

Abstract
The nucleus is highly organized, such that factors involved in the transcription and processing of distinct classes of RNA are confined within specific nuclear bodies1,2. One example is the nuclear speckle, which is defined by high concentrations of protein and noncoding RNA regulators of pre-mRNA splicing3. What functional role, if any, speckles might play in the process of mRNA splicing is unclear4,5. Here we show that genes localized near nuclear speckles display higher spliceosome concentrations, increased spliceosome binding to their pre-mRNAs and higher co-transcriptional splicing levels than genes that are located farther from nuclear speckles. Gene organization around nuclear speckles is dynamic between cell types, and changes in speckle proximity lead to differences in splicing efficiency. Finally, directed recruitment of a pre-mRNA to nuclear speckles is sufficient to increase mRNA splicing levels. Together, our results integrate the long-standing observations of nuclear speckles with the biochemistry of mRNA splicing and demonstrate a crucial role for dynamic three-dimensional spatial organization of genomic DNA in driving spliceosome concentrations and controlling the efficiency of mRNA splicing.
The nucleus is organized such that DNA, RNA and protein molecules involved in transcription and processing of distinct RNA classes (for example, ribosomal RNA, histone mRNAs, small nuclear RNAs (snRNAs) and mRNAs) are spatially organized within or near specific nuclear bodies1,2 (for example, nucleolus, histone locus body, Cajal body and nuclear speckles). Although it has long been speculated that nuclear bodies may play a crucial part in RNA biogenesis, such a role has not been directly demonstrated6–8. In theory, nuclear bodies could represent structures that are crucial for transcription and/or processing of specialized classes of RNA (that is, structure enables function). Alternatively, they could represent an emergent property whereby regions of shared regulation self-assemble in three-dimensional (3D) space (that is, function results in structure).
To explore this question, we focused on the relationship between nuclear structure and mRNA splicing. In higher eukaryotes, splicing involves the removal of intronic sequences from genes transcribed by RNA polymerase II (PolII) to generate mature mRNA. This process is predominantly co-transcriptional such that nascent pre-mRNAs are spliced as they are transcribed9. Incomplete splicing produces mRNAs that are degraded by nonsense-mediated decay and results in decreased protein levels10. Owing to its central importance, splicing must be highly efficient to ensure the fidelity of mRNA and protein production, and disruption of mRNA splicing is associated with many human diseases11.
Early studies that visualized the localization of splicing factors—including proteins (for example, SRRM1 and SF3a66) and noncoding RNAs (for example, U1 and U2)12,13–observed that these factors were enriched within specific 3D territories called nuclear speckles14,15. Because of this preferential localization, speckles were initially thought to represent the site of splicing3,13,16. However, this proposal was challenged by subsequent observations that DNA and nascent pre-mRNAs are not primarily located near speckles17–20. Moreover, speckles are enriched for ‘inactive’ spliceosome components3,21–23 that diffuse away from speckles22,24 to bind nascent pre-mRNAs and catalyse the splicing reaction21,22,25–28. These observations led to the prevailing notion that speckles act as storage assemblies of inactive spliceosomes3–5. Additional models of nuclear speckles in splicing have been proposed1,3,7,16,29–32, including speckles acting as hubs that facilitate transcription and splicing of specific genes16,33,34, retaining incompletely spliced transcripts31 or buffering the nucleoplasmic concentration of spliceosomes29,30. However, these models are largely based on correlative observations and have not been directly tested. Accordingly, although speckles were initially described more than 40 years ago14,15,35, what functional role, if any, they play in the process of splicing is unclear.
Recently, we and others identified that speckles represent major structural hubs that organize interchromosomal contacts corresponding to genomic regions that contain highly transcribed PolII genes36–38 and their associated pre-mRNAs29,30,39. On the basis of these observations, we sought to revisit the role of speckles in splicing. Specifically, we propose that organization of highly transcribed PolII genes on the periphery of speckles increases the concentration of spliceosomes at these pre-mRNAs, thereby increasing their splicing efficiency. Here we demonstrate an essential role for 3D organization of genomic DNA in controlling the efficiency of splicing.
snRNAs are enriched at mRNAs near speckles
We previously identified DNA regions that preferentially localize in proximity to nuclear speckles (speckle hubs)37. The frequency of co-occurrence between each genomic DNA region and these speckle hubs in data from split-pool recognition of interactions by tag extension (SPRITE) defined a continuous metric that is correlated with distance to nuclear speckles (speckle proximity score)37. To explore DNA localization relative to nuclear speckles (Fig. 1a), we compared speckle proximity scores (calculated from SPRITE data) and distance to nuclear speckles (measured by microscopy: sequential fluorescence in situ hybridization (seqFISH+)) in mouse embryonic stem (ES) cells (Methods). We observed that DNA regions that exhibit high speckle proximity scores (for example, Foxj1 and Nrxn2) were preferentially located adjacent to the periphery of SF3a66-segmented foci, a protein marker of nuclear speckles (Fig. 1b). Conversely, DNA regions with low speckle proximity scores on the same chromosomes (for example, Efemp1 and Zfand5) were located farther away from SF3a66 foci (Fig. 1b). In a comparison of 2,460 paired genomic regions, the speckle proximity score and the DNA distance to SF3a66 foci were inversely correlated (r = −0.72) (Fig. 1c). Moreover, speckle proximity scores were highly reproducible across multiple independent SPRITE replicates (Extended Data Fig. 1a–d) and correlated with speckle proximity measurements generated by tyramide signal amplification and sequencing (TSA–seq) (Extended Data Fig. 1e,f). These results demonstrate that speckle proximity strongly correlates with genomic distance to nuclear speckles when measured using multiple independent approaches. We refer to genomic regions with the highest 5% of speckle proximity scores as ‘speckle close’ and those with the lowest 5% as ‘speckle far’ (Methods).
Fig. 1 |. snRNAs preferentially bind pre-mRNAs of genes that are close to speckles.
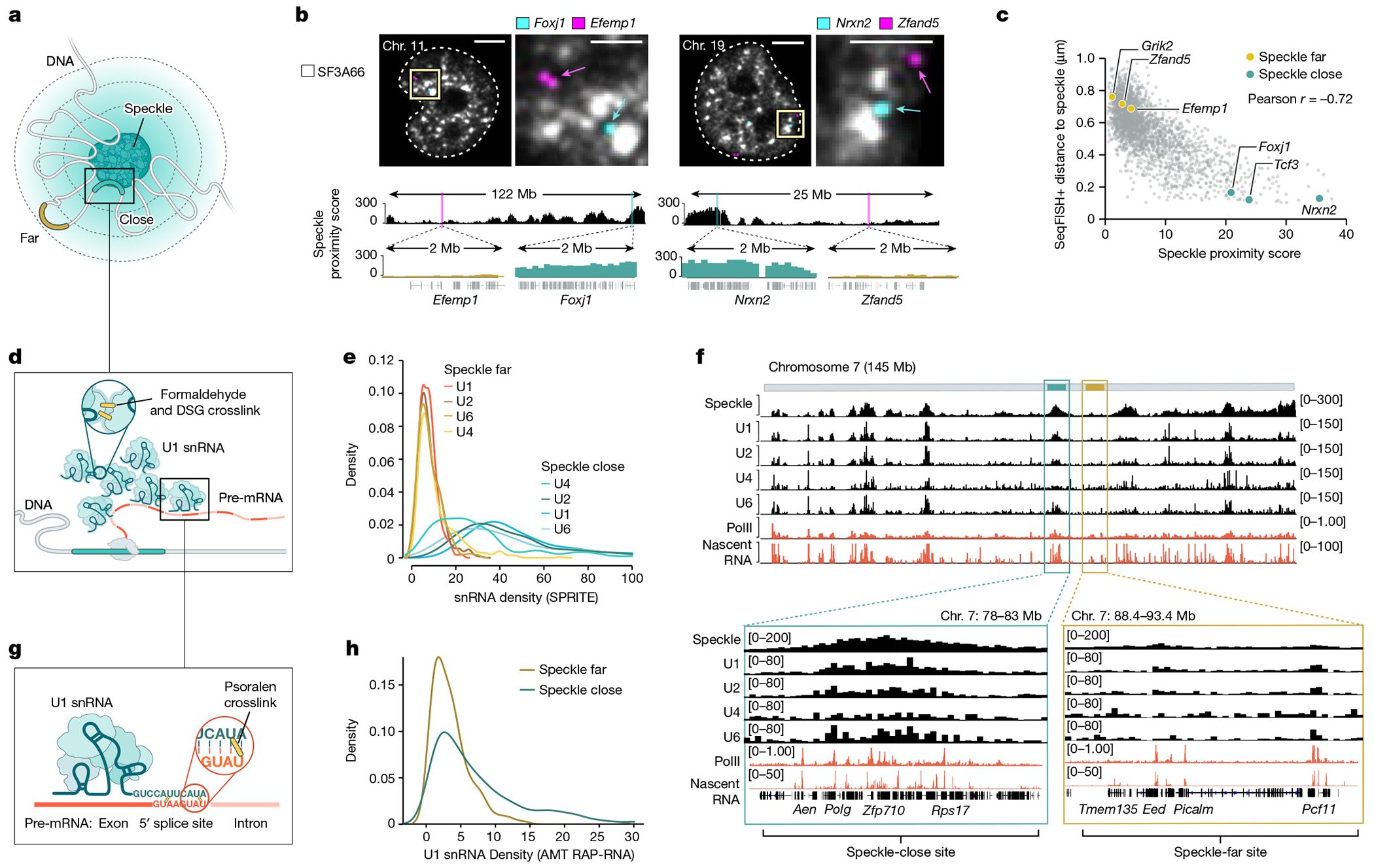
a, Schematic of DNA regions close to (blue) or far from (yellow) nuclear speckles. b, Top, two reconstructed images of DNA seqFISH+ and immunofluorescence (SF3a66) in mouse ES cells comparing speckle-close (Foxj1 and Nrxn2) and speckle-far (Efemp1 and Zfand5) genes. Images are maximum-intensity z projected for a 1 μm section. White lines represent nuclear segmentation. Scale bars, 2.5 μm (zoom-in) or 5μm (zoom-out). Bottom, speckle proximity scores from SPRITE data for the corresponding genomic regions at 100-kb resolution. Zoom-in regions show speckle proximity scores for a specific genomic region (2 Mb) visualized by seqFISH+. n = 446 cells from two seqFISH+biological replicates from ref. 38. c, Genome-wide comparison of seqFISH+ distance to the periphery of a speckle (determined by microscopy) and SPRITE speckle proximity score (determined by sequencing) for 2,460 paired regions. d, Schematic of U1–DNA contacts measured by SPRITE. Formaldehyde and DSG crosslinked nucleic acids and proteins and SPRITE measure the number of molecules within each crosslinked complex. e, Density of U1, U2, U4 and U6 snRNA contacts across 100-kb genomic bins for speckle-close and speckle-far genomic regions. The distributions are quantile-normalized to have the same range as U1 to enable visualization of all snRNAs on the same scale. f, Speckle proximity scores at 100-kb resolution across chromosome 7 (top) and zoom-in views at 100-kb resolution (bottom) for a speckle hub, U1, U2, U4 and U6 snRNAs. PolII-S2P chromatin immunoprecipitation with sequencing (ChIP–seq) and nascent RNA data (10 min of 5EU) densities at 1-kb resolution. g, Schematic of direct RNA–RNA interactions by RAP-RNA43. Psoralen forms direct crosslinks between RNA–RNA hybrids, and affinity purification selectively captures U1 and its directly hybridized pre-mRNAs. h, U1 density over each 5’ splice site within a pre-mRNA measured by RAP-RNA and binned within 100-kb ChIP–seq genomic bins corresponding to speckle-close and speckle-far regions. Illustrations in a, d and g created by Inna-Marie Strazhnik, Caltech.
Having defined genome-wide proximity to nuclear speckles, we explored the localization of the spliceosome across the genome. The spliceosome is the molecular machinery that carries out splicing and consists of U-rich snRNAs and associated proteins40. Although there are different conformational and catalytic states of the spliceosome, in this context, we use the term to refer to snRNAs that bind directly to pre-mRNAs and initiate the splicing reaction41. We considered two possible models of spliceosome association with nascent pre-mRNAs on chromatin: mRNA-directed recruitment or speckle-proximity recruitment. In the mRNA-directed recruitment model, the spliceosome is directly recruited to nascent pre-mRNAs (either through association with PolII or through binding to the pre-mRNA). In this model, the concentration of spliceosomes associating with a transcribed region on chromatin would be proportional to its transcription level (pre-mRNA abundance). Alternatively, in the speckle-proximity recruitment model, spliceosomes are recruited to nascent pre-mRNAs based on their spatial position relative to nuclear speckles. In this model, the concentration of spliceosomes associating with genomic regions that are located closer to speckles would be higher than those that are located farther from speckles independent of the transcription level of the individual pre-mRNA.
To test these two models, we mapped the localization of U1, U2, U4 and U6 snRNAs across the genome using RNA & DNA SPRITE (RD-SPRITE; Fig. 1d). As expected, these snRNAs were enriched over genomic DNA regions that are actively transcribed into pre-mRNA. However, rather than simply reflecting pre-mRNA levels, as would be predicted by the mRNA-directed recruitment model, regions that are close to nuclear speckles displayed about tenfold higher enrichment of snRNAs (Fig. 1e). This increased snRNA density was observed even when focusing only on genomic regions that are transcribed at comparable levels (Extended Data Fig. 2a–i) and when controlling for the number of splice sites per gene (Extended Data Fig. 3a–g), gene length (Extended Data Fig. 3h–l) and gene density (Extended Data Fig. 2f–i) within a genomic region. For example, two neighbouring genomic regions on mouse chromosome 7 that are transcribed at comparable levels, but are located at different distances relative to speckles, displayed about a fourfold difference in snRNA levels (Fig. 1f). These results indicate that spliceosome concentrations are highest at nascent pre-mRNAs that are in proximity to nuclear speckles.
Although proximity to speckles is associated with increased spliceosome concentrations, this finding alone does not indicate that speckle-proximity drives snRNA loading. For example, if the spliceosome concentration mediated through the pre-mRNA is sufficiently high that splice sites are saturated, the additional increase observed at genes close to the speckle would have no impact on spliceosome binding and function. Because RD-SPRITE utilizes protein–protein crosslinking (formaldehyde and disuccinimidyl glutarate (DSG)) to map RNA–DNA contacts, this approach does not measure direct snRNA binding to pre-mRNAs37,39 (Fig. 1d). To measure the number of spliceosomes that directly bind to nascent pre-mRNAs, we used psoralen-mediated crosslinking (which forms covalent crosslinks only between directly hybridized nucleic acids42) to map U1 interactions with pre-mRNAs (Fig. 1g). We have previously shown that this approach is highly specific at mapping U1 binding to 5′ splice sites at exon–intron junctions43. We re-analysed our data and computed the frequency of U1 binding over each 5′ splice site and binned these frequencies into 100-kb windows to compare U1 binding to speckle proximity. We observed higher levels of U1 binding to pre-mRNAs transcribed from speckle-close genes than those transcribed from speckle-far genes (Fig. 1h). Moreover, genomic regions that are enriched for U1 binding showed an approximate threefold increase in speckle proximity (Extended Data Fig. 3m). We observed the same effect even when normalizing for the number of splice sites per genomic bin (Extended Data Fig. 3n) or when directly comparing the distribution of counts for each individual junction (2.6-fold increase, chi-square P < 0.0001; (Methods).
Together, these results indicate that the proximity of genomic DNA regions to nuclear speckles is associated with increased concentrations of spliceosomes and spliceosome engagement on pre-mRNA.
Splicing is highest near speckles
We reasoned that increased concentrations of spliceosome components (enzyme) at nascent pre-mRNAs (substrate) located proximal to nuclear speckles would lead to increased co-transcriptional splicing efficiencies (that is, the proportion of spliced products to total mRNA produced; Fig. 2a) relative to pre-mRNAs that are located farther from the speckle.
Fig. 2 |. Co-transcriptional splicing efficiency varies based on the proximity to nuclear speckles.
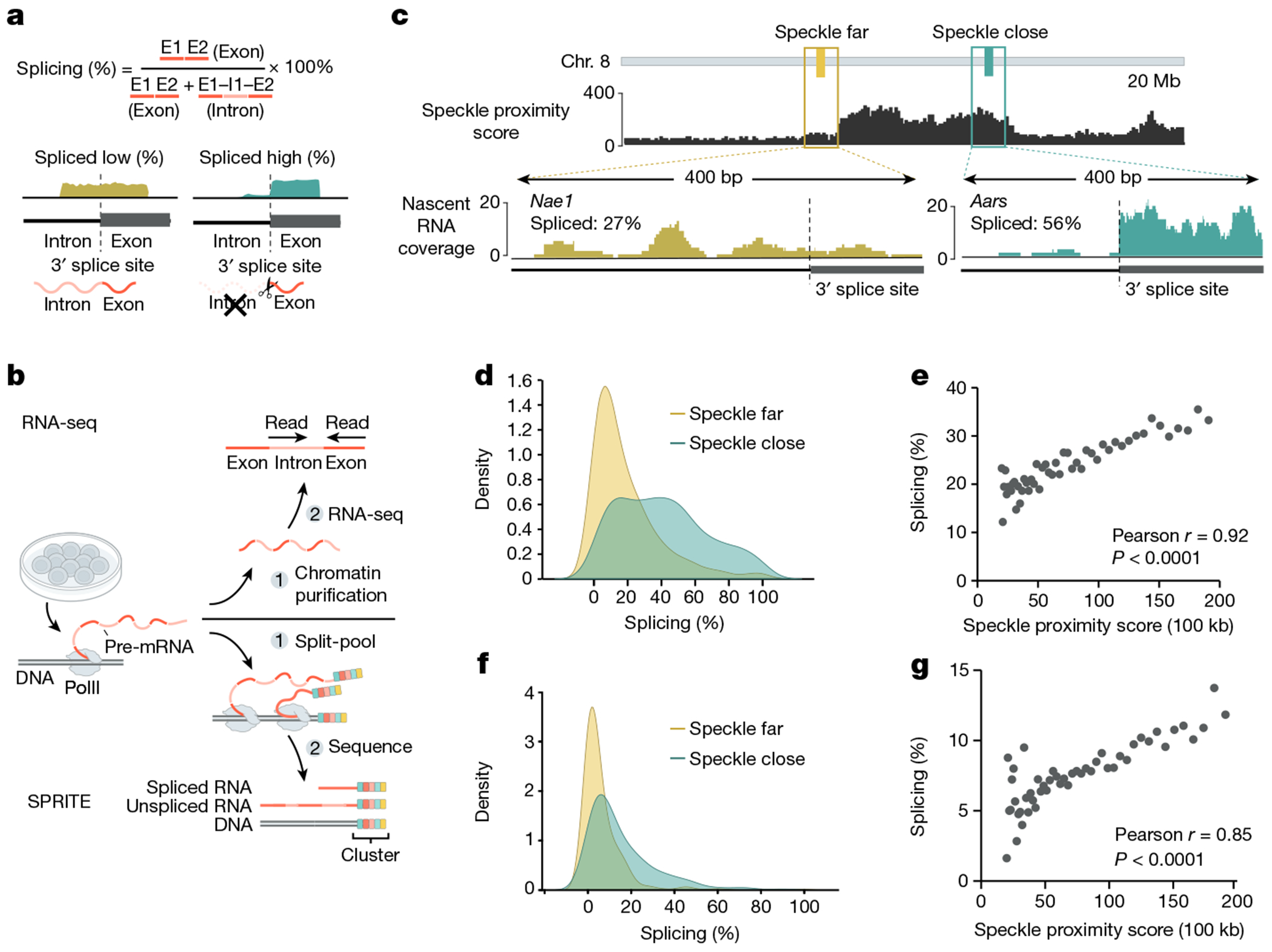
a, Schematic of nascent RNA splicing efficiency calculation. Splicing efficiency of a gene is calculated by taking the ratio of exon to total pre-mRNA counts from RNA-seq (exons + introns). b, Schematic of nascent RNA-seq and SPRITE methods used to measure splicing efficiency. c, Top, SPRITE speckle proximity score for a 20-Mb region on mouse chromosome 8. Bottom, nascent RNA coverage from chromatin RNA-seq for a speckle-far (Nae1) and speckle-close (Aars) gene around a single 3’ splice site. Per cent spliced across entire gene is indicated. d, Density plot from chromatin RNA-seq of per cent spliced for genes located within speckle-close or speckle-far 100-kb genomic regions (461 speckle-close genes and 460 speckle-far genes). e, SPRITE speckle proximity score (x axis) and per cent spliced for genes from nascent RNA-seq within each bin (y axis) across 50 bins. Each point or bin contains at least 20 genes and reflects the average splicing for that bin. f, Density plot of per cent spliced within 100-kb genomic intervals from SPRITE for speckle-close and speckle-far regions (312 speckle-close and 311 speckle-far 100-kb regions). g, SPRITE speckle proximity score (x axis) and per cent spliced within genomic bins from SPRITE (y axis) across 50 bins. Each point or bin contains at least 20 regions and reflects the average splicing for that bin. Illustrations in a and b created by Inna-Marie Strazhnik, Caltech.
