

Abstract
Alois Alzheimer described the first patient with Alzheimer’s disease (AD) in 1907 and today AD is the most frequently diagnosed of dementias. AD is a multi-factorial neurodegenerative disorder with familial, life style and comorbidity influences impacting a global population of more than 47 million with a projected escalation by 2050 to exceed 130 million. In the USA the AD demographic encompasses approximately six million individuals, expected to increase to surpass 13 million by 2050, and the antecedent phase of AD, recognized as mild cognitive impairment (MCI), involves nearly 12 million individuals. The economic outlay for the management of AD and AD-related cognitive decline is estimated at approximately 355 billion USD. In addition, the intensifying prevalence of AD cases in countries with modest to intermediate income countries further enhances the urgency for more therapeutically and cost-effective treatments and for improving the quality of life for patients and their families. This narrative review evaluates the pathophysiological basis of AD with an initial focus on the therapeutic efficacy and limitations of the existing drugs that provide symptomatic relief: acetylcholinesterase inhibitors (AChEI) donepezil, galantamine, rivastigmine, and the N-methyl-D-aspartate receptor (NMDA) receptor allosteric modulator, memantine. The hypothesis that amyloid-β (Aβ) and tau are appropriate targets for drugs and have the potential to halt the progress of AD is critically analyzed with a particular focus on clinical trial data with anti-Aβ monoclonal antibodies (MABs), namely, aducanumab, lecanemab and donanemab. This review challenges the dogma that targeting Aβ will benefit the majority of subjects with AD that the anti-Aβ MABs are unlikely to be the “magic bullet”. A comparison of the benefits and disadvantages of the different classes of drugs forms the basis for determining new directions for research and alternative drug targets that are undergoing pre-clinical and clinical assessments. In addition, we discuss and stress the importance of the treatment of the co-morbidities, including hypertension, diabetes, obesity and depression that are known to increase the risk of developing AD.
Keywords: Alzheimer’s disease, acetylcholinesterase inhibitors, donepezil, N-methyl-Daspartate receptor, memantine, amyloid protein, monoclonal antibody, lecanemab
1 Introduction
Dementia is a multifaceted neurological disorder that covers a spectrum of cognitive impairments primary affecting memory, thinking, attention, behavior, and the ability to perform everyday tasks. Demographically, it disproportionally affects older adults, with the risk significantly increasing after the age of 65. While various types of dementia exist, such as vascular dementia, including CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy), the Notch 3 gene mutation that manifests as cerebral arteriopathy, Lewy body dementia, and frontotemporal dementia, it is Alzheimer’s Disease (AD) stands out as the most prevalent form, accounting for approximately 60% of all dementia cases. Despite the diversity among dementia subtypes, AD is the primary culprit, imposing profound emotional and economic burdens on individuals, families, and healthcare systems worldwide, especially in rapidly aging populations. As such, as reflected in Supplementary Figure S1, the search for the cause(s) and effective treatment(s) of AD has intensified over the years since its first description by Alois Alzheimer in 1907 (Alzheimer, 1907; Alzheimer et al., 1995, also see Alzheimer’s, 2020).
A major difficulty in developing effective therapeutic agents is linked to the long pre-clinical phase between the early stages of the brain pathology and the detection of cognitive decline (McGeer, et al., 2018). Clinically, AD has both a familial (FAD) and a sporadic (SAD) occurrence, with FAD accounting for approximately 5% of cases (Ertekin-Taner, 2007). Given these complexities, it is not surprising that the determination of the most effective treatment for AD as well as optimizing and providing early treatment for individuals with AD has proved difficult–a challenge that to date has not been met (Ferrari and Sorbi, 2021).
AD can be distinguished from other types of dementia by its closer association in its early stage with short term memory impairment, and, later by a buildup of amyloid protein (Aβ) in the brain. The greater majority of cases of AD are seen in subjects over the age of 65 and referred to as Late Onset AD (LOAD), or sporadic AD (SAD); however, approximately 5%–10% of cases are seen in younger patients and referred to as Early Onset AD (EOAD), or familial AD (FAD, FD), which is seen in patients as young as 30 years of age and may have an atypical clinical presentation (Sirkis et al., 2022). Unlike LOAD patients, EOAD has a strong genetic determination and autosomal-dominant inheritance linked primarily to three genes: Amyloid Precursor Protein (APP), Presenilin-1 (PSEN1), and Presenilin-2 (PSEN2), which code for presenilin-1 (PS-1), and presenilin-2 (PS-2) respectively, with mutations in PSEN1 being more frequent (Cacace et al., 2016). Presenilin are a family of transmembrane proteins that make up the catalytic component of γ-secretase, an enzyme which cleaves more than 140 substrates, including APP (Bagaria, et al., 2022; Hur, 2022)–see section 3.3. However, as a further complication to the understanding the pathophysiological basis of AD and effective treatment(s), genome-wide association studies (GWAS) have identified over 50 loci linked to AD and notably associated with three genes: the clusterin (CLU) gene; the PICALM (phosphatidylinositol-binding clathrin assembly) gene; and the complement component (3b/4b) receptor one on chromosome 1 (CR1) (reviewed by Sims et al., 2020). EOAD also has a nonmendelian link to the apolipoprotein E4, ApoE4, allele (Reitz et al., 2020), with polymorphisms associated with lipid metabolism, immunity and endocytosis pathways that contribute to sporadic AD (Barber et al., 2017), and offer additional targets for optimizing the treatment of AD (reviewed by Femminella et al. (2021)–see also section 3.5.
An analysis of data from the UK biobank of over 350,000 participants aged under 65 with dementia identified 39 risk factors that included not only CVD, diabetes and depression, but also vitamin D deficiency, benzodiazepine and alcohol use, smoking, a low level of physical activity, lower grip strength, environmental factors, depression, and sleep problems (Hendriks et al., 2023). These data, as also reflected in Figure 1, emphasize the importance of a greater focus on identifying the key modifiable risk factors that play an important role in both EOAD and LOAD to prevent disease development, as was stressed in the 2020 Lancet Commission report (Livingston et al., 2020). The importance of reducing these modifiable risk factors has been emphasized by many others (see also Wang et al., 2018; Zhang et al., 2021).
FIGURE 1.

Flowchart of Alzheimer’s Dementia pathogenesis and targets for drug intervention. MAB, monoclonal antibody; BAC1, beta-secretase 1; NMDA-receptor, N-Methyl-D-Aspartate receptor; TK, tyrosine kinase; NK, natural killer. This figure was created with BioRender.com.
In very rare instances AD may result from a prion-like transfer of Aβ that seeds the buildup of plaques, as has been reported in 8 cases and linked to the subjects being treated with cadaver-derived growth hormone (Banerjee, et al., 2024). These cases complement earlier evidence of a prion-like transfer (Jaunmuktane et al., 2015; Purro et al., 2018), and was also the title of a 1984 article in the New England Journal of Medicine ‘Some speculations about prions, amyloid, and Alzheimer’s disease’ (Prusiner, 1984). Collectively, these findings suggest that under certain rare circumstances AD is transferable, although confirmation is required (Jucker and Walker, 2024).
Fully effective cures or disease-modifying drugs for AD are lacking and currently approved drugs belong to three classes; 1/acetylcholinesterase inhibitors (AChEIs), 2/NMDA receptor modulators (memantine), and 3/anti-amyloid monoclonal antibodies (anti-Aβ MABs), however, other putative cellular pathways involved in the pathology have been identified, and are summarized in Figure 1. Also included is a brief review of NSAIDs, which may reduce neuroinflammation, anti-diabetes drugs such as metformin and glucagon-like peptide receptor agonists (GLP-1 RAs), and anti-hypertensive drugs, whose effects are to reduce the impact of diabetes and obesity co-morbidities that enhance the risk of developing AD. Recognizing the limitation of the anti-Aβ-MABs other potential targets are discussed including neuroinflammation, which is discussed in the context that a very early stimulus for AD is the activation of microglia. Of historical significance it was McGeer et al. (1987) who first reported the presence of the class II major histocompatibility antigen, HLA-DR, on microglia in the hippocampus and linked HLA-DR to AD and the presence of Aβ plaques as well as a negative correlation with cortical choline acetyltransferase. Later, Mattiace et al. (1990) demonstrated that HLA-DR was constitutively expressed in white matter, but in grey matter it was induced as a result of the disease buildup of Aβ.
1.1 Tests for diagnosing AD and determining the effectiveness of drugs
Tests for both the diagnosis and for the determination of the development and effectiveness of drugs are of critical importance for treating AD as well as important aids in determining the effectiveness of drugs. However, there is no single definitive test for the diagnosis of AD, and a combination of diagnostic tools, medical history review, cognitive and functional assessments, as well as brain imaging, cerebrospinal fluid analysis, and blood tests are collectively employed (Schachter and Davis, 2000; Tsoi et al., 2015). The main cognitive and functional tests employed include the Mini-Mental State Examination (MMSE); the Montreal Cognitive Assessment (MoCA); and the Alzheimer’s Disease Assessment Scale–Cognitive Subscale (ADAS-Cog); Addenbrooke’s Cognitive Examination (ACE) A summary of the advantages and limitations of these tests as well key references is provided in Supplementary Table S1. The ADAS-Cog test is designed to specifically measure the cognitive performance of subjects who already carry a diagnosis of AD and is a widely used primary outcome measure in both clinical research and drug trials to track the progression of moderate to severe AD (Rosen et al., 1984). However, ADAS-Cog is not a suitable assessment tool for broader screening or for detecting mild cognitive impairment (MCI) and the subtle cognitive changes associated with the early stages of cognitive decline; in contrast MMSE, MoCA and ACE are broad screening tools that assess a wider range of cognitive functions used in a wide range of dementias (Podhorna et al., 2016).
Along with cognitive tests, several biomarkers have value in the diagnosis of AD–see Figure 2 and Supplementary Table S2. Biomarkers can be classified as linked to Aβ deposition, pathologic tau and neurodegeneration (Jack et al. (2018). Assessing CSF is a relatively invasive and carries risk, therefore blood biomarkers that could track AD progression would be preferable; however, there are limitations related to quantification and reproducibility as only limited amounts of brain proteins diffuse into the bloodstream (Blennow and Zetterberg, 2015). For instance, although the longitudinal data has indicated an association between elevated plasma tau levels and subsequent cognitive decline, the slight increase in plasma tau may not be diagnostically significant (Mattsson et al., 2016; Blennow and Zetterberg, 2018). However, in a recent study of 786 patients a commercially available kit for p-tau217 provided comparable diagnostic evidence to that obtained from CSF biomarkers (Ashton et al., 2024). Furthermore, an analysis of blood samples in the UK Biobank identified four other plasma proteins linked to AD (Guo et al., 2024). Replication of these findings would prove beneficial to using blood tests to aid in the diagnosis of AD.
FIGURE 2.

Depiction of a neuron illustrating key CSF biomarkers useful in diagnosing AD in addition to a potential synaptic biomarker candidate, neurogranin. Total tau (T-tau) shows the extent of neuronal damage but lacks specificity for AD. Phosphorylated Tau (P-tau) represents tau proteins with relatively AD-specific modifications. The CSF Aβ42 level is reduced in AD despite the concurrent increase in amyloid (Aβ) deposition in the brain, which is a characteristic of AD pathology. The Aβ42/40 ratio compensates for individual variabilities, thus offering a more standard measure of amyloid pathology. This figure was created with BioRender.com.
In conclusion, current data indicates that CSF biomarkers have a greater diagnostic value than plasma biomarkers; however, improvements in the kits available for assessment of plasma biomarkers may change this conclusion and thereby enhance prospects for earlier diagnosis, assessment of therapeutic treatment and, perhaps, prevention of AD.
2 Methods
A critical review of the published literature concerning the treatment of AD was conducted using PubMed and Scopus searches directed at:
(i) The pathophysiological basis of AD as a framework for the identification of drug targets.
(ii) The pharmacological properties of the drugs that have been used to treat AD
(iii) Evaluation of the clinical trial data that supports the use of the different classes of drugs, alone or in combination.
(iv) Discussion of the controversies over the recent approval of monoclonal antibodies directed at beta amyloid plaques.
(v) Discussion of the potential for new targets for drugs, including cell therapy, which are directed at reducing neuroinflammation.
The resulting narrative review paper is supported by over 300 citations that cover the pathophysiological basis for the drugs that have been developed and are currently used to treat AD as well as a critical evaluation of their effectiveness and insights as to other potential targets for new drug development.
This narrative review was the result of searches in the PubMed and Scopus databases databases (see Supplementary Figures S1-4 for search details). Publications were selected based on their study design and use of statistical evidence to support their conclusions.
3 Hypotheses and drug targets
Although several hypotheses have been offered to explain the pathophysiological basis for AD it is likely that there are multiple triggers. As of 2019, the largest proportion of clinical trials (23.3%) have addressed the amyloid hypothesis, followed by the neurotransmitter hypothesis (including both acetylcholine and glutamate) (19%), mitochondrial dysfunction (17%), neurovascular (7.9%), exercise (6%), the neuroinflammation hypothesis (4.6%), diabetes (2.3%), links to virus infection (0.5%) (Liu et al., 2019). In this review, we focus on the following hypotheses: (i) cholinergic, (ii) glutamate, (iii) amyloid, (iv) tau, (v) vascular hypothesis, and finally (vi) viruses and the benefit of vaccinations. Interest in the role of virus infections has increased following COVID-19 as many people with “Long COVID” appear to suffer neurological dysfunction and cognitive decline (Venkataramani and Winkler, 2022). Other hypotheses such as those linked to diabetes (de la Monte and Wands, 2008) and mitochondrial dysfunction (Cardoso et al., 2004; Swerdlow et al., 2010) are considered tangentially as linked to the putative benefits of drugs, such as the anti-diabetes drug metformin. For instance, a clinical trial, Metformin in Alzheimer’s Dementia Prevention (MAP), NCT04098666, will be completed in 2026. Metformin has putative effects on mitochondrial function that have been argued to underly a neuroprotective action, and are, at least in part, supported by epidemiological data and human genetic studies linking metformin to the NADH:Ubiquinone Oxidoreductase Subunit A2 (NDUFA2) gene and mitochondrial complex 1 (El-Mir et al., 2008; Campbell et al., 2018; Zheng et al., 2022); however, the contribution of a mitochondrial action of metformin as a basis for its therapeutic benefits have been challenged on the basis of the high concentrations used in in vitro studies (He and Wondisford, 2015; Fontaine, 2018). Other drugs that are used to treat type 2 diabetes (T2D), such as the GLP-1 RAs, also reduce cognitive decline suggesting that the primary benefit of anti-diabetes drugs is via improved glycemic control and reducing the pathophysiological sequalae of metabolic dysregulation, (Nørgaard et al., 2022; Nowell et al., 2023). Epidemiological data also suggests that anti-inflammatory NSAIDs reduce the risk of AD; however, the data is controversial.
3.1 Cholinergic hypothesis
A deficiency of acetylcholine (ACh) in the brain as the pathophysiological basis of AD was first proposed in 1976 and based on the observation that choline acetyltransferase, the enzyme responsible for the synthesis of ACh, was greatly reduced in the amygdala, cortex and hippocampus in postmortem brains from patients with AD compared to brains from non-AD subjects (Davies and Maloney, 1976). Cholinergic neurons, particularly in the basal forebrain, play critical roles in memory, attention, and learning (Whitehouse et al., 1982) and degeneration of ACh-producing neurons in AD patients affects neuronal communication, resulting in memory deficits (Hasselmo, 2006; Chen et al., 2022). As summarized in Figure 3 and based on co-immunoprecipitation data acetylcholinesterase (AChE), the enzyme responsible for the degradation of ACh, binds to and interacts with PS-1 in the same intracellular compartment in CNS neurons (Silveyra et al., 2008).
FIGURE 3.

The Acetylcholine Hypothesis and role of AChE Inhibition: Decreased levels of acetylcholine (ACh) contribute to cognitive decline; however, the inhibition of acetylcholinesterase (AChE) with acetylcholinesterase inhibitors (AChEIs) prevents the breakdown of ACh, thus resulting in elevated synaptic ACh levels. The increase in available ACh is associated with improved cognitive function. AChE also interacts with the enzyme presenilin-1 PS-1, which plays a crucial role in Aβ production including the regulation of γ-secretase, and its association with AChE underscores a significant cholinergic-amyloid link in the pathophysiology of AD. Mutations in the PSENI gene result in the enhanced production of Aβ (Silveyra et al., 2008). It has been reported that AChE enhances both transcription and production of PS-1, decreasing γ-secretase activity and reducing the processing of APP (Campanari et al., 2014). Thus, the loss of the regulation of PS-1 by AChE and a subsequent increase in γ-secretase activity will increase Aβ. This figure was created with BioRender.com.
3.1.1 Enhancing availability of ACh - Acetylcholinesterase inhibitors
The first and the main class of drugs currently used for AD are the cholinesterase inhibitors (AChEI) that have varying specificity for AChEI versus pseudo-AChE (also known as butyrl ChE [BuChE]) - see Figure 3, and are widely used for the symptomatic treatment of mild-moderate AD. In the brain, ACh is degraded by both AChE and BuChE (Nordberg et al., 2013), with AChE present in the nerve synaptic junctions, and BuChE in glial cells (Mesulam et al., 2002). In the healthy brain, AChE activity is the main enzyme that breaks down ACh, while in the brains of AD subjects, AChE activity is decreased and BuChE activity is increased to compensate for the reduced AChE activity (Perry et al., 2003). Four AChEIs have been approved for the treatment of AD and have variable specificity for AChE versus BuChE. Tacrine was the first to be approved in 1993; however, it was withdrawn in 2013 due to frequent reports of elevated liver enzymes and fatal liver toxicity (Watkins et al., 1994). The three remaining ACEIs are donepezil, rivastigmine, and galantamine. In addition to actions as an AChEI, galantamine also acts as a positive allosteric modulator of nicotinic acetylcholine receptors and potentiates cholinergic neurotransmission (Wang and Reddy, 2017).
All of the currently available AChEIs suffer from the same common gastrointestinal side effects (diarrhea and vomiting) related to their systemic effects on cholinergic transmission, although reportedly less for donepezil (Sugimoto et al., 2000). A summary of the AChEIs, including key references for trial data and meta-analysis is provided in Table 2 together with key information for other drugs that are used to treat AD. Donepezil is the most widely used of the available AChEIs and is used for mild-moderate and severe AD and also combined with the N-methyl-D-aspartate receptor (NMDAR) modulator, memantine (see section, 3.2). Although meta-analysis and several studies conclude that donepezil improves cognitive scores, it is important to realize that AD is a “fluid disease” and measuring cognitive scores for only 24 weeks, or a year, might not be sufficient to accurately determine whether there is slowing of the progression of AD: comparable data from long-term treatment is needed not only for donepezil but all drugs used for AD.
TABLE 2.
Summary of drugs and drug targets for the treatment of Alzheimer’s disease.
