

Abstract
Genome editing with CRISPR RNA-guided endonucleases generates DNA breaks that are resolved by cellular DNA repair machinery. However, analogous methods to manipulate RNA remain unavailable. Here, we show that site-specific RNA breaks generated with type III CRISPR complexes are repaired in human cells, and this repair can be used for programmable deletions in human transcripts to restore gene function. Collectively, this work establishes a technology for precise RNA manipulation with potential therapeutic applications.
One-Sentence Summary:
CRISPR-guided RNA breaks are repaired in human cells, and this RNA repair can be used for programmable editing of human transcriptomes.
CRISPR-guided endonucleases have enabled programable DNA cleavage and the development of new therapeutics (1, 2). The first generation of CRISPR genome editing used Cas9 nuclease to make double-stranded DNA breaks that are repaired by the cell, leading to site-specific edits (3). These technologies have enriched our mechanistic understanding of DNA repair, and new insights into repair are used to improve methods for genome editing (4).
While DNA editing has the potential to cure genetic diseases, it can result in unintended changes to the genome (5–8) and toxic cellular stress responses (9, 10). In contrast, RNA editing can alter the cellular program without changing the DNA. However, options for editing RNA are limited. Cas13, a type VI CRISPR-guided endoribonuclease, has been used for RNA knockdown, but target recognition also activates collateral nuclease activity that degrades non-target RNAs (11, 12). Nuclease-inactivated mutants of Cas13 (dCas13) have been used for exon skipping (13), trans-splicing of synthetic RNA payloads (14), or sequence-specific delivery of base-editing enzymes for adenosine-to-inosine or cytosine-to-uracil conversions (15, 16). However, programmable deletions in RNA, analogous to those introduced by DNA editors, have not been described.
To address the need for versatile and facile RNA manipulation, we have developed a technology for sequence-specific RNA editing in living cells. This technology uses type III-A CRISPR complexes for programmable cleavage of target RNA. Unlike Cas13, type III complexes exclusively cleave the target RNA in six nucleotide increments (17, 18). We repurpose this cleavage activity to excise segments from human transcripts and show that resulting RNA fragments are repaired, which produces programmable deletions in target RNA. We apply this method to remove a non-sense mutation common in cystic fibrosis patients, which rescues protein expression and demonstrates the therapeutic potential of this technique. Overall, this work establishes a technology for RNA manipulation in living cells and provides a model for discovering new RNA repair pathways.
Programmable deletions in human transcripts
The programmable RNase activity of type III-A complexes has been used for RNA knockdown (19–21), but we hypothesized that cells repair CRISPR-guided RNA breaks, which could be leveraged for RNA editing (Fig. 1A). To test this hypothesis, we targeted two human transcripts, PPIB and PARK7, that are highly expressed in 293T cells (fig. S1A). Mutations in PPIB are associated with severe osteogenesis imperfecta, while mutations in PARK7 are linked to Parkinson’s disease (22). We transfected 293T cells with plasmids that express type III CRISPR-associated proteins from Streptococcus thermophilus (Csm2 – Csm5, Cas10, and Cas6) (19) fused to a nuclear localization signal (NLS) and RNA guides complementary to PPIB or PARK7 mRNAs (SthCsm complex; fig. S1).
Fig. 1. Programmable deletions of RNA with RNA-guided type III-A CRISPR complexes.
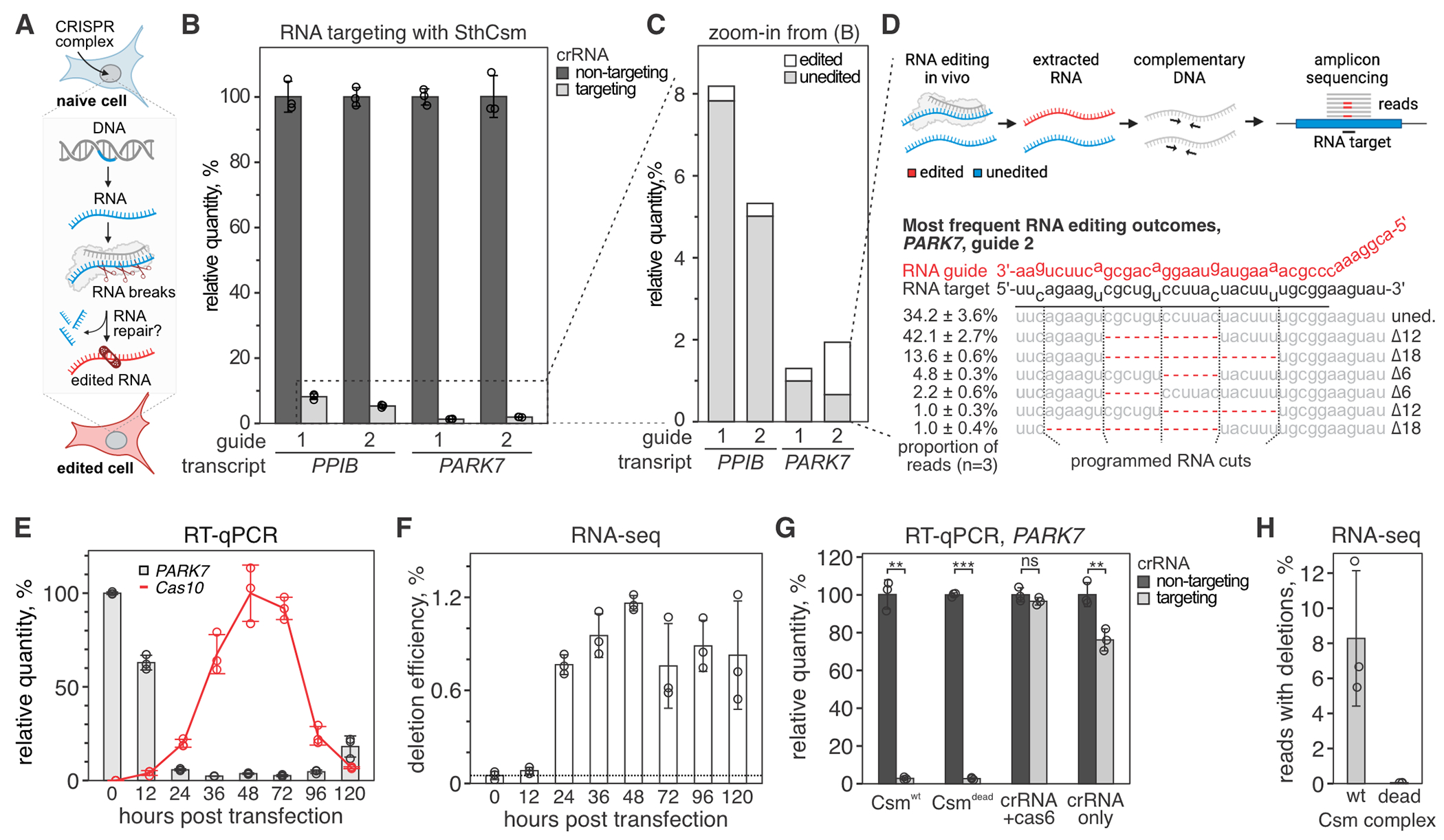
(A) Diagram of RNA editing in eukaryotic cells through sequence-specific RNA cleavage and RNA repair. (B) Human cells (293T) were transfected with plasmids encoding for NLS-tagged type III CRISPR complex of Streptococcus thermophilus (SthCsm) and RNA guides targeting PPIB or PARK7 messenger RNAs. Target transcripts were quantified with RT-qPCR, and the qPCR signal was normalized to ACTB and a non-targeting guide RNA control. Data is shown as mean ± SD of three biological replicates. (C) Zoom-in from panel B. Deep sequencing was used to quantify the proportion of signal that is derived from edited RNA. (D) Top: schematics of deep sequencing approach used to quantify RNA editing. Bottom: top five most frequent RNA editing outcomes in PARK7 transcript (guide 2). Dotted lines indicate the positions of RNA breaks by the SthCsm complex. Data is shown as mean ± SD. (E) Kinetics of PARK7 mRNA knockdown (guide 2) vs. NLS-tagged SthCsm complex expression. PARK7 qPCR signal was normalized to ACTB and 0 h time point. Cas10 expression was normalized to ACTB and maximum expression level at 48 h. Data is shown as mean ± SD of three biological replicates. (F) PARK7 qPCR products in (E) were sequenced, and deletion efficiency was calculated as [relative quantity] × [fraction of reads with deletions]. (G) PARK7 knockdown efficiencies with the “wildtype” NLS-tagged SthCsm complex (Csmwt), catalytically inactive NLS-tagged SthCsm complex (Csmdead), crRNA expressed only with Cas6 gene (no csm genes), and cRNA alone were measured using RT-qPCR. Welch’s t-test was used to compare samples expressing targeting and non-targeting crRNA. ** - p < 0.01, *** - p < 0.001, ns – non-significant. (H) RT-qPCR products in (G) were sequenced, and a fraction of reads with programmable deletions was quantified.
