

Abstract
A 3D bioprinted neurovascular unit (NVU) model is developed to study glioblastoma (GBM) tumor growth in a brain‐like microenvironment. The NVU model includes human primary astrocytes, pericytes and brain microvascular endothelial cells, and patient‐derived glioblastoma cells (JHH‐520) are used for this study. Fluorescence reporters are used with confocal high content imaging to quantitate real‐time microvascular network formation and tumor growth. Extensive validation of the NVU‐GBM model includes immunostaining for brain relevant cellular markers and extracellular matrix components; single cell RNA sequencing (scRNAseq) to establish physiologically relevant transcriptomics changes; and secretion of NVU and GBM‐relevant cytokines. The scRNAseq reveals changes in gene expression and cytokines secretion associated with wound healing/angiogenesis, including the appearance of an endothelial mesenchymal transition cell population. The NVU‐GBM model is used to test 18 chemotherapeutics and anti‐cancer drugs to assess the pharmacological relevance of the model and robustness for high throughput screening.
Keywords: 3D Bioprinting, glioblastomas, high‐throughput screening, neurovascular unit, transcriptomics
A high‐throughput amenable 3D bioprinted neurovascular unit (NVU) creates a patient‐derived glioblastoma (GBM) growth assay using human primary brain cells and patient‐derived GBM cells in a 96‐well plate with real‐time readouts of angiogenesis and GBM growth for drug screening. Rigorous validation of the NVU model uses scRNAseq analysis, confocal microscopy, and pharmacological relevance tests for drug discovery and development.

1. Introduction
Current in vitro cellular assays used for drug discovery and development lack the physiological relevance necessary to reliably predict clinical outcomes. In the last decade, three‐dimensional (3D) organotypic cellular models that capture the physiological complexity of tissues and organs have been developed as predictive assays for efficacy and toxicity drug testing. The advent of stem cells and bioengineering technologies have enabled the development of a wide range of 3D organotypic models, included but not limited to spheroids developed from single or multiple cell sources,[ 1 , 2 ] stem cell‐derived organoids,[ 3 , 4 ] and tissue‐ and multiorgan‐on‐chip.[ 5 ] More recently, 3D tissue bioprinting (3DTBP) has emerged as a versatile additive manufacturing technology to assemble tissue biomimetics in a well with controlled spatial arrangements and desired cell type composition. 3DTBP is becoming a promising tool to assemble complex tissue models due to its flexibility and reproducibility for large‐scale production.[ 6 , 7 ] 3DTBP is an additive manufacturing process in which cells are suspended in a polymeric bioink, and droplet‐ or extrusion‐based techniques are used to create a customized xyz spatial pattern with selected cell types. 3DTBP has been successfully used for the bioprinting of multicellular constructs recapitulating different types of human tissues such as bone,[ 8 , 9 ] cartilage,[ 10 ] muscle,[ 11 ] brain,[ 12 ] liver,[ 13 ] blood vessels,[ 14 ] retina,[ 15 ] and skin.[ 16 ] 3DTBP has also been shown to be a versatile tool to create physiologically relevant disease tissue models, including modeling cancers by creating a patho‐physiologically relevant tumor microenvironment to study of tumor biology and drug mechanism of action[ 17 ] in the context of a biomimetic tumor niche.
Glioblastoma (GBM) is an adult brain cancer with high recurrence and poor prognosis.[ 18 ] Its genetic heterogeneity has been shown to produce drug resistance and relapse. The growth of GBM depends on its microenvironment, including cell–cell, cell–vasculature, and cell‐extra cellular matrix (ECM) interactions. Further complicating the treatment of GBM is the challenge of drug penetration through the blood–brain‐barrier (BBB). For these reasons, treatments for GBM have remained elusive and there still is a high unmet medical need to find effective therapies for GBM. The conventional drug screening platforms for GBM use immortalized cell lines such as U87, U251, and T98G in 2D monocultures which poorly reflect the GBM genetic heterogeneity and tumor microenvironment.[ 19 ] Patient‐derived GBM spheroids have recently been generated that recapitulated the heterogeneity of the tumor and therefore are more physiologically relevant in vitro models for translational GBM research.[ 20 , 21 ] However, even in assays using patient‐derived cells, there remains a lack of other essential components of tumor growth such as microvascular, ECM, and immune cells.[ 22 ] To increase the physiological relevance of GBM cellular models for drug development, GBM tumoroids have been generated that include vascular components by mixing with,[ 23 ] by co‐culturing the GBM spheroids with vascular endothelial cells[ 24 ] or culturing the self‐assembled GBM spheroids onto the microfluidic BBB chip.[ 25 ] Complex GBM‐vascular models were also developed using 3DTBP by seeding the GBM spheroids onto the bioprinted vascular network formed by human umbilical vein endothelial cells (HUVECs),[ 26 ] or by bioprinting the GBM core surrounded with HUVECs on a glass slide.[ 27 ] These vascularized tumor models provide GBM‐vasculature models to study tumor‐promoted angiogenesis and reproduce the results from clinical therapies.[ 28 ] However, none of these assay systems were developed using all human primary cells, and they were not in a well‐based microplate format to enable high‐throughput drug screening (HTS).
A critical hurdle in the development of complex 3D tissue models and their use as drug screening platforms is to establish their biological validity and robustness and reproducibility in a multi‐well plate platform that is compatible with common laboratory automation equipment used in HTS laboratories. Here, we describe the development of a 3D bioprinted model of the neurovascular unit (NVU) in a 96‐well plate to create a patient‐derived GBM growth assay in an NVU microenvironment that is amenable for HTS. This NVU‐GBM model was validated by immunostaining fluorescence microscopy followed by the analysis of single‐cell RNA sequencing (scRNAseq) data for the transcriptomics between the cells in 2D monolayers and 3D NVU and between 3D NVU and NVU‐GBM. The reproducibility and HTS compatibility of this 96‐well bioprinted NVU model were demonstrated by implementing a focused screen of 18 selected compounds on tested in 4‐point dose responses to assess anti‐GBM potency and efficacy, as well as effects on the microvasculature network integrity. In summary, a bioprinted human NVU‐GBM model was created using primary cells and patient‐derived GBM spheroid cells, which shows features of clinical GBM, and it is robust for HTS in a 96‐well multiwell plate and drug screening. The biofabrication approach described here is modular and in future iterations, it will enable the incorporation of additional relevant cells like neurons and microglia cells.
2. Results
2.1. Bioprinting a 3D NVU Model
A ring‐shaped geometry was chosen for the bioprinted NVU design to achieve an optimal printing time of ≈10 min for a whole 96‐well plate to minimize dehydration of the printed hydrogel. Furthermore, the design facilitated using fluorescence microscopy to quantitate inward and outward angiogenesis from the bioprinted ring structure where de novo tube formation of endothelial cells (ECs) labeled with green fluorescent protein (GFP) occurred two days after bioprinting (Figure 1A). To build the basic components of NVU, we used a mixture of GFP‐labeled primary human brain endothelial cells (BMVEC‐GFP), astrocytes, and pericytes in a gelatin–fibrinogen hydrogel as a bioink where the gelatin enabled the bioprinting and the fibrinogen promoted tube formation. After bioprinting, the ring structure was covered with acellular gelatin–fibrinogen hydrogel for supporting and maintaining the bioprinted structure and angiogenesis. One‐day treatment of thrombin (2 U ml−1) in the growth media fully polymerized fibrinogen while the gelatin dissolved at 37 °C. Media containing vascular endothelial growth factor (VEGF) and angiopoietin‐1 (ANG‐1) helped de novo tube formation and stabilization of the microvessel network, respectively. We first evaluated the technical repeatability of the bioprinted geometry by measuring roundness and circle area from the GFP fluorescence signal using the high‐content imaging Harmony software (see Experimental Section for details). The coefficient of variation (CV) was calculated from 238 wells, both intra and interplate. The CVs for both measurements demonstrated high uniformity (CV circular area = 8%; CV roundness = 3%) (Figure 1B,C). We next evaluated inward and outward tube formation using real‐time fluorescence microscopy from the BMVEC‐GFP cells. Inward and outward angiogenic tube formation was detected 3 days after bioprinting (Figure 1D). The quantification of the total length of inward and outward angiogenesis based on the BMVEC‐GFP fluorescence signal measured with the confocal high‐content reader confirmed the continuous angiogenesis growth and expanding vascular network during the culturing period of 24 days (Figure 1E). We finally evaluated the morphology of individual cell types using immune staining, including a cluster of differentiation 31 (CD31) as a BMVEC marker, alpha‐smooth muscle actin (αSMA) as a pericyte marker and s100beta (s100β) as an astrocyte marker (Figure 1F). The co‐localization of αSMA expression and GFP confirmed pericyte growth in alignment with the micro‐vessels. Astrocytes were spread out and migrated into the acellular area in the ring center and made direct contacts to the micro‐vessels. Further histological analysis with hematoxylin and eosin (H&E) staining confirmed the integrity of the original bioprinted structure and revealed the spider‐like morphology of astrocytes with its endfeet contacting on the microvessels (Figure 1G).
Figure 1.