| Drug class | Drugs | Status and use | Properties and common side effects | Key references and notes |
|---|---|---|---|---|
| AChE inhibitors (AChEIs) | Tacrine (CognexR), donepezil (AriceptR), rivastigmine (ExelonR), galantamine (ReminylR) | Used for both mild to moderate AD. | All AChEIs inhibit both AChE and butyrl (pseudo) BuChE but with different affinities | Tacrine |
| Tacrine approved in 1993 but withdrawn in 2021 due to liver toxicity; donepezil, approved in 1996 | Tacrine is a non-specific AChEI; donepezil has ∼1000 selectivity for AChE and also has a long plasma half-life (60–90 h) that facilitates daily dosing and also close to 100% bioavailability; rivastigmine, is a non-specific AChEI, which in addition to oral formulations is also available as a transdermal patch; galantamine in addition to AChEI actions is also a positive allosteric inhibitor of nicotinic receptors | Gracon et al., 1998; Jarrott, 2017; Watkins et al., 1994; Blackard et al., 1998; Samuels and Davis, 1997 | ||
| Meta-analysis by Birks and Harvey, 2018, concluded that donepezil use resulted in better scores on ADL (activity of daily living) | The common side-effects are similar for all AChEIs and mainly | Donepezil | ||
| Donepezil is widely used and in combination with the NMDA receptor modulator, memantine for symptomatic relief, and in in 2001 for severe AD. | GI-related, but reportedly less for donepezil–see Ali et al., 2015 | Atri, 2019; Birks and Harvey, 2018; Marucci et al., 2021; Fish 2011 | ||
| The third AChEI to be approved was rivastigmine in 1997, and the fourth, galantamine, was approved in 2001 | Sugimoto et al. (2000) | |||
| Rivastigmine | ||||
| Birks and Harvey, 2018 | ||||
| Marucci et al., 2021 | ||||
| Multum, 2019 | ||||
| Galantamine | ||||
| Wang et al., 2007 | ||||
| Mohammad et al., 2017; Brodaty et al., 2005; Kavanagh et al., 2011; Jiang et al., 2015 | ||||
| NMDA Receptor Antagonist | Memantine (AxuraR, EbixaR, NamendaR) | A low-affinity uncompetitive antagonist of NMDAR. Approved by FDA in 2003 for symptomatic relief of mild to moderate AD and based on the results of two clinical trials (Kavirajan, 2009) Memantine is available in oral formulations including extended-release (ER). Memantine has high bioavailability (approaching100%) and a plasma half-life of 60–70 h. Frequently used in combination with donepezil | Well-tolerated with headache, blurred vision, dizziness as infrequent side effects (Kuns et al., 2024) | (I). Affinity for NMDAR versus other receptors |
| Memantine is safe with co-morbidities (diabetes, and co-administration with metformin or glyburide); co-use with AChEIs is safe | Chen and Lipton, 2005; Johnson and Kotermanski, 2006; Seeman et al., 2008. (ii). Supportive clinical trial data: Kavirajan et al., 2009; Periclou et al., 2004; Noetzli and Eap, 2013 | |||
| Based on meta-analysis (Blanco-Silveste et al. (2018) discontinuation rates for memantine are higher than for placebo | Matsunaga et al., 2015 | |||
| Kishi et al., 2017. (iii) Cochrane Report: McShane et al., 2019 | ||||
| (iii) Use with co-morbidities | ||||
| Freudenthaler et al. (1998), Periclou et al. (2004), Shua-Haim et al. (2008) | ||||
| Combination therapy AChEI plus NMDAR antagonist (donepezil + memantine) | Donepezil + memantine. (NamzaricR) | Fixed dose combinations approved in 2014 for patients with moderate to severe AD. | Overall, the evidence suggests that side-effects of combination therapy is more effective than monotherapy and that side effects are no greater than for monotherapy treatment with either AChEI or memantine (Kuns et al., 2024) | (i). Positive data based on ADCS-ASL scores (Tariot et al., 2004; Grossberg et al., 2013) |
| Meta-analysis suggests combination many be more effective for non-AD dementias | (ii). Contradictory data: Atri et al., 2008; Porseinsson et al., 2008; Howard et al., 2012; Chen et al., 2017a) | |||
| Also see | ||||
| Deardorff and Grossberg, 2016; Calhoun et al., 2018; Saint-Laurent Thibault et al., 2015 | ||||
| (iii). Meta-analysis for combination therapy AD versus other dementias | ||||
| Blanco-Silvente et al., 2018; Knight et al., 2018; Veroniki et al., 2022 | ||||
| MABs directed at amyloid (Aβ) proteins | Bapineuzumab, solanezumab | First generation chimeric humanized zumabs failed clinical trials | Infusion reactions are the most common SEs with an incidence of ∼25% | (i). Based on Aβ accumulation in post-mortem brains |
| (Anti- Aβ MABs) | crenezumab | Aducanumab (a numab–fully human MAB) approved with considerable controversy in 2021, but Biomega announced availability will end in late 2024 | Reports of cerebral edema in addition to cost of drug and associated costs of CSF and MRI monitoring for ARIAs may limit use and wider global use | Glenner and Wong, 1984; Hardy and Allsop, 1991; Hardy and Higgins, 1992 |
| aducanumab (AduhelmR) | Lecanemab approved in 2023 and based on positive data from CLARITY-AD trial. Based on TRAILBLAZER-ALZ 2 RCT approval of docanemab anticipated in late 2024 | Insufficient data to know long-term therapeutic efficacy and effects of anti-Aβ MABs | Also see: Spirling et al., 2011 | |
| lecanemab (LeqembiR) | Considerable controversy over the approval and effectiveness of anti-Aβ MABs | Mahase 2021a, b, c | ||
| gantenerumab | Rahman et al., 2023 | |||
| docanemab (LY3002813, or N3pG) | Wojtnik-Kulesza et al. (2023) | |||
| Controversies | ||||
| Teich and Arancio 2012; Ackley et al., 2021; Reiss et al., 2021; Richards et al., 2021; Herrup, 2022; Whitehouse and Saini, 2022; Kaur et al., 2024 | ||||
| Additional and longer clinical trials with lecanemab are ongoing (van Dyck et al., 2023) - results should help clarify how beneficial this class of drugs are re. long-term treatment of AD and whether anti- Aβ MABs slow cognitive decline | ||||
| β-secretase (BACE1) inhibitors) | Atabecestat; verubecestat | Failed Phase 2/3 clinical trials | Low clinical efficacy with cognitive decline greater than with placebo and concerns over psychiatric side effects. High incidence of side effects linked to ‘on target’ effects of β-secretase on proteins other than APP. | Henley et al., 2019 |
| Lanabecestat | Egan et al., 2019; Wessels et al., 2020 | |||
| Targets APP (amyloid precursor protein) | See also McDade et al., 2021 | |||
| γ-secretase | Selective modulators (GSMs) of γ-secretase predicted to have fewer side effects than BACE1 inhibitors | None tested | Hur (2022) | |
| Tau-inhibitors | Semoriinemab, tilavonemab, gosuranemab target the N-terminal region of tau | Failed to show significant benefits as in the TANGO trial with the humanized MAB, gosuranemab | Insufficient data to determine significance of side effects in humans; studies in mice did not show significant issues | Hoskin t al., 2019; Shulman et al., 2023; Sperling, 2023 |
| RNA-bases anti-sense oligonucleotide targeting tau (IONIS in partnership with Roche). Phase 1b study in progress (NCT03186989) | Data suggests that more specific targets are needed and/or combined therapy with multiple targets | Teng et al., 2022 | ||
| Panza and Lozupone (2022) | ||||
| September 2023 IONIS entered into an agreement with Roche for the further development of antisense RNA therapies for the treatment of AD and Huntington’s Disease (https://ir.ionispharma.com/news-releases/news-release-details/ionis-enters-agreement-roche-two-novel-rna-targeted-programs) | ||||
| NSAIDs | Numerous NSAIDs including aspirin, naproxen, ibuprofen, and coxibs (celecoxib) | Early positive data based on retrospective studies not supported by later meta-analysis and Cochrane Review in 2012 that concluded there was no evidence to support either the use of aspirin, NSAIDs, selective COX-2 inhibitors (coxibs), or steroids for the prevention or treatment of AD. | Chronic use of NSAIDs linked to risk of increase in GI and cardiovascular morbidity and mortality, and elevated cardiovascular risk for coxibs–Schjerning et al., 2020 | (i). Supportive: McGeer et al., 1990, 1996; Szekely et al., 2004 |
| The results of the 2019 clinical trial, INTREPAD, with the NSAID, naproxen were negative | In t’Veld et al., 2001; Pasinetti 2002 | |||
| (ii). Cochrane Review Jaturapatporn et al., 2012 | ||||
| (ii). INTREPAD data (with naproxen)–no benefits in AD (Meyer et al., 2019) | ||||
| (iii). NSAIDs and coxib use and elevated risk in elderly patients (Wehling, 2014) | ||||
| Anti-diabetes drugs | Metformin and GLP-1 receptor agonists | Support provided by pre-clinical and retrospective clinical data as well genetic analysis. Diabetes increases the risk of AD and benefits of drugs may be secondary to improving metabolic control in patients | Side effects with metformin are primarily GI. | El-Mir et al., 2008 |
| Metformin in Alzheimer’s–a Phase II/III trial (NCT04098666) with long-acting metformin in non-diabetes subjects with early and late MCI. Results expected in late 2026 | GLP-1 receptor agonists frequently cause nausea, vomiting, loss of appetite | Campbell et al., 2018 | ||
| Zheng et al., 2022 | ||||
| Nogaard et al., 2022 | ||||
| Nowell, et al. (2023) | ||||
| ApoE and statins | Gene therapy to target ApoE4 homozygotes | Lexeo Therapeutics, Phase I/II Clinical Trial, NCT03634007, on-going | Data on gene therapy clinical trial expected in late 2024 | Controversy re statins are beneficial in AD Wagstaff et al., 2003; Zandi et al., 2005 |
| See Rosenberg et al., 2018 | Using an adeno-associated virus gene transfer vector that expresses the cDNA coding for human APOE2 | Statins may reduce risk of AD, cognitive decline and mortality Kostapanos and Elisaf, 2017; Jeong et al., 2021; Nowak et al., 2022; Olmastroni et al., 2022; Murphy et al., 2023 | ||
| Targeting APOE4–see Hunsberger et al., 2019; Vecchio et al., 2022 | ||||
| Expression of ApoE4 damages pericyte function and integrity of BBB | ||||
| Armulik et al., 2010; Nishitsuji et al., 2011; Halliday et al., 2016; Zhou et al., 2022) | ||||
| Viruses and Vaccinations | Several viruses have been linked to increasing the risk of AD including Herpes Simplex (HSV-1), SARS-CoV-2 | Viral infections linked to increase in neuroinflammation, and increases in β- and γ-secretase activities and enhancing APP processing and tau kinases | Vaccinations have been shown to reduce the risk of AD (Wu et al., 2022) | Wozniak et al., 2007 |
| A prospective study (n = 49), with the Bacillus Calmette–Guérin (BCG) vaccine for tuberculosis has provided positive data that vaccines against AD (Dow et al., 2022) | De Vlieger et al., 2022 | |||
| Kitazawa et al. (2011) | ||||
| Patients with “Long COVID” experience sleep disruption, fatigue, anxiety, depression and what has been referred to as “brain-fog” (inability to focus, loss of memory, and difficulty to conduct normal activities) that can persist for months (Venkataramani and Winkler, 2022). Whether protection is provided by COVID-19 vaccinations remains to be analysed |
In summary, although widely used for the symptomatic relief of mild-moderate AD, ACHEIs, either alone or in combination with the NMDAR modulator, memantine, have limited effectiveness as disease-modifying drugs. They are not a cure and their use can be associated with troublesome side-effects. A number of trials (see Amenta et al., 2001) have investigated alternative approaches to correcting cholinergic transmission, notably the use of choline precursors including choline and phosphatidylcholine (lecithin) CDP-choline, alphaglyceryl-phosphoryl-choline (α-GPC), choline alphoscerate, and phosphatidylserine. The results from one trial with CDP-choline indicated improved cognitive evaluation scales and arguably slowed the progression of AD (Secades and Frontera, 1995). These data suggest that the use of choline precursors should be re-examined, possibly in combination with AChEIs, to determine whether treatment decreases neuroinflammation and Aβ-associated neurotoxicity as has been shown in pre-clinical studies (Alvarez et al., 1999; Munafò et al., 2024).
3.2 Glutamate toxicity hypothesis
The glutamate hypothesis is based on evidence that there is a reduction in the binding of l-[3H] glutamate in the postmortem brains of subjects with AD (Greenamyre et al., 1987; Maragos et al., 1987) thus linking a defect in glutaminergic neurotransmission, the principle excitatory pathway in the brain, to AD (Maragos et al., 1987; Hladky and Barrand, 2022). As summarized in Figure 4 glutaminergic transmission and synaptic NMDARs are important for synaptic plasticity, LTP and neuronal survival, and also play an essential role in memory (see Abraham et al., 2019), whereas the activation of extra-synaptic NMDARs is associated with cell death and AD. This association form the basis for the use of the NMDAR antagonist, memantine, for the treatment of AD (Hardingham et al., 2002; Hardingham, 2006; Hardingham and Bading, 2010).
FIGURE 4.

The excitatory neurotransmitter glutamate plays an essential role in synaptic plasticity, a process that refers to the ability of synapses to strengthen, or weaken, in response to neuronal activity. Glutamate-mediated synaptic plasticity involves two key processes: long-term potentiation (LTP) and long-term depression (LTD) (Riedel et al., 2003; Abraham et al., 2019). In brief, LTP requires the activation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR) and N-methyl-D-aspartate receptors (NMDAR). Activation of the NMDAR is initially prevented due to intracellular Mg2+ blocking the cation channel, however, when glutamate activates the AMPAR this results in the entry of Na+ and depolarization of the neuron (Step 1 in Figure 5) and removal of the Mg2+ block (Step 2 in Figure 5), allowing activation of the NMDAR and the intracellular entry of both Na+ and Ca2+ (Vargas-Caballero and Robinson, 2004; Blanke and VanDongen, 2009). The increase in intracellular Ca2+ initiates a signaling cascade that involves the enzyme calcium/calmodulin-dependent protein kinase II (CaMKII) and triggers the translocation of intracellular AMPARs to the postsynaptic membrane (Step 4a in Figure 4) and phosphorylates AMPAR (Step 4b in Figure 4) (see Sumi and Horada, 2020, for details of the signaling pathway). Collectively, these events result in the strengthening of the neuronal signal and ultimately lead to the development of LTP and memory formation and learning (Wang, 2017), including changes in gene transcription that affect receptor density and result in changes in neuron function. However, the excessive release and presence of glutamate in the neuronal synapse results in elevated levels of intracellular calcium and prolonged cell depolarization (Choi, 1987). This, in turn, results in the generation of increased levels of reactive oxygen species (ROS), causing neuronal damage and cell death (Savolainen et al., 1995). Excess levels of glutamate promote microglia-mediated neuroinflammation, further damaging adjacent neurons (Qin and Crews, 2012; Lee V. M. et al., 2021). This supports the argument that the combination of microglia-mediated inflammation and oxidative stress results in neural damage, synaptic dysfunction and impairment of LTP leading to AD. Elevated Aβ is also associated with hyperactivity of NMDA-mediated currents and neurotoxicity, thus providing a link between the role of Aβ plaques and defective glutaminergic neurotransmission (Harkany et al., 2000; Domingues et al., 2007). This figure was created with BioRender.com.
3.2.1 Memantine–A NMDAR antagonist
Memantine was approved by the FDA in 2003 for the treatment of moderate to severe AD and based on the evidence that excessive activation of the NMDAR triggered neuronal toxicity and apoptosis (Zeevalk and Nicklas, 1992; Bonfoco et al., 1995; Thomas and Grossberg, 2009). Memantine binds to the NMDAR in a voltage dependent manner and, since it is a low-affinity uncompetitive antagonist and open-channel blocker, the risk of memantine binding to receptors in the non-depolarized state is low and therefore does not interfere with Long Term Potentiation (LTP) (Bresink et al., 1996). Unlike ketamine, a high affinity NMDAR antagonist acting on allosteric/dizocilpine sites, memantine blocks NMDAR but dissociates rapidly, thus avoiding prolonged receptor blockade and associated negative side effects such as interruption of learning and memory formation processes (Folch et al., 2018). Although memantine has been reported to bind with variable affinity to other sites including dopamine cholinergic, serotoninergic, and also sigma receptors, the contributions of these actions to its therapeutic actions are unknown and it is its action on the NMDAR that is thought to be the major contributor to reducing neuronal excitotoxicity (Seeman et al., 2008).
Although early studies and including meta-analysis indicated that monotherapy with memantine demonstrated greater efficacy than placebo in improving cognitive function a 2019 Cochrane report that included data up to 25 March 2018 from double-blind, placebo-controlled, randomized trials concluded that there was only a small clinical benefit with the use of monotherapy memantine for moderate-severe AD, but not mild-moderate AD (See Table 2; McShane et al., 2019). In terms of safety outcomes, there was no significant difference in all-cause discontinuation between memantine and placebo groups but the memantine-treated group was more likely to develop dizziness (RR = 1.53, 95% CIs = 1.02–2.28, p = 0.04). As pointed out in the meta-analysis report by Blanco-Silveste et al. (2018) an effective treatment should have a lower discontinuation rate than placebo as this would indicate that an improvement in symptoms outweighs side effects. Common side effects of memantine are headache, confusion, diarrhea, and constipation.
In summary, despite limitations, questions over effectiveness, and side effects, memantine either alone or in combination with an AChEI, is widely used for the symptomatic treatment for moderate to severe AD.
3.2.2 Combination therapy AChEI plus NMDAR antagonist (donepezil + memantine)
To enhance symptomatic relief a fixed dose combination of memantine ER/donepezil was approved by the FDA in 2014 for patients with moderate to severe AD. Despite contradictory results (see Table 2), the overall conclusion is that combination therapy is more effective than monotherapy in delaying cognitive and functional decline as well as delaying the requirement for nursing home care (see Lopez et al., 2009).
3.3 Amyloid hypothesis
The most widely promoted hypotheses to explain the pathogenesis of AD is that neuronal damage is caused by aberrations in the processing of APP and the accumulation of Aβ due to a failure of the brain to clear Aβ (see Figure 5). Aβ was first isolated from the postmortem brains of subjects with AD and also Down’s Syndrome by Glenner and Wong in 1984, and in 1991, with the discovery of a mutation in the APP gene, (now associated with FAD) the amyloid hypothesis was proposed by Hardy and Allsop (Hardy and Allsop, 1991). Importantly, the presence of the ApoE4 allele significantly increases the risk of late-onset AD and increases the neurotoxicity of Aβ proteins (Kaplitt et al., 1996; Farrer et al., 1997).
FIGURE 5.

Non-amyloidogenic and amyloidogenic pathways and β-secretase (BACE-1) inhibition. This figure illustrates the non-amyloidogenic (A) and amyloidogenic (B) pathways of amyloid precursor protein (APP) processing. Light green circle represents normal PSEN-1 protein at the γ-secretase complex. Dark brown circle represents mutated PSEN-1 protein at the γ-secretase complex, which increases production of longer and aggregation-prone beta amyloids. Aβ42 is rich in hydrophobic amino acids such as isoleucine, phenylalanine, and valine. Specifically, the hydrophobic side chains at the positions 41 and 42 increase the propensity of Aβ42 to aggregate (Kim and Hecht, 2005). This promotes the formation of the β-sheet structures characteristic of aggregated amyloid proteins. In a normal physiologic state (Figure 5A), APP is processed by α-secretase and then by β-secretase (Butterfield et al., 2013). However, when there is reduced α-secretase together with increased β-secretase activity, the amyloidogenic pathway is induced (Figure 5B). As Aβ42 increases, aggregates of Aβ form monomers that then develop into small oligomer clusters that form more stable Aβ fibrils, highly ordered crossed beta-sheet structures that align perpendicular to the fibril axis forming an extended rigid fiber (Chen et al., 2017). As more and more Aβ proteins are produced, the fibrils form a tighter and more stable structure due to the hydrophobic interactions between the amino acids side chains that stabilize into a senile plaque in the extracellular space (Chen et al., 2017). BACE-1 inhibitors (purple) target beta-secretase, reducing activity along the amyloidogenic pathway. This eventually results in the decrease in the production of the amyloid-beta (Aβ) peptides associated with Alzheimer’s disease. This figure was created with BioRender.com.
APP is a transmembrane protein that is present in many types of cells, including neurons, and is known to play a major role in neuron synaptogenesis (Wang et al., 2009). In healthy humans without evidence of cognitive decline, Aβ is produced by the cleavage of APP by two major types of secretases: α- and β-secretase (Figure 5). When APP is cleaved by α-secretase, two fragments are generated: (i) soluble amyloid precursor protein alpha (sAPPα) and (ii) C83 (a membrane-bound C-terminal fragment) (Chow et al., 2010; Hare J, 2010). sAPPα has a role in neuroprotection and maintaining synaptic function and plasticity, which are essential for neuronal communication, learning, and memory formation (Mattson, 1997). In addition, a rat model of AD has shown that sAPPα promotes neurogenesis, which is also important for learning and memory formation (Huber et al., 1997). Most importantly, sAPPα inhibits aggregation of Aβ peptides by shifting the APP processing pathway into an anti-amyloidogenic pathway, thus preventing formation of toxic amyloid plaques generated by β-secretase. Cleavage of APP by β-secretase generates two different fragments: (i) soluble amyloid precursor protein beta (sAPPβ) and (ii) C99 (also called β-CTF), a membrane-bound fragment that is further processed by γ-secretase (Qiang et al., 2017) to yield several different beta-amyloid peptides, of which Aβ42 is the most common, and the basis of the amyloidogenic pathway (de Paula et al., 2009). γ-secretase is a protein complex made of many subunits including PSEN1, PSEN2, nicastrin, anterior pharynx-defective 1 (APH-1), and presenilin enhancer 2 (PEN-2) (Zhang et al., 2014). As mentioned previously, although a mutation in the PSEN1 gene is more commonly associated with earlier onset of FAD, a mutation in PSEN2 can also contribute. In fact, both PSEN1 and PSEN2 play important roles in forming γ-secretase’s catalytic subunit that if mutated, leads to alternations in the APP cleavage process and results in increased production of longer and more aggregation-prone forms of Aβ (Kabir et al., 2020).
Co-morbidities, such as insulin resistance in patients with T2D, enhance the buildup of Aβ plaques and advanced glycation end-products (AGEs) can modify Aβ peptides and accelerate aggregation of soluble Aβ peptides (Vitek et al., 1994). There is a strong link between insulin deficiency, insulin resistance, diabetes, and AD, with some research referring to AD as ‘type 3 diabetes’ (Steen et al., 2005; de La Monte and Ward, 2008). In addition, patients who are subjected to elevated oxidative stress, or inflammation, such as during frequent infection (also see section 3.6), are prone to developing abnormal Aβ plaques (Sharma and Kim, 2023), and Aβ42 itself has pro-oxidant activity (Ganguly et al., 2017; Butterfield and Boyd-Kimball, 2018). As previously discussed, studies have shown that Aβ42 levels decrease in CSF but increase in plasma (Teunissen et al., 2018), implying the measuring of plasma Aβ42 to monitor AD progression; however, the data has questioned its diagnostic utility.
Normally, Aβ is rapidly cleared by microglia, which act as the phagocytic cells in the CNS (Liu et al., 2021). However, in elderly people, the ability of microglia to phagocytose and degrade Aβ is decreased, resulting in decreased clearance and accumulation of Aβ inside neurons, eventually triggering inflammation, reduced neuronal communication, tau tangles, and ultimately death and degeneration (Lee and Landreth, 2010; Mucke and Selkoe, 2012; Bloom, 2014).
The argument that the buildup of amyloid plaques is the cause of AD has been vigorously disputed (Kepp, 2017), and this skepticism is supported by disappointments in the results of clinical trials with anti-Aβ MABs (Reiss et al., 2021). Skepticism supports the need to explore outside of the amyloid hypothesis for alternative targets (Karran and Hardy, 2014; Tse and Herrup, 2017; Herrup, 2022; Kaur et al., 2024). Amyloid plaques may start 20–30 years prior to evidence of cognitive function thereby raising issues over early detection, when to initiate treatment, and the potential of significant side-effects arising from long-term chronic treatment with drugs (Jansen et al., 2015). In addition, a significant percentage of patients with dementia are amyloid negative (Beach et al., 2012; Serrano-Pozo et al., 2014); in contrast, there are reports that elderly people with normal cognitive function have elevated levels of Aβ plaques (Aizenstein et al., 2008; Kepp, 2017)
β-secretase (BACE-1) inhibitors have been developed to decrease the level of Aβ proteins. However, Phase 2/3 clinical trials of subjects with mild-moderate AD with atabecestat and verubecestat were stopped due to low clinical efficacy with cognitive decline greater than in the placebo group (Egan et al., 2019; Henley et al., 2019). Similarly, lanabecestat also failed to slow cognitive decline and raised concerns over psychiatric adverse events (Wessels et al., 2020). Conceivably the failure of BACE1 inhibitors is linked to β-secretase targeting not only APP but other proteins, thus contributing to toxic side effects (Bazzari and Bazzari, 2022). It is argued that targeting γ-secretase with specific modulators (GSMs) will provide better specificity (Hur, 2022).
3.3.1 Anti-amyloid (anti-Aβ) monoclonal antibodies (MABs)
AChEIs and NMDARIs provide only symptomatic relief to patients with AD whereas the argument for targeting amyloid Aβ aggregates in the brain is that this will terminate the downstream pathophysiological sequalae and potentially reverse the disease process. Passive immunization of APP transgenic mice with the MAB, mAb158, which is highly selective for protofibrils, showed improvements in learning and memory, although there was a minimal effect on amyloid burden (Lord A et al., 2009). These data suggest that soluble amyloid protein oligomers, rather than insoluble amyloid plaques, induce neural toxicity in AD patients and MABs that selectively target soluble oligomers should have better outcomes in patients with AD (Lord et al., 2009; Tolar et al., 2020). Several anti-Aβ MABs have been developed and tested and include first generation bapineuzumab, solanezumab, and crenezumab, the latter being highly homologous to solanezumab, and second generation aducanumab, lecanemab, gantenerumab, and donanemab. Table 1 provides a summary of the six MABs that have entered clinical trials. Only two appear to offer therapeutic benefits, albeit with considerable controversy, and only one, as of April 2024 has been approved–namely, lecanemab. The following discussion will focus on lecanemab, donanemab (currently under review), and aducanumab (provisionally approved but withdrawn from the market in early 2024). Table 2 includes a comprehensive summary of drug targets including additional information on aducanumab, lecanemab, donanemab
TABLE 1.
Monoclonal Antibodies (MABs) Developed to Target Amyloid Plaques. Abbreviations: ARIA-E − Amyloid-Related Imaging Abnormalities with Edema; ARIA-H microhaemorrhages, or small hemorrhages and hemosiderosis; Clinical Dementia Rating Sum of Boxes (CDR-SB), is a scale that assesses both function and cognition; Institute for Clinical and Economical Review–ICER; IV–intravenous administration; SC–subcutaneous administration.
| MAB | Status | Results | Negative effects | Controversies | Key references |
|---|---|---|---|---|---|
| Aducanumab | 2 trials of patients with MCI and confirmed amyloid pathology aged 50–85: 1638 subjects in EMERGE and 647 in ENGAGE. | Primary objective met in EMERGE but not in ENGAGE–based on Clinical Dementia Rating Sum of Boxes (CDR-SB) | i. ARIAs, were experienced by more than 40% of patients taking aducanumab and dose-dependent, of whom 7.5% were symptomatic (Sevigny et al., 2016). ARIASs were more frequent in patients carrying the ApoE4 allele, and most frequent for those who carry the ApoE4 4/4 versus 4/3 or 3/3 alleles (Kwan et al., 2020) | i. Three FDA resigned alleging absence of evidence of effectiveness | Mahase 2021a, b, c |
| IV (human MAB derived from a blood lymphocyte library of elderly people without any evidence of cognitive impairment)Biogen | FDA approved June 2021 | ii. Conflicting evidence from 2 trials presented at the November 2020 meeting of the FDA’s Peripheral and CNS Drug Advisory Committee meeting with some data favouring the placebo (Dr. Krudys) | ii, 2021 not approved in Europe | Budd Haeberlein et al. (2022) | |
| Has strong avidity for epitope-rich insoluble amyloid plaques and oligomers rather than amyloid monomers. spares amyloid monomers, which have putative protective actions half-life of approximately 25 days (Beshir et al., 2022) | July 2021 use restricted to MCI. | iii. June 2022 Biogen withdraws review from Health Canada | Arndt et al. (2018) | ||
| In early 2024 Biomega announces availability of aducanumab will end in late 2024 | Tolar et al. (2020) | ||||
| Decourt et al., 2021; Haddad et al., 2022 | |||||
| Whitehouse and Saini (2022) | |||||
| Rahman et al. (2023) | |||||
| Wojtunik-Kulesza et al. (2023) | |||||
| Ackley et al., 2021 | |||||
| Richard et al. (2022) | |||||
| Bapineuzumab (humanized MAB) | 2012: Failed two Phase III trials | Failed to produce significant cognitive improvements; despite lowering Aβ, and also phosphorylate tau in CSF. | First MAB to be associated with ARIA-E | Miles et al. (2013) | |
| Pfizer and Johnson and Johnson | Discontinued 2013 | 6% developed aseptic meningitis | Abushouk et al., 2017 | ||
| SC | |||||
| Binds to the N-terminal of Aβ residues 1–5 | |||||
| Donanemab (humanized) | TRAILBLAZER-ALZ Phase III trials | Significant improvement noted at 76 weeks | Mintun et al., 2021 | ||
| Eli-Lilly | Sims et al. (2023) | ||||
| IV | |||||
| Selectively targets amyloid plaques. See also Table 2 | |||||
| Gantenerumab (human) | 2017 Failed in Phase III | Reduced Aβ plaques but did not slow cognitive decline at 116 weeks | Ostrowitzki et al., 2017 | ||
| Hoffman-La-Roche | 2023 GRADUATE I and II trials | Bateman et al. (2023) | |||
| SC | |||||
| Targets insoluble plaques | |||||
| Lecanemab (human) | approved by the FDA in July 2023 | CLARITY AD trial with 1795 participants for 18 months. Moderately less decline based on CDR-SB scale | ARIA-E and | April 2023 ICER report raised concerns about cost-effectiveness of lecanemab for AD. | Van Dyck et al. (2023) |
| IV | ARIA-H and concerns over neuroinflammation and brain shrinkage, edema and some deaths | Longer trials also recommended | Couzin-Frankel & Piller (2022) | ||
| Biogen and Eisai | ARIA higher in ApoE4 patients | Piller (2022) | |||
| Targets both oligomers and plaques | Couzin-Frankel, (2023) | ||||
| See also Table 2 | |||||
| Solanezumab (humanized from mouse) | Phase III EXPEDITION 1, EXPEDITION 2. showed + ve results, but failed in EXPEDITION 3. Also failed in The Anti-Amyloid Treatment in Asymptomatic Alzheimer’s (A4) study | After 240 weeks there was no slowing of cognitive decline in preclinical Alzheimer’s disease | ARIA-E >1% in each group. ARIA-H in ∼30% and similar in placebo group | Hoffman La Roche withdraws support in 2019 for continuation of trials with crenezumab | Sperling et al. (2023) |
| Eli-Lilly | Similarly, the 2014 phase II studies BLAZE and ABBY failed to show significant improvement with crenezumab | In 2022 NIH stated that crenezumab failed for treatment of early onset AD. | Cummings et al. (2018) | ||
| High affinity (picomolar) binding to monomeric aβ (amino acid sequence KLVFFAED) | |||||
| Highly homologous to crenezumab | |||||
| Genentech | |||||
| IV |
The failure of several clinical trials, deaths of a number of subjects in the trials, controversy over the approval process, and the cost of the anti-Aβ MABs has added to earlier skepticism that these MABs will proce to be “game-changers” in the treatment of AD (see Kaur et al., 2024; Table 2). Furthermore, PET analysis suggests as many as 25% of patients diagnosed with mild to moderate AD are amyloid negative AD (Beach et al., 2012; Serrano-Pozo et al., 2014; Herrup, 2015). In addition, racial differences, such as reports of significantly lower CSF levels of the biomarkers T-tau and p-tau18 in African-Americans diagnosed with AD (Morris et al., 2019), complicate the assessment of the therapeutic efficacy of treatment and require careful design of clinical trials to ensure appropriate ethnic representation. Another concern is evidence that Aβ may play an important role in water homeostasis in the brain via association with the aquaporin AQP4 water channel, expressed primarily in the CSF and brain interface, which is upregulated in the brains of patients with AD (Moftakhar et al., 2010; Lehrer and Rheinstein, 2023; Maroli, 2023). This putative link between Aβ and the regulation of water homeostasis could explain the frequent occurrence of amyloid-related imaging abnormalities (ARIA) in patients treated with MABs.
