

Abstract
Background
Many cardiomyopathy‐associated FLNC pathogenic variants are heterozygous truncations, and FLNC pathogenic variants are associated with arrhythmias. Arrhythmia triggers in filaminopathy are incompletely understood.
Methods and Results
We describe an individual with biallelic FLNC pathogenic variants, p.Arg650X and c.970‐4A>G, with peripartum cardiomyopathy and ventricular arrhythmias. We also describe clinical findings in probands with FLNC variants including Val2715fs87X, Glu2458Serfs71X, Phe106Leu, and c.970‐4A>G with hypertrophic and dilated cardiomyopathy, atrial fibrillation, and ventricular tachycardia. Induced pluripotent stem cell‐derived cardiomyocytes (iPSC‐CMs) were generated. The FLNC truncation, Arg650X/c.970‐4A>G, showed a marked reduction in filamin C protein consistent with biallelic loss of function mutations. To assess loss of filamin C, gene editing of a healthy control iPSC line was used to generate a homozygous FLNC disruption in the actin binding domain. Because filamin C has been linked to protein quality control, we assessed the necessity of filamin C in iPSC‐CMs for response to the proteasome inhibitor bortezomib. After exposure to low‐dose bortezomib, FLNC‐null iPSC‐CMs showed an increase in the chaperone proteins BAG3, HSP70 (heat shock protein 70), and HSPB8 (small heat shock protein B8) and in the autophagy marker LC3I/II. FLNC null iPSC‐CMs had prolonged electric field potential, which was further prolonged in the presence of low‐dose bortezomib. FLNC null engineered heart tissues had impaired function after low‐dose bortezomib.
Conclusions
FLNC pathogenic variants associate with a predisposition to arrhythmias, which can be modeled in iPSC‐CMs. Reduction of filamin C prolonged field potential, a surrogate for action potential, and with bortezomib‐induced proteasome inhibition, reduced filamin C led to greater arrhythmia potential and impaired function.
Keywords: arrhythmia, autophagy, BAG3, filamin C, proteostasis
Subject Categories: Clinical Studies, Cardiomyopathy, Sudden Cardiac Death
Nonstandard Abbreviations and Acronyms
- ABD
actin binding domain
- DCM
dilated cardiomyopathy
- EHT
engineered heart tissue
- FLNC
filamin C
- iPSC‐CM
induced pluripotent stem cell‐derived cardiomyocyte
- NSVT
nonsustained ventricular tachycardia
Research Perspective.
What Is New?
Filamin C is important for protein quality control and in FLNC‐null induced pluripotent stem cell‐cardiomyocytes, bortezomib, a proteosome inhibitor, caused an increase in the chaperone proteins and autophagy markers.
Low dose bortezomib treatment prolonged field potential duration in FLNC‐null induced pluripotent stem cell‐cardiomyocytes and intensified functional deficits in FLNC‐null engineered heart tissues.
What Question Should Be Addressed Next?
Further study is needed to understand the mechanism that hearts lacking filamin C to proteotoxic stress and how disruption of FLNC ultimately leads to cardiomyopathy with increased risk of arrhythmia.
Pathogenic variants in FLNC, the gene encoding the actin binding protein filamin C, lead to cardiomyopathy and myofibrillar myopathy. 1 Multiple cardiomyopathy subtypes have been described in association with FLNC mutations, including dilated, hypertrophic, restrictive, and arrhythmogenic. 2 , 3 , 4 The myofibrillar myopathy associated with FLNC mutations affects distal or proximal skeletal muscles and features intracellular aggregates in myofibers. 5 With FLNC mutations, cardiomyopathy can occur in the absence or presence of skeletal muscle myopathy. FLNC variants associated with myofibrillar myopathy include both missense and truncating mutations, and the inheritance pattern is primarily autosomal dominant with some cases of recessive inheritance. 1 , 6 Cardiomyopathy‐associated FLNC variants are most commonly associated with dominant inheritance, and the most readily interpretable cardiomyopathy‐FLNC variants are premature truncations.
