem. 2013;288(48):34809–25. 10.1074/jbc.M113.486340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tan K, Song HW, Wilkinson MF. RHOX10 drives mouse spermatogonial stem cell establishment through a transcription factor signaling cascade. Cell Rep. 2021;36(3):109423. 10.1016/j.celrep.2021.109423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu Z, Dandekar D, O’Shaughnessy PJ, De Gendt K, Verhoeven G, Wilkinson MF. Androgen-induced Rhox homeobox genes modulate the expression of AR-regulated genes. Mol Endocrinol. 2010;24(1):60–75. 10.1210/me.2009-0303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.MacLean JA 2nd, Hayashi K, Turner TT, Wilkinson MF. The Rhox5 homeobox gene regulates the region-specific expression of its paralogs in the rodent epididymis. Biol Reprod. 2012;86(6):189. 10.1095/biolreprod.112.099184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bhardwaj A, Song HW, Beildeck M, et al. DNA demethylation-dependent AR recruitment and GATA factors drive Rhox5 homeobox gene transcription in the epididymis. Mol Endocrinol. 2012;26(4):538–49. 10.1210/me.2011-1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Song HW, Bettegowda A, Lake BB, et al.The Homeobox Transcription Factor RHOX10 Drives Mouse Spermatogonial Stem Cell Establishment. Cell Rep. 2016;17(1):149–164. 10.1016/jxelrep.2016.08.090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Busada JT, Velte EK, Serra N, et al. Rhox13 is required for a quantitatively normal first wave of spermatogenesis in mice. Reproduction. 2016;152(5):379–88. 10.1530/REP-16-0268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takasaki N, Rankin T, Dean J. Normal gonadal development in mice lacking GPBOX, a homeobox protein expressed in germ cells at the onset of sexual dimorphism. Mol Cell Biol. 2001;21(23):8197–202. 10.1128/MCB.21.23.8197-8202.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Welborn JP, Davis MG, Ebers SD, et al. Rhox8 Ablation in the Sertoli Cells Using a Tissue-Specific RNAi Approach Results in Impaired Male Fertility in Mice. Biol Reprod. 2015;93(1):8. 10.1095/biolreprod.114.124834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Song HW, Bettegowda A, Oliver D, et al. shRNA off-target effects in vivo: impaired endogenous siRNA expression and spermatogenic defects. PLoS One. 2015;10(3):e0118549. 10.1371/journal.pone.0118549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Song HW, Dann CT, McCarrey JR, Meistrich ML, Cornwall GA, Wilkinson MF. Dynamic expression pattern and subcellular localization of the Rhox10 homeobox transcription factor during early germ cell development. Reproduction. 2012;143(5):611–24. 10.1530/REP-11-0479 [DOI] [PubMed] [Google Scholar]
- 59.Tan K, Song HW, Wilkinson MF. Single-cell RNAseq analysis of testicular germ and somatic cell development during the perinatal period. Development. 2020;147(3). 10.1242/dev.183251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tan K, Kim ME, Song HW, et al. The Rhox gene cluster suppresses germline LINE1 transposition. Proc Natl Acad Sci U S A. 2021;118(23). 10.1073/pnas.2024785118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Newkirk SJ, Lee S, Grandi FC, et al. Intact piRNA pathway prevents L1 mobilization in male meiosis. Proc Natl Acad Sci U S A. 2017;114(28):E5635–E5644. 10.1073/pnas.1701069114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.MacLean JA 2nd, Rao MK, Doyle KM, Richards JS, Wilkinson MF. Regulation of the Rhox5 homeobox gene in primary granulosa cells: preovulatory expression and dependence on SP1/SP3 and GABP. Biol Reprod. 2005;73(6):1126–34. 10.1095/biolreprod.105.042747 [DOI] [PubMed] [Google Scholar]
- 63.Coufal NG, Garcia-Perez JL, Peng GE, et al. L1 retrotransposition in human neural progenitor cells. Nature. 2009;460(7259):1127–31. 10.1038/nature08248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee E, Iskow R, Yang L, et al. Landscape of somatic retrotransposition in human cancers. Science. 2012;337(6097):967–71. 10.1126/science.1222077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Costoya JA, Hobbs RM, Barna M, et al. Essential role of Plzf in maintenance of spermatogonial stem cells. Nat Genet. 2004;36(6):653–9. 10.1038/ng1367 [DOI] [PubMed] [Google Scholar]
- 66.Buaas FW, Kirsh AL, Sharma M, et al. Plzf is required in adult male germ cells for stem cell self-renewal. Nat Genet. 2004;36(6):647–52. 10.1038/ng1366 [DOI] [PubMed] [Google Scholar]