Two types of ARIA are seen with the use of anti- Aβ MABs. ARIA-E is a vasogenic edema, whereas ARIA-H involves microhemorrhages and hemosiderosis (Barakos et al., 2022). ARIA-H is usually preceded by cerebral amyloid angiopathy as the amyloid proteins weaken the blood vessel endothelium and increase the risk of bleeding (Goos et al., 2010). Both forms of ARIA are usually transient and minimally symptomatic; however, when symptomatic, ARIA presents with headaches, dizziness, confusion, visual disturbance, nausea, seizures, and even death, as has been associated with the use of the recently approved MAB for AD, lecanemab (Piller, 2022; Withington and Turner, 2022). It is conceivable that a refinement in the epitope recognized by the MAB may reduce the incidence of ARIAs and enhance plaque removal. In a mouse model of AD targeting Aβ(p3-42) promotes plaque removal, whereas targeting Aβ(p1-42) was associated with a higher number of microhemorrhages (Demattos et al., 2012).
Abucanumab was provided accelerated approval by the FDA in June 2021 and the first new drug approved for AD since 2003 (Rahman et al., 2023). Approval was based on two Phase III trials, EMERGE and ENGAGE, wherein a total of 1,643 and 1,647 subjects with MCI or mild AD were enrolled to take either low-dose or high-dose aducanumab once every 4 weeks for a period of 18 months; lower doses were given to subjects carrying the ApoE4 allele due the higher risk of ARIA. In July 2021 the FDA restricted use to patients with MCI (see Table 1; Mahase, 2021a; b). In part, the controversy relates to the differences in the data from the EMERGE versus ENGAGE trials. Data from the EMERGE trial showed that aducanumab improved several cognitive and functional assessment scores (Cummings et al., 2021). However, data from the ENGAGE trial showed no benefit and, of note, it was the data from the higher dose and recruitment of additional patients in the EMERGE trial that provided the positive benefit (Knopman et al., 2021; Budd Haeberlein et al., 2022). A meta-analysis reported that based on the PET data aducanumab treatment lowered amyloid load and increased p181-tau in the CSF. However, although statistically significantly the improvement in the ADAS-Cog was small and no change was noted in MMSE (Avgerinos et al., 2021).
Whitehouse and Saini (2022) argued that based on questionable efficacy and the risk of significant side effects, the approval of abucanumab should be withdrawn. Furthermore, neither the European Medicines Regulatory Network nor the UK regulatory agency granted approval for aducanumab (Mahase, 2021c). In early 2024, Biogen announced that it would stop developing and marketing aducanumab (Alzheimer’s, 2024) and contributing factors likely included the decision of the US Centers for Medicare and Medicaid Services (CMS) to restrict reimbursement to participants in a CMS-approved clinical trial (Cummings, 2023; Wojtunik-Kulesza, et al., 2023). Additional factors that also apply to similar anti-Aβ MABs include the estimated annual $28,000 cost of the drug, and the extra costs needed for monthly CSF analysis and MRI reports, the later required for monitoring signs of ARIAs–potentially the most serious side effects of anti-Aβ MABs (Sperling et al., 2011; Wojtunik-Kulesza, et al., 2023).
Lecanemab was granted accelerated approval by the FDA on January 2023 and based on data from the CLARITY AD Phase III trial, full approval was granted in July 2023 for AD subjects with MCI. Lecanemab, like aducanumab, is a humanized IgG1 monoclonal antibody that selectively binds to soluble amyloid protofibrils and initiates the clearance of both protofibrils and amyloid plaques (McDade et al., 2022). CLARITY AD was an 18-month trial that involved 1,795 patients with MCI or mild dementia who received weekly IV infusion of 10 mg/kg of lecanemab; all the participants had confirmed pre-trial evidence of amyloid deposits observed by PET scan. End of trial data demonstrated, compared to placebo, a significant reduction in amyloid plaque burden, a 27% slowing of cognitive decline measured by CDR-SOB, 37% slowing of the decline of ADCS-MCI, and based on ADAS-Cog measurements, a 26% reduction in the decline of cognition (Van Dyck et al., 2023). A news release from Biogen and Eisai on the 29 November 2022 reported that subjects treated with lecanemab had reduced pTau181 and T-tau in the CSF together with a reduction in tau pathology, and lower levels of Glial Fibrillary Acidic Protein (GFAP) (a marker for astrocyte activation during neuroinflammation) and neurogranin were observed in the plasma (Biogen, 2022). Data from longer trials are required to determine how beneficial anti-Aβ MABs are for the long-term treatment of AD and whether they slow cognitive decline (van Dyck et al., 2023).
Although the data from the CLARITY-AD trial have been interpreted as positive it is important to note that the side effects associated with lecanemab were comparable to those attributed to aducanumab. The most common side effects were infusion-related reactions with approximately 40% of patients requiring acetaminophen or antihistamine prior to infusions. Subjects receiving lacenemab experienced more ARIA-E or ARIA-H (17%) compared to the placebo group (9%), more commonly in those carrying the ApoE4 gene allele, particularly homozygotes. Importantly, AD patients who were receiving thrombolytics were at higher risk of ARIA-H. Other concerns, as expressed by the European Alzheimer’s Disease Consortium Executive Committee relate to the cost of the drug estimated at $26,500/year, and access currently limited to those with MCI but not those with moderate to severe AD (Jönsson et al., 2023). Kwan et al. (2020) raised the issue that data was needed patients who were Aβ positive but asymptomatic for AD. Additional concerns have been expressed regarding the trial’s racial demographics, with approximately 77% of participants white, followed by 17% Asian and only 2.6% Black (and among participants from the U.S., 94.5% white with the rest either Asian or Black) (Van Dyck et al., 2023). Concerns over the race/ethnicity makeup have been raised for clinical trials with other drugs (Turner et al., 2022). Collectively, these concerns add to the questions raised in the Anti-Amyloid Treatment in Asymptomatic Alzheimer’s (A4) study (Stopping AD before Symptoms Begin) as to screening for AD and when treatment should begin (Sperling et al., 2014), the results of which were not positive for the MAB, solanezumab, despite following patients for 240 weeks (Sperling et al., 2023). Results of the TRAILBLAZER-ALZ 2 RCT with donanemab were released in July 2023, with comparable positive outcomes to lecanemab, albeit also with limited clinical benefits, similar limitations, and questions over whether these MABs halt the progression of AD or improve quality of life for those receiving the drugs (Sims et al., 2022; O’Leary, 2023).
In conclusion, as of early 2024, it is clearly premature to predict whether the anti-Aβ MABs will prove to be a provide the “magic bullet” for the treatment of AD (Kaur et al., 2024). As stated in a July 2023 editorial in JAMA the beneficial effects of both lecanemab and donanemab are modest, and ongoing research is crucial to fully understand the clinical effectiveness and long-term safety profile, particularly concerning potential adverse effects. Collectively, although the data show that anti-Aβ MABs do slow the rate of functional and cognitive decline in some patients, the results also add to the evidence that amyloid is not the only factor responsible for the progression of AD (Kepp, 2017; Widera et al., 2023).
3.4 Tau hypothesis
As summarized in Figure 6 the tau hypothesis is based on observations that AD is associated not only with an accumulation of Aβ but also aggregates of misfolded tau protein (see Lee et al., 2001; Frost et al., 2009). In AD, aggregates of Aβ are implicated in causing hyperphosphorylation of tau proteins (Gong and Iqbal, 2008), which interact with the nuclear pore complex producing structural disruption and functional loss culminating in neurotoxicity (Eftekharzadeh et al., 2019). Tau, which in the human brain exists in six isoforms, is a microtubule-associated protein that stabilizes the microtubules that serve as the highway for transporting cellular components.
FIGURE 6.

Impact of hyperphosphorylated tau on Microtubule Stability. This figure illustrates the detrimental effect of hyperphosphorylated tau proteins on microtubules. In the normal state (A), tau stabilizes microtubules via a balance between negatively and positively charged residues in their monomers (Mukrasch et al., 2007; Jho et al., 2010). In the absence of any pathological conditions, the balance between these positively and negatively charged molecules keeps tau proteins attached to microtubules where they assist in essential molecular transport. However, when tau becomes hyperphosphorylated in the diseased state (B), via the src family tyrosine kinase, fyn (Lee et al., 2004), tau proteins become less and less positive weakening the electrostatic force between tau and microtubules (Fischer et al., 2009). This results in microtubule disintegration impairing ubiquitin proteasome-mediated autophagic clearance of Aβ (Cowan et al., 2010; Weng and He, 2021). In addition, tau neurofibrillary tangles within the neuron physically obstruct the movement of cellular components along the microtubules; the density of neurofibrillary tangles and severity of the pathology correlate with the level of cognitive impairment (Cowan et al., 2010; Nelson et al., 2012). In the normal state (A), tau stabilizes microtubules, which are essential for cellular structure and transport. However, when tau becomes hyperphosphorylated in the diseased state (B), it loses its stabilizing ability, resulting in microtubule disintegration. This disruption compromises cellular structure and function, contributing to the pathogenesis of Alzheimer’s disease. Figures C and D compare the role of tau in the normal brain (C) with the AD brain (D). D shows microtubule disintegration, neurofibrillary tangle formation, and amyloid plaque deposition and accumulation. This figure was created with BioRender.com.
3.4.1 Targeting tau
There is considerable interest in targeting tau to treat AD, but despite positive pre-clinical data, at present no approach has been approved for use in patients (Soeda and Takashima, 2020). The greater number of studies involve MABs, and the TANGO trial with the humanized MAB, gosuranemab, which like other anti-tau MABs (semorinemab and tilavonemab) directed at the N-terminal region of tau, showed no significant benefits (see Table 2; Monteiro, et al., 2023). AV-1980R/A, which targets the N-terminus of tau, has also shown promise in cynomolgus Monkeys (Macaca fascicularis) with a robust anti-tau antibody response, which, in theory, should reduce tau tangles (Hovakimyan et al., 2022). Subject to positive data from trials in humans this MAB could be used in patients at risk of AD.
A different approach to targeting tau is to use an antisense RNA that targets microtubule-associated protein tau and this is the focus of a randomized, double-blind, placebo-controlled clinical trial (NCT03186989) with BIIB080 (also known as IONIS -MAPTRx) (https://clinicaltrials.gov/).
3.5 Vascular hypothesis
The vascular hypothesis, as originally proposed in 1993 by De La Torre and Mussalvand, states that any disruption of blood supply that compromises cerebral perfusion will result in microglial activation and the build-up of neurofibrillary tangles and elevate the risk of a decline in cognitive function. This hypothesis is supported by epidemiological data as well as ultrastructural data from postmortem brains of subjects with AD that show extensive pathological changes in cerebral capillaries.
Cerebral hypoperfusion not only results in hypoxia and reduced nutrient delivery to the brain but also hinders the adequate clearance of metabolic waste products, including Aβ proteins. The primary route for Aβ clearance from the brain is via the glymphatic system - a combination of astrocyte (a type of glial cell) and lymphatic system that serves as a perivascular transit network linking the CSF and interstitial solutes that serves as the brain’s clearance system (Iliff et al., 2012). As illustrated in Figure 7, CSF produced in the choroid plexus passes through the subarachnoid space and crosses the periarterial space to the interstitial fluid space via aquaporin-4 (AQP4) channels present on astrocytes. Dysfunction of the glymphatic system has been associated with neurodegenerative disease (de Leon et al., 2017; Lv et al., 2021). Ablation of the meningeal glymphatic system in mice results in the accumulation of Aβ proteins (Da Mesquita et al., 2018a; 2018b).
FIGURE 7.

The Vascular Hypothesis. The Blood-Brain Barrier and ApoE4: There are many waste products in the interstitial space, including Aβ and tau proteins and neurofibril tangles. With normal cerebral vascular function and pulsatile blood flow, the direction of CSF flow is from the periarterial space to the interstitial space to perivenous space. While the interstitial fluid crosses the AQP4 channel on the astrocytes of the perivenous space, the waste products in the interstitial space get carried along to the perivalvular space and to the lymphatic system. However, for subjects with vascular abnormalities, such as due to atherosclerosis, the decreased pulsatile blood flow drives CSF flow from the periarterial to interstitial to perivenous space, thus resulting in accumulation of beta amyloid and tau protein in the interstitial space. The presence of amyloid plaques in turn causes additional vascular dysfunction by damaging nearby blood vessels and disrupting the regulation of blood flow in the brain (Thomas et al., 1996). Subjects who carry the ApoE4 genotype have a reduced ability to clear Aβ, which then accumulates in brain microvessels and parenchyma (Martel et al., 1997), and is then associated with reduced cerebral blood flow and metabolism across multiple cortical regions, increasing the risk of hypoxic brain injury (Small et al., 1995; Mielke et al., 1998; Kim et al., 2013). In addition, cognitively normal ApoE4 carriers show significant age-related deficits in cerebral perfusion as they age, which increases the risk of AD (Thambisetty et al., 2010; Liu et al., 2013). Carrying the ApoE4 also heightens the risk of pericyte dysfunction thereby reducing the critical role of pericytes in maintaining the integrity of the blood brain barrier (BBB) thereby allowing Aβ and other inflammatory molecules to penetrate the CNS, inducing neuroinflammation and accelerating AD in part via inhibition of the anti-inflammatory effects of the TREM2-DAP12 complex on microglia (Armulik et al., 2010; Nishitsuji et al., 2011; Halliday et al., 2016; Fitz et al., 2021; Iannucci et al., 2021; Zhou et al., 2023). Data from ApoE4 carriers shows that the breakdown in the BBB starts in the medial temporal lobe, which is the part of the brain critical for cognitive function (Montagne et al., 2020). In consequence, inflammation and oxidative stress associated with both vascular dysfunction and Aβ worsen the damage caused by each factor, creating a feedback loop that accelerates AD progression. This figure was created with BioRender.com.
ApoE, a glycoprotein that is instrumental in cholesterol transport, participates in lipid metabolism, and aids in the removal of Aβ proteins (Martel et al., 1997; Mahley and Rall, 2000; Marais, 2019). Humans express three ApoE genotypes, with ApoE3 being the most prevalent genotype, and ApoE2 the most protective genotype for AD, with higher risk in ApoE2/4 and ApoE3/4 heterozygotes (McCorkindale et al., 2022). Risk is also higher in females who carry ApoE4 (Riedel et al., 2016). Approximately 25% of the population express the ApoE4 genotype and it is a major contributor to LOAD (Crean et al., 2011; Di Battista et al., 2016; Montagne et al., 2020), and lowers the clearance of Aβ (Ma et al., 2018; Zhou et al., 2023), increases the transcription of AP-1, promotes tau phosphorylation (Huang et al., 2017; Barthélemy et al., 2020), and neuroinflammation (Fitz et al., 2021; Iannucci et al., 2021). Targeting ApoE4 offers another approach to the treatment of AD and a gene therapy trial directed at ApoE4 is currently being pursued (see Table 2).
A number of studies link atherosclerosis to dementia and AD; however, it is unclear whether the enhanced risk applies to all subjects with atherosclerosis. For example, a meta-analysis reported a link to carotid artery intima-media thickness (CMIT) and the risk of AD (Xie et al., 2020). Dolan et al. (2010) also concluded that the risk of AD was enhanced in those with cerebral atherosclerosis, but no significant risk was noted in those with peripheral atherosclerosis including coronary artery disease. Similarly, Dearborn et al. (2017) reported that intracranial atherosclerosis was associated with MCI and dementia, but not specifically with AD; however, the ApoE4 genotype is associated with an elevated risk of atherosclerosis and AD (Davignon, 2005). Gender also affects the risk of atherosclerosis: women are also at higher risk of developing AD and higher for those who have the ApoE4 genotype (Mielke et al., 2014; Riedel et al., 2016). Nonetheless, the between lipid levels and AD and benefits of lipid lowering drugs has been questioned (see Table 2; Reitz et al., 2004). Matsuzaki et al. (2011) demonstrated an association between hyperlipidemia and neuritic plaques seen in AD but it did not show any relationship with the levels of neurofibrillary tangles. A 2023 report of a cohort study of over 15,500 subjects with dementia and an average age of approximately 80 reported that statin use had positive effects of cognition as based on MMSE scores and favouring those taking simvastatin (Petek et al., 2023). A 2024 population-based cohort study in Hong Kong that statin use decreased the risk of dementia in patients with heart failure by ∼20% (Ren et al., 2024), and the most recent American Diabetes Association (ADA) guidelines recommend that to avoid cognitive risk in the age group 40–75, LDL levels should not exceed 70 mg/dL (1.8 mmol/L) (see also Chou et al., 2022).
Hypertension, which affects approximately 1.3 billion people worldwide, is a significant risk factor associated with AD. Pathological changes in the vasculature result in endothelial dysfunction, a reduction in vasoprotective factors including nitric oxide (NO), elevated levels of ROS, the promotion of vasoconstriction and atherosclerotic plaque formation, and a heightened risk of the formation of thrombi (Brandes, 2014). High mid-life, but not late-life, blood pressure has been correlated with AD (Gabin et al., 2017) but lower blood pressure in those older than 75 is likely a secondary phenomenon not directly linked to AD (Skoog et al., 1998). Data suggests that angiotensin receptor blockers may be the most beneficial antihypertensive drugs for patients with AD and hypertension inferring a contributing role for aberrant angiotensin-signaling in the pathogenesis of AD (Adesuyan et al., 2022, see Table 2).
There is also a racial link between expression of the ApoE4 genotype and Aβ protein levels in the brain as well as the CSF biomarkers T-tau, which are reported to be lower in African Americans who are diagnosed with AD (Morris et al., 2019). Furthermore, subjects expressing ApoE4 who also have other risk factors such as hypertension, atherosclerosis, and hypercholesterolemia, experience reduced protective benefits from anti-hypertensive drug therapy against AD (Hajjar et al., 2002; Stampfer, 2006; de Oliveira et al., 2018). Risk is further heightened by body mass index (BMI) and leptin signaling (Blautzik et al., 2018). Collectively, these findings highlight the importance of targeting modifiable risk factors as has been emphasised by several reports (see Femminella et al., 2018; Wang et al., 2018; Livingston et al., 2020; Zhang et al., 2021).
3.6 Virus hypothesis
As a consequence of COVID-19 and in part because therapies for other targets such as acetylcholine and glutamate excitotoxity have given ambivalent results, there has been renewed interest in the role of viral infections, and not just SARS-CoV-2, in the development of AD, and also the potential preventive benefit of vaccinations (De Vlieger et al., 2022). (SCOPUS data are presented Supplementary Figures S2, 3 for the role of viruses and vaccinations.)
As early as 1952 links were made between the role of viral infections and AD (Sjogren et al., 1952). In 1982, it was proposed that recurring infections with human herpesvirus (HSV)-1 might be involved in the development and the progression of AD (Ball, 1982). Several other viruses including SARS-CoV-2, HIV, and spirochetal Gram-negative bacterial infections such as syphilis have been linked with the development of AD, however, a key question is: “How does a viral infection initiate or worsen AD?”. Two possible answers are summarized in Figure 8. According to the direct infection hypothesis (Figure 8A), the virus directly enters the CNS and causes neuronal death or activates an antiviral response (Seaks and Wilcock, 2020), causing neuroinflammation and AD pathology (De Vlieger et al., 2022). More frequent re-infection produces the greater cumulative damage seen in AD patients (Itzhaki et al., 2016). The indirect infection theory suggests that the virus does not necessarily need to enter the CNS but rather a peripheral viral infection induces a systemic inflammation that can cause AD pathology (De Vlieger et al., 2022). An additional mechanism (Figure 8B) involves extracellular vesicles (EVs). EVs are small sized vesicles that transport components between cells, including proteins, lipids, nucleic acids, and misfolded proteins such as Aβ and tau proteins (Hill A.F., 2019), thereby supporting the hypothesis of a prion-like transmissible process as a cause for AD proposed in 1984 (Prusiner, 1984).
FIGURE 8.

This figure depicts two potential pathways whereby viruses may trigger Alzheimer’s Disease (AD) pathology. (A). The direct invasion of the virus into the Central Nervous System (CNS) or the induction of systemic inflammation by peripheral viral infection, even in the absence of direct CNS invasion. Both routes converge in the development of neuroinflammation, increased Aβ production, tau hyperphosphorylation, and neurodegeneration, collectively resulting in AD symptomatology. (B). The virus utilizes extracellular vesicles (EVs) as a protective mechanism, allowing it to evade the immune system by encapsulating itself within EVs during its transit into the CNS. In addition, EVs serve as carriers for viral proteins, viral nucleic acids, and pro-inflammatory cytokines, and avoid detection by the immune system, before passing through the BBB to enter the CNS and cause infection and neuroinflammation (Yates et al., 2019; Horn and Maclean, 2021; Liu et al., 2021). The injection of EVs containing proinflammatory mediators into mice has been shown to increase activation of astrocytes and microglia, and thus supports a link between viral infections, EVs and neuroinflammation (Li et al., 2018). This figure was created with BioRender.com.
Once the virus enters the CNS, an inflammatory response occurs resulting in neuroinflammation, Aβ aggregation, and formation of neurofibrillary tangles. The process becomes amplified if the microglia fail to clear the virus, enhancing the of release inflammatory cytokines and oxidative compounds (De Vlieger et al., 2022). Data from mice show that proinflammatory cytokines induce the expression of interferon-induced transmembrane protein 3 (Ifitm3) in neurons; Ifitm3 is a transmembrane molecule known to increase the activity of γ-secretase and thereby raise levels of Aβ proteins (Hur et al., 2020). The proinflammatory molecules released as a result of viral infection shift the APP processing towards the amyloidogenic pathway. Viral infection also increases the activity of Tau kinases and increases the formation of neurofibrillary tau tangles (Kitazawa et al., 2011). Viral infection is also associated with increased quinolinic acid (QA) (De Vlieger et al., 2022), produced by microglia and activated macrophages (Braidy et al., 2009). In the nematode Caenorhabditis elegans, QA acts as a neurotoxin and has been shown to contribute to neurodegeneration via the activation of NMDA receptors, increasing cytosolic calcium and ROS levels and ultimately depleting ATP (da Silveira et al., 2018). Collectively, these data prompt further investigations of the link between viral infections, neuroinflammation and the development of AD.