To focus on pre-mRNA splicing that occurs near the DNA locus from which they are transcribed (which we refer to as co-transcriptional splicing), we analysed nascent RNA that is associated with chromatin using a stringent biochemical purification procedure44 (Fig. 2b). Using these data, we computed the splicing efficiency for each gene, which accounts for transcription levels by taking the ratio of spliced reads to total pre-mRNA reads (spliced reads plus unspliced reads) (Fig. 2a). Overall, genes located closest to nuclear speckles showed a >2-fold higher splicing efficiency than genes farthest from nuclear speckles (41.0% compared with 19.1%) (Fig. 2c,d). More generally, we observed a strong correlation between speckle proximity and splicing efficiency in mouse ES cells (r = 0.92, P < 0.0001; Fig. 2e and Extended Data Fig. 4a). Notably, there was a similar increase in splicing efficiency at speckle-proximal genes when measuring nascent RNA purified after 10 min of metabolic incorporation of 5-ethynyl uridine (5EU) in mouse ES cells (r = 0.95, P < 0.0001; Extended Data Fig. 4b–d). This result demonstrates that this effect does not depend on the method used to measure nascent RNA. To ensure that these differences in splicing efficiency are not due to differences in transcription levels, gene lengths or number of splice junctions per gene, we analysed sets of genomic regions that were comparable for each of these features (Methods). The results showed increases in splicing efficiency at speckle-close genes in all cases (Extended Data Fig. 4e–s).
To further validate this effect and exclude the possibility that the observed splicing differences might reflect mature mRNA in our biochemical purification samples, we used an orthogonal method to measure mRNA levels on chromatin. Specifically, we used RD-SPRITE to analyse splicing ratios of RNAs45 exclusively when they were associated with the DNA of their own nascent locus (Fig. 2b). We then computed splicing efficiency as the fraction of exons over the total number of exons and introns. Consistent with the chromatin and 5EU-purified RNA sequencing (RNA-seq) data, we observed about 3-fold higher splicing in speckle-close regions (16.1%) than in speckle-far regions (5.5%) (Fig. 2f). Furthermore, we observed a strong correlation between the splicing efficiency per gene and its speckle proximity score (r = 0.85, P < 0.0001; Fig. 2g and Extended Data Fig. 4a). More generally, genes that have higher speckle proximity scores also showed higher splicing efficiencies in other cell types, including mouse myocytes (Spearman r = 0.64, P < 0.0001) and H1 human ES cells (Spearman r = 0.70, P < 0.0001) (Extended Data Fig. 4t,u).
Together, these results indicate that pre-mRNA splicing efficiency is highest for speckle-associated genes and that this increased splicing efficiency occurs while the pre-mRNA is bound at its nascent locus.
Gene distance to speckle drives splicing
Genes differ in multiple ways beyond their nuclear speckle proximity (for example, promoter type and activity, gene length, splice site strength, alternative splicing patterns and sequence-specific features). Therefore, it is possible that the observed increase in splicing efficiency is due to other gene-specific features that might also correlate with speckle proximity.
To account for potential gene-specific features that might affect splicing, we generated a splicing reporter that contains an exon–intron–exon minigene fused in-frame to a GFP that is translated when spliced but not when unspliced (Fig. 3a). To account for potential splicing-independent effects that might influence GFP levels (for example, transcription, nuclear export or polyadenylation), we linked this spliced GFP reporter to a bidirectionally transcribed BFP reporter that does not contain an intron and therefore does not require splicing for expression. In this system, if splicing is affected, then we would observe a difference between GFP and BFP levels. However, if splicing is unaffected, then the levels between GFP and BFP levels would be comparable (Fig. 3a).
Fig. 3 |. Expression of a gene from a genomic locus in proximity to nuclear speckles leads to increased mRNA splicing.
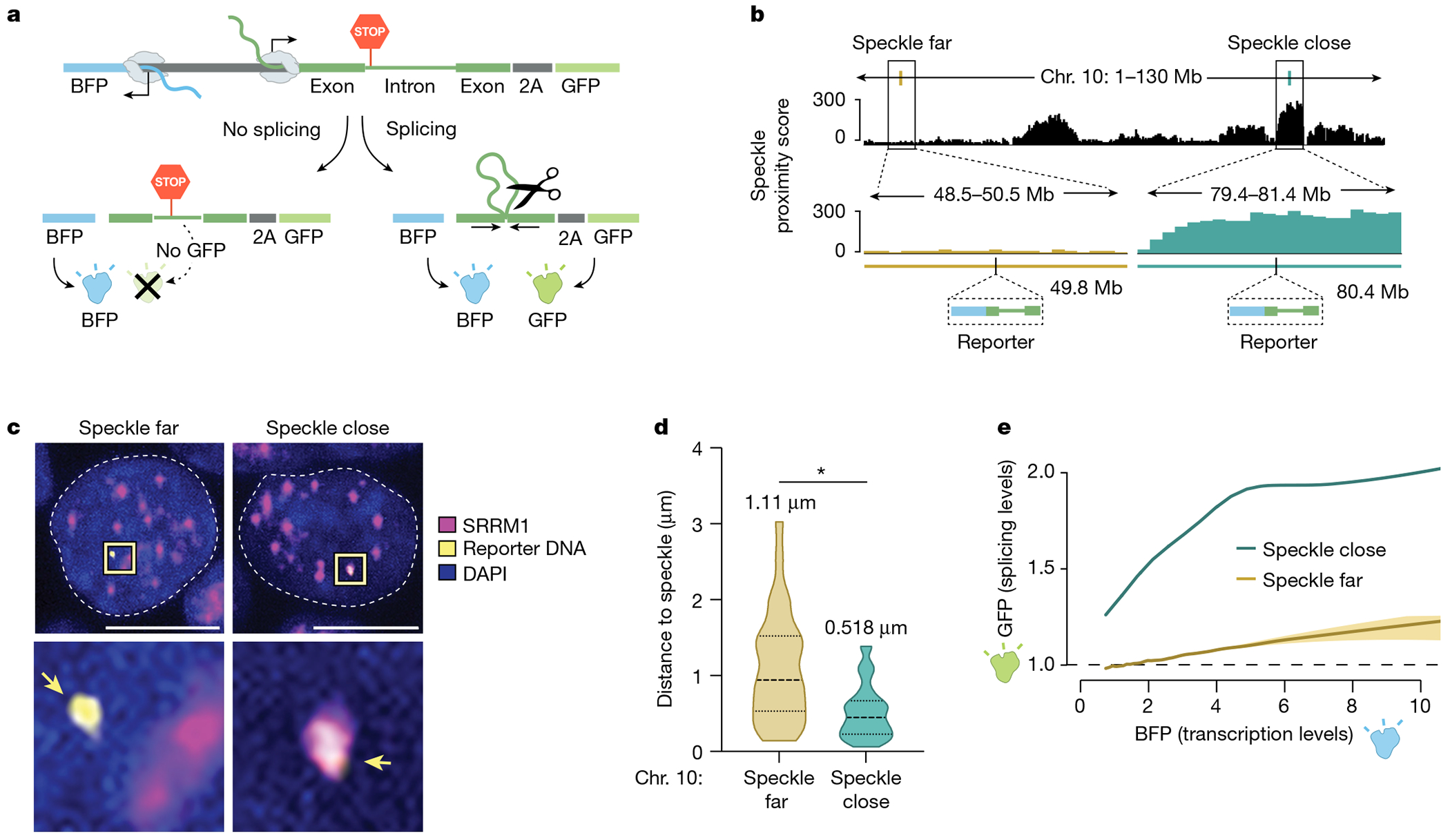
a, Schematic of the bidirectional reporter assay using a fluorescence-based readout. The splicing reporter contains an exon–intron–exon minigene fused in-frame to a GFP that is translated when spliced but not when unspliced. The spliced GFP reporter is linked to a bidirectionally transcribed BFP reporter that is expressed and translated regardless of whether it is spliced. b, SPRITE speckle proximity score (100-kb bin) for the two genomic regions on mouse chromosome 10 where the reporter was integrated. c, Representative images and zoom-in images of SRRM1 immunofluorescence combined with DNA FISH for cells containing the two integrated reporters. Scale bar, 10 μm. n = 85 cells over two biological replicates. d, Violin plots of the distance of DNA FISH spots of speckle-far and speckle-close integrated loci to the nearest nuclear speckle (immunofluorescence of SRRM1) across multiple cells (n = 28 speckle-far and n = 57 speckle-close). Mean distance is displayed above each distribution. *P < 0.0001. e, GFP expression (fluorescence intensity) as a function of BFP transcription levels for speckle-close and speckle-far integrated loci. We estimated the variation of these measurements using a bootstrap procedure from ten random bootstraps generated from these data (Methods). n = 744,019 cells analysed for speckle-close and n = 158,971 cells analysed for speckle-far. P value from two-sided t-test. Illustrations in a and b created by Inna-Marie Strazhnik, Caltech.
We used CRISPR–Cas9 to integrate this bidirectional reporter into two different genomic locations in mouse ES cells corresponding to a speckle-close (Tcf3 locus in Fig. 1c) and speckle-far region (Grik2 locus in Fig. 1c) located on the same chromosome (Fig. 3b). To ensure that genomic integration of the reporter does not affect speckle proximity, we imaged the genomic DNA of the reporter gene (using DNA FISH) together with nuclear speckles (immunofluorescence of SRRM1). The reporter integrated within a speckle-close region consistently showed closer speckle proximity than the reporter integrated into a speckle-far region (difference in distance to speckle = 0.5 μm; Fig. 3c). Notably, these integrated reporters showed comparable differences in their average speckle distance as observed when visualizing their endogenous loci (Figs. 1c and 3d and Extended Data Fig. 5a–c).
Next, we determined the splicing efficiency of the reporter at each integrated location by quantitatively measuring the levels of GFP (splicing reporter) relative to BFP (splicing-independent reporter) within >100,000 individual BFP-expressing cells using flow cytometry. GFP levels were significantly increased relative to BFP levels in cells in which the reporter was integrated close to speckles, but not in cells in which the reporter was integrated far from speckles (Fig. 3e and Extended Data Fig. 5d). This increase was consistently observed regardless of the level of BFP expressed within each individual cell.
These results indicate that a gene transcribed from a genomic location proximal to nuclear speckles is more efficiently spliced than the same gene transcribed from a genomic region located farther from nuclear speckles.
DNA location and splicing vary by cell type
The location of a gene relative to nuclear speckles is associated with increased splicing efficiency, and genomic DNA organization around speckles has been reported to change between distinct cell types46,47. Therefore, we explored whether dynamic organization around nuclear speckles might be a mechanism for dynamic regulation of splicing efficiency across cell types.
To that end, we compared genomic DNA organization around nuclear speckles in two distinct mouse cell types with different gene expression programs: mouse ES cells and mouse myocytes. Specifically, we generated SPRITE maps in differentiated mouse myocytes and compared speckle proximity scores for each genomic region between myocytes and ES cells (Fig. 4a and Extended Data Fig. 6a–e). About 8% of the genome was speckle-proximal in both mouse ES and myocytes, with around 46% of these regions showing preferential localization in ES cells and approximately 14% showing preferential localization in myocytes (Extended Data Fig. 6f,g). Consistent with the fact that speckle proximity is correlated with PolII density37,46,47, genomic regions that were preferentially speckle-proximal in one of the two cell types corresponded to genomic regions that contained the largest differences in RNA PolII density between myocytes and ES cells (Spearman correlation = 0.53, P = 0.0001; Fig. 4b,c and Extended Data Fig. 6h,i).
Fig. 4 |. Differential gene positioning around speckles leads to different splicing efficiencies across cell types.
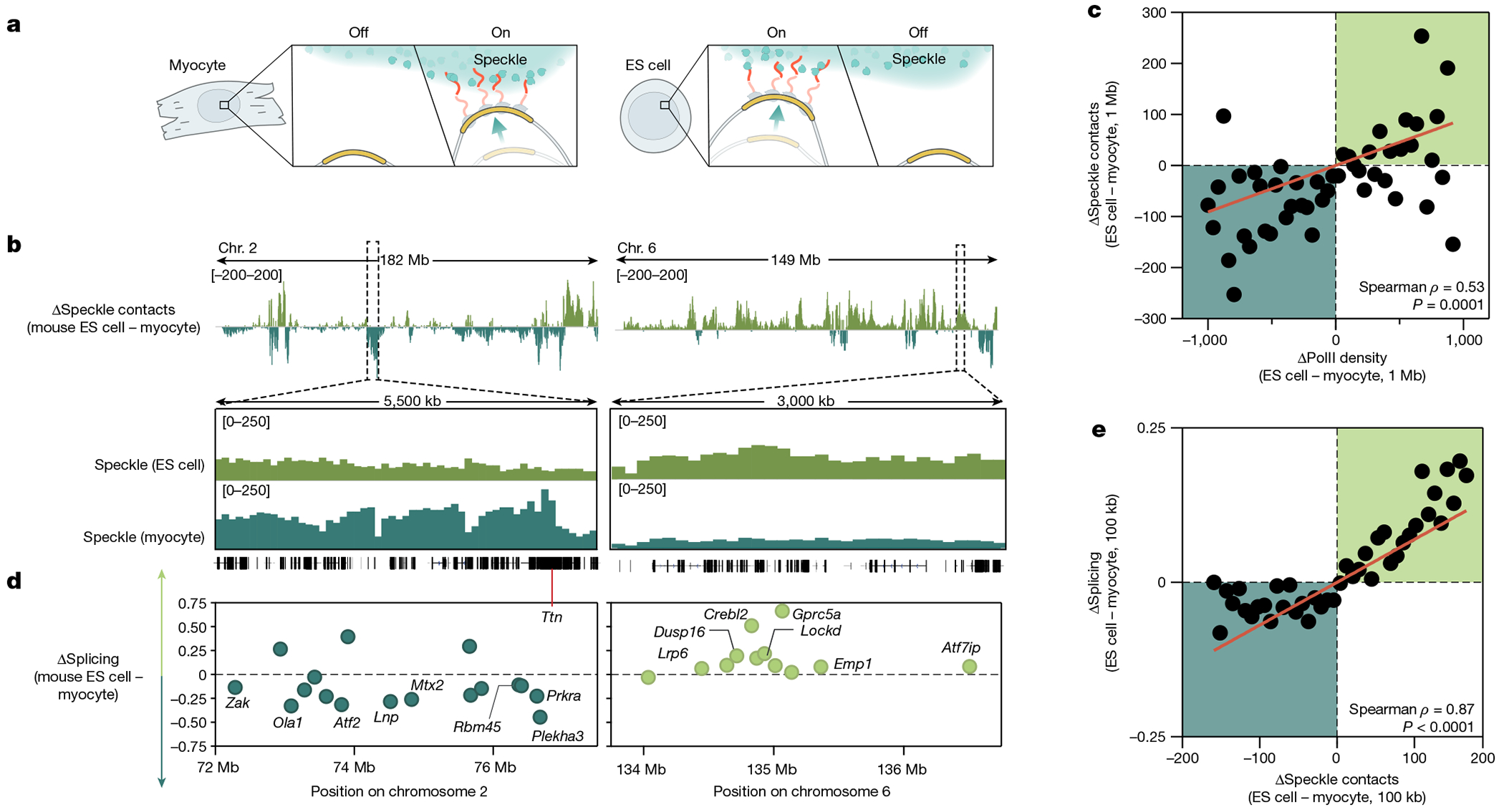
a, Schematic of the differential localization of genomic DNA relative to nuclear speckles and its dependence on PolII activity. b, Top, difference in speckle proximity score between mouse ES cells and myocytes for chromosomes 2 and 6. Bottom, 5.5-Mb and 3-Mb zoom-in regions of speckle proximity scores around the Ttn (myocyte preference) and Crebl2 (mouse ES cell preference) loci, respectively. c, Difference in PolII-S2P density (x axis) versus difference in SPRITE speckle proximity score (y axis) between ES cells and myocytes at 1-Mb resolution. n = 48 bins each containing at least 10 regions. d, Difference in splicing between mouse ES cells and myocytes for genomic regions expressed in both cell types for the same zoom-in regions as in c. Individual genes are labelled. To calculate the change in splicing efficiency between cell types, we only included genes expressed in both cell types. e, Difference in speckle proximity score (x axis) versus difference in splicing (y axis) between ES cells and myocytes at 100-kb resolution. n = 41 bins each containing at least 20 regions. P value is two-tailed (c,e). Illustration in a created by Inna-Marie Strazhnik, Caltech.
We next explored whether these changes in speckle proximity correspond to changes in mRNA splicing efficiency. Indeed, genes located within genomic regions that displayed the largest changes in speckle proximity showed the largest changes in splicing efficiency between cell types (Spearman correlation = 0.87, P < 0.0001; Fig. 4d,e). For example, a cluster of genes on chromosome 6 that are expressed in both ES cells and myocytes but located within a genomic region that is preferentially localized near speckles in ES cells displayed higher levels of splicing when transcribed in ES cells than in myocytes (Fig. 4d). Conversely, skeletal-muscle-specific genes that are transcriptionally induced during myogenic differentiation, such as titin (encoded by Ttn), are located proximal to speckles in myocytes but away from speckles in ES cells (Fig. 4b and Extended Data Fig. 6j). Notably, genes within the genomic locus containing Ttn that are transcribed in both ES cells and myocytes showed higher levels of splicing in myocytes than in ES cells (Fig. 4d).
Together, these results demonstrate that changes in gene organization relative to nuclear speckles correspond to changes in splicing efficiency in distinct cell types.
Driving mRNA to speckles boosts splicing
Although splicing efficiency of the same gene (endogenous and/or integrated reporter) differs based on its location relative to nuclear speckles, genomic location relative to speckles is also correlated with PolII density. Accordingly, changes observed in speckle proximity, PolII density and splicing efficiency are confounded, which makes it difficult to establish a direct causal relationship between speckle proximity and splicing efficiency.
To address this challenge, we developed a system that enables directed recruitment of a pre-mRNA to the speckle in a manner that is decoupled from PolII density, transcription or other potential chromatin features. Specifically, we utilized our bidirectional reporter containing two linked reporters–a GFP that is produced only when spliced and a BFP that is produced independently of splicing activity–and transiently expressed this reporter from a plasmid (Fig. 5a). In the intron of the reporter, we embedded a MS2 bacteriophage RNA hairpin that binds with high affinity to the MS2 bacteriophage coat protein (MCP)48. We used this system to localize the pre-mRNA reporter to specific nuclear locations by co-expressing the splicing reporter together with specific MCP fusion proteins that are known to localize at different locations within the nucleus (Fig. 5b). Specifically, we expressed SRRM1 and SRSF1, two proteins that localize within nuclear speckles49,50. SRRM1 is primarily localized in nuclear speckles (punctate), whereas SRSF1 exhibits both speckle (punctate) and nucleoplasmic (diffuse) localization. As controls, we expressed two non-speckle proteins: SRSF3 (a splicing protein that is not enriched within nuclear speckles but localized throughout the nucleoplasm)51 and LBR (a protein that is anchored in the nuclear membrane and associates with the transcriptionally inactive nuclear lamina)52.