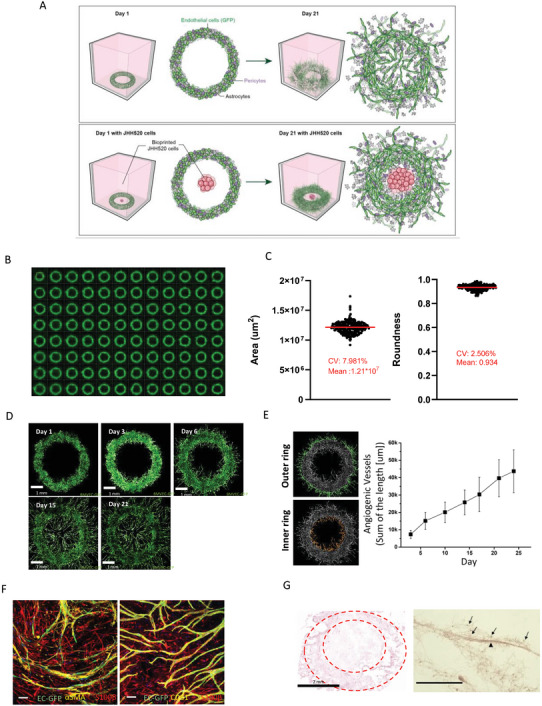
Bioprinting a Neurovascular Unit (NVU) model. A) Schematic cartoon describing the Biofabrication approach used to assemble an NVU and NVU‐glioblastoma model. B) Representative image of the bioprinted NVUs in a 96‐well plate. C) The mean and CV on area and roundness from 283 wells with bioprinted NVUs were calculated to assess the uniformity between NVUs. D) Time course images of vasculogenesis and angiogenesis formation using GFP expressing human brain endothelial cells (scale bar = 1 mm). E) Quantitation of angiogenesis at both outer‐ring (green) and inner‐ring area (orange) area across the culturing period from real time fluorescence microscopy images. F) Cell identity of the NVU by immunostaining and fluorescence microscopy: αSMA as a pericyte marker, s100β as an astrocyte marker, CD31 as an endothelial cell marker (scale bar = 100 µm). G) The H&E staining of NVU, the area surrounded by a dot line indicated the area of the printed structure. Astrocytes are indicated by arrows and the microvessel is labeled with an arrowhead (scale bar = 75 µm).
2.2. Bioprinting a 3D NVU‐GBM Model
For modeling GBM tumor growth on the NVU, we bioprinted the patient‐derived GBM cells JHH520 expressing mCherry at the center area of the bioprinted NVU ring structure (Figure 1A and Figure 2A). By printing the tumor in the center of the ring structure, any factors secreted from the tumor will be evenly diffused within the geland NVU. We used the BMVEC‐GFP fluorescence signal to quantitate the area of inward and outward angiogenesis growth by measuring the total tube length from the ring structure, and the size of the GBM tumor by measuring the total area of mCherry fluorescence signal. The results show that there was an increase in the outer‐ring area microvessels grew in the NVUJHH520 co‐culture group (Figure 2B), we also observed that inward angiogenesis was significantly decreased by the inclusion of GBM, after 2 weeks of culture (Figure S1A, Supporting Information). We observed the GBM tumor growth up to day 21, and GBM growth was slower when the NVU was included, compared to when the GBM was bioprinted in an acellular hydrogel without NVU (Figure 2C). To further investigate the interactions between the GBM and the NVU, we removed the additional VEGF and ANG‐1 added to the culture media at day 13, and quantitated angiogenesis of the NVU with or without tumor cells. Under the basal VEGF condition (2.5 ng ml−1), angiogenesis growth was supported by the GBM tumor cells, but the vascular network degenerated in the absence of GBM cells, suggesting proangiogenic effects of GBM (Figure 2D). This data suggests that outward angiogenesis growth results from proangiogenic cues from the GBM cells, while reduced inward angiogenesis could potentially be a consequence of physical space constraints (Figure S1B, Supporting Information) or direct local interactions between the NVU and GBM cells. Interestingly, in the inner area of the ring structure, the GFP signal was detected within the GBM tumor area as a puncta‐like expression pattern, disconnected from the ring structure (Figure 2E). This data showed that GBM cells appear to support vessel formation but also disrupt BMVECs from the vasculature network.[ 29 ] The H&E staining in Figure 2F confirmed the tumor growth in the central area of the ring structure and the appearance of tube‐like structures extending from the tumor cell aggregates. The extended tube‐like structure from the tumor aggregates could be evidence of tumor vascular mimicry.[ 30 ] Tube‐like structures with tumors were also detected using fluorescence imaging in the NVU model. Figure 2G shows fluorescence microscopy images of the tumor cells in the acellular hydrogel and NVU structure at day 12, respectively. The tube‐like structure can be seen in the red channel only when the NVU is present, suggesting vascular mimicry of the tumor cells.
Figure 2.
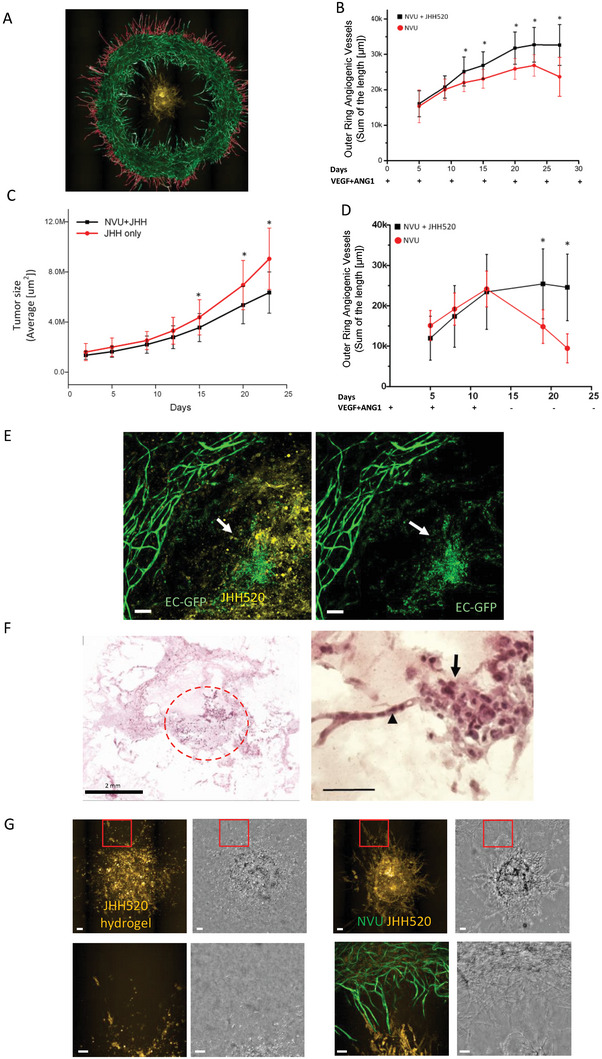
Glioblastoma model by co‐culture patient‐derived JHH520 glioma cells with NVU. A) Representative fluorescence microscopy image of an NVU (green) with JHH520 glioma cells (yellow), and a mask used to quantitate angiogenetic vessel at outer‐ring area. B) Plots comparing the increase of total angiogenic vessel length in the outer ring area between the NVU tumor model and NVU, with time. C) Plot comparing the growth of tumor area with and without co‐culture in NVU. D) Plot illustrating the influence of tumor secretory factors on the total length of angiogenetic vessel in the outer‐ring area. In these experiments, exogenous VEGF was removed at day 13. E) Fluorescence image illustrating the loss of vessel integrity around the tumor area. The left panel includes fluorescence from tumor cells in yellow. The arrow indicates endothelial cells from tumor‐disrupted vasculature (Scale bar = 100 µm). F) H&E staining of NVU‐JHH520 tumor model. Tumor cells are indicated by the circle (left panel);, and vascular mimicry tube‐like structures are indicated by the arrowhead (right panel) (scale bar = 75 µm). G) Fluorescence images showing details of the structure of the vasculature and tumor in close contact. The lower pictures are the zoom‐in images from the selected red square area of the upper pictures. The morphology of the bioprinted JHH520 with and without co‐culture of NVU (scale bar = 100 µm). Asterisks mean significant difference. At least 19 samples were used in Figure 2B–D.
2.3. Single Cell RNA Sequencing Analysis and Comparison of Transcriptomic Changes between 2D Monoculture and 3D NVU
We investigate transcriptomic changes that may occur when the cells are grown separately in 2D compared to when are grown in the NVU, with and without the presence of GBM cells to further validate and assess the physiological relevance of the bioprinted NVU‐GBM model. Based on PCA and UMAP clustering, a total of 19 cell clusters were annotated in the data sets (Figure 3A, Table S1, Supporting Information), within five major cell populations identified using described expression of canonical cell‐specific markers (Table S2, Supporting Information): Endothelial cells (PECAM1, CDH5, vWF, CLDN5),[ 30 , 31 , 32 , 33 ] Pericytes (CD248, ACTA2),[ 34 , 35 ] Astrocytes (SPARC, SLC1A2, SOX9),[ 36 , 37 ] EndMT cells (PLCG2,CCNL2) which are endothelial cells undergoing Endothelial–Mesenchymal Transition (EMT)[ 38 ] and Glioma cells (SOX2, TP53, RB1)[ 39 , 40 , 41 ] (Figure 3B; Figure S2A, Supporting Information).
Figure 3.
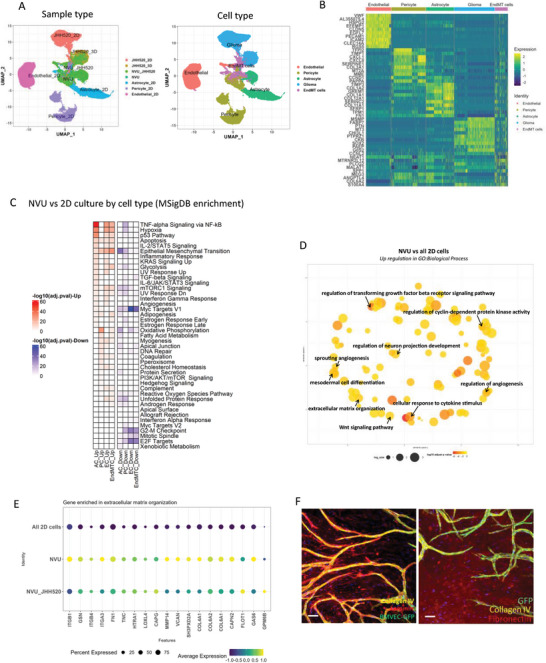
sc RNAseq analysis and comparison of transcriptomic changes between cells in 2D monoculture and 3D NVU. A) UMAP of cell distribution based on sample types and cell types. B) Gene expression profile among each cell type. C) The MSigDB enrichment on the biomechanical process between 3D NVU and 2D cells. D) The upregulated GO: Biological process between 3D NVU and 2D cells. E) The activation of genes in ECM‐related signaling pathways in NVU. F) Immuno‐fluorescent staining of ECM proteins. Scale bar = 100 µm.