To quantify target transcripts, we designed RT-qPCR primers that flank target regions (fig. S1). Expression of the NLS-tagged SthCsm complex results in >90% reduction of target transcript levels compared to cells transfected with a non-complementary guide RNA (Fig. 1B), which is similar to the knockdown efficiencies reported for other guides (19). We hypothesized that the remaining target RNA was either not cleaved, cleaved and repaired without modification, or cleaved and repaired with indels at the target site (Fig. 1C). To test this hypothesis, we deep-sequenced PPIB and PARK7 amplicons. We detected 6, 12, 18, or 24 nucleotide deletions in each of the two RNA targets, which are consistent with the six-nucleotide cleavage pattern of the SthCsm complex and not seen in controls transfected with a non-targeting guide (Fig. 1D). SthCsm-dependent deletions were detected in 4.4 ± 0.6% (guide 1) and 5.9 ± 0.6% (guide 2) of PPIB amplicons, and 23.9 ± 2.3% (guide 1) and 65.8 ± 3.6% (guide 2) of PARK7 amplicons (Fig. 1D, fig. S1). These data demonstrate that most of the target RNA is degraded, but some of the RNA is repaired in human cells.
To measure the kinetics of RNA repair, we extracted RNA from 293T cells at various time points after transfection with SthCsm plasmid targeting PPIB or PARK7 transcripts. RT-qPCR and amplicon-seq demonstrated that transcripts with RNA deletions were detectable at 12 h post-transfection, accumulated over time, peaked at 48 h, and decreased with decreasing expression of the SthCsm (Fig. 1, E and F; fig. S2). The loss of SthCsm expression is likely due to plasmid loss with cell division. The RNA knockdown plateaued at 36 h, but the quantity of edited transcripts rose until 48 h, suggesting an accumulation of edited transcripts that cannot be re-cleaved. The decrease of edited RNA after 48 h suggests that the accumulation is capped by the RNA half-life, which was estimated as 11.4 h for Ppib and 7.8 h for Park7 mouse transcripts (23).
To confirm that RNA deletions in PPIB and PARK7 are produced by RNA cleavage, we used an inactive SthCsm complex with D33A amino acid substitution in the Csm3 ribonuclease (18). Recent work demonstrated robust knockdown of nuclear transcripts (i.e., XIST, MALAT1, and NEAT1) with the SthCsm complex. However, no knockdown is detected with inactive SthCsm complexes (19). To our surprise, targeting transcripts that are exported from the nucleus (i.e., PPIB and PARK7) with catalytically inactive NLS-tagged Csm complex (NLS-Csmdead) resulted in a robust knockdown that was identical to the knockdown with nuclease-active Csm complexes (NLS-Csmwt; P = 0.88) (Fig. 1G; fig. S2). Expression of the crRNA alone or crRNA together with crRNA-processing protein Cas6 resulted in far less knockdown, demonstrating that the Csm proteins are necessary for efficient knockdown. While knockdown efficiencies are similar for NLS-Csmdead and NLS-Csmwt, only the nuclease active complex produced programmable RNA excision (Fig. 1H). Sequencing target RNA after knockdown identified the characteristic deletions (increments of six) only in cells transfected with the NLS-Csmwt plasmid (fig. S2).
RTCB ligase repairs type III CRISPR-mediated RNA breaks
RNA cleavage by type III CRISPR complexes leaves a 2’,3’-cyclic phosphate (2’,3’>P) and a 5’-hydroxyl (5’-OH) at each cut site (17). Mammalian RNA 2′,3′-cyclic phosphate and 5′-OH ligase (RTCB) joins 2′,3′>P and 5′-OH termini when introns are excised during tRNA splicing and non-conventional splicing of XBP1 mRNA in the unfolded protein response (24–27) (Fig. 2A). To test if RTCB ligase repairs transcripts cleaved by SthCsm in human cells, we used Cas9 nuclease to knockdown RTCB in 293T cells (fig. S3, see Supplementary Text for details), and targeted PARK7 transcript in cells with or without the RTCB depletion (Fig. 2, B and C). In wildtype 293T cells, 2.3 ± 0.1% of the target RNA was detected after transfection with SthCsm plasmid, while target RNA was ~1.5-fold lower (1.5 ± 0.3%; P = 0.029) in RTCB-depleted cells.
Fig. 2. RTCB ligase repairs RNA cleaved by the type III CRISPR complex.
