

Abstract
Asgardarchaeota harbour many eukaryotic signature proteins and are widely considered to represent the closest archaeal relatives of eukaryotes. Whether similarities between Asgard archaea and eukaryotes extend to their viromes remains unknown. Here we present 20 metagenome-assembled genomes of Asgardarchaeota from deep-sea sediments of the basin off the Shimokita Peninsula, Japan. By combining a CRISPR spacer search of metagenomic sequences with phylogenomic analysis, we identify three family-level groups of viruses associated with Asgard archaea. The first group, verdandiviruses, includes tailed viruses of the class Caudoviricetes (realm Duplodnaviria); the second, skuldviruses, consists of viruses with predicted icosahedral capsids of the realm Varidnaviria; and the third group, wyrdviruses, is related to spindle-shaped viruses previously identified in other archaea. More than 90% of the proteins encoded by these viruses of Asgard archaea show no sequence similarity to proteins encoded by other known viruses. Nevertheless, all three proposed families consist of viruses typical of prokaryotes, providing no indication of specific evolutionary relationships between viruses infecting Asgard archaea and eukaryotes. Verdandiviruses and skuldviruses are likely to be lytic, whereas wyrdviruses potentially establish chronic infection and are released without host cell lysis. All three groups of viruses are predicted to play important roles in controlling Asgard archaea populations in deep-sea ecosystems.
Asgard archaea are an expansive group of metabolically versatile archaea that thrive primarily in anoxic sediments around the globe1–9. Based on phylogenomic analyses, Asgard archaea were originally classified into multiple phylum-level lineages, including Lokiarchaeota, Thorarchaeota, Odinarchaeota, Heimdallarchaeota, Helarchaeota, Sifarchaeota, Wukongarchaeota and several others, most of which were named after Norse gods1,5,7,9–12. Recently, taxonomic rank normalization using relative evolutionary divergence has suggested that Asgard archaea represent a phylum, tentatively named Asgardarchaeota, including the classes Lokiarchaeia, Thorarchaeia, Odinarchaeia, Heimdallarchaeia, Sifarchaeia, Hermodarchaeia, Sifarchaeia, Baldrarchaeia, Wukongarchaeia and Jordarchaeia, with the other lineages classified as lower-rank taxa within the classes8,13. The vast majority of Asgard archaea have been discovered through metagenomics, whereas only one species has been isolated and successfully grown in the laboratory14. Asgard archaea gained prominence due to their inferred key role in the origin of eukaryotes15. Indeed, Heimdallarchaeia form a sister group to eukaryotes in most phylogenetic analyses1,5 although alternative phylogenies have also been presented16,17. Compared with other archaea, Asgard archaea encode a substantially expanded set of eukaryotic signature proteins, including many proteins implicated in membrane trafficking, vesicle formation and transport, cytoskeleton formation, the ubiquitin network and other processes characteristic of eukaryotes1,5. A tantalizing question is whether eukaryotes also inherited viruses and other types of mobile genetic elements (MGEs) from the Asgard archaea.
Viruses infecting archaea are remarkably diverse, in terms of both their genome sequences and virion structures18–20. Some archaeal viruses, in particular those with icosahedral virions, are evolutionarily related to bacterial and eukaryotic viruses but the majority of archaeal virus groups are specific to archaea, with no identifiable relatives in the two other domains. Archaea-specific viruses often have odd-shaped virions that resemble lemons, champagne bottles or droplets19. Most archaeal viruses have, thus far, been isolated from hyperthermophilic or halophilic hosts, with only a handful of virus species described for methanogenic and ammonia-oxidizing mesophilic archaea18. No viruses infecting Asgard archaea have been isolated, primarily due to the inherent difficulty in propagation of Asgard archaeal hosts. Nevertheless, analysis of CRISPR–Cas loci in the genomes of Asgard archaea revealed a remarkable diversity of defence systems in these organisms21, implying a rich Asgard archaeal virome. CRISPR arrays are archives of past encounters with viruses and other MGEs, which can be harnessed to uncover the associations between viruses and their hosts. Indeed, matching the CRISPR spacers from a known organism to viruses with unknown hosts is widely used in host assignment for viruses discovered by metagenomics and, arguably, is the most straightforward and efficient approach to identification of the hosts of viruses infecting prokaryotes22.
Here we harness CRISPR spacer sequences from the sequenced Asgard archaeal genomes to search for viruses infecting these organisms, and describe three distinct family-level groups of Asgard-associated viruses, all of which display typical features of viruses infecting bacteria or archaea.
Results
To search for viruses infecting Asgard archaea, we sequenced 12 metagenomes constructed from total environmental DNA directly extracted from subseafloor sediments originating off the Shimokita Peninsula, Japan (site C9001, water depth 1,180 m)23 and representing different sediment depths ranging 0.91–363.3 m below the seafloor (mbsf). Asgard archaeal metagenome-assembled genomes (MAGs) were assembled from seven of the 12 samples. A total of 20 Asgardarchaeota MAGs, including 12 Lokiarchaeia, two Thorarchaeia and six Heimdallarchaeia, were analysed in this study (Fig. 1), with an estimated completeness of 65.95 ± 17.93, estimated quality of 54.68 ± 15.78 and guanine-cytosine content of 32.78% ± 3.99% (Supplementary Table 1).
Fig. 1 |. Phylogenomic tree of Asgardarchaeota.
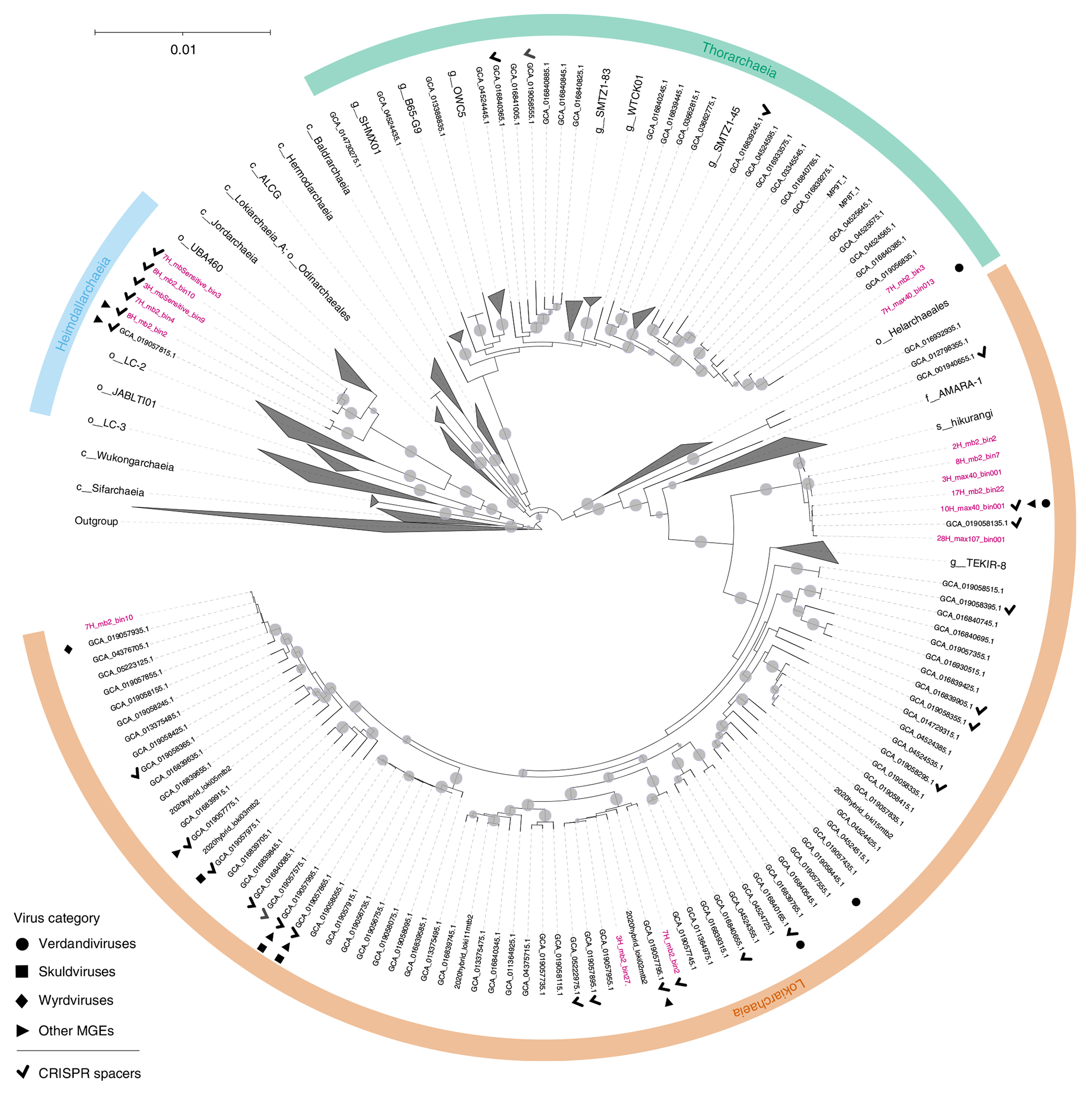
The alignment is based on a concatenated set of 53 protein markers (subsampled to 42 sites each, resulting in a total alignment length of 2,058 sites) from 339 taxa. These taxa encompass 276 Asgardarchaeota MAGs, including 16 recovered in this study (highlighted in pink; MAGs 3H_mb2_bin20, 4H_max40_bin02,1 4H_mb2_bin40 and 7H_mb2_bin7 were removed after the alignment trimming step due to low completeness), and 63 non-Asgardarchaeota species representatives from GTDB release RS95 as an outgroup. Maximum-likelihood analysis was performed using IQ-TREE under the LG + C10 + F + G + PMSF model. The tree is rooted on the non-Asgardarchaeota group. Nodes with bootstrap statistical support >90% are indicated by grey dots. MAGs containing viral contigs and CRISPR arrays are indicated by different symbols, with the key provided in the bottom left corner. GCA_019058445.1 contains a defective verdandivirus-derived provirus (Extended Data Fig. 2).
To assign putative viral genomes to Asgard archaeal hosts, we compiled a dataset of CRISPR spacers from Asgard archaeal MAGs assembled in this study as well as those reported previously (Fig. 2 and Supplementary Table 2). Analysis of Asgard CRISPR arrays allowed us to define Asgard-specific CRISPR repeat sequences, which were used to identify additional CRISPR arrays from the contigs obtained from our sediment samples (Fig. 2a and Methods). In total, our CRISPR spacer dataset (Fig. 2b and Supplementary Data 1) included 2,532 spacers assigned to different Asgardarchaota lineages, with Lokiarchaeia contributing the highest number (Fig. 2c). All spacers were then used to search for protospacers in the 20 assembled MAGs and putative virus genomes from our dataset, as well as contigs from GenBank and JGI sequence databases. In total, 14 contigs could be assigned to Asgard archaeal hosts based on CRISPR spacer matches with an estimated false positive rate of 0.00276 (Methods and Extended Data Fig. 1). By contrast, none of the contigs was targeted by spacers from the CRISPRHost database (675,911 spacers) and only two, probably false positive, spacers were identified in the CRISPR Spacer Database and Exploration Tool (11,674,395 spacers; Methods).
Fig. 2 |. Description of the Asgardarchaeota CRISPR spacer dataset.