- 67.Puszyk W, Down T, Grimwade D, et al. The epigenetic regulator PLZF represses L1 retrotransposition in germ and progenitor cells. EMBO J. 2013;32(13):1941–52. 10.1038/emboj.2013.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fischer S, Kohlhase J, Bohm D, et al. Biallelic loss of function of the promyelocytic leukaemia zinc finger (PLZF) gene causes severe skeletal defects and genital hypoplasia. J Med Genet. 2008;45(11):731–7. 10.1136/jmg.2008.059451 [DOI] [PubMed] [Google Scholar]
- 69.Goodier JL, Kazazian HH Jr. Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell. 2008;135(1):23–35. 10.1016/jxell.2008.09.022 [DOI] [PubMed] [Google Scholar]
- 70.Doulatov S, Notta F, Rice KL, et al. PLZF is a regulator of homeostatic and cytokine-induced myeloid development. Gene Dev. 2009;23(17):2076–2087. 10.1101/gad.1788109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boward B, Wu T, Dalton S. Concise review: control of cell fate through cell cycle and pluripotency networks. Stem Cells. 2016;34(6):1427–36. 10.1002/stem.2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81(3):323–30. 10.1016/0092-8674(95)90385-2 [DOI] [PubMed] [Google Scholar]
- 73.Spiller CM, Wilhelm D, Koopman P. Retinoblastoma 1 protein modulates XY germ cell entry into G1/G0 arrest during fetal development in mice. Biol Reprod. 2010;82(2):433–43. 10.1095/biolreprod.109.078691 [DOI] [PubMed] [Google Scholar]
- 74.Du G, Oatley MJ, Law NC, Robbins C, Wu X, Oatley JM. Proper timing of a quiescence period in precursor prospermatogonia is required for stem cell pool establishment in the male germline. Development. 2021;148(9). 10.1242/dev.194571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang QE, Gwost I, Oatley MJ, Oatley JM. Retinoblastoma protein (RB1) controls fate determination in stem cells and progenitors of the mouse male germline. Biol Reprod. 2013;89(5):113. 10.1095/biolreprod.113.113159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hu YC, de Rooij DG, Page DC. Tumor suppressor gene Rb is required for self-renewal of spermatogonial stem cells in mice. Proc Natl Acad Sci U S A. 2013;110(31):12685–90. 10.1073/pnas.1311548110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Montoya-Durango DE, Liu Y, Teneng I, et al. Epigenetic control of mammalian LINE-1 retrotransposon by retinoblastoma proteins. Mutat Res. 2009;665(1-2):20–8. 10.1016/j.mrfmmm.2009.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Petrie K, Guidez F, Zhu J, et al. Retinoblastoma protein and the leukemia-associated PLZF transcription factor interact to repress target gene promoters. Oncogene. 2008;27(39):5260–6. 10.1038/onc.2008.159 [DOI] [PubMed] [Google Scholar]
- 79.Jetten AM. GLIS1-3 transcription factors: critical roles in the regulation of multiple physiological processes and diseases. Cell Mol Life Sci. 2018;75(19):3473–3494. 10.1007/s00018-018-2841-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jetten AM, Scoville DW, Kang HS. GLIS1-3: Links to primary cilium, reprogramming, stem cell renewal, and disease. Cells. 2022;11(11). 10.3390/cells11111833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kang HS, Chen LY, Lichti-Kaiser K, et al. Transcription factor GLIS3: A new and critical regulator of postnatal stages of mouse spermatogenesis. Stem Cells. 2016;34(11):2772–2783. 10.1002/stem.2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ungewitter EK, Rotgers E, Kang HS, et al. Loss of Glis3 causes dysregulation of retrotransposon silencing and germ cell demise in fetal mouse testis. Sci Rep. 2018;8(1):9662. 10.1038/s41598-018-27843-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Goertz MJ, Wu ZR, Gallardo TD, Hamra FK, Castrillon DH. Foxo1 is required in mouse spermatogonial stem cells for their maintenance and the initiation of spermatogenesis. J Clin Invest. 2011;121(9):3456–3466. 10.1172/Jci57984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kang HS, Kim YS, ZeRuth G, et al. Transcription factor Glis3, a novel critical player in the regulation of pancreatic beta-cell development and insulin gene expression. Mol Cell Biol. 2009;29(24):6366–79. 10.1128/MCB.01259-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Beak JY, Kang HS, Kim YS, Jetten AM. Kruppel-like zinc finger protein Glis3 promotes osteoblast differentiation by regulating FGF18 expression. J Bone Miner Res. 2007;22(8):1234–44. 10.1359/jbmr.070503 [DOI] [PubMed] [Google Scholar]