Fig. 5 |. Pre-mRNA organization around nuclear speckles drives splicing efficiency.
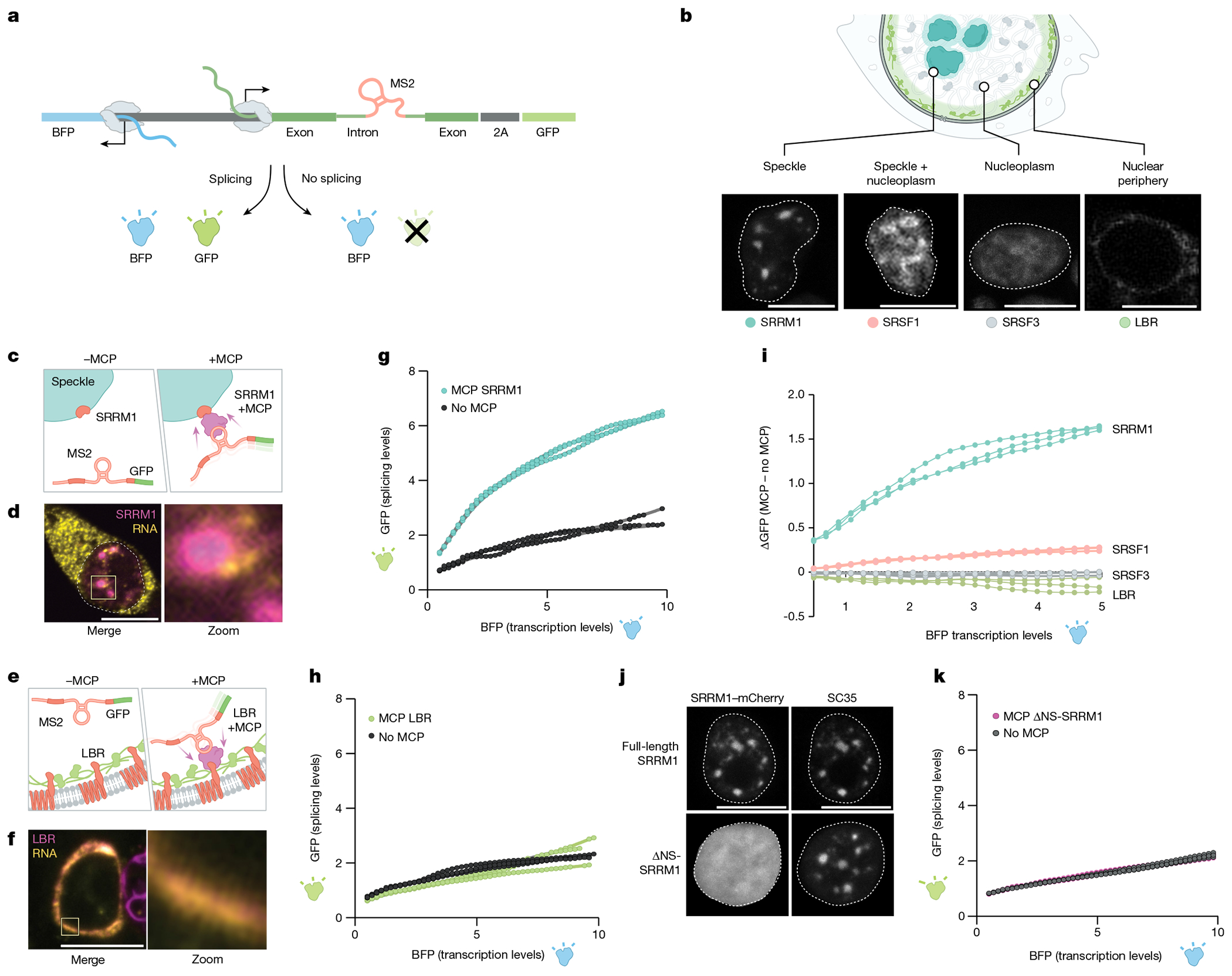
a, Schematic of the pre-mRNA splicing assay using a bidirectional GFP and BFP fluorescence-based readout. The MS2 stem–loop is embedded within the intron. GFP is expressed only when the reporter is spliced and was measured by FACS. b, Top, schematic of specific nuclear locations (speckle, speckle + nucleoplasm, nucleoplasm and nuclear periphery). Bottom, mCherry fluorescence of their corresponding proteins (SRRM1, SRSF1; SRSF3 and LBR). The nucleus is outlined in white. n = 3 biological replicates. c, Schematic of SRRM1 tagged with mCherry with or without a MCP tag. The MCP protein binds to the complementary MS2 stem–loop embedded within the intron of the pre-mRNA reporter. d, Single-molecule RNA FISH and zoom-in images of the localization of SRRM1 and MCP with the mCherry reporter. Nucleus is outlined in white. n = 3 biological replicates. e, Schematic of LBR tagged with mCherry with or without a MCP tag. f, Single-molecule RNA FISH and zoom-in images of the localization of LBR and MCP with the mCherry. n = 3 biological replicates. g, Fluorescence intensity of GFP (y axis) versus BFP (x axis) for three replicates of SRRM1 ± MCP. h, Fluorescence intensity of GFP (y axis) versus BFP (x axis) for three replicates of LBR ± MCP. i, Difference of GFP expression between constructs with or without MCP (y axis) versus BFP fluorescence intensity (x axis) for all constructs tested. Three replicates plotted for each sample. j, Fluorescence microscopy for mCherry±SRRM1 (top left) and mCherry ΔNS-SRRM1 (bottom left) with co-immunofluorescence for SC35 (top right and bottom right). n = 3 biological replicates. k, GFP levels (y axis) versus fluorescence intensity (levels) of BFP (x axis) (bottom) for three replicates of SRRM1 ΔNS-SRRM1 ± MCP. Scale bars, 10 μm (b,d,f,j). Illustrations in a–c and e created by Inna-Marie Strazhnik, Caltech.
We transfected each of these proteins fused to MCP and mCherry (to directly visualize localization). Fluorescence microscopy analyses confirmed that each protein localized in the nucleus as expected (Fig. 5b and Extended Data Fig. 7a–d). SRRM1–MCP and SRSF1–MCP co-localized with endogenous SC35, a well-characterized marker of nuclear speckles (Fig. 5j and Extended Data Fig. 7a ,b). By contrast, SRSF3 localized diffusely throughout the nucleus and LBR localized to the periphery of the nucleus (Fig. 5b and Extended Data Fig. 7c,d). Results from RNA FISH coupled with fluorescence microscopy of mCherry confirmed that the MS2-containing reporter RNA preferentially co-localized with nuclear speckles when co-expressed with SRRM1–MCP and co-localized at the nuclear periphery when co-expressed with LBR–MCP (Fig. 5c–f and Supplementary Video 1).
Having demonstrated the ability to drive recruitment of a mRNA to a specific nuclear location, we sought to test the impacts of nuclear speckle localization on splicing efficiency. To establish the baseline splicing efficiency and to account for non-MCP-dependent effects on GFP expression—including transfection and specific protein-dependent effects—we expressed each protein without MCP. We quantified splicing efficiency by measuring the difference in GFP fluorescence with and without MCP for each protein construct (ΔGFP) relative to BFP levels. Recruitment of the reporter specifically to the speckle protein SRRM1 or SRSF1 resulted in a nonlinear increase in GFP levels (splicing) relative to BFP levels (nonlinear four parameter logistic regression R2 = 0.98; Fig. 5g and Extended Data Fig. 7e,f,i–k). To ensure that this observed effect is specifically due to nuclear speckle recruitment, we recruited this MS2–RNA to the diffusely localized splicing protein SRSF3 or to the nuclear lamina using LBR. In both cases, these conditions had no impact on GFP levels (Fig. 5h–i and Extended Data Fig. 7g–k).
To ensure that this positional effect also occurs across different introns containing different splice sites and intron architectures, we generated two additional reporter constructs. For these constructs, we replaced the intron sequence within the spliced GFP with either an intron sequence derived from CORO1B that contains a strong splice site and has high GC content (referred to as a levelled intron29) or an intron derived from FRG1 that contains a weak splice site and low GC content (referred to as a differential intron29; Extended Data Fig. 8a,b). In both cases, GFP levels relative to BFP increased when recruited to nuclear speckles through SRRM1 but were not affected when recruited to the nuclear lamina through LBR (Extended Data Fig. 8c–f). Although there was a significant increase in the splicing efficiency of both intron sequences when recruited to nuclear speckles, we observed a smaller effect size for the differential intron architecture (FRG1), which may reflect the presence of a weaker splice site (which is known to affect overall levels of splicing53). Moreover, these observations further confirm that speckle proximity affects splicing efficiency and not other aspects of mRNA processing (for example, export).
To ensure that this effect is specifically due to nuclear speckle localization, we expressed a truncated form of SRRM1 that lacks the domain responsible for nuclear speckle localization but has been previously shown to retain its catalytic domain required for RNA processing54 (ΔNS-SRRM1; Fig. 5j). We confirmed that ΔNS-SRRM1 no longer localized within nuclear speckles (Fig. 5j and Extended Data Fig. 7l). Notably, expression of ΔNS-SRRM1 led to loss of the MCP-dependent increase in splicing efficiency (ΔGFP) and instead showed a response similar to that observed for other non-speckle-associated proteins (Fig. 5k and Extended Data Fig. 7m).
Together, these data demonstrate that directed recruitment of a pre-mRNA to nuclear speckles, but not to other nuclear locations, is sufficient to increase mRNA splicing efficiency.
Discussion
Together, our results integrate the long-standing observations of nuclear speckles with the biochemistry of mRNA splicing. We propose a model whereby nuclear speckles consist of high concentrations of splicing factors that diffuse away from speckles to engage pre-mRNAs3,4,25. When a nascent pre-mRNA is located closer to a speckle, there is a reduced volume through which these splicing factors need to diffuse to interact with the pre-mRNA. This decrease in diffusion volume creates a higher concentration of splicing factors in the vicinity of speckle-close genes and results in increased spliceosome binding to these pre-mRNAs and conversion into spliced mRNA (Fig. 6). Whereas speckle proximity affects the concentration of splicing factors bound to a pre-mRNA, differences in pre-mRNA sequence features (for example, splice site strength) would affect splicing factor activity when bound to a pre-mRNA53. In this way, these two components would be expected to have different kinetic effects on splicing, with speckle proximity affecting the proportion of a pre-mRNA bound and splicing activity affecting the maximum output of the splicing reaction when a pre-mRNA is saturated (Extended Data Fig. 9).
Fig. 6 |. Integrated model of how gene organization around nuclear speckles affects splicing.
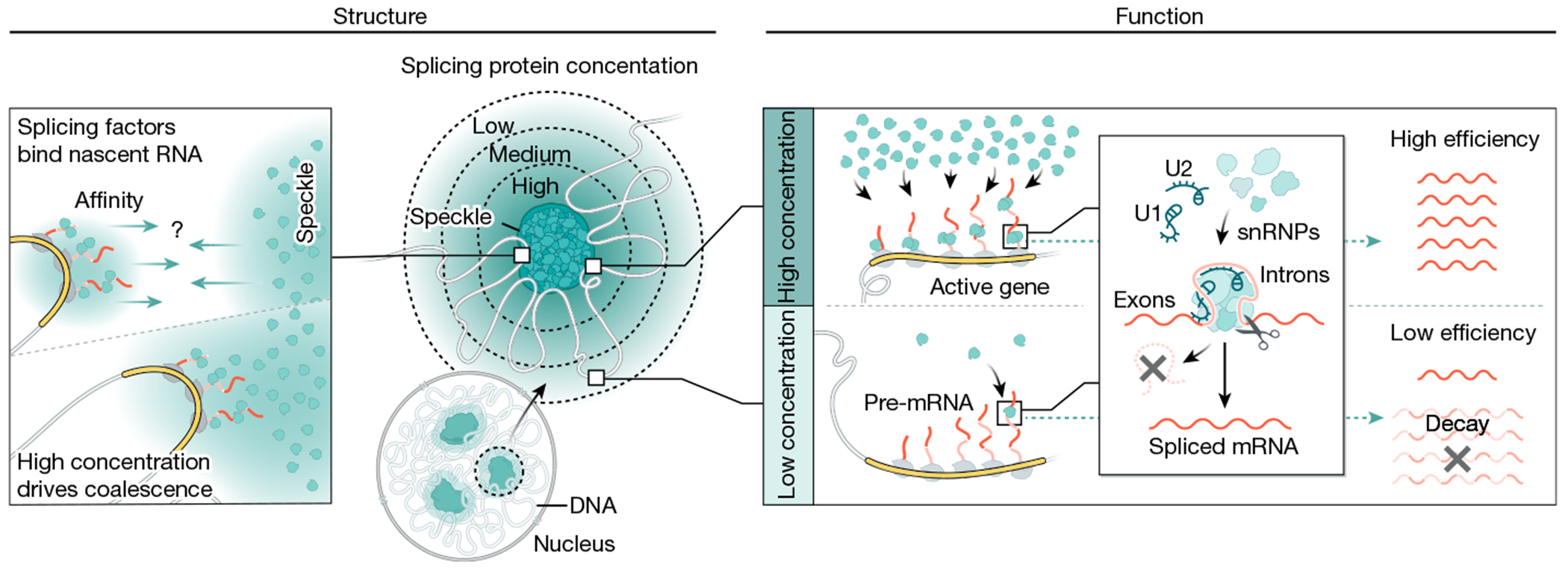
Model of how 3D genome organization drives mRNA splicing. Because nascent pre-mRNAs have high affinity for splicing factors and because PolII-dense regions contain the highest concentrations of nascent pre-mRNAs, these genomic regions can achieve multivalent contacts with splicing factors that are enriched within nuclear speckles. Because nuclear speckles contain the highest concentration of these factors within the nucleus, these multivalent contacts may drive coalescence (self-assembly) of these genomic DNA sites with the nuclear speckle. Genomic regions and pre-mRNAs close to nuclear speckles have higher levels of spliceosomes than regions farther away. Locally concentrating pre-mRNAs, genomic DNA and spliceosomes at speckle-proximal regions leads to increased splicing efficiency, whereas a speckle-far gene transcribed at the same level is not spliced as efficiently. Model created by Inna-Marie Strazhnik, Caltech.
Because speckle proximity is correlated with PolII density and genes are differentially organized relative to speckles on the basis of transcriptional activity, high levels of transcription may act to organize genomic DNA closer to nuclear speckles. It remains to be determined whether actively transcribed loci reposition towards existing nuclear speckles or whether actively transcribed loci can seed the assembly of new speckles. In both scenarios, because nascent pre-mRNAs have high affinity for splicing factors (including SR proteins and other RNA-binding proteins) and PolII-dense regions contain the highest concentrations of nascent pre-mRNAs, these genomic regions would achieve multivalent contacts with splicing factors that are enriched within nuclear speckles. These multivalent contacts may in turn drive coalescence (self-assembly) of these genomic DNA sites with the nuclear speckle55 (Fig. 6). Indeed, this self-assembly concept explains how newly transcribed ribosomal DNA genes and snRNA gene loci coalesce into the nucleolus and Cajal bodies, respectively2,55. Although RNA PolII density is associated with speckle proximity37, not all highly transcribed genes in a cell type are organized around the speckle. Because differential splicing efficiency would affect mRNA and protein levels in a cell, changes in genome organization relative to speckles would lead to changes in splicing efficiencies, thereby creating another dimension of gene expression control.
mRNA splicing and PolII transcription are kinetically coupled56 such that increasing the transcription of a gene leads to a nonlinear increase in its splicing efficiency (referred to as ‘economy of scale’ splicing57). Although individual splicing proteins can associate with the C-terminal domain of PolII58, direct binding of splicing factors to PolII would predict a linear relationship between transcription and splicing and therefore cannot fully explain this coupling. Moreover, PolII is not sufficient to stimulate splicing efficiency in cellular extracts59. This finding implies that there must be some additional cellular mechanism required to functionally couple transcription and splicing in cells. Indeed, our results suggest that this mechanism may be differential gene organization relative to nuclear speckles. Specifically, high levels of PolII transcription would act to reposition genomic DNA into proximity with nuclear speckles and increase splicing efficiency at these genes. Consistent with this notion, it was previously observed that increasing transcription of an individual reporter gene leads to nonlinear increases in its splicing efficiency, and this coincides with an increased proximity between the gene locus and nuclear speckles57. Because the increase in spliceosome concentration achieved at DNA regions positioned at nuclear speckles would exceed the proportional concentration of the pre-mRNAs transcribed at that locus, this model would explain the observed nonlinear increase in splicing efficiency that is achieved when a gene is recruited to the nuclear speckle. In this way, spatial organization around nuclear speckles may act to couple PolII transcription and mRNA splicing efficiency.
More generally, our results indicate a new mechanism by which nuclear organization can coordinate regulatory processes in the nucleus and ensure strong nonlinear control. Beyond speckles, there are many other bodies that similarly organize RNA-processing enzymes with their co-transcriptional DNA and RNA targets1,2,39,55. These compartments include nascent rRNA loci and rRNA-processing factors (for example, small nucleolar RNAs and nucleolin) within the nucleolus, histone mRNAs and histone-processing factors (for example, U7 snRNA) in histone locus bodies, and snRNAs and their processing factors (for example, small Cajal body-specific RNAs) within Cajal bodies. In each of these examples, these nuclear bodies organize around active transcription of the genes that they process39. Our results indicate that this structural arrangement may be an important and shared role for coordinating the co-transcriptional efficiency of RNA processing. Specifically, assembling genomic DNA encoding nascent pre-RNAs and their associated regulatory factors within the nucleus could act to increase the local concentration of these factors and therefore couple the efficiency of RNA processing to transcription of these specialized RNAs. This organization would enable localization of these RNA-processing enzymes at their targets as they are being produced. The importance of ensuring precise and efficient co-transcriptional processing and coordinating these processes in space and time may explain why all known classes of RNA processing are associated with specialized nuclear bodies and why disruption of nuclear bodies is a common hallmark in various human diseases60.
Methods
Cell lines and cell culture conditions
We used the following cell lines in this study: (1) male ES cells (pSM33 ES cell line) derived from a 129 × castaneous F1 mouse cross; (2) two male ES cell lines, in which we integrated a bidirectional fluorescent splicing reporter (BFP and GFP) at two different loci (speckle-close and speckle-far integration lines); in these cells, BFP is constitutively on and GFP is expressed on the basis of whether splicing is completed or not; (3) MM14 mouse myocytes (gift from B. Wold and B. Williams); (4) male H1 human ES cells (gift from R. Maehr and K. Mohan Parsi); and (5) HEK293T, a female human embryonic kidney cell line (American Type Culture Collection, CRL-3216, RRID: CVCL_0063). Authentication of cell lines was performed using SPRITE (mouse ES cells, MM14 mouse myocytes and H1 human ES cells), RNA-seq (mouse ES cells) and DNA FISH for integrated loci (integrated reporter in mouse ES cells), all of which gave results consistent with their respective cellular identities. The cells were not tested for mycoplasma contamination.
Mouse ES cell culturing conditions.
Mouse ES cells were grown on plates coated with 0.2% gelatin and 3.5 μg ml−1 laminin in serum-free 2i/LIF medium composed as follows: 1:1 mix of DMEM/F-12 (Gibco) and neurobasal (Gibco) supplemented with 1× N2 (Gibco), 0.5× B-27 (Gibco 17504-044), 2 mg ml−1 bovine insulin (Sigma), 1.37 μg ml−1 progesterone (Sigma), 5 mg ml−1 BSA fraction V (Gibco), 0.1 mM 2-mercaptoethanol (Sigma), 5 ng ml−1 mouse LIF (GlobalStem), 0.1 μM PD0325901 (Selleck Chem) and 0.3 μM CHIR99021 (Selleck Chem).
Myoblast cell culture and differentiation.