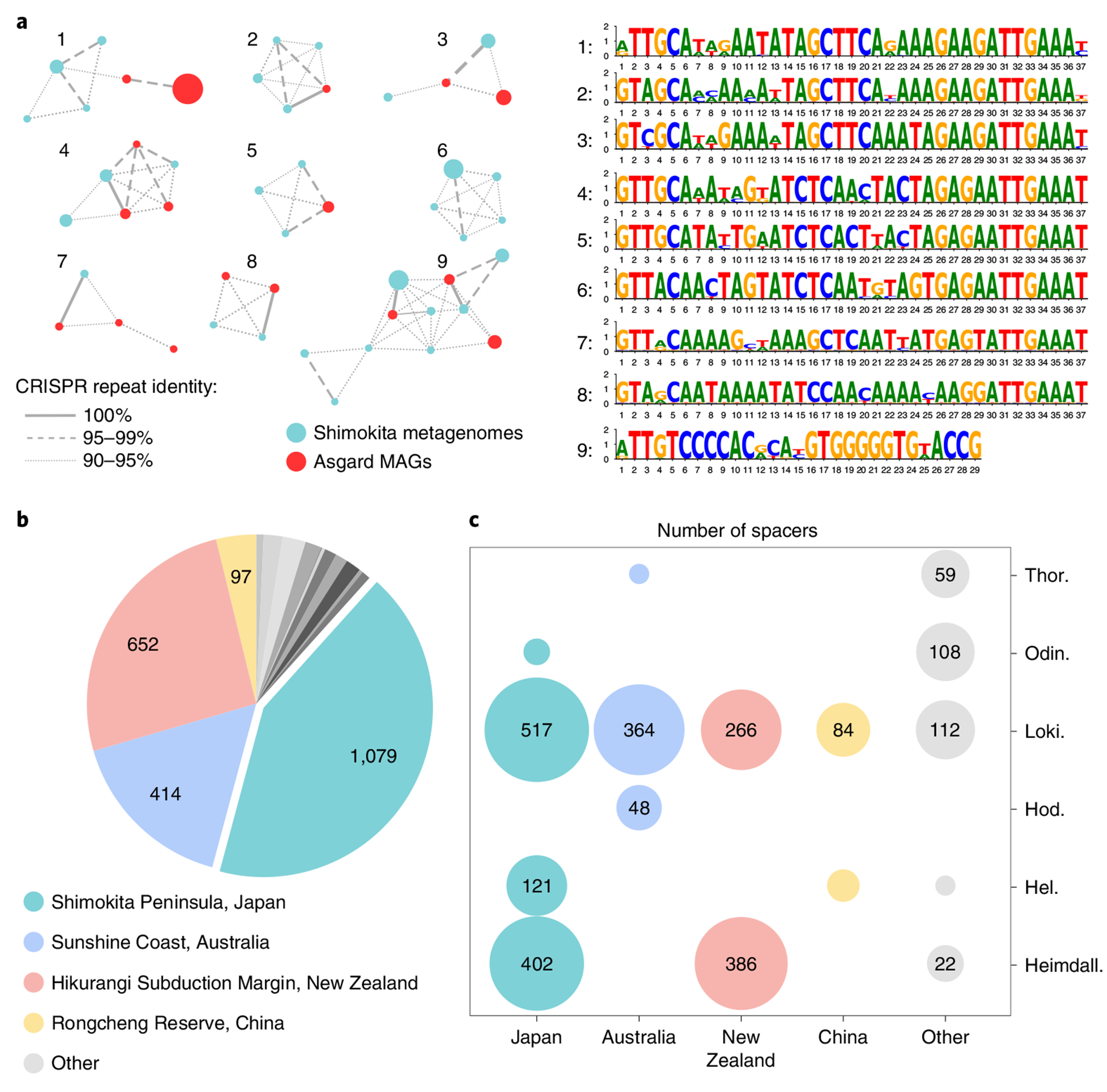
a, Similarity of CRISPR repeats from metagenomic data (teal) and from Asgardarchaeota MAGs (red). Unique CRISPR repeat sequences are shown as nodes in the network. Node diameter is proportional to the number of spacers associated with the repeat sequence in the dataset. Identical CRISPR repeats from both metagenomic data and Asgard MAGs are connected by solid lines; dashed and dotted lines connect CRISPR repeat sequences with 95–99% and 90–95% identities, respectively. Consensus sequences for the nine major clusters are shown on the right. b, Sources of Asgardarchaeota CRISPR spacers. The number of spacers in the dataset originating from different geographical sites is indicated on the pie chart. c, Taxonomic distribution of Asgardarchaeota MAGs with CRISPR arrays for different isolation sites. Circle size is proportional to the number of spacers in the dataset.
Eight of the putative viral genomes originated from the metagenomes sequenced in this study (from depths ranging 0.91–87.7 mbsf) whereas six additional genomes were recovered from datasets sequenced previously, including anoxic subseafloor sediment samples from two Pacific Ocean sites (the Hikurangi Subduction Margin (144.3 mbsf)8 and Cascadia Margin (2 mbsf))24 and one Indian Ocean site (Sumatra Forearc (1.60–6.07 mbsf; PRJNA367446)). Each genome was targeted (≥90% identity between spacer and protospacer) by one to six CRISPR spacers assigned to putative Asgard classes Lokiarchaeia, Thorarchaeia and Heimdallarchaeia (Fig. 1 and Supplementary Tables 3 and 4). Based on the conservation of viral hallmark proteins, including major capsid proteins (MCPs), seven of the genomes were unequivocally identified as belonging to viruses from three unrelated groups (Figs. 3–5) whereas the remaining seven could represent either unknown viruses or other types of MGE (Supplementary Fig. 1 and below). The viral dataset was further enriched by searching the JGI and GenBank sequence databases for related contigs. As a result, we collected 21 contigs representing three groups of Asgard archaeal viruses originating from seven geographically remote locations (Fig. 6a). Further analyses using CRISPR-based (SpacePHARER) and kmer-based (WiSH and PHIST) methods confirmed the association of these 21 contigs with Asgard archaeal hosts (Methods and Supplementary Tables 3 and 5). Network analysis of these viral genomes, together with the bacterial and archaeal virus genomes available in RefSeq, showed that the three Asgard virus groups are disconnected from each other as well as from other known viruses (Fig. 6b).
Fig. 3 |. Diversity of verdandiviruses.
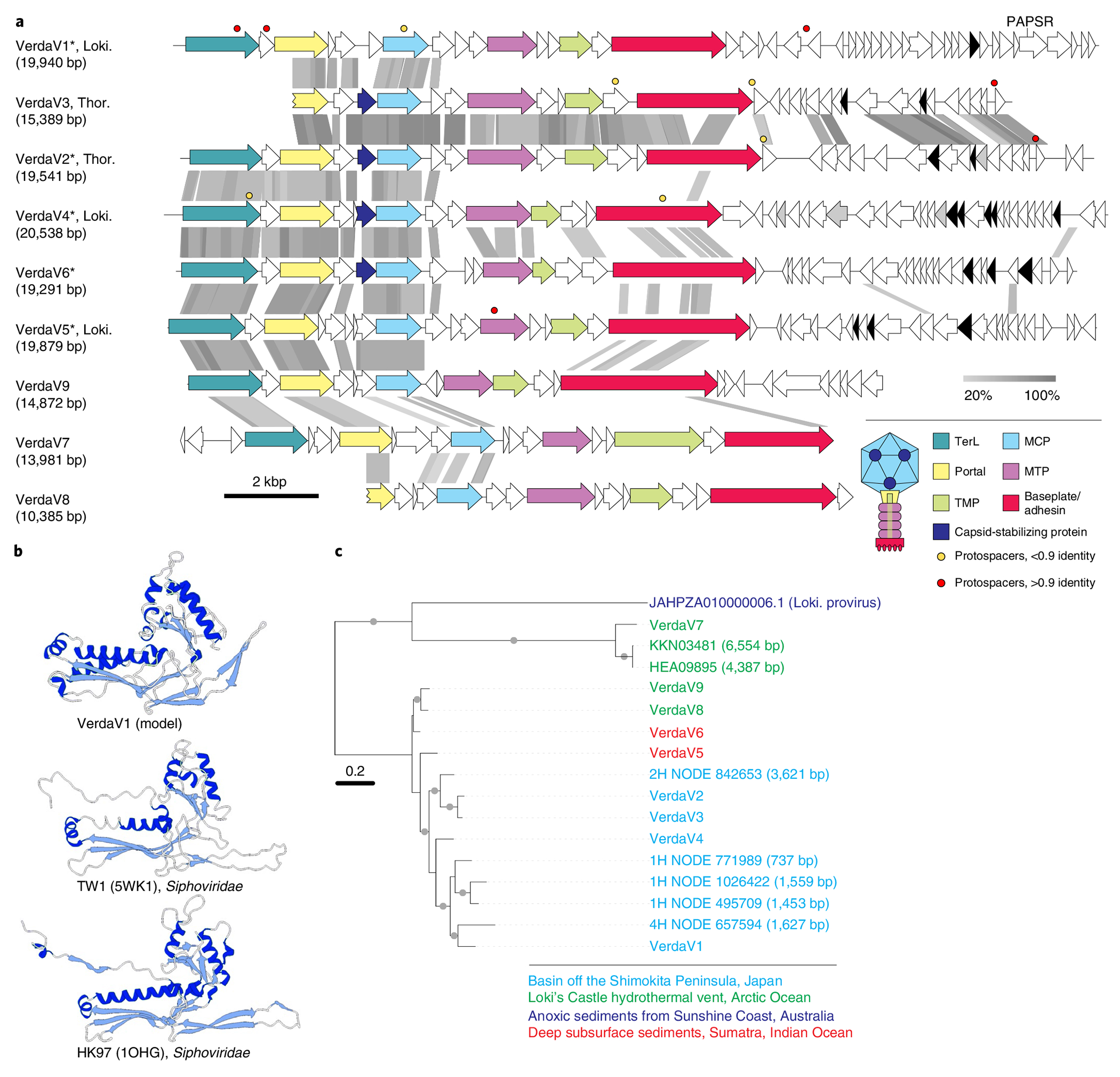
a, Genome maps of verdandiviruses. Homologous genes are shown using the same colours, with the key provided at the bottom. Also shown is the deduced schematic organization of the verdandivirus virion, with colours matching those of genes encoding the corresponding proteins. Genes encoding putative DNA-binding proteins with Zn-binding and HTH domains coloured in black and grey, respectively. Coloured circles indicate the positions of protospacers. Grey shading connects genes displaying sequence similarity at the protein level, with the percentage of sequence identity indicated by different shades of grey (see scale at the bottom). Asterisks denote complete genomes assembled as circular contigs. PAPSR, phosphoadenosine phosphosulfate reductase; TMP, tail tape measure protein; MTP, major tail protein. b, Comparison of the structural model of the MCP of verdnadivirus AsTV-10H2 with the corresponding structures of siphoviruses TW1 and HK97. The models are coloured according to their secondary structure: α-helices, dark blue; β-strands, light blue. c, Maximum-likelihood phylogeny of MCPs encoded by verdandiviruses. Taxa are coloured based on the source of origin (key at the bottom). The tree was constructed using the automatic optimal model selection (LG + G4) and is midpoint rooted. The scale bar represents the number of substitutions per site. Circles at nodes denote aLRT SH-like branch support values >90%.
Fig. 5 |. Diversity of wyrdviruses.
