

Abstract
Mixed methods studies of human disease that combine surveillance, biomarker, and qualitative data can help elucidate what drives epidemiological trends. Viral genetic data are rarely coupled with other types of data due to legal and ethical concerns about patient privacy. We developed a novel approach to integrate phylogenetic and qualitative methods in order to better target HIV prevention efforts. The overall aim of our mixed methods study was to characterize HIV transmission clusters. We combined surveillance data with HIV genomic data to identify cases whose viruses share enough similarities to suggest a recent common source of infection or participation in linked transmission chains. Cases were recruited through a multi-phase process to obtain consent for recruitment to semi-structured interviews. Through linkage of viral genetic sequences with epidemiological data, we identified individuals in large transmission clusters, which then served as a sampling frame for the interviews. In this article, we describe the multi-phase process and the limitations and challenges encountered. Our approach contributes to the mixed methods research field by demonstrating that phylogenetic analysis and surveillance data can be harnessed to generate a sampling frame for subsequent qualitative data collection, using an explanatory sequential design. The process we developed also respected protections of patient confidentiality. The novel method we devised may offer an opportunity to implement a sampling frame that allows for the recruitment and interview of individuals in high-transmission clusters to better understand what contributes to spread of other infectious diseases, including COVID-19.
In the health sciences, “mixed methods” research has long been at least an implicit part of many researchers’ practice (Andrew & Halcomb, 2009), but the language used to describe the mixing, and the particular methods mixed, have been highly variable (Creswell, Klassen, Plano Clark, & Clegg Smith, 2011). Critical engagement with mixed methods as a formal approach to research in the field is growing (Creswell, 2009), and is increasingly of interest to the U.S. National Institutes of Health (NIH) (Creswell, 2010; Plano Clark, 2010). As a major funder of HIV research, NIH priorities impact the field, and quantitative and qualitative components are now often incorporated within larger research projects, including clinical trials. A famous example is the VOICE trial, which tested multiple pharmaceutical methods of preventing HIV in women. VOICE failed to produce evidence of effectiveness for any of the study arms despite good self-reported adherence during the trial; when drug concentration levels in participants’ blood were measured, however, results suggested widespread non-adherence (Marrazzo et al., 2015). A qualitative post-trial sub-study was conducted to explore participants’ reasons for over-reporting adherence (Montgomery et al., 2017).
The drug concentration levels measurements in the VOICE study are an example of biomarkers, which can be defined simply as measurable indicators of the physiological state of an organism. Oft-used biomarkers in HIV-related research include infection status (HIV or other sexually-transmitted infections) and drug levels (e.g., to assess medication adherence, toxicity). In mixed methods studies, these are frequently coupled with surveys or semi-structured interviews (Alary et al., 2015; Koester et al., 2015). One potentially useful biomarker that has not been frequently incorporated in mixed methods research is viral genetic data. Previously, processing times and cost to obtain such data were often prohibitive, but technological advances now make the use of this particular type of biomarker more feasible than ever before. The availability of such data, however, raises new kinds of questions and concerns in terms of research design and ethics. As such, the incorporation of genetic data to mixed methods studies requires careful consideration and, potentially, new kinds of partnerships and research practices.
This manuscript documents the novel approach we developed to mix the analysis of genetic data with qualitative methods, in order to explore whether this could offer greater opportunities for targeting HIV prevention. We adopted an explanatory sequential design (Fetters, Curry, & Creswell, 2013), in which previously collected viral genetic samples were subjected to phylogenetic analysis, and the findings informed sampling for semi-structured interviews from within so-called “transmission clusters” (more on this below). The two phases were connected, or integrated, not only at the point of selecting and recruiting participants for the qualitative component (Ivankova, Creswell, & Stick, 2006), but in other dimensions as well (e.g., team dimension, as our team was intentionally chosen to represent various disciplines; at the theoretical dimension through framing that employed the Social Ecological Model) (Fetters & Molina-Azorin, 2017). While use of the explanatory sequential design in HIV-related mixed methods projects was common at the time this research was conceived, to the best of our knowledge, no previously published studies mixed the particular methods we employed, in the way we devised. Indeed, phylogenetic analysis had not typically been mixed with other methods at all. Even today, publications from studies that bring phylogenetic analysis together with other methods do not discuss their study design using mixed methods terminology (see below).
The overall aim of our mixed-methods study was to characterize HIV transmission clusters in San Francisco. With this paper, our goal is multi-fold. First, we aim to paint a general picture of the kinds of challenges we feel would be relevant to any mixed methods study incorporating similar biomarkers (e.g., construction of workable, multi-party partnerships, respect for participant privacy, feasibility of recruitment). Second, we offer detailed information on the method itself so that other researchers can attempt to replicate or adapt it to ethically use sensitive genetic data in a mixed methods approach. Third, we hope to encourage more scholarly conversations regarding the use of mixed-methods in phylogenetics studies. In what follows, we outline the potential value of our novel approach, offer brief background on phylogenetics and its use in HIV research (including previous, mixed methods work), explain how our approach is different, and identify two main challenges it faced from the outset (ethical implementation and recruitment feasibility). Then we detail the methods and provide recruitment results, along with a brief example of thematic findings from one cluster to better illustrate how the method works. In the Discussion, we highlight surprises, lessons learned, and reflect briefly on the potential applicability of the method to research on other infections, including SARS-CoV-2.
Contribution to HIV Prevention Research
Before detailing our mixed methods technique and how it differs from previous work, we note why we believe devising a novel approach is worth the effort—that is, how we imagine it would contribute to HIV prevention research. In general, the hope is that this particular mixed method approach would produce data that, when integrated, would achieve an “expansive” fit (Fetters et al., 2013), enriching our understanding of the dynamics that structure HIV transmission within particular clusters by revealing previously unknown patterns among cluster members. Those findings could then help enhance the impact of prevention efforts. For instance, though it was a decade ago (Grant et al., 2010) that science demonstrated the effectiveness of HIV pre-exposure prophylaxis (PrEP—a daily pill taken to prevent HIV), this is still far from universal knowledge. If our research found that a large proportion of people in a particular cluster had never heard of PrEP, this could indicate that public health campaigns are missing important segments of their intended audience. On the other hand, a finding that certain locations are implicated in transmission clusters would suggest targeted prevention strategies, such as offering testing services and prevention materials at such epidemiological “hotspots,” could be of value. Finally, if homelessness or substance abuse formed the behavioral context of rapidly growing clusters, then a narrow focus on providing PrEP or HIV testing would be unlikely to be ideally effective. In short, by more fully characterizing transmission clusters, our approach could identify key drivers of the epidemic and help direct HIV prevention resources to their most efficacious use.
Brief Background on HIV and Viral Genetic Data
As noted above, in general, technological advances have made genetic analysis faster and cheaper, and therefore more feasible to incorporate in mixed methods studies. Regarding HIV specifically, there is another contributor to this feasibility. Since 2016 U.S. guidelines for HIV treatment have recommended antiretroviral (ARV) drug resistance testing at time of diagnosis (Günthard et al., 2016), and this includes analyzing viral genetic sequences (i.e., the order of the virus’s genetic information). Thus, greater availability of HIV-related biomarkers is a by-product of appropriate treatment for the virus. However, even before this, viral sequences had been used in phylogenetic analysis, which seeks to understand the evolutionary development and diversification of the virus. Often the goal has been to identify transmission clusters, defined as groups of cases whose viruses share enough similarities to suggest a recent common source of infection or participation in linked chains of transmission. Epidemiological linkage data are displayed in phylogenetic “trees” (a diagram that shows the evolutionary interrelations of a group of genes from viruses derived from a common source), to visualize the transmission clusters. This analytical tool has long been used in HIV research (Leitner, Escanilla, Franzén, Uhlén, & Albert, 1996), though only relatively recently have researchers begun to explore incorporating socio-demographic information as a way to seek new possibilities for intervention, most specifically at the transmission cluster level.