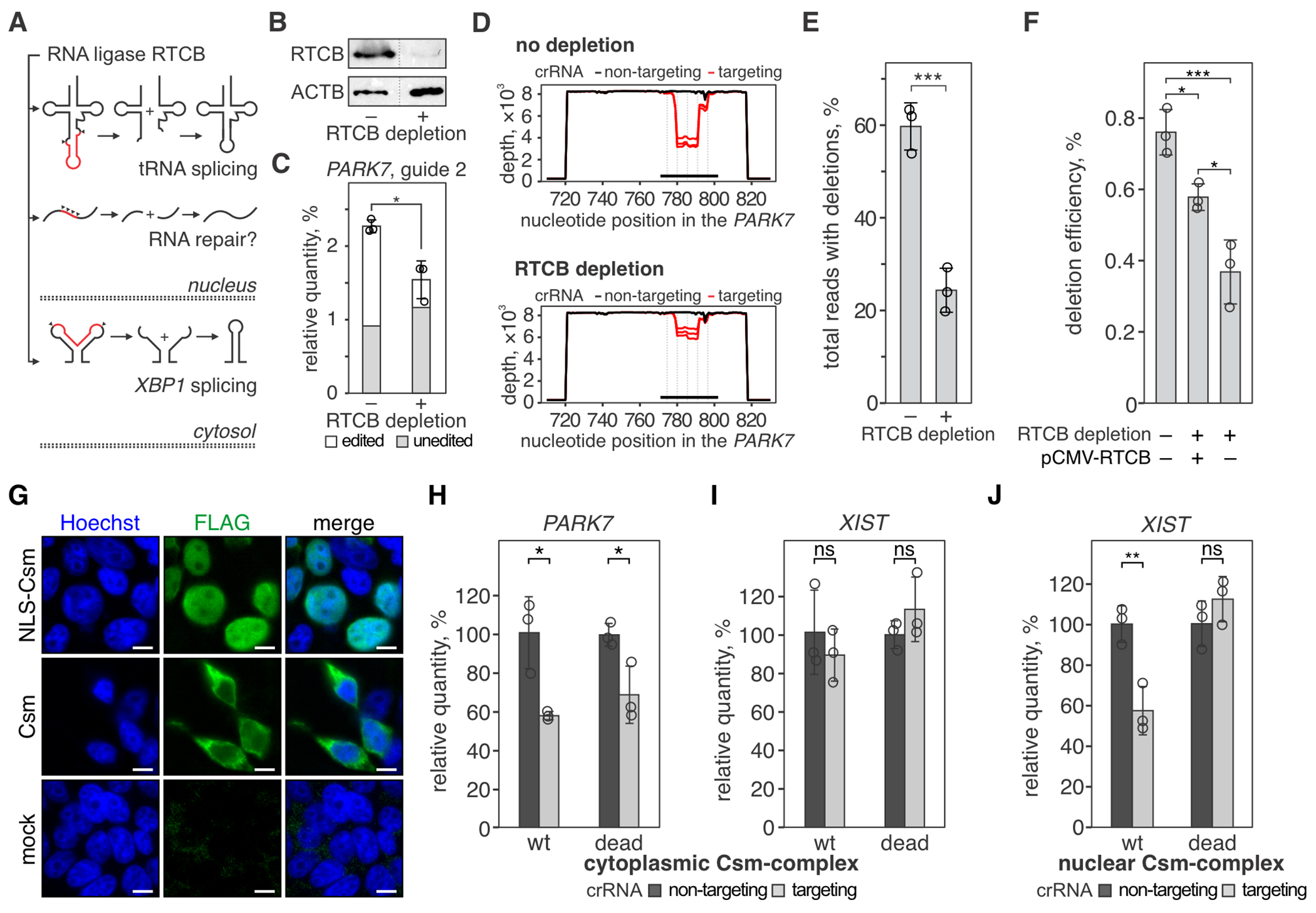
(A) Human RNA ligase RTCB joins RNA ends that are produced in tRNA splicing (top) and non-canonical XBP1 mRNA splicing (bottom) during the unfolded protein response. We hypothesized that the ligase activity of RTCB is involved in the RNA repair of CRISPR-guided RNA breaks (middle). (B) Western blot with anti-RTCB or anti-ACTB (loading control) antibodies was performed with lysates of 293T cells with (+) or without (−) RTCB depletion. See the uncropped images in fig. S3. (C) The PARK7 transcript was targeted with SthCsm (guide 2) in 293T cells with (+) or without (−) RTCB depletion. The PARK7 transcript was quantified with RT-qPCR and normalized to ACTB and non-targeting guide RNA. Data are shown as the mean ± standard deviation of three biological replicates. *P < 0.05, ***P < 0.001; one-way analysis of variance (ANOVA) with Tukey HSD post-hoc comparisons. (D) qPCR products in (C) were sequenced, and resulting reads were aligned to the reference sequence of the PARK7 transcript (NM_007262, GenBank). Graphs show sequencing depth (y-axes) at the amplified region of the transcript (x-axes). Every line shows a biological replicate (n = 3). The horizontal black bar indicates a region complementary to the guide RNA of the SthCsm complex. Vertical dotted lines mark predicted positions of RNA breaks. (E) Quantification of deletions in the target region of the PARK7 transcript. Data is shown as the mean ± standard deviation of three biological replicates. Welch’s t-test was used to compare mean values. *** p < 0.001. (F) RTCB deficient cells were transfected with a plasmid expressing RTCB (pCMV-RTCB), and the efficiency of CRISPR RNA-guided programmable deletions in PARK7 was quantified as in panels C-E. * p < 0.05; OneWelch’s t-test. (G) Immunostaining of cells expressing Flag-tagged Csm complex with (NLS-Csm) or without (Csm) NLS-tag. Scale bars are 10 μm. (H-J) Knockdown of PARK7 and XIST transcripts with cytoplasmic SthCsm (no NLS) (H, I) and knockdown of XIST with nuclear Csm (NLS-tagged) (J) was quantified with RT-qPCR and normalized to ACTB and non-targeting control. “wt” – nuclease-active SthCsm, “dead”– catalytically inactivated SthCsm (Csm3D33A mutation). *P < 0.05, ns – non-significant; Welch’s t-test. Data is shown as mean ± SD (n = 3).
Deep-sequencing of PARK7 amplicons showed that the proportion of reads with deletions is significantly decreased in cells with depleted RTCB (Fig. 2D). In samples with wildtype levels of RTCB, deletions in target sequence were detected in 59.7 ± 5.1% of the reads, while the frequency of these deletions was 24.4 ± 4.8% (P < 0.001) in cells with depleted RTCB (Fig. 2E). Complementation of RTCB with a plasmid encoding RTCB cDNA increased the abundance of ligated PARK7 transcript ~1.5-fold compared to an untransfected control (Fig. 2F; P = 0.02). While the complementation of RNA repair was not complete (~76%), RTCB activity may be affected by the transfection efficiency and non-physiological expression levels. Overall, the reduction of RNA repair with knockdown and complementation with RTCB overexpression support the role of RTCB in the repair of CRISPR-guided RNA breaks.
To determine if repair of CRISPR-guided RNA breaks happens in the nucleus or cytoplasm, we removed the NLS tags from the SthCsm complex and targeted nuclear (XIST) or exported (PPIB and PARK7) transcripts. SthCsm complexes without the NLS tag localized in the cytoplasm (Fig. 2G) and depleted PPIB (31.2 ± 6.2% knockdown, P = 0.023) (fig. S4) and PARK7 (41.9 ± 2.1% knockdown, P = 0.017) (Fig. 2H). However, RNA retained in the nucleus (i.e., XIST) was not depleted by the untagged Csm complex (P = 0.47) (Fig. 2I). Deep sequencing detected programmable deletions in 1.4 ± 0.2% of PPIB and 9.1 ± 0.9% of PARK7 reads, demonstrating that transcripts cleaved in the cytoplasm are repaired in the cytoplasm (fig. S4, C–E). Knockdown of PPIB and PARK7 with cytoplasmic Csm complex was less efficient (~30-40%) than with NLS-tagged Csm complex (>90%). However, this does not mean that RNA cleavage in the cytoplasm is less efficient. In this experiment, we extracted total cellular RNA that contained both cytoplasmic RNA and nascent RNA from the nucleus. The latter is inaccessible to the cytoplasmic Csm complex and contributed to the qPCR signal.