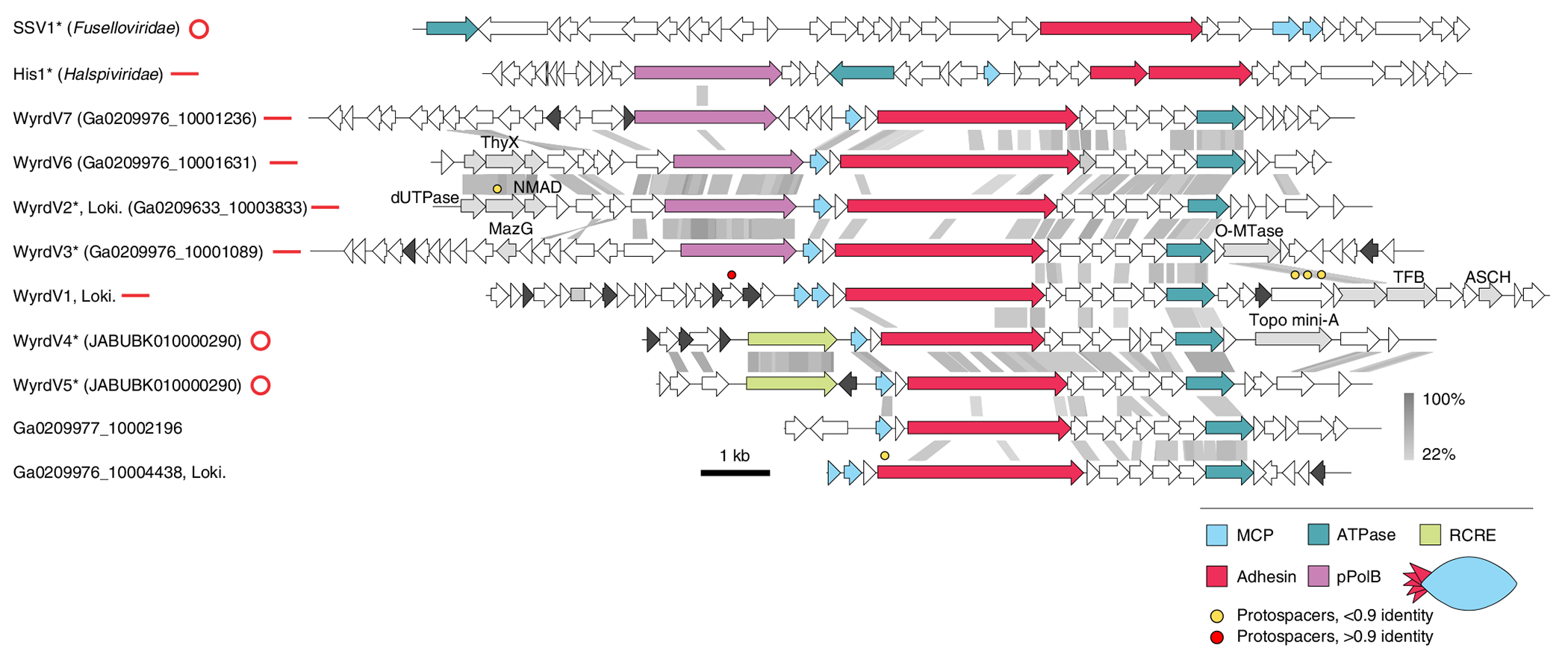
Genome maps of wyrdviruses, fusellovirus SSV1 and halspivirus His1. Homologous genes are shown using the same colours, with the key provided at the bottom. Also shown is the deduced schematic organization of the skuldvirus virion with colours marching those of the genes encoding the corresponding proteins. Coloured circles indicate the positions of protospacers. Genes encoding putative DNA-binding proteins with Zn-binding and HTH domains are coloured black and grey, respectively. Grey shading connects genes displaying sequence similarity at the protein level, with the percentage of sequence identity indicated by different shades of grey (see scale on the right). Asterisks denote complete genomes assembled as circular contigs or linear contigs with terminal inverted repeats. The topology of each genome is shown next to the corresponding name: circles for circular genomes, lines for linear genomes. ThyX, thymidylate synthase X; NMAD, nucleotide modification-associated domain 1 protein; O-MTase, carbohydrate-specific methyltransferase; TFB, transcription initiation factor B; ASCH, ASC-1 homology domain; pPolB, protein-primed family B DNA polymerase. Genome maps of SSV1 and His1 viruses are shown for comparison.
Fig. 6 |. Asgard archaeal viruses and MGEs.
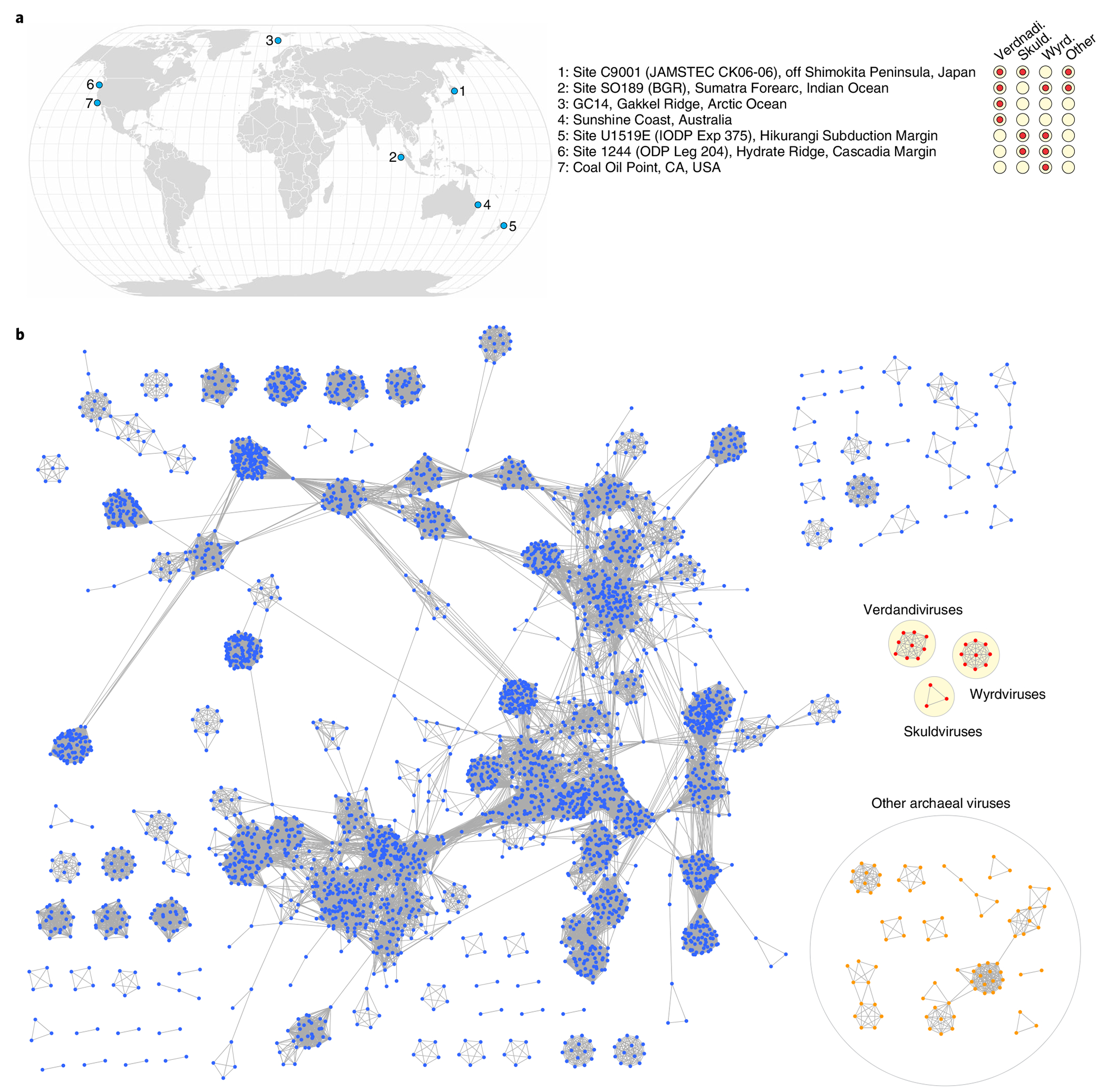
a, Geographical distribution of Asgard archaeal viruses and MGEs. Filled circles next to geographical locations signify the presence of corresponding virus groups in sediment samples from that location. b, Network-based analysis of shared protein clusters among Asgard archaeal viruses and prokaryotic double-stranded DNA viruses. Nodes represent viral genomes and edges represent the strength of connectivity between each genome based on shared protein clusters. Nodes representing genomes of the three groups of Asgard archaeal viruses are in shown in red, with the three groups circled within a yellow background. Nodes corresponding to other bacterial and archaeal viruses are shown in blue and orange, respectively.
Verdandiviruses are tailed viruses of the Caudoviricetes.
Three of the genomes, VerdaV1, VerdaV2 and VerdaV3, assembled from the Shimokita dataset, encode the hallmark proteins specific to viruses of the class Caudoviricetes, the most widespread, environmentally abundant and genetically diverse group of viruses25,26. Members of the Caudoviricetes infect bacteria and archaea and have characteristic virions consisting of an icosahedral capsid and a helical tail attached to one of the capsid vertices. Caudoviricetes, together with eukaryotic herpesviruses, form the realm Duplodnaviria27. Similar to previously characterized bacterial and archaeal Caudoviricetes18, each of the three virus genomes encodes the HK97-like MCP, a large subunit of the terminase (genome-packaging ATPase-nuclease), the portal protein and several other structural proteins, including tail components (Fig. 3a). Structural modelling of the VerdaV1 MCP using RoseTTAFold28 yielded a model with the canonical HK97-like fold (Fig. 3b).
The VerdaV1 (19.9 kb) and VerdaV2 (19.5 kb) genomes were assembled as circular contigs (Supplementary Table 3), suggesting that they correspond to complete, terminally redundant virus genomes, whereas VerdaV3 (15.4 kb) probably represents a partial genome. Comparative genomics analysis showed that VerdaV1, VerdaV2 and VerdaV3 belong to the same virus group (Fig. 3a), which we refer to as ‘verdandiviruses’ (for Verdandi, one of the three Norns, the most powerful beings in Norse mythology that govern the lives of gods and mortals). Given that the three genomes were identified in different sediment depths offshore of Shimokita Peninsula (at 18.7, 59.5 and 87.7 mbsf; Supplementary Table 3), we addressed the possibility that related viruses could also be detected in samples from other depths. To this end, we performed BLASTP searches (E-value cutoff of 1 × 10−5) queried with the corresponding MCP sequences against the assembled sequences from samples retrieved along the depth gradient. The analysis yielded six additional viral contigs and showed that related MCPs (and viruses) are also present in sediment samples from 0.9, 9.3 and 30.8 mbsf, indicating a broad distribution of verdandiviruses through the sediment column. One of the retrieved contigs (VerdaV4. 20.6 kb) was found to be circular and thus also represents a complete virus genome. Furthermore, searches against the GenBank and JGI sequence databases yielded eight hits to virus-like contigs affiliated to Asgard archaea (Supplementary Table 3). Five of the MCPs were encoded within large contigs (>10 kb), including two (Ga0114925_10000341 and Ga0114923_10001063 (VerdaV5 and VerdaV6, respectively)) circular examples. Notably, VerdaV5 was also targeted by a CRISPR spacer from a recently described Asgardarchaeota MAG1, further supporting the host assignment for the Verdandivirus group (Fig. 3a and Supplementary Table 3). Finally, we identified a partial provirus related to verdandiviruses integrated in a large genomic contig of Lokiarchaeia (Extended Data Fig. 3). The same contig contained cellular genes unequivocally belonging to Asgardarchaeota (98–99% protein identity), including those encoding ribosomal proteins. Verdandivirus contigs were recovered from four geographically remote sampling sites (Fig. 6a), and maximum-likelihood phylogenetic analysis of the MCPs revealed an overall biogeographic clustering of verdandiviruses (Fig. 3c).
Although virus-encoded homologues could be identified for only 2% (seven out of 298) of verdandivirus proteins by BLASTP searches (E value = 1 × 10−5), functional annotation for some of the other proteins was enabled by sensitive profile–profile comparisons in which HK97-like MCP and a large subunit of the terminase (TerL) MCPs were readily identified with highly significant scores (Supplementary Fig. 2 and Supplementary Table 5). Verdandiviruses display considerable sequence conservation within the capsid formation and genome-packaging modules (Fig. 3a). Notably, upstream of the MCP all viruses carry a gene encoding a putative capsid stabilization protein distantly related to the corresponding protein of marine siphoviruses—for example, TW1 (ref. 29)—which might be important for maintaining capsid integrity under the high hydrostatic pressures of deep-sea ecosystems. Verdandiviruses encode tail proteins most closely similar to those of siphoviruses, including the major tail protein, tail tape measure protein and a baseplate hub protein with an adhesin domain, suggesting that verdandiviruses possess flexible, non-contractile tails. In most of these viruses, the tape measure protein is relatively short (median length 259 amino acids (aa)), suggesting that the tails themselves are also short. Assuming ~1.5 Å of tail length per amino acid residue of tape measure protein30,31, the median tail length of verdandiviruses is predicted to be ~39 nm. The genes encoding tail proteins display low sequence conservation, whereas gene contents downstream of the tail modules are distinct in most of the identified viruses (Fig. 3a). Differences in tail modules might correspond to different host ranges of the corresponding viruses. Indeed, whereas VerdaV1 and VerdaV5 were matched by CRISPR spacers from Lokiarchaeia, VerdaV2 and VerdaV3 were predicted to infect Thorarchaeia (Supplementary Table 3). The distinct conservation patterns of the capsid and tail modules probably reflect modular evolution of these Asgard archaeal virus genomes, a common trait in bacterial and archaeal members of the Caudoviricetes32,33.
Verdandiviruses do not encode any proteins implicated in virus genome replication, and thus can be predicted to be fully reliant on the host for this process. Complete dependence on the host replication machinery is common among archaeal viruses with small and midsized genomes34, in particular, for the tailed viruses of haloarchaea and methanogens in the families Saparoviridae, Suolaviridae, Leisingerviridae and Anaerodiviridae35. Verdandiviruses encode predicted DNA-binding proteins with Zn-finger and helix-turn-helix (HTH) motifs, which could participate in recruitment of host replication and transcription machineries. Indeed, in the case of hyperthermophilic archaeal virus SIRV2, a viral HTH protein has been shown to recruit the host DNA sliding clamp protein, a known interaction partner for many other components of the host replisome36.