Utility of phylogenetics during the COVID-19 pandemic
The current COVID-19 pandemic also demonstrates the value of phylogenetics in both the research and public health arenas. Viral sequencing of SARS-CoV-2 has been used to explore possible origins and reservoirs for the novel coronavirus (e.g., Shereen et al., 2020; Rothan & Byrareddy, 2020; Zhang et al., 2020), as well as its evolution in people (e.g., Stefanelli et al., 2020). Genomic sequencing of SARS-CoV-2 led to the recognition of the emergence of variants that are potentially more transmissible and cause more severe disease, such as B.1.1.7 initially identified in the United Kingdom (Rambaut et al., 2020), B.1.351 initially identified in South Africa (Tegally et al., 2020) and B.1.1.28 initially identified in Brazil (Candido et al., 2020). Phylogenetic analysis has also been used to characterize COVID-19 clusters, superspreader events and transmission dynamics, both retrospectively (Chan et al., 2020; Deng et al., 2020; Gonzalez-Reiche et al., 2020; Lemieux et al., 2021) and prospectively (Meredith et al., 2020).
Phylogenetic Analysis in Mixed Methods Research
As noted previously, the push to combine phylogenetic data sets with clinical, epidemiological, and behavioral information is recent (Delva, Leventhal, & Helleringer, 2016; Frost & Pillay, 2015; German, Grabowski, & Beyrer, 2017). This has previously been pursued in two basic ways. In one approach, researchers typically access two existing pools of data, which may have accumulated over long periods of time: viral samples, and information drawn from sources like HIV case surveillance, contact tracing/partner notification, or clinical records. After conducting phylogenetic analysis on the viral samples and producing hypothesized transmission clusters, researchers “tag” sequences in the clusters with additional details about the individuals, for example, gender, race/ethnicity, exposure category, number of contacts reported, results of other lab work (Avila et al., 2014; Chan et al., 2015; Dennis et al., 2015; Jovanović et al., 2019; Levy et al., 2011; Lin et al., 2013; Little et al., 2014; Oster et al., 2011; Pasquale et al., 2016; Poon et al., 2015; Wertheim et al., 2017). Because interest in HIV transmission clusters often drives the research, social and demographic information are usually only added for clustered individuals. The limitation of this approach is that, as researchers do not generally attempt to re-locate cluster members, only previously-collected data can be added. In essence, this approach entails retrospectively building a bridge between two existing data sources that may not have been envisioned as complementary at the time of their creation. Although none of the studies cited here used this terminology, they might technically be described as using an explanatory sequential design (Fetters et al., 2013). The quantitative/phylogenetic analysis did occur first, and “qualitative data collection and analysis” followed—but only in the sense that qualitative data were retrieved after clusters are produced; scant “analysis” (if any) was performed on the non-genetic data separately.
An alternate, more prospective approach has sometimes been taken in research aiming to reconstruct and characterize specific local transmission networks. For these studies, participants recruited from particular locales consent to contributing two types of data at roughly the same time: viral samples for genotyping and demographic and risk behavior data, most often via computer-assisted self-interviews or questionnaires (Dennis et al., 2018; Grabowski et al., 2014; Jafa et al., 2009; Kharsany et al., 2014; Kiwuwa-Muyingo et al., 2017; Lee et al., 2009; Robertson et al., 2014). In contrast to the first group of studies discussed, this research is characterized by a convergent design, with the explicit intention of merging (Fetters et al., 2013) the two data sets. Using this approach, more (and more varied) non-genetic data were collected from research participants and hence were available to subsequently add to transmission clusters; however, sampling for the qualitative data collection was not driven by cluster membership because clusters had not been identified at the time data collection took place. A final detail bears mention about these previous efforts to incorporate phylogenetic analysis in a mixed methods approach: we could only locate one prior mixed-method HIV transmission cluster research study that included semi-structured interviews in the study design (Robertson et al., 2014). In this study, however, sampling for those interviews was not guided by cluster membership.
As methodologists will appreciate, these subtleties are significant because in any study, the type of data collected and the way participants are sampled have profound implications for the types of questions the resulting data set is capable of answering. Since the aim of our study was to discover whether deeply contextualized social aspects of HIV transmission clusters might reveal new opportunities for HIV prevention, two elements of research design were crucial: 1) that our sample for qualitative data collection be recruited with reference to cluster membership, and 2) that we collect data capable of sufficiently exploring cluster members’ experience. Hence, we developed an approach that uses HIV transmission clusters as a sampling frame for semi-structured interviews to study HIV transmission clusters in San Francisco. (We also included any patients with new infections and a few patients not from a cluster to keep interviewers blinded to whether a particular participant was in a cluster; the same recruitment procedures were used for these patients, but their data are not included in this paper). Our approach combines two elements of previous work in a novel way: it combines the sampling by cluster frequently employed in the first group of studies mentioned above with the strategy of collecting additional data directly from research participants used in the second group. Our use of semi-structured interviewing is also novel, allowing a more holistic and in-depth exploration of participants’ perceptions and experiences than is possible through questionnaires or other structured methods (Arnold & Lane, 2011). Findings from thematic analysis of interview data by cluster will be reported in a separate manuscript.
Recruitment Challenges Using Surveillance Data
This method raised two important questions, acknowledged from the outset. First: would enough cluster members be locatable/contactable to render this approach feasible? Second: could we devise ethical ways to identify and recruit potential interviewees (especially regarding respect for privacy)? The latter concern has increasingly been raised with reference to phylogenetic research (Coltart et al., 2018; Mutenherwa, Wassenaar, & de Oliveira, 2019; Schairer et al., 2017). This paper details the procedures we developed, and answers these two questions.
Methods
Overview
Collaborating Investigator Institutions
This study is a collaborative effort between the University of California San Francisco (UCSF) and the San Francisco Department of Public Health (SFDPH). The AIDS Research Institute (ARI)-UCSF Laboratory of Clinical Virology, which conducts HIV-1 drug resistance testing for publicly-funded and community-based clinics in San Francisco, provided viral sequence data exclusively for the purposes of phylogenetic analysis for this study.
Identifying & Selecting Clusters
The parameters used for identifying transmission clusters have been described previously (Truong et al., 2015). In brief, HIV transmission cluster membership was based on full protease and partial reverse transcriptase sequences. Transmission clusters were defined as having Shimodaira-Hasegawa node support greater than 0.90 and mean pairwise genetic distance less than 0.03 substitutions per site. Sequences from transmission clusters were matched to the San Francisco HIV/AIDS Case Registry to obtain data on demographic and risk characteristics. Purposeful selection of transmission clusters for possible inclusion in the qualitative component of the study was conducted between 2015 and 2017 (described below).
Recruitment
A multi-phase recruitment process followed, focusing on individuals who had contributed sequences to selected clusters. The process respected individual patient confidentiality while allowing the SFDPH-based recruiter (hereafter, “recruiter”) and qualitative team at UCSF (hereafter, UCSF) to jointly manage recruitment and ensure that potential participants met the following eligibility criteria: at least 16 years of age,1 cognitive capacity to provide informed consent; and their ability to understand and respond to interview questions in English (because translating the lengthy qualitative interview guide and survey to the multiple major foreign languages spoken by San Francisco residents was not feasible), commute to interview location, and complete a 2-hour interview. The UCSF team scheduled and conducted the interviews. Given the extensive communication and potentially sensitive information involved in this research, special attention was devoted to the design of study-specific data-transfer architecture, which included a searchable, de-identified database to track recruitment efforts, and a separately-stored spreadsheet to transfer names and contact information of consenting participants (described in detail below). This research was approved by the UCSF Institutional Review Board.
Detailed Methods
We designed the procedures below to identify and select clusters for recruitment (Phase 1–2), and recruit and interview selected individuals from those clusters (Phase 3–5), as shown in Figure 1. A critical aspect of our collaboration was that SFDPH utilized existing patient contact information to work directly with the potential participants, thereby safeguarding their anonymity until specific consent to release their names and contact information to UCSF could be obtained.