We first looked at transcriptional changes for each cell type between 2D monolayer cultures and NVU, and corresponding gene ontology enrichments. We identified three types of endothelial cell populations present in 2D monoculture and 3D NVU growth conditions (EC_1, EC_2, EC_3; Table S1a, Supporting Information). EC_1 (73%) and EC_2 (26%) were the majority of the EC types in the 2D monoculture but were not detected in the 3D NVU. EC_3 population was the only one found in the NVU (100% of EC cells), but it was a minority of ECs (1.4%) in 2D monoculture (Table S1A, Figure S3A, Supporting Information). The Differentiated Expression Gene (DEG) on the characteristic of ECs demonstrated that EC_3 has high expression of genes associated with angiogenetic tip cells,[ 42 , 43 ] clinical capillary, and BBB‐related[ 32 ] (Figure S3B, Supporting Information).
Four sub‐clusters of astrocytes were detected in 2D mono‐ and 3D NVU cultures and the distribution of these cell populations was distinct between 2D monocultures and NVU. The ratio of each subtype of astrocytes in the 2D mono culture was: AC_1 (69%), AC_2 (30%), and for the 3D NVU it was mostly AC_4 (99%) which did not exist in the 2D mono‐culture (Table S1B, Figure S4A, Supporting Information). DotPlot from Seurat package analysis showed that the AC_4 had high expression of mature astrocyte genes[ 44 , 45 ] and astrocyte endfeet relate genes,[ 46 ] which indicated the AC_4 was a more physiological relevant type of astrocyte (Figure S4B, Supporting Information). Astrocyte endfeets could indeed be observed clearly by the histological sample, indicating the high correlation between the gene expression and phenotype in our bioprinted NVU model (Figure 1G).
We identified 5 sub populations of pericytes, and their relative amounts also changed from the 2D monoculture to the 3D NVU.[ 34 ] PC_1 (58.3%), PC_2 (2.5%), PC_3 (14.3%), PC_4 (24.5%), in 2D monoculture; and PC_5 (98.9%) in the 3D NVU (Table S1C, Figure S5A, Supporting Information). CCND1, COL6A2, and CD63 were highly expressed in PC_5, the largest pericyte population in the NVU, shows the functional impact of endothelial direct contact on pericyte proliferation[ 47 ] (Figure S5B, Supporting Information). DotPlot from Seurat package analysis showed pericytes in NVU have upregulated gene sets in myogenesis (ITGB1, TAGLN, SPARC, MYH9, etc.) alpha‐SMA (ACTA2), and myosin light chain9 (MLY9) suggesting that pericytes were activated and transformed into smooth muscle‐like cells in the NVU model, as we would expect (Figure S5B, Supporting Information).
Endothelial cells are known to have plasticity, that is, the ability to change their phenotypes in response to environmental cues, during vasculature development.[ 38 ] In our 3D NVU samples, scRNAseq identified a cell population with high expression of cell plasticity‐related gene (e.g., PLCG2, CCNL2),[ 48 , 49 , 50 ] and low endothelial markers (e.g., PECAM1, VWF, CDH2, CDH5) (Figure 3B). The identity of this cell type has been described as endothelial cells undergoing an endothelial to mesenchymal transition (EndMT) process.[ 38 , 51 , 52 , 53 ] We classified these cells as “EndMT cells” in our dataset. Two EndMT cell clusters were identified in our datasets as EndMTC_1, and EndMTC_2 (Table S1D, Figure S6A, Supporting Information). EndMTC_1 is more abundant in the 3D NVU, with high expression of the S100 family of genes, which are involved in cell proliferation and differentiation. EndMTC_2 has high expression of the long non‐coding RNAs (NEAT1, MALAT1, MEG8) which are involved in regulating vascular integrity (Figure S6A, Supporting Information).[ 54 , 55 ] Through analysis of gene expression feature with EndMTC_1 and EndMTC_2, we discovered that the EndMTC_1 is early stage and EndMTC_2 is late stage in the EndMT process (Figure S6B, Supporting Information).
A gene set enrichment analysis was performed at the sample level to evaluate the overall transcriptomics changes between cells in 3D NVU compared to 2D monolayer cultures (Figure 3C), followed by GO Biological Process analysis (Figure 3D). The overall DEG analysis (Figure 3C) found that there is an upregulation of inflammatory‐related genes in 3D astrocytes, maturation, and formation of junctions in endothelial cells and pericytes, myogenesis‐related genes in pericyte, and epithelial mesenchymal transition in EndMT cells and endothelial cells. We also noticed that there is an upregulation of hypoxia‐related genes in endothelial cells, astrocytes, and EndMT cells, which is consistent with angiogenesis occurring. An enrichment for biological processes associated with angiogenesis, extracellular matrix organization, cellular responses to cytokine stimulation, and upregulation of the Wnt signaling pathway was also identified in 3D NVU (Figure 3D). These results confirm the upregulation of pathways in the NVU that are consistent with angiogenesis and BBB development, highlighting the increased clinical relevance of our NVU model.[ 35 , 56 , 57 , 58 , 59 , 60 ] We specifically focus on ECM genes because they represent the formation of a tissue‐specific microenvironment. Figure 3E shows an expression of a set of genes involved in extracellular organization and interactions between cells and the ECM, including ITGB1, FN1, and ColVI.[ 61 , 62 , 63 ] We used immunofluorescence imaging to confirm the expression of ECM proteins like collagen IV, laminin, and fibronectin in the NVU model (Figure 3F). As expected, collagen IV and laminin had a vascular basement expression pattern following the endothelial vessels, while fibronectin was expressed broadly in the tissue (Figure 3F).
2.4. Transcriptomic Comparison of Cells Making the NVU Cells When JHH520 Tumor Cells are Added to the NVU Model
We next explored the differences in gene expression when JHH520 glioma cells were added to the NVU model. The addition of tumor cells to the NVU did not change the identity and composition of the cell subpopulations in the NVU, as shown in the UMAP in Figure 4A. However, there were changes in the gene expression profiles of each cell type in the NVU by the addition of glioma cells, Figure S2B–F,S7 (Supporting Information). The gene enrichment analysis of the global differentially expressed genes showed a glioma cell‐induced upregulation of cell metabolism (oxidative phosphorylation) in ECs and EndMT cells, mesenchymal transition genes in endothelial cells, KRAS signaling pathway in pericytes, and interferon‐driven immune responses in astrocytes and EndMT cells (Figure 4B). We also detected the downregulation of migration (DLL4, NRP1, SPARC, etc.), angiogenesis (CLDN5, CDH5, KDR, etc.), and extracellular matrix organization (FN1, TIMP1, TIMP3, PECAM1, etc.) related genes in endothelial cells (Figure S7, Supporting Information), which might be related to the phenomena of tumor infiltration to the vessel area causing the breakdown of the vessel in our 21 days of NVU‐JHH520 culture sample (Figure 2E). The upregulated inflammatory response detected by the transcriptomics analysis was confirmed with an observed increase in secretion at IL6, IL8, CCL2, CX3CL1, GM‐CSF, CCL20, and PDGF‐AA (Figure 4C). We observed a temporal pattern of cytokine secretion in the NVU and NVU‐JHH520 bioprinted tissues: IL6, IL8, and CCL2 were highly secreted in the first week of culture and their levels gradually decreased with time or stayed constant, confirming initial inflammatory responses and angiogenesis in wound healing‐like process. On the other hand, CCL20, CX3CL1, VEGF‐C, PDGF‐AA, and GM‐CSF increased with culture time, which indicated the tumor‐related cytokine secretion and inflammatory response. The analysis of genes involved in cytokine‐mediated pathway is shown in Figure 4D, which reveals the increase of cytokine‐related genes in 3D culture environments compared 2D monocultures.
Figure 4.
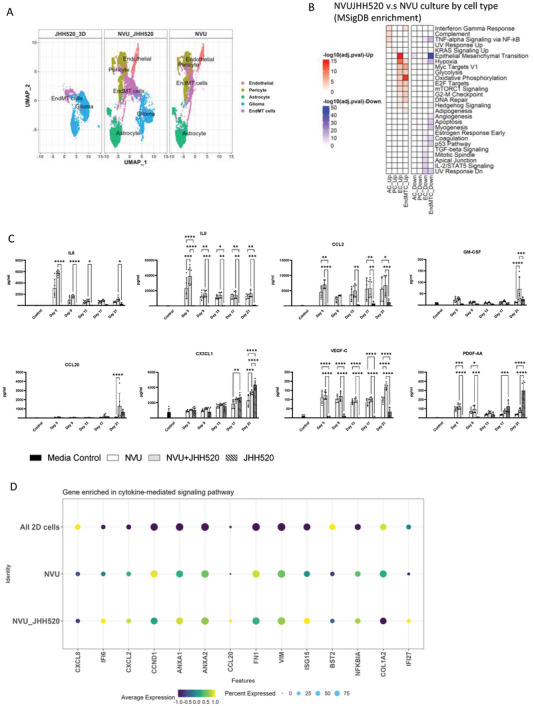
scRNAseq gene analysis and comparison of transcriptomic changes in cells of the NVU with JHH520 glioma cells. A) UMAP plots showing cell cluster distribution and gene expression for cells in the NVU, between NVU, NVUJHH520, and 3D monoculture of JHH520. B) The MSigDB enrichment on the biomechanical process of NVU between the comparison of NVU‐JHH520 co‐culture to NVU. C) The time‐dependent cytokine secretion data for NVU, NVU‐JHH520, and 3D monoculture of JHH520. D) Dotplot for genes enriched in cytokine‐mediated signaling pathway in all NVU cells cultured in 2D, NVU, and NVU‐JHH520 coculture.