Ventricular arrhythmias are associated frequently with FLNC cardiomyopathy and include nonsustained and sustained ventricular tachycardia. Notably, ventricular arrhythmias occur even with mildly reduced left ventricular dysfunction or in minimally hypertrophied left ventricles, indicating that risk assessment strategies should take into consideration FLNC genotype. 7 , 8 , 9 In the myofibrillar myopathies, intracellular aggregates are a characteristic finding in skeletal muscle; however, these aggregates are not readily apparent in cardiomyopathic hearts. 3 , 10 , 11 The presence of intracellular aggregates in skeletal muscle and the lack of finding aggregates in the heart might reflect the limited sampling of the heart. Alternatively, the lack of detectable aggregates could derive from intrinsic differences between heart and skeletal muscle, or more likely the nature of specific FLNC variants.
In heart and skeletal muscle, filamin C is enriched near Z discs, and there is a concentration of filamin C protein at the plasma membrane. In the heart, filamin C is also found at intercalated discs. 12 , 13 Filamin C has an actin binding domain at its amino terminus, followed by 24 immunoglobulin‐like repeat domains. Between the 15th and 16th repeat domains is a region that is variably spliced with inclusion or exclusion of exon 31. 14 This splicing event encodes an additional flexible hinge into the filamin C protein. Exon 31‐containing transcripts are increased in failed hearts. 14 The final immunoglobulin‐like repeat domain mediates dimerization of filamin C, and a premature truncation in this domain, W2710X, was first identified in German families with myofibrillar myopathy and associated cardiomyopathy. 15 , 16
Mice engineered with the equivalent of human W2710X, called W2711X in mice, express both the truncated filamin C protein and the full‐length form in skeletal muscle, demonstrating that truncated filamin C protein escapes nonsense‐mediated decay in skeletal muscle. 17 Heterozygous W2711X mice develop an exercise‐induced myopathic process characterized by intracellular aggregates in skeletal myofibers. 17 Mice with homozygous W2711X similarly develop skeletal muscle myopathy with a faster time course to disease. In contrast to the W2711X mice, homozygous deletion of the last eight exons of Flnc (Δ41‐48), which removes the last 5 immunoglobulin domains, results in early postnatal lethality from respiratory muscle weakness. 18 Unlike the W2711X mice, there is little expression of filamin C protein in the Δ41‐48 muscle, supporting the idea that some FLNC truncations may be subject to nonsense‐mediated decay or protein instability. Multiple lines of evidence support a role for filamin C in regulating protein homeostasis, and this is especially important in striated muscle. 19 Filamins, including filamin C, can undergo unfolding of specific immunoglobulin domains under mechanical and other stressors. 20 BAG3 and small heat shock proteins interact with filamin C to mediate the chaperone‐assisted selective autophagy pathway to remove damaged and misfolded proteins. 21 , 22 , 23 BAG3 is essential to stabilize the interaction with heat shock factor proteins and to maintain proteostasis. 24
Here we describe multiple FLNC variants and a range of cardiomyopathy outcomes. Induced pluripotent stem cell‐derived cardiomyocytes (iPSC‐CMs) were generated from individuals with FLNC truncating pathogenic variants with focus on the severe reduction in filamin C seen in a patient with biallelic FLNC truncating muations. To better understand the effect of reduced filamin C protein, we used gene editing to generate a truncation in the actin binding domain of filamin C in iPSC‐CMs. We found that FLNC‐null iPSC‐CMs displayed increased sensitivity to proteotoxic stress with an altered response in heat shock proteins and components of the autophagy pathway. To model clinically relevant arrhythmias associated with FLNC mutations, we assessed extracellular field potential as a reflection of action potential, and we identified prolonged field potential duration in FLNC null iPSC‐CMs. Furthermore, when subjected to proteotoxic stress with bortezomib, field potential prolonged even more, providing a link between proteostasis and arrhythmia susceptibility. In engineered heart tissues lacking filamin C, bortezomib produced functional impairment. Together, these findings identify that reduction in filamin C leads to impaired proteostasis and enhanced arrhythmogenesis.
Methods
The authors declare that all supporting data are available with the article and its supplemental files.
Data Access and Responsibility
All data are contained within the manuscript.