Figure 1:
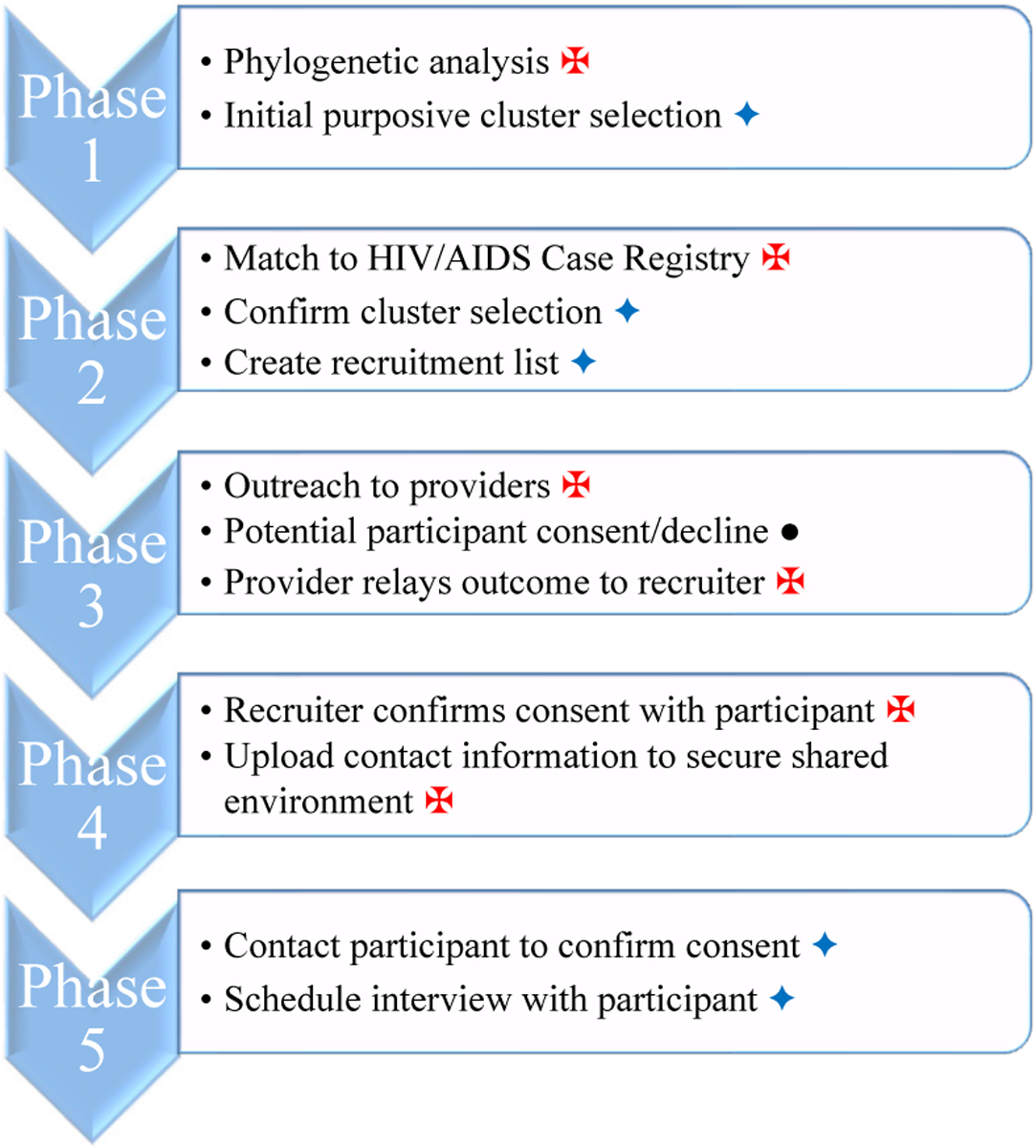
Phases of sample selection and participant recruitment for qualitative interviews conducted by UCSF and SFDPH, San Francisco, 2015–2017.
Phase 1: Identifying Initial Clusters
SFDPH-based personnel analyzed viral genetic data provided by the ARI-UCSF Laboratory of Clinical Research to identify HIV transmission clusters. The SFDPH analyst annotated each sequence within the transmission clusters. This provided a unique but anonymous identifier for each sequence and for each contributing individual. The latter was necessary because roughly one-quarter of patients had contributed multiple sequences to the data set; we included all sequences (rather than only the earliest) in the phylogenetic analysis. Additional information about each sequence included mode of transmission and gender (see Table 1). Annotated, anonymized clusters were uploaded to a shared, secure online platform, hosted by UCSF, to which the SFDPH and UCSF study teams both had access.
Table 1:
Cluster characteristics and sample size during Phases 1 and 2, and data collection for qualitative interviews, San Francisco, 2015–2017.
| Phase 1 | Phase 2 | Data Collection | |||||
|---|---|---|---|---|---|---|---|
| Cluster ID | Number of Sequences | Matched Individuals N | HIV Transmission Mode | Female Gender | Recruitable Individuals† n (%)1 | Race/Ethnicity | Interviewed Individuals n (%)2 |
| A | 29 | 20* | 18 MSM 2 MSM-PWID |
0 CW 0 TW |
10 (50) | 6 PC 4 white |
6 (60) |
| B | 17 | 14* | 5 PWID 4 MSM 4 MSM-PWID 1 HET |
2 CW 4 TW |
9 (64) | 8 PC 1 white |
3 (33) |
| C | 16 | 15* | 12 MSM 3 MSM-PWID |
0 CW 0 TW |
8 (53) | 4 PC 4 white |
3 (38) |
| D | 16 | 11 | 5 HET 3 PWID 2 MSM 1 NIR |
6 CW 0 TW |
8 (73) | 8 PC 0 white |
1 (13) |
| E | 16 | 13 | 12 MSM 1 MSM-PWID |
0 CW 1 TW |
8 (62) | 5 PC 3 white |
3 (38) |
| F | 16 | 9* | 3 PWID 3 HET 2 NRR 1 NIR |
5 CW 0 TW |
7 (78) | 6 PC 1 white |
2 (29) |
| G | 14 | 11 | 6 MSM-PWID 2 MSM 2 PWID 1 HET |
1 CW 2 TW |
9 (82) | 5 PC 4 white |
3 (33) |
| H | 12 | 11 | 9 MSM 1 MSM-PWID 1 PWID |
0 CW 0 TW |
9 (82) | 3 PC 6 white |
3 (33) |
Contained one sequence that was unmatchable (out of SFDPH jurisdiction), hence not counted
= number of matched individuals less the number deceased, lost to follow-up, out of jurisdiction
Number and percent are out of the total number of matched individuals in the cluster
Number and percent are out of the total number of recruitable individuals the cluster
MSM: men who have sex with men
PWID: person who injects drugs
MSM-PWID: MSM who inject drugs
HET: heterosexual
NIR: no identified risk
NRR: no risk reported
CW: cis-women
TW: transwomen
PC: persons of color
UCSF reviewed the data seeking the largest clusters for possible inclusion in the qualitative component, and determining how many clusters to select for recruitment. Clusters composed of more than ten sequences were selected. Several factors contributed to the determination of this minimum cluster size. First, our objective was to interview a meaningful proportion (20–40%) of persons in all selected clusters and we assumed the participation rate for interviews would not exceed 50%. Second, it was anticipated that some persons in clusters would be deceased, lost to follow-up (LTFU), or fall outside of SFDPH’s jurisdiction (OOJ), and therefore be ineligible to participate. These considerations collectively suggested that very small clusters would be unlikely to yield sufficient interviews.
Further, recognizing the possibility of multiple contributions from individual patients, among clusters with ten or more sequences, those with the greatest number of unique individuals were assigned the highest priority for recruitment. UCSF uploaded the list of selected clusters to the secure online platform.
Phase 2: Selecting Clusters
SFDPH-based personnel accessed the HIV/AIDS Case Registry to obtain additional demographic and risk data on the cases in clusters prioritized for recruitment. Data included sample collection date, HIV diagnosis date, age at diagnosis (given as a range), race/ethnicity, and whether the individual was not “recruitable” due to being deceased, LTFU, or OOJ. The revised dataset was uploaded into the secure online platform.
Although all cases were still anonymized at this stage, these additional data allowed UCSF to review the overall diversity of the pool of possible participants, as well as determine the number of recruitable individuals in each cluster. Using this information, UCSF generated a final recruitment spreadsheet and uploaded it to the secure, online systems.
Phase 3: Identifying and Informing Potential Study Participants
The SFDPH’s HIV/AIDS Case Registry contains all the information needed to contact the selected patients directly. Nevertheless, the investigators deemed it prudent to start the recruitment process by first notifying medical providers about the selected patients. Hearing about the study first from providers or their office staff would help to minimize the impression of direct intrusion by the study into the patients’ private medical circumstances. The procedure would also allow providers to advise the recruiter to refrain pursuing any patient who should not be approached.