All complete verdandivirus genomes encompass arrays of short genes encoding predicted small, poorly conserved proteins, some of which are adjacent to genes encoding predicted transcriptional regulators (Fig. 3a). Similar to many bacterial viruses of the Caudoviricetes37,38, some of these small, fast-evolving genes could encode antidefense—in particular, anti-CRISPR proteins.
Skuldviruses: tailless icosahedral viruses of Asgard archaea.
One of the contigs from Asgard archaeal MAGs assembled from anoxic sediments from the Hikurangi Subduction Margin8, SkuldV1, targeted by two Asgard archaeal spacers (Supplementary Table 3), encodes a double jelly-roll (DJR) MCP (Fig. 4a), a hallmark of viruses of the realm Varidnaviria, an expansive assemblage of viruses evolutionarily and structurally unrelated to duplodnaviruses27. Varidnaviruses are environmentally abundant and infect hosts from all domains of life27,39. Sequence searches with the SkuldV1 MCP led to the identification of two additional contigs, one from our dataset (SkuldV2) and the other from the GenBank database (JABUBK010000319, hereinafter referred to as SkuldV3). The latter contig was obtained from subseafloor sediments from the Cascadia Margin (Ocean Drilling Programme, site 1244) in the Pacific Ocean24 and is targeted by four spacers from our dataset. SkuldV1 and SkuldV3 were assembled as circular contigs and thus correspond to complete virus genomes (Fig. 4a). In addition to the DJR MCP, the vast majority of varidnaviruses encode genome-packaging ATPases of the FtsK-HerA superfamily40. A homologue of such ATPase was identified in all three SkuldV genomes, indicating that they are bona fide members of the realm Varidnaviria. We propose referring to this group of viruses as ‘Skuldviruses’ (for Skuld, another of the Norns).
Fig. 4 |. Diversity of skuldviruses.
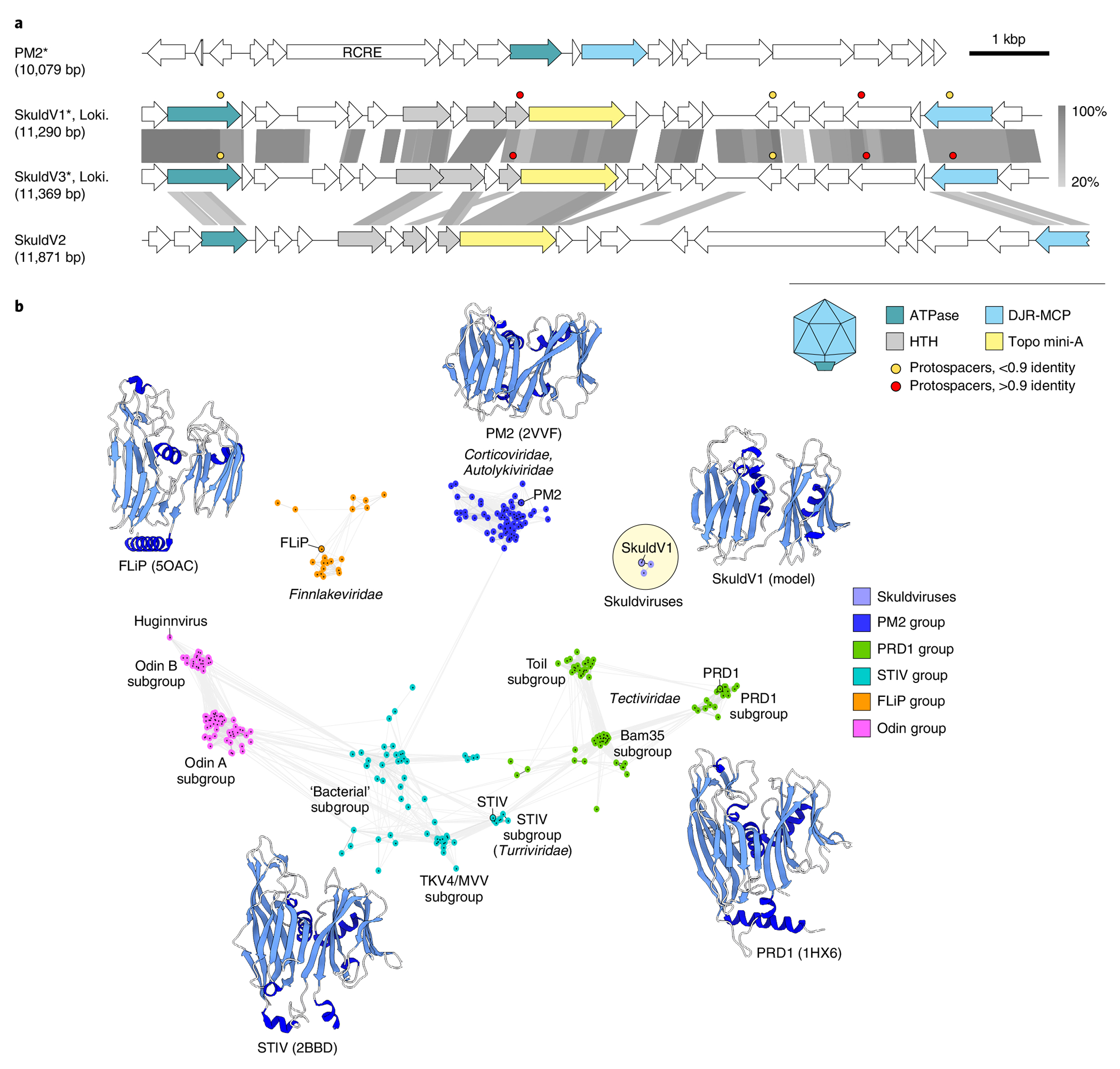
a, Genome maps of skuldviruses and PM2 virus. Homologous genes are shown using the same colours, with the key provided at the bottom. Also shown is the deduced schematic organization of the skuldvirus virion, with colours matching those of genes encoding the corresponding proteins. Coloured circles indicate the positions of protospacers. Grey shading connects genes displaying sequence similarity at the protein level, with the percentage of sequence identity indicated by different shades of grey (scale on the right). Asterisks denote complete genomes assembled as circular contigs. RCRE, rolling circle replication endonuclease. Genome map of corticovirus PM2 is shown for comparison. b, Sequence similarity network of prokaryotic virus DJR MCPs. Protein sequences were clustered by pairwise sequence similarity using CLANS. Lines connect sequences with CLANS P≤ 1 × 10−4. CLANS uses P values of BLASTP comparisons calculated from Poisson distribution of high-scoring segment pairs. Different previously defined groups42 of DJR MCP are shown as clouds of differentially coloured circles, with the key provided on the right. Note that the Odin group has been named as such previously because some of the MCPs in this cluster originated from Odinarchaeia bin42. The position of Huginnvirus described by Tamarit et al.66 is indicated. Subgroups PRD1, Toil and Bam35 are named after corresponding members of the family Tectiviridae. Skuldviruses are highlighted within a yellow circle. When available, MCP structures of viruses representing each group are shown next to the corresponding cluster (PDB accession numbers given in parentheses). The Skuldvirus cluster is represented by a structural model of the SkuldV1 MCP. Models are coloured according to their secondary structure: α-helices, dark blue; β-strands, light blue.
The vast majority of skuldvirus proteins (97%, 66 of the 68 proteins) show no similarity to proteins encoded by other known viruses. Network analysis using CLANS41 showed that the skuldvirus MCPs form a cluster separate from the previously characterized42 groups of DJR MCPs (Fig. 4b). Nevertheless, profile–profile comparisons showed that they are most closely related to the corresponding proteins of prokaryotic viruses of the families Corticoviridae (bacteriophage PM2: HHsearch probability of 98.3), Turriviridae (archaeal virus STIV: HHsearch probability of 97.8) and Tectiviridae (bacteriophage PRD1: HHsearch probability of 96.2) (Supplementary Fig. 3), whereas eukaryotic viruses with DJR MCPs were recovered with considerably lower scores (Phycodnaviridae, Pyramimonas orientalis virus: HHsearch probability of 83.4). Structural comparison of the SkuldV1 MCP model obtained using RoseTTAFold28 (Fig. 4b) further showed that it is most similar to the MCP of Pseudoalteromonas phage PM2 (ref. 43), a prototype of the Corticoviridae44 that is widespread in marine ecosystems45. Nevertheless, skuldviruses do not share genes with other known viruses other than those encoding the MCP and the genome-packaging ATPase. Corticoviruses, turriviruses and tectiviruses belong to the class Tectiliviricetes27. We propose that skuldviruses represent a separate virus family within the Tectiliviricetes.
Corticoviruses employ a rolling circle mechanism for genome replication and encode characteristic HUH superfamily endonucleases46. No such genes or other putative replication genes were identified in skuldviruses. Instead, all three skuldviruses encode a protein related to the A subunit of type IIB topoisomerases, such as topoisomerase VI. The latter enzyme consists of two distinct subunits, A and B, with the catalytic tyrosine residue responsible for DNA nicking located in the A subunit. Standalone A subunits, dubbed Topo mini-A, have recently been discovered in diverse bacterial and archaeal MGEs47 but the functions of these proteins remain unknown. In the maximum-likelihood phylogeny of Topo mini-A homologues, skuldvirus proteins form a clade with homologues from methanogenic and ammonia-oxidizing archaea (Extended Data Fig. 4). Skuldviral Topo mini-A might function as a replication protein, possibly initiating the rolling circle replication of the circular skuldvirus genomes in a manner analogous to the HUH endonuclease.
Wyrdviruses are related to spindle-shaped archaeal viruses.
One of the contigs from Asgard archaeal MAGs assembled from the Hikurangi Subduction Margin8, WyrdV1 (15,570 base pairs (bp)), targeted by one CRISPR spacer from our collection, was found to encode two homologues of the MCPs specific to spindle-shaped archaeal viruses48. In particular, the closest homologue was found in haloarchaeal virus His1 (family Halspiviridae; Extended Data Fig. 5)49. His1-like MCPs are ~80 aa in length and contain two hydrophobic segments predicted to be membrane-spanning domains48 and in all spindle-shaped viruses playing key roles in virion structure and assembly50. BLASTP searches using the WyrdV1 MCP as a query against the JGI and GenBank databases identified eight additional contigs (Fig. 5). All these contigs shared several genes encoding a morphogenetic module, including the MCP and receptor-binding adhesin, which in fuselloviruses and halspiviruses is located at one of the pointed ends of the virion51,52. In addition, all these viruses encode a AAA+ ATPase which, in profile–profile comparisons, showed the highest similarity to the morphogenesis (pI) proteins of bacterial filamentous phages (order Tubulavirales)53. This ATPase is responsible for extrusion of the viral genome through the cellular membrane during virion assembly54. We propose referring to this group of viruses as ‘wyrdviruses’ (for Wyrd (Urðr), the third Norn). The homology between the tubulaviral and wyrdviral ATPases suggests that virion assembly of wyrdviruses is mechanistically similar to the extrusion of filamentous bacteriophages. Indeed, spindle-shaped viruses infecting other archaea are released from the host without causing cell lysis through a budding-like mechanism50,55,56. Notably, some spindle-shaped viruses encode a single MCP (for example, halspiviruses) whereas others encode two paralogous MCP (for example, fuselloviruses). Similarly, wyrdviruses encode either one or two MCP paralogs (Fig. 5).