MM14 mouse skeletal myoblasts were passaged at 50–60% confluency every 1–2 days according to a protocol from the Wold Laboratory (https://www.encodeproject.org/documents/a5f5c35a-cdda-4a45-9742-22e69ff50c9c/@@download/attachment/C2C12_Wold_protocol.pdf). Undifferentiated myoblasts were grown in growth medium (20% FBS). Myogenic differentiation was initiated after reaching confluence by switching the cells to medium containing 2% horse serum supplemented with insulin. Differentiation was performed for 60 h by rinsing fully confluent cells once with PBS and adding 25 ml of low-serum differentiation medium. Fresh differentiation medium was changed every 24 h up to the 48 h time point. At 12 h afterwards, cells were crosslinked using SPRITE crosslinking procedures39,61.
Human cell culture.
Human H1 ES cells were maintained on Matrigel matrix (Corning, 354277) in feeder-free medium using mTeSR1 (Stemcell Tech, 85850). Every 4–5 days, cells were passaged using ReLeSR reagent (Stemcell Tech, 05872).
HEK293T cells were cultured in complete medium consisting of DMEM (Gibco, Thermo Fisher Scientific) supplemented with 10% FBS (Seradigm Premium grade HI FBS, VWR), 1× penicillin–streptomycin (Gibco, Thermo Fisher Scientific), 1× MEM non-essential amino acids (Gibco, Thermo Fisher Scientific), 1 mM sodium pyruvate (Gibco, Thermo Fisher Scientific) and maintained at 37 °C under 5% CO2. For maintenance, 800,000 cells were seeded into 10 ml of complete medium every 3–4 days in 10 cm dishes.
Generation of SPRITE samples
We generated DNA SPRITE maps in mouse myocytes derived from differentiated MM14 mouse myoblast cells and computed genome-wide nuclear speckle distances from >14 million SPRITE clusters (Extended Data Fig. 6a–d). The DNA SPRITE was performed using our previous protocol61 with one minor modification, which included diluting mouse myocyte cells twofold and keeping the DNAse concentration the same to reduce DNA fragment size to a range amenable to sequencing (200–1,000 bp) due to the difficulty of digesting myocyte DNA. We also performed RD-SPRITE maps in mouse ES cells and human ES cells using our previously published SPRITE protocol39,61. The RD-SPRITE protocol was performed in the same manner for mouse and human ES cells. In brief, cells underwent trypsinization for detachment and were subsequently crosslinked in suspension at room temperature using 2 mM DSG for 45 min. This was followed by a 10-min treatment with 3% formaldehyde to preserve RNA and DNA interactions in situ. To quench the formaldehyde crosslinker, 2.5 M glycine was added, reaching a final concentration of 0.5 M, for 5 min. The cells were then centrifuged, resuspended in 1× PBS + 0.5% RNase-free BSA (American-Bio, AB01243-00050) through 3 washes and subsequently flash-frozen at −80 °C for storage. The inclusion of RNase-free BSA was crucial to prevent RNA degradation, and RNase inhibitor (at a 1:40 ratio, using NEB Murine RNase Inhibitor or ThermoFisher Ribolock) was incorporated into all lysis buffers and subsequent steps to further protect against RNA degradation. Following lysis, the cells underwent sonication at 4–5 W of power for 1 min (with pulses of 0.7 s on and 3.3 s off) using a Branson sonicator. Chromatin was then fragmented using DNase digestion to achieve DNA fragments of approximately 150 bp to 1 kb in length. RNA integrity was assayed to ensure RNA average sizes of at least 1,000 nucleotides in length. After split-pool barcoding and sequencing, we computed genome-wide nuclear speckle distances from >4 million SPRITE clusters for mouse ES cells and >3 million SPRITE clusters for human ES cells.
SPRITE cluster size calculations
DNA SPRITE and RD-SPRITE were performed as previously described39. Unless stated otherwise, all analyses were based on SPRITE clusters of size 2–1,000 reads. These cluster sizes were chosen to be consistent with the analysis in our previous papers, in which we showed that many known structures such as topologically associating domains (TADs), compartments, RNA–DNA and RNA–RNA interactions, among others, occur within SPRITE clusters containing 2–1,000 reads.
Computing genome-wide speckle proximity scores from SPRITE data
To compute genome-wide speckle proximity scores by SPRITE, we used a two-step procedure: (1) we defined the active hub corresponding to interchromosomal contacts and (2) we computed the continuous speckle proximity score for each genomic locus.
Defining active hub regions.
To compute active hub regions from DNA SPRITE data, we computed an ICE-normalized, genome-wide DNA–DNA contact map at 1-Mb resolution and removed all intrachromosomal contacts to generate an interchromosomal contact matrix. We computed an interaction P value for each pairwise region within this interchromosomal contact matrix using a one-tailed binomial test, whereby the expected frequency assumes a uniform distribution of interchromosomal contacts. We retained interchromosomal regions that had a P value lower than a significance threshold (the precise significance threshold used was varied for each dataset to account for differences in sequencing depth and total number of contacts). To ensure accurate identification of interchromosomal contacts, we only retained interchromosomal contacts that were significant across three consecutive genomic bins. Using these sets of pairwise interchromosomal contacts, we clustered these interactions into a hub such that all the regions within a hub are connected to each other. The result of this procedure is a set of hubs in which each contains a set of genomic DNA regions that interact among themselves but do not interact across the hubs. For mouse ES cells, we previously found that this approach led to two hubs (clusters of regions) and defined these as the inactive hub (nucleolar hub) and active hub (speckle hub) based on gene expression and noncoding RNA localization37,39,45. In this paper, we used the speckle hub regions (Gene Expression Omnibus (GEO) database accession identifier GSE114242, samples GSM3154187–GSM3154193) defined in our previous paper37. These hubs were defined solely based on DNA contact frequencies. However, the enrichment of RNAs within these hubs enabled us to recognize these hubs as speckle or nucleolar. Similarly, for myocyte and human data, the speckle hub was selected from the resulting clusters based on gene expression.
Computing continuous speckle proximity scores.
Using these speckle hub regions, we computed a continuous speckle proximity score for each genomic region. Specifically, for each genomic region, we identified all SPRITE clusters containing the genomic region and at least one speckle hub region that was present on a distinct chromosome (if it overlapped only with a speckle hub region on the same chromosome, we did not count it to avoid counting contacts that might be due to other intrachromosomal structures). We weighted each overlapping cluster based on its cluster size (defined as 1/(cluster size − 1)). The speckle proximity score is the sum of all weighted scores across all overlapping clusters. In this way, genomic regions with a larger number of SPRITE clusters connecting it to an interchromosomal speckle hub would have a higher score than those with fewer. We previously showed that this continuous metric is correlated with the distance between each genomic region and nuclear speckles37.
Computing distance to speckle using seqFISH+ data
We analysed DNA seqFISH+ and immunofluorescence data previously generated in mouse ES cells (embryonic day 14)38 and available from Zenodo (https://zenodo.org/record/3735329#.Y1t7Xuxuf0o). We used this previous dataset and analysis to define distance to speckles for each of 2,460 genomic loci. This speckle distance calculation involved a three-step procedure: (1) segmentation of nuclear speckles, (2) DNA FISH spot detection and (3) computing the distance between speckles and DNA FISH spots in single cells. Note that these analyses were performed and described in a previous paper38, and we outline the procedures here simply for completeness.
Segmentation of nuclear speckles.
Nuclear speckles were identified through segmentation of SF3a66 immunofluorescence images as previously described38. Within the nucleus of each cell, we computed the intensity of the SF3a66 immunofluorescence signal for each voxel (x,y,z position). We converted these intensities into z scores by subtracting the mean immunofluorescence intensity across the entire nucleus and dividing this by the standard deviation of values. We thresholded voxels containing a z score > 2 (intensity value exceeding 2 standard deviations of the mean signal within the nucleus). We then merged adjacent voxels that exceeded this threshold. We previously showed that this ‘thresholding approach’ to segmentation is more robust to small differences in features sizes than other segmentation approaches (for example, Otsu’s thresholding). A visual example of the results of this segmentation is shown in Supplementary Fig. 1.
DNA FISH spot detection.
We identified the location of each genomic DNA locus by using the Laplacian of Gaussians filter to enhance spot detection, reduce noise and sharpen spot edges to define regions of rapid intensity change, which are indicative of the edges of FISH spots.
The Laplacian of Gaussians filter used sigma = 1 and the scipy.ndimage.gaussian_filter function in Python (v.3.7.13). We then binarized the image by retaining all voxels that exceed a selected threshold. The precise threshold used varied and was selected through an automated procedure that accounts for the signal present in the first hybridization round. Using this thresholded and binarized image, we segmented the binarized voxels into a merged volume using a 3D local maxima finder.
Computing the distance between DNA and speckles.
We computed the distance between DNA regions and the segmented nuclear speckles by computing the centre position of the segmented DNA region using a 3D radial centre algorithm (DNA position). We then computed the sphere outlining the outer edge of each segmented nuclear speckle (sphere of the speckle). For each DNA and speckle, we computed the Euclidian distance between the DNA position and each position on the sphere of the speckle. The score was computed as the minimum distance relative to any location on the sphere. We then computed this score between a given DNA position and all segmented speckles and retained the minimum score between a DNA position and any speckle region as the speckle distance score.
The result of this procedure is a speckle distance for each genomic locus measured. We repeated this procedure across each of the 446 single cells and across the 2,460 1-Mb tiled genomic regions probed by DNA FISH. We computed the average of these distances across all cells to plot the mean speckle distance across all loci shown in Fig. 1. The calculated micrometre distances of DNA loci to SF3a66 nuclear speckle regions are available in Supplementary Table 3 of our previous study38 and Supplementary Table 1 of this study.
Comparison of SPRITE and seqFISH+
To compare SPRITE and seqFISH+ immunofluorescence measurements, we used SPRITE speckle proximity scores from contact maps binned at 1-Mb resolution, focusing only on SPRITE clusters containing 2–1,000 reads and downweighting for cluster size (described above). The distance for seqFISH+ represented the average of minimum distance between the genomic DNA spot and the periphery of the SF3a66 domain. When a DNA region and speckle are close, the seqFISH+ distance is expected to be low and the SPRITE speckle proximity score is expected to be high. We then computed a Spearman rank correlation between SPRITE and seqFISH+ measurements across all 2,460 genomic positions that were probed by seqFISH+. A juyptr notebook containing the code and datasets to perform this comparison is available at GitHub (https://github.com/GuttmanLab/speckle).
Comparing speckle proximity score measured by SPRITE and TSA–seq in H1 human ES cells
To measure the correlation between SPRITE speckle proximity score and SON TSA–seq, we downloaded TSA–seq data generated for H1 human ES cells from a previous study47. We computed speckle proximity scores from our SPRITE data by computing the speckle hub in our H1 dataset at 1-Mb resolution (as previously described37) and then computing the weighted contact frequency of each genomic bin (at 100-kb resolution) contacting the speckle hub. We used our SPRITE speckle proximity scores for H1 human ES cells at 100-kb resolution and compared these to the average TSA-seq speckle score of genes located within the same 100-kb bins throughout the genome.
Comparing SPRITE datasets
To map and compare speckle proximity scores (mouse ES cells versus myocytes; human SPRITE datasets) in each cell type, we performed a quantile normalization of the speckle hub contacts for each cell line to account for differences in coverage for each SPRITE.
To assess the significance of differences in speckle proximity between myocytes and ES cells, we began by collecting the observed speckle distance values from myocyte and ES cell SPRITE clusters. Subsequently, we combined these two sets of cluster files to create a unified myocyte–ES cell cluster file. To establish a baseline for comparison, we introduced randomly sampled cluster interactions by randomly permuting these interactions 100 times for every 100-kb genomic bin. For each permutation, we calculated the fold change for each shuffled score. We then compared the observed fold change to the distribution of fold changes generated by these permutations.
If the observed fold change was greater than that of 99% of the permutations, we considered it significant in either cell type 1 or cell type 2. Notably, cases in which the fold change was not significant could be due to speckles remaining close in both cases or distant in both cases. To address this possibility, we computed the median speckle proximity from a representative speckle-close region of mouse chromosome 2 (ref. 37) and identified 100-kb regions in both myocytes and ES cells that were equal to or above this median value, designating them as speckle-close regions for each cell type.
Last, we cross-referenced our list of significantly fold-change values with our merged speckle list, classifying the interactions as ES cell preferred, myocyte preferred, shared speckle region or neither based on their relationship to speckle proximity.
5EU nascent RNA labelling and capture
Mouse ES cells were cultured as described above, lifted with TVP, washed and suspended in 2i/LIF medium supplemented with 1 mM 5EU (Jena) for 10 min with shaking at 750 r.p.m. on a Thermomixer (Eppendorf). Cells were then pelleted for extraction. A link to the 5EU–seq protocol can be found on the Guttman Laboratory website at https://guttmanlab.caltech.edu/files/2024/02/5EU-RNA-seq.pdf.
Total RNA was collected using a RNeasy Minikit (Qiagen). 5EU-labelled RNA was biotinylated by mixing samples with water, 100 mM HEPES, 1 mM biotin picolyl azide (Click Chemistry Tools), Ribo RNase inhibitor, premixed 2 mM CuSO4 and 10 mM THPTA, and finally 12 mM sodium ascorbate. Biotinylated RNA was then captured as follows: MyOne Streptavidin C1 Dynabeads (ThermoFisher Scientific) were first washed 3 times in urea buffer (10 mM HEPES, pH 7.5, 10 mM EDTA, 0.5 M LiCl, 0.5% Triton X-100, 0.2% SDS, 0.1% sodium deoxycholate, 2.5 mM TCEP and 4 M urea) followed by 3 additional washes in M2 buffer (20 mM Tris, pH 7.5, 50 mM NaCl, 0.2% Triton X-100, 0.2% sodium deoxycholate and 0.2% NP-40). Washed beads were mixed with 3 parts 4 M urea buffer and 1 part biotinylated RNA and incubated for 60 min at 900 r.p.m. in a thermomixer at room temperature. After magnetic separation, beads were washed 3 times with M2 buffer followed by 3 washes with urea buffer at 37 C at 750 r.p.m. for 5 min. RNA was eluted from beads in 2 sequential elutions by incubating with elution buffer (5.7 M guanidine thiocyanate and 1% N-lauroylsarcosine; both Sigma) at 65 °C for 2 min, repeating with more elution buffer for a second elution. The elutions were pooled, diluted with urea buffer, incubated with pre-washed streptavidin beads, washed and eluted for 2 additional rounds exactly as described above for a total of 3 sequential captures. Final elutions were pooled, cleaned with Zymo RNA Clean and Concentrate following the manufacturer’s protocols.
Captured RNA was used for library construction as previously described62.
snRNA enrichment calculation from RNA and DNA SPRITE
We computed RNA–DNA contacts frequencies for U1, U2, U4 and U6 snRNAs in 1-Mb or 100-kb bins across the genome, weighted by cluster size. Specifically, we took all SPRITE clusters containing U1 (or U2, U4 or U6, respectively) and counted the number of reads within these clusters that overlap each 1-Mb or 100-kb genomic bin. We weighted each read count by the cluster size that it was observed in. We summed these weighted scores across all U1-containing clusters to generate a U1 contact profile genome-wide. For the same 1-Mb and 100-kb bins, we computed speckle proximity scores for each genomic bin as described above.
To calculate transcription rate, we used data generated from mouse ES cells labelled for 10 min with 5EU and sequenced (described above). We quantified nascent RNA expression by aligning reads to mm10 using kallisto-bustools63 to two references separately: a cDNA reference (for exon reads and exon–exon junction reads) and a genomic DNA reference genome (for exon–intron and intron reads). We subsequently normalized the counts per gene by its length and focused our subsequent analyses only on genes with nascent RPKMs with a value of at least 1.
To compare snRNA enrichment and speckle frequency genome-wide, we defined speckle-far regions as the genomic regions corresponding to the lowest 5% of speckle proximity scores and speckle-close regions as the top 5% of genomic regions. To normalize all snRNA values to the same distribution to enable us to compare them to each other and to display them on the same scales, we performed quantile normalization on the U1–U6 snRNA contact frequencies.
Because speckle proximity is correlated with the density of RNA PolII, we wanted to ensure that the observed increases in snRNA density were not simply due to increased transcription or nascent pre-mRNAs in these regions. To do this, we focused on genomic bins that have comparable transcribed gene density. Specifically, we counted the total number of reads contained within each genomic bin observed within the nascent 5EU dataset. This metric integrates both the level of transcription per gene and the density of genes contained within an individual genomic bin. We then compared genomic bins containing comparable integrated transcription levels between speckle-close and speckle-far regions. We filtered the genomic regions into five bins based on percentiles of transcription density for speckle-close and speckle-far regions. The resulting analysis involved plotting snRNA density within these matched regions of nascent RNA transcription density.
Additionally, we controlled directly for transcription level by comparing only regions of equivalent expression. Specifically, we thresholded regions corresponding to low, medium or high expression. To do this, we defined three bins of expression: high (>7.5 reads per kilobase mapped reads (RPKM)), medium (2.5–7.5 RPKM) and low (1–2.5 RPKM). Density plots for speckle-close and speckle-far regions, for each snRNA, and for each expression level were plotted using the seaborn kde function.
To compute snRNA enrichment for speckle-close and speckle-far regions containing the exact same densities of splice junctions, we computed the number of junctions per 100-kb bin across the genome. We randomly sampled these regions to analyse an identical number and identical distribution of junction densities between speckle-close and speckle-far regions (Extended Data Fig. 3a). We filtered for regions with similar nascent expression and the same distribution of junction counts and plotted contact frequencies of U1, U2, U4 and U6 snRNAs for the corresponding 100-kb bins, weighted by cluster size.
U1 snRNA enrichment calculation from psoralen crosslinking (AMT RAP-RNA)
To compute direct U1 snRNA–pre-mRNA binding, we re-analysed data that we previously generated using RAP-RNA on U1 after crosslinking a psoralen derivative (AMT)43 (GEO identifiers GSM1348350 (input RNA AMT) and GSM1348348 (U1 AMT RAP-RNA)). In this procedure, cells are treated with a psoralen crosslinker to form direct crosslinks between directly base-pair-hybridized RNA–RNA sequences. Affinity capture for U1 snRNA and sequencing of associated RNAs identifies the RNAs that were directly bound to U1. To normalize for transcript abundance, input RNA libraries were sequenced in parallel.
To control for U1 occupancy on pre-mRNAs of varying expression, the enrichment of U1 snRNAs over each 5’ splice site of a pre-mRNA was computed by counting the number of U1 reads that fell within a 200 bp of the 5’ splice site and subtracting the read coverage over this region observed in the input, in which the input sample reflects mRNA levels. To exclude junctions that are not well covered and junctions that have other artefacts that lead to strong read pile-ups in the input (for example, repeats), we excluded all junctions containing zero or negative values (which represent junctions with equal or fewer U1 reads than input reads) and summed the normalized counts across 100-kb genomic intervals. We note that this is a conservative approach because we observed that the distribution of zero and negative values are preferentially enriched within speckle-far relative to speckle-close regions, which may reflect lower U1 engagement on these junctions. Focusing only on junctions containing a positive (≥1) score, we computed the number of counts for each individual junction within speckle-close and speckle-far regions and observed a clear shift towards higher coverage in speckle-close relative to speckle-far junctions. Because the counts for each junction are relatively low, we binned junction counts into the same 100-kb bins computed as above. We plotted the density for all speckle-close and speckle-far regions for the U1 snRNA using the seaborn kde function. Finally, to ensure that these differences do not reflect differences in number of junctions within each 100-kb bin, we plotted the enrichment per bin as a function of number of junctions and observed a clear separation for each size.