This recruitment process called for the SFDPH-based recruiter to use the HIV/AIDS Case Registry data to identify the medical provider of each individual selected for recruitment and contact the provider through telephone or email using approved scripts/materials and following privacy procedures compliant with the Health Insurance Portability and Accountability Act of 1996 (HIPAA). The recruiter described the study to the provider and either requested that the provider inform the patient about the opportunity to participate or obtained the provider’s assent to have the recruiter approach the patient directly. Providers who opted to inform their patients would discuss the study with their patients, ascertain their interest in learning more about the study, and relay the response back to the recruiter. Patients who wished to receive further information about the study would verbally consent to contact by the recruiter. In cases where providers assented to have the recruiter directly contact and introduce the study to their patients, the recruiter used the contact information already available to the recruiter in the registry.
Phase 4: Initial Consent to Be Contacted by UCSF
The recruiter called all selected patients who had been approached by their providers or whose providers assented to the contact to explain the study in greater detail, specifically indicating that the UCSF study involved a 2-hour, recorded interview that required the recruiter to pass patients’ contact information to UCSF. If interested, they were asked to consent for the release of their name and contact information to the UCSF team. Only then was contact information for consenting participants added to the recruitment spreadsheet on the secure online platform. The recruiter also alerted the UCSF interviewers immediately by email that new contact information had been added to the recruitment spreadsheet.
Phase 5: Study Recruitment & Scheduling
The UCSF study team accessed the secure online spreadsheet to obtain names and contact information for potential interviewees as they became available. UCSF interviewers contacted potential participants to answer any remaining questions and schedule qualitative interviews.
Qualitative data collection
Interviews took place in a private location and were conducted by one of two trained qualitative researchers on the study team. The interviews lasted between 1–2 hours, and were audio recorded. They employed a narrative approach to the initial, semi-structured portion (to enhance interviewees’ ability to account for their lived experience) and a structured survey at the end (to ensure a small number of standard data points were covered with each participant). Topics covered included pre- and post-diagnosis conceptions of risk, prevention strategies, sexual practices, and patterns of drug use, as well as the context around seroconversion, HIV diagnosis, and ARV treatment. Participants received an incentive of $75 as compensation for time and travel.
Results
Phase 1: Identifying Initial Clusters
The study’s initial dataset contained 7,253 viral sequences, from which our analysis identified 1,392 clusters. Nine of those clusters contained more than ten sequences. UCSF deemed the largest eight clusters likely sufficient to satisfy needed sample size. These eight clusters ranged in size from 12–29 sequences, contributed by 9–20 unique individuals who could be matched to records in the case registry (again, clusters with the greatest number of unique individuals were prioritized for recruitment). Of the 104 individuals in the identified clusters, 83 were cis-men, 14 were cis-women, and seven were transwomen. In four of the clusters, the mode of HIV transmission reported for the majority of members was sex among men who have sex with men (MSM). In one cluster, transmission was largely MSM and people who injection drugs (PWID); three clusters contained mixed HIV transmission modes. Collectively, less than 60% of the transmission for cases within the chosen eight clusters was attributed to MSM. More details on the clusters selected for recruitment are shown in Table 1.
Phase 2: Selecting Clusters
Cluster members known to be deceased, LTFU or OOJ were removed. The remaining clusters contained between seven and ten individuals for whom recruitment efforts could be undertaken. On average, 53% of the sequences in clusters translated to recruitable individuals. To provide one detailed example, the second-largest cluster (B in Table 1) contained 17 sequences. Two patients had each contributed two sequences, and one sequence was classified as OOJ, leaving 14 matched individuals. Of these, three were LTFU and two more were OOJ. Thus, the recruitment list for that cluster contained nine individuals (9/17=53%). Overall, persons of color comprised two-thirds of the pool of potential participants. The finalized recruitment list included 68 individuals.
Phase 3: Identifying and Informing Potential Study Participants
A review of medical records by the recruiter revealed that five of the 68 possible participants were ineligible because they relocated outside the study area, were non-English speaking, incarcerated, or suffering from severe mental health issues. Initial study letters or emails were sent to the medical providers of the remaining 63 individuals. It was often necessary to contact multiple providers per case. On average, outreach attempts were made to 1.74 providers per potential participant (range 1–5). The average number of providers contacted for individuals who completed interviews was 1.67 (range 1–3). Of the 110 providers contacted in the course of recruitment, half practiced in a public hospital setting (n=55) and nearly one-third practiced in public clinics (n=32). The remaining providers were in private practice at a large health maintenance organization (n=10) or another location (n=6), or at the Veteran’s Administration (n=4).
One public clinic providing care for two patients being recruited declined to participate in the study. In addition, one medical provider did not respond to outreach; we interpreted this as a passive decline. Through discussions between the recruiter and providers who did respond, an additional 17 participants were found to be ineligible, predominantly due to clinicians’ assessment of patients’ cognitive function or mental health, or having dropped out of care (LTFU). In 35 cases, providers authorized the recruiter to contact patients directly about the research. In eight cases, providers or other personnel at the practice location approached the patient about the study; four of these patients consented to be contacted by the recruiter and four declined.
Phase 4: Initial Consent to Be Contacted by UCSF
The study recruiter was able to contact all but one of the 39 individuals who entered this phase. Of those contacted, five were ineligible (because they had relocated outside study area, or were non-English speaking), five declined to participate (not interested, too busy), and 28 consented to release contact information to UCSF. No identifying information about any patient was ever shared with UCSF before a patient provided verbal consent to release their contact information, a primary aim for ethical recruitment of our participants.
Phase 5: Study Recruitment & Scheduling
UCSF attempted to contact 28 potential participants; two were unreachable or unresponsive. Of the 26 potential participants reached by UCSF, two declined (re-located out of the study area; no longer interested) and 24 were scheduled for an interview at UCSF.
Interview Data Collection:
The 24 scheduled participants were successfully interviewed. Overall, an average of 35% of recruitable participants in each selected cluster was interviewed. Interviewees included 20 men (including 14 MSM), three cis-women, and one transwoman; nearly two-thirds were persons of color. The number and characteristics of individuals interviewed, organized by clusters, are displayed in Table 1. Thematic analysis of interview data (Ryan & Bernard, 2003) is underway. As the qualitative data were collected with the intent of “merging” them with the phylogenetic trees, this integration will occur after thematic analysis is complete. Narratives from each cluster will be analyzed for areas of convergence and divergence, and trends will be compared across clusters. More detail on data analysis and the findings produced will be reported in future manuscripts.
Findings by Cluster:
As noted, our hope is that integrating our phylogenetic and qualitative data produces an expansion in our understanding of the dynamics structuring transmission clusters. Though full thematic findings of the interviews are beyond the scope of this manuscript, and integration is not yet complete, a brief example may help to demonstrate the “value added” of the method detailed here. Cluster B (see Table 1) contained 17 sequences, contributed by 14 unique individuals, nine of whom were available to recruit. This was an ethnically diverse cluster, composed of Black, Hispanic, Native American, white, and multi-racial individuals. In terms of modes of transmission, cases had been attributed to PWID, MSM, a combination MSM-PWID (used when an individual reports both MSM and injection drug use), and heterosexual transmission. There were cis-men, cis-women, and transgender women in the cluster (the database counts transwomen as MSM). This is what was known about Cluster B from phylogenetic analysis and surveillance data.
Unfortunately, during recruitment, four of the nine potential participants were found to have been lost to medical follow up or suffer from mental health issues their physicians considered serious enough to preclude their participation; one had been incarcerated; and an additional individual declined participation, thus we were able to interview only three cluster members. We spoke with a Black cis-woman infected through heterosexual transmission, a multi-racial cis-male infected through MSM, and a Black transgender woman infected through MSM-PWID (we’ll call them Renee, Jordan, and Bella, respectively). Given the multiple ways these participants’ cases appear to differ, one might puzzle over the dynamics linking the cluster. The qualitative findings from these interviews, however, add more context to the phylogenetic assessment, illustrating how surveillance data can be correct and still insufficient to understand the full story behind chains of transmission.
Renee did report believing she’d contracted HIV from a male partner, but their joint substance use, which she characterized as addiction, was the crucial context that she considered the real source of her risk. She noted that this partner “was cheatin’ on me with other people, so I was faithful but they weren’t.” When asked to confirm that this had been a stable partner, she responded, “If you call it that. I mean it’s— [pause] we were on drugs, we’re all doin’ drugs. I mean, what stability is that?” She also intimated that her substance use had influenced the types of partners she had: “And now when I’m clean, and I look back and I see some of these people – What the hell was I thinkin’? Look at him! What was I thinking?”