Herpes Simplex Virus type 1 (HSV-1), has been extensively studied for its role in AD. Upon initial infection, HSV-1 stays latent in the brain and in the trigeminal ganglia (Jamieson et al., 1991). During periods of acute stress or immunocompromised states, it can reactivate, resulting in viral shedding and initiating inflammation in nervous tissue leading to the release of ROS from microglial cells. HSV-1 infection itself may shift APP processing into the amyloidogenic pathway by increasing the expression of β-secretase and nicastrin, a component of γ-secretase, thus increasing the production of amyloid proteins (Figure 5) (Wozniak et al., 2007). HSV-1 may play a role in the abnormal phosphorylation of tau proteins that then form neurofibrillary tangles and cause AD (Alvarez et al., 2012). Supportive data shows recurrent HSV-1 infections in mice result in oxidative stress in the brain (Protto, et al., 2020). HSV-1 also interferes with proteasomal degradation of damaged DNA by downregulating Ku80 protein involved in the repair of double-strand DNA breaks, eventually resulting in neuronal death (De Chiara et al., 2010).
COVID-19 has also been linked to AD. Even though it is still too early to determine the long-term harmful effects of SARS-CoV-2, several studies report signs of AD after infection, including cognitive and motor difficulties, and memory loss issues (Helms et al., 2020; Bliddal et al., 2021). Patients who experience “Long COVID” describe sleep disruption, fatigue, anxiety, depression and a “brain-fog” (inability to focus, loss of memory, and difficulty conducting normal activities) that can persist for months (Venkataramani and Winkler, 2022). SARS-CoV-2, like HSV-1, induces neuroinflammation by either direct invasion into the CNS or through peripheral infection causing systemic inflammation (De Vlieger et al., 2022). Yang et al. (2021) reported that the gene expression pattern of the microglia subpopulation induced by COVID-19 overlaps with that of microglia seen in AD patients. In addition, increased levels of the AD biomarkers Aβ, T-tau, P-tau, and neurogranin were significantly increased in the EV of COVID-infected patients (De Vlieger et al., 2022). The fact that Aβ40 and 42 levels and P-tau are increased in the EV suggests that SARS-CoV-2 contributes to the accumulation of Aβ in infected patients as well as phosphorylation of tau proteins, thus increasing the risk of developing AD (Sun et al., 2021). Brain imaging data from a UK brain-bank of pre and post COVID infection of patients showed reductions in global brain size, thickness of grey matter, and cognitive function, plus damage to olfactory tissue, indicating that long COVID accelerates the onset of AD and can exacerbate the key symptoms (Douaud et al., 2022).
Vaccinations have also been shown to reduce the risk of AD (Wu et al., 2022). Both influenza and pneumonia vaccinations reduce the risk of AD (Lehrer, 2022), with a 46-month follow up study showing that influenza vaccination decreased the risk of AD by 3.4% (Bukhbinder et al., 2022). Similarly, a recent meta-analysis indicates vaccinations against Tdap (tetanus, diphtheria, and pertussis), influenza, hepatitis A and B, and typhoid) reduce the risk for dementia (Wu et al., 2022). Other herpetic vaccinations, for instance for shingles (varicella-zoster) have been associated with a 15% reduced risk of AD and other neurodegenerative diseases (Lehrer and Rheinstein, 2022). A prospective study (albeit proof of concept with n = 49) with the Bacillus Calmette–Guérin (BCG) vaccine for tuberculosis has also provided positive data that will stimulate additional interest in the role of vaccines as a protection against AD (Dow et al., 2022). Whether benefits are seen with COVID-19 vaccinations remains to be analysed.
Acyclovir, a first-line treatment for HSV-1 infection, has been shown to reduce tau phosphorylation and Aβ deposition in HSV-1 infected Vero cells (Wozniak et al., 2011). Hui et al. (2020) found the combination of acyclovir and dexamethasone protective against cognitive impairment in mice injected with Aβ. Finally, vaccines against common viral infections associated with increasing the risk of AD have been developed and tested (Lehrer and Rheinstein, 2022).
4 Summary and conclusion
Despite the availability of drugs that target the different pathways contributing to the development of AD, the problem remains that the pre-clinical changes in brain pathology can occur decades before neuronal dysfunction and neurodegeneration become evident (Jack et al., 2010). Furthermore, prior to the introduction of the anti-amyloid MABs, the drugs available to treat AD (AChEIs and the NMDAR modulator, memantine) only provide symptomatic relief without proven efficacy to slow or reverse progression. Worse, such drugs may be ineffective if prescribed at the wrong stage of the disease and reduce compliance as a result of drug-related side-effects (Buchhave et al., 2012). Early diagnosis of MCI and AD is therefore essential so that timely intervention can be initiated, including the reduction of modifiable risk factors with appropriate therapeutic intervention, or lifestyle modifications that may also reduce the risk of unnecessary drug-specific side effects, to allow patients and families time to better plan their futures (Rasmussen and Langerman, 2019).
The approval of the anti-Aβ MABs abucanumab in June 2021, and lecanemab in July 2023 raised hopes that a magic bullet directed at the cause of the disease was now available, rather than, as for AChEIs and NMDAR modulators, merely the symptoms of AD. Although the link between familial AD and Aβ plaques is accepted anti-Aβ MABs are unlikely to benefit the majority of subjects with AD. On the positive side the data from the CLARITY trials indicated that lecanemab delayed cognitive decline inferring an approximate 6-month extension in the quality of life for patients with mild-to-moderate AD, however, there are concerns over long term use and potentially serious side-effects linked to the frequency of ARIAs. Thus, qualified conclusions about the impact of anti-Aβ MABs will require more and longer clinical trials and several years of use by ethnically diverse groups of patients with AD. Questions have also been raised as whether the modest benefits will preclude use after considering the costs of treatment and related assessments, such as frequent MRI assessment and CSF tests, and the need for a highly vigilant post-marketing surveillance (Watt et al., 2023). Ito et al. (2021) provided an estimated annual cost of drug therapy at $16,000 USDA in the US; however, that was based on 2021 costs prior to the introduction of the MABs, and the cost will vary from country to country. Collectively, these issues support arguments that although we can accept that Aβ plaques are neurotoxic it does not necessarily follow that the amyloid cascade should be accepted as dogma and causality (Herrup, 2022), and alternative hypotheses and therapeutic targets need to be vigorously pursued (Tse and Herrup, 2017).
The role of systemic inflammation as an important contributory factor to the initiation of neuroinflammation and AD deserves greater attention including the targeting of reactive microglia (Heneka et al., 2015; Lee V. M. et al., 2021). Early diagnosis together with combination therapy that includes an anti-inflammatory preferably selective for microglia would reduce neuroinflammation (Wang et al., 2023). As early as 1990 the potential of NSAIDs to offset the development of AD was proposed based on the lower prevalence of AD in patients with rheumatoid arthritis and their use of anti-inflammatory drugs (McGeer et al., 1990). Data from 17 retrospective studies also provided support (McGeer et al., 1996). Despite continuing interest (see Supplementary Figures S4) subsequent studies of NSAIDS have proved contradictory and controversial (see Table 2 for a summary). Furthermore, the chronic use of NSAIDs is associated with the risk of GI ulcers and hemorrhage, while both NSAIDs and coxibs also increase cardiovascular risk, especially in elderly patients (Wehling, 2014; Schjerning et al., 2020). Interest in the gut microbiota and its role in chronic systemic and neuroinflammation has stimulated research as to whether a leaky intestinal barrier may promote an inflammatory response that contributes to the development of AD (Mou et al., 2022). Interestingly, sodium oligomannate (GV-971), derived from brown algae and approved in China for AD, targets neuroinflammation triggered by gut bacteria (Wang et al., 2019; Xiao et al., 2021); however, data from additional studies are required for validation.
The repurposing of drugs approved for other diseases is an active field for exploration. As reflected in Figure 1, small molecule tyrosine kinase (TK) inhibitors originally developed for cancer, such as the multi-target TK inhibitor, ponatinib, and the Bruton kinase inhibitor, ibrutinib, have been suggested as having potential therapeutic efficacy to reduce neuroinflammation (Chen et al., 2019; Tan et al., 2019; Li et al., 2023). Encouragingly, a combination of ponatinib with the pan caspase inhibitor, emricasan, was found to target apoptosis and necroptosis, and reduce ischemia/reperfusion injury in the rat brain (Tian et al., 2018). Similarly, in silico Genome-Wide Association Studies (GWAS) analysis has identified dabrafenib, the B Raf kinase (TK) inhibitor originally developed for the treatment of malignant melanoma, as a candidate apoptosis inhibitor to protect against neurotoxicity and Parkinson’s Disease (Uenaka et al., 2018; Okamoto, 2019).
In conclusion, recognizing that there are multiple contributory factors including familial and life style that can result in the development of AD a “one-size fits all” approach to treatment is inappropriate and in addition to an early recognition and reduction of modifiable risk factors that would also help reduce systemic inflammation, new drug targets need to be explored and studied with a particular focus on early-stage intervention and metabolic contributions (Chakrabarti et al., 2015). Drug development, plus early detection and intervention, will be facilitated if biomarker testing for AD from blood tests can be validated. Besides p-tau (Ashton et al., 2024) other proteins including GFAP, neurofilament light (Nfl), growth differentiation factor-15, and latent-transforming growth factor beta-binding protein 2, have been reported to be potential blood bio-markers for AD (Guo et al., 2024; Qiang et al., 2024). Advances in pre-clinical models of AD are also required. Animal models, notably with rodents, have been the mainstay for drug testing and a large number of transgenic mouse models are available; however, there are significant limitations to the data obtained from the study of small animals (McKean et al., 2021). An ex vivo model that captures all of the features of the human pathology is required and an important advance was made when a human brain 3D organoid was generated using induced pluripotent stem cells (iPSCs) from subjects with AD (Raja et al., 2016). The use of 3D human stem cell models of AD should greatly facilitate drug discovery and development (Arber et al., 2017; Centeno et al., 2018).
Acknowledgments
Figures 1–8 were created using BioRender.com.
Funding Statement
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Publication costs were provided by a grant from the Department of Medical Education, Weill Cornell Medicine-Qatar.
Author contributions
AK: Conceptualization, Writing–original draft, Writing–review and editing. JA: Writing–review and editing. RM: Writing–review and editing. CT: Conceptualization, Writing–original draft, Writing–review and editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2024.1399121/full#supplementary-material
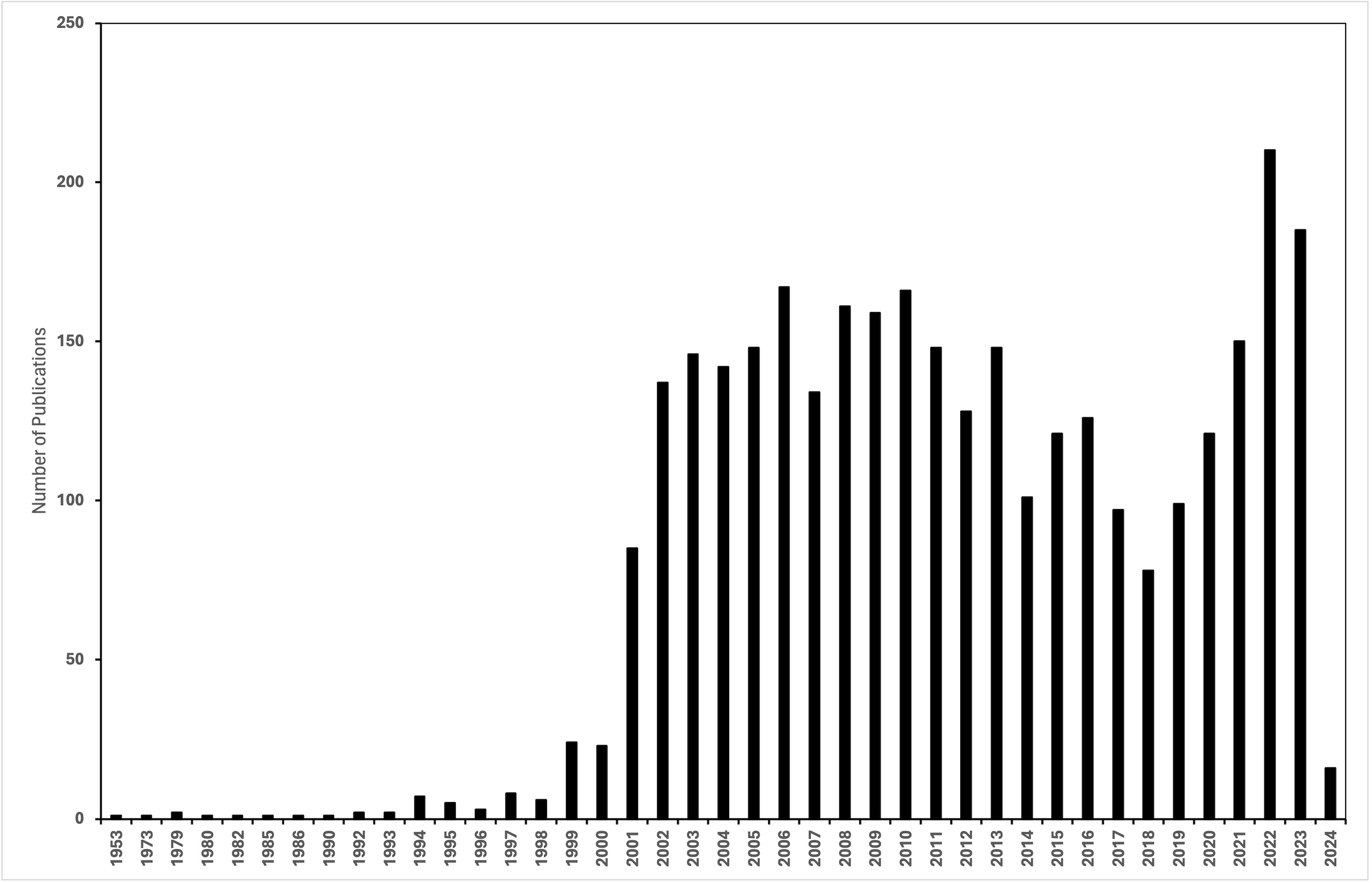{kind=link}
{kind=link}
{kind=link}
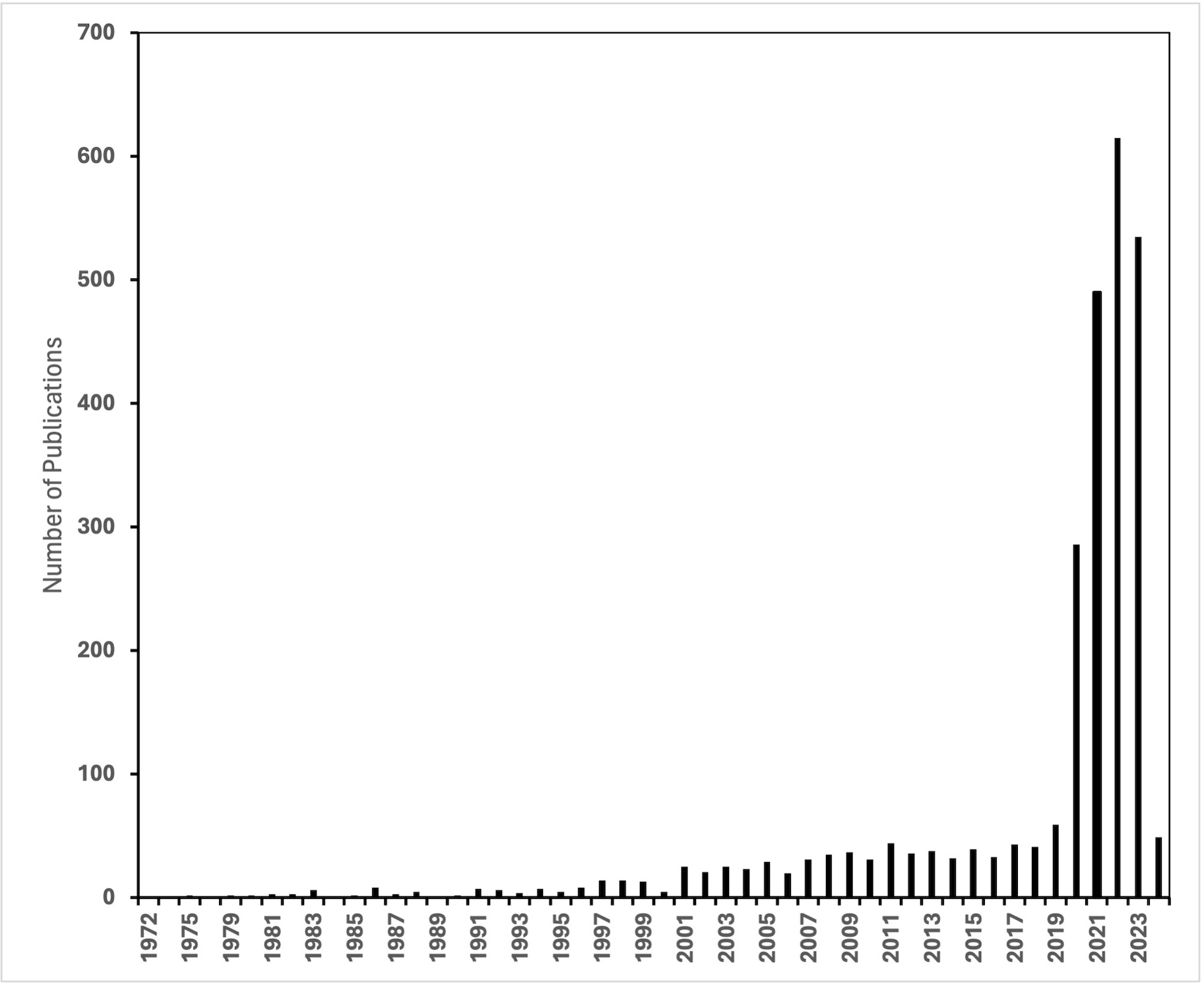{kind=link}
References
- Abraham W. C., Jones O. D., Glanzman D. L. (2019). Is plasticity of synapses the mechanism of long-term memory storage? NPJ Sci. Learn 4, 9. 10.1038/s41539-019-0048-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackley S. F., Zimmerman S. C., Brenowitz W. D., Tchetgen E. J., Gold A. L., Manly J. J., et al. (2021). Effect of reductions in amyloid levels on cognitive change in randomized trials: instrumental variable meta-analysis. BMJ 372, n156. Erratum in: BMJ. 2022 Aug 30, 378, o2094. 10.1136/bmj.n156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adesuyan M., Jani Y. H., Alsugeir D., Cheung E. C. L., Chui C. S. L., Howard R., et al. (2022). Antihypertensive agents and incident Alzheimer's disease: a systematic review and meta-analysis of observational studies. J. Prev. Alzheimers Dis. 9 (4), 715–724. 10.14283/jpad.2022.77 [DOI] [PubMed] [Google Scholar]
- Aizenstein H. J., Nebes R. D., Saxton J. A., Price J. C., Mathis C. A., Tsopelas N. D., et al. (2008). Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch. Neurol. 65 (11), 1509–1517. 10.1001/archneur.65.11.1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali T. B., Schleret T. R., Reilly B. M., Chen W. Y., Abagyan R. (2015). Adverse effects of cholinesterase inhibitors in dementia, according to the pharmacovigilance databases of the united-states and Canada. PloS one 10 (12), e0144337. 10.1371/journal.pone.0144337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez G., Aldudo J., Alonso M., Santana S., Valdivieso F. (2012). Herpes simplex virus type 1 induces nuclear accumulation of hyperphosphorylated tau in neuronal cells. J. Neurosci. Res. 90 (5), 1020–1029. 10.1002/jnr.23003 [DOI] [PubMed] [Google Scholar]
- Alvarez X. A., Sampedro C., Lozano R., Cacabelos R. (1999). Citicoline protects hippocampal neurons against apoptosis induced by brain beta-amyloid deposits plus cerebral hypoperfusion in rats. Methods Find. Exp. Clin. Pharmacol. 21 (8), 535–540. 10.1358/mf.1999.21.8.794835 [DOI] [PubMed] [Google Scholar]
- Alzheimer A. (1907). Uber eigenartige Erkrankung der Hirnrinde. All. Z. Psychiatr. 64, 146–148. [Google Scholar]
- Alzheimer A., Stelzmann R. A., Schnitzlein H. N., Murtagh F. R. (1995). An English translation of Alzheimer's 1907 paper, "Uber eine eigenartige Erkankung der Hirnrinde. Clin. Anat. 8 (6), 429–431. 10.1002/ca.980080612 [DOI] [PubMed] [Google Scholar]
- Alzheimer’s (2020). Alzheimer’s disease facts and figures. Alzheimer’s Dement. 16, 391–440. 10.1002/alz.12068 [DOI] [Google Scholar]
- Alzheimer s (2024). Alzheimer’s association. Available at: https://www.alz.org/alzheimers-dementia/treatments/aducanumab#:∼:text=Aducanumab%20to%20Be%20Discontinued%20as%20an%20Alzheimer's%20Treatment,-Aducanumab%20to%20Be&text=Aducanumab%20(Aduhelm%C2%AE)%2C%20which,manufacturer%20(Biogen)%20in%202024 (Accessed February 5, 2024).