Finally, even when directly comparing the distribution of counts for each individual junction within speckle-close and speckle-far regions, we observed a clear shift towards higher coverage in speckle-close relative to speckle-far junctions. To explore this, we computed the normalized U1 counts (U1 input) for each junction and used all junctions containing a positive (≥1) score and split them into speckle-close and speckle-far regions. We then asked whether the distribution of positive counts were similar or if there was a skew towards larger values in the speckle-close junctions. To do this, we computed the number of junctions containing each discrete integer score (1, 2, 3, 4, 5 and ≥6) and compared this count distribution between speckle-close and speckle-far regions using a chi-square test of association. We observed a chi-square P < 0.0001 (chi-square test statistic = 67.63, degrees of freedom = 5). For example, we observed a 2.6-fold increase in the proportion of junctions containing a score of ≥6 within speckle-close relative to speckle-far regions.
Although the distribution of U1 scores within 100-kb regions were significantly higher for speckle-close versus speckle-far regions, the effect sizes were smaller than observed when analysed using SPRITE. However, this difference probably reflects the known reduced dynamic range of the AMT dataset. To explore this aspect, we analysed this dataset in an orthogonal way. We defined all 100-kb genomic bins that have enriched numbers of U1 binding at 5’ splice sites by summing the U1 counts for each genomic bin and then generating 100 random permutation bins containing the same number of junctions. For each permutation, we sampled from the distribution of all observed junctions. The idea here was to ask what the distribution of scores would look like if we have n junctions within a bin and we constructed these n junctions at random. We then retained only genomic regions that exceeded these permuted values such that the probability of observing a count as high as the observed in the 100 random permutations was less than 5%. We then took these significantly enriched genomic windows and plotted the distribution of speckle proximity scores compared with the total speckle proximity score distribution. We observed a striking increase in speckle proximity score at enriched U1 regions relative to all genomic regions, which provided confirmation that pre-mRNAs transcribed from speckle-close regions are enriched for direct U1 binding.
Splicing efficiency calculations from various RNA-seq methods
Total chromatin RNA-seq data64 were re-analysed from our previous study (GEO identifier GSM2123095) and re-aligned using the kallisto-bustools workflow63 to two references separately: a cDNA reference (for exon reads and exon–exon junction reads) and a genomic DNA reference genome (for exon–intron and intron reads). The same alignment procedure was done for the newly generated 5EU nascent RNA dataset. The splicing efficiency metric was computed as the fraction of normalized exon counts over normalized intron + exon (total) counts. We filtered for speckle-close and speckle-far regions as described above and plotted the distribution of per cent splicing using the seaborn kde function. For the continuous distribution plot, we plotted all speckle proximity scores (x axis) versus the average splicing ratio in each of 50 bins, in which each bin contains at least 20 genes.
To calculate the splicing efficiency for genes of similar expression in mouse ES cells, we first computed the normalized expression of genes (≥2 exons per gene) by dividing the total counts by the length of the gene. This normalized expression was rank-normalized from 0 to 1, and the top 20% of expressed genes were compared. This corresponded to 15 speckle-far genes and 96 speckle-close genes. For all genes ≥2 exons, this corresponded to 392 speckle-far genes and 394 speckle-close genes. The empirical cumulative distribution function for expression and splicing efficiency were plotted using the seaborn ecdfplot function.
Splicing efficiency calculation from RD-SPRITE
Because RD-SPRITE captures interactions occurring between DNA and RNA, we reasoned that any mRNA that was in a SPRITE cluster with its own DNA locus corresponded to nascent chromatin associated RNA. Indeed, we previously showed that this approach accurately captures and quantifies nascent pre-mRNA levels45. Using these clusters, we computed splicing efficiency based on the total number of exon reads in a nascent genomic bin divided by the total number of exon and intron reads (total pre-mRNA reads) within that same bin. To ensure that we had broad coverage to estimate this frequency, we filtered for genomic regions that contained at least 50 RNA reads (exons + introns). In both RD-SPRITE datasets analysed (mouse ESCs and human ESCs), we filtered for speckle-close and speckle-far regions as described above and plotted the distribution of per cent splicing using the seaborn kde function. For the continuous distribution plot, we plotted all speckle proximity scores (x axis) versus the average splicing ratio in each of 50 bins, in which each bin contains at least 3 genomic regions.
Splicing analysis of C2C12 myotubes from nuclear RNA-seq
Single-cell SPLiT-seq65 RNA-seq data from mouse C2C12 myoblasts were obtained from GEO accession identifier GSE168776 (ref. 66). Sequencing reads from the seven short-read sequencing sublibraries (sample identifiers GSM5169184, GSM5169185, GSM5169186, GSM5169187, GSM5169188, GSM5169189 and GSM5169190) associated with that accession identifier were used for analysis. The kb-python (v.0.28.0), kallisto (v.0.50.0) and bustools (v.0.43.0) software63 were used to process the dataset as follows. The ‘kb ref’ (with--workflow=nac) command was used to generate a kallisto index of nascent and mature RNA transcripts prepared from the GRCm39 genome reference. The ‘kb count’ command was used to map reads to the index and to generate three cell-by-gene count matrices containing unique molecular identifier (UMI) counts. The three matrices correspond to UMI counts from nascent, mature and ambiguous reads. Nascent reads are those that span an intronic region and are therefore considered unspliced, mature reads are those that span an exon–exon splice junction and are therefore considered spliced, and ambiguous reads are those that are contained entirely within an exon (and hence could be assigned to either unspliced or spliced RNA transcripts). The three count matrices were subsetted to contain only the quantifications from the random hexamer primed reads from the wells containing differentiated C2C12 myoblast (that is, myotube) nuclei as determined by the final 8 bp of the reads in the R2 read files. Pseudobulk analysis was performed by adding up the UMI counts across all rows in the final count matrices to obtain a single mature, nascent and ambiguous count for each gene. UMIs assigned to more than one gene were not considered.
Difference in splicing efficiency calculations
Although there is a clear relationship between speckle proximity and splicing efficiency when measured by multiple distinct metrics (that is, chromatin RNA-seq, 5EU nascent RNA-seq and RD-SPRITE), the raw splicing efficiency can differ according to the assay used. This does not affect our analyses when comparing samples within a cell type, but would lead to systematic issues when comparing between cell types. To account for this possibility and enable comparison of splicing efficiency measurements between cell types, we rank-normalized the splicing efficiencies of all expressed genes (that contain at least one intron) from 0 to 1. Subsequently, we calculated the difference in splicing efficiency per gene by subtracting the normalized splicing efficiencies between the two specific cell types: mouse ES cells and mouse myocytes. We plotted the difference in the normalized splicing efficiency (mouse ES cell – myocyte) versus the difference in normalized SPRITE speckle proximity score (at 100-kb resolution) for 50 bins. We analysed bins that contained at least 20 regions.
Difference in speckle proximity score versus difference in PolII density calculations
We compared the change in speckle proximity score between mouse ES cells and mouse myocytes versus the change in S2 PolII density in the same cell types. Specifically, we calculated the difference in speckle proximity score per 1-Mb bin by subtracting the normalized splicing efficiencies between the two specific cell types. We rank normalized the S2 PolII density between the two cell types at 1 Mb resolution so that we could compare PolII occupancy across the entire genomic segment, rather than per gene. We plotted the difference in normalized SPRITE speckle proximity score (at 1-Mb resolution) versus the difference in normalized PolII density (mouse ES cell – myocyte) for 50 bins. We analysed bins that contained at least ten regions.
Generation of MS2 bidirectional reporter plasmid (GFP and BFP)
The bidirectional splicing reporter was derived from an existing expression plasmid carrying a bidirectional promoter driving expression of eGFP and mRuby (gift from M. Elowitz). mRuby was replaced with BFP using the restriction sites SalI and MluI.
To place the reporter Irf7 gene upstream of self-cleaving peptide 2A (P2A) and eGFP in a plasmid containing these cassettes (gift from D. Majumdar), Gblocks from IDT encoding exons 5–6 of mouse Irf7 (ENMUST00000026571.10) were designed to include the endogenous intron and Gibson assembly overhang sequences and assembled together. The Gblock also included a Kozak sequence and ATG start codon upstream of exon 5 (ref. 67).
To combine these pieces, the restriction enzymes AflII and ClaI were used to generate a vector backbone from the modified bidirectional expression plasmid. The IRF7 splicing reporter (including P2A–GFP) cassette was PCR-amplified with these same restriction enzyme sites flanking the amplicon. Once digested, the PCR fragment was ligated into a MSCV vector (PIG, Addgene)68 to generate the splicing reporter. This splicing reporter has a stop codon embedded within the intron, thereby only when the reporter is spliced will eGFP be translated.
The same cloning strategy was used for CORO1B (exons 4–5, NC_000011.10: c67443809–67435510, Homo sapiens chromosome 11) and FRG1 (exons 3–4, NG_008142.1, Homo sapiens FSHD region gene 1) minigenes derived from a previous study29. The forward primer was designed to include a BstBI restriction site, the Kozak sequence and the ATG start codon, whereas the reverse primer included an AscI restriction site. Genomic DNA was amplified using these primers, gel-purified, double-digested with the appropriate restriction enzymes and then ligated into the splicing reporter backbone without MS2.
Finally, we performed site-directed mutagenesis to insert a single MS2 stem–loop sequence downstream of the predicted U1 binding site and upstream of the branch point recognition site of the intron to avoid interfering with splicing. We introduced the MS2 stem–loop into the intron to enable recruitment of the nascent pre-mRNA splicing reporter specifically to MCP-tagged proteins. We co-transfected the MS2 and tagged protein constructs into HEK293T cells. Splicing, as measured by GFP fluorescence, was assayed 24 and 48 h after transfection by flow cytometry (Macsquant) and analysed using FlowJo analysis software. Transfections were performed using BioT transfection reagent (Bioland) according to the manufacturer’s recommendations. Transfected constructs included SRRM1, SRSF1, SRSF3 and LBR; all constructs were fused to a C-terminal mCherry tag. Constructs harbouring the MCP tag were fused to two tandem repeats of the MCP peptide at the amino terminus.
Integration of reporter construct into specific genomic DNA regions using CHoP-In
To distinguish the specific impact of splicing efficiency from other variables such as transcription and export efficiency, we integrated the bidirectional reporter plasmid into a genomic DNA region that is speckle-close or speckle-far in mouse ES cells. These cell lines allowed us to interrogate the relationship between speckle proximity and splicing efficiency for the same gene at two different nuclear locations. We achieved this through a CRISPR–Cas9-based method known as CHoP-In69. For each genomic region (Tcf3 locus and Grik2 locus), gRNAs were designed (Tcf3: cggaacatgtctcccgccgc; Grik2: gccagcgagagcgc aagtga) and cloned into a gRNA expression vector. Recombination templates were generated by PCR amplification of our bidirectional splicing reporter and attaching above gRNAs, including their PAM sequences in orientations allowing for integration.
These recombination templates, gRNA expression plasmids and a wild-type Cas9 expression plasmid that also confers puromycin resistance were co-transfected into mouse ES cells using a Neon electroporator (ThermoFisher). Cells were selected using 1 μg ml−1 puromycin for 48 h and then expanded. FACS was used to isolate cells that were positive for BFP.
The integration of the CHoP-In template at the specific genomic site for each gene was verified by amplifying the insert using primers sets that flanked the integration site and confirming the presence of an amplicon for which the size reflected that of the recombination template.
The GFP in the bidirectional reporter was used as an indicator of splicing levels, with BFP serving as a measure of splicing independent (for example, transcription) effects. These fluorescent levels were measured on a Sony MA900 or a Macquant Vyb.
To analyse only cells in which integration was successful, we FACS to sort for BFP-positive cells and measured the levels of GFP and BFP per cell (Supplementary Fig. 2a). This was done because every successful integrant is expected to express BFP (transcription) but not necessarily GFP (splicing). We plotted GFP as a function of BFP and fit a Lowess curve to the observed values. To determine whether the two distributions were distinct, we estimated the variance of these distributions by performing a bootstrap procedure. Specifically, we randomly sampled BFP-positive cells with replacement from the speckle-close and speckle-far populations. For each permutation, we fit a Lowess curve and repeated this process ten times. We then plotted the entire range of values of the Lowess curve for each of the ten randomized samples.
Immunofluorescence followed by DNA FISH
To confirm distance to speckles of the integrated reporters, we performed DNA FISH and immunofluorescence. Specifically, immunofluorescence and DNA FISH were performed using a 96-well glass-bottom plate. The protocol was adapted from a previous study38. The wells were initially cleaned with 100% ethanol and allowed to air-dry for 20 min. Then they were coated with a solution of 10 μg ml−1 of PDL and left to incubate at room temperature for 2 h. After this, the wells were washed with 1× PBS and subsequently coated with human laminin. The plate was sealed and incubated at 37 °C for more than 1 h. Cells were detached using trypsin, neutralized with medium and suspended as single cells. These cells were then seeded onto the 96-well plate and cultured in 2i medium with 1% FBS for 8 h. After 8 h, a 4% formaldehyde solution was added to fix the cells for 10 min. The fixed cells were then washed twice with 1× PBS and stored in 70% ethanol at −20 °C for at least overnight.
The next day, permeabilization and pre-treatments were carried out. Initially, cells were permeabilized using a solution of 0.5% Triton X-100 in 1× PBS for 15 min at room temperature. Afterwards, the cells were washed with 1× PBS 3 times. Subsequently, blocking was performed with a custom blocking solution containing 1× PBS, 1% BSA, 0.3% Triton X-100, 0.1% dextran sulfate and 0.5 mg ml−1 of salmon sperm DNA. Immunofluorescence was performed initially for SRRM1 using a 1:200 dilution in the blocking solution, incubating at room temperature for 2 h. Following this, the cells were washed 3 times with 1× PBS with Tween, and secondary antibodies and anti-rabbit AlexaFluor488 were applied using a 1:500 dilution for 40 min at room temperature. DAPI solution was used for washing, and day 1 imaging was carried out to capture DAPI and SRRM1 signals as the DNA FISH heating steps would remove the speckle signal.
After imaging and calibrating the plate/LSM980 scope to ensure the same position for analysis by DNA FISH the next day, post-fixation was conducted. Freshly made 4% paraformaldehyde in 1× PBS was used for post-fixation, lasting for 5 min at room temperature. The sample was then washed with 1× PBS 6 times and incubated for 15 min. Post-fixation with 1.5 mM BS(PEG)5 and 1× PBS for 20 min at room temperature followed, and the sample was washed 3 times with 100 mM Tris-HCl pH 7.4, with each wash lasting 5 min. The final wash consisted of 3 rinses with 1× PBS before leaving it to air-dry.
The next step involved treating the sample with a 100-fold diluted RNaseA/T1 and 1× PBS for 1 h at 37°C, followed by a wash with 1× PBS. Subsequently, the sample was incubated with a 50% denaturation buffer at room temperature for 15 min, consisting of 2× SSC and 50% formamide.
For the DNA FISH portion, the primary probe hybridization was initiated by washing the sample multiple times with 2× SSC. Subsequently, a 40% hybridization buffer with 10 mM of 35-mer primary probes targeting specific regions was applied, and the sample was incubated for more than 24 h at 37 °C in the dark within a humidity chamber (see Supplementary Table 6 for probe sequences). The hybridization buffer comprised 40% formamide, 10% dextran sulfate and 2.25× SSC.
On the following day, the sample was washed twice with 40% wash buffer, followed by another wash with 40% wash buffer and a 15-min incubation at room temperature in the dark. After that, the sample was washed 3 times with 2× SSC. A 10% ethylene carbonate buffer was introduced, along with 50 nM readout probes labelled with AlexaFluor 647. The sample was washed twice with 12.5% wash buffer, once with 4× SSC and then washed with DAPI solution and anti-bleaching buffer base. Finally, anti-bleaching buffer was added before imaging on a LSM980, using the same saved positions as day 1, but now including the AlexaFluor 647 channel.
We measured the distance of integrated genomic loci by identifying the centroid of the DNA FISH spot and manually computing the micrometre distance in Fiji between the centroid and the periphery of the nearest SRRM1 spot. At least 25 cells were quantified for each condition.
Plasmid generation for the MS2–MCP assay
mCherry-fused, MCP-tagged expression plasmid.
The Gateway destination plasmid pCAG-NSTF-DEST-V5 (gift from P. McDonel) was digested with SrfI and AgeI to generate a vector backbone fragment. A Gblock from IDT encoding a portion of the ccdB survival cassette, an attR2 recombination sequence, a V5 tag and mCherry was digested with these same restriction enzymes. This insert was ligated to the vector fragment to add mCherry in-frame, generating the −MCP Gateway destination vector.
To generate the +MCP version, NheI and AscI restriction enzymes were used to remove the NSTF (N-terminal SpyTag-TEV-Flag) cassette and to replace it with 2× MCP amplified from another plasmid (gift from J.Jachowicz). These destination vectors were used in Gateway LR recombination reactions with entry clones for each protein of interest. Entry clones were obtained from DNASU.
ΔNS-SRRM1 entry clone.
The SRRM1 entry clone from DNASU was modified using a Q5 site directed mutagenesis kit (New England Biolabs) to delete the predicted region responsible for nuclear speckle localization as annotated using UniProt (https://www.uniprot.org/uniprotkb/Q8IYB3/entry). The resulting clone lacked one additional amino acid at the C terminus as determined by Sanger sequencing of the clone and alignment with the predicted sequence.
Imaging analysis for MS2–MCP reporter assay
RNA FISH.
To visualize RNA localization in MCP–MS2 recruitment assays, we co-transfected HEK293 cells with splicing reporter and domain recruitment constructs then performed single-molecule RNA FISH as previously described70. At 24 h after transfection, we rinsed samples once with 1× PBS then fixed in 4% buffered formaldehyde for 10 min at room temperature. Following fixation, we rinsed the samples twice with 1× PBS then permeabilized in 70% ethanol overnight at 4 °C. For hybridization, we rinsed the samples once with wash buffer (10% formamide 2× SSC) then added hybridization buffer (10% formamide, 10% dextran sulfate and 2× SSC) containing RNA FISH probes targeting GFP RNA. These probes were provided by A. Raj (University of Pennsylvania). After adding the hybridization solution, we covered samples with glass coverslips and hybridized them overnight at 37°C in a humidified container. Following hybridization, we rinsed the samples once with wash buffer to remove coverslips and then washed twice for 30 min at 37 °C. We added 50 μg ml−1 DAPI to the second wash to stain nuclei. Following washes, we rinsed the samples twice with 2× SSC, added SlowFade Diamond Antifade solution and proceeded with imaging on a Nikon spinning-disk confocal equipped with Andor Zyla 4.2P sCMOS camera, Nikon LUNF-XL laser unit, and Yokogawa CSU-W1 with 50 μm disk patterns. For each sample, we selected at least 10 positions on the basis of DAPI signal and acquired z stacks at 0.5 μm intervals using a 60× oil objective.
Immunofluorescence.
We fixed cells on coverslips with 4% formaldehyde in PBS for 15 min at room temperature and permeabilized with 0.5% Triton X-100 in PBS for 10 min at room temperature. After washing twice with PBS containing 0.05% Tween (PBST) and blocking with 2% BSA in PBST for 30 min, we incubated cells with primary antibodies for anti-SC35 antibody at 1:200 dilution (Abcam, ab11826) overnight at 4°C in 1% BSA in PBST. After overnight incubation at 4 °C, we washed cells 3 times in 1× PBST and incubated for 1 h at room temperature with secondary antibodies labelled with Alexa fluorophores (Invitrogen) diluted in 1× PBST (1:500). Next, we washed coverslips three times in PBST, rinsed them in PBS and then double-distilled H2O, mounted them with ProLong Gold with DAPI (Invitrogen, P36935) and stored them at 4 °C until image acquisition.
Image analysis
To quantify RNA recruitment to nuclear lamina or speckles, we used Cellpose (https://github.com/mouseland/cellpose) to segment nuclear boundaries based on the DAPI signal and used the smFISH pipeline from the Raj Laboratory (https://github.com/arjunrajlaboratory/rajlabimagetools) to localize intranuclear reporter RNA70,71. We then quantified mCherry fluorescence intensity at the position of each reporter RNA molecule. To account for heterogeneity in mCherry expression across cells, we calculated the rank pixel intensity to measure relative RNA–mCherry co-localization across conditions. We note that expression heterogeneity precluded us from segmenting speckle domains consistently across cells. In addition, to account for heterogeneity in co-transfection efficiency, we had a blinded author manually select non-mitotic cells co-transfected with both the splicing reporter and the domain recruitment construct.