In an analogous way, while most sexual experiences Jordan discussed during his interview involved cis-women, he did report behavior that would have been classified as MSM in surveillance data (see Truong et al. 2019 for a discussion of whether such classifications enhance or complicate our understanding of the epidemic). However, this happened only under very particular circumstances: during his own period of substance abuse, specifically of methamphetamine, he related having sexual contact with transgender women. Although he also used crack, he explicitly differentiated the effect the two drugs had on his sexual behavior:
“I can get weird on drugs … the drugs will make you [do] whatever.… I’ve lowered my standards with crack, but I’ve never done some weird – hung out with – let a gay dude touch my penis so I could get a hit– or something like that….[but with meth I had] sex with some trannies in there. I actually lived with a chick for a little while who had the full thing, the full – you know, she had tools. So I wouldn’t call her my girlfriend, but I did have sex with her many times, ten to fifteen times. I lived with her for a couple months. But then it just was getting too weird.”
Finally, like Renee and Jordan, in her interview Bella reported behavior that accorded with the surveillance data associated with her case. Unlike them, the surveillance data captured what she considered the most significant elements of her risk fairly well (with the important exception that she didn’t consider herself a man but was still classified as one, through use of the category MSM). Bella had gone through a period of substance abuse, during which she’d performed transactional sex and had other sexual relationships, very often with cis-men who identified as heterosexual. She explained, “Then I started shootin’ up [meth] and then the rush was even different. So then everything become sexual, everything.” Bella believed she knew which of her partners she’d acquired the infection from: “I risked myself, messin’ around with somebody that I knew that messed around with somebody who had [HIV], and he told me he put on a condom and he didn’t.” However, she also recognized that this had not been her only potential exposure. Like Renee, she looked back and lamented the encounters she’d had with “scrungy guys” she wouldn’t choose as partners when sober, but “when I was high, I didn’t care … [a]bout messin’ around with guys, sleepin’ around, suckin’ dick, doin’ whatever, I didn’t care. ‘Cause I was high. So it didn’t matter.”
For these three participants, then, drug dependency and sex were intertwined as HIV risk factors, even though this was not apparent from the surveillance data attached to each case. Their stories also overlapped in other ways. All had experienced periods of housing instability and homelessness, and one San Francisco neighborhood served as an important backdrop in all three cases. In the case of Cluster B, merging semi-structured interview data with phylogenetics and surveillance data offers a previously-unseen glimpse into the processes by which diverse people came together in particular ways, at a specific time and place. This is the context necessary to understand the dynamics of HIV exposure and acquisition within this heterogeneous transmission cluster.
Discussion
We describe the process of integrating phylogenetic and qualitative approaches with the aim of achieving greater insight into HIV transmission clusters in San Francisco, and through these to improve HIV prevention. The procedures we developed linked viral genetic sequences with epidemiological data to identify individuals in large transmission clusters, which then served as a sampling frame for semi-structured interviews. This approach required collaboration across multiple entities responsible for maintaining different and sensitive datasets. Sharing sensitive patient data with UCSF investigators without prior patient consent was not possible given legal protection of surveillance data, HIPAA regulations and standard ethical practice. Hence, our procedures called for sharing only de-identified data across organizations until patients gave specific verbal consent to the recruiter to release contact information to UCSF. Data sharing, with or without identifiers, was always conducted through secure data systems. Recruitment data show that we could develop and implement feasible procedures while protecting patient confidentiality.
Approaching Providers First
The nature of this research, focused as it was on transmission clusters, required participation of specific individuals, making it infeasible to rely on recruitment methods often used for in-depth interviews, e.g., study fliers or online banner ads. After all, even patients who believe they know how they acquired the virus likely do not know how many other transmission events that source might have been involved in. Hence, surveillance and genetic data offered the only route to identify patients in large transmission clusters. Given these are sensitive datasets of private patient medical information, we opted to reach possible participants by approaching their providers first through our SFDPH collaborators. This approach allowed providers to decline the recruitment of any individual for whom they considered participation would be detrimental and provided potential participants an additional layer of protection. Provider responsiveness, however, was the most significant barrier to contacting patients. To address this challenge, we modified recruitment procedures after the study was under way to allow providers to assent to direct contact of patients by the recruiter, a change suggested by providers themselves. This procedure facilitated contact with some patients, though an important barrier remained. Since we chose to obtain the assent for direct outreach on an opt-in basis only, it could not be used to recruit patients whose providers did not respond to our outreach. A similar procedure on an opt-out basis, that is alerting providers that a particular patient had been selected for recruitment and would receive direct outreach from recruiters if the provider failed to respond within a given time frame, might have reduced this barrier, but whether it could be ethically employed requires careful consideration.
Fortunately, once the recruiter was able to contact potential participants, the large majority consented both to share the contact information with UCSF and to participate in the study. Moreover, most who agreed to be interviewed completed the interview.
Cluster Size Consequences
Our experience with these procedures showed that even when starting from clusters with the largest number of sequences, the number of recruitable individuals per cluster quickly shrank, whether due to multiple sequences per person, or removal of OOJ and LTFU cases. While such culling, along with the more limited determination of ineligibility through medical records review, greatly reduced cluster sizes, these procedures resulted in the recruiter conducted time-intensive provider outreach only for potential participants with a realistic chance of participation.
The relatively small size and diversity of the largest clusters in our dataset was unexpected. Based on findings of other studies, we had predicted the largest clusters would include upwards of 30 sequences (Brenner et al., 2011; Lewis, Hughes, Rambaut, Pozniak, & Leigh Brown, 2008). As reported, no cluster in our dataset contained more than 29 sequences and 20 individuals. Furthermore, as this study focused on HIV transmission clusters in San Francisco where 74% of people living with HIV are MSM, 60% are white, and 92% are male, one might expect the largest clusters to be composed primarily of white MSM (San Francisco Department of Public Health, 2016). However, in the large clusters from which we recruited interviewees, MSM as mode of transmission was underrepresented while people of color and cis-women were overrepresented, compared to people known to be living with HIV in San Francisco (San Francisco Department of Public Health, 2016). Transwomen were represented in roughly the proportion one would expect, based on surveillance data. The under- and overrepresentation of certain key populations within these large clusters raises questions about local HIV transmission chains that the larger phylogenetic study will be well-positioned to address. One aspect of our study to consider is that sequence data came from a laboratory that provides services for publicly-funded facilities. Hence, sequences from patients obtaining care at private facilities were not included. If white MSM are more likely to access care at private facilities, this may partly explain the difference between the sociodemographic composition of individuals contributing viral sequences and the characteristics of San Francisco residents living with HIV.
Contributions of Clusters to HIV Prevention
The narratives provided in the interviews will attest to whether the contexts in which individuals acquire HIV share similarities within clusters, and whether the qualitative data we gathered can be used to inform HIV prevention interventions. We provided a brief example, based on interviews with three members of a highly diverse cluster, which illustrated the way cis-gender men and women and transwomen were linked through similar behavior/mental health conditions (substance use/addiction), although this was not apparent from the surveillance data attached to each case and resulted in different modes of transmission. We believe this suggests that, at a minimum, qualitative data can usefully “thicken” (à la Geertz, 1973), the “description” of the HIV epidemic offered by phylogenetic trees.
To be fair, finding a relationship between drug use and sex (transactional and not) is, of course, not novel in the HIV literature. Neither is the increased HIV risk experienced by the unhoused. But here we would like to make two points. First, just because the mixed method approach documented here did not uncover new correlations or practices in this one, brief example does not mean it can’t. Second, findings need not be novel to be valuable if the knowledge can be applied. A nuanced understanding of the context surrounding HIV transmission events (even if that context echoes previous research findings) can inform prevention planning by public health officials and help prioritize efforts when resources are constrained.