- Amenta F., Parnetti L., Gallai V., Wallin A. (2001). Treatment of cognitive dysfunction associated with Alzheimer's disease with cholinergic precursors. Ineffective treatments or inappropriate approaches? Mech. Ageing Dev. 122 (16), 2025–2040. 10.1016/s0047-6374(01)00310-4 [DOI] [PubMed] [Google Scholar]
- Andreasen N., Hesse C., Davidsson P., Minthon L., Wallin A., Winblad B., et al. (1999). Cerebrospinal fluid beta-amyloid(1-42) in Alzheimer disease: differences between early- and late-onset Alzheimer disease and stability during the course of disease. Arch. Neurol. 56 (6), 673–680. 10.1001/archneur.56.6.673 [DOI] [PubMed] [Google Scholar]
- Arber C., Lovejoy C., Wray S. (2017). Stem cell models of Alzheimer's disease: progress and challenges. Alzheimers Res. Ther. 9 (1), 42. 10.1186/s13195-017-0268-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arighi A., Arcaro M., Fumagalli G. G., Carandini T., Pietroboni A. M., Sacchi L., et al. (2002). Aquaporin-4 cerebrospinal fluid levels are higher in neurodegenerative dementia: looking at glymphatic system dysregulation. Alzheimers Res. Ther. 14 (1), 135. 10.1186/s13195-022-01077-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A., Genové G., Mäe M., Nisancioglu M. H., Wallgard E., Niaudet C., et al. (2010). Pericytes regulate the blood-brain barrier. Nature 468 (7323), 557–561. 10.1038/nature09522 [DOI] [PubMed] [Google Scholar]
- Arndt J. W., Qian F., Smith B. A., Quan C., Kilambi K. P., Bush M. W., et al. (2018). Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci. Rep. 8 (1), 6412. 10.1038/s41598-018-24501-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton N. J., Brum W. S., Di Molfetta G., Benedet A. L., Arslan B., Jonaitis E., et al. (2024). Diagnostic accuracy of a plasma phosphorylated tau 217 immunoassay for alzheimer disease pathology. JAMA Neurol. 81, 255–263. 10.1001/jamaneurol.2023.5319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton N. J., Janelidze S., Mattsson-Carlgren N., Binette A. P., Strandberg O., Brum W. S., et al. (2022). Differential roles of Aβ42/40, p-tau231 and p-tau217 for Alzheimer's trial selection and disease monitoring. Nat. Med. 28 (12), 2555–2562. 10.1038/s41591-022-02074-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atri A. (2019). Current and future treatments in Alzheimer's disease. Semin. Neurol. 39 (2), 227–240. 10.1055/s-0039-1678581 [DOI] [PubMed] [Google Scholar]
- Atri A., Shaughnessy L. W., Locascio J. J., Growdon J. H., et al. (2008). Long-term course and effectiveness of combination therapy in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 22 (3), 209–221. 10.1097/WAD.0b013e31816653bc [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avgerinos K. I., Ferrucci L., Kapogiannis D. (2021). Effects of monoclonal antibodies against amyloid-β on clinical and biomarker outcomes and adverse event risks: a systematic review and meta-analysis of phase III RCTs in Alzheimer's disease. Ageing Res. Rev. 68, 101339. 10.1016/j.arr.2021.101339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagaria J., Bagyinszky E., An S. S. A. (2022). Genetics, functions, and clinical impact of presenilin-1 (PSEN1) gene. Gene. Int. J. Mol. Sci. 23 (18), 10970. 10.3390/ijms231810970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball M. J. (1982). Limbic predilection in Alzheimer dementia: is reactivated herpesvirus involved? Can. J. Neurol. Sci. 9 (3), 303–306. 10.1017/s0317167100044115 [DOI] [PubMed] [Google Scholar]
- Banerjee G., Farmer S. F., Hyare H., Jaunmuktane Z., Mead S., Ryan N. S., et al. (2024). Iatrogenic Alzheimer's disease in recipients of cadaveric pituitary-derived growth hormone. Nat. Med. 30, 394–402. 10.1038/s41591-023-02729-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barakos J., Purcell D., Suhy J., Chalkias S., Burkett P., Marsica Grassi C., et al. (2022). Detection and management of amyloid-related imaging abnormalities in patients with Alzheimer's disease treated with anti-amyloid beta therapy. J. Prev. Alzheimers Dis. 9 (2), 211–220. 10.14283/jpad.2022.21 [DOI] [PubMed] [Google Scholar]
- Barber I. S., Braae A., Clement N., Patel T., Guetta-Baranes T., Brookes K., et al. (2017). Mutation analysis of sporadic early-onset Alzheimer's disease using the NeuroX array. Neurobiol. Aging 49, 215.e1–215.e8. 10.1016/j.neurobiolaging.2016.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthélemy N. R., Horie K., Sato C., Bateman R. J. (2020). Blood plasma phosphorylated-tau isoforms track CNS change in Alzheimer's disease. J. Ex.p Med. 217 (11), e20200861. 10.1084/jem.20200861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzari F. H., Bazzari A. H. (2022). BACE1 inhibitors for Alzheimer's disease: the past, present and any future? Molecules 27 (24), 8823. 10.3390/molecules27248823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach T. G., Monsell S. E., Phillips L. E., Kukull W. (2012). Accuracy of the clinical diagnosis of alzheimer disease at national Institute on aging alzheimer disease centers, 2005-2010. J. Neuropathol. Exp. Neurol. 71 (4), 266–273. 10.1097/NEN.0b013e31824b211b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beishon L. C., Batterham A. P., Quinn T. J., Nelson C. P., Panerai R. B., Robinson T., et al. (2019). Addenbrooke's Cognitive Examination III (ACE-III) and mini-ACE for the detection of dementia and mild cognitive impairment. Cochrane database Syst. Rev. 12 (12), CD013282. 10.1002/14651858.CD013282.pub2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergström S., Remnestål J., Yousef J., Olofsson J., Markaki I., Carvalho S., et al. (2021). Multi-cohort profiling reveals elevated CSF levels of brain-enriched proteins in Alzheimer's disease. Ann. Clin. Transl. Neurol. 8 (7), 1456–1470. 10.1002/acn3.51402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beshir S. A., Aadithsoorya A. M., Parveen A., Goh S. S. L., Hussain N., Menon V. B. (2022). Aducanumab therapy to treat Alzheimer's disease: a narrative review. Int. J. Alzheimers Dis. 2022, 9343514. 10.1155/2022/9343514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biogen (2022). Eisai-presents-full-results-lecanemab-phase-3-confirmatory. Available at: https://investors.biogen.com/news-releases/news-release-details/eisai-presents-full-results-lecanemab-phase-3-confirmatory.
- Birks J. S., Harvey R. J. (2018). Donepezil for dementia due to Alzheimer's disease. Cochrane database Syst. Rev. 6 (6), CD001190. 10.1002/14651858.CD001190.pub3 [DOI] [PubMed] [Google Scholar]
- Blackard W. G., Jr, Sood G. K., Crowe D. R., Fallon M. B. (1998). Tacrine. A cause of fatal hepatotoxicity? J. Clin. Gastroenterol. 26 (1), 57–59. 10.1097/00004836-199801000-00015 [DOI] [PubMed] [Google Scholar]
- Blanco-Silvente L., Capellà D., Garre-Olmo J., Vilalta-Franch J., Castells X. (2018). Predictors of discontinuation, efficacy, and safety of memantine treatment for Alzheimer's disease: meta-analysis and meta-regression of 18 randomized clinical trials involving 5004 patients. BMC Geriatr. 18 (1), 168. 10.1186/s12877-018-0857-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanke M. L., VanDongen A. M. J. (2009). “Activation mechanisms of the NMDA receptor,” in Biology of the NMDA receptor. Editor Van Dongen A. M. (Boca Raton (FL): CRC Press/Taylor and Francis; ). [PubMed] [Google Scholar]
- Blautzik J., Kotz S., Brendel M., Sauerbeck J., Vettermann F., Winter Y., et al. (2018). Relationship between body mass index, ApoE4 status, and PET-based amyloid and neurodegeneration markers in amyloid-positive subjects with normal cognition or mild cognitive impairment. J. Alzheimer's Dis. 65 (3), 781–791. 10.3233/JAD-170064 [DOI] [PubMed] [Google Scholar]
- Blennow K., Hampel H. (2003). CSF markers for incipient Alzheimer's disease. Lancet. Neurology 2 (10), 605–613. 10.1016/s1474-4422(03)00530-1 [DOI] [PubMed] [Google Scholar]
- Blennow K., Zetterberg H. (2015). Understanding biomarkers of neurodegeneration: ultrasensitive detection techniques pave the way for mechanistic understanding. Nat. Med. 21 (3), 217–219. 10.1038/nm.3810 [DOI] [PubMed] [Google Scholar]
- Blennow K., Zetterberg H. (2018). The past and the future of Alzheimer's disease fluid biomarkers. J. Alzheimers Dis. 62 (3), 1125–1140. 10.3233/JAD-170773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliddal S., Banasik K., Pedersen O. B., Nissen J., Cantwell L., Schwinn M., et al. (2021). Acute and persistent symptoms in non-hospitalized PCR-confirmed COVID-19 patients. Sci. Rep. 11 (1), 13153. 10.1038/s41598-021-92045-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom G. S. (2014). Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71 (4), 505–508. 10.1001/jamaneurol.2013.5847 [DOI] [PubMed] [Google Scholar]
- Bonfoco E., Krainc D., Ankarcrona M., Nicotera P., Lipton S. A. (1995). Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc. Natl. Acad. Sci. U. S. A. 92 (16), 7162–7166. 10.1073/pnas.92.16.7162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braidy N., Grant R., Adams S., Brew B. J., Guillemin G. J. (2009). Mechanism for quinolinic acid cytotoxicity in human astrocytes and neurons. Neurotox. Res. 16 (1), 77–86. 10.1007/s12640-009-9051-z [DOI] [PubMed] [Google Scholar]
- Brandes R. P. (2014). Endothelial dysfunction and hypertension. Hypertension 64 (5), 924–928. 10.1161/HYPERTENSIONAHA.114.03575 [DOI] [PubMed] [Google Scholar]
- Bresink I., Benke T. A., Collett V. J., Seal A. J., Parsons C. G., Henley J. M., et al. (1996). Effects of memantine on recombinant rat NMDA receptors expressed in HEK 293 cells. Br. J. Pharmacol. 119 (2), 195–204. 10.1111/j.1476-5381.1996.tb15971.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodaty H., Corey-Bloom J., Potocnik F. C., Truyen L., Gold M., Damaraju C. R. (2005). Galantamine prolonged-release formulation in the treatment of mild to moderate Alzheimer's disease. Dement. Geriatr. Cogn. Disord. 20 (2-3), 120–132. 10.1159/000086613 [DOI] [PubMed] [Google Scholar]
- Buchhave P., Minthon L., Zetterberg H., Wallin Å. K., Blennow K., Hansson O. (2012). Cerebrospinal fluid levels of β-amyloid 1-42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch. Gen. Psychiatry 69 (1), 98–106. 10.1001/archgenpsychiatry.2011.155 [DOI] [PubMed] [Google Scholar]
- Budd Haeberlein S., Aisen P. S., Barkhof F., Chalkias S., Chen T., Cohen S., et al. (2022). Two randomized phase 3 studies of aducanumab in early Alzheimer's disease. J. Prev. Alzheimers Dis. 9 (2), 197–210. 10.14283/jpad.2022.30 [DOI] [PubMed] [Google Scholar]
- Bukhbinder A. S., Ling Y., Hasan O., Jiang X., Kim Y., Phelps K. N., et al. (2022). Risk of Alzheimer's disease following influenza vaccination: a claims-based cohort study using propensity score matching. J. Alzheimers Dis. 88 (3), 1061–1074. 10.3233/JAD-220361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield D. A., Boyd-Kimball D. (2018). Oxidative stress, amyloid-β peptide, and altered key molecular pathways in the pathogenesis and progression of Alzheimer's disease. J. Alzheimers Dis. 62 (3), 1345–1367. 10.3233/JAD-170543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield D. A., Swomley A. M., Sultana R. (2013). Amyloid β-peptide (1-42)-induced oxidative stress in Alzheimer disease: importance in disease pathogenesis and progression. Antioxid. Redox. Signal. 19 (8), 823–835. 10.1089/ars.2012.5027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacace R., Sleegers K., Van Broeckhoven C. (2016). Molecular genetics of early-onset Alzheimer's disease revisited. Alzheimers Dement. 12 (6), 733–748. 10.1016/j.jalz.2016.01.012 [DOI] [PubMed] [Google Scholar]
- Calhoun A., King C., Khoury R., Grossberg G. T. (2018). An evaluation of memantine ER + donepezil for the treatment of Alzheimer's disease. Expert Opin. Pharmacother. 19 (15), 1711–1717. 10.1080/14656566.2018.1519022 [DOI] [PubMed] [Google Scholar]
- Campanari M. L., García-Ayllón M. S., Belbin O., Galcerán J., Lleó A., Sáez-Valero J. (2014). Acetylcholinesterase modulates presenilin-1 levels and γ-secretase activity. J. Alzheimers Dis. 41 (3), 911–924. 10.3233/JAD-140426 [DOI] [PubMed] [Google Scholar]
- Campbell J. M., Stephenson M. D., de Courten B., Chapman I., Bellman S. M., Aromataris E. (2018). Metformin use associated with reduced risk of dementia in patients with diabetes: a systematic review and meta-analysis. J. Alzheimers Dis. 65 (4), 1225–1236. 10.3233/JAD-180263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso S. M., Santana I., Swerdlow R. H., Oliveira C. R. (2004). Mitochondria dysfunction of Alzheimer's disease cybrids enhances Abeta toxicity. J. Neurochem. 89 (6), 1417–1426. 10.1111/j.1471-4159.2004.02438.x [DOI] [PubMed] [Google Scholar]
- Centeno E. G. Z., Cimarosti H., Bithell A. (2018). 2D versus 3D human induced pluripotent stem cell-derived cultures for neurodegenerative disease modelling. Mol. Neurodegener. 13 (1), 27. 10.1186/s13024-018-0258-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S., Khemka V. K., Banerjee A., Chatterjee G., Ganguly A., Biswas A. (2015). Metabolic risk factors of sporadic Alzheimer's disease: implications in the pathology, pathogenesis and treatment. Aging Dis. 6 (4), 282–299. 10.14336/AD.2014.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G. F., Xu T. H., Yan Y., Zhou Y. R., Jiang Y., Melcher K., et al. (2017b). Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 38 (9), 1205–1235. 10.1038/aps.2017.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. S., Lipton S. A. (2005). Pharmacological implications of two distinct mechanisms of interaction of memantine with N-methyl-D-aspartate-gated channels. J. Pharmacol. Exp. Ther. 314 (3), 961–971. 10.1124/jpet.105.085142 [DOI] [PubMed] [Google Scholar]
- Chen R., Chan P. T., Chu H., Lin Y. C., Chang P. C., Chen C. Y., et al. (2017a). Treatment effects between monotherapy of donepezil versus combination with memantine for Alzheimer disease: a meta-analysis. PLoS One 12 (8), e0183586. 10.1371/journal.pone.0183586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Liu G., Chen J., Hu A., Zhang L., Sun W., et al. (2019). Ponatinib protects mice from lethal influenza infection by suppressing cytokine storm. Front. Immunol. 10, 1393. 10.3389/fimmu.2019.01393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z. R., Huang J. B., Yang S. L., Hong F. F. (2022). Role of cholinergic signaling in Alzheimer's disease. Molecules 27 (6), 1816. 10.3390/molecules27061816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi D. W. (1987). Ionic dependence of glutamate neurotoxicity. J. Neurosci. 7 (2), 369–379. 10.1523/JNEUROSCI.07-02-00369.1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou R., Cantor A., Dana T., Wagner J., Ahmed A. Y., Fu R., et al. (2022). Screening for hepatitis C virus infection in adolescents and adults: updated evidence report and systematic review for the US preventive Services task force. JAMA 328 (8), 976–771. 10.1001/jama.2019.20788 [DOI] [PubMed] [Google Scholar]
- Chow V. W., Mattson M. P., Wong P. C., Gleichmann M. (2010). An overview of APP processing enzymes and products. Neuromolecular. Med. 12 (1), 1–12. 10.1007/s12017-009-8104-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan C. M., Bossing T., Page A., Shepherd D., Mudher A. (2010). Soluble hyper-phosphorylated tau causes microtubule breakdown and functionally compromises normal tau in vivo . Acta. Neuropathol. 120 (5), 593–604. 10.1007/s00401-010-0716-8 [DOI] [PubMed] [Google Scholar]
- Crean S., Ward A., Mercaldi C. J., Collins J. M., Cook M. N., Baker N. L., et al. (2011). Apolipoprotein E ε4 prevalence in Alzheimer's disease patients varies across global populations: a systematic literature review and meta-analysis. Dement. Geriatr. Cogn. Disord. 31 (1), 20–30. 10.1159/000321984 [DOI] [PubMed] [Google Scholar]
- Cummings J. (2023). Anti-amyloid monoclonal antibodies are transformative treatments that redefine Alzheimer's disease therapeutics. Drugs 83 (7), 569–576. 10.1007/s40265-023-01858-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings J., Aisen P., Lemere C., Atri A., Sabbagh M., Salloway S. (2021). Aducanumab produced a clinically meaningful benefit in association with amyloid lowering. Alzheimers Res. Ther. 13 (1), 98. 10.1186/s13195-021-00838-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Mesquita S., Fu Z., Kipnis J. (2018a). The meningeal lymphatic system: a new player in neurophysiology. Neuron 100 (2), 375–388. 10.1016/j.neuron.2018.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Mesquita S., Louveau A., Vaccari A., Smirnov I., Cornelison R. C., Kingsmore K. M., et al. (2018b). Functional aspects of meningeal lymphatics in ageing and Alzheimer's disease. Nature 560 (7717), 185–191. 10.1038/s41586-018-0368-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darin J., Mokhtar G.El K. N. I., Jenny M. C., Naji T. (2012). Aspirin, steroidal and non-steroidal anti-inflammatory drugs for the treatment of Alzheimer's disease. Cochrane Database Syst. Rev. (2), CD006378. 10.1002/14651858.CD006378.pub2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silveira T. L., Zamberlan D. C., Arantes L. P., Machado M. L., da Silva T. C., Câmara D. F., et al. (2018). Quinolinic acid and glutamatergic neurodegeneration in Caenorhabditis elegans . Neurotoxicology 67, 94–101. 10.1016/j.neuro.2018.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dautzenberg G., Lijmer J., Beekman A. (2020). Diagnostic accuracy of the Montreal Cognitive Assessment (MoCA) for cognitive screening in old age psychiatry: determining cutoff scores in clinical practice. Avoiding spectrum bias caused by healthy controls. Int. J. Geriatr. Psychiatry 35 (3), 261–269. 10.1002/gps.5227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies P., Maloney A. J. (1976). Selective loss of central cholinergic neurons in Alzheimer's disease. Lancet 2 (8000), 1403. 10.1016/s0140-6736(76)91936-x [DOI] [PubMed] [Google Scholar]
- Davignon J. (2005). Apolipoprotein E and atherosclerosis: beyond lipid effect. Arterioscler. Thromb. Vasc. Biol. 25 (2), 267–269. 10.1161/01.ATV.0000154570.50696.2c [DOI] [PubMed] [Google Scholar]
- Dearborn J. L., Zhang Y., Qiao Y., Suri M. F. K., Liu L., Gottesman R. F., et al. (2017). Intracranial atherosclerosis and dementia: the atherosclerosis risk in communities (ARIC) study. Neurology 88 (16), 1556–1563. 10.1212/WNL.0000000000003837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deardorff W. J., Grossberg G. T. A. (2016). A fixed-dose combination of memantine extended-release and donepezil in the treatment of moderate-to-severe Alzheimer's disease. Drug Des. devel. Ther. 10, 3267–3279. 10.2147/DDDT.S86463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Chiara G., Marcocci M. E., Civitelli L., Argnani R., Piacentini R., Ripoli C., et al. (2010). APP processing induced by herpes simplex virus type 1 (HSV-1) yields several APP fragments in human and rat neuronal cells. PloS One 5 (11), e13989. 10.1371/journal.pone.0013989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decourt B., Boumelhem F., Pope E. D., Shi J., Mari Z., Sabbagh M. N. (2021). Critical appraisal of amyloid lowering agents in AD. Curr. Neurol. Neurosci. Rep. 21 (8), 39. 10.1007/s11910-021-01125-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky S. T., Scheff S. W. (1990). Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann. Neurology 27 (5), 457–464. 10.1002/ana.410270502 [DOI] [PubMed] [Google Scholar]
- de la Monte S. M., Wands J. R. (2008). Alzheimer's disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2 (6), 1101–1113. 10.1177/193229680800200619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre J. C., Mussivand T. (1993). Can disturbed brain microcirculation cause Alzheimer's disease? Neurological Res. 15 (3), 146–153. 10.1080/01616412.1993.11740127 [DOI] [PubMed] [Google Scholar]
- de Leon M. J., Li Y., Okamura N., Tsui W. H., Saint-Louis L. A., Glodzik L., et al. (2017). Cerebrospinal fluid clearance in alzheimer disease measured with dynamic PET. J. Nucl. Med. 58 (9), 1471–1476. 10.2967/jnumed.116.187211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demattos R. B., Lu J., Tang Y., Racke M. M., Delong C. A., Tzaferis J. A., et al. (2012). A plaque-specific antibody clears existing β-amyloid plaques in Alzheimer's disease mice. Neuron 76 (5), 908–920. 10.1016/j.neuron.2012.10.029 [DOI] [PubMed] [Google Scholar]
- de Oliveira F. F., Chen E. S., Smith M. C., Bertolucci P. H. F. (2018). Pharmacogenetics of angiotensin-converting enzyme inhibitors in patients with Alzheimer's disease dementia. Curr. Alzheimer Res. 15 (4), 386–398. 10.2174/1567205014666171016101816 [DOI] [PubMed] [Google Scholar]
- de Paula V. J. R., Guimarães F. M., Diniz B. S., Forlenza O. V. (2009). Neurobiological pathways to Alzheimer's disease: amyloid-beta, TAU protein or both? Dement. Neuropsychol. 3 (3), 188–194. 10.1590/S1980-57642009DN30300003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vlieger L., Vandenbroucke R. E., Van Hoecke L. (2022). Recent insights into viral infections as a trigger and accelerator in Alzheimer's disease. Drug Discov. Today 27 (11), 103340. 10.1016/j.drudis.2022.103340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Battista A. M., Heinsinger N. M., Rebeck G. W. (2016). Alzheimer's disease genetic risk factor APOE-ε4 also affects normal brain function. Curr. Alzheimer Res. 13 (11), 1200–1207. 10.2174/1567205013666160401115127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan H., Crain B., Troncoso J., Resnick S. M., Zonderman A. B., Obrien R. J. (2010). Atherosclerosis, dementia, and alzheimer disease in the Baltimore longitudinal study of aging cohort. Ann. Neurol. 68 (2), 231–240. 10.1002/ana.22055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingues A., Almeida S., da Cruz e Silva E. F., Oliveira C. R., Rego A. C. (2007). Toxicity of beta-amyloid in HEK293 cells expressing NR1/NR2A or NR1/NR2B N-methyl-D-aspartate receptor subunits. Neurochem. Int. 50 (6), 872–880. 10.1016/j.neuint.2007.03.001 [DOI] [PubMed] [Google Scholar]
- Douaud G., Lee S., Alfaro-Almagro F., Arthofer C., Wang C., McCarthy P., et al. (2022). SARS-CoV-2 is associated with changes in brain structure in UK Biobank. Nature 604 (7907), 697–707. 10.1038/s41586-022-04569-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow C. T., Greenblatt C. L., Chan E. D., Dow J. F. (2022). Evaluation of BCG vaccination and plasma amyloid: a prospective, pilot study with implications for Alzheimer’s disease. Microorganisms 10 (2), 424. 10.3390/microorganisms10020424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond E., Pires G., MacMurray C., Askenazi M., Nayak S., Bourdon M., et al. (2020). Phosphorylated tau interactome in the human Alzheimer's disease brain. Brain 143 (9), 2803–2817. 10.1093/brain/awaa223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumurgier J., Schraen S., Gabelle A., Vercruysse O., Bombois S., Laplanche J. L., et al. (2015). Cerebrospinal fluid amyloid-β 42/40 ratio in clinical setting of memory centers: a multicentric study. Alzheimers Res. Ther. 7 (1), 30. 10.1186/s13195-015-0114-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eftekharzadeh B., Daigle J. G., Kapinos L. E., Coyne A., Schiantarelli J., Carlomagno Y., et al. (2019). Tau protein disrupts nucleocytoplasmic transport in Alzheimer's disease. Neuron 99 (5), 925–940.e7. 10.1016/j.neuron.2018.07.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan M. F., Kost J., Voss T., Mukai Y., Aisen P. S., Cummings J. L., et al. (2019). Randomized trial of verubecestat for prodromal Alzheimer's disease. N. Engl. J. Med. 380 (15), 1408–1420. 10.1056/NEJMoa1812840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Mir M. Y., Detaille D., R-Villanueva G., Delgado-Esteban M., Guigas B., Attia S., et al. (2008). Neuroprotective role of antidiabetic drug metformin against apoptotic cell death in primary cortical neurons. J. Mol. Neurosci. 34 (1), 77–87. 10.1007/s12031-007-9002-1 [DOI] [PubMed] [Google Scholar]