Similar to halspiviriruses and thaspiviruses, two of the contigs—Ga0209633_10003833 and Ga0209976_10001089, which we refer to as WyrdV2 and WyrdV3, respectively—contained terminal inverted repeats, indicating that these are (nearly) complete genomes. By contrast, contigs JABUBK010000290 and JABUBK010000290, herein referred to as WyrdV4 and WyrdV5, respectively, contained direct terminal repeats, indicative of the completeness and circular structure of the genomes, resembling fuselloviruses. Consistent with the different genome structures, WyrdV2 and WyrdV3 (and halspiviruses) encode protein-primed family B DNA polymerases whereas WyrdV4 and WyrdV5 encode rolling circle replication initiation endonucleases of the HUH superfamily (Fig. 5). Notably WyrdV4, in addition, encodes Topo mini-A. The latter does not cluster with homologues from skuldviruses, but instead forms a clade with the Topo mini-A from the unassigned Asgard archaeal MGEs 10H_0 and 7H_42 (Extended Data Fig. 3). Contigs Ga0209976_10001631 (WyrdV6) and Ga0209976_10001236 (WyrdV7) also encode DNA polymerases and are probably linear, nearly complete viral genomes (Fig. 5). Remarkably, the DNA polymerase encoded by WyrdV7 is not orthologous to those of WyrdV2, WyrdV3 and WyrdV6 (Fig. 5). The latter group is most closely related (~30% identity) to the corresponding protein of an uncultured virus (MW522971) associated with Altiarchaeota57, whereas the former protein is most similar to the DNA polymerase encoded by the spindle-shaped virus infecting marine Nitrososphaeria55 (Supplementary Table 3). Ga0209977_10002196 and Ga0209976_10004438 are partial genomes that lack the region encompassing the replication modules. Notably, WyrdV1 lacks either the polymerase or the rolling circle endonuclease gene and, instead, at the equivalent locus contains an array of short genes, some of which might encode replication initiators. Similar dramatic variation in gene content has previously been observed only in haloarchaeal viruses of the family Pleolipoviridae, where members of three genera encode non-homologous genome replication proteins58. Our present observations further illuminate the remarkable plasticity of the genome replication modules and genome structures in relatively closely related archaeal viruses, in general, and in wyrdviruses in particular.
Enigmatic MGEs and auxiliary genes of Asgard archaeal viruses.
In addition to the three groups of viruses, we identified seven other contigs (7H_11, 8H_18, 8H_67, 10H_0, 7H_42, Ga0114923_10000127 and Ga0209976_10000148) targeted by Asgard archaeal CRISPR spacers (Supplementary Table 3). Four of these contigs, 7H_11, 8H_18, 10H_0 and Ga0114923_10000127, were assembled as circular molecules and probably represent complete MGE genomes of 8,776, 8,776, 84,544 and 58,806 bp, respectively, whereas Ga0209976_10000148 (48,997 bp), 7H_42 (44,162 bp) and 8H_67 (13,282 bp) appeared to be partial. The seven contigs do not encode identifiable homologues of MCPs of known viruses and are likely to represent either unknown viruses or non-viral MGEs, such as plasmids (see Supplementary text for description of Asgard archaeal MGEs). Notably, these MGEs appear to be unrelated to those reported recently in Heimdallarchaeia59.
Some Asgard archaeal MGEs and viruses encode auxiliary functions, including metabolic genes, such as phosphoadenosine phosphosulfate (PAPS) reductase, which could facilitate sulfur metabolism and/or synthesis of sulfur-containing amino acids60; and enzymes involved in nucleotide metabolism, including dUTPase, thymidylate synthase X and nucleoside pyrophosphohydrolase MazG, which might function in disarming antiviral systems triggered by nucleotide-based alarmones, such as ppGpp (Supplementary text).
Discussion
Here we describe three previously undetected, distinct groups of viruses associated with Asgard archaea of the lineages Lokiarchaeia and Thorarchaeia. Each group was identified in marine sediment samples from geographically remote sites (Fig. 6), suggesting a wide distribution of these viruses in Asgard archaea-inhabited ecosystems. In addition, we recovered seven CRISPR-targeted MGEs associated with Lokiarchaeia, Thorarchaeia and Heimdallarchaeia that might represent distinct viruses with structural and morphogenetic proteins unrelated to those of any known viruses or, perhaps more likely, plasmids. Although it is clear that verdandiviruses, skuldviruses and wyrdviruses do not comprise the complete Asgard virome, they provide important insights into the diversity and evolution of the Asgard viruses. All three virus groups are sufficiently distinct from previously characterized viruses to be considered as founding representatives of three previously undescribed families. Wyrdviruses appear to be evolutionarily related to spindle-shaped viruses and thus belong to one of the archaea-specific groups of viruses19 not known in bacteria or eukaryotes. In archaea, spindle-shaped viruses are widely distributed and infect hyperthermophilic, halophilic and ammonia-oxidizing hosts from different phyla48,55. Thus, wyrdviruses further expand the reach of spindle-shaped viruses to Asgard archaea, supporting the notion that this group of viruses was associated with the last archaeal common ancestor (LACA)61. By contrast, verdandiviruses and skuldviruses encode HK97-like and DJR MCPs, respectively and, accordingly, at the highest taxonomic level belong to the realms Duplodnaviria and Varidnaviria. Viruses of both realms have deep evolutionary origins and were proposed to have been present in both LACA and the last universal cellular ancestor61. The current work further supports this inference. Although members of both Duplodnaviria and Varidnaviria also infect eukaryotes, analyses of the verdandivirus and skuldvirus genome and protein sequences unequivocally show that they are more closely, even if distantly, related to their respective prokaryotic relatives. Thus, no putative direct ancestors of eukaryotic viruses were detected. Further exploration of the Asgard archaeal virome is needed to determine whether any of the virus groups associated with extant eukaryotes originate from Asgard viruses.
All prokaryotic members of the realms Duplodnaviria and Varidnaviria are lytic viruses, which are released from the host cells by lysis (although some alternate lysis with lysogeny)62–64. Thus, verdandiviruses and skuldviruses are also likely to kill their hosts at the end of the infection cycle, thereby promoting the turnover of Asgard archaea and nutrient cycling in deep-sea ecosystems. This possibility is consistent with previous results showing that viruses in deep-sea sediments lyse archaea more rapidly compared with bacteria65. By contrast, the mechanism of virion release employed by spindle-shaped viruses does not involve cell lysis, with virions continuously released from chronically infected cells55,56. Infection of marine Nitrososphaeria with spindle-shaped virus NSV1 resulted in inhibition of host growth and was accompanied by severe reduction in the rate of ammonia oxidation and nitrite reduction55. Thus, infection dynamics and the impact of wyrdviruses are likely to be quite different from those of verdandiviruses and skuldviruses. Finally, some Asgard archaeal viruses carry auxiliary metabolic genes, such as the gene encoding PAPS reductase, which might boost the metabolism of infected cells.
The present work is accompanied by two other studies, by Tamarit et al.66 and Rambo et al.67, respectively, describing the identification of other Asgard archaeal viruses. Rambo et al. describe several distinct groups of viruses, all members of the class Caudoviricetes. The complete genomes of these viruses are considerably larger than those of verdandiviruses described here and have distinct gene contents, justifying their placements into separate families. The study by Tamarit et al. describes two viruses, Huginnvirus and Muninnvirus, related to icosahedral and spindle-shaped viruses, respectively. Huginnvirus is only distantly related to skuldviruses described here, with the two groups of viruses forming disconnected clusters in the MCP network (Fig. 4b). Muninnvirus is distantly related to wyrdviruses associated with Lokiarchaeia from our study, but infects a host from the Asgard archaeal class Odinarchaeia. Thus, viruses described in the three studies complement each other by representing distinct virus groups. Collectively, these findings provide valuable insights into the virome of Asgard archaea and open the door for understanding virus–host interactions in deep-sea ecosystems. Undoubtedly, many more Asgard archaeal virus groups remain to be discovered, which should clarify the contribution of these viruses to the evolution of the extant eukaryotic virome.
Methods
Site description and sampling.
A total of 365 m of sediment cores were recovered from Hole C9001 C of Site C9001 (water depth 1,180 m) located at the forearc basin off the Shimokita Peninsula, Japan (41° 10.638′ N, 142° 12.08′ E) by the drilling vessel Chikyu during the JAMSTEC CK06-06 cruise in 2006. Coring procedure, subsampling for molecular analyses, profiles of lithology, age model, porewater inorganic chemistry, organic chemistry and cell abundance, and summary of the molecular microbiology in sediments at site C9001 were reported previously23,68 (and references therein). Subsampled whole round cores (WRCs) for microbiology were stored at −80 °C.
Sample descriptions.
A total of 12 sediment samples at depths of 0.9, 9.3, 18.5, 30.8, 48.3, 59.5, 68.8, 87.7, 116.4, 154.3, 254.7 and 363.3 mbsf were used in this study. These representatives were chosen based on porewater chemical profiles, lithostratigraphic properties, molecular biology data on core sediments and F430 contents as follows. Note that ten samples—at depths of 0.9, 9.3, 18.5, 30.8, 48.3, 59.5, 116.4, 154.3, 254.3 and 363.3 mbsf—were previously analysed by small subunit ribosomal RNA gene tag sequencing with a 454 FLX Titanium sequencer, and details are reported in ref. 23.
At 0.9 mbsf, the uppermost section of the core column was chosen; at 9.3 mbsf, the region just beneath the sulfate–methane transition zone was chosen; at 18.5 mbsf, the highest relative abundance of Lokiarchaeia was found in the previous SSU rRNA gene tag sequencing; at 48.3 mbsf, relatively higher abundance of the South African gold mine miscellaneous euryarchaeal group in the archaeal community was observed; at 59.5 mbsf, the predominance of a lineage in Nanoarchaeia (formerly Woesearchaeota) was observed. For sediment, at a depth of 68.8 mbsf the sample harboured anomalously high F430 concentrations, and thus the sample from a depth of 87.7 mbsf was used as reference. Sediment at 116.4 mbsf consisted of ash/pumice whereas other samples used in this study were pelagic clay sediments, and a predominance of Bathyarchaeia was observed. Samples from 154.3, 254.3 and 363.3 mbsf were selected in 100-m depth intervals from the bottom of this drilling hole23,68.
Shotgun metagenomics and SSU rRNA gene tag sequencing.
DNA extraction and shotgun metagenome library construction are described in ref. 69. Briefly, total environmental DNA was extracted from approximately 5 g of sediment subsampled from the inner part of WRCs using the DNeasy PowerMax Soil Kit (Qiagen), with one minor modification70. Next, further purification was performed with NucleoSpin gDNA Clean-up (MACHERY-NAGEL). Purified DNA was used for Illumina sequencing library construction using a KAPA Hyper Prep Kit (for Illumina) (KAPA Biosystems). Sequencing was performed on an Illumina HiSeq 2500 platform (San Diego), and 250-bp paired-end reads were generated.
Assembly, binning and Asgardarchaeota phylogeny.
Contigs from each sample were assembled using MetaSpades v.0.7.12-r1039 (ref. 71) and binned with UniteM (https://github.com/dparks1134/unitem). MAG sets were generated after dereplication of bins using DAS_Tool v.1.1.2 (ref. 72) and quality check with CheckM73. MAGs with an estimated quality (completeness – fourfold contamination) <30% were excluded in downstream analysis. Taxonomic assignments of all MAGs were performed using the ‘classify’ function in GTDBtk74.
Phylogenetic analysis included 20 Asgardarchaeota MAGs, together with 255 published Asgardarchaeota and 64 non-Asgardarchaeota archaeal genomes. A marker set consisting of 53 ribosomal proteins of the 239 genomes was identified and independently aligned using the ‘identify’ and ‘align’ functions in GTDBtk74. Individual multiple sequence alignments were then concatenated and trimmed in GTDBtk using ‘trim_msa’ (−min_perc_aa 0.4). Maximum-likelihood phylogenies of Asgardarchaeota MAGs were initially estimated by FastTree v.2.1.11 (ref. 75) with default settings, and subsequently inferred using IQ-Tree v.1.6.984 (ref. 76) under the LG + C10 + F + G + PMSF model with 100 bootstraps. The final consensus tree was visualized and beautified in iTOL77.
Assembly and analysis of viral contigs.
Potential viral and MGE contigs were assembled from metagenomic reads by the MetaViralSPAdes pipeline, with default parameters78. Direct and inverted terminal repeats were identified by BLASTN79. Open reading frames were identified with Prokka80 and annotated using HHsearch81 against Pfam, PDB, SCOPe, CDD and viral protein sequence databases. Minimum information about uncultured virus genomes82 assembled in the course of this work is provided in Supplementary Table 7. Read depth, along with the assembled viral genomes, is shown in Supplementary Fig. 4. To identify similar contigs in public databases, we searched sequences of MCPs in GenBank and JGI databases (BLASTP, E-value cutoff 1 × 10−5, 70% query coverage). Genomes of assembled viral genomes were compared and visualized using EasyFig83 with the tBLASTx option. Network analysis of viral genomes was performed using vConTACT v.2.0.9.19 with default parameters against the Viral RefSeq v.201 reference database84. The virus network was visualized with Cytoscape85.