- 86.Pastor WA, Stroud H, Nee K, et al. MORC1 represses transposable elements in the mouse male germline. Nat Commun. 2014;5:5795. 10.1038/ncomms6795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kang YK. SETDB1 in Early Embryos and Embryonic Stem Cells. Curr Issues Mol Biol. 2015;17:1–10 [PubMed] [Google Scholar]
- 88.Mochizuki K, Tandot Y, Sekinaka T, et al. SETDB1 is essential for mouse primordial germ cell fate determination by ensuring BMP signaling. Development. 2018;145(23). 10.1242/dev.164160 [DOI] [PubMed] [Google Scholar]
- 89.Liu S, Brind’Amour J, Karimi MM,et al.Setdb1 is required for germline development and silencing of H3K9me3-marked endogenous retroviruses in primordial germ cells. Gene Dev. 2014;28(18):2041–2055. 10.1101/gad.244848.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Markouli M, Strepkos D, Piperi C. Structure, Activity and Function of the SETDB1 Protein Methyltransferase. Life (Basel). 2021;11(8). 10.3390/life11080817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fei Q, Yang XQ, Jiang H, et al. SETDB1 modulates PRC2 activity at developmental genes independently of H3K9 trimethylation in mouse ES cells. Genome Res. 2015;25(9):1325–1335. 10.1101/gr.177576.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.An J, Zhang X, Qin J, et al. The histone methyltransferase ESET is required for the survival of spermatogonial stem/progenitor cells in mice. Cell Death Dis. 2014;5. 10.1038/cddis.2014.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Toscani A, Mettus RV, Coupland R, et al. Arrest of spermatogenesis and defective breast development in mice lacking A-myb. Nature. 1997;386(6626):713–7. 10.1038/386713a0 [DOI] [PubMed] [Google Scholar]
- 94.Bolcun-Filas E, Bannister LA, Barash A, et al. A-MYB (MYBL1) transcription factor is a master regulator of male meiosis. Development. 2011;138(15):3319–30. 10.1242/dev.067645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li XZ, Roy CK, Dong X, et al. An ancient transcription factor initiates the burst of piRNA production during early meiosis in mouse testes. Mol Cell. 2013;50(1):67–81. 10.1016/j.molcel.2013.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Maezawa S, Sakashita A, Yukawa M, et al. Super-enhancer switching drives a burst in gene expression at the mitosis-to-meiosis transition. Nat Struct Mol Biol. 2020;27(10):978–988. 10.1038/s41594-020-0488-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Whyte WA, Orlando DA, Hnisz D, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–319. 10.1016/j.cell.2013.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Aravin A, Gaidatzis D, Pfeffer S, et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature. 2006;442(7099):203–7. 10.1038/nature04916 [DOI] [PubMed] [Google Scholar]
- 99.Girard A, Sachidanandam R, Hannon GJ, Carmell MA. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature. 2006;442(7099):199–202. 10.1038/nature04917 [DOI] [PubMed] [Google Scholar]
- 100.Tiwari B, Jones AE, Caillet CJ, Das S, Royer SK, Abrams JM. p53 directly represses human LINE1 transposons. Genes Dev. 2020. 10.1101/gad.343186.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wallace NA, Belancio VP, Deininger PL. L1 mobile element expression causes multiple types of toxicity. Gene. 2008;419(1-2):75–81. 10.1016/j.gene.2008.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gasior SL, Wakeman TP, Xu B, Deininger PL. The human LINE-1 retrotransposon creates DNA double-strand breaks. J Mol Biol. 2006;357(5):1383–93. 10.1016/j.jmb.2006.01.089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Faulkner GJ, Kimura Y, Daub CO, et al. The regulated retrotransposon transcriptome of mammalian cells. Nat Genet. 2009;41(5):563–71. 10.1038/ng.368 [DOI] [PubMed] [Google Scholar]
- 104.Dewannieux M, Esnault C, Heidmann T. LINE-mediated retrotransposition of marked Alu sequences. Nat Genet. 2003;35(1):41–8. 10.1038/ng1223 [DOI] [PubMed] [Google Scholar]
- 105.Hancks DC, Goodier JL, Mandal PK, Cheung LE, Kazazian HH Jr. Retrotransposition of marked SVA elements by human L1s in cultured cells. Hum Mol Genet. 2011;20(17):3386–400. 10.1093/hmg/ddr245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Raiz J, Damert A, Chira S, et al. The non-autonomous retrotransposon SVA is trans-mobilized by the human LINE-1 protein machinery. Nucleic Acids Res. 2012;40(4):1666–83. 10.1093/nar/gkr863 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.