- Ertekin-Taner N. (2007). Genetics of Alzheimer's disease: a centennial review. Neurol. Clin. 25 (3), 611–667. 10.1016/j.ncl.2007.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer L. A., Cupples L. A., Haines J. L., Hyman B., Kukull W. A., Mayeux R., et al. (1997). Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278 (16), 1349–1356. PMID: 9343467. 10.1001/jama.278.16.1349 [DOI] [PubMed] [Google Scholar]
- Femminella G. D., Harold D., Scott J., Williams J., Edison P. Alzheimer’s Disease Neuroimaging Initiative (2021). The differential influence of immune, endocytotic, and lipid metabolism genes on amyloid deposition and neurodegeneration in subjects at risk of Alzheimer's disease. J. Alzheimers Dis. 79 (1), 127–139. 10.3233/JAD-200578 [DOI] [PubMed] [Google Scholar]
- Femminella G. D., Taylor-Davies G., Scott J., Edison P. Alzheimer’s Disease Neuroimaging Initiative (2018). Do cardiometabolic risk factors influence amyloid, tau, and neuronal function in APOE4 carriers and non-carriers in Alzheimer's disease trajectory? J. Alzheimer's Dis. 64 (3), 981–993. 10.3233/JAD-180365 [DOI] [PubMed] [Google Scholar]
- Ferrari C., Sorbi S. (2021). The complexity of Alzheimer's disease: an evolving puzzle. Physiol. Rev. 101 (3), 1047–1081. 10.1152/physrev.00015.2020 [DOI] [PubMed] [Google Scholar]
- Fischer D., Mukrasch M. D., Biernat J., Bibow S., Blackledge M., Griesinger C., et al. (2009). Conformational changes specific for pseudophosphorylation at serine 262 selectively impair binding of tau to microtubules. Biochemistry 48 (42), 10047–10055. 10.1021/bi901090m [DOI] [PubMed] [Google Scholar]
- Fish J. (2011) Encyclopedia of clinical neuropsychology. Berlin, Germany: Srpinger, 111–112. 10.1007/978-0-387-79948-3_1791 [DOI] [Google Scholar]
- Fitz N. F., Nam K. N., Wolfe C. M., Letronne F., Playso B. E., Iordanova B. E., et al. (2021). Phospholipids of APOE lipoproteins activate microglia in an isoform-specific manner in preclinical models of Alzheimer's disease. Nat. Commun. 12 (1), 3416. 10.1038/s41467-021-23762-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J., Busquets O., Ettcheto M., Sánchez-López E., Castro-Torres R. D., Verdaguer E., et al. (2018). Memantine for the treatment of dementia: a review on its current and future applications. J. Alzheimers Dis. 62 (3), 1223–1240. 10.3233/JAD-170672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine E. (2018). Metformin-induced mitochondrial complex I inhibition: facts, uncertainties, and consequences. Front. Endocrinol. (Lausanne) 9, 753. 10.3389/fendo.2018.00753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenthaler S., Meineke I., Schreeb K. H., Boakye E., Gundert-Remy U., Gleiter C. H. (1998). Influence of urine pH and urinary flow on the renal excretion of memantine. Br. J. Clin. Pharmacol. 46 (6), 541–546. 10.1046/j.1365-2125.1998.00819.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost B., Jacks R. L., Diamond M. I. (2009). Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 284 (19), 12845–12852. 10.1074/jbc.M808759200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabin J. M., Tambs K., Saltvedt I., Sund E., Holmen J. (2017). Association between blood pressure and Alzheimer disease measured up to 27 years prior to diagnosis: the HUNT Study. Alzheimers Res. Ther. 9 (1), 37. 10.1186/s13195-017-0262-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly G., Chakrabarti S., Chatterjee U., Saso L. (2017). Proteinopathy, oxidative stress and mitochondrial dysfunction: cross talk in Alzheimer's disease and Parkinson's disease. Drug Des. devel. Ther. 11, 797–810. 10.2147/DDDT.S130514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner G. G., Wong C. W. (1984). Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120 (3), 885–890. 10.1016/s0006-291x(84)80190-4 [DOI] [PubMed] [Google Scholar]
- Gong C. X., Iqba L. K. (2008). Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Curr. Med. Chem. 15 (23), 2321–2328. 10.2174/092986708785909111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goos J. D., Henneman W. J., Sluimer J. D., Vrenken H., Sluimer I. C., Barkhof F., et al. (2010). Incidence of cerebral microbleeds: a longitudinal study in a memory clinic population. Neurology 74 (24), 1954–1960. 10.1212/WNL.0b013e3181e396ea [DOI] [PubMed] [Google Scholar]
- Gracon S. I., Knapp M. J., Berghoff W. G., Pierce M., DeJong R., Lobbestael S. J., et al. (1998). Safety of tacrine: clinical trials, treatment IND, and postmarketing experience. Alzheimer. Dis. Assoc. Disord. 12 (2), 93–101. 10.1097/00002093-199806000-00007 [DOI] [PubMed] [Google Scholar]
- Graham N. L., Emery T., Hodges J. R. (2004). Distinctive cognitive profiles in Alzheimer's disease and subcortical vascular dementia. J. Neurol. Neurosurg. Psychiatry. 75 (1), 61–71. PMID: 14707310. [PMC free article] [PubMed] [Google Scholar]
- Greenamyre J. T., Penney J. B., D'Amato C. J., Young A. B. (1987). Dementia of the Alzheimer's type: changes in hippocampal L-[3H]glutamate binding. J. Neurochem. 48 (2), 543–551. 10.1111/j.1471-4159.1987.tb04127.x [DOI] [PubMed] [Google Scholar]
- Grossberg G. T., Manes F., Allegri R. F., Gutiérrez-Robledo L. M., Gloger S., Xie L., et al. (2013). The safety, tolerability, and efficacy of once-daily memantine (28 mg): a multinational, randomized, double-blind, placebo-controlled trial in patients with moderate-to-severe Alzheimer's disease taking cholinesterase inhibitors. CNS Drugs 27 (6), 469–478. 10.1007/s40263-013-0077-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y., You J., Zhang Y., Liu W. S., Huang Y. Y., Zhang Y. R., et al. (2024). Plasma proteomic profiles predict future dementia in healthy adults. Nat. Aging. 4, 247–260. 10.1038/s43587-023-00565-0 [DOI] [PubMed] [Google Scholar]
- Haddad H. W., Malone G. W., Comardelle N. J., Degueure A. E., Kaye A. M., Kaye A. D. (2022). Aducanumab, a novel anti-amyloid monoclonal antibody, for the treatment of Alzheimer's disease: a comprehensive review. Health Psychol. Res. 10 (1), 31925. 10.52965/001c.31925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajjar I., Schumpert J., Hirth V., Wieland D., Eleazer G. P. (2002). The impact of the use of statins on the prevalence of dementia and the progression of cognitive impairment. J. Gerontol. A. Biol. Sci. Med. Sci. 57 (7), M414–M418. 10.1093/gerona/57.7.m414 [DOI] [PubMed] [Google Scholar]
- Halliday M. R., Rege S. V., Ma Q., Zhao Z., Miller C. A., Winkler E. A., et al. (2016). Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer's disease. J. Cereb. Blood Flow. Metab. 36 (1), 216–227. 10.1038/jcbfm.2015.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham G. E. (2006). Pro-survival signalling from the NMDA receptor. Biochem. Soc. Trans. 34 (Pt 5), 936–938. 10.1042/BST0340936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham G. E., Bading H. (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 11 (10), 682–696. 10.1038/nrn2911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham G. E., Fukunaga Y., Bading H. (2002). Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 5 (5), 405–414. 10.1038/nn835 [DOI] [PubMed] [Google Scholar]
- Hardy J., Allsop D. (1991). Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol. Sci. 12 (10), 383–388. 10.1016/0165-6147(91)90609-v [DOI] [PubMed] [Google Scholar]
- Hardy J. A., Higgins G. A. (1992). Alzheimer's disease: the amyloid cascade hypothesis. Science 256 (5054), 184–185. 10.1126/science.1566067 [DOI] [PubMed] [Google Scholar]
- Hare J. (2010). Trafficking of amyloid β-precursor protein products C83 and C99 on the endocytic pathway. Biochem. Biophys. Res. Commun. 401 (2), 219–224. 10.1016/j.bbrc.2010.09.033 [DOI] [PubMed] [Google Scholar]
- Harkany T., Abrahám I., Timmerman W., Laskay G., Tóth B., Sasvári M., et al. (2000). beta-amyloid neurotoxicity is mediated by a glutamate-triggered excitotoxic cascade in rat nucleus basalis. Eur. J. Neurosci. 12 (8), 2735–2745. 10.1046/j.1460-9568.2000.00164.x [DOI] [PubMed] [Google Scholar]
- Hasselmo M. E. (2006). The role of acetylcholine in learning and memory. Curr. Opin. Neurobiol. 16 (6), 710–715. 10.1016/j.conb.2006.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L., Wondisford F. E. (2015). Metformin action: concentrations matter. Cell Metab. 21 (2), 159–162. 10.1016/j.cmet.2015.01.003 [DOI] [PubMed] [Google Scholar]
- Hellwig K., Kvartsberg H., Portelius E., Andreasson U., Oberstein T. J., Lewczuk P., et al. (2015). Neurogranin and YKL-40: independent markers of synaptic degeneration and neuroinflammation in Alzheimer's disease. Alzheimers Res. Ther. 7, 74. 10.1186/s13195-015-0161-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms J., Kremer S., Merdji H., Clere-Jehl R., Schenck M., Kummerlen C., et al. (2020). Neurologic features in severe SARS-CoV-2 infection. N. Engl. J. Med. 382 (23), 2268–2270. 10.1056/NEJMc2008597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks S., Ranson J. M., Peetoom K., Lourida I., Tai X. Y., de Vugt M., et al. (2023). Risk factors for young-onset dementia in the UK biobank. JAMA Neurol. 81, 134–142. 10.1001/jamaneurol.2023.4929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka M. T., Carson M. J., El Khoury J., Landreth G. E., Brosseron F., Feinstein D. L., et al. (2015). Neuroinflammation in Alzheimer's disease. Lancet Neurol. 14 (4), 388–405. 10.1016/S1474-4422(15)70016-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley D., Raghavan N., Sperling R., Aisen P., Raman R., Romano G. (2019). Preliminary results of a trial of atabecestat in preclinical Alzheimer's disease. N. Engl. J. Med. 11 (15), 1483–1485. 10.1056/NEJMc1813435 [DOI] [PubMed] [Google Scholar]
- Herrup K. (2015). The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 18 (6), 794–799. 10.1038/nn.4017 [DOI] [PubMed] [Google Scholar]
- Herrup K. (2022). Fallacies in neuroscience: the Alzheimer's edition. eNeuro 9 (1). 10.1523/ENEURO.0530-21.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill A. F. (2019). Extracellular vesicles and neurodegenerative diseases. J. Neurosci. 39 (47), 9269–9273. 10.1523/JNEUROSCI.0147-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hladky S. B., Barrand M. A. (2022). The glymphatic hypothesis: the theory and the evidence. Fluids Barriers CNS 19 (1), 9. 10.1186/s12987-021-00282-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn M. D., MacLean A. G. (2021). Extracellular vesicles as a means of viral immune evasion, CNS invasion, and glia-induced neurodegeneration. Front. Cell. Neurosci. 15, 695899. 10.3389/fncel.2021.695899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskin J. L., Sabbagh M. N., Al-Hasan Y., Decourt B. (2019). Tau immunotherapies for Alzheimer's disease. Expert. Opin. Investig. Drugs. 28 (6), 545–554. 10.1080/13543784.2019.1619694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovakimyan A., Zagorski K., Chailyan G., Antonyan T., Melikyan L., Petrushina I., et al. (2022). Immunogenicity of MultiTEP platform technology-based Tau vaccine in non-human primates. NPJ Vaccines 7 (1), 117. 10.1038/s41541-022-00544-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard R., McShane R., Lindesay J., Ritchie C., Baldwin A., Barber R., et al. (2012). Donepezil and memantine for moderate-to-severe Alzheimer's disease. N. Engl. J. Med. 366 (10), 893–903. 10.1056/NEJMoa1106668 [DOI] [PubMed] [Google Scholar]
- Huang Y. A., Zhou B., Wernig M., Südhof T. C. (2017). ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and Aβ secretion. Cell 168 (3), 427–441. 10.1016/j.cell.2016.12.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber G., Bailly Y., Martin J. R., Mariani J., Brugg B. (1997). Synaptic beta-amyloid precursor proteins increase with learning capacity in rats. Neuroscience 80 (2), 313–320. 10.1016/s0306-4522(97)00120-6 [DOI] [PubMed] [Google Scholar]
- Hui Z., Zhijun Y., Yushan Y., Liping C., Yiying Z., Difan Z., et al. (2020). The combination of acyclovir and dexamethasone protects against Alzheimer's disease-related cognitive impairments in mice. Psychopharmacology 237, 1851–1860. 10.1007/s00213-020-05503-1 [DOI] [PubMed] [Google Scholar]
- Hunsberger H. C., Pinky P. D., Smith W., Suppiramaniam V., Reed M. N. (2019). The role of APOE4 in Alzheimer's disease: strategies for future therapeutic interventions. Neuronal Signal 3 (2), NS20180203. 10.1042/NS20180203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur J. Y. (2022). γ-Secretase in Alzheimer's disease. Exp. Mol. Med. 54 (4), 433–446. 10.1038/s12276-022-00754-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur J. Y., Frost G. R., Wu X., Crump C., Pan S. J., Wong E., et al. (2020). The innate immunity protein IFITM3 modulates γ-secretase in Alzheimer's disease. Nature 586 (7831), 735–740. 10.1038/s41586-020-2681-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannucci J., Sen A., Grammas P. (2021). Isoform-specific effects of apolipoprotein E on markers of inflammation and toxicity in brain glia and neuronal cells in vitro . Curr. Issues Mol. Biol. 43 (1), 215–225. 10.3390/cimb43010018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff J. J., Wang M., Liao Y., Plogg B. A., Peng W., Gundersen G. A., et al. (2012). A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 4 (147), 147ra111. 10.1126/scitranslmed.3003748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- In t Veld B. A., Ruitenberg A., Hofman A., Launer L. J., van Duijn C. M., Stijnen T., et al. (2001). Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N. Engl. J. Med. 345 (21), 1515–1521. 10.1056/NEJMoa010178 [DOI] [PubMed] [Google Scholar]
- Ito K., Chapman R., Pearson S. D., Tafazzoli A., Yaffe K., Gurwitz J. H. (2021). Evaluation of the cost-effectiveness of drug treatment for alzheimer disease in a simulation model that includes caregiver and societal factors. JAMA Netw. Open 4 (10), e2129392. 10.1001/jamanetworkopen.2021.29392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhaki R. F., Lathe R., Balin B. J., Ball M. J., Bearer E. L., Braak H., et al. (2016). Microbes and Alzheimer's disease. J. Alzheimers Dis. 51 (4), 979–984. 10.3233/JAD-160152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C. R., Jr., Knopman D. S., Jagust W. J., Shaw L. M., Aisen P. S., Weiner M. W., et al. (2010). Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 9 (1), 119–128. 10.1016/S1474-4422(09)70299-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C. R. Jr., Bennett D. A., Blennow K., Carrillo M. C., Dunn B., Haeberlein S. B., et al. (2018). NIA-AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 14 (4), 535–562. 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson G. A., Maitland N. J., Wilcock G. K., Craske J., Itzhaki R. F. (1991). Latent herpes simplex virus type 1 in normal and Alzheimer's disease brains. J. Med. Virol. 33 (4), 224–227. 10.1002/jmv.1890330403 [DOI] [PubMed] [Google Scholar]
- Jansen W. J., Ossenkoppele R., Knol D. L., Tijms B. M., Scheltens P., Verhey F. R., et al. (2015). Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA 313 (19), 1924–1938. 10.1001/jama.2015.4668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrott B. (2017). Tacrine: in vivo veritas. Pharmacol. Res. 116, 29–31. 10.1016/j.phrs.2016.12.033 [DOI] [PubMed] [Google Scholar]
- Jaturapatporn D., Isaac M. G., McCleery J., Tabet N. (2012). Aspirin, steroidal and non-steroidal anti-inflammatory drugs for the treatment of Alzheimer's disease. Cochrane Database Syst. Rev. 2, CD006378. 10.1002/14651858.CD006378.pub2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaunmuktane Z., Mead S., Ellis M., Wadsworth J. D., Nicoll A. J., Kenny J., et al. (2015). Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 525 (7568), 247–250. 10.1038/nature15369 [DOI] [PubMed] [Google Scholar]
- Jeong S. M., Shin D. W., Yoo T. G., Cho M. H., Jang W., Lee J., et al. (2021). Association between statin use and Alzheimer's disease with dose response relationship. Sci. Rep. 11 (1), 15280. 10.1038/s41598-021-94803-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jho Y. S., Zhulina E. B., Kim M. W., Pincus P. A. (2010). Monte Carlo simulations of tau proteins: effect of phosphorylation. Biophys. J. 99 (8), 2387–2397. 10.1016/j.bpj.2010.06.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia X., Wang Z., Huang F., Su C., Du W., Jiang H., et al. (2021). A comparison of the Mini-Mental State Examination (MMSE) with the Montreal Cognitive Assessment (MoCA) for mild cognitive impairment screening in Chinese middle-aged and older population: a cross-sectional study. BMC Psychiatry 21 (1), 485. 10.1186/s12888-021-03495-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D., Yang X., Li M., Wang Y., Wang Y. (2015). Efficacy and safety of galantamine treatment for patients with Alzheimer's disease: a meta-analysis of randomized controlled trials. J. Neural. Transm. (Vienna). 122 (8), 1157–1166. 10.1007/s00702-014-1358-0 [DOI] [PubMed] [Google Scholar]
- Johnson J. W., Kotermanski S. E. (2006). Mechanism of action of memantine. Curr. Opin. Pharmacol. 6 (1), 61–67. 10.1016/j.coph.2005.09.007 [DOI] [PubMed] [Google Scholar]
- Jönsson L., Wimo A., Handels R., Johansson G., Boada M., Engelborghs S., et al. (2023). The affordability of lecanemab, an amyloid-targeting therapy for Alzheimer's disease: an EADC-EC viewpoint. Lancet Reg. Health Eur. 229, 100657. 10.1016/j.lanepe.2023.100657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M., Walker L. C. (2024). Evidence for iatrogenic transmission of Alzheimer’s disease. Nat. Med. 30, 344–345. 10.1038/s41591-023-02768-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabir M. T., Uddin M. S., Setu J. R., Ashraf G. M., Bin-Jumah M. N., Abdel-Daim M. M. (2020). Exploring the role of PSEN mutations in the pathogenesis of Alzheimer's disease. Neurotox. Res. 38 (4), 833–849. 10.1007/s12640-020-00232-x [DOI] [PubMed] [Google Scholar]
- Kandimalla R. J., Prabhakar S., Wani W. Y., Kaushal A., Gupta N., Sharma D. R., et al. (2013). CSF p-Tau levels in the prediction of Alzheimer's disease. Biol. Open 2 (11), 1119–1124. 10.1242/bio.20135447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplitt M., Gouras G. K., Makimura H., Jovanovic J., Sweeney D., Greengard P., et al. (1996). Apolipoprotein E, A beta-amyloid, and the molecular pathology of Alzheimer's disease. Therapeutic implications. Ann. N. Y. Acad. Sci. 802, 42–49. 10.1111/j.1749-6632.1996.tb32597.x [DOI] [PubMed] [Google Scholar]
- Karran E., Hardy J. (2014). Antiamyloid therapy for Alzheimer's disease--are we on the right road? N. Engl. J. Med. 370 (4), 377–378. 10.1056/NEJMe1313943 [DOI] [PubMed] [Google Scholar]
- Kaur U., Reddy J., Tiwari A., Chakrabarti S., Chakrabarti S. S. (2024). Lecanemab: more questions than answers. Clin. Drug Investig. 44 (1), 1–10. 10.1007/s40261-023-01331-1 [DOI] [PubMed] [Google Scholar]
- Kavanagh S., Gaudig M., Van Baelen B., Adami M., Delgado A., Guzman C., et al. (2011). Galantamine and behavior in Alzheimer disease: analysis of four trials. Acta. Neurol. Scand. 124 (5), 302–308. 10.1111/j.1600-0404.2011.01525.x [DOI] [PubMed] [Google Scholar]
- Kavirajan H. (2009). Memantine: a comprehensive review of safety and efficacy. Expert Opin. Drug Saf. 8 (1), 89–109. 10.1517/14740330802528420 [DOI] [PubMed] [Google Scholar]
- Kepp K. P. (2017). Ten challenges of the amyloid hypothesis of Alzheimer's disease. J. Alzheimers Dis. 55 (2), 447–457. 10.3233/JAD-160550 [DOI] [PubMed] [Google Scholar]
- Kim S. M., Kim M. J., Rhee H. Y., Ryu C. W., Kim E. J., Petersen E. T., et al. (2013). Regional cerebral perfusion in patients with Alzheimer's disease and mild cognitive impairment: effect of APOE epsilon4 allele. Neuroradiology 55 (1), 25–34. 10.1007/s00234-012-1077-x [DOI] [PubMed] [Google Scholar]
- Kim W., Hecht M. H. (2005). Sequence determinants of enhanced amyloidogenicity of Alzheimer A{beta}42 peptide relative to A{beta}40. J. Biol. Chem. 280 (41), 35069–35076. 10.1074/jbc.M505763200 [DOI] [PubMed] [Google Scholar]
- Kishi T., Matsunaga S., Oya K., Nomura I., Ikuta T., Iwata N. (2017). Memantine for Alzheimer's disease: an updated systematic review and meta-analysis. J. Alzheimers Dis. 60 (2), 401–425. 10.3233/JAD-170424 [DOI] [PubMed] [Google Scholar]
- Kitazawa M., Cheng D., Tsukamoto M. R., Koike M. A., Wes P. D., Vasilevko V., et al. (2011). Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer's disease model. J. Immunol. 187 (12), 6539–6549. 10.4049/jimmunol.1100620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight R., Khondoker M., Magill N., Stewart R., Landau S. (2018). A systematic review and meta-analysis of the effectiveness of acetylcholinesterase inhibitors and memantine in treating the cognitive symptoms of dementia. Dement. Geriatr. Cogn. Disord. 45 (3-4), 131–151. 10.1159/000486546 [DOI] [PubMed] [Google Scholar]
- Knopman D. S., Jones D. T., Greicius M. D. (2021). Failure to demonstrate efficacy of aducanumab: an analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement. 17 (4), 696–701. 10.1002/alz.12213 [DOI] [PubMed] [Google Scholar]
- Kostapanos M. S., Elisaf M. S. (2017). Statins and mortality: the untold story. Br. J. Clin. Pharmacol. 83 (5), 938–941. 10.1111/bcp.13202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuns B., Rosani A., Patel P., Varghese D. (2024). “Memantine,” in StatPearls (St. Petersburg, Florida, United States: StatPearls Publishing; ). [PubMed] [Google Scholar]
- Kwan A. T. H., Arfaie S., Therriault J., Rosa-Neto P., Gauthier S. (2020). Lessons learnt from the second generation of anti-amyloid monoclonal antibodies clinical trials. Dement. Geriatr. Cogn. Disord. 49 (4), 334–348. 10.1159/000511506 [DOI] [PubMed] [Google Scholar]
- Larner A. J., Mitchell A. J. (2014). A meta-analysis of the accuracy of the Addenbrooke's Cognitive Examination (ACE) and the Addenbrooke's Cognitive Examination-Revised (ACE-R) in the detection of dementia. Int. psychogeriatrics 26 (4), 555–563. 10.1017/S1041610213002329 [DOI] [PubMed] [Google Scholar]
- Lee C., Landreth G. (2010). The role of microglia in amyloid clearance from the AD brain. J. Neural. Transm. (Vienna). 117 (8), 949–960. 10.1007/s00702-010-0433-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G., Thangavel R., Sharma V. M., Litersky J. M., Bhaskar K., Fang S. M., et al. (2004). Phosphorylation of tau by fyn: implications for Alzheimer's disease. J. Neurosci. 24 (9), 2304–2312. 10.1523/JNEUROSCI.4162-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. C., Kim S. J., Hong S., Kim Y. S. (2019). Diagnosis of Alzheimer’s disease utilizing amyloid and tau as fluid biomarkers. Exp. Mol. Med. 51, 1–10. 10.1038/s12276-019-0250-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. H., Cha M., Lee B. H. (2021a). Crosstalk between neuron and glial cells in oxidative injury and neuroprotection. Int. J. Mol. Sci. 22 (24), 13315. 10.3390/ijms222413315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee V. M., Goedert M., Trojanowski J. Q. (2001). Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24, 1121–1159. 10.1146/annurev.neuro.24.1.1121 [DOI] [PubMed] [Google Scholar]
- Lee V. M., Goedert M., Trojanowski J. Q. (2021b). Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24, 1121–1159. 10.1146/annurev.neuro.24.1.1121 [DOI] [PubMed] [Google Scholar]
- Lehrer S., Rheinstein P. H. (2022). Vaccination reduces risk of Alzheimer's disease, Parkinson's disease and other neurodegenerative disorders. Discov. Med. 34 (172), 97–101. PMID: 36281030. [PMC free article] [PubMed] [Google Scholar]