Collection of the CRISPR spacer dataset.
CRISPR arrays were detected in metagenomic contigs and 255 published Asgardarchaeota MAGs using minced v.0.4.2 (ref. 86) and CRISPRDetect87. CRISPR arrays from metagenomic contigs were assigned to ‘Asgard’ and ‘non-Asgard’ groups based on CRISPR repeat similarity to previously characterized Asgard CRISPR repeats, with 90% identity cutoff over the full length of the repeat21. CRISPR repeat sequences from both metagenomic contigs and Asgard MAGs were then clustered together using an all-against-all BLASTN search (E-value cutoff 1 × 10−5, 90% identity, word size 7), and the result of clustering was visualized in Cytoscape85. Consensus sequences for the nine major clusters of CRISPR repeats were constructed using the python package Logomaker88.
Assignment of CRISPR arrays to Asgardarchaeota.
To assemble the CRISPR spacer database we relied on two categories of CRISPR loci: (1) ‘reference’ sequences of manually curated Asgard CRISPR loci from Makarova et al.21 and CRISPR loci from Asgard archaeal MAGs; and (2) ‘metagenomic’ CRISPR loci from contigs originating from Shimokita Peninsula subseafloor sediments. CRISPR loci from the latter category were assigned to Asgard archaea if the identity of CRISPR repeat to the reference Asgard archaeal CRISPRs from the former category was >90% and protein sequences encoded on these CRISPR-containing contigs (when present) had the best BLASTP hit to Asgard archaeal proteins. All reference contigs were downloaded from GenBank without annotation. We used prodigal and CRISPR−Cas++ to identify cas genes in contigs with CRISPR arrays. Protein sequences were compared to the nr database with BLASTP to confirm Asgard assignment of the contig with CRISPR.
Verdandiviruses were targeted by spacers from five reference and four metagenomic CRISPR loci (Extended Data Fig. 1). The length of reference contigs varied from 3 to 126 kbp. Five reference contigs and one metagenomic contig contained cas genes, with the most conserved Cas protein (Cas1) being assigned to Asgard archaea (Lokiarachaeia, Helarchaeia) by the best BLASTP hit with 72–74% identity. Skuldviruses were targeted by spacers from three reference and three metagenomic CRISPR loci, whereas wyrdviruses were targeted by spacers from two reference and three metagenomic CRISPR loci (Extended Data Fig. 1). CRISPR repeats of metagenomic contigs targeting the three groups of viruses were 97% identical to those from the reference category21.
Asgard archaeal CRISPR repeats.
Predicted Asgard archaeal CRISPR arrays contained distinct repeats that could be clustered into nine groups with 90% identity of sequences within groups (Fig. 2a). Comparison of Asgard archaeal CRISPR repeats with those in the public RepeatTyper database produced significant results only for group 9 repeat, which was identified as belonging to CRISPR-Cas type I-E. The closest repeat to group 9 was from Thermobaculum terrenum, with two mismatches (27/29). Repeats from group 4 partially matched to Desulfosarcina (29/37). Other repeat groups showed no matches in the CRISPR–Cas++ database (https://crisprcas.i2bc.paris-saclay.fr/).
Structural modelling and network analysis of MCPs.
Structural modelling of the representative verdnadivirus and skuldvirus MCPs was performed with RoseTTAFold28. The MCPs of skuldviruses were compared with DJR MCPs of other known prokaryotic viruses using CLANS41, an implementation of the Fruchterman–Reingold force-directed layout algorithm that treats protein sequences as point masses in a virtual multidimensional space in which they attract or repel each other based on the strength of their pairwise similarities (CLANS P values). The reference dataset of DJR MCP was obtained from ref. 42. Sequences were clustered using CLANS with BLASTP option (E value = 1 × 10−4)41.
Phylogenetic analysis of viral proteins.
Sequences were aligned using MAFFT in ‘Auto’ mode89. For phylogenetic analysis, uninformative positions were removed using TrimAl with a gap threshold of 0.2 for verdandiviral MCPs and the gappyout option for Topo mini-A90. The final alignments contained 323 and 277 positions, respectively. Maximum-likelihood trees were inferred using IQ-TREE v.2 (ref. 76). The best-fitting substitution models were selected by IQ-TREE, and were LG + G4 and LG + R5 for verdandiviral MCPs and Topo mini-A, respectively. The trees were visualized with iTOL77. Phylogenetic trees and underlying alignments in editable format are provided in Supplementary Data 2.
CRISPR-based virus–host assignments.
Spacer sequences from Asgard archaeal CRISPR arrays were matched to metagenomic contigs and published Asgard MAGs (BLASTN, E value = 1 × 10−5, 90% identity, word size 7), and to viral genomes available on the IGV database (default parameters) at https://img.jgi.doe.gov/cgi-bin/m/main.cgi?section=WorkspaceBlast&page=viralform. To assess the false positive rate of BLASTN we used a control set of viral contigs from human metagenomes (~400,000 sequences)91–94. Seven spacers out of 2,532 (false positive rate = 0.00276) were matched to a control dataset by BLASTN with the same parameters (E value = 1 × 10−5, 90% identity, word size 7). For each spacer we calculated the difference between the identity of best match in the test and control datasets. Spacers with <10% identity difference (n = 5) were removed as potential low-complexity spacers. SpacePHARER95, with false discovery rate < 0.001, was used for a sensitive search of protospacers with lower nucleotide identity to support the BLASTN results. CRISPRHost and CRISPR Spacer Database and Exploration Tool96 were used to match large collections of prokaryotic spacers to identified Asgard MGE sequences.
Kmer-based virus–host assignments.
WIsH97 and PHIST98, kmer-based methods that compare 8-mer (WIsH) and 25-mer (PHIST) nucleotide frequencies of viruses and potential hosts, were applied to validate prediction of CRISPR-based virus–host assignment. Unlike other kmer-based methods, PHIST and WIsH can predict virus–host interactions for user-provided host sequences rather than precomputed host databases. For the potential host dataset, we combined 195 MAGs of Asgardarchaeota, 267 non-Asgard MAGs reconstructed from Shimokita Peninsula metagenomes, 2,143 RefSeq archaeal genomes and 1,077 representative bacterial genomes (Supplementary Table 8). For the virus dataset we used MGE sequences targeted by Asgard spacers. Viral contigs from human metagenomes were used as a negative dataset to calculate null parameters for models of hosts for WIsH.
WIsH confirmed Asgard archaeal hosts for all MGE sequences targeted by Asgard archaeal spacers, with P values ranging from 1.18 × 10−6 for WyrdV1 to 1.32 × 10−1 for VerdaV2 (Supplementary Table 5). In the 25-mer-based PHIST analysis we considered only those predictions based on >10 kmer. Using these parameters, hosts were predicted for 17 out of 28 (60%) contigs. Of these, 11 (65%) contigs, representing all groups of viruses and MGEs described in this work, were predicted as being associated with Asgard archaea. However, the remaining six, all verdandiviruses, were affiliated to Thermoplasmatota MAG 2H_mb2_bin16 assembled from the Shimokita Peninsula dataset. We note that the latter prediction probably resulted from the inclusion of a small (~2 kbp) verdandivirus-like contig in Thermoplasmatota MAG 2H_mb2_bin16, which is probably a binning artefact because the full-length verdandivirus contig was assigned to Lokiarachaeia MAG. Indeed, we could not confirm Thermoplasmatota assignment as a verdandivirus host with CRISPR spacer analysis.
To further verify the validity of our CRISPR matches, we checked the predicted Asgard archaeal viruses and MGEs for the presence of non-Asgard protospacers using large spacer collections of CRISPRHost and CRISPR Spacer Database and Exploration Tool (CRISPR_SDET). CRISPRHost did not find any protospacers with the default parameters. CRISPR_SDET search was performed with 90% identity threshold. Two protospacers were found: verdandivirus VerdaV4 was assigned to Floccifex porci, a Firmicutes bacterium from the pig gut metagenome; two mobile elements from the ‘Other’ group, JGI-127 and JGI-148, were assigned to Treponema sp., a pathogenic bacterium of humans and other mammals. Given that neither of these bacteria resides in deep-sea sediments, the two spacers are likely to represent false positives.
Extended Data
Extended Data Fig. 1 |. Matches between asgardarchaeal CRISPR spacers and viruses.
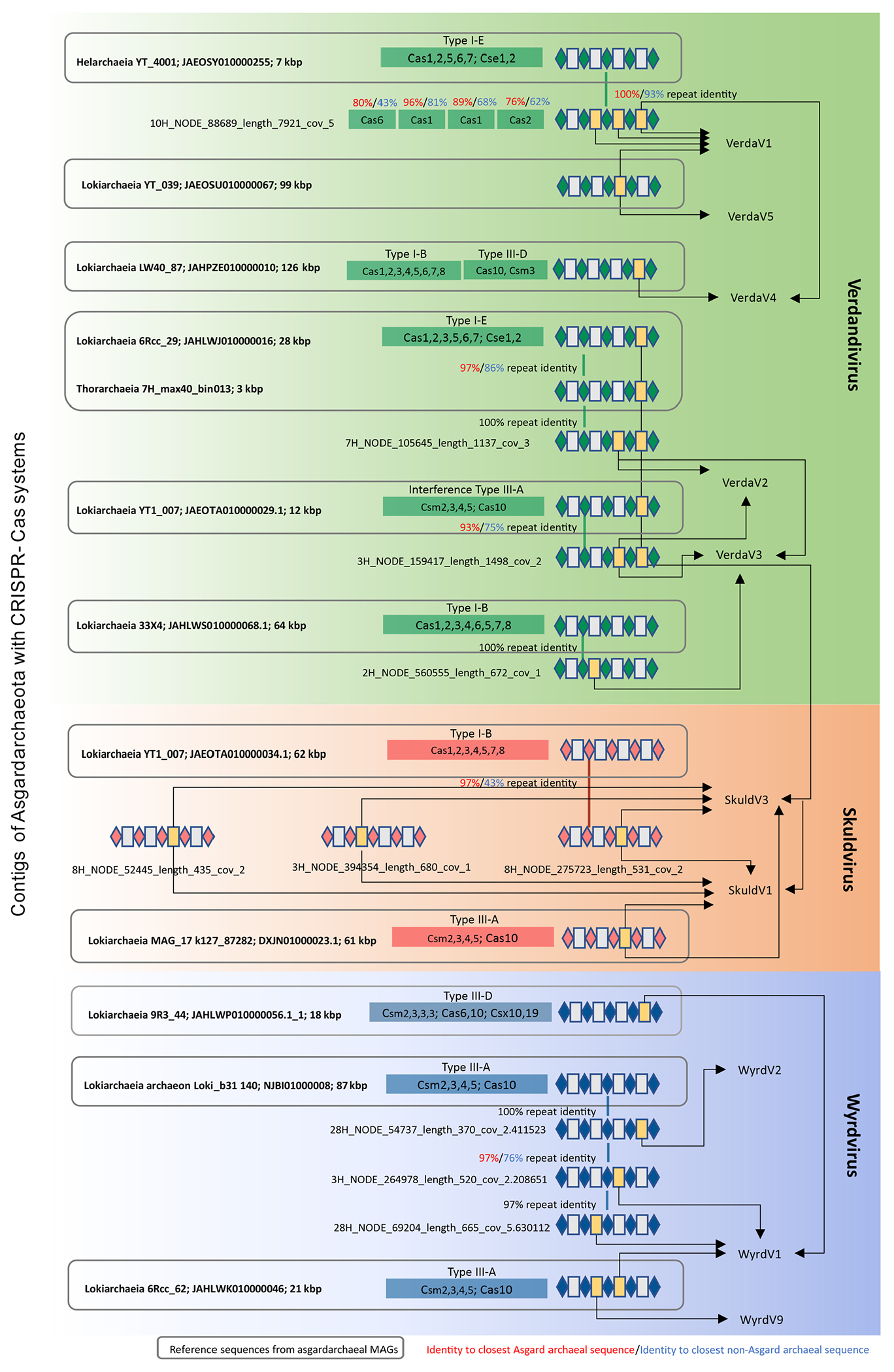
The figure is divided into three blocks (green, red and blue) corresponding to the three groups of asgardarchaeal viruses, namely, verdandiviruses, skuldviruses and wyrdviruses. CRISPR repeats and spacers are indicated as diamonds and boxes, respectively. Spacers matching asgardarchaeal viruses are shown in yellow and are connected to the names of targeted viruses with arrows. Thick vertical lines connect related repeats and the exact pairwise sequence identities are indicated.