Because of the sequence and length (GUACAUCUGGUCCAUCCU UCCUAGCUGCGUCCUGGUGGCGC AGGUGUGGGGGAUCGGCAGGU GCCUACCACUAUGCUGUCUAUUACAG; 88 nucleotides) the intron in our splicing reporter, we were unable to design smFISH probes that selectively target nascent RNA. Instead, we used a probe set targeting exons present in both nascent and mature RNA. Because only nascent (unspliced) RNA contain the MS2 hairpin, our results probably underestimate the extent of reporter RNA recruitment.
Overexpression of MS2–MCP constructs in HEK293T cells
To test whether directed recruitment of pre-mRNA to nuclear speckles is sufficient to increase splicing efficiency, we performed our MS2–MCP experiments in a transient overexpression system in human HEK293T cells. Specifically, we wanted to extract the components from other confounding factors associated with speckle proximity and splicing efficiency in the endogenous context (for example, PolII activity, gene architecture and chromatin structure). This type of sufficiency experiment is traditionally performed by purifying the relevant components and ‘reconstituting’ the system. However, because the mechanism we are describing relies on spatial positioning within the nucleus, a traditional reconstitution experiment would not work.
For the MS2–MCP experiments that required a wide range of protein expression, human HEK293T cells were used instead of mouse ES cells because they can be efficiently transfected such that most cells express high levels of the multiple plasmids required simultaneously. By comparison, mouse ES cells are more difficult to transfect and therefore more difficult to obtain the multiple constructs required into a large number of cells at the same time. In practice, we could not achieve strong expression of these components within ES cells to perform these experiments. Transient transfection also enabled investigation of the effect of varying levels of transcription (with and without recruitment) on splicing efficiency.
Normalization of mCherry–fusion proteins
For each construct, we obtained two types of fluorescence data: one from FACS and the other from microscopy. To determine the appropriate percentile of mCherry expression to use, we selected the 25th percentile of mCherry expression in our FACS analysis. We found that the same 25th percentile of mCherry expression in our microscopy data corresponded to cells with the correct nuclear localization. Because excessive fusion protein expression can lead to improper localization within the nucleus, we excluded these values from our analysis.
GFP expression as a function of BFP
For each construct (±MCP), we sorted on BFP (any amount of BFP over background) and mCherry (25th percentile of mCherry expression, as noted above). We sorted on BFP because it should always be expressed and because splicing (GFP) might not be present (Supplementary Fig. 2b). As a control, we also sorted cells that contained constructs expressing GFP only, BFP only or mCherry only to ensure there was no spillover of the fluorescence detection between constructs. Additionally, we sorted untransfected cells to set a baseline threshold to filter out cells with background autofluorescence. To that end, because the range of expression is variable (for example, owing to differences in transfection efficiency), we thresholded cells that contained the same range of BFP fluorescence intensity (between 0 and 5 for ΔGFP comparisons of all constructs and 0 to 10 for SRRM1 and LBR constructs). The upper threshold of 5 for BFP fluorescence was chosen because that represents the upper bound of BFP expression for the protein construct with the overall lowest levels of expression. To analyse the relationship between GFP and BFP levels, we fit a Lowess curve across BFP versus GFP for all cells. We performed this analysis for each individual replicate sample and for the +MCP and −MCP sample individually. We plotted the Lowess curve for each replicate. 2D FACS scatterplots for each protein construct is included in Supplementary Fig. 3.
Difference in GFP expression calculations
To compute differences between +MCP and −MCP values, we thresholded cells that contained BFP fluorescence intensity between 0 and 5, and took the average GFP fluorescence intensity for 50 equally spaced BFP bins. For each x value (50 BFP bins), the difference in average GFP fluorescence was computed between MCP and no MCP constructs. The average difference of at least three replicates were plotted separately for all constructs and the Lowess curve for the ΔGFP of each sample was fit to each individual sample.
Nonlinear regression statistics
Data from each construct (ΔGFP for SRRM1, SRSF1, SRSF3 and LBR) were fitted using a four-parameter logistic curve, and goodness of fit was calculated using GraphPad Prism 9 software.
Data visualization
Scatter plots were generated using GraphPad Prism (v.9.5.1), and kernel density plots were generated using the Seaborn package (v.0.13.2). Pandas (v.2.2.1) was used for processing data before visualization. Sequencing data were visualized using IGV (v.2.9.4).
Statistics and reproducibility
Data are presented as the mean ± s.e.m. or as indicated in the figure legends. Statistical analyses were performed using two-sided z-tests. Methods and details on individual statistical analyses and tests can be found in the respective figure legends. The number of times individual experiments were replicated is noted in the respective figure legends. For SPRITE experiments, one replicate mouse myocyte and two replicates for H1 human ES cells were performed.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Correlation between speckle proximity scores between SPRITE datasets and TSA-seq for SON.
A. Chromosome wide view of speckle proximity score at 1 Mb-resolution for three replicates of SPRITE datasets in mouse ES cells. Two collected in Quinodoz et al Cell 2021 and a third dataset collected for this manuscript. Speckle hub regions highlighted on chromosomes in red. Gene density track on bottom. Correlation of SPRITE experiments between: B. RD SPRITE Cell 2021 (Replicate 1) and RD SPRITE Cell 2021 (Replicate 2) (spearman r = 0.94, p < 0.0001, P value is two-tailed). C. RD SPRITE Cell 2021 (Replicate 1) and Bhat et al 2024 (spearman r = 0.90, p < 0.0001, P value is two-tailed). D. RD SPRITE Cell 2021 (Replicate 2) and Bhat et al 2024 (spearman r = 0.87, p < 0.0001, P value is two-tailed). E. Correlation of SPRITE and TSA-seq for speckle protein, SON, in H1 hESCs (spearman r = 0.75, p < 0.0001, P value is two-tailed). F. Chromosome wide view of speckle proximity score (top track) and TSA-seq (middle track, values > 0 shown) at 100-kb resolution for H1 hESCs. Gene density shown on bottom.
Extended Data Fig. 2 |. snRNA density for differently expressed genomic regions and different nascent transcription density.
A. To ensure that splicing factor difference were not due to expression differences between speckle close and speckle far genes, we divided genes up based on expression ranges: high expression (RPKM = 7.5-20), medium expression (RPKM = 2.5-7.5), low expression (RPKM = 1-2.5). The distribution of expression within these ranges were the same for speckle close and speckle far genes. The number of 100-kb regions analyzed are 8 regions each for high expression speckle close and far, 70 regions for medium expression speckle close and 28 for medium expression speckle far, and 194 for low expression speckle close and 62 for low expression speckle far. In the box plot, the center line represents the median, boxes show the interquartile range, whiskers show the range of values. B. U1 snRNA density is plotted for high (top), medium (middle), and low expression genes (bottom). C. U2 snRNA density is plotted for high (top), medium (middle), and low expression genes (bottom). D. U4 snRNA density is plotted for high (top), medium (middle), and low expression genes (bottom). E. U6 snRNA density is plotted for high (top), medium (middle), and low expression genes (bottom). (F-I): To ensure that splicing factor difference were not due to density of nascent transcription differences between speckle close and speckle far genes, we divided genes up based on transcription density ranges based on the number of nascent RNA reads from 5EU spanning each 100-kb bin. The number of 100-kb regions analyzed are 693 top 20% speckle close and 25 top 20% speckle far, 282 of 60–80% speckle close and 68 of 60–80% speckle far, 101 of 40–60% speckle close and 228 of 40–60% speckle far, 29 of 20–40% speckle close and 428 of 20–40% speckle far, and 7 of bottom 20% speckle close and 362 of bottom 20% speckle far. F. U1 snRNA density is plotted for top 20%, 60–80%, 40–60%, 20–40%, and bottom 20% of nascent transcription density. G. U2 snRNA density is plotted for top 20%, 60–80%, 40–60%, 20–40%, and bottom 20% of nascent transcription density. H. U4 snRNA density is plotted for top 20%, 60–80%, 40–60%, 20–40%, and bottom 20% of nascent transcription density. I. U6 snRNA density is plotted for top 20%, 60–80%, 40–60%, 20–40%, and bottom 20% of nascent transcription density.
Extended Data Fig. 3 |. snRNA density for junction matched genomic regions, genomic regions harboring genes of different lengths, and U1 AMT RAP-RNA enrichment for junction matched genomic regions.
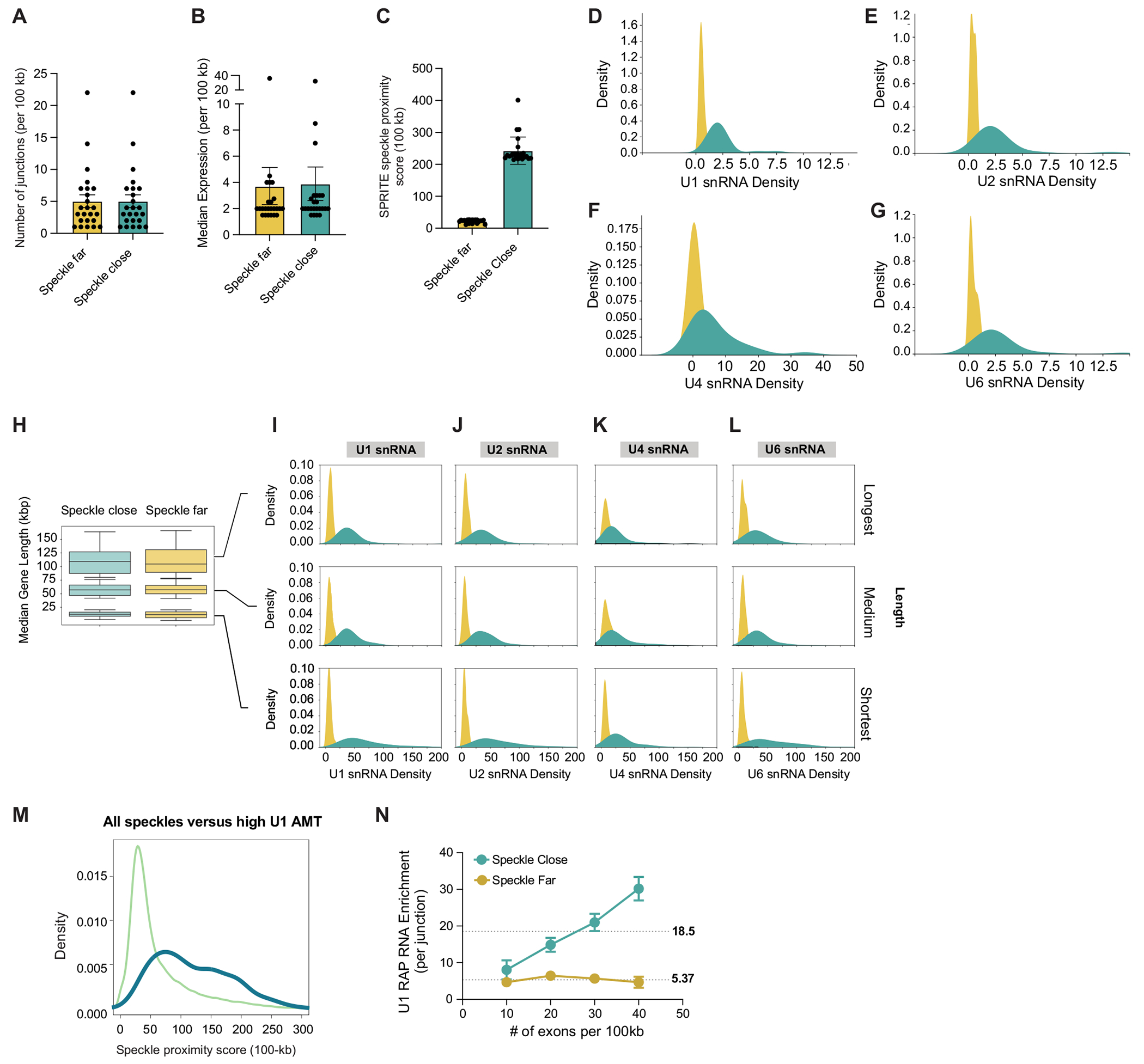
A. An identical number of regions with an identical number of junctions (179 regions each for speckle close and speckle far regions)were randomly sampled to compare regions with equivalent junction density (See Methods). B. The expression levels were matched to compare the regions in A with similar mean expression per 100-kb bin. C. SPRITE speckle proximity score of filtered speckle close and speckle far regions analyzed in panel A. D. U1 snRNA density is plotted for junction and expression-controlled regions. E. U2 snRNA density is plotted for junction and expression-controlled regions. F. U4 snRNA density is plotted for junction and expression-controlled regions. G. U6 snRNA density is plotted for junction and expression-controlled regions. H. To ensure that splicing factor difference were not due to gene length differences between speckle close and speckle far genes, we divided genes up based on gene length ranges: longest genes (60th to 80th percentile), medium length range genes (40th to 60th percentile), shortest genes (bottom 20%). The distribution of length within these ranges were the same for speckle close and speckle far genes. For the regions with the longest genes, 53 speckle close and 84 speckle far 100-kb regions analyzes. For the regions with the medium length genes, 73 speckle close and 63 speckle far 100-kb regions analyzed. For the regions with the shortest genes, 178 speckle close and 102 speckle far 100-kb regions analyzed. In the box plot, the center line represents the median, boxes show the interquartile range, whiskers show the range of values. I. U1 snRNA density is plotted for longest (top), medium (middle), and shortest length genes (bottom). J. U2 snRNA density is plotted for longest (top), medium (middle), and shortest length genes (bottom). K. U4 snRNA density is plotted for longest (top), medium (middle), and shortest length genes (bottom). L. U6 snRNA density is plotted for longest (top), medium (middle), and shorted length genes (bottom). M. Density plot showing speckle proximity score (100-kb) for genomic regions enriched for U1 binding. N. U1 RAP RNA enrichment per junction (y-axis) versus number of exons per 100-kb genomic bin for speckle close and speckle far regions. Dotted lines are mean U1 enrichment values and error is SEM. Number of regions per point: n = 97, 91, 28, and 12 for speckle far regions exon number = 10, 20, 30 and 40, respectively; n = 18, 68, 70, and 47 for speckle close regions exon number = 10, 20, 30 and 40, respectively.
Extended Data Fig. 4 |. Higher splicing efficiency in speckle close regions across measurements, cell-types, and when comparing to genes of similar expression, length, and junction density to speckle far regions.
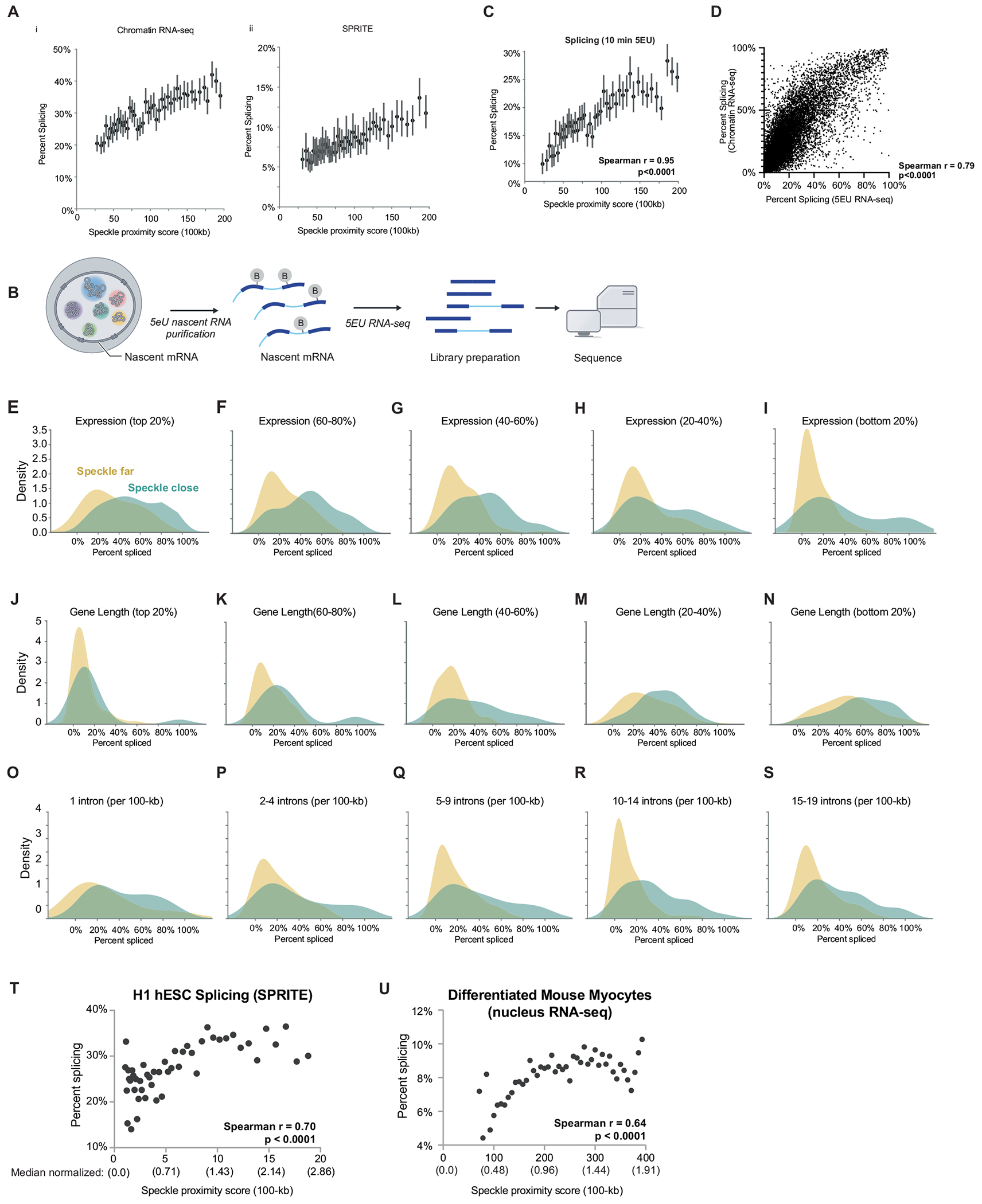
A. i. SPRITE speckle proximity score at 100-kb resolution (x axis) in mESCs and per cent spliced (from chromatin RNA-seq). 50 bins across all contact frequencies were taken and bins with speckle proximity scores between 0 and 200 are shown. Data are presented as mean values and bars represent 95% confidence interval. ii. SPRITE speckle proximity score at 100-kb resolution (x axis) in mESCs and per cent spliced (from SPRITE). 50 bins across all contact frequencies were taken and bins with speckle proximity scores between 0 and 200 are shown. Data are presented as mean values and bars represent 95% confidence interval. B. Schematic of 5EU labeling and nascent RNA sequencing pipeline. C. SPRITE speckle proximity score at 100-kb resolution (x axis) in mESCs and per cent spliced (from 5EU RNA-seq). 50 bins across all contact frequencies were taken and bins with speckle proximity scores between 0 and 200 are shown. Bars represent 95% confidence interval. Spearman r correlation = 0.95, p < 0.0001, P value is two-tailed. D. Correlation of splicing efficiency between previously published chromatin RNA-seq and newly generated 5EU RNA-seq (this paper; Spearman r correlation = 0.79, p < 0.0001), P value is two-tailed. (E-S) Splicing efficiency for speckle close and speckle far regions normalized for with genes that are: E. The top expressed genes (within 80–100% of expressed genes). 96 speckle close and 15 speckle far genes analzyed. F. Within 60–80% of expressed genes. 95 speckle close and 53 speckle far genes analyzed. G. Within 40–60% of expressed genes. 74 speckle close and 62 speckle far genes analyzed. H. Within 20–40% of expressed genes. 78 speckle close and 90 speckle far genes analyzed. I. The least expressed genes (0–20% of expressed genes). 51 speckle close and 173 speckle far genes analyzed. J. The longest genes (80–100% of genes lengths). 30 speckle close and 143 speckle far genes analyzed. K. 60–80% of gene lengths. 57 speckle close and 86 speckle far genes analyzed. L. 40–60% of gene lengths. 59 speckle close and 72 speckle far genes analyzed. M. 20–40% of gene lengths. 101 speckle close and 56 speckle far genes analyzed. N. The shortest genes (0–20% of gene lengths). 147 speckle close and 36 speckle far genes analyzed. O. 2 exons (single intron) per 100-kb region. 15 speckle close and 13 speckle far genes analyzed. P. 3–5 exons per 100-kb region. 50 speckle close and 119 speckle far genes analyzed. Q. 6–10 exons per 100-kb region. 51 speckle close and 202 speckle far genes analyzed. R. 11–15 exons per 100-kb region. 74 speckle close and 153 speckle far genes analyzed. S. 16–20 exons per 100-kb region. 95 speckle close and 78 speckle far genes analyzed. T. SPRITE speckle proximity score at 100-kb resolution (x axis) in H1-hESCs and per cent spliced within genomic bins from SPRITE (y axis) across 50 bins. Spearman r correlation = 0.70, p < 0.0001, P value is two-tailed. Median normalized speckle proximity scores are reported under each raw speckle hub contact value. Median value for H1 hESC = 7.0. U. SPRITE speckle proximity score at 100-kb resolution (x axis) in myocytes and per cent spliced (from nuclear RNA-seq) across 50 bins. Pearson r correlation = 0.64, p < 0.0001, P value is two-tailed. RD SPRITE data was not collected in myocytes for technical reasons. Median normalized speckle proximity scores are reported under each raw speckle hub contact value. Median value for mouse myocytes = 209. The range of speckle proximity scores vary between H1 hESC (1–20) and mouse myocytes (~75–400) due to the myocyte SPRITE data being sequenced more deeply.