NLS-tagged Csm complex localized in the nucleus (Fig. 2G) and knocked down 42.4 ± 11.9% of the nuclear XIST transcript (P = 0.009; Fig. 2J). Deep sequencing of the XIST amplicons identified site-specific deletions (Fig. 3, A and B), indicating that cleaved XIST RNA is repaired in the nucleus. Collectively, these results demonstrate that RNA repair happens both in the nucleus and in the cytoplasm of human cells.
Fig. 3. Repair of concurrent RNA breaks results in large RNA excisions.
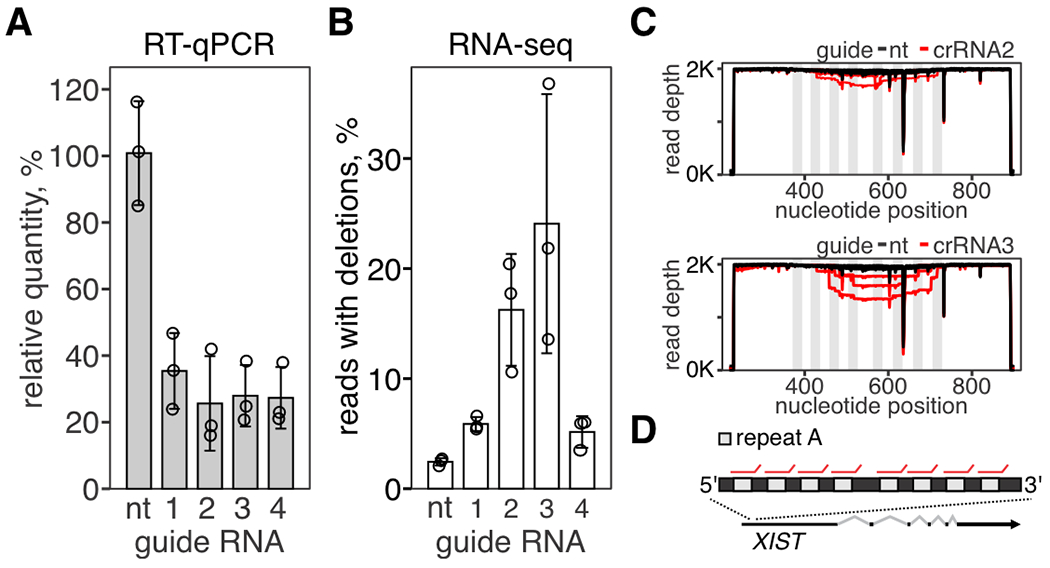
(A) Repetitive region (repeat A) in XIST transcript was targeted with SthCsm complex with four different guide RNAs. Knockdown of XIST was quantified with RT-qPCR. Data is shown as the mean of three biological replicates ± SD. B) Amplicon-seq was used to quantify programmed RNA deletions in XIST. C) Repeat A was amplified and deep-sequenced. Reads were aligned to the reference sequence (NR_001564.2, GenBank). Graphs show sequencing depth (y-axes) at the amplified region of the transcript (x-axes). Every line shows a biological replicate (n = 3). Vertical light gray rectangles indicate the position of repeats targeted by Csm complexes. D) Repeat A architecture in XIST lncRNA. Red lines show binding sites for Csm complexes.
We hypothesize that human transcripts bound by inactive SthCsm complex (~350 kDa) fail RNA quality control during nuclear export or that the bound complex blocks translation in the cytoplasm, which results in the decay of the target RNA (28). In agreement with the latter possibility, targeting PPIB and PARK7 in the cytoplasm with nuclease-inactive SthCsm (Csmdead) resulted in significant RNA knockdown (Fig. 2H, fig. S4), but targeting nuclear transcript XIST with NLS-Csmdead did not change its relative quantity (Fig. 2J, P = 0.26).
Multiple RNA breaks produce large RNA excisions
Human XIST is a long non-coding RNA that contains eight copies of a repetitive sequence (repeat A) required for XIST-mediated inactivation of the X-chromosome (29, 30). Previous work has targeted repeat A with GFP-fused nuclease-inactive SthCsm complex for live imaging of XIST RNA (19). We hypothesized that targeting repeat A in XIST with catalytically active SthCsm complexes will result in simultaneous cleavage at multiple locations, and repair of these cleavages will generate large deletions. We tested four different guide RNAs that target repeats in XIST (Fig. 3, A and B). Sequencing of amplicons that span the repetitive region identified the characteristic 6, 12, and 18 nucleotide deletions but also large deletions consistent with multiple cleavages at the repeated sequence (Fig. 3, C and D). These data indicate that simultaneous cleavages at multiple locations within the same transcript can be used for large RNA deletions.
Programmable excision of premature stop codons in RNA restores protein expression
The ClinVar database documents more than 40,000 non-sense mutations (i.e., stop codons) in the human population (22). We hypothesized that break-and-repair RNA editing can be used to delete premature stop codons and restore the expression of the encoded proteins (Fig. 4A). To test this, we developed a reporter plasmid that encodes for GFP fused to an N-terminal flag-hemagglutinin (flag-HA) epitope. Between the epitope and the gfp sequence, we inserted a stop codon to terminate translation (stop-GFP; Fig. 4B). Transfection of the stop-GFP plasmid in 293T cells produced no detectable fluorescence. A plasmid with the same sequence but no stop codon produced a robust GFP signal 48 h post-transfection (Fig. 4C). To cut out the stop codon, we designed six guides for SthCsm complex that tile across the target RNA sequence in three-nucleotide increments (Fig. 4B). Transfection of the stop-GFP plasmid together with three of the six SthCsm complexes (crRNA 3, 4, and 6) partially restored expression of the GFP (Fig. 4C; fig. S5A). To confirm that transcripts edited with SthCsm produce functional GFP, we used site-directed mutagenesis to make the same mutations in the stop-GFP reporter plasmid (fig. S5). Transfection of plasmids containing the 12 or 18 nucleotide deletions resulted in the same levels of fluorescence as transfection of the positive control plasmid (fig. S5D).
Fig. 4. Programmable excision of stop codons restores protein expression.