Limitations
Attention to characteristics of potential research participants is always required, especially when conducting purposive sampling. Working from transmission clusters, however, requires even greater reflexivity (German et al., 2017). Researchers must be cognizant of who is contributing viral sequence specimens to the data set, and who is not. As mentioned above, the source for viral sequence samples analyzed in this study may have impacted the sociodemographic composition of the clusters we identified. Likewise, individuals with undiagnosed HIV, as well as people living with HIV who have not had resistance testing done, did not contribute to our data set. To the extent that any of the above dispositions are more likely among certain types of people than others, selection bias may be in effect. It is essential to bear in mind this potential bias, because all such excluded individuals could have been involved in the actual transmission chains that produced the clusters depicted in our phylogenetic tree, even though these cases were not in our dataset and therefore could not be recruited.
In addition, although all patients were included in the cluster data, the interview data will be biased by not having cases classified as LTFU and OOJ that were removed from our pool; these individuals may have moved away or simply dropped out of care. Similarly, cases that did not meet eligibility criteria will contribute to bias. None of the patients failed to meet the 16 years of age criterion. However, the selected clusters included a few non-English speakers, as well as a few patients with insufficient mental capacity to provide informed consent and complete an interview. Unfortunately the contexts in which these individuals practiced HIV prevention and acquired HIV remain outside our understanding of transmission within the clusters we studied.
Thus, we will need to be mindful that the stories told in interviews conducted from our sample cannot not be considered “the whole story” of a cluster. This circumstance, however, is often the case with qualitative research, which does not typically claim to be exhaustive and generalizable (Pope & Mays, 1995; Tracy, 2010). In that sense, the limitations of this sampling method may not differ significantly from more traditional qualitative approaches. That being noted, the technical expertise and resources required to employ this sampling method are considerable, and the conditions under which sampling from clusters is practicable, e.g., in places where there is a high enough sampling fraction (Grabowski, Herbeck, & Poon, 2018) are narrower than, for instance, convenience sampling (Volz, Koopman, Ward, Brown, & Frost, 2012).
Conclusion
The procedures described here take up the challenge of pairing phylogenetics with other kinds of data in a way that differs from that used in research to date. As described above, some previous studies have made excellent use of existing data, combining phylogenetic analysis with information from epidemiological surveillance or contact tracing while other studies have taken a valuable prospective approach. However, the methods used in previous research meant that the broader circumstances surrounding the infection of individuals within particular transmission clusters could not be explored. In contrast, our method samples from transmission clusters and collects data spanning temporal, topical, and emotional breadth through semi-structured interviewing. This difference has important ramifications, as already noted, for the kinds of questions the resulting data set can answer. It also, however, has ethical implications. As Coltart et al. (2018) noted in their review of ethical issues in phylogenetic research on HIV, these analyses can inadvertently create the perception that certain groups are responsible for spreading the virus, and may lead to their (further) stigmatization, especially if structural factors are omitted from analysis. They further indicated, however, that researching “structural factors and their effect on HIV transmission risk can decrease the blame mentality and create an alternative understanding of how to reduce HIV transmission and which individuals or groups are most at risk and why” (Coltart et al., 2018, e659).
Analysis of the collected qualitative data will enable assessment of cluster-level patterns with reference to precisely this structural context, and thus may contribute to improved tailoring of prevention and treatment efforts among populations at risk for or diagnosed with HIV. This approach also has relevance beyond our study. Sampling from transmission clusters can be done for many types of subsequent data collection (i.e., not only semi-structured interviews), which may expand the ways those clusters can be characterized and understood. Alternatively, the combination of sampling and qualitative data collection could be incorporated in research on infectious diseases other than HIV where greater knowledge of transmission context could be helpful (e.g., Hepatitis C, tuberculosis) (Grabowski et al., 2018).
The proliferation of genetic data begs the question of whether the novel method detailed here could be used to learn more about the context of human-to-human transmission, particularly in fast-growing clusters associated with COVID-19 outbreaks. At the moment, the answer appears to be “maybe,” largely because debate is ongoing among the scientific community about the best way to conduct phylogenetic analysis on COVID-19 samples, and what the results mean (Forster et al., 2020; Mavian et al., 2020). As consensus emerges on the phylogenetic methods, the demand for testing will likely have generated a large number of viral sequences for analysis, making it possible to identify clusters quickly. The unfortunately high number of infections will be associated with a significant amount of surveillance data, as states and localities ramp up contact tracing efforts (Fortin, 2020). As with HIV, however, the data collected will not provide the nuanced context surrounding infection that a semi-structured interview can elicit. Thus, the method we devised may offer an opportunity for researchers to carefully implement a sampling frame that will allow them to recruit and interview individuals in high-transmission clusters to better understand what contributes to spread. However, it will still be necessary to attend to issues like the ones outlined above. While COVID-19 may be less (or at least differently) stigmatized than HIV and other sexually transmitted infections, both survivors of the virus (Maslin Nir, 2020) and people of Chinese descent (in the U.S.) are reporting negative, marginalizing reactions from others (Tavernise & Oppel, 2020). Viral genetic data are personal, private health information, therefore concerns about privacy are still salient. Research partnerships like the one described for this study are likely to be necessary, and laws and ethical considerations already in place will need to be observed when using surveillance data and contacting patients. Overall, we would concur with the voices encouraging collaboration to speed results and urging great care be taken with research findings (Chookajorn, 2020).
Contributions to the Field of Mixed Methods Research
One contribution to the field is the demonstration that phylogenetic analysis and surveillance data can be harnessed to generate a sampling frame for subsequent qualitative data collection, using an explanatory sequential design. The novel aspect of our approach is primarily that semi-structured interviews with cluster members were planned and collected prospectively. This is in contrast to previous studies that had access only to data collected for other reasons, and/or that are typically very “coarse” and closed-ended. Another contribution of relevance to mixed methods investigators with interest in phylogenetics is that we provided a detailed account of how well each step in our approach worked, including the limitations and challenges encountered. We offered concrete lessons learned, highlighting certain steps in our approach that could be improved upon, such as engagement of providers. A third contribution is simply to apply the constructs and terminology of the mixed methods community to research that includes phylogenetic analysis (both our own and previous studies). Our literature review suggested that these scholarly communities are rarely in dialog; this may be partly because they speak such specialized languages (e.g., “explanatory sequential design” and “Shimodaira-Hasegawa node support”). Particularly in the context of the COVID-19 pandemic, we hope to have encouraged other researchers to consider experimenting with sampling from clusters as a systematic approach to achieve a more nuanced understanding of viral transmission networks.
Acknowledgments
NIH R01 MH096642 (PI: Hong-Ha M. Truong)
List of Abbreviations:
- AIDS
Acquired Immune Deficiency Syndrome
- ARV
Antiretroviral
- CW
Cis-women
- HET
Heterosexual
- HIPAA
Health Insurance Portability and Accountability Act of 1996
- HIV
Human immunodeficiency virus
- LTFU
Lost to follow-up
- MSM
Men who have sex with men
- MSM-PWID
MSM who inject drugs
- NIR
No identified risk
- NRR
No risk reported
- OOJ
Outside of (SFDPH’s) jurisdiction
- PC
Persons of color
- PWID
Person who injects drugs
- SFDPH
San Francisco Department of Public Health
- TW
Transwomen
- UCSF
University of California San Francisco
Footnotes
The age of consent for receiving pregnancy, STI and HIV care without parental/guardian involvement in California is 12 years old. Hence, 16 year-olds would be able to consent (rather than merely assent) to participate in this study.