Extended Data Fig. 2 |. Partial provirus integrated within a genomic contig of Lokiarchaeia.
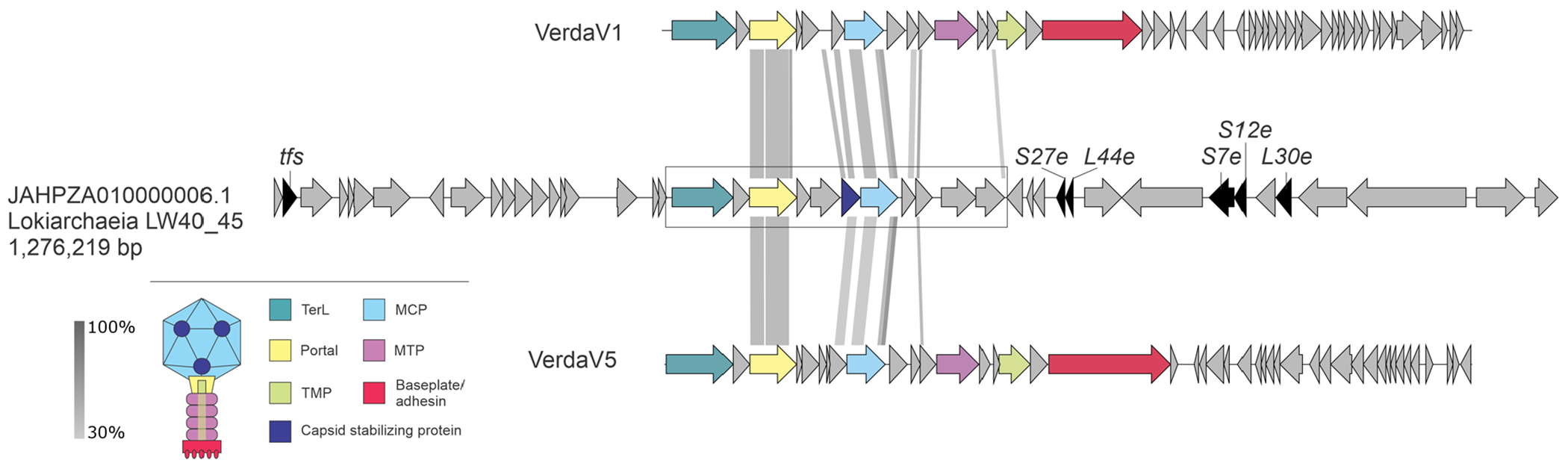
The verdandivirus-derived region is boxed. Homologous genes are shown using the same colors and the key is provided on the left of the figure. Housekeeping cellular genes are shown in black and include those encoding transcription factor S (TFS) and ribosomal proteins S7e, S12e, S27e, L30e and L44e. Grey shading connects genes displaying sequence similarity at the protein level, with the percent of sequence identity depicted with different shades of grey.
Extended Data Fig. 3 |. Maximum likelihood phylogenetic tree of Topo mini-A proteins.
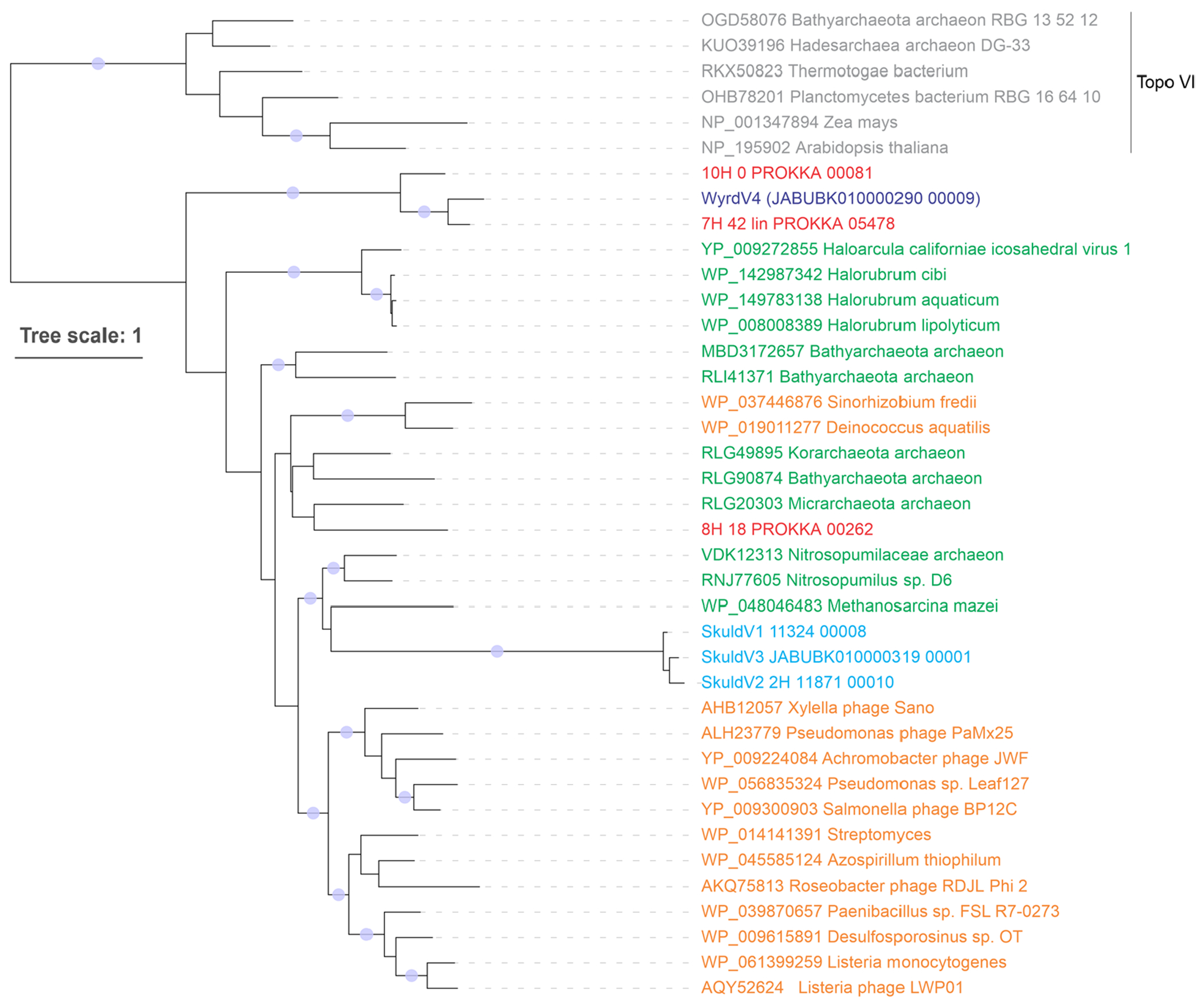
Proteins of skuldviruses and wyrdviruses are shown in cyan and dark blue, respectively, whereas those encoded by other asgardarchaeal MGE are shown in red. Other Topo mini-A homologs encoded by archaea and bacteria (or their corresponding viruses) are shown in green and orange, respectively. Topo VI proteins were used as an outgroup and are colored grey. The tree was constructed using the automatic optimal model selection (LG+R5). The scale bar represents the number of substitutions per site. Circles at the nodes denote aLRT SH-like branch support values larger than 90%.
Extended Data Fig. 4 |. Sequence alignment of the major capsid proteins of selected wyrdviruses, fusellovirus SSV1 and halspivirus His1.
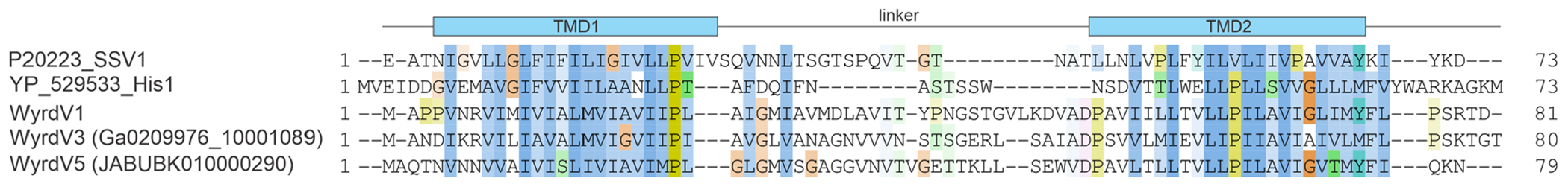
When available, proteins are identified with their accession numbers followed by a virus name. TMD, transmembrane domain.
Supplementary Material
Acknowledgements
We thank the crews, technical staff and shipboard scientists of the DV Chikyu for the operation and sampling during cruise CK06-06 in 2006. We thank M. Hirai and Y. Takaki for library construction, sequencing and data deposition of subseafloor samples off Shimokita. The work in the M.K. laboratory is supported by grants from l’Agence Nationale de la Recherche (nos. ANR-20-CE20-0009-02 and ANR-21-CE11-0001-01) and Ville de Paris (Emergence(s) project MEMREMA). S.M. was supported by the Metchnikov fellowship from Campus France and Russian Science Foundation (grant no. 19-74-20130). N.Y. and E.V.K. are supported by the Intramural Research Program of the National Institutes of Health of the USA (National Library of Medicine). The work of C.R. and J.S. is funded by the Australian Research Council Future Fellow Award (no. FT170100213, to CR). T.N. was partly supported by MEXT KAKENHI (grant nos. JP19H05684 within JP19H05679 (Post-Koch Ecology) and 16H06429, 16K21723 and 16H06437 (NeoVirology)).
Footnotes
Reporting summary. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Code availability
No custom code was used.
Competing interests
The authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41564-022-01144-6.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41564-022-01144-6.
Data availability
The raw reads, as well as assembled virus and MGE genome sequences from the metagenomes described in this study, are available at NCBI under BioProject no. PRJDB12054, BioSample accession nos SAMD00394285–SAMD00394296. Accession numbers of the 20 MAGs assembled in the course of this study are listed in Supplementary Table 1. Accession numbers of the virus and MGE genome sequence are listed in Supplementary Table 3. Source data are provided with this paper.