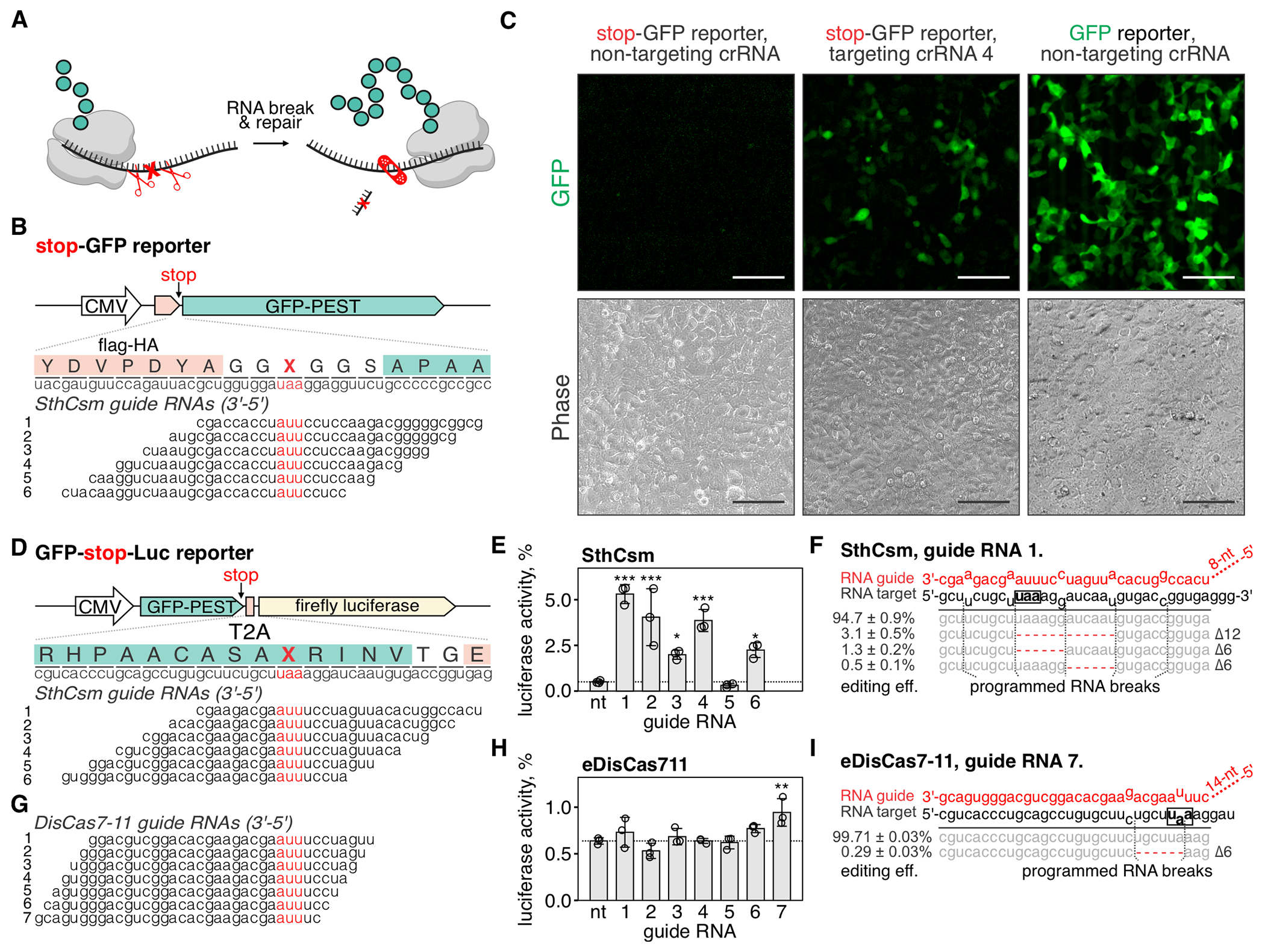
(A) Schematic representation of the proposed approach for deleting premature stop codons in human transcripts. (B) Top: schematic diagram of the stop-GFP reporter plasmid. Bottom: Six guide RNAs for SthCsm complex were designed to excise the stop codon in the gfp transcript. Underlined (red) sections of target RNA are expected to be deleted. Vertical red ticks indicate predicted sites for RNA breaks. (C) Cells were transfected with plasmids for the stop-GFP reporter and SthCsm with a non-targeting guide (left), stop-GFP reporter and SthCsm with a targeting guide (middle), or GFP reporter and SthCsm with the non-targeting guide (right). Fluorescence microscopy was used to image cells 48 h post-transfection. Scale bars – 50 μm. (D) Top: schematic diagram of the GFP-stop-Luc reporter plasmid. Bottom: Six crRNAs for SthCsm complex were designed to delete the stop codon at the 3’-end of the gfp gene. (E) Luciferase activity was measured in cell lysates 48 hours after transfection with GFP-stop-Luc and SthCsm plasmids. Luciferase activity is normalized to a control transfected with a reporter plasmid without the stop codon. Data are shown as mean ± SD of three replicates. Means were compared using one-way ANOVA, and samples with targeting guide RNAs were compared to the non-targeting control using one-tailed Dunnett’s test. *P < 0.5, **P < 0.1, ***P < 0.001. (F) Most frequent RNA editing outcomes in the sample with the most efficient rescue of luciferase activity in (B) (guide 1). Editing efficiency was quantified as mean ± SD of three biological replicates. The black box shows the stop codon that was targeted by type III CRISPR complexes. (G, H, I). The same as (D-F), but with eDisCas7-11.
To quantify the rescue of reporter expression, we developed a bicistronic plasmid that expresses GFP and firefly luciferase linked with a viral 2A peptide. At the 3’-end of the gfp gene, we inserted a stop codon, which permits GFP, but not luciferase expression (GFP-stop-Luc). To cut out the stop codon, we designed six SthCsm guide RNAs that tile across the stop codon (Fig. 4D). In cells expressing SthCsm complexes that target the stop codon, we detected luciferase activity with five of the six tested crRNAs, which ranged from 2.0 ± 0.2 % with crRNA 3 to 5.3 ± 0.5% with crRNA 1 (Fig. 4E). Sequencing of the target RNA amplicons confirmed that samples with rescued luciferase activity contain RNA deletions eliminating the stop codon (Fig. 4F; fig. S5).
Type III-E CRISPR systems encode single polyprotein effectors that process CRISPR RNA and cleave the target RNA at two sites six nucleotides apart (31, 32). These effectors are simple and compact and have the potential to produce a single editing outcome, which makes them attractive for use in RNA editing. We expressed type III-E effector from Desulfonema ishimotonii fused to the NLS tag (DisCas7-11) in 293T cells with seven guide RNAs tiled across the stop codon of the GFP-stop-Luc reporter (Fig. 4G). Only crRNA 7 resulted in a significant increase of the luciferase signal (0.9 ± 0.2%; P < 0.001), which was less efficient than SthCsm (fig. S5G). In addition to editing, we quantified knockdown efficiencies of the target RNA using RT-qPCR. The RNA knockdown efficiency with SthCsm ranged from 36.8 ± 7.2 % to 54.0 ± 2.8 %, while knockdowns with DisCas7-11 were less than half as efficient (12.2 ± 5.9% with guide 6 and 16.5 ± 11.2% with guide 7) (fig. S5I). Analysis of sequenced amplicons identified six nucleotide deletions in 0.04 ± 0.01% of the reads at the site targeted with DisCas7-11 (fig. S5H).
An enhanced version of the DisCas7-11 complex (eDisCas7-11) has been recently engineered for CRISPR-assisted trans-splicing by adding positively charged amino acids to regions of the protein that are physically close to the bound target RNA (33). Targeting the GFP-stop-Luc reporter with eDisCas7-11 resulted in a more efficient knockdown (52.3 ± 3.8% with guide 6 and 47.7 ± 7.8% with guide 7; fig. S5I), rescue of 0.9 ± 0.1% of luciferase activity (P = 0.003; Fig. 4H), and programmed RNA excisions with 0.29 ± 0.03% efficiency (Fig. 4I).