References
- Andrew S & Halcomb EJ (2009). Mixed methods research for nursing and the health sciences. Chichester, U.K., Blackwell Publishing, Ltd. doi: 10.1002/9781444316490 [DOI] [Google Scholar]
- Alary M, Béhanzin L, Guédou F, Béhanzin L, Guédou F, Geraldo N, … Giguère K (2015). P18.05 Contribution of biomarkers to the validation of self-reported condom use in a treatment as prevention and a pre-exposure prophylaxis demonstration study among female sex workers. Sexually Transmitted Infections, 91, A242–A243. [Google Scholar]
- Arnold E, & Lane S (2011). Qualitative Research in Transfusion Medicine. Transfusion Medicine, 21, 291–300. [DOI] [PubMed] [Google Scholar]
- Avila D, Keiser O, Egger M, Kouyos R, Boni J, Yerly S, … Spycher BD (2014). Social meets molecular: Combining phylogenetic and latent class analyses to understand HIV-1 transmission in Switzerland. Am J Epidemiol, 179(12), 1514–1525. [DOI] [PubMed] [Google Scholar]
- Brenner BG, Roger M, Stephens D, Moisi D, Hardy I, Weinberg J, … Group, t. M. P. C. S. (2011). Transmission Clustering Drives the Onward Spread of the HIV Epidemic Among Men Who Have Sex With Men in Quebec. J Infect Dis, 204 (7), 1115–1119. doi:doi: 10.1093/infdis/jir468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candido DS, Claro IM, de Jesus JG, Souza WM, Moreira FRR, Dellicour S, … Faria NR (2020). Evolution and epidemic spread of SARS-CoV-2 in Brazil. Science, 369(6508), 1255–1260. doi: 10.1126/science.abd2161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JF-W, Yuan S, Kok K-H, To KK-W, Chu H, Yang J, … Yuen K-Y (2020). A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet, 395, 514–523. doi: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PA, Hogan JW, Huang A, DeLong A, Salemi M, Mayer KH, & Kantor R (2015). Phylogenetic Investigation of a Statewide HIV-1 Epidemic Reveals Ongoing and Active Transmission Networks Among Men Who Have Sex With Men. JAIDS Journal of Acquired Immune Deficiency Syndromes, 70(4), 428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chookajorn T (2020). Evolving COVID-19 conundrum and its impact. Proceedings of the National Academy of Sciences, 117 (23) 12520–12521. doi: 10.1073/pnas.2007076117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coltart CEM, Hoppe A, Parker M, Dawson L, Amon JJ, Simwinga M, … Group, o. b. o. t. E. i. H. P. W. (2018). Ethical considerations in global HIV phylogenetic research. The Lancet HIV, 5(11), e656–e666. doi: 10.1016/S2352-3018(18)30134-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creswell JW (2009. ). Mapping the Field of Mixed Methods Research. Journal of Mixed Methods Research, 3(2), 95–108. doi: 10.1177/1558689808330883 [DOI] [Google Scholar]
- Creswell JW (2010). Mapping the Developing Landscape of Mixed Methods Research. In Teddlie ATC (Ed.), (pp. 45–68). Thousand Oaks, CA: SAGE Publications, Inc. [Google Scholar]
- Creswell JW, Klassen AC, Plano Clark VL, & Clegg Smith K (2011). Best Practices for Mixed Methods Research in the Health Sciences. Retrieved from Bethesda (Maryland): [Google Scholar]
- Delva W, Leventhal GE, & Helleringer S (2016). Connecting the dots: network data and models in HIV epidemiology. AIDS, 30(13), 2009–2020. doi: 10.1097/qad.0000000000001184 [DOI] [PubMed] [Google Scholar]
- Deng X, Gu W, Federman S, du Plessis L, Pybus OG, Faria NR, … Chiu CY (2020). Genomic surveillance reveals multiple introductions of SARS-CoV-2 into Northern California. Science, 369(6503), 582–587. doi: 10.1126/science.abb9263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis AM, Cohen MS, Rucinski KB, Rutstein SE, Powers KA, Pasquale DK, … Miller WC (2018). HIV-1 transmission among persons with acute HIV-1 infection in Malawi: demographic, behavioral and phylogenetic relationships. Clinical Infectious Diseases, ciy1006. doi: 10.1093/cid/ciy1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis AM, Hué S, Pasquale D, Napravnik S, Sebastian J, Miller WC, & Eron J (2015). HIV Transmission Patterns Among Immigrant Latinos Illuminated by the Integration of Phylogenetic and Migration Data J. AIDS Research and Human Retroviruses, 31(10), 973–980. doi:doi: 10.1089/aid.2015.0089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetters MD, Curry LA, & Creswell JW (2013). Achieving integration in mixed methods designs-principles and practices. Health Services Research, 48(6 Pt 2), 2134–2156. doi: 10.1111/1475-6773.12117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetters MD, & Molina-Azorin JF (2017). The Journal of Mixed Methods Research Starts a New Decade: The Mixed Methods Research Integration Trilogy and Its Dimensions. Journal of Mixed Methods Research, 11(3), 291–307. doi: 10.1177/1558689817714066 [DOI] [Google Scholar]
- Forster P, Forster L, Renfrew C, & Forster M (2020). Phylogenetic network analysis of SARS-CoV-2 genomes. Proceedings of the National Academy of Sciences, 117(17): 9241–9243. doi: 10.1073/pnas.2004999117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost SDW, & Pillay D (2015). Understanding Drivers of Phylogenetic Clustering in Molecular Epidemiological Studies of HIV. The Journal of Infectious Diseases, 211, 856–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geertz C (1973). The Interpretation of Cultures. New York: Basic Books. [Google Scholar]
- German D, Grabowski MK, & Beyrer C (2017). Enhanced use of phylogenetic data to inform public health approaches to HIV among men who have sex with men. Sexual health, 14(1), 89–XX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Reiche AS, Hernandez MM, Sullivan MJ, Ciferri B, Alshammary H, Obla A, … van Bakel H (2020). Introductions and early spread of SARS-CoV-2 in the New York City area. Science, 369(6501), 297–301. doi: 10.1126/science.abc1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski MK, Herbeck JT, & Poon AFY (2018). Genetic Cluster Analysis for HIV Prevention. Current HIV/AIDS Reports, 15(2), 182–189. doi: 10.1007/s11904-018-0384-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski MK, Lessler J, Redd AD, Kagaayi J, Laeyendecker O, Ndyanabo A, … the Rakai Health Sciences, P. (2014). The Role of Viral Introductions in Sustaining Community-Based HIV Epidemics in Rural Uganda: Evidence from Spatial Clustering, Phylogenetics, and Egocentric Transmission Models. PLoS Medicine, 11(3), e1001610. doi: 10.1371/journal.pmed.1001610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant RM, Lama JR, Anderson PL, McMahan V, Liu AY, Vargas L, … Glidden DV (2010). Preexposure Chemoprophylaxis for HIV Prevention in Men Who Have Sex with Men. N Engl J Med, 363:2587–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günthard H, Saag M, Benson C, del Rio C, Eron J, Gallant J, … Volberding P (2016). Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults: 2016 Recommendations of the International Antiviral Society-USA Panel. JAMA, 316(2), 191–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivankova NV, Creswell JW, & Stick SL (2006). Using Mixed-Methods Sequential Explanatory Design: From Theory to Practice. Field Methods, 18(1), 3–20. doi: 10.1177/1525822x05282260 [DOI] [Google Scholar]
- Jafa K, McElroy P, Fitzpatrick L, Borkowf CB, MacGowan R, Margolis A, … Sullivan PS (2009). HIV Transmission in a State Prison System, 1988–2005. PLOS ONE, 4(5), e5416. doi: 10.1371/journal.pone.0005416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanović L, Šiljić M, Ćirković V, Salemović D, Pešić-Pavlović I, Todorović M, … Stanojević M (2019). Exploring Evolutionary and Transmission Dynamics of HIV Epidemic in Serbia: Bridging Socio-Demographic With Phylogenetic Approach. Frontiers in Microbiology, 10(287). doi: 10.3389/fmicb.2019.00287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharsany AB, Buthelezi TJ, Frohlich JA, Yende-Zuma N, Samsunder N, Mahlase G, … Karim QA (2014). HIV infection in high school students in rural South Africa: role of transmissions among students. AIDS Res Hum Retroviruses, 30(10), 956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiwuwa-Muyingo S, Nazziwa J, Ssemwanga D, Ilmonen P, Njai H, Ndembi N, … Kaleebu P (2017). HIV-1 transmission networks in high risk fishing communities on the shores of lake victoria in Uganda: A phylogenetic and epidemiological approach. PLoS ONE, 12(10), e0185818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koester KA, Liu A, Eden C, Amico KR, McMahan V, Goicochea P, … Grant RM (2015). Acceptability of drug detection monitoring among participants in an open-label pre-exposure prophylaxis study. AIDS Care, 27(10), 1199–1204. doi: 10.1080/09540121.2015.1039958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Tam D, Tan Y, Mak W, Wong K, Chen J, & Yam W (2009). An exploratory study on the social and genotypic clustering of HIV infection in men having sex with men. AIDS, 23(13), 1755–1764. doi:DOI: 10.1097/QAD.0b013e32832dc025 [DOI] [PubMed] [Google Scholar]