Dissertation
A portion of this work was published as a PhD dissertation. 25
Generation and Differentiation of iPSC Lines
Human iPSCs were generated from human skin fibroblasts (Coriell, sample GM03348), urine‐derived cells or peripheral blood mononuclear cells and reprogramed by electroporation with pCXLE‐hOCT3/4‐shp53‐F (Addgene plasmid 27077), pCXLE‐hSK (Addgene plasmid 27078), and pCXLE‐hUL (Addgene plasmid 27080) as described previously. 26 , 27 iPSCs were maintained on Matrigel™‐coated 6‐well plates in mTeSR‐1 (Stem Cell Technologies, Cat#85850) and passaged approximately every 5 days. iPSC‐CMs were differentiated using Wnt modulation as described previously. 28 Differentiation was conducted in CDM3 (RPMI 1640 with l‐glutamine, 213 μg/mL l‐ascorbic acid 2‐phosphate, 500 μg/mL recombinant human albumin). 28 When cells reached ~95% confluency, they were treated with 6 to 10 μM CHIR99021 for 24 hours and allowed to recover for 24 hours. Cells were then treated with 2 μM Wnt‐C59 for 48 hours and the media was changed with CDM3 every 2 days. After differentiation (~day 6–12) when cells were visibly beating, cells were passed through a 100 μm cell strainer and purified by MACS‐based cardiomyocyte enrichment (Miltenyi, Cat#130‐110‐188), counted, and replated. iPSC‐CMs were maintained by changing chemically defined cardiomyocyte differentiation media (RPMI, Cat#11875‐119 supplemented with CDM3) every other day. 28 Cells (2 × 106) were collected from each differentiation and tested for cardiomyocyte purity by staining for cardiac troponin T (BD, Cat#565744) and assessing by flow cytometry (BD, Acuri C6 Plus flow cytometer). Differentiations were >90% TNNT2 positive (Figure S1).
Gene Editing/Off Target Analysis, Genotyping
The guide RNA targeting FLNC exon 1 was designed using CRISPOR 29 (Table S1). iPSCs at 70% confluency were digested with TrypLE (Thermo, Cat#12563011), resuspended, and treated with 3 μg of guide RNA with pSpCas9(BB)‐2A‐GFP (Addgene plasmid #48138) control, or no DNA control in resuspension buffer (Neon, Cat#MPK10096) and replated onto a 6‐well Matrigel‐coated plate with 10% CloneR (Stem Cell, Cat#5888) and 2 μM Thiazovivin Rock Inhibitor (Sigma, Cat#SML1045) in mTESR. After 24 hours, cells were treated with 0.15 μg/mL puromycin (Thermo, Cat#A1113803), and then switched to puromycin at 0.2 μg/mL from 48 to 72 hours after guide electroporation. After 14 days, colonies were isolated manually. Individual clones were genotyped by Sanger sequencing. Clones with Sanger sequencing suggestive of successful editing were further subjected to amplicon sequencing using the same primers to verify the purity of clones (Amplicon EZ sequencing, Azenta Life Sciences). Analysis of potential off‐target mutations was conducted using primers targeting regions identified by CRISPOR. These regions were amplified and subjected to Sanger sequencing to confirm absence of inadvertent mutations (Tables S2 and S3). Chromosomal analysis was conducted using the hPSC Genetic Analysis kit (Stem Cell, Cat#07550).
Quantitative Polymerase Chain Reaction
iPSC‐CMs (day 30) were washed with cold 1X PBS and collected. RNA was isolated in TRIzol (Thermo, Cat#15596‐018) following the manufacturer's instructions. Isopropanol was used to precipitate RNA, which was collected in ultrapure water. The NanoDrop 2000 (Thermo) was used to measure RNA concentration. One microgram RNA was used to make complementary DNA (cDNA). Fifty nanograms of cDNA, Amplitaq gold 360 Master Mix (Thermo, Cat#4398881), and primer sets amplifying the 5′UTR to exon 1 and exon 48 to the 3′UTR were used to conduct quantitative polymerase chain reaction on the Thermocycler (Biorad) (Table S1). Gene expression was measured to calculate the ΔΔCq values by averaging Cq values of technical replicates, normalizing to MYBPC3, and normalizing the ΔCq value of each sample to the average ΔCq value for the unedited control.
Immunoblotting, Immunofluorescence Microscopy, and Antibodies
iPSC‐CMs (day 30) were washed with cold 1X PBS and collected in cell lysis buffer containing protease (Sigma, Cat#11836170001) and phosphatase inhibitor (Sigma, Cat# 4906837001) cocktails using a cell scraper. Samples were centrifuged at 7500 rpm for 5 minutes at 4 °C and the supernatant was collected. Bradford assay was conducted by mixing sample