Programmed RNA excision with DisCas7-11 and eDisCas7-11 is less efficient compared to targeting with SthCsm. The low frequency of deletions with DisCas7-11 is consistent with the inefficiency of the DisCas7-11 nuclease in biochemical assays and the low frequency of two simultaneous breaks in the target RNA (31, 32). Collectively, these data demonstrate the potential of type III-E effectors to produce a single RNA editing outcome, but further engineering of the Cas7-11 nuclease will be important to repurpose this enzyme for RNA excision.
Programmable excision of non-sense mutation in the CFTR transcript
After testing fluorescent and luminescent reporters, we sought to determine the therapeutic potential of CRISPR-guided RNA excision by targeting a clinically relevant nonsense mutation (c.3846G>A; W1282X) in the CFTR mRNA. The truncated CFTRW1282X protein is partially functional, but the premature stop codon triggers nonsense-mediated decay (NMD) of the CFTRW1282X mRNA in the cytoplasm (34, 35). The decay of CFTR mRNA results in insufficient levels of CFTR protein, leading to the development of cystic fibrosis. We hypothesized that targeting the CFTRW1282X transcript with NLS-tagged SthCsm would remove the W1282X stop codon in the nucleus, and the repaired RNA would escape NMD in the cytoplasm, rescuing protein expression and potentially rescuing disease phenotype.
To screen for crRNAs that guide efficient excision of W1282X in the CFTR, we created a luciferase-based NMD-reporter plasmid building off previously published designs (Fig. 5A) (36). We screened eight crRNAs and identified one (crRNA 5) that guides the most efficient removal of the premature termination codon in the fLuc-CFTRW1282X reporter (Fig. 5, B and C; fig. S6A). The co-transfection of fLuc-CFTRW1282X and a plasmid encoding for Csm complex with crRNA 5 restored the C-terminus of CFTR protein compared to the cells that received non-targeting Csm complex (Fig. 5D, fig. S6B). In addition, expression of the Csm complex and crRNA 5 increased luciferase activity by 11.1% (P = 0.024) compared to the non-targeting control, indicating escape from the NMD (Fig. 5E).
Fig. 5. Programmable excision of a non-sense mutation in the CFTR transcript restores translation.
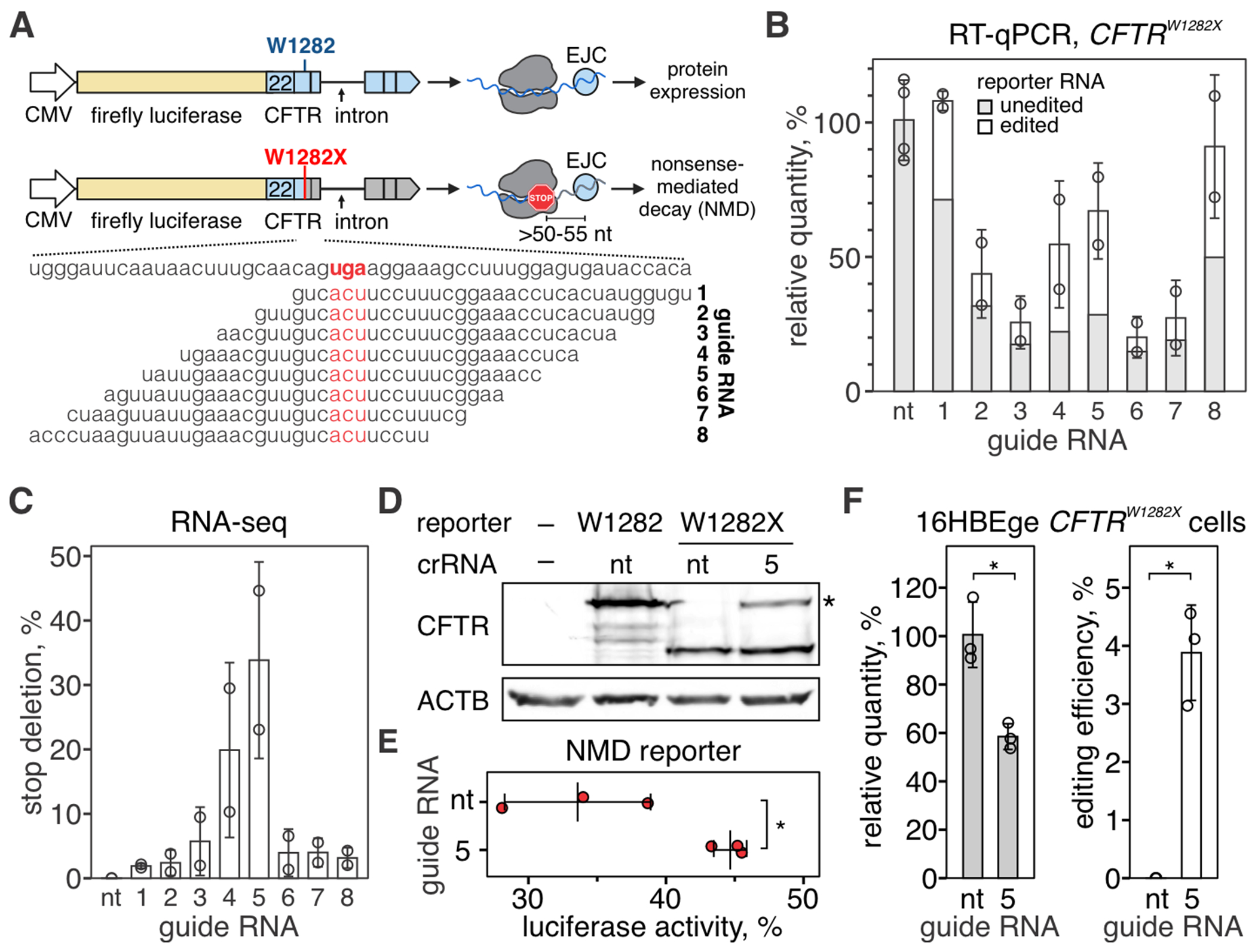
(A) Diagram of a luciferase-based reporter for CFTRW1282X mutation (fLuc-CFTR, top) and guide RNA design (bottom). Top: Firefly luciferase was genetically linked to exons 22-27 of the CFTR cDNA, and synthetic intron sequence was inserted between exons 24 and 25. See Methods for additional details. Bottom: Eight crRNAs were tiled across the mutation (W1282X) to guide the excision of the stop codon (UGA, highlighted with red). (B) RT-qPCR was used to quantify fLuc-CFTR transcript targeted with Csm complexes. Amplicons were deep-sequenced to quantify edited vs. unedited reporter RNA. See fig. S6A for sequencing depth plots. (C) Quantification of deletions that remove stop codon (W1282X). (D) Western blot with antibodies against CFTR amino acid residues 1204-1211 with lysates from 293T cells expressing fLuc-CFTR or fLuc-CFTRW1282X) and Csm complexes with non-targeting (nt) guide RNA or CFTR-targeting guide RNA 5. See fig. S6B for uncropped images. (E) Quantification of luciferase activity in 293T cells transfected with fLuc-CFTRW1282X and Csm complexes with non-targeting guide RNA or targeting guide RNA 5. Middle bar shows mean of three biological replicates (red dots). Error bars show mean ± SD. *P < 0.05, Welch’s t-test. (F) Left: RT-qPCR was used to quantify CFTR transcript in HBE16ge CFTRW1282X cells transfected with plasmids encoding for Csm complexes with non-targeting guide RNA (nt) or CFTR-targeting guide RNA 5. Right: qPCR amplicons were deep-sequenced, and deletions removing W1282X codon were quantified. See fig. S6C for depth plot. Data is shown as the mean of three biological replicates ± SD. *P < 0.05, Welch’s t-test.