Extended Data Fig. 5 |. Integrated reporter maintains endogenous speckle distances.
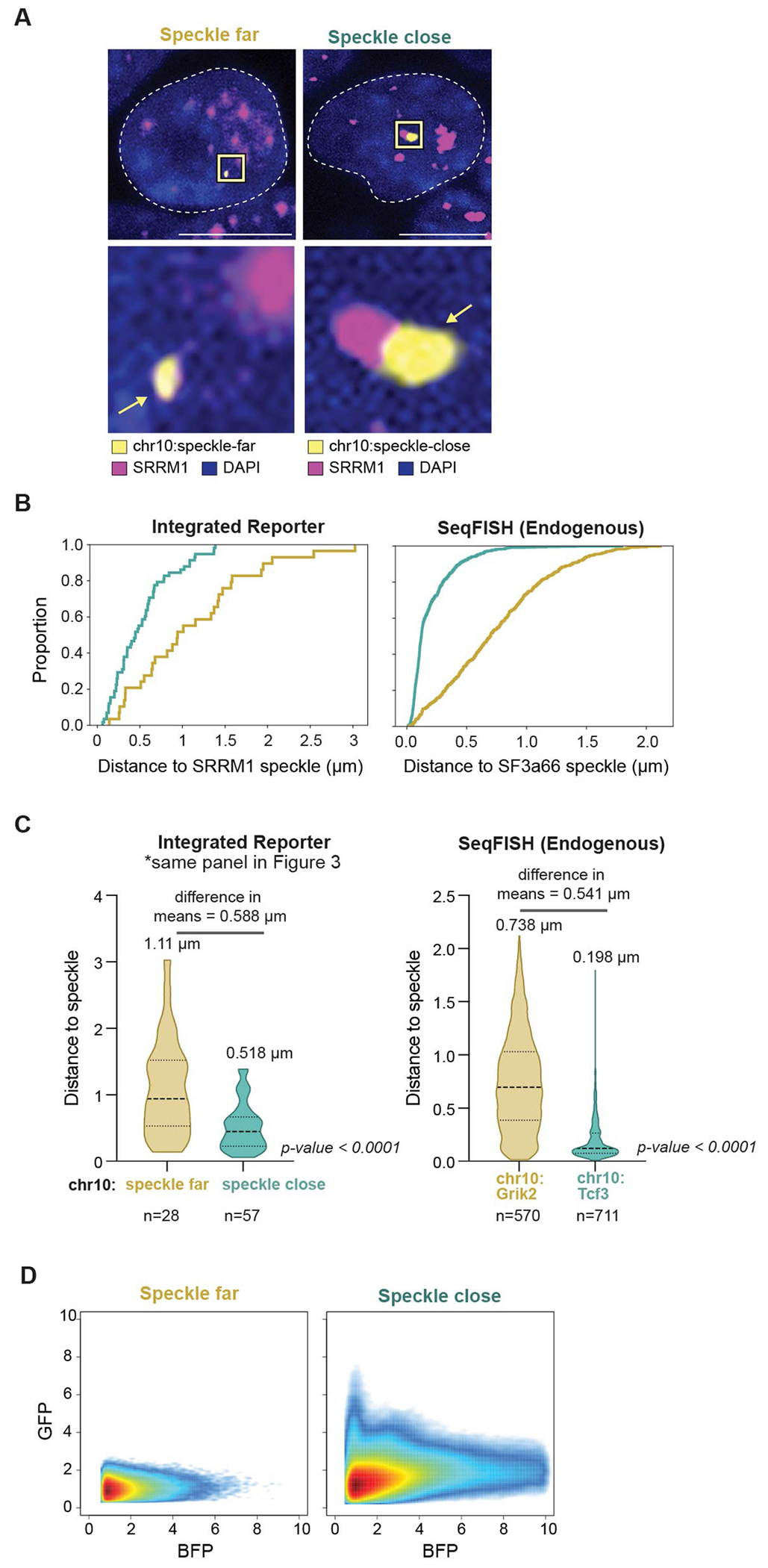
A. Representative images and zoom-ins of SRRM1 immunofluorescence combined with DNA FISH for the integrated reporter mini-gene. SRRM1 in magenta, reporter DNA in yellow and DAPI. Scale bar is 10 μm. n = 85 cells from 2 biological replicates. B. ECDF plots showing distance of DNA FISH spots of integrated location to the nearest nuclear speckle in the integrated cell lines (left) or distances computed from DNA seqFISH (right). C. Violin plots showing distance of DNA FISH spots of integrated location to the nearest nuclear speckle in the integrated cell lines (left) or distances computed from DNA seqFISH (right). Same data used as in 3C. Difference in means between speckle close and speckle far regions calculated for integrated loci and endogenous loci are represented above the distributions. D. 2D FACS plots showing GFP splicing levels as a function of BFP transcription levels between speckle close and speckle far integrated cell lines.
Extended Data Fig. 6 |. SPRITE analysis of myocyte cells and comparison to mES cells.
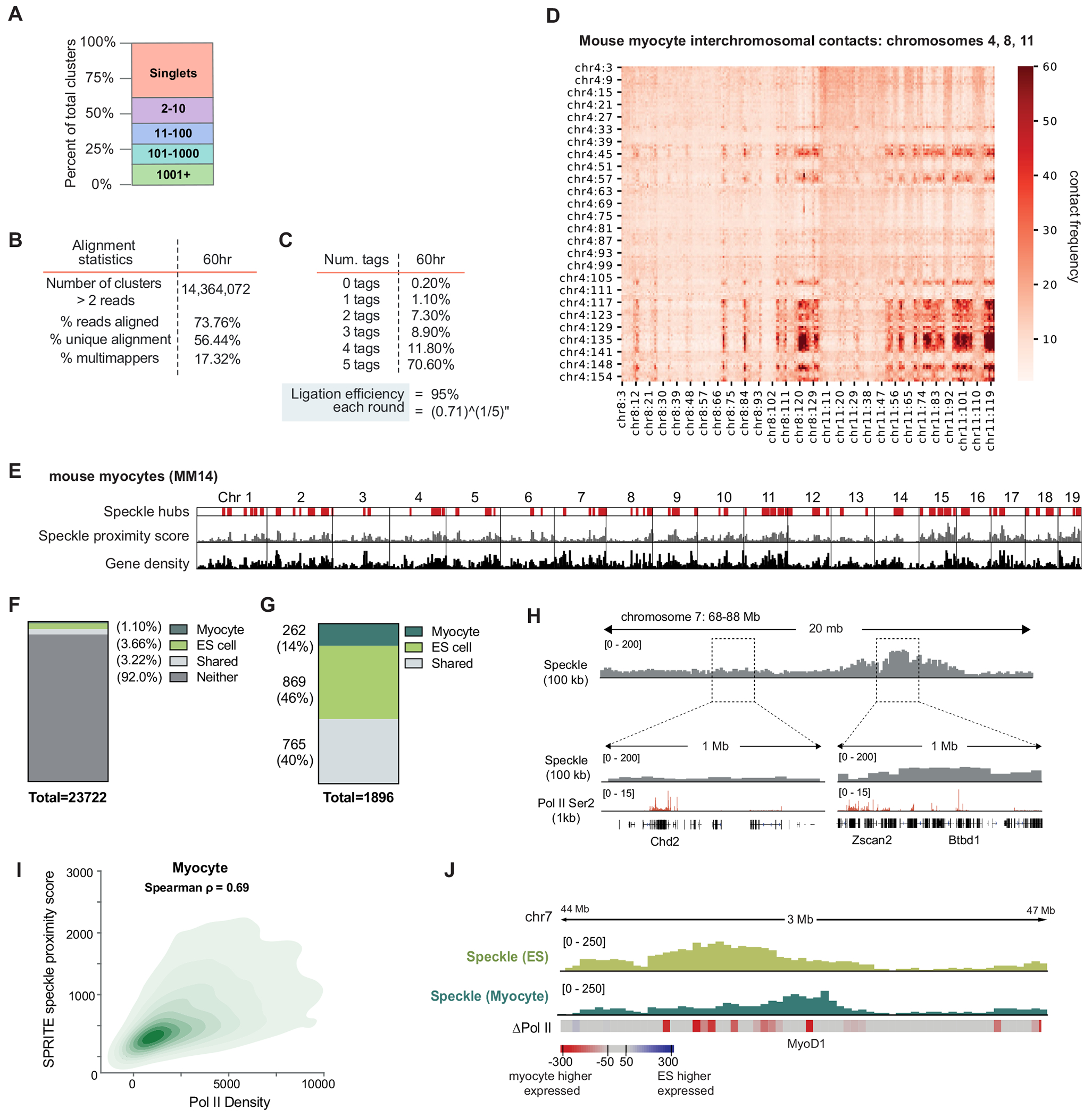
A. Distribution of SPRITE cluster sizes for myocyte SPRITE. The percentage of reads was calculated for different SPRITE cluster sizes (1, 2–10, 11–100, 101–1000, and over 1001 reads) and reported as the percentage of total reads. Cluster size is defined as the number of reads with the same barcode. B. Alignment statistics. C. A summary of ligation efficiency statistics to confirm tags have successfully ligated to each DNA molecule. D. Mouse myocyte interchromosomal contacts on chromosomes 4, 8, 11. E. Speckle hubs in mouse myocytes highlighted in red on chromosome track. Genome wide distribution of SPRITE speckle proximity scores (100-kb resolution). Gene density track on bottom. F. Distribution of SPRITE speckle proximity scores (100-kb resolution) for normalized mES and myocyte cell SPRITE. G. Distribution of number of genomic regions categorized as speckle hubs in myocyte, ES cells, both, or neither. H. SPRITE speckle proximity score at 100-kb resolution for a 20-Mb region on chromosome 7 in mouse myocytes. Pol II-S2P ChIP-seq density at 1-kb resolution. I. Ser2-P Pol II density (x axis) and normalized speckle proximity score (100-kb resolution) for myocytes. Spearman correlation = 0.69; p < 0.0001, P value is two-tailed. Similar to previous observations in other cellular contexts, we observed that DNA regions located close to speckles correspond to genomic regions containing high-density of RNA Pol II in differentiated myocytes. J. ES cell speckle proximity score (light green) and skeletal muscle speckle proximity score (dark green) for genomic locus near MyoD1 (expressed in myocyte). ΔPol II refers to difference in Ser2P-Pol II ChIP seq signal between mES cells and myocytes at 100-kb resolution, red is high in myocyte and blue high in ES.
Extended Data Fig. 7 |. pre-mRNA organization around nuclear speckles drives splicing efficiency.
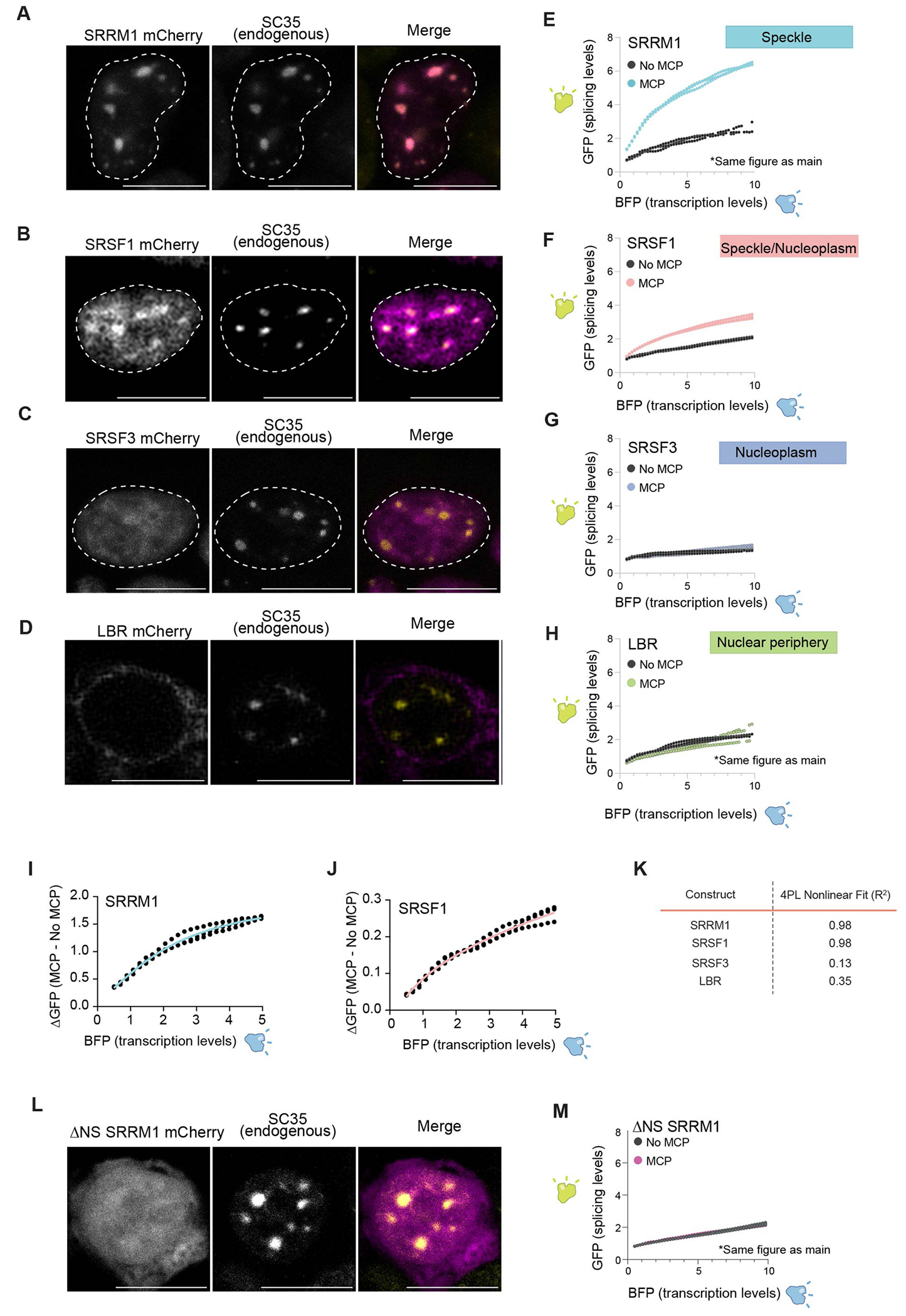
(A-D) Whole cell imaging of each protein with SC35 immunofluorescence and overlay with nucleus outlined in white for: A. SRRM1. B. SRSF1. C. SRSF3. D. LBR. Scale bars are 10 μm. Experiment was performed three times. (E-H) GFP fluorescence (splicing levels) (y axis) versus BFP fluorescence intensity for constructs with MCP or without MCP for: E. SRRM1. F. SRSF1. G. SRSF3. H. LBR. I. Difference in GFP splicing levels between SRRM1 MCP and no MCP with a four-parameter nonlinear regression. J. Difference in GFP splicing levels between SRSF1 MCP and no MCP with a four-parameter nonlinear regression. K. Four parameter logistic nonlinear fits for SRRM1, SRSF1, SRSF3, and LBR. L. Whole cell imaging of ΔNS SRRM1 with SC35 immunofluorescence overlay. Scale bar is 10 μm. Experiment was performed three times. M. GFP fluorescence (splicing levels) (y axis) versus BFP fluorescence intensity for constructs with MCP or without MCP for ΔNS SRRM1.
Extended Data Fig. 8 |. Differential versus leveled intron architectures also display speckle dependent splicing efficiency.
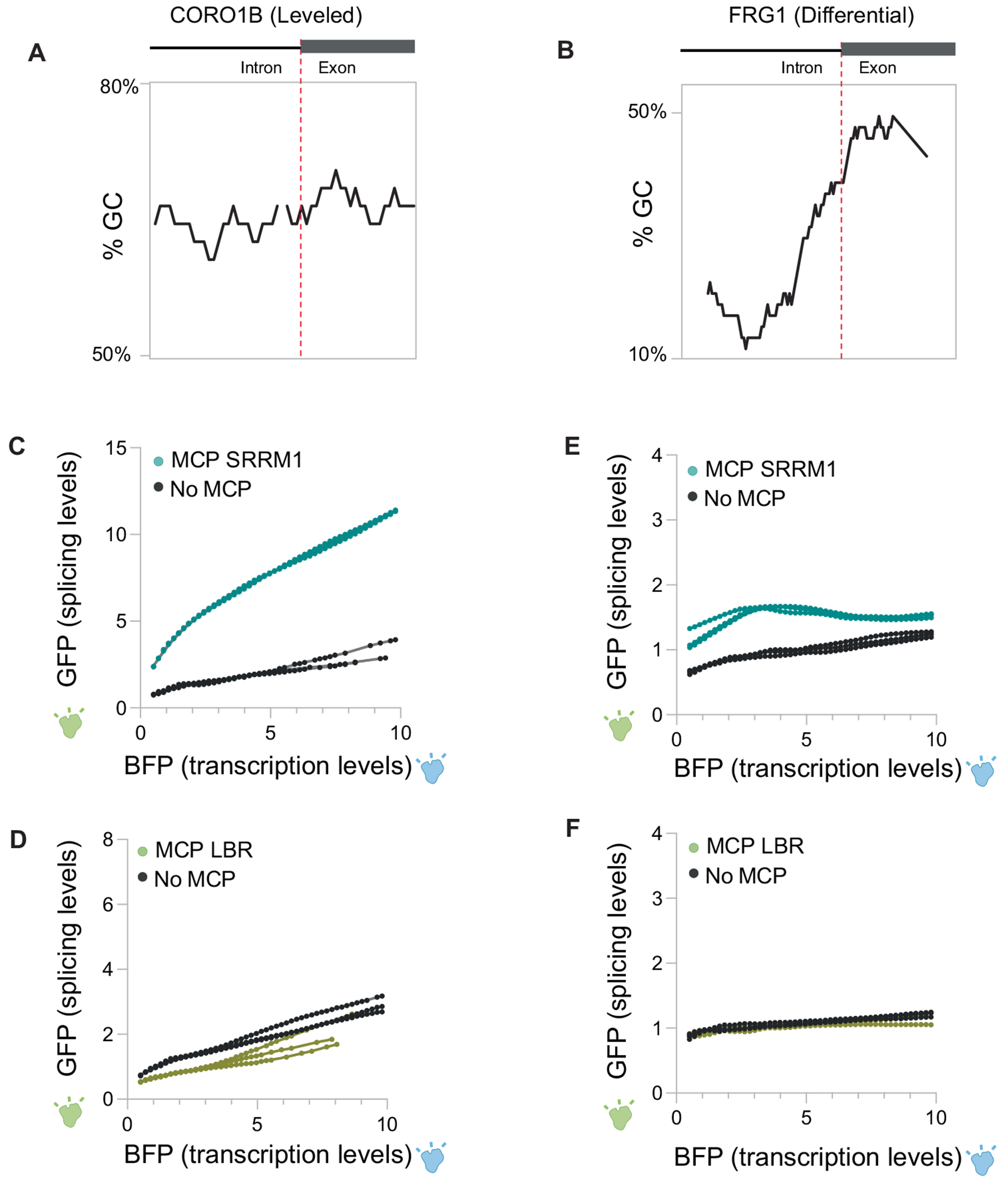
A. Schematic of CORO1B (leveled) intron and mapped %GC content across intron and exon boundary. B. Schematic of FRG1 (differential) intron and mapped %GC content across intron and exon boundary. C. GFP levels (y axis) versus fluorescence intensity (levels) of BFP (x axis) (bottom) for three replicates of SRRM1+/− MCP co-transfected with CORO1B splicing reporter. D. GFP levels (y axis) versus fluorescence intensity (levels) of BFP (x axis) (bottom) for three replicates of LBR+/− MCP co-transfected with CORO1B splicing reporter. E. GFP levels (y axis) versus fluorescence intensity (levels) of BFP (x axis) (bottom) for three replicates of SRRM1+/− MCP co-transfected with FRG1 splicing reporter. F. GFP levels (y axis) versus fluorescence intensity (levels) of BFP (x axis) (bottom) for three replicates of LBR+/− MCP co-transfected with FRG1 splicing reporter.