2.5. Transcriptomic Comparison of GBM JHH520 Cells in NVU‐JHH520 Compared to Monocultures
We also compared the transcriptomic differences in GBM cells as 2D cultures (JHH520_2D), to 3D monoculture in a hydrogel (JHH520_3D), or 3D co‐culture with NVU(NVU_JHH520). Five subtypes of glioblastoma (GBM) were identified (Figure 5A), and the GBM_5 was the largest population in the 3D environment, both hydrogel and NVU, among all subtypes (Table S1E, Supporting Information). By comparing all subtypes of GBMs to clinical GBM samples, the GBM_5 has the highest expression of clinically relevant genes (Figure 5C; Figure S8A, Supporting Information). There were still some transcriptomics differences in GBM_5 in hydrogel versus NVU. We noticed there was a high expression in glycolysis‐related genes in in the NVU co‐culture, which might related the higher proliferation of GBM cells and the progression of the tumor in NVU (Figure 5B,D; Figure S8B, Supporting Information).[ 64 ] We further assessed whether the GBM cells are grown in a 3D environment, with or without vasculature, there is further expression and release of cytokines, as was seen when NVU was formed. DotPlot analysis showed an upregulation of genes related to cytokine signaling pathways when the glioma cells were grown in the NVU, but not in the hydrogel, compared to cultures in 2D (Figure 5E; Figure S8C, Supporting Information). We also analyzed angiogenesis‐related genes in JHH520 glioma cells cultured in different environments.[ 65 ] The data in Figure 5E shows that angiogenetic‐related genes were more upregulated in JHH520 when cocultured with NVU compared to cultures in 2D or 3D in hydrogel without NVU which might relate to the tube formation by cancer cells seen by histology (Figure 2F).[ 66 ]
Figure 5.
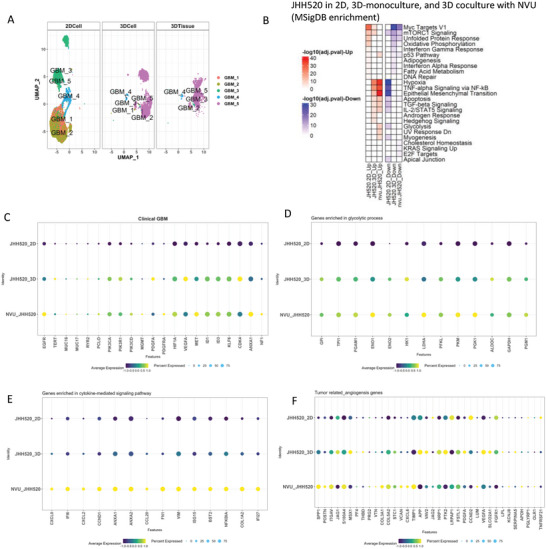
scRNAseq gene analysis and comparison of transcriptomic changes in JHH520 glioma cells grown as 2D monocultures, JHH520 3D hydrogel monocultures (JHH520 3D) and JHH520 coculture with NVU (NVU‐JHH520). A) UMAP plots of GBM cell cluster distribution. B) MSigDB enrichment plot of biomechanical processes. Dotplots gene expression analysis for C) clinical GBM genes; D) glycolytic process genes E) genes related to cytokine‐mediated signaling pathways; and F) tumor‐related angiogenesis genes.
2.6. A Tumor Growth/Angiogenesis Assay with the NVU‐JHH520 Tumor Model for Drug Screening
The robustness and pharmacological relevance of the tumor growth and angiogenesis assays developed with the bioprinted NVU‐JHH520 tissue model were assessed for drug screening by testing a set of 18 compounds in dose responses. These compounds inhibited different targets and mechanisms of action and were selected based on what gene targets were upregulated in the GBM cells in the NVU model (Figure 5C), including EGFR inhibitors, MAPK inhibitors, cyclin‐dependent kinase (CDK) inhibitors, compounds targeting the tumor microenviroment such as including angiogenesis blockers like VEGFR and TGFβR inhibtors, and matrix metalloproteinase inhibitors, drugs approved for brain tumors like Lomustine and Temezolomide, and other compounds being investigated for the treatment of glioblastomas like Eflornithine and Marizomib. Compounds were tested in four‐point dose responses (40, 6.67, 1.1, and 0.2 µm). A screenshot of the GFP and RFP fluorescence signal from a whole plate from the screen is shown in Figure 6A. The effect of the compounds on the total length of outward angiogenesis and the size of the GBM tumor area measured as described in the Experimental Section were used as assay readouts for the screen. The percentage in the change of the vasculature or tumor area was calculated as the ratio in signal between days 13 and 21, the length of the compound's treatment, from the compounds over the change in signal from the control wells treated with DMSO, the solvent used to solubilize the compounds (see Experimental Section for details). Overall, we observed that the tested compounds were more potent at disrupting the vasculature than reducing tumor growth, and we did not observe any compounds that were able to selectively arrest tumor growth without affecting vasculature. The proteasome inhibitor Bortezomib inhibited ≈95% of the vasculature and tumor area (Table S3, Supporting Information), and all the VEGFR inhibitors decreased vasculature even at the lowest compound concentration (Figure 6B,C). Only the VEGFR inhibitors Anlotinib, Cabozantinib, and Regorafenib inhibited tumor size by more than 80% at 40 µm (Table S3, Supporting Information). The two CDK inhibitors showed distinct pharmacologies: Although they both have almost 90% of inhibition on tumor and vessel growth at the highest compound concentration tested, the CDK 1/2/5/9 inhibitor, Dinaciclib, was more potent in inhibiting vasculature and tumor growth, similar to the Bortezomib treatment, whereas the CDK 4/6 inhibitor, Abemaciclib, showed low potency (Figure 6B,C). The MEK1 inhibitor, Trametinib, which had shown to arrest cell growth in JHH520 in 2D mono‐culture and in animal models,[ 29 ] showed 50% inhibition of tumor growth and 90% inhibition of vascular growth at the highest dose of the compound. Like Trametinib, the EGFR inhibitor Neratinib had a greater effect on the vasculature (90% area inhibition) than the tumor reduction (42% area inhibition) at the compound concentration of 40 µm. Both CSF‐1R inhibitor, Pexidartinib, and CT‐L β5/C‐L β1/T‐L β2 proteasome inhibitor, Marizomib, treatments resulted in more than 90% reduction in both tumor growth and vascular area, at 40 µm of compound concentration. There was no significant change in both tumor growth and vascular area at the highest compound concentration for DNA alkylating agents (Lomustine and Temezolomide), ornithine decarboxylases inhibitors (ODC, Eflornithine), smoothened inhibitors (Smo, Glasdegib), and matrix metalloproteinase inhibitors (Marimastat). Interestingly, a 117% increase of vascular area was observed with the treatment of TGFβR1 inhibitor, Galunisertib, at 40 µm of compound concentration, which might be related to previously observed effects of TGFβ inhibitors in vascular angiogenesis.[ 67 ]
Figure 6.
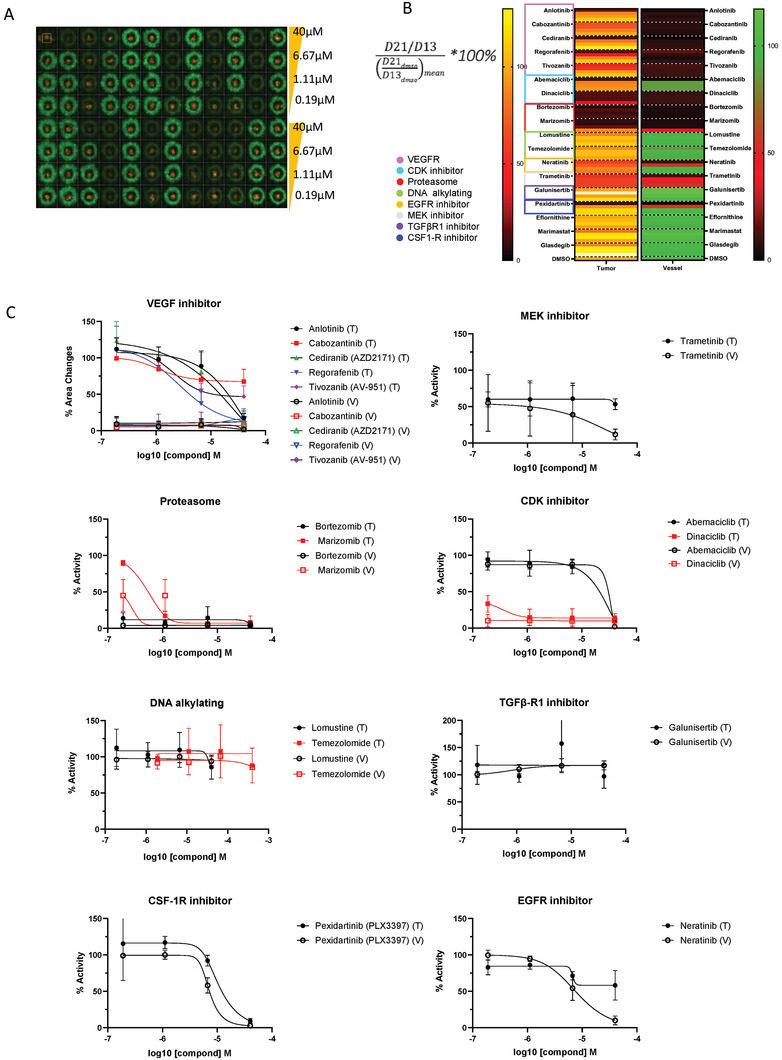
Screening of a focus collection of drugs with the NVU‐JHH520 glioblastoma model. A) The representative fluorescence microscopy image of a whole 96‐well microplate with NVU (green)‐JHH520 (yellow) model in each well, 21 days after bioprinting and after 1‐week of compound treatment. Each plate included 18 drugs (each in 4‐dose responses, rows A‐D and E‐H. Columns 1 and 12 include wells with no drug treatment and including 0.4% DMSO to mimic the amount of solvent in drug‐containing wells B) A heatmap showing %changes on vascular outer ring vessel length (from green signal) and tumor area (yellow signal) after compound treatment, as calculated by the formula shown. C) Dose‐response plots for both % changes in outer ring vessel length (V) and tumor area (T). Error bars are SD and n = 4.