Finally, we used immortalized human bronchial epithelium cells with the CFTRW1282X allele (16HBEge CFTRW1282X) to test if CRISPR-guided RNA excision can remove the W1282X codon in the endogenous CFTR transcript. RT-qPCR and amplicon-sequencing identified editing of 3.9 ± 0.8% CFTR transcripts (Fig. 5F, fig. S6C). Future studies will be needed to measure the impact of this correction on cellular physiology and determine the RNA editing levels required for therapeutic outcomes.
Discussion
Precise DNA manipulation has been enabled by breaking DNA at sequence-specific locations and repairing the resulting fragments. In a previous study, we applied the concept of break-and-repair to engineer viral RNA genomes in vitro (37). Here, we extend this approach to make RNA deletions in human cells, which is facilitated by the repair of programmable RNA breaks made using type III CRISPR complexes.
We anticipate that the deletion of toxic mutants, complemented by fractional repair, could be therapeutic. Alternatively, premature stop codons that trigger NMD result in low to no protein (e.g. CFTRW1282X mutation), and excision of premature stop codons could rescue protein expression. Other potential applications of site-specific RNA excision include editing non-coding RNAs (38), removing disease-causing trinucleotide expansions (39), or correcting intronic mutations that dysregulate splicing (40).
Repair of RNAs is understudied, and the biological significance of this process is often dismissed due to the high turnover of RNA in the cell. We hypothesize that end-modifying enzymes, additional ligases, nucleases (e.g., exosome complex), and other RNA processing factors regulate this process, and tuning RNA processing has the potential to improve the efficiency of RNA editing. We have established reporter assays for RNA repair, which enable the discovery of new RNA repair pathways and rapid screening for conditions that favor RNA repair to improve the efficiency of RNA editing.
We show that Csm-mediated knockdown of cytoplasmic transcripts does not require target RNA cleavage (Figs. 1G and 2H). However, cleavage is required for site-specific RNA deletion (Fig. 1H). This observation suggests that the efficiency of RNA repair is an underestimate because most of the target RNA is degraded by RNA quality control pathways before it can be repaired. Single-molecule studies of enzymatic activities of the Csm complex from Streptococcus thermophilus (used in this work) demonstrate that cleavage happens within seconds, but then the complex retains cleavage products for over ~80 minutes (41). These data suggest that RNA release, rather than cleavage or ligation, may be the rate-limiting step in RNA editing.
The kinetics of Csm complex dissociation is on the same timescale as mRNA transport to the cytoplasm (10-30 min for β-actin mRNA) (42), and it is possible that the Csm complex stalls ribosomes and triggers RNA decay before it releases target RNA for repair (43). These observations suggest that programmable ribonucleases that cleave and release the target quickly or cleave RNA outside of the bound sequence (i.e., Cas13) will enable more efficient editing.
Orthologs of Cas9 and Cas12 cleave complementary RNA targets, enabling programmable RNA manipulation with single-subunit effectors (44–50). RNA cleavage by Cas9 or Cas12 likely produces a 5’-phosphate and a 3’-hydroxyl, similar to DNA cleavage (51, 52). These ends are not compatible with RTCB-mediated ligation, but other cellular RNA repair pathways may enable Cas9- and Cas12-mediated RNA editing (53).
Unlike DNA editing, which often depends on adjacent sequence motifs (i.e., PAM), type III CRISPR systems only require complementarity between the RNA guide and the RNA target (18), which improves target site versatility. The frequency and distribution of RNA repair outcomes varied across RNA targets that we tested, which may suggest that local sequence context, secondary structure, RNA modifications, or other factors may influence the efficiency of target binding, target cleavage, or repair. Screens like those performed for Cas13 (54, 55) will help establish predictive models to enhance the efficiencies of RNA editing and advance this technology for therapeutic applications.
The initial efficiency of editing with Cas9 was ~3-30% for indels and ~0.5-0.7% efficiency for homology-directed repair (56). A decade later, Cas9-based technologies are precise, efficient, versatile, and clinically relevant (57). Here, we demonstrate programmable break-and-repair editing of RNA, which reveals an important aspect of RNA biology that has not been explored. We anticipate that efficiencies will improve and that these technologies will lead to new insights about the RNA damage response and enable discoveries of previously unrecognized mechanisms of RNA repair.
Supplementary Material
Acknowledgments:
Base-calling of Nanopore sequencing data was performed with the Tempest High Performance Computing System, which is maintained by University Information Technology Research Cyberinfrastructure at Montana State University. We thank Dr. Matthew Taylor for the generous use of the fluorescent microscope. Some diagrams and schematics were created with BioRender.com.
Funding:
National Institutes of Health grant 5K99AI171893-02 (to A. Nemudryi)
National Institutes of Health grant R35GM134867 (to B.W.)
National Science Foundation grant 2331325 (to B.W.)
Footnotes
Competing interests: B.W. is the founder of SurGene LLC. B.W., A. Nemudryi, and A. Nemudraia are inventors of the patent applications US 63/523,592 and US 63/534,305 pertaining to use of type III CRISPR-Cas system for sequence-specific editing of RNA filed by Montana State University.
Data and materials availability:
Raw sequencing data were deposited to the NCBI Sequence Read Archive under accession number PRJNA1099688 (58). The code used to analyze sequencing data and generate figures has been deposited to Zenodo (59). Plasmids generated in this study are available through Addgene and by request from the authors.