References
- 1.Liu Y et al. Expanded diversity of Asgard archaea and their relationships with eukaryotes. Nature 593, 553–557 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dombrowski N, Teske AP & Baker BJ Expansive microbial metabolic versatility and biodiversity in dynamic Guaymas Basin hydrothermal sediments. Nat. Commun 9, 4999 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wong HL et al. Disentangling the drivers of functional complexity at the metagenomic level in Shark Bay microbial mat microbiomes. ISME J. 12, 2619–2639 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Y et al. Comparative genomic inference suggests mixotrophic lifestyle for Thorarchaeota. ISME J. 12, 1021–1031 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaremba-Niedzwiedzka K et al. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature 541, 353–358 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Seitz KW, Lazar CS, Hinrichs KU, Teske AP & Baker BJ Genomic reconstruction of a novel, deeply branched sediment archaeal phylum with pathways for acetogenesis and sulfur reduction. ISME J. 10, 1696–1705 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spang A et al. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 521, 173–179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun J et al. Recoding of stop codons expands the metabolic potential of two novel Asgardarchaeota lineages. ISME Commun. 1, 30 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seitz KW et al. Asgard archaea capable of anaerobic hydrocarbon cycling. Nat. Commun 10, 1822 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farag IF, Zhao R & Biddle JF “Sifarchaeota,” a novel Asgard phylum from Costa Rican sediment capable of polysaccharide degradation and anaerobic methylotrophy. Appl. Environ. Microbiol 87, e02584–20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang JW et al. Newly discovered Asgard archaea Hermodarchaeota potentially degrade alkanes and aromatics via alkyl/benzyl-succinate synthase and benzoyl-CoA pathway. ISME J. 15, 1826–1843 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cai M et al. Diverse Asgard archaea including the novel phylum Gerdarchaeota participate in organic matter degradation. Sci. China Life Sci 63, 886–897 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Rinke C et al. A standardized archaeal taxonomy for the Genome Taxonomy Database. Nat. Microbiol 6, 946–959 (2021). [DOI] [PubMed] [Google Scholar]
- 14.Imachi H et al. Isolation of an archaeon at the prokaryote-eukaryote interface. Nature 577, 519–525 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopez-Garcia P & Moreira D The Syntrophy hypothesis for the origin of eukaryotes revisited. Nat. Microbiol 5, 655–667 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Da Cunha V, Gaia M, Nasir A & Forterre P Asgard archaea do not close the debate about the universal tree of life topology. PLoS Genet. 14, e1007215 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Da Cunha V, Gaia M, Gadelle D, Nasir A & Forterre P Lokiarchaea are close relatives of Euryarchaeota, not bridging the gap between prokaryotes and eukaryotes. PLoS Genet. 13, e1006810 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baquero DP et al. Structure and assembly of archaeal viruses. Adv. Virus Res 108, 127–164 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Prangishvili D et al. The enigmatic archaeal virosphere. Nat. Rev. Microbiol 15, 724–739 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Dellas N, Snyder JC, Bolduc B & Young MJ Archaeal viruses: diversity, replication, and structure. Annu. Rev. Virol 1, 399–426 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Makarova KS et al. Unprecedented diversity of unique CRISPR-Cas-related systems and Cas1 homologs in Asgard archaea. CRISPR J. 3, 156–163 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coclet C & Roux S Global overview and major challenges of host prediction methods for uncultivated phages. Curr. Opin. Virol 49, 117–126 (2021). [DOI] [PubMed] [Google Scholar]
- 23.Nunoura T et al. Variance and potential niche separation of microbial communities in subseafloor sediments off Shimokita Peninsula, Japan. Environ. Microbiol 18, 1889–1906 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Glass JB et al. Microbial metabolism and adaptations in Atribacteria-dominated methane hydrate sediments. Environ. Microbiol 23, 4646–4660 (2021). [DOI] [PubMed] [Google Scholar]
- 25.Dion MB, Oechslin F & Moineau S Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol 18, 125–138 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Iranzo J, Krupovic M & Koonin EV The double-stranded DNA virosphere as a modular hierarchical network of gene sharing. mBio 7, e00978–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koonin EV et al. Global organization and proposed megataxonomy of the virus world. Microbiol. Mol. Biol. Rev 84, e00061–19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baek M et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Z et al. Structure of the marine siphovirus TW1: evolution of capsid-stabilizing proteins and tail spikes. Structure 26, 238–248 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Hendrix RW Tail length determination in double-stranded DNA bacteriophages. Curr. Top. Microbiol. Immunol 136, 21–29 (1988). [DOI] [PubMed] [Google Scholar]
- 31.Mahony J et al. Functional and structural dissection of the tape measure protein of lactococcal phage TP901-1. Sci. Rep 6, 36667 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pope WH et al. Whole genome comparison of a large collection of mycobacteriophages reveals a continuum of phage genetic diversity. eLife 4, e06416 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krupovic M, Forterre P & Bamford DH Comparative analysis of the mosaic genomes of tailed archaeal viruses and proviruses suggests common themes for virion architecture and assembly with tailed viruses of bacteria. J. Mol. Biol 397, 144–160 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Krupovic M, Cvirkaite-Krupovic V, Iranzo J, Prangishvili D & Koonin EV Viruses of archaea: structural, functional, environmental and evolutionary genomics. Virus Res. 244, 181–193 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y et al. Diversity, taxonomy, and evolution of archaeal viruses of the class Caudoviricetes. PLoS Biol. 19, e3001442 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gardner AF, Bell SD, White MF, Prangishvili D & Krupovic M Protein-protein interactions leading to recruitment of the host DNA sliding clamp by the hyperthermophilic Sulfolobus islandicus rod-shaped virus 2. J. Virol 88, 7105–7108 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gussow AB et al. Machine-learning approach expands the repertoire of anti-CRISPR protein families. Nat. Commun 11, 3784 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Y & Bondy-Denomy J Anti-CRISPRs go viral: the infection biology of CRISPR-Cas inhibitors. Cell Host Microbe 29, 704–714 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krupovic M & Bamford DH Virus evolution: how far does the double beta-barrel viral lineage extend? Nat. Rev. Microbiol 6, 941–948 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Hong C et al. A structural model of the genome packaging process in a membrane-containing double stranded DNA virus. PLoS Biol. 12, e1002024 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frickey T & Lupas A CLANS: a Java application for visualizing protein families based on pairwise similarity. Bioinformatics 20, 3702–3704 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Yutin N, Bäckström D, Ettema TJG, Krupovic M & Koonin EV Vast diversity of prokaryotic virus genomes encoding double jelly-roll major capsid proteins uncovered by genomic and metagenomic sequence analysis. Virol. J 15, 67 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abrescia NG et al. Insights into virus evolution and membrane biogenesis from the structure of the marine lipid-containing bacteriophage PM2. Mol. Cell 31, 749–761 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Oksanen HM, ICTV Report Consortium. ICTV Virus Taxonomy Profile: Corticoviridae. J. Gen. Virol 98, 888–889 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krupovic M & Bamford DH Putative prophages related to lytic tailless marine dsDNA phage PM2 are widespread in the genomes of aquatic bacteria. BMC Genomics 8, 236 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kazlauskas D, Varsani A, Koonin EV & Krupovic M Multiple origins of prokaryotic and eukaryotic single-stranded DNA viruses from bacterial and archaeal plasmids. Nat. Commun 10, 3425 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takahashi TS et al. Expanding the type IIB DNA topoisomerase family: identification of new topoisomerase and topoisomerase-like proteins in mobile genetic elements. NAR Genom. Bioinform 2, lqz021 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krupovic M, Quemin ER, Bamford DH, Forterre P & Prangishvili D Unification of the globally distributed spindle-shaped viruses of the Archaea. J. Virol 88, 2354–2358 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bath C, Cukalac T, Porter K & Dyall-Smith ML His1 and His2 are distantly related, spindle-shaped haloviruses belonging to the novel virus group, Salterprovirus. Virology 350, 228–239 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Wang F et al. Spindle-shaped archaeal viruses evolved from rod-shaped ancestors to package a larger genome. Cell 185, 1297–1307.e11 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hong C et al. Lemon-shaped halo archaeal virus His1 with uniform tail but variable capsid structure. Proc. Natl Acad. Sci. USA 112, 2449–2454 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quemin ER et al. Sulfolobus spindle-shaped virus 1 contains glycosylated capsid proteins, a cellular chromatin protein, and host-derived lipids. J. Virol 89, 11681–11691 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roux S et al. Cryptic inoviruses revealed as pervasive in bacteria and archaea across Earth’s biomes. Nat. Microbiol 4, 1895–1906 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Straus SK & Bo HE Filamentous bacteriophage proteins and assembly. Subcell. Biochem 88, 261–279 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Kim JG et al. Spindle-shaped viruses infect marine ammonia-oxidizing thaumarchaea. Proc. Natl Acad. Sci. USA 116, 15645–15650 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quemin ER et al. Eukaryotic-like virus budding in Archaea. mBio 7, e01439–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rahlff J et al. Lytic archaeal viruses infect abundant primary producers in Earth’s crust. Nat. Commun 12, 4642 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bamford DH et al. ICTV Virus Taxonomy Profile: Pleolipoviridae. J. Gen. Virol 98, 2916–2917 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu F et al. Unique mobile elements and scalable gene flow at the prokaryote–eukaryote boundary revealed by circularized Asgard archaea genomes. Nat. Microbiol 7, 200–212 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Summer EJ, Gill JJ, Upton C, Gonzalez CF & Young R Role of phages in the pathogenesis of Burkholderia, or ‘Where are the toxin genes in Burkholderia phages?’. Curr. Opin. Microbiol 10, 410–417 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krupovic M, Dolja VV & Koonin EV The LUCA and its complex virome. Nat. Rev. Microbiol 18, 661–670 (2020). [DOI] [PubMed] [Google Scholar]
- 62.Cahill J & Young R Phage lysis: multiple genes for multiple barriers. Adv. Virus Res 103, 33–70 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Snyder JC & Young MJ Lytic viruses infecting organisms from the three domains of life. Biochem. Soc. Trans 41, 309–313 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Krupovic M, Daugelavicius R & Bamford DH A novel lysis system in PM2, a lipid-containing marine double-stranded DNA bacteriophage. Mol. Microbiol 64, 1635–1648 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Danovaro R et al. Virus-mediated archaeal hecatomb in the deep seafloor. Sci. Adv 2, e1600492 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tamarit D et al. A closed Candidatus Odinarchaeum chromosome exposes Asgard archaeal viruses. Nat. Microbiol 10.1038/s41564-022-01122-y (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rambo IM, de Anda V, Langwig MV & Baker BJ Genomes of six viruses that infect Asgard archaea from deep-sea sediments. Nat. Microbiol 10.1038/s41564-022-01150-8 (2022). [DOI] [PubMed] [Google Scholar]
- 68.Kaneko M et al. Insights into the methanogenic population and potential in subsurface marine sediments based on coenzyme F430 as a function-specific compound analysis. JACS Au 1, 1743–1751 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hirai M et al. Library construction from subnanogram DNA for pelagic sea water and deep-sea sediments. Microbes Environ. 32, 336–343 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hiraoka S et al. Microbial community and geochemical analyses of trans-trench sediments for understanding the roles of hadal environments. ISME J. 14, 740–756 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nurk S, Meleshko D, Korobeynikov A & Pevzner PA metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sieber CMK et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol 3, 836–843 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P & Tyson GW CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chaumeil PA, Mussig AJ, Hugenholtz P & Parks DH GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Price MN, Dehal PS & Arkin AP FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nguyen LT, Schmidt HA, von Haeseler A & Minh BQ IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol 32, 268–274 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Letunic I & Bork P Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Antipov D, Raiko M, Lapidus A & Pevzner PA Metaviral SPAdes: assembly of viruses from metagenomic data. Bioinformatics 36, 4126–4129 (2020). [DOI] [PubMed] [Google Scholar]
- 79.Altschul SF, Gish W, Miller W, Myers EW & Lipman DJ Basic local alignment search tool. J. Mol. Biol 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- 80.Seemann T Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014). [DOI] [PubMed] [Google Scholar]
- 81.Steinegger M et al. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics 20, 473 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Roux S et al. Minimum Information about an Uncultivated Virus Genome (MIUViG). Nat. Biotechnol 37, 29–37 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sullivan MJ, Petty NK & Beatson SA Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bin Jang H et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol 37, 632–639 (2019). [DOI] [PubMed] [Google Scholar]
- 85.Shannon P et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bland C et al. CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 8, 209 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Biswas A, Staals RH, Morales SE, Fineran PC & Brown CM CRISPRDetect: a flexible algorithm to define CRISPR arrays. BMC Genomics 17, 356 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tareen A & Kinney JB Logomaker: beautiful sequence logos in Python. Bioinformatics 36, 2272–2274 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Katoh K, Rozewicki J & Yamada KD MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform 20, 1160–1166 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Capella-Gutierrez S, Silla-Martinez JM & Gabaldon T trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nayfach S et al. Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome. Nat. Microbiol 6, 960–970 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Camarillo-Guerrero LF, Almeida A, Rangel-Pineros G, Finn RD & Lawley TD Massive expansion of human gut bacteriophage diversity. Cell 184, 1098–1109 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gregory AC et al. The gut virome database reveals age-dependent patterns of virome diversity in the human gut. Cell Host Microbe 28, 724–740 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shkoporov AN et al. The human gut virome is highly diverse, stable, and individual specific. Cell Host Microbe 26, 527–541 (2019). [DOI] [PubMed] [Google Scholar]
- 95.Zhang R et al. SpacePHARER: sensitive identification of phages from CRISPR spacers in prokaryotic hosts. Bioinformatics 37, 3364–3366 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dion MB et al. Streamlining CRISPR spacer-based bacterial host predictions to decipher the viral dark matter. Nucleic Acids Res. 49, 3127–3138 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Galiez C, Siebert M, Enault F, Vincent J & Soding J WIsH: who is the host? Predicting prokaryotic hosts from metagenomic phage contigs. Bioinformatics 33, 3113–3114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zielezinski A, Deorowicz S & Gudys A PHIST: fast and accurate prediction of prokaryotic hosts from metagenomic viral sequences. Bioinformatics 38, 1447–1449 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw reads, as well as assembled virus and MGE genome sequences from the metagenomes described in this study, are available at NCBI under BioProject no. PRJDB12054, BioSample accession nos SAMD00394285–SAMD00394296. Accession numbers of the 20 MAGs assembled in the course of this study are listed in Supplementary Table 1. Accession numbers of the virus and MGE genome sequence are listed in Supplementary Table 3. Source data are provided with this paper.