3. Discussion
The goal of this work was to create a bioengineered brain‐like vascularized tumor microenvironment (TME) model for GBM which captured physiologically relevant tumor‐TME interactions, including vasculature, and was robust and amenable to drug screening. We used commercially available primary human brain cells and a patient‐derived GBM cell to create the NVU‐GBM tissue model and designed a bioprinted NVU ring structure that allowed for the radial formation of angiogenesis and facilitated quantitation of vessel formation and GBM tumor growth in a 96‐well plate format for drug testing. The bioprinting technologies provide the versatility of customized experimental designs. In our case, the bioprinted ring structure made it possible to distinguish vasculogenesis and angiogenesis of the endothelial cells. In addition, by using this design, any factors produced by the tumor cells in the center of the bioprinted NVU ring can evenly diffuse the surrounding NVU cells. In our NVU, the individual cell types successfully self‐assembled to form microvessels and deposited brain‐relevant ECMs to further create a TME. Sc‐RNAseq together with immunostaning for cell and ECM markers and cytokine secretion analysis was used to help assess the physiology of the bioprinted NVU model. Perhaps not surprisingly, the NVU was formed in a wound healing‐like environment, including the induction of an EndMT cell population derived from the original endothelial cells to form the microvessels, seen in pathological states of vasculature regeneration. Extensive brain‐relevant ECM deposition, including perivascular Collagen IV and laminin, and a transient inflammatory environment was also detected. There was only a small number of endothelial cells with the canonical markers identified in the NVU and they all expressed the marker of angiogenetic tip cells, which indicate the presence of active angiogenesis in the NVU.[ 42 , 68 ] The large population of EndMT cells are likely responsible for the vasculature development and angiogenesis.[ 69 ] The EndMT cells lost some canonical EC markers such as CD31 while they had upregulated mesenchymal cell makers such as S100A4 and α‐SMA.[ 70 ] The induction of EMT transition may cause by the inflammatory environment created and was revealed in our scRNAseq data, including up‐ regulation of TNFα, TGFβ, and IL6 signaling (Figure 3D).[ 71 ] The upregulation of genes related to inflammatory response, together with the secretion of early inflammatory cytokines like IL6, IL8, and CCL2, further supports that the bioprinted fibrin‐based hydrogel creates a fibrotic, wound‐healing microenvironment that favors angiogenesis.[ 45 , 72 ] These inflammatory cytokines could be essential for the migration, tuberization, or ECM deposition in a fibrin gel environment, which mimics the wound healing process.[ 73 , 74 ] Most of the inflammatory cytokines return to steady levels 1‐week after bioprinting, however, some cytokines keep increasing during the three weeks culturing period, including CCL20, CX3CL1, VEGFC, and PDGF‐AA. The secretion of those cytokines might be directly related to tumor cells and involved in the regulation of immunoevasion,[ 75 , 76 , 77 ] angiogenesis,[ 78 ] and the resistance to tumor apoptosis.[ 79 ] It is well known that ECM can trigger signal transduction pathways in cells.[ 80 ] In our bioprinted model, the cells contributed their own ECM to the surrounding environment, and it is no surprising that the JHH520 cultured in a 3D environment has more clinically relevant gene expression compared to the 2D mono cell culture. As clinical GBM heavily relies on the communication with GBM parenchyma for maintaining the immunosuppression and progression,[ 81 , 82 ] the increase of expression on glycolysis, cytokine signaling, angiogenesis‐related gene seen in GBM cells in the NVU co‐culture demonstrates that this bioprinted GBM models has clinical similarities.
This bioprinted NVU‐GBM model enabled the quantitative measurements of angiogenesis and tumor growth in a 96 well microplate format and screening of drugs in dose‐responses to test anti‐tumor activity and effects on the vasculature. A similar approach to the Biofabrication of a GBM model was previously introduced by Yi et al., in which both human umbilical vein endothelial cells and tumor cells were bioprinted on a glass slide using a bioink made from decellularized pig's brain tissue. Their model allowed the study of the tumor morphological changes as well as tumor‐vessel interaction,[ 27 , 83 ] however, the low throughput of the assay system made it challenging to use for drug screening, and the vascular morphology was unclear in the context of the hydrogel, making angiogenesis quantification challenging. In our study, we made significant improvements in modeling GBM for drug testing, including clear vascular morphology and increased throughput. Eighteen compounds with different anti‐cancer mechanisms of action were selected based on either our transcriptomics data, targets involved in tumor‐TME interactions or clinical relevance for glioblastoma. In our model, most of the compounds tested were much more potent disrupting the vasculature than inhibiting tumor growth. Not surprisingly, given our data indicating the dependence of vasculature formation on VEGF, VEGR inhibitors disrupted angiogenesis. However, their effect on tumor growth was minimal, indicating that any clinical effects of these compounds will be by limiting blood supply to the tumors, which is not captured in this assay. In fact, bevacizumab, an anti‐VEGF antibody is clinically used for the treatment of GBM when the tumor progresses. The CDK inhibitors were chosen based on the high expression on CDK4 in GBM populations in the NVU. Both CDK inhibitors, Abemaciclib and Dinaciclib decreased both tumor and vasculature dramatically, albeit with different potency, likely due to the different polypharmacology: Abemaciclib[ 84 ] is a CDK4/6 inhibitor and had a weaker effect on tumor growth and vasculature integrity; Dinaciclib[ 85 ] is a CDK 1/2/5/9 inhibitor and was a general cytotoxic compound with very potent effects in both tumor growth and vasculogenesis. EGFR plays a critical role in promoting tumor motility and proliferation,[ 86 , 87 ] and we also observed upregulation of EGFR in JHH520 in the gene expression analysis. Treatment with the EGFR inhibitor, Neratinib produced some reduction of tumor cell growth but had a stronger effect on vessel integrity. Current chemotherapy options for GBM such as nitrosoureas (Lomustine) and the imidazotetrazine‐class monofunctional DNA alkylating agent, temozolomide (TMZ). Neither of these compounds had any effect in our model, for both tumor and vasculature. The lack of effect for TMZ might be due to the very short half‐life of TMZ in vitro[ 88 ] and not reaching an efficacious dose in our model. The lack of effect seen with Lomustine cannot be readily explained, but it could be because of the general resistance of the patient GBM cells used to this compound.[ 29 ] Other compounds targeting ECM remodeling like MMP inhibitors were not active suggesting in our model, perhaps several proteases regulate ECM remodeling to allow angiogenesis and cancer cell mobility, and inhibiting a subset is not sufficient to stop cancer cell growth. Finally, Glasdegib (PF‐04449913) is a SMO inhibitor that is clinical trials as a rational therapeutic agent for patients with newly diagnosed Glioblastoma because it inhibits the Sonic Hedgehog pathway, which interferes with cancer stem cells and endothelial migration. This compound did not have an effect in either the vasculature or tumor growth in our model, perhaps not surprisingly because the Hedgehog signaling pathway was slightly upregulated in endothelial cells within the NVU‐GBM co‐cultured sample, and no upregulation was seen in the GBM cells in the NVU.
The challenge remains to engineer a cancer model that captures all the interactions between the tumor and the TME in an HTS‐compatible format for drug screening. There remain many tissue components to be added to increase the physiological relevance of this NVU model. To keep a balance between physiological complexity and HTS comparability, this NVU model was built in a static 3D culture system that lacks vascular perfusion to increase nutrient supply, BBB function, which limits the lifetime of the in vitro cellular system. However, our model recapitulated some important features of TME and demonstrated a similar response on therapeutic compounds compared to its preclinical results. The inclusion of immune cells and perfusion of NVU will be a critical next step to improve tissue integrity, viability, and physiological relevance. Moreover, the inclusion of the neurons in the NVU will also be needed to study neuron‐vascular and neuron‐GBM interactions to better assess drug responses. Interestingly, we observed upregulations of the neuronal development related gene expressions in the GO: Biological analysis of NVU during the comparison with 2D mono‐cell culture despite the lack of neurons. This data supports the idea that the conditions developed for the NVU formation create a microenvironment for neuronal cell differentiation and growth. Finally, further refinements will also be needed for quantitative measurements using high‐throughput (HT) image‐based analysis. Although the angiogenic vessels are detectable using the current software, it frequently separated one continuous angiogenetic vessel into several segment depending on the vasculature diameter, which limited the quantification and only the total length of angiogenetic vessels could be used quantitate the vascular network.
In summary, the 3D bioprinted NVU described in this study provides an initial basic system of the NVU to facilitate increased physiological complexity in an HTS format. The modular tissue engineering approach developed makes for a powerful tool to study the role of individual cellular components and ultimately to develop personalized models with patient‐derived human induced pluripotent stem cells for developing personalized medicine.