Extended Data Fig. 9 |. Integrated model for how spliceosome activity and proximity to nuclear speckles impact kinetics of splicing.
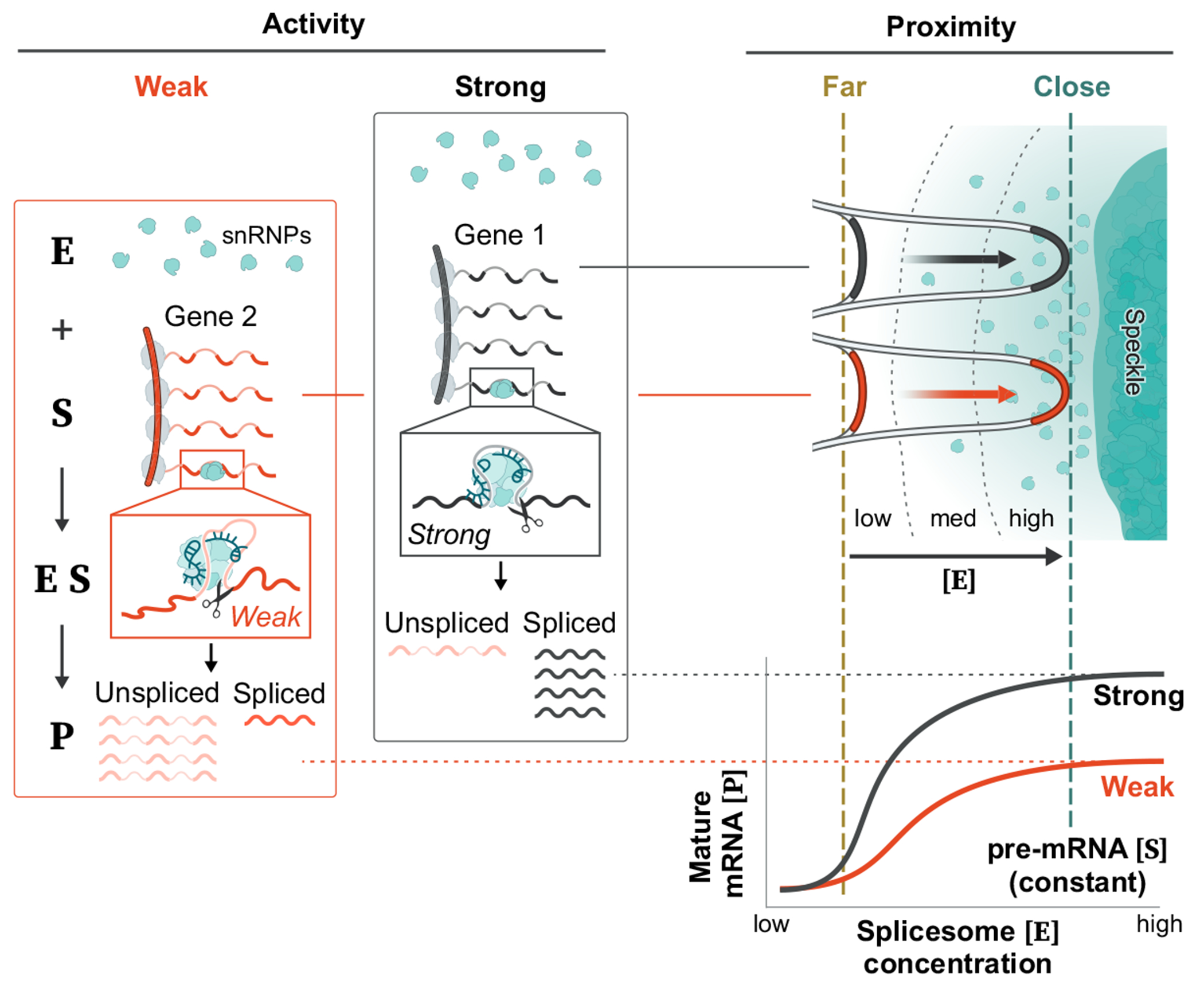
There are two components impacting the kinetics of splicing – spliceosome concentration and spliceosome activity. (i) Proximity to nuclear speckles impacts the concentration of spliceosomes at a given pre-mRNA, such that genes that are close to speckles will have higher spliceosome concentration than genes that are far from speckles. (ii) In contrast, splice site strength is defined by the activity of the spliceosome at the splice site53. In this way, spliceosomes engaged at ‘strong’ splice sites would have higher activity while ‘weak’ splice sites would have lower activity. These two components would be expected to have different effects on the kinetics of splicing. Specifically, modulating activity (splice site strength) would be expected to impact the maximum output of the reaction. Conversely, modulating concentration (speckle proximity) would be expected to impact the efficiency of each reaction.
Supplementary Material
Acknowledgements
We thank D.Honson, E.Detmar and D. Perez for experimental help; B.Riviere, L.Pachter and N.Ollikainen for computational help; M.Flynn for the bidirectional reporter plasmid; L.Cai, M.Elowitz and A.Raj for reagents; F.Ding, H.Yin, J.Jachowicz, L.Frankiw and Y.Luo for discussions; B.Yeh for discussions about splicing efficiency calculations; I.Antoshechkin for sequencing; G.Spigola for microscopy advice; A.Lin for sequencing advice; B.Yeh, D.Honson and K.Leslie for critical comments on the manuscript; R.Maehr and K.Mohan Parsi for H1 ES cell lines; B.Wold and B.Williams for myocyte cell lines; and S.Hiley for editing. Illustrations in Figs. 1a,c–d,e,g 2a,b, 3a,b, 4a, 5a–c and 6 and Extended Data Figs. 4b and 9 were created by I.-M.Strazhnik, Caltech. Imaging was performed at the Biological Imaging Facility with the support of the Caltech Beckman Institute and the Arnold and Mabel Beckman Foundation. This work was funded by NIH T32 GM 7616-40, NIH NRSA CA247447, the UCLA-Caltech Medical Scientist Training Program, a Chen Graduate Innovator Grant, and the Josephine De Karman Fellowship Trust (P.B.); and a HHMI Gilliam Fellowship, NSF GRFP Fellowship, and the HHMI Hanna H. Gray Fellows Program (S.A.Q.). This work was funded by the NIH 4DN program (U01 DK127420), NIH Directors’ Transformative Research Award (R01 DA053178), the NYSCF, CZI Ben Barres Early Career Acceleration Award, and funds from Caltech.
Footnotes
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-024-07429-6.
Code availability
Additional scripts and data are available at GitHub (https://github.com/GuttmanLab/speckle).
Competing interests S.A.Q. and M.G. are inventors on a patent covering the SPRITE method.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-024-07429-6.
Data availability
Sequencing datasets have been deposited into the GEO with accession identifier GSE247833.
References
- 1.Bhat P, Honson D & Guttman M Nuclear compartmentalization as a mechanism of quantitative control of gene expression. Nat. Rev. Mol. Cell Biol 22, 653–670 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Dundr M & Misteli T Biogenesis of nuclear bodies. Cold Spring Harb. Perspect. Biol 10.1101/cshperspect.a000711 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spector DL & Lamond AI Nuclear speckles. Cold Spring Harb. Perspect. Biol 3, a000646 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mattaj IW Splicing in space. Nature 10.1038/372727a0 (1994). [DOI] [PubMed] [Google Scholar]
- 5.Lewis JD & Tollervey D Like attracts like: getting RNA processing together in the nucleus. Science 288, 1385–1389 (2000). [DOI] [PubMed] [Google Scholar]
- 6.Shachar S & Misteli T Causes and consequences of nuclear gene positioning. J. Cell Sci 130, 1501–1508 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belmont AS Nuclear compartments: an incomplete primer to nuclear compartments, bodies, and genome organization relative to nuclear architecture. Cold Spring Harb. Perspect. Biol 14, a041268 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Putnam A, Thomas L & Seydoux G RNA granules: functional compartments or incidental condensates? Genes Dev. 37, 354–376 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herzel L, Ottoz DSM, Alpert T & Neugebauer KM Splicing and transcription touch base: co-transcriptional spliceosome assembly and function. Nat. Rev. Mol. Cell Biol 10.1038/nrm.2017.63 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maquat LE Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol 5, 89–99 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Scotti MM & Swanson MS RNA mis-splicing in disease. Nat. Rev. Genet 17, 19–32 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang S & Spector DL U1 and U2 small nuclear RNAs are present in nuclear speckles. Proc. Natl Acad. Sci. USA 89, 305–308 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu XD & Maniatis T Factor required for mammalian spliceosome assembly is localized to discrete regions in the nucleus. Nature 343, 437–441 (1990). [DOI] [PubMed] [Google Scholar]
- 14.Spector DL, Schrier WH & Busch H Immunoelectron microscopic localization of snRNPs. Biol. Cell 49, 1–10 (1983). [DOI] [PubMed] [Google Scholar]
- 15.Lerner EA, Lerner MR, Janeway CAJ & Steitz JA Monoclonal antibodies to nucleic acid-containing cellular constituents: probes for molecular biology and autoimmune disease. Proc. Natl Acad. Sci. USA 78, 2737–2741 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hall LL, Smith KP, Byron M & Lawrence JB Molecular anatomy of a speckle. Anat. Rec. A Discov. Mol. Cell. Evol. Biol 10.1002/ar.a.20336 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cmarko D et al. Ultrastructural analysis of transcription and splicing in the cell nucleus after bromo-UTP microinjection. Mol. Biol. Cell 10, 211–223 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fakan S, Leser G & Martin TE Ultrastructural distribution of nuclear ribonucleoproteins as visualized by immunocytochemistry on thin sections. J. Cell Biol 98, 358–363 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang G, Taneja KL, Singer RH & Green MR Localization of pre-mRNA splicing in mammalian nuclei. Nature 372, 809–812 (1994). [DOI] [PubMed] [Google Scholar]
- 20.Huang S, Deerinck TJ, Ellisman MH & Spector DL In vivo analysis of the stability and transport of nuclear poly(A)+ RNA. J. Cell Biol 126, 877–899 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Misteli T, Cáceres JF & Spector DL The dynamics of a pre-mRNA splicing factor in living cells. Nature 387, 523–527 (1997). [DOI] [PubMed] [Google Scholar]
- 22.Jiménez-García LF & Spector DL In vivo evidence that transcription and splicing are coordinated by a recruiting mechanism. Cell 73, 47–59 (1993). [DOI] [PubMed] [Google Scholar]
- 23.Sacco-Bubulya P & Spector DL Disassembly of interchromatin granule clusters alters the coordination of transcription and pre-mRNA splicing. J. Cell Biol 156, 425–436 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Misteli T & Spector DL Serine/threonine phosphatase 1 modulates the subnuclear distribution of pre-mRNA splicing factors. Mol. Biol. Cell 7, 1559–1572 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang S & Spector DL Intron-dependent recruitment of pre-mRNA splicing factors to sites of transcription. J. Cell Biol 133, 719–732 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Misteli T & Spector DL RNA polymerase II targets pre-mRNA splicing factors to transcription sites in vivo. Mol. Cell 3, 697–705 (1999). [DOI] [PubMed] [Google Scholar]
- 27.Xie SQ, Martin S, Guillot PV, Bentley DL & Pombo A Splicing speckles are not reservoirs of RNA polymerase II, but contain an inactive form, phosphorylated on serine2 residues of the C-terminal domain. Mol. Biol. Cell 17, 1723–1733 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Misteli T et al. Serine phosphorylation of SR proteins is required for their recruitment to sites of transcription in vivo. J. Cell Biol 143, 297–307 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tammer L et al. Gene architecture directs splicing outcome in separate nuclear spatial regions. Mol. Cell 82, 1021–1034.e8 (2022). [DOI] [PubMed] [Google Scholar]
- 30.Barutcu AR et al. Systematic mapping of nuclear domain-associated transcripts reveals speckles and lamina as hubs of functionally distinct retained introns. Mol. Cell 82, 1035–1052.e9 (2022). [DOI] [PubMed] [Google Scholar]
- 31.Wang K et al. Intronless mRNAs transit through nuclear speckles to gain export competence. J. Cell Biol 217, 3912–3929 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim J, Venkata NC, Hernandez Gonzalez GA, Khanna N & Belmont AS Gene expression amplification by nuclear speckle association. J. Cell Biol 219, e201904046 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhong X-Y, Wang P, Han J, Rosenfeld MG & Fu X-D SR proteins in vertical integration of gene expression from transcription to RNA processing to translation. Mol. Cell 35, 1–10 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lawrence JB & Clemson CM Gene associations: true romance or chance meeting in a nuclear neighborhood? J. Cell Biol 182, 1035–1038 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perraud M, Gioud M & Monier JC [Intranuclear structures of monkey kidney cells recognised by immunofluorescence and immuno-electron microscopy using anti-ribonucleoprotein antibodies (author’s transl)]. Ann. Immunol 130C, 635–647 (1979). [PubMed] [Google Scholar]
- 36.Chen Y et al. Mapping 3D genome organization relative to nuclear compartments using TSA-seq as a cytological ruler. J. Cell Biol 217, 4025–4048 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quinodoz SA et al. Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell 174, 744–757.e24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takei Y et al. Integrated spatial genomics reveals global architecture of single nuclei. Nature 590, 344–350 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quinodoz SA et al. RNA promotes the formation of spatial compartments in the nucleus. Cell 184, 5775–5790.e30 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilkinson ME, Charenton C & Nagai K RNA splicing by the spliceosome. Annu. Rev. Biochem 89, 359–388 (2020). [DOI] [PubMed] [Google Scholar]
- 41.Will CL & Lührmann R Spliceosome structure and function. Cold Spring Harb. Perspect. Biol 3, a003707 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Calvet JP & Pederson T Base-pairing interactions between small nuclear RNAs and nuclear RNA precursors as revealed by psoralen cross-linking in vivo. Cell 26, 363–370 (1981). [DOI] [PubMed] [Google Scholar]
- 43.Engreitz JM et al. RNA–RNA interactions enable specific targeting of noncoding RNAs to nascent pre-mRNAs and chromatin sites. Cell 159, 188–199 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wuarin J & Schibler U Physical isolation of nascent RNA chains transcribed by RNA polymerase II: evidence for cotranscriptional splicing. Mol. Cell. Biol 14, 7219–7225 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goronzy IN et al. Simultaneous mapping of 3D structure and nascent RNAs argues against nuclear compartments that preclude transcription. Cell Rep. 41, 111730 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takei Y. et al. Single-cell nuclear architecture across cell types in the mouse brain. Science 374, 586–594 (2021). [DOI] [PubMed] [Google Scholar]
- 47.Zhang L. et al. TSA-seq reveals a largely conserved genome organization relative to nuclear speckles with small position changes tightly correlated with gene expression changes. Genome Res. 31, 251–264 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pichon X, Robert M-C, Bertrand E, Singer RH & Tutucci E New generations of MS2 variants and MCP fusions to detect single mRNAs in living eukaryotic cells. Methods Mol. Biol 2166, 121–144 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tripathi V. et al. SRSF1 regulates the assembly of pre-mRNA processing factors in nuclear speckles. Mol. Biol. Cell 23, 3694–3706 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ilik i. A. et al. SON and SRRM2 are essential for nuclear speckle formation. eLife 9, e60579 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thul PJ et al. A subcellular map of the human proteome. Science 356, eaal3321 (2017). [DOI] [PubMed] [Google Scholar]
- 52.Olins AL, Rhodes G, Welch DBM, Zwerger M & Olins DE Lamin B receptor: multi-tasking at the nuclear envelope. Nucleus 1, 53–70 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reed R & Maniatis T A role for exon sequences and splice-site proximity in splice-site selection. Cell 46, 681–690 (1986). [DOI] [PubMed] [Google Scholar]
- 54.Szymczyna BR et al. Structure and function of the PWI motif: a novel nucleic acid-binding domain that facilitates pre-mRNA processing. Genes Dev. 17, 461–475 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quinodoz SA & Guttman M Essential roles for RNA in shaping nuclear organization. Cold Spring Harb. Perspect. Biol 14, a039719 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCracken S. et al. The C-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature 385, 357–360 (1997). [DOI] [PubMed] [Google Scholar]
- 57.Ding F & Elowitz MB Constitutive splicing and economies of scale in gene expression. Nat. Struct. Mol. Biol 26, 424–432 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yuryev A. et al. The C-terminal domain of the largest subunit of RNA polymerase II interacts with a novel set of serine/arginine-rich proteins. Proc. Natl Acad. Sci. USA 93, 6975–6980 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lazarev D & Manley JL Concurrent splicing and transcription are not sufficient to enhance splicing efficiency. RNA 13, 1546–1557 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zimber A, Nguyen Q-D & Gespach C Nuclear bodies and compartments: functional roles and cellular signalling in health and disease. Cell Signal. 16, 1085–1104 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Quinodoz SA et al. SPRITE: a genome-wide method for mapping higher-order 3D interactions in the nucleus using combinatorial split-and-pool barcoding. Nat. Protoc 17, 36–75 (2022). [DOI] [PubMed] [Google Scholar]
- 62.Banerjee AK et al. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell 183, 1325–1339.e21 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Melsted P. et al. Modular, efficient and constant-memory single-cell RNA-seq preprocessing. Nat. Biotechnol 39, 813–818 (2021). [DOI] [PubMed] [Google Scholar]
- 64.Engreitz JM et al. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 539, 452–455 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang S. et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang S. et al. Mapping and modeling the genomic basis of differential RNA isoform expression at single-cell resolution with LR-Split-seq. Genome Biol. 22, 286 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Majumdar DS et al. Programmed delayed splicing: a mechanism for timed inflammatory gene expression. Preprint at bioRxiv 10.1101/443796 (2018). [DOI] [Google Scholar]
- 68.Mayr C & Bartel DP Widespread shortening of 3’ UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138, 673–684 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Manna PT, Davis LJ & Robinson MS Fast and cloning-free CRISPR/Cas9-mediated genomic editing in mammalian cells. Traffic 20, 974–982 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A & Tyagi S Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 5, 877–879 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stringer C, Wang T, Michaelos M & Pachitariu M Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods 18, 100–106 (2021). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing datasets have been deposited into the GEO with accession identifier GSE247833.