- Leitner T, Escanilla D, Franzén C, Uhlén M, & Albert J (1996). Accurate reconstruction of a known HIV-1 transmission history by phylogenetic tree analysis. Proceedings of the National Academy of Sciences, 93(20), 10864–10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux JE, Siddle KJ, Shaw BM, Loreth C, Schaffner SF, Gladden-Young A, … MacInnis BL (2021). Phylogenetic analysis of SARS-CoV-2 in Boston highlights the impact of superspreading events. Science, 371(6529). doi: 10.1126/science.abe3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy I, Mor Z, Anis E, Maayan S, Leshem E, Pollack S, … Grossman Z (2011). Men Who Have Sex With Men, Risk Behavior, and HIV Infection: Integrative Analysis of Clinical, Epidemiological, and Laboratory Databases. Clinical Infectious Diseases, 52(11), 1363–1370. doi: 10.1093/cid/cir244 [DOI] [PubMed] [Google Scholar]
- Lewis F, Hughes GJ, Rambaut A, Pozniak A, & Leigh Brown AJ (2008). Episodic Sexual Transmission of HIV Revealed by Molecular Phylodynamics. PLoS Medicine, 5(3), e50. doi: 10.1371/journal.pmed.0050050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, He N, Zhou S, Ding Y, Qiu D, Zhang T, & Wong FY (2013). Behavioral and molecular tracing of risky sexual contacts in a sample of Chinese HIV-infected men who have sex with men. Am J Epidemiol, 177(4), 343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little SJ, Kosakovsky Pond SL, Anderson CM, Young JA, Wertheim JO, Mehta SR, … Smith DM (2014). Using HIV networks to inform real time prevention interventions. PLoS ONE, 9(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrazzo JM, Ramjee G Richardson, B.A., Gomez, K., Mgodi, N., Nair, G. … Chirenje, Z. M. (2015). N Engl J Med, 372(6):509–518 doi: 10.1056/NEJMoa1402269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslin Nir S (2020). For Some Survivors, the Stigma of Illness Far Outlasts Its Symptoms. New York Times, May 21, Section A, Page 16. [Google Scholar]
- Mavian C, Pond SK, Marini S, Magalis BR, Vandamme AM, Dellicour S, … & Salemi M (2020). Sampling bias and incorrect rooting make phylogenetic network tracing of SARS-COV-2 infections unreliable. Proceedings of the National Academy of Sciences, 117(23), 12522–12523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith LW, Hamilton WL, Warne B, Houldcroft CJ, Hosmillo M, Jahun AS, … Goodfellow I (2020). Rapid implementation of SARS-CoV-2 sequencing to investigate cases of health-care associated COVID-19: a prospective genomic surveillance study. The Lancet Infectious Diseases, 20(11), 1263–1271. doi: 10.1016/S1473-3099(20)30562-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery ET, Mensch B, Musara P, Hartmann M, Woeber K, Etima J, van der Straten A (2017). “Misreporting of Product Adherence in the MTN-003/VOICE Trial for HIV Prevention in Africa: Participants’ Explanations for Dishonesty.” AIDS Behav, 21(2): 481–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutenherwa F, Wassenaar DR, & de Oliveira T (2019). Experts’ Perspectives on Key Ethical Issues Associated With HIV Phylogenetics as Applied in HIV Transmission Dynamics Research. Journal of Empirical Research on Human Research Ethics, 14(1), 61–77. doi: 10.1177/1556264618809608 [DOI] [PubMed] [Google Scholar]
- Oster AM, Pieniazek D, Zhang X, Switzer WM, Ziebell RA, Mena LA, … Heffelfinger JD (2011). Demographic but not geographic insularity in HIV transmission among young black MSM. AIDS, 25(17), 2157–2165. doi: 10.1097/QAD.0b013e32834bfde9 [DOI] [PubMed] [Google Scholar]
- Pasquale DK, Doherty IA, Sampson LA, Leone PA, Sebastian J, Ledford SL, … Dennis AM (2016). HIV Transmission Hotspot Detection Combining Sexual and Phylogenetic Network Analysis. Paper presented at the Conference on Retroviruses and Opportunistic Infections (CROI). [Google Scholar]
- Plano Clark VL (2010). The adoption and practice of mixed methods: U.S. trends in federally funded health-related research. Qualitative Inquiry, 6(6), 428–440. [Google Scholar]
- Poon AF, Joy JB, Woods CK, Shurgold S, Colley G, Brumme CJ, … Harrigan PR (2015). The impact of clinical, demographic and risk factors on rates of HIV transmission: a population-based phylogenetic analysis in British Columbia, Canada. J Infect Dis, 211(6), 926–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope C, & Mays N (1995). Reaching the parts other methods cannot reach: an introduction to qualitative methods in health and health services research. BMJ: British Medical Journal, 311(6996), 42–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, Loman N, Pybus O, Barclay W, Barrett J, Carabelli A, … Volz E (2020). Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563
- Robertson AM, Garfein RS, Wagner KD, Mehta SR, Magis-Rodriguez C, Cuevas-Mota J, … Strathdee SA (2014). Evaluating the impact of Mexico’s drug policy reforms on people who inject drugs in Tijuana, B.C., Mexico, and San Diego, CA, United States: a binational mixed methods research agenda. Harm Reduct J, 11(4), 1477–7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothan HA & Byrareddy SN (2020). The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J Autoimmun, 109:102433. doi: 10.1016/j.jaut.2020.102433. Epub 2020 Feb 26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan GW, & Bernard HR (2003). Techniques to Identify Themes. Field Methods, 15, 85–109. [Google Scholar]
- San Francisco Department of Public Health, P. H. D. (2016). HIV Epidemiology: Annual Report 2015. Retrieved from San Francisco, CA: [Google Scholar]
- Schairer C, Mehta SR, Vinterbo SA, Hoenigl M, Kalichman M, & Little S (2017). Perceptions of molecular epidemiology studies of HIV among stakeholders. Journal of Public Health Research, 6(3). doi:DOI 10.4081/jphr.2017.992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shereen MA, Khan S, Kazmi A, Bashir N, & Siddique R (2020). COVID-19 infection: Origin, transmission, and characteristics of human coronaviruses. Journal of Advanced Research, 24: 91–98. doi: 10.1016/j.jare.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavernise S, & Oppel RA (2020). Spit On, Yelled At, Attacked: Chinese-Americans Fear for Safety. March 24, Section A, Page 1.
- Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Fonseca V, Giandhari J, … de Oliveira T (2020). Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv, 2020.2012.2021.20248640. doi: 10.1101/2020.12.21.20248640 [DOI] [Google Scholar]
- Tracy SJ (2010). Qualitative Quality: Eight “Big-Tent” Criteria for Excellent Qualitative Research. Qualitative Inquiry, 16(10), 837–851. doi:DOI: 10.1177/1077800410383121 [DOI] [Google Scholar]
- Truong HM, Louie B, Liegler T, McFarland W, … Scheer S (2015). Recent Infection, Sexually Transmitted Infections and Transmission Clusters Frequently Observed Among Persons Newly-Diagnosed with HIV in San Francisco. Journal of acquired immune deficiency syndromes (1999), 69(5), 606–609. doi: 10.1097/qai.0000000000000681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong HM, O’Keefe KJ, Pipkin S, Liegler T, Scheer S, & McFarland W (2019). How are transgender women acquiring HIV? Insights from phylogenetic transmission clusters. AIDS, 33(13), 2073–2079 10.1097/QAD.0000000000002318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volz EM, Koopman JS, Ward MJ, Brown AL, & Frost SDW (2012). Simple Epidemiological Dynamics Explain Phylogenetic Clustering of HIV from Patients with Recent Infection. PLOS Computational Biology, 8(6), e1002552. doi: 10.1371/journal.pcbi.1002552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertheim JO, Kosakovsky Pond SL, Forgione LA, Mehta SR, Murrell B, Shah S, …. Torian LV (2017). Social and Genetic Networks of HIV-1 Transmission in New York City. PLOS Pathogens, 13(1), e1006000. doi: 10.1371/journal.ppat.1006000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Wu Q, & Zhang Z (2020). Probable pangolin Origin of SARS-CoV-2 Associated with the COVID-19 Outbreak. Current Biology 30(7): 1346–1351.e2. doi: 10.1016/j.cub.2020.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]