4. Experimental Section
Cell Culture
GFP tagged Human primary brain microvascular endothelial cell (BMVEC‐GFP; cAP‐0002GFP), brain astrocytes (cAP‐0031), and brain microvascular pericyte (cAP‐0030) were purchased from Angioproteomie (Boston, MA). The cells were cultured in flasks pretreated with quick coat solution (Angioproteomie, cAP‐01) for at least 5 minutes. BMVECs‐GFP media includes VascuLife VEGF Endothelial Medium Complete Kit (Lifeline CellTechnology, Frederick, MD; LL‐0003) with the replacement of the fetal bovine serum (FBS) into 50 ml of the CDI iCell supplement (final concentration is 10% CDI iCell supplement; FUJIFILM Cellular Dynamics, Inc., Madison, WI; M1019). The astrocyte media was DMEM GlutaMax (ThermoFisher Scientific, Waltham, MA; 10 569 010), 10% FBS (ThermoFisher Scientific, Waltham, MA; A3840002), and 1X N2 supplement (ThermoFisher Scientific, Waltham, MA; 17 502 001). The pericytes were cultured with pericyte growth media (Angioproteomie, cAP‐09). The patient‐derived glioblastoma (JHH520) was a gift from Dr. Gregory J. Riggins.[ 89 ] The cells were maintained in a suspension culture with NeuroCult NS‐A Proliferation Kit (StemCell Technologies, 0 5751) with an additional 20 ng ml−1 EGF (StemCell Technologies, 78 006), 10 ng ml−1 FGF (StemCell Technologies, 78 003), and 0.0002% Heparin (StemCell Technologies, 0 7980). The glioblastoma JHH‐520 cells were labeled with mCherry using a lentivirus transfection kit (Vectalys, 0009VCT – ILV‐EF1a‐mCherry‐IRES‐Puro) with 7 M.O.I for 4.5 hours. All cells were cultured at 37 °C in a 5% CO2 and the media was refreshed every other day. Human primary cells were trypsinized with 0.05% trypsin (ThermoFisher Scientific, 25 300) before bioprinting.
Bioink Preparation for 3D Tissue Bioprinting
The fibrinogen (Sigma‐Aldrich, St. Louis, MO; F3879) solutions for the bioink were diluted from 15 mg ml−1 stock solution (by adding the fibrinogen powder in DPBS (ThermoFisher Scientific, Waltham, MA; 14190144)) into the final concentrations of either 5 or 2.5 mg ml−1 in DPBS. The 18.75 U ml−1 Aprotinin (Sigma‐Aldrich; A4529) stock solution was added to the fibrinogen solution to reach the final concentration of 0.075 U ml−1 aprotinin followed by sterilizing the solution with filter membrane (Millipore; SCGP00525). Gelatin powder (Sigma, G1890) was added to the fibrinogen solution for a final concentration of 6% gelatin. The 6% gelatin solution was prepared by directly adding the gelatin powder into DPBS. These three types (A: 5 mg ml−1 fibrinogen with 6% gelatin and aprotinin. B: 2.5 mg ml−1 fibrinogen with 6% gelatin and aprotinin. C: 6% gelatin) of the Bioink were kept as a liquid state at a 37 °C before bioprinting.
The 3D NVUs were bioprinted in a 96‐well plate (Ibidi GmbH, Germany; 89 626) using a RegenHU bioprinter (RegenHU Ltd., Switzerland; R‐GEN 200). BMVECs‐GFP, astrocytes, and pericytes were mixed at a 6:6:2 (million cells ml−1) ratio in bioink‐B. The bioink‐B was loaded into a 5‐ml glass syringe (RegenHU Ltd., Switzerland). A ring pattern was printed through with a 21G needle (inner diameter of 0.514 mm) on the 96‐well plate of which the well surface was pre‐treated by O2‐plasma. For modeling GBM, a dot pattern was printed at the center of the ring structure of NVU with JHH520 cells in 3 million cells ml−1 concentration in bioink‐C via 26G needle (inner diameter of 0.26 mm). After bioprinting, 100 ul of acellular 5 mg ml−1 bioink‐A was added into the well to fill the unprinted area of each well. The plate was incubated at 4 °C for 5 min to solidify the bioink. On Day 0 (day of bioprinting), 250 µl culture media (containing 2 U ml−1 thrombin) was added to each well, and the plate was left at room temperature for 3 hours to give sufficient time of the fibrin gel formation. A media change was performed on Day 1 to remove the thrombin and was refreshed every other day.
Cell Culture Media for 3D Bioprinted NVU
The culture media (1:1 AE) was prepared by 1:1 mixing of endothelial culture media (with 5% CDI iCell supplement) and serum‐free astrocyte media together with the supplement of VEGF (R&D Systems, 293‐VE), Recombinant Human Angiopoietin‐1 Protein (ANG1, R&D Systems, 923‐AN) and aprotinin (Sigma‐Aldrich; A4529). The final concentrations of the supplements were 21.25 ng ml−1 VEGF, 100 ng ml−1 ANG1, and 0.075 U ml−1 aprotinin. After bioprinting, thrombin (Sigma, T6884) was added in the 1:1 AE media with the final concentration of 2 U ml−1 for the 24 hours. During incubation time, growth media was refreshed every other day. Aprotinin was removed from the media at the beginning of the final week of the tissue culture (day 15–day 21). For the GBM NVU model, the final concentration of the CDI iCell supplement was reduced to 1% to allow the growth of JHH520 cells and maintain vasculature assembly.
High Content Imaging
Fluorescence cell imaging was done with an Opera Phenix High‐Content Screening System. Images were taken with a 10× objective with confocal acquisition mode, (z = 270 µm, 15 µm step−1), and the maximum intensity projections of the entire well and plates were reconstructed and analyzed with Harmony software (PerkinElmer). The vascular area and angiogenetic vessels were analyzed based on the GFP signal, the tumor size was detected by mCherry signal, and the original printing structure was determined by bright‐field image. The measurement of area and roundness at Figure 1 was based on the GFP channel, which the signal area was captured and calculated by software. For the angiogenetic vessel, the area of the original printing structure was detected and captured by a bright‐field image with the texture filter and was used as the imaging mask for separating the angiogenetic vessels from the printing structure based on the GFP channel. For tumor size, the mCherry signaling area was captured for calculation.
Immunofluorescence
The printed 3D NVU was harvested at Day 21 and fixed in 4% PFA in DPBS (diluted from 32% PFA solution (Fisher Scientific, 50‐980‐495)) overnight at 4 °C. The tissue was permeated with 3% Triton X‐100 (ThermoFisher Scientific, 85 111), blocked with 5% normal goat serum (ThermoFisher Scientific, 31 872), and incubated with the primary antibody at 4 °C overnight. CD31 (Dako, GA61061‐2) was used as the vascular endothelial cell marker with 1:100 dilution. S100β (Abcam, ab52642) was used as an astrocyte marker with 1:100 dilution, and αSmooth muscle actin (αSMA; Abcam, ab202368) was used as pericyte marker with 1:100 dilution. Goat anti‐mouse/rabbit antibodies with either 568 or 647 nm wavelengths were used for the secondary antibody and incubated at room temperature for 3 hours. The images were taken with a Lecia confocal microscope (Lecia, DMi8). For extracellular matrix staining, Day 21 samples were used and treated with the same protocol described as above. Collagen IV (Dako, M078501‐2), laminin (Dako, Z0097), and fibronectin (Abcam, ab2413) at a 1:100 dilution was used as the primary antibody and incubated at 4 °C overnight followed by the incubation of the second antibodies at room temperature for 3 hours. Images were taken under a Lecia confocal microscope.
Single Cell RNA Sequencing
Sixteen samples of cells were collected and sent to NHLBI genomic core for single‐cell RNA sequencing using 10× Chromium Single Cell 3′ platform (Pleasanton, CA) with 10× Chromium Next GEM Single Cell 3ʹ Reagent Kits v3.1. For 2D cell culture, the cells dissociated with 0.05% trypsin (ThermoFisher Scientific, 25 300) and kept on the ice until sequencing. For 3D bioprinted tissue, the tissues were chopped into small pieces and incubated with serum‐free culture media containing 450 U collagenase (Gibco, 17018–029) at 37 °C for 30 min with gental rock for collecting cells. The libraries were sequenced on an Illumina Novaseq S2 for the experiment. The raw data were demultiplexed and mapped to a human reference genome (GRCh38‐2020‐A) using CellRanger 6.0.6 (10× Genomics) with standard default pipeline parameters. The raw count matrix for each sample was imported into an R pipeline using the Seurat v4 package. Low‐quality cells (<200 genes, <400 UMI, <0.8 gene complexity (log10GenesPerUMI) and >0.2 mitochondrial ratio) were filtered out from the analysis. Additionally, we kept only genes that are expressed in 10 or more cells and doublet cells further removed by running the Doublet Finder R package. The filtered data then normalized, scaled, and log‐transformed with Seurat packages using the SCTransform method. Principal component analysis (PCA) as well as UMAP were performed using the first 40 principal components and the resolution parameter as 0.4.
Cell Type Identification
The main cell types were identified by annotation of DEG between clusters using the Seurat FindAllMarkers function. The whole dataset also run with a singleR package for cell type prediction using Human Cell Atlas database. Each cluster further annotated manually by using cell‐type‐specific single‐cell signatures from respective cell atlases and curated publication. Labels were added to the main object as primarycelltype and subcelltype identities.
Differential Analysis
Differential Gene Expression Analysis (DEG) was assessed by comparing between different cluster identities (“seurat cluster”,’sampletype’, “primarycelltye”, “subcelltype”) with Seurat FindAllMarkers, and FindMarkers functions. The visualization plot was performed using R packages (EnhancedVolcano and ggplot2). The lists of DEG were saved for Enrichment analysis (g: GOSt and Enrichr analysis). The significance cutoff for Enrichment analysis was 0.25 Log2FC (≈1.2 foldchange) with an adjusted p‐value threshold 0.05.
Enrichment Pathway Analysis
The enrichment analysis was ran using R package EnrichR (libraries: MSigDB_Hallmark_2020, GO_Biological_Process_2021), gProfiler (g: GOSt Gene Ontology database), and clusterprofiler (library Go.db 2.1) packages with selected DEG list from Differential analysis (Described above). Revigoonline tool was used to summarize GO term enrichment results.[ 90 ] The enriched gene lists from each enrichment category were then calculated the combined expression with AddModuleScore and FoldChange function in Seurat packages for visualization. Nichenet analysis[ 91 ] was also performed to predict ligand‐receptor activity change between different culture conditions. The online STRING 11.5 (https://string‐db.org/) was also used for the enrichment pathway analysis section.
Visualization
All plots were generated using Seurat visualization functions, ggplot2, Complexheatmap, and EnhancedVolcano R packages.