References and Notes
- 1.Jinek M et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gasiunas G, Barrangou R, Horvath P, Siksnys V, Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Sciences 109, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ran FA et al. Genome engineering using the CRISPR-Cas9 system. Nature Protocols 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Overbeek M et al. DNA Repair Profiling Reveals Nonrandom Outcomes at Cas9-Mediated Breaks. Mol Cell 63, 633–646 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Kosicki M, Tomberg K, Bradley A, Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nature Biotechnology 36, 765–771 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu Y et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31, 822–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsu PD et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31, 827–832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pattanayak V et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol 31, 839–843 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J, CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nature Medicine 24, 927–930 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Ihry RJ et al. p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nature Medicine 24, 939–946 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Abudayyeh OO et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353, aaf5573 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abudayyeh OO et al. RNA targeting with CRISPR-Cas13. Nature 550, 280–284 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Konermann S et al. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 173, 665–676.e614 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borrajo J et al. Programmable multi-kilobase RNA editing using CRISPR-mediated trans-splicing. bioRxiv, (2023). [Google Scholar]
- 15.Cox DBT et al. RNA editing with CRISPR-Cas13. Science 358, 1019–1027 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abudayyeh OO et al. A cytosine deaminase for programmable single-base RNA editing. Science 365, 382–386 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hale CR et al. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 139, 945–956 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamulaitis G et al. Programmable RNA shredding by the type III-A CRISPR-Cas system of Streptococcus thermophilus. Mol Cell 56, 506–517 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Colognori D, Trinidad M, Doudna JA, Precise transcript targeting by CRISPR-Csm complexes. Nature Biotechnology, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woodside WT et al. Type III-A CRISPR systems as a versatile gene knockdown technology. Rna 28, 1074–1088 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fricke T et al. Targeted RNA Knockdown by a Type III CRISPR-Cas Complex in Zebrafish. The CRISPR Journal 3, 299–313 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. https://www.ncbi.nlm.nih.gov/clinvar . [Google Scholar]
- 23.Sharova LV et al. Database for mRNA Half-Life of 19 977 Genes Obtained by DNA Microarray Analysis of Pluripotent and Differentiating Mouse Embryonic Stem Cells. DNA Research 16, 45–58 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu Y, Liang F-X, Wang X, A Synthetic Biology Approach Identifies the Mammalian UPR RNA Ligase RtcB. Molecular Cell 55, 758–770 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jurkin J et al. The mammalian tRNA ligase complex mediates splicing of XBP1 mRNA and controls antibody secretion in plasma cells. EMBO J 33, 2922–2936 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Popow J et al. HSPC117 is the essential subunit of a human tRNA splicing ligase complex. Science 331, 760–764 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Kroupova A et al. Molecular architecture of the human tRNA ligase complex. eLife 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maquat LE, Carmichael GG, Quality Control of mRNA Function. Cell 104, 173–176 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Wutz A, Rasmussen TP, Jaenisch R, Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nature Genetics 30, 167–174 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Minks J, Baldry SEL, Yang C, Cotton AM, Brown CJ, XIST-induced silencing of flanking genes is achieved by additive action of repeat a monomers in human somatic cells. Epigenetics & Chromatin 6, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Beljouw SPB et al. The gRAMP CRISPR-Cas effector is an RNA endonuclease complexed with a caspase-like peptidase. Science 373, 1349–1353 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Özcan A et al. Programmable RNA targeting with the single-protein CRISPR effector Cas7-11. Nature 597, 720–725 (2021). [DOI] [PubMed] [Google Scholar]
- 33.Schmitt-Ulms C et al. Programmable RNA writing with trans-splicing. bioRxiv, (2024). [Google Scholar]
- 34.Rowe SM et al. Restoration of W1282X CFTR Activity by Enhanced Expression. American Journal of Respiratory Cell and Molecular Biology 37, 347–356 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagy E, Maquat LE, A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends in Biochemical Sciences 23, 198–199 (1998). [DOI] [PubMed] [Google Scholar]
- 36.Kim YJ et al. Gene-specific nonsense-mediated mRNA decay targeting for cystic fibrosis therapy. Nature Communications 13, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nemudryi A et al. CRISPR-based engineering of RNA viruses. Science Advances 9, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nemeth K, Bayraktar R, Ferracin M, Calin GA, Non-coding RNAs in disease: from mechanisms to therapeutics. Nature Reviews Genetics 25, 211–232 (2023). [DOI] [PubMed] [Google Scholar]
- 39.Batra R et al. Elimination of Toxic Microsatellite Repeat Expansion RNA by RNA-Targeting Cas9. Cell 170, 899–912.e810 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vaz-Drago R, Custódio N, Carmo-Fonseca M, Deep intronic mutations and human disease. Human Genetics 136, 1093–1111 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Irmisch P, Mogila I, Samatanga B, Tamulaitis G, Seidel R, Retention of the RNA ends provides the molecular memory for maintaining the activation of the Csm complex. Nucleic Acids Research, (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ben-Ari Y. a. et al. The life of an mRNA in space and time. Journal of Cell Science 123, 1761–1774 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.D’Orazio KN, Green R, Ribosome states signal RNA quality control. Molecular Cell 81, 1372–1383 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dugar G et al. CRISPR RNA-Dependent Binding and Cleavage of Endogenous RNAs by the Campylobacter jejuni Cas9. Molecular Cell 69, 893–905.e897 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Connell MR et al. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature 516, 263–266 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rousseau BA, Hou Z, Gramelspacher MJ, Zhang Y, Programmable RNA Cleavage and Recognition by a Natural CRISPR-Cas9 System from Neisseria meningitidis. Molecular Cell 69, 906–914.e904 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strutt SC, Torrez RM, Kaya E, Negrete OA, Doudna JA, RNA-dependent RNA targeting by CRISPR-Cas9. eLife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bravo JPK et al. RNA targeting unleashes indiscriminate nuclease activity of CRISPR–Cas12a2. Nature 613, 582–587 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dmytrenko O et al. Cas12a2 elicits abortive infection through RNA-triggered destruction of dsDNA. Nature 613, 588–594 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yan WX et al. Functionally diverse type V CRISPR-Cas systems. Science 363, 88–91 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jinek M et al. Structures of Cas9 Endonucleases Reveal RNA-Mediated Conformational Activation. Science 343, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamano T et al. Crystal Structure of Cpf1 in Complex with Guide RNA and Target DNA. Cell 165, 949–962 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuan Y et al. Chemoproteomic discovery of a human RNA ligase. Nature Communications 14, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wessels H-H et al. Massively parallel Cas13 screens reveal principles for guide RNA design. Nature Biotechnology 38, 722–727 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wessels H-H et al. Prediction of on-target and off-target activity of CRISPR–Cas13d guide RNAs using deep learning. Nature Biotechnology, (2023). [DOI] [PubMed] [Google Scholar]
- 56.Cong L et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Villiger L et al. CRISPR technologies for genome, epigenome and transcriptome editing. Nature Reviews Molecular Cell Biology, (2024). [DOI] [PubMed] [Google Scholar]
- 58.Nemudraia A, Nemudryi A, Wiedenheft B, Amplicon sequencing of human mRNA and reporter mRNA targeted with type III-A CRISPR complex of Streptococcus thermophilus, NCBI; (2024); https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1099688/ [Google Scholar]
- 59.Nemudraia A, Nemudryi A, Wiedenheft B, Code for generating figures and analyzing amplicon sequencing of human mRNA and reporter mRNA targeted with type III-A CRISPR complex from Streptococcus thermophiles, Zenodo; (2024); doi: 10.5281/zenodo.10966960 [DOI] [Google Scholar]
- 60.Shen MW et al. Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 563, 646–651 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kato K et al. Structure and engineering of the type III-E CRISPR-Cas7-11 effector complex. Cell 185, 2324–2337.e2316 (2022). [DOI] [PubMed] [Google Scholar]
- 62.Conant D et al. Inference of CRISPR Edits from Sanger Trace Data. The CRISPR Journal 5, 123–130 (2022). [DOI] [PubMed] [Google Scholar]
- 63.Kim D et al. The Architecture of SARS-CoV-2 Transcriptome. Cell 181, 914–921.e910 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lin Y-C et al. Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nature Communications 5, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing data were deposited to the NCBI Sequence Read Archive under accession number PRJNA1099688 (58). The code used to analyze sequencing data and generate figures has been deposited to Zenodo (59). Plasmids generated in this study are available through Addgene and by request from the authors.