- Lehrer S., Rheinstein P. H. (2023). Alignment of human aquaporin 4 and ß-amyloid proteins may indicate involvement of ß-amyloid in brain water homeostasis and prevention of brain edema. Chronic Dis. Transl. Med. 9 (2), 177–181. PMID: 37305107; PMCID: PMC10249176. 10.1002/cdt3.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. J., Wang B., Kodali M. C., Chen C., Kim E., Patters B. J., et al. (2018). In vivo evidence for the contribution of peripheral circulating inflammatory exosomes to neuroinflammation. J. Neuroinflammation 15, 8. 10.1186/s12974-017-1038-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Lu C., Zhao Z., Lu D., Zheng G. (2023). Uncovering neuroinflammation-related modules and potential repurposing drugs for Alzheimer's disease through multi-omics data integrative analysis. Front. Aging Neurosci. 15, 1161405. 10.3389/fnagi.2023.1161405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. C., Liu C. C., Kanekiyo T., Xu H., Bu G. (2013). Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9 (2), 106–118. 10.1038/nrneurol.2012.263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P. P., Xie Y., Meng X. Y., Kang J. S. (2019). History and progress of hypotheses and clinical trials for Alzheimer's disease. Signal Transduct. Target Ther. 4, 29. 10.1038/s41392-019-0063-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Hossinger A., Heumüller S. E., Hornberger A., Buravlova O., Konstantoulea K., et al. (2021). Highly efficient intercellular spreading of protein misfolding mediated by viral ligand-receptor interactions. Nat. Commun. 12 (1), 5739. 10.1038/s41467-021-25855-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston G., Huntley J., Sommerlad A., Ames D., Ballard C., Banerjee S., et al. (2020). Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 396 (10248), 413–446. 10.1016/S0140-6736(20)30367-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez O. L., Becker J. T., Wahed A. S., Saxton J., Sweet R. A., Wolk D. A., et al. (2009). Long-term effects of the concomitant use of memantine with cholinesterase inhibition in Alzheimer disease. J. Neurol. Neurosurg. Psychiatry 80 (6), 600–607. 10.1136/jnnp.2008.158964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord A., Gumucio A., Englund H., Sehlin D., Sundquist V. S., Söderberg L., et al. (2009). An amyloid-beta protofibril-selective antibody prevents amyloid formation in a mouse model of Alzheimer's disease. Neurobiol. Dis. 36 (3), 425–434. 10.1016/j.nbd.2009.08.007 [DOI] [PubMed] [Google Scholar]
- Lv T., Zhao B., Hu Q., Zhang X. (2021). The glymphatic system: a novel therapeutic target for stroke treatment. Front. Aging Neurosci. 13, 689098. 10.3389/fnagi.2021.689098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q., Zhao Z., Sagare A. P., Wu Y., Wang M., Owens N. C., et al. (2018). Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-β42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol. Neurodegener. 13 (1), 57. 10.1186/s13024-018-0286-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahase E. (2021a). Aducanumab: European agency rejects Alzheimer's drug over efficacy and safety concerns. BMJ 375, n3127. 10.1136/bmj.n3127 [DOI] [PubMed] [Google Scholar]
- Mahase E. (2021b). FDA approves controversial Alzheimer's drug despite uncertainty over effectiveness. BMJ 373, n1462. 10.1136/bmj.n1462 [DOI] [PubMed] [Google Scholar]
- Mahase E. (2021c). Three FDA advisory panel members resign over approval of Alzheimer's drug. BMJ 373, n1503. 10.1136/bmj.n1503 [DOI] [PubMed] [Google Scholar]
- Mahley R. W., Rall S. C., Jr (2000). Apolipoprotein E: far more than a lipid transport protein. Annu. Rev. Genomics Hum. Genet. 1, 507–537. 10.1146/annurev.genom.1.1.507 [DOI] [PubMed] [Google Scholar]
- Maragos W. F., Chu D. C., Young A. B., D'Amato C. J., Penney J. B., Jr. (1987). Loss of hippocampal [3H]TCP binding in Alzheimer's disease. Neurosci. Lett. 74 (3), 371–376. 10.1016/0304-3940(87)90326-0 [DOI] [PubMed] [Google Scholar]
- Marais A. D. (2019). Apolipoprotein E in lipoprotein metabolism, health and cardiovascular disease. Pathology 51 (2), 165–176. 10.1016/j.pathol.2018.11.002 [DOI] [PubMed] [Google Scholar]
- Maroli N. (2023). Aquaporin-4 mediated aggregation of Alzheimer’s amyloid β-peptide. ACS Chem. Neurosci. 14 (15), 2683–2698DOI. 10.1021/acschemneuro.3c00233 [DOI] [PubMed] [Google Scholar]
- Martel C. L., Mackic J. B., Matsubara E., Governale S., Miguel C., Miao W., et al. (1997). Isoform-specific effects of apolipoproteins E2, E3, and E4 on cerebral capillary sequestration and blood-brain barrier transport of circulating Alzheimer's amyloid beta. J. Neurochem. 69 (5), 1995–2004. 10.1046/j.1471-4159.1997.69051995.x [DOI] [PubMed] [Google Scholar]
- Marucci G., Buccioni M., Ben D. D., Lambertucci C., Volpini R., Amenta F. (2021). Efficacy of acetylcholinesterase inhibitors in Alzheimer's disease. Neuropharmacology 190, 108352. 10.1016/j.neuropharm.2020.108352 [DOI] [PubMed] [Google Scholar]
- Matsunaga S., Kishi T., Iwata N. (2015). Memantine monotherapy for Alzheimer's disease: a systematic review and meta-analysis. PLoS One 10 (4), e0123289. 10.1371/journal.pone.0123289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki T., Sasaki K., Hata J., Hirakawa Y., Fujimi K., Ninomiya T., et al. (2011). Association of Alzheimer disease pathology with abnormal lipid metabolism: the Hisayama Study. Neuropharmacology 190, 108352. 10.1016/j.neuropharm.2020.108352 [DOI] [PubMed] [Google Scholar]
- Mattiace L. A., Davies P., Dickson D. W. (1990). Detection of HLA-DR on microglia in the human brain is a function of both clinical and technical factors. Am. J. Pathol. 136 (5), 1101–1114. PMID: 1693471. [PMC free article] [PubMed] [Google Scholar]
- Mattson M. P. (1997). Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol. Rev. 77 (4), 1081–1132. 10.1152/physrev.1997.77.4.1081 [DOI] [PubMed] [Google Scholar]
- Mattsson N., Zetterberg H., Janelidze S., Insel P. S., Andreasson U., Stomrud E., et al. (2016). Plasma tau in Alzheimer disease. Neurology 87 (17), 1827–1835. 10.1212/WNL.0000000000003246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCorkindale A. N., Mundell H. D., Guennewig B., Sutherland G. T. (2022). Vascular Dysfunction Is Central to Alzheimer's Disease Pathogenesis in APOE e4 Carriers. Int. J. Mol. Sci. 23 (13), 7106. 10.3390/ijms23137106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDade E., Cummings J. L., Dhadda S., Swanson C. J., Reyderman L., Kanekiyo M., et al. (2022). Lecanemab in patients with early Alzheimer's disease: detailed results on biomarker, cognitive, and clinical effects from the randomized and open-label extension of the phase 2 proof-of-concept study. Alzheimers Res. Ther. 14 (1), 191. 10.1186/s13195-022-01124-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDade E., Voytyuk I., Aisen P., Bateman R. J., Carrillo M. C., De Strooper B., et al. (2021). The case for low-level BACE1 inhibition for the prevention of Alzheimer disease. Nat. Rev. Neurol. 17 (11), 703–714. 10.1038/s41582-021-00545-1 [DOI] [PubMed] [Google Scholar]
- McGeer P. L., Guo J. P., Lee M., Kennedy K., McGeer E. G. (2018). Alzheimer's disease can Be spared by nonsteroidal anti-inflammatory drugs. J. Alzheimers Dis. 62 (3), 1219–1222. 10.3233/JAD-170706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer P. L., Itagaki S., Tago H., McGeer E. G. (1987). Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci. Lett. 79 (1-2), 195–200. 10.1016/0304-3940(87)90696-3 [DOI] [PubMed] [Google Scholar]
- McGeer P. L., McGeer E., Rogers J., Sibley J. (1990). Anti-inflammatory drugs and Alzheimer disease. Lancet London, Engl. 335 (8696), 1037. 10.1016/0140-6736(90)91101-f [DOI] [PubMed] [Google Scholar]
- McGeer P. L., Schulzer M., McGeer E. G. (1996). Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiologic studies. Neurology 47 (2), 425–432. 10.1212/WNL.47.2.425 [DOI] [PubMed] [Google Scholar]
- Mckean N. E., Handley R. R., Snell R. G. (2021). A review of the current mammalian models of Alzheimer's disease and challenges that need to Be overcome. Int. J. Mol. Sci. 22 (23), 13168. 10.3390/ijms222313168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShane R., Westby M. J., Roberts E., Minakaran N., Schneider L., Farrimond L. E., et al. (2019). Memantine for dementia. Cochrane Database Syst. Rev. 3 (3), CD003154. 10.1002/14651858.CD003154.pub6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam M., Guillozet A., Shaw P., Quinn B. (2002). Widely spread butyrylcholinesterase can hydrolyze acetylcholine in the normal and Alzheimer brain. Neurobiol. Dis. 9 (1), 88–93. 10.1006/nbdi.2001.0462 [DOI] [PubMed] [Google Scholar]
- Meyer P. F., Tremblay-Mercier J., Leoutsakos J., Madjar C., Lafaille-Magnan M. E., Savard M., et al. (2019). INTREPAD: a randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology 92 (18), e2070–e2080. 10.1212/WNL.0000000000007232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielke M. M., Vemuri P., Rocca W. A., Quinn B. (2014). Clinical epidemiology of Alzheimer's disease: assessing sex and gender differences. Neurobiol. Dis. 9 (1), 88–93. 10.1006/nbdi.2001.0462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielke R., Zerres K., Uhlhaas S., Kessler J., Heiss W. D. (1998). Apolipoprotein E polymorphism influences the cerebral metabolic pattern in Alzheimer's disease. Neurosci. Lett. 254 (1), 49–52. 10.1016/s0304-3940(98)00673-9 [DOI] [PubMed] [Google Scholar]
- Miles L. A., Crespi G. A., Doughty L., Parker M. W. (2013). Bapineuzumab captures the N-terminus of the Alzheimer's disease amyloid-beta peptide in a helical conformation. Sci. Rep. 3, 1302. 10.1038/srep01302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moftakhar P., Lynch M. D., Pomakian J. L., Vinters H. V. (2010). Aquaporin expression in the brains of patients with or without cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 69 (12), 1201–1209. 10.1097/NEN.0b013e3181fd252c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad D., Chan P., Bradley J., Lanctôt K., Herrmann N. (2017). Acetylcholinesterase inhibitors for treating dementia symptoms - a safety evaluation. Expert Opin. Drug Saf. 16 (9), 1009–1019. 10.1080/14740338.2017.1351540 [DOI] [PubMed] [Google Scholar]
- Montagne A., Nation D. A., Sagare A. P., Barisano G., Sweeney M. D., Chakhoyan A., et al. (2020). APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 581 (7806), 71–76. 10.1038/s41586-020-2247-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro C., Toth B., Brunstein F., Bobbala A., Datta S., Ceniceros R., et al. (2023). Randomized phase II study of the safety and efficacy of semorinemab in participants with mild-to-moderate alzheimer disease: lauriet. Lauriet. Neurol. 101 (14), e1391–e1401. 10.1212/WNL.0000000000207663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris J. C., Schindler S. E., McCue L. M., Moulder K. L., Benzinger T. L. S., Cruchaga C., et al. (2019). Assessment of racial disparities in biomarkers for alzheimer disease. JAMA Neurol. 76 (3), 264–273. 10.1001/jamaneurol.2018.4249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motter R., Vigo-Pelfrey C., Kholodenko D., Barbour R., Johnson-Wood K., Galasko D., et al. (1995). Reduction of beta-amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer's disease. Ann. Neurol. 38 (4), 643–648. 10.1002/ana.410380413 [DOI] [PubMed] [Google Scholar]
- Mou Y., Du Y., Zhou L., Yue J., Hu X., Liu Y., et al. (2022). Gut microbiota interact with the brain through systemic chronic inflammation: implications on neuroinflammation, neurodegeneration, and aging. Front. Immunol. 13, 796288. 10.3389/fimmu.2022.796288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L., Selkoe D. J. (2012). Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb. Perspec.t Med. 2 (7), a006338. 10.1101/cshperspect.a006338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukrasch M. D., von Bergen M., Biernat J., Fischer D., Griesinger C., Mandelkow E., et al. (2007). The "jaws" of the tau-microtubule interaction. J. Biol. Chem. 282 (16), 12230–12239. 10.1074/jbc.M607159200 [DOI] [PubMed] [Google Scholar]
- Müller S. A., Shmueli M. D., Feng X., Tüshaus J., Schumacher N., Clark R., et al. (2023). The Alzheimer’s disease-linked protease BACE1 modulates neuronal IL-6 signaling through shedding of the receptor gp130. Mol. Neurodegener. 18 (1), 13. 10.1186/s13024-023-00596-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munafò A., Cantone A. F., Di Benedetto G., Torrisi S. A., Burgaletto C., Bellanca C. M., et al. (2024). Pharmacological enhancement of cholinergic neurotransmission alleviates neuroinflammation and improves functional outcomes in a triple transgenic mouse model of Alzheimer's disease. Front. Pharmacol. 15, 1386224. 10.3389/fphar.2024.1386224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy C., Dyer A. H., Lawlor B., Kennelly S. P. NILVAD study group (2023). What is the impact of ongoing statin use on cognitive decline and dementia progression in older adults with mild-moderate Alzheimer disease? PLoS One 18 (5), e0285529. 10.1371/journal.pone.0285529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasreddine Z. S., Phillips N. A., Bédirian V., Charbonneau S., Whitehead V., Collin I., et al. (2005). The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J. Am. Geriatr. Soc. 53 (4), 695–699. 10.1111/j.1532-5415.2005.53221.x [DOI] [PubMed] [Google Scholar]
- Nelson P. T., Alafuzoff I., Bigio E. H., Bouras C., Braak H., Cairns N. J., et al. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 71 (5), 362–381. 10.1097/NEN.0b013e31825018f7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwenhuis-Mark R. E. (2010). The death knoll for the MMSE: has it outlived its purpose? J. Geriatr. Psychiatry Neurol. 23 (3), 151–157. 10.1177/0891988710363714 [DOI] [PubMed] [Google Scholar]
- Nishitsuji K., Hosono T., Nakamura T., Bu G., Michikawa M. (2011). Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. J. Biol. Chem. 286 (20), 17536–17542. 10.1074/jbc.M111.225532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noetzli M., Eap C. B. (2013). Pharmacodynamic, pharmacokinetic and pharmacogenetic aspects of drugs used in the treatment of Alzheimer's disease. Clin. Pharmacokinet. 52 (4), 225–241. 10.1007/s40262-013-0038-9 [DOI] [PubMed] [Google Scholar]
- Nordberg A., Ballard C., Bullock R., Darreh-Shori T., Somogyi M. (2013). A review of butyrylcholinesterase as a therapeutic target in the treatment of Alzheimer's disease. Prim. Care. Companion. CNS Disord. 15 (2), 12r01412. 10.4088/PCC.12r01412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nørgaard C. H., Friedrich S., Hansen C. T., Gerds T., Ballard C., Møller D. V., et al. (2022). Treatment with glucagon-like peptide-1 receptor agonists and incidence of dementia: data from pooled double-blind randomized controlled trials and nationwide disease and prescription registers. Alzheimers Dement. (N Y). 8 (1), e12268. 10.1002/trc2.12268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak M. M., Niemczyk M., Florczyk M., Kurzyna M., Pączek L. (2022). Effect of statins on all-cause mortality in adults: a systematic review and meta-analysis of propensity score-matched studies. J. Clin. Med. 11 (19), 5643. 10.3390/jcm11195643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowell J., Blunt E., Gupta D., Edison P. (2023). Antidiabetic agents as a novel treatment for Alzheimer's and Parkinson's disease. Ageing Res. Rev. 89, 101979. 10.1016/j.arr.2023.101979 [DOI] [PubMed] [Google Scholar]
- Okamoto T. (2019). Parkinson's Disease: amantadine, zonisamide, dabrafenib. Brain Nerve 71 (9), 953–959. Japanese. 10.11477/mf.1416201387 [DOI] [PubMed] [Google Scholar]
- O’Leary K. (2023). Seeing through the fog of long COVID. Nat. Med. 29, 2973. 10.1038/s41591-023-02713-w [DOI] [PubMed] [Google Scholar]
- Olmastroni E., Molari G., De Beni N., Colpani O., Galimberti F., Gazzotti M., et al. (2022). Statin use and risk of dementia or Alzheimer's disease: a systematic review and meta-analysis of observational studies. Eur. J. Prev. Cardiol. 29 (5), 804–814. 10.1093/eurjpc/zwab208 [DOI] [PubMed] [Google Scholar]
- Olsson B., Lautner R., Andreasson U., Öhrfelt A., Portelius E., Bjerke M., et al. (2016). CSF and blood biomarkers for the diagnosis of Alzheimer's disease: a systematic review and meta-analysis. Lancet Neurol. 15 (7), 673–684. 10.1016/S1474-4422(16)00070-3 [DOI] [PubMed] [Google Scholar]
- Panza F., Lozupone M. (2022). The challenges of anti-tau therapeutics in Alzheimer disease. Nat. Rev. Neurol. 18, 577–578. 10.1038/s41582-022-00702-0 [DOI] [PubMed] [Google Scholar]
- Pasinetti G. M. (2002). Cyclooxygenase as a target for the antiamyloidogenic activities of nonsteroidal anti-inflammatory drugs in Alzheimer's disease. Neurosignals 11 (5), 293–297. 10.1159/000067428 [DOI] [PubMed] [Google Scholar]
- Periclou A. P., Ventura D., Sherman T., Rao N., Abramowitz W. T. (2004). Lack of pharmacokinetic or pharmacodynamic interaction between memantine and donepezil. Ann. Pharmacother. 38 (9), 1389–1394. 10.1345/aph.1D638 [DOI] [PubMed] [Google Scholar]
- Perry E., McKeith I., Ballard C. (2003). Butyrylcholinesterase and progression of cognitive deficits in dementia with Lewy bodies. Neurology 60 (11), 1852–1853. 10.1212/01.wnl.0000068336.84399.9e [DOI] [PubMed] [Google Scholar]
- Petek B., Häbel H., Xu H., Villa-Lopez M., Kalar I., Hoang M. T., et al. (2023). Statins and cognitive decline in patients with Alzheimer's and mixed dementia: a longitudinal registry-based cohort study. Alzheimers Res. Ther. 15 (1), 220. 10.1186/s13195-023-01360-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piller C. (2022). Second death linked to potential antibody treatment for Alzheimer's disease. Woman's brain hemorrhage while receiving Eisai's widely heralded lecanemab heightens concerns overs its safety. Science. 10.1126/science.adf9701 [DOI] [Google Scholar]
- Podhorna J., Krahnke T., Shear M., Harrison J. E. Alzheimer’s Disease Neuroimaging Initiative (2016). Alzheimer's Disease Assessment Scale-Cognitive subscale variants in mild cognitive impairment and mild Alzheimer's disease: change over time and the effect of enrichment strategies. Alzheimers Res. Ther. 8, 8. 10.1186/s13195-016-0170-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porsteinsson A. P., Grossberg G. T., Mintzer J., Olin J. T., Memantine M. E. M. (2008). Memantine treatment in patients with mild to moderate Alzheimer's disease already receiving a cholinesterase inhibitor: a randomized, double-blind, placebo-controlled trial. Curr. Alzheimer. Res. 5 (1), 83–89. 10.2174/156720508783884576 [DOI] [PubMed] [Google Scholar]
- Portelius E., Tran A. J., Andreasson U., Persson R., Brinkmalm G., Zetterberg H., et al. (2007). Characterization of amyloid beta peptides in cerebrospinal fluid by an automated immunoprecipitation procedure followed by mass spectrometry. J. Proteome. Res. 6 (11), 4433–4439. 10.1021/pr0703627 [DOI] [PubMed] [Google Scholar]
- Portelius E., Zetterberg H., Skillbäck T., Törnqvist U., Andreasson U., Trojanowski J. Q., et al. (2015). Cerebrospinal fluid neurogranin: relation to cognition and neurodegeneration in Alzheimer's disease. Brain 138 (Pt 11), 3373–3385. 10.1093/brain/awv267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts C., Richardson J., Bond R. B., Price R. K., Mulvenna M. D., Zvolsky P., et al. (2022). Reliability of Addenbrooke's Cognitive Examination III in differentiating between dementia, mild cognitive impairment and older adults who have not reported cognitive problems. Eur. J. Ageing 19, 495–507. 10.1007/s10433-021-00652-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protto V., Tramutola A., Fabiani M., Marcocci M. E., Napoletani G., Iavarone F., et al. (2020). Multiple herpes simplex virus-1 (HSV-1) reactivations induce protein oxidative damage in mouse brain: novel mechanisms for Alzheimer's disease progression. Microorganisms 8 (7), 972. 10.3390/microorganisms8070972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner S. B. (1984). Some speculations about prions, amyloid, and Alzheimer's disease. N. Engl. J. Med. 310 (10), 661–663. 10.1056/NEJM198403083101021 [DOI] [PubMed] [Google Scholar]
- Purro S. A., Farrow M. A., Linehan J., Nazari T., Thomas D. X., Chen Z., et al. (2018). Transmission of amyloid-β protein pathology from cadaveric pituitary growth hormone. Nature 564 (7736), 415–419. 10.1038/s41586-018-0790-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang W., Yau W. M., Lu J. X., Collinge J., Tycko R. (2017). Structural variation in amyloid-β fibrils from Alzheimer's disease clinical subtypes. Nature 541 (7636), 217–221. 10.1038/nature20814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang Y. X., You J., He X. Y., Guo Y., Deng Y. T., Gao P. Y., et al. (2024). Plasma metabolic profiles predict future dementia and dementia subtypes: a prospective analysis of 274,160 participants. Alzheimers Res. Ther. 16 (1), 16. 10.1186/s13195-023-01379-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L., Crews F. T. (2012). NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J. Neuroinflammation 9, 5. 10.1186/1742-2094-9-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman A., Hossen M. A., Chowdhury M. F. I., Bari S., Tamanna N., Sultana S. S., et al. (2023). Aducanumab for the treatment of Alzheimer's disease: a systematic review. Psychogeriatrics 23 (3), 512–522. 10.1111/psyg.12944 [DOI] [PubMed] [Google Scholar]
- Raja W. K., Mungenast A. E., Lin Y. T., Ko T., Abdurrob F., Seo J., et al. (2016). Self-organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate Alzheimer's disease phenotypes. PLoS One 11 (9), e0161969. 10.1371/journal.pone.0161969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen J., Langerman H. (2019). Alzheimer's disease - why we need early diagnosis. Degener. Neurol. Neuromuscul. Dis. 9, 123–130. 10.2147/DNND.S228939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P. H., Mani G., Park B. S., Jacques J., Murdoch G., Whetsell W., Jr., et al. (2005). Differential loss of synaptic proteins in Alzheimer's disease: implications for synaptic dysfunction. J. Alzheimers. Dis. 7 (2), 103–117. ; discussion 173-80. 10.3233/jad-2005-7203 [DOI] [PubMed] [Google Scholar]
- Reiss A. B., Montufar N., DeLeon J., Pinkhasov A., Gomolin I. H., Glass A. D., et al. (2021). Alzheimer disease clinical trials targeting amyloid: lessons learned from success in mice and failure in humans. Neurologist 26 (2), 52–61. 10.1097/NRL.0000000000000320 [DOI] [PubMed] [Google Scholar]
- Reitz C., Rogaeva E., Beecham G. W. (2020). Late-onset vs nonmendelian early-onset Alzheimer disease: a distinction without a difference? Neurol. Genet. 6 (5), e512. 10.1212/NXG.0000000000000512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz C., Tang M. X., Luchsinger J., Mayeux R. (2004). Relation of plasma lipids to Alzheimer disease and vascular dementia. Arch. Neurol. 61 (5), 705–714. 10.1001/archneur.61.5.705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Q. W., Teng T. H. K., Tse Y. K., Tsang C. T. W., Yu S. Y., Wu M. Z., et al. (2024). Statins and risks of dementia among patients with heart failure: a population-based retrospective cohort study in Hong Kong. Lancet Reg. Health West Pac 44, 101006. 10.1016/j.lanwpc.2023.101006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Represa A., Deloulme J. C., Sensenbrenner M., Ben-Ari Y., Baudier J. (1990). Neurogranin: immunocytochemical localization of a brain-specific protein kinase C substrate. J. Neurosci. 10 (12), 3782–3792. 10.1523/JNEUROSCI.10-12-03782.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard E., den Brok M. G. H. E., van Gool W. A. (2021). Bayes analysis supports null hypothesis of anti-amyloid beta therapy in Alzheimer's disease. Alzheimers Dement. 17 (6), 1051–1055. 10.1002/alz.12379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel B. C., Thompson P. M., Brinton R. D. (2016). Age, APOE and sex: triad of risk of Alzheimer's disease. J. Steroid Biochem. Mol. Biol. 160, 134–147. 10.1016/j.jsbmb.2016.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel G., Platt B., Micheau J. (2003). Glutamate receptor function in learning and memory. Behav. Brain Res. 140 (1-2), 1–47. 10.1016/s0166-4328(02)00272-3 [DOI] [PubMed] [Google Scholar]