Cytokine Measurements
The culture media of bioprinted NVUs collected at days 5, 9, 13, 17, and 21 were stored at −80 °C. A multiplex Luminex Assay (R&D Systems, Cat#LXSAHM‐21) was used to quantitate cytokines. The measurements were done at Frederick National Laboratory for Cancer Research followed by the kit's protocol. Generally, all samples were at least twofold dilution with the Calibrator Diluent RD6‐52. The calibration curve was by mixing the provided standard cocktail with Calibrator Diluent RD6‐52, and a threefold dilution series was preformed to make the curve. Fifty microliters sample/standard was added to each well followed by adding another 50 µl of diluted microparticle cocktail and incubate at RT for 2 hours under 800 rpm shaking. Three times wash with wash buffer was preformed after 2 hours of incubation. After washing, the 50 µl of the biotin‐antibody cocktail was added and incubated at RT for 1 hour on the shaker at 800 rpm. The three times wash with wash buffer was preformed again after the biotin‐antibody cocktail incubation and 50 µl of diluted streptavidin‐PE was added in each well and incubated at RT for 30 minutes under 800 rpm shaking. Another three times wash with wash buffer was preformed after streptavidin‐PE incubation and an additional 100 µl wash buffer was added to each well for Luminex analyzer measurement.
Compound Treatment
Eighteen compounds were serially diluted at a 1/4 ratio in DMSO starting from a 10 mm solution in DMSO. An intermediate dilution with culture media with a 1/25 ratio was made followed by the final dilution with culture media at 1/10 ratio. The compound treatment started at day 15 after printing and continue to day 21. The media with the compound was refreshed every other day. The area of the vessel and tumor was detected through the Opera Phenix High‐Content Screening System and calculated through the Harmony software separately. In order to reduce the well‐to‐well and plate‐to‐plate variation, the data was present as the percentage of size change from day 15 to day 21 compared to DMSO samples (Figure 6b). Data from four different batches with sample numbers at least three were collected in this experiment.
Statistics
ANOVA and Bonferroni post‐hoc multiple comparison test and Student's t‐test for a pair comparison were used as the statistical method and p < 0.05 was considered as significantly different. Standard deviation was used for plotting the error bar.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
The authors thank Jon Inglefield and Yanyu Wang from the Clinical Support Laboratory, Clinical Services Program, Leidos Biomedical Research, Inc., Frederick National Laboratory for Cancer Research for performing the cytokine analysis. This research was supported by the Intramural Research Program and Cure Acceleration Program at NCATS, NIH.
Tung Y.‐T., Chen Y.‐C., Derr K., Wilson K., Song M. J., Ferrer M., A 3D Bioprinted Human Neurovascular Unit Model of Glioblastoma Tumor Growth. Adv. Healthcare Mater. 2024, 13, 2302831. 10.1002/adhm.202302831
Contributor Information
Min Jae Song, Email: minjae.song@nih.gov.
Marc Ferrer, Email: marc.ferrer@nih.gov.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Friedrich J., Seidel C., Ebner R., Kunz‐Schughart L. A., Nat. Protoc. 2009, 4, 309. [DOI] [PubMed] [Google Scholar]
- 2. Banerjee D., Singh Y. P., Datta P., Ozbolat V., O'Donnell A., Yeo M., Ozbolat I. T., Biomaterials 2022, 291, 121881. [DOI] [PubMed] [Google Scholar]
- 3. Kim J., Koo B.‐K., Knoblich J. A., Nat. Rev. Mol. Cell Biol. 2020, 21, 571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chiaradia I., Lancaster M. A., Nat. Neurosci. 2020, 23, 1496. [DOI] [PubMed] [Google Scholar]
- 5. Song L., Yuan X., Jones Z., Griffin K., Zhou Y., Ma T., Li Y., Sci. Rep. 2019, 9, 5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Germain N., Dhayer M., Dekiouk S., Marchetti P., Int. J. Mol. Sci. 2022, 23, 3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Murphy S. V., De Coppi P., Atala A., Nat. Biomed. Eng. 2020, 4, 370. [DOI] [PubMed] [Google Scholar]
- 8. Duarte Campos D. F., Blaeser A., Korsten A., Neuss S., Jakel J., Vogt M., Fischer H., Tissue Eng. Part A 2015, 21, 740. [DOI] [PubMed] [Google Scholar]
- 9. Im S., Choe G., Seok J. M., Yeo S. J., Lee J. H., Kim W. D., Lee J. Y., Park S. A., Int. J. Biol. Macromol. 2022, 205, 520. [DOI] [PubMed] [Google Scholar]
- 10. Daly A. C., Critchley S. E., Rencsok E. M., Kelly D. J., Biofabrication 2016, 8, 045002. [DOI] [PubMed] [Google Scholar]
- 11. Kim J. H., Seol Y. J., Ko I. K., Kang H. W., Lee Y. K., Yoo J. J., Atala A., Lee S. J., Sci. Rep. 2018, 8, 12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sharma R., Smits I. P. M., De La Vega L., Lee C., Willerth S. M., Front. Bioeng. Biotechnol. 2020, 8, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kryou C., Leva V., Chatzipetrou M., Zergioti I., Bioengineering 2019, 6, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schöneberg J., De Lorenzi F., Theek B., Blaeser A., Rommel D., Kuehne A. J., Kießling F., Fischer H., Sci. Rep. 2018, 8, 10430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Song M. J., Quinn R., Nguyen E., Hampton C., Sharma R., Park T. S., Koster C., Voss T., Tristan C., Weber C., Singh A., Dejene R., Bose D., Chen Y. C., Derr P., Derr K., Michael S., Barone F., Chen G., Boehm M., Maminishkis A., Singec I., Ferrer M., Bharti K., Nat. Methods 2023, 20, 149. [DOI] [PubMed] [Google Scholar]
- 16. Derr K., Zou J., Luo K., Song M. J., Sittampalam G. S., Zhou C., Michael S., Ferrer M., Derr P., Tissue Eng., Part C 2019, 25, 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Augustine R., Kalva S. N., Ahmad R., Zahid A. A., Hasan S., Nayeem A., McClements L., Hasan A., Transl. Oncol. 2021, 14, 101015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wen P. Y., Weller M., Lee E. Q., Alexander B. M., Barnholtz‐Sloan J. S., Barthel F. P., Batchelor T. T., Bindra R. S., Chang S. M., Chiocca E. A., Neuro‐Oncology 2020, 22, 1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee J., Kotliarova S., Kotliarov Y., Li A. G., Su Q., Donin N. M., Pastorino S., Purow B. W., Christopher N., Zhang W., Park J. K., Fine H. A., Cancer Cell 2006, 9, 391. [DOI] [PubMed] [Google Scholar]
- 20. Hubert C. G., Rivera M., Spangler L. C., Wu Q. L., Mack S. C., Prager B. C., Couce M., McLendon R. E., Sloan A. E., Rich J. N., Cancer Res. 2016, 76, 2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jacob F., Salinas R. D., Zhang D. Y., Nguyen P. T. T., Schnoll J. G., Wong S. Z. H., Thokala R., Sheikh S., Saxena D., Prokop S., Liu D. A., Qian X., Petrov D., Lucas T., Chen H. I., Dorsey J. F., Christian K. M., Binder Z. A., Nasrallah M., Brem S., O'Rourke D. M., Ming G. L., Song H., Cell 2020, 180, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang C., Jin M., Zhao J., Chen J., Jin W., Am. J. Cancer Res. 2020, 10, 2242. [PMC free article] [PubMed] [Google Scholar]
- 23. Tatla A. S., Justin A. W., Watts C., Markaki A. E., Sci. Rep. 2021, 11, 19550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McCoy M. G., Nyanyo D., Hung C. K., Goerger J. P., Zipfel W. R., Williams R. M., Nishimura N., Fischbach C., Sci. Rep. 2019, 9, 9069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marino A., Battaglini M., Carmignani A., Pignatelli F., De Pasquale D., Tricinci O., Ciofani G., APL Bioeng. 2023, 7, 36103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Han S., Kim S., Chen Z., Shin H. K., Lee S. Y., Moon H. E., Paek S. H., Park S., Int. J. Mol. Sci. 2020, 21, 2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yi H. G., Jeong Y. H., Kim Y., Choi Y. J., Moon H. E., Park S. H., Kang K. S., Bae M., Jang J., Youn H., Paek S. H., Cho D. W., Nat. Biomed. Eng. 2019, 3, 509. [DOI] [PubMed] [Google Scholar]
- 28. Piantino M., Figarol A., Matsusaki M., Front. Toxicol. 2021, 3, 656254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wilson K. M., Mathews‐Griner L. A., Williamson T., Guha R., Chen L., Shinn P., McKnight C., Michael S., Klumpp‐Thomas C., Binder Z. A., SLAS Technol. 2019, 24, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Angara K., Borin T. F., Arbab A. S., Transl. Oncol. 2017, 10, 650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Garcia F. J., Sun N., Lee H., Godlewski B., Mathys H., Galani K., Zhou B., Jiang X., Ng A. P., Mantero J., Tsai L. H., Bennett D. A., Sahin M., Kellis M., Heiman M., Nature 2022, 603, 893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ross J. M., Kim C., Allen D., Crouch E. E., Narsinh K., Cooke D. L., Abla A. A., Nowakowski T. J., Winkler E. A., Front. Physiol. 2020, 11, 600767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Orsenigo F., Conze L. L., Jauhiainen S., Corada M., Lazzaroni F., Malinverno M., Sundell V., Cunha S. I., Brannstrom J., Globisch M. A., Maderna C., Lampugnani M. G., Magnusson P. U., Dejana E., Elife 2020, 9, e61413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Smyth L. C. D., Rustenhoven J., Scotter E. L., Schweder P., Faull R. L. M., Park T. I. H., Dragunow M., J. Chem. Neuroanat. 2018, 92, 48. [DOI] [PubMed] [Google Scholar]
- 35. Brandt M. M., van Dijk C. G. M., Maringanti R., Chrifi I., Kramann R., Verhaar M. C., Duncker D. J., Mokry M., Cheng C., Sci. Rep. 2019, 9, 15586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jurga A. M., Paleczna M., Kadluczka J., Kuter K. Z., Biomolecules 2021, 11, 1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bjørnsen L. P., Hadera M. G., Zhou Y., Danbolt N. C., Sonnewald U., J. Neurochem. 2014, 128, 641. [DOI] [PubMed] [Google Scholar]
- 38. Dejana E., Hirschi K. K., Simons M., Nat. Commun. 2017, 8, 14361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Szopa W., Burley T. A., Kramer‐Marek G., Kaspera W., Biomed. Res. Int. 2017, 2017, 8013575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hassn Mesrati M., Behrooz A. B., Abuhamad A. Y., Syahir A., Cells 2020, 9, 1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ludwig K., Kornblum H. I., J. Neurooncol. 2017, 134, 505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rocha S. F., Schiller M., Jing D., Li H., Butz S., Vestweber D., Biljes D., Drexler H. C., Nieminen‐Kelha M., Vajkoczy P., Adams S., Benedito R., Adams R. H., Circ. Res. 2014, 115, 581. [DOI] [PubMed] [Google Scholar]
- 43. Gerhardt H., Golding M., Fruttiger M., Ruhrberg C., Lundkvist A., Abramsson A., Jeltsch M., Mitchell C., Alitalo K., Shima D., Betsholtz C., J. Cell Biol. 2003, 161, 1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lattke M., Goldstone R., Ellis J. K., Boeing S., Jurado‐Arjona J., Marichal N., MacRae J. I., Berninger B., Guillemot F., Nat. Commun. 2021, 12, 4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rueda‐Carrasco J., Martin‐Bermejo M. J., Pereyra G., Mateo M. I., Borroto A., Brosseron F., Kummer M. P., Schwartz S., Lopez‐Atalaya J. P., Alarcon B., Esteve P., Heneka M. T., Bovolenta P., EMBO Rep. 2021, 22, e51696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Simon M. J., Wang M. X., Murchison C. F., Roese N. E., Boespflug E. L., Woltjer R. L., Iliff J. J., Sci. Rep. 2018, 8, 12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brandt M. M., Van Dijk C. G., Maringanti R., Chrifi I., Kramann R., Verhaar M. C., Duncker D. J., Mokry M., Cheng C., Sci. Rep. 2019, 9, 15586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chan J. M., Quintanal‐Villalonga A., Gao V. R., Xie Y., Allaj V., Chaudhary O., Masilionis I., Egger J., Chow A., Walle T., Cancer Cell 2021, 39, 1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhuo L., Gong J., Yang R., Sheng Y., Zhou L., Kong X., Cao K., Biochem. Biophys. Res. Commun. 2009, 390, 451. [DOI] [PubMed] [Google Scholar]
- 50. Rathbone S. R., Glossop J. R., Gough J. E., Cartmell S. H., J. Mech. Behav. Biomed. Mater. 2012, 11, 82. [DOI] [PubMed] [Google Scholar]
- 51. Aird W. C., Cold Spring Harbor Perspect. Med. 2012, 2, a006429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xiong J., Kawagishi H., Yan Y., Liu J., Wells Q. S., Edmunds L. R., Fergusson M. M., Yu Z. X., Rovira, Brittain E. L., Wolfgang M. J., Jurczak M. J., Fessel J. P., Finkel T., Mol. Cell 2018, 69, 689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Choi K. J., Nam J. K., Kim J. H., Choi S. H., Lee Y. J., Exp. Mol. Med. 2020, 52, 781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xiang Y., Zhang Y., Tang Y., Li Q., Cell. Physiol. Biochem. 2017, 42, 357 [DOI] [PubMed] [Google Scholar]
- 55. Zhou Z. W., Zheng L. J., Ren X., Li A. P., Zhou W. S., Brain Res. 2019, 1707, 90. [DOI] [PubMed] [Google Scholar]
- 56. Banks W. A., Kovac A., Morofuji Y., J. Cereb. Blood Flow Metab. 2018, 38, 1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Reis M., Liebner S., Exp. Cell Res. 2013, 319, 1317. [DOI] [PubMed] [Google Scholar]
- 58. Fontaine M., Herkenne S., Ek O., Paquot A., Boeckx A., Paques C., Nivelles O., Thiry M., Struman I., Int. J. Mol. Sci. 2021, 23, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Heymans M., Figueiredo R., Dehouck L., Francisco D., Sano Y., Shimizu F., Kanda T., Bruggmann R., Engelhardt B., Winter P., Gosselet F., Culot M., Fluids Barriers CNS 2020, 17, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. McCord M., Mukouyama Y. S., Gilbert M. R., Jackson S., Front. Cell. Neurosci. 2017, 11, 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wu D., Xu Y., Ding T., Zu Y., Yang C., Yu L., Cell Res. 2017, 27, 1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gregorio I., Braghetta P., Bonaldo P., Cescon M., Dis. Models Mech. 2018, 11, dmm032946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Soles A., Selimovic A., Sbrocco K., Ghannoum F., Hamel K., Moncada E. L., Gilliat S., Cvetanovic M., Int. J. Mol. Sci. 2023, 24, 7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Stanke K. M., Wilson C., Kidambi S., Front. Mol. Biosci. 2021, 8, 752404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li X.‐y., Ma W.‐N., Su L.‐x., Shen Y., Zhang L., Shao Y., Wang D., Wang Z., Wen M.‐Z., Yang X.‐t., Front. Cell Dev. Biol. 2022, 10, 805507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wechman S. L., Emdad L., Sarkar D., Das S. K., Fisher P. B., Adv. Cancer Res. 2020, 148, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jarad M., Kuczynski E. A., Morrison J., Viloria‐Petit A. M., Coomber B. L., BMC Cell Biol. 2017, 18, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. del Toro R., Prahst C., Mathivet T., Siegfried G., Kaminker J. S., Larrivee B., Breant C., Duarte A., Takakura N., Fukamizu A., Blood 2010, 116, 4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fang J. S., Hultgren N. W., Hughes C. C. W., Front. Cell Dev. Biol. 2021, 9, 702021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Piera‐Velazquez S., Jimenez S. A., Physiol. Rev. 2019, 99, 1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pérez L., Muñoz‐Durango N., Riedel C. A., Echeverría C., Kalergis A. M., Cabello‐Verrugio C., Simon F., Cytokine Growth Factor Rev. 2017, 33, 41. [DOI] [PubMed] [Google Scholar]
- 72. Jumeau C., Awad F., Assrawi E., Cobret L., Duquesnoy P., Giurgea I., Valeyre D., Grateau G., Amselem S., Bernaudin J. F., Karabina S. A., PLoS One 2019, 14, e0217005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Johnson B. Z., Stevenson A. W., Prele C. M., Fear M. W., Wood F. M., Biomedicines 2020, 8, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rennekampff H. O., Hansbrough J. F., Kiessig V., Dore C., Sticherling M., Schroder J. M., J. Surg. Res. 2000, 93, 41. [DOI] [PubMed] [Google Scholar]
- 75. Labani‐Motlagh A., Ashja‐Mahdavi M., Loskog A., Front. Immunol. 2020, 11, 940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mojic M., Takeda K., Hayakawa Y., Int. J. Mol. Sci. 2017, 19, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tormoen G. W., Crittenden M. R., Gough M. J., Adv. Radiat. Oncol. 2018, 3, 520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lugano R., Ramachandran M., Dimberg A., Cell. Mol. Life Sci. 2020, 77, 1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Khan S., Mahalingam R., Sen S., Martinez‐Ledesma E., Khan A., Gandy K., Lang F. F., Sulman E. P., Alfaro‐Munoz K. D., Majd N. K., Balasubramaniyan V., de Groot J. F., Cancers 2021, 13, 5284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hastings J. F., Skhinas J. N., Fey D., Croucher D. R., Cox T. R., Br. J. Pharmacol. 2019, 176, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hambardzumyan D., Gutmann D. H., Kettenmann H., Nat. Neurosci. 2016, 19, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kälin R. E., Cai L., Li Y., Zhao D., Zhang H., Cheng J., Zhang W., Wu Y., Eisenhut K., Janssen P., Cell Syst. 2021, 12, 248. [DOI] [PubMed] [Google Scholar]
- 83. Tang M., Xie Q., Gimple R. C., Zhong Z., Tam T., Tian J., Kidwell R. L., Wu Q., Prager B. C., Qiu Z., Yu A., Zhu Z., Mesci P., Jing H., Schimelman J., Wang P., Lee D., Lorenzini M. H., Dixit D., Zhao L., Bhargava S., Miller T. E., Wan X., Tang J., Sun B., Cravatt B. F., Muotri A. R., Chen S., Rich J. N., Cell Res. 2020, 30, 833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lubanska D., Porter L., Drugs R$D 2017, 17, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Saqub H., Proetsch‐Gugerbauer H., Bezrookove V., Nosrati M., Vaquero E. M., de Semir D., Ice R. J., McAllister S., Soroceanu L., Kashani‐Sabet M., Osorio R., Dar A. A., Sci. Rep. 2020, 10, 18489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Pudełek M., Król K., Catapano J., Wróbel T., Czyż J., Ryszawy D., Int. J. Mol. Sci. 2020, 21, 3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Keller S., Schmidt M. H. H., Int. J. Mol. Sci. 2017, 18, 1295.28629170 [Google Scholar]
- 88. Liu H.‐L., Huang C.‐Y., Chen J.‐Y., Wang H.‐Y. J., Chen P.‐Y., Wei K.‐C., PLoS One 2014, 9, e114311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wilson K. M., Mathews‐Griner L. A., Williamson T., Guha R., Chen L., Shinn P., McKnight C., Michael S., Klumpp‐Thomas C., Binder Z. A., Ferrer M., Gallia G. L., Thomas C. J., Riggins G. J., SLAS Technol. 2019, 24, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Supek F., Bošnjak M., Škunca N., Šmuc T., PLoS One 2011, 6, e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Browaeys R., Saelens W., Saeys Y., Nat. Methods 2020, 17, 159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.