- Riemenschneider M., Wagenpfeil S., Vanderstichele H., Otto M., Wiltfang J., Kretzschmar H., et al. (2003). Phospho-tau/total tau ratio in cerebrospinal fluid discriminates Creutzfeldt-Jakob disease from other dementias. Mol. Psychiatry. 8 (3), 343–347. 10.1038/sj.mp.4001220 [DOI] [PubMed] [Google Scholar]
- Rosen W. G., Mohs R. C., Davis K. L. (1984). A new rating scale for Alzheimer's disease. Am. J. Psychiatry. 141 (11), 1356–1364. 10.1176/ajp.141.11.1356 [DOI] [PubMed] [Google Scholar]
- Rosenberg J. B., Kaplitt M. G., De B. P., Chen A., Flagiello T., Salami C., et al. (2018). AAVrh.10-Mediated APOE2 central nervous system gene therapy for APOE4-associated Alzheimer's disease. Hum. Gene Ther. Clin. Dev. 29 (1), 24–47. 10.1089/humc.2017.231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Laurent Thibault C., Özer Stillman I., Chen S., Getsios D., Proskorovsky I., Hernandez L., et al. (2015). Cost-utility analysis of memantine extended release added to cholinesterase inhibitors compared to cholinesterase inhibitor monotherapy for the treatment of moderate-to-severe dementia of the Alzheimer's type in the US. J. Med. Econ. 18 (11), 930–943. 10.3111/13696998.2015.1063501 [DOI] [PubMed] [Google Scholar]
- Samuels S. C., Davis K. L. (1997). A risk-benefit assessment of tacrine in the treatment of Alzheimer's disease. Drug. Saf. 16 (1), 66–77. 10.2165/00002018-199716010-00005 [DOI] [PubMed] [Google Scholar]
- Savolainen K. M., Loikkanen J., Naarala J. (1995). Amplification of glutamate-induced oxidative stress. Toxicol. Lett. 82-83, 399–405. 10.1016/0378-4274(95)03490-0 [DOI] [PubMed] [Google Scholar]
- Schachter A. S., Davis K. L. (2000). Alzheimer's disease. Dialogues Clin. Neurosci. 2 (2), 91–100. 10.31887/DCNS.2000.2.2/asschachter [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schjerning A. M., McGettigan P., Gislason G. (2020). Cardiovascular effects and safety of (non-aspirin) NSAIDs. Nat. Rev. Cardiol. 17 (9), 574–584. 10.1038/s41569-020-0366-z [DOI] [PubMed] [Google Scholar]
- Seaks C. E., Wilcock D. M. (2020). Infectious hypothesis of Alzheimer disease. PLoS Pathog. 16 (11), e1008596. 10.1371/journal.ppat.1008596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secades J. J., Frontera G. (1995). CDP-choline: pharmacological and clinical review. Methods Find. Exp. Clin. Pharmacol. 17 (Suppl. B), 1–54. PMID: 8709678. [PubMed] [Google Scholar]
- Seeman P., Caruso C., Lasaga M. (2008). Memantine agonist action at dopamine D2High receptors. Synapse 62 (2), 149–153. 10.1002/syn.20472 [DOI] [PubMed] [Google Scholar]
- Serrano-Pozo A., Qian J., Monsell S. E., Blacker D., Gómez-Isla T., Betensky R. A., et al. (2014). Mild to moderate Alzheimer dementia with insufficient neuropathological changes. Ann. Neurol. 75 (4), 597–601. 10.1002/ana.24125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevigny J., Chiao P., Bussière T., Weinreb P. H., Williams L., Maier M., et al. (2017). The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature 546 (7659), 50–56. 10.1038/nature19323 [DOI] [PubMed] [Google Scholar]
- Sharma C., Kim S. R. (2023). Oxidative stress: culprit or consequence in Alzheimer's amyloidopathy. Neural Regen. Res. 18 (9), 1948–1949. 10.4103/1673-5374.367843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shua-Haim J., Smith J., Picard F., Sedek G., Athalye S., Pommier F., et al. (2008). Steady-state pharmacokinetics of rivastigmine in patients with mild to moderate Alzheimer's disease not affected by co-administration of memantine: an open-label, crossover, single-centre study. Clin. Drug Investig. 28 (6), 361–374. 10.2165/00044011-200828060-00004 [DOI] [PubMed] [Google Scholar]
- Shulman M., Kong J., O'Gorman J., Ratti E., Rajagovindan R., Viollet L., et al. (2023). TANGO: a placebo-controlled randomized phase 2 study of efficacy and safety of the anti-tau monoclonal antibody gosuranemab in early Alzheimer's disease. Nat. Aging 3, 1591–1601. 10.1038/s43587-023-00523-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silveyra M. X., Evin G., Montenegro M. F., Vidal C. J., Martínez S., Culvenor J. G., et al. (2008). Presenilin 1 interacts with acetylcholinesterase and alters its enzymatic activity and glycosylation. Mol. Cell Biol. 28 (9), 2908–2919. 10.1128/MCB.02065-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims J. R., Zimmer J. A., Evans C. D., Lu M., Ardayfio P., Sparks J., et al. (2022). Donanemab in early symptomatic alzheimer disease: the TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA 330 (6), 512–527. 10.1001/jama.2023.13239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims R., Hill M., Williams J. (2020). The multiplex model of the genetics of Alzheimer's disease. Nat. Neurosci. 23 (3), 311–322. 10.1038/s41593-020-0599-5 [DOI] [PubMed] [Google Scholar]
- Sirkis D. W., Bonham L. W., Johnson T. P., La Joie R., Yokoyama J. S. (2022). Dissecting the clinical heterogeneity of early-onset Alzheimer’s disease. Mol. Psychiatry. 27 (6), 2674–2688. 10.1038/s41380-022-01531-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjogren T., Sjogren H., Lindgren A. G. (1952). Morbus Alzheimer and morbus Pick; a genetic, clinical and patho-anatomical study. Acta. Psychiatr. Neurol. Scand. Suppl. 82, 1–152. PMID: 13171126. [PubMed] [Google Scholar]
- Skillbäck T., Rosén C., Asztely F., Mattsson N., Blennow K., Zetterberg H. (2014). Diagnostic performance of cerebrospinal fluid total tau and phosphorylated tau in Creutzfeldt-Jakob disease: results from the Swedish Mortality Registry. JAMA Neurol. 71 (4), 476–483. 10.1001/jamaneurol.2013.6455 [DOI] [PubMed] [Google Scholar]
- Skinner J., Carvalho J. O., Potter G. G., Thames A., Zelinski E., Crane P. K., et al. (2012). The Alzheimer's Disease Assessment Scale-Cognitive-Plus (ADAS-Cog-Plus): an expansion of the ADAS-Cog to improve responsiveness in MCI. Brain Imaging Behav. 6 (4), 489–501. 10.1007/s11682-012-9166-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoog I., Andreasson L. A., Landahl S., Lernfelt B. (1998). A population-based study on blood pressure and brain atrophy in 85-year-olds. Hypertension 32 (3), 404–409. 10.1161/01.hyp.32.3.404 [DOI] [PubMed] [Google Scholar]
- Small G. W., Mazziotta J. C., Collins M. T., Baxter L. R., Phelps M. E., Mandelkern M. A., et al. (1995). Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA 273 (12), 942–947. PMID: 7884953. 10.1001/jama.273.12.942 [DOI] [PubMed] [Google Scholar]
- Soeda Y., Takashima A. (2020). New insights into drug discovery targeting tau protein. Front. Mol. Neurosci. 13, 590896. 10.3389/fnmol.2020.590896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R. A., Donohue M. C., Raman R., Rafii M. S., Johnson K., Masters C. L., et al. (2023). Trial of solanezumab in preclinical Alzheimer's disease. N. Engl. J. Med. 389 (12), 1096–1107. 10.1056/NEJMoa2305032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R. A., Jack C. R. Jr., Black S. E., Frosch M. P., Greenberg S. M., Hyman B. T., et al. (2011). Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer's Association Research Roundtable Workgroup. Alzheimers Dement. 7 (4), 367–385. 10.1016/j.jalz.2011.05.2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R. A., Rentz D. M., Johnson K. A., Karlawish J., Donohue M., Salmon D. P., et al. (2014). The A4 study: stopping AD before symptoms begin? Sci. Transl. Med. 6 (228), 228fs13. 10.1126/scitranslmed.3007941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stampfer M. J. (2006). Cardiovascular disease and Alzheimer's disease: common links. J. Intern. Med. 260 (3), 211–223. 10.1111/j.1365-2796.2006.01687.x [DOI] [PubMed] [Google Scholar]
- Steen E., Terry B. M., Rivera E. J., Cannon J. L., Neely T. R., Tavares R., et al. (2005). Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease--is this type 3 diabetes? J. Alzheimers Dis. 7 (1), 63–80. 10.3233/jad-2005-7107 [DOI] [PubMed] [Google Scholar]
- Stern A. M., Sperling R. A. (2023). Tangles, not TANGO: targeting tau aggregates. Nat. Aging. 3, 1472–1473. 10.1038/s43587-023-00526-7 [DOI] [PubMed] [Google Scholar]
- Sugimoto H., Yamanishi Y., Iimura Y., Kawakami Y. (2000). Donepezil hydrochloride (E2020) and other acetylcholinesterase inhibitors. Curr. Med. Chem. 7 (3), 303–339. 10.2174/0929867003375191 [DOI] [PubMed] [Google Scholar]
- Sumi T., Harada K. (2020). Mechanism underlying hippocampal long-term potentiation and depression based on competition between endocytosis and exocytosis of AMPA receptors. Sci. Rep. 10 (1), 14711. 10.1038/s41598-020-71528-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B., Tang N., Peluso M. J., Iyer N. S., Torres L., Donatelli J. L., et al. (2021). Characterization and biomarker analyses of post-COVID-19 complications and neurological manifestations. Cells 10 (2), 386. 10.3390/cells10020386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow R. H., Burns J. M., Khan S. M. (2010). The Alzheimer's disease mitochondrial cascade hypothesis. J. Alzheimers Dis. 20 (Suppl. 2), S265–S279. 10.3233/JAD-2010-100339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szekely C. A., Thorne J. E., Zandi P. P., Ek M., Messias E., Breitner J. C., et al. (2004). Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer's disease: a systematic review. Neuroepidemiology 23 (4), 159–169. 10.1159/000078501 [DOI] [PubMed] [Google Scholar]
- Tan F. H., Putoczki T. L., Stylli S. S., Luwor R. B. (2019). Ponatinib: a novel multi-tyrosine kinase inhibitor against human malignancies. Onco. Targets Ther. 12, 635–645. 10.2147/OTT.S189391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapiola T., Alafuzoff I., Herukka S. K., Parkkinen L., Hartikainen P., Soininen H., et al. (2009). Cerebrospinal fluid {beta}-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch. Neurol. 66 (3), 382–389. 10.1001/archneurol.2008.596 [DOI] [PubMed] [Google Scholar]
- Tariot P. N., Farlow M. R., Grossberg G. T., Graham S. M., McDonald S., Gergel I., et al. (2004). Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA 291 (3), 317–324. 10.1001/jama.291.3.317 [DOI] [PubMed] [Google Scholar]
- Teich A. F., Arancio O. (2012). Is the amyloid hypothesis of Alzheimer's disease therapeutically relevant? Biochem. J. 446 (2), 165–177. 10.1042/BJ20120653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng E., Manser P. T., Pickthorn K., Brunstein F., Blendstrup M., Sanabria Bohorquez S., et al. (2022). Safety and efficacy of semorinemab in individuals with prodromal to mild alzheimer disease: a randomized clinical trial. JAMA Neurol. 79 (8), 758–767. 10.1001/jamaneurol.2022.1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teunissen C. E., Chiu M. J., Yang C. C., Yang S. Y., Scheltens P., Zetterberg H., et al. (2018). Plasma amyloid-β (Aβ42) correlates with cerebrospinal fluid Aβ42 in Alzheimer's disease. J. Alzheimers Dis. 62 (4), 1857–1863. 10.3233/JAD-170784 [DOI] [PubMed] [Google Scholar]
- Thambisetty M., Beason-Held L., An Y., Kraut M. A., Resnick S. M. (2010). APOE epsilon4 genotype and longitudinal changes in cerebral blood flow in normal aging. Arch. Neurol. 67 (1), 93–98. 10.1001/archneurol.2009.913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S. J., Grossberg G. T. (2009). Memantine: a review of studies into its safety and efficacy in treating Alzheimer's disease and other dementias. Clin. Interv. Aging 4, 367–377. 10.2147/cia.s6666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas T., Thomas G., McLendon C., Sutton T., Mullan M. (1996). beta-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature 380 (6570), 168–171. 10.1038/380168a0 [DOI] [PubMed] [Google Scholar]
- Tian J., Guo S., Chen H., Peng J. J., Jia M. M., Li N. S., et al. (2018). Combination of emricasan with ponatinib synergistically reduces ischemia/reperfusion injury in rat brain through simultaneous prevention of apoptosis and necroptosis. Transl. Stroke Res. 9 (4), 382–392. 10.1007/s12975-017-0581-z [DOI] [PubMed] [Google Scholar]
- Tolar M., Abushakra S., Hey J. A., Porsteinsson A., Sabbagh M. (2020). Aducanumab, gantenerumab, BAN2401, and ALZ-801-the first wave of amyloid-targeting drugs for Alzheimer's disease with potential for near term approval. Alzheimers Res. Ther. 12 (1), 95. 10.1186/s13195-020-00663-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse K. H., Herrup K. (2017). Re-imagining Alzheimer's disease - the diminishing importance of amyloid and a glimpse of what lies ahead. J. Neurochem. 143 (4), 432–444. 10.1111/jnc.14079 [DOI] [PubMed] [Google Scholar]
- Tsoi K. K., Chan J. Y., Hirai H. W., Wong S. Y., Kwok T. C. (2015). Cognitive tests to detect dementia: a systematic review and meta-analysis. JAMA Intern. Med. 175 (9), 1450–1458. 10.1001/jamainternmed.2015.2152 [DOI] [PubMed] [Google Scholar]
- Turner B. E., Steinberg J. R., Weeks B. T., Rodriguez F., Cullen M. R. (2022). Race/ethnicity reporting and representation in US clinical trials: a cohort study. Lancet Reg. Health. 11, 100252. 10.1016/j.lana.2022.100252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uenaka T., Satake W., Cha P. C., Hayakawa H., Baba K., Jiang S., et al. (2018). In silico drug screening by using genome-wide association study data repurposed dabrafenib, an anti-melanoma drug, for Parkinson's disease. Hum. Mol. Genet. 27 (22), 3974–3985. 10.1093/hmg/ddy279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungar L., Altmann A., Greicius M. D. (2014). Apolipoprotein E, gender, and Alzheimer's disease: an overlooked, but potent and promising interaction. Brain Imaging Behav. 8 (2), 262–273. 10.1007/s11682-013-9272-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dyck C. H., Swanson C. J., Aisen P., Bateman R. J., Chen C., Gee M., et al. (2023). Lecanemab in early Alzheimer's disease. N. Engl. J. Med. 388 (1), 9–21. 10.1056/nejmoa2212948 [DOI] [PubMed] [Google Scholar]
- Vargas-Caballero M., Robinson H. P. (2004). Fast and slow voltage-dependent dynamics of magnesium block in the NMDA receptor: the asymmetric trapping block model. J. Neurosci. 24 (27), 6171–6180. 10.1523/JNEUROSCI.1380-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecchio F. L., Bisceglia P., Imbimbo B. P., Lozupone M., Latino R. R., Resta E., et al. (2022). Are apolipoprotein E fragments a promising new therapeutic target for Alzheimer's disease? Ther. Adv. Chronic Dis. 13, 20406223221081605. 10.1177/20406223221081605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataramani V., Winkler F. (2022). Cognitive deficits in long covid-19. N. Engl. J. Med. 387 (19), 1813–1815. 10.1056/NEJMcibr2210069 [DOI] [PubMed] [Google Scholar]
- Veroniki A. A., Ashoor H. M., Rios P., Seitidis G., Stewart L., Clarke M., et al. (2022). Comparative safety and efficacy of cognitive enhancers for Alzheimer's dementia: a systematic review with individual patient data network meta-analysis. BMJ Open 12 (4), e053012. 10.1136/bmjopen-2021-053012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitek M. P., Bhattacharya K., Glendening J. M., Stopa E., Vlassara H., Bucala R., et al. (1994). Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc. Natl. Acad. Sci. U. S. A. 91 (11), 4766–4770. 10.1073/pnas.91.11.4766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagstaff L. R., Mitton M. W., Arvik B. M., Doraiswamy P. M. (2003). Statin-associated memory loss: analysis of 60 case reports and review of the literature. Pharmacotherapy 23 (7), 871–880. 10.1592/phco.23.7.871.32720 [DOI] [PubMed] [Google Scholar]
- Wang C., Zong S., Cui X., Wang X., Wu S., Wang L., et al. (2023). The effects of microglia-associated neuroinflammation on Alzheimer's disease. Front. Immunol. 22 (14), 1117172. 10.3389/fimmu.2023.1117172 [DOI] [Google Scholar]
- Wang D., Noda Y., Zhou Y., Mouri A., Mizoguchi H., Nitta A., et al. (2007). The allosteric potentiation of nicotinic acetylcholine receptors by galantamine ameliorates the cognitive dysfunction in beta amyloid25-35 i.c.v.-injected mice: involvement of dopaminergic systems. Neuropsychopharmacology 32 (6), 1261–1271. 10.1038/sj.npp.1301256 [DOI] [PubMed] [Google Scholar]
- Wang J. H., Wu Y. J., Tee B. L., Lo R. Y. (2018). Medical comorbidity in Alzheimer's disease: a nested case-control study. J. Alzheimers Dis. 63 (2), 773–781. 10.3233/JAD-170786 [DOI] [PubMed] [Google Scholar]
- Wang R., Reddy P. H. (2017). Role of glutamate and NMDA receptors in Alzheimer's disease. J. Alzheimers Dis. 57 (4), 1041–1048. 10.3233/JAD-160763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Sun G., Feng T., Zhang J., Huang X., Wang T., et al. (2019). Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer's disease progression. Cell Res. 29 (10), 787–803. 10.1038/s41422-019-0216-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Wang B., Yang L., Guo Q., Aithmitti N., Songyang Z., et al. (2009). Presynaptic and postsynaptic interaction of the amyloid precursor protein promotes peripheral and central synaptogenesis. J. Neurosci. 29 (35), 10788–10801. 10.1523/JNEUROSCI.2132-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins P. B., Zimmerman H. J., Knapp M. J., Gracon S. I., Lewis K. W. (1994). Hepatotoxic effects of tacrine administration in patients with Alzheimer's disease. JAMA 271 (13), 992–998. PMID: 8139084. 10.1001/jama.271.13.992 [DOI] [PubMed] [Google Scholar]
- Watt J. A., Isaranuwatchai W., Grossman L., Straus S. E. (2023). Disease-modifying drugs for Alzheimer disease: implications for people in Canada. CMAJ 195 (42), E1446–E1448. 10.1503/cmaj.230595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehling M. (2014). Non-steroidal anti-inflammatory drug use in chronic pain conditions with special emphasis on the elderly and patients with relevant comorbidities: management and mitigation of risks and adverse effects. Eur. J. Clin. Pharmacol. 70 (10), 1159–1172. 10.1007/s00228-014-1734-6 [DOI] [PubMed] [Google Scholar]
- Wellington H., Paterson R. W., Portelius E., Törnqvist U., Magdalinou N., Fox N. C., et al. (2016). Increased CSF neurogranin concentration is specific to Alzheimer disease. Neurology 86 (9), 829–835. 10.1212/WNL.0000000000002423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng F. L., He L. (2021). Disrupted ubiquitin proteasome system underlying tau accumulation in Alzheimer's disease. Neurobiol. Aging. 99, 79–85. 10.1016/j.neurobiolaging.2020.11.015 [DOI] [PubMed] [Google Scholar]
- Wessels A. M., Tariot P. N., Zimmer J. A., Selzler K. J., Bragg S. M., Andersen S. W., et al. (2020). Efficacy and safety of lanabecestat for treatment of early and mild alzheimer disease: the AMARANTH and DAYBREAK-ALZ randomized clinical trials. JAMA Neurol. 77 (2), 199–209. 10.1001/jamaneurol.2019.3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse P. J., Price D. L., Struble R. G., Clark A. W., Coyle J. T., Delon M. R. (1982). Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science 215 (4537), 1237–1239. 10.1126/science.7058341 [DOI] [PubMed] [Google Scholar]
- Whitehouse P. J., Saini V. (2022). Making the case for the accelerated withdrawal of aducanumab. J. Alzheimers Dis. 87 (3), 999–1001. 10.3233/JAD-220264 [DOI] [PubMed] [Google Scholar]
- Widera E. W., Brangman S. A., Chin N. A. (2023). Ushering in a new era of alzheimer disease therapy. JAMA 330 (6), 503–504. 10.1001/jama.2023.11701 [DOI] [PubMed] [Google Scholar]
- Withington C. G., Turner R. S. (2022). Amyloid-related imaging abnormalities with anti-amyloid antibodies for the treatment of dementia due to Alzheimer's disease. Front. Neurol. 13, 862369. 10.3389/fneur.2022.862369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtunik-Kulesza K., Rudkowska M., Orzeł-Sajdłowska A. (2023). Aducanumab-hope or disappointment for Alzheimer's disease. Int. J. Mol. Sci. 24 (5), 4367. 10.3390/ijms24054367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak M. A., Frost A. L., Preston C. M., Itzhaki R. F. (2011). Antivirals reduce the formation of key Alzheimer's disease molecules in cell cultures acutely infected with herpes simplex virus type 1. PLoS One 6 (10), e25152. 10.1371/journal.pone.0025152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak M. A., Itzhaki R. F., Shipley S. J., Dobson C. B. (2007). Herpes simplex virus infection causes cellular beta-amyloid accumulation and secretase upregulation. Neurosci. Lett. 429 (2-3), 95–100. 10.1016/j.neulet.2007.09.077 [DOI] [PubMed] [Google Scholar]
- Wu X., Yang H., He S., Xia T., Chen D., Zhou Y., et al. (2022). Adult vaccination as a protective factor for dementia: a meta-analysis and systematic review of population-based observational studies. Front. Immunol. 13, 872542. 10.3389/fimmu.2022.872542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S., Chan P., Wang T., Hong Z., Wang S., Kuang W., et al. (2021). A 36-week multicenter, randomized, double-blind, placebo-controlled, parallel-group, phase 3 clinical trial of sodium oligomannate for mild-to-moderate Alzheimer's dementia. Alzheimers Res. Ther. 13 (1), 62. 10.1186/s13195-021-00795-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie B., Shi X., Xing Y., Tang Y. (2020). Association between atherosclerosis and Alzheimer's disease: a systematic review and meta-analysis. Brain Behav. 10 (4), e01601. 10.1002/brb3.1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A. C., Kern F., Losada P. M., Agam M. R., Maat C. A., Schmartz G. P., et al. (2021). Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature 595 (7868), 565–571. 10.1038/s41586-021-03710-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates A. G., Anthony D. C., Ruitenberg M. J., Couch Y. (2019). Systemic immune response to traumatic CNS injuries-are extracellular vesicles the missing link? Front. Immunol. 10, 2723. 10.3389/fimmu.2019.02723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandi P. P., Sparks D. L., Khachaturian A. S., Tschanz J., Norton M., Steinberg M., et al. (2005). Do statins reduce risk of incident dementia and Alzheimer disease? The Cache County Study. Arch. Gen. Psychiatry 62 (2), 217–224. 10.1001/archpsyc.62.2.217 [DOI] [PubMed] [Google Scholar]
- Zeevalk G. D., Nicklas W. J. (1992). Evidence that the loss of the voltage-dependent Mg2+ block at the N-methyl-D-aspartate receptor underlies receptor activation during inhibition of neuronal metabolism. J. Neurochem. 59 (4), 1211–1220. 10.1111/j.1471-4159.1992.tb08430.x [DOI] [PubMed] [Google Scholar]
- Zhang X., Li Y., Xu H., Zhang Y. W. (2014). The γ-secretase complex: from structure to function. Front. Cell Neurosci. 11 (8), 427. 10.3389/fncel.2014.00427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. X., Tian Y., Wang Z. T., Ma Y. H., Tan L., Yu J. T. (2021). The epidemiology of Alzheimer's disease modifiable risk factors and prevention. J. Prev. Alzheimers Dis. 8 (3), 313–321. 10.14283/jpad.2021.15 [DOI] [PubMed] [Google Scholar]
- Zheng J., Xu M., Walker V., Yuan J., Korologou-Linden R., Robinson J., et al. (2022). Evaluating the efficacy and mechanism of metformin targets on reducing Alzheimer's disease risk in the general population: a Mendelian randomisation study. Diabetologia 65 (10), 1664–1675. 10.1007/s00125-022-05743-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Shi Q., Zhang X., Gu L., Li J., Quan S., et al. (2023). ApoE4-mediated blood-brain barrier damage in Alzheimer's disease: progress and prospects. Brain Res. Bull. 199, 110670. 10.1016/j.brainresbull.2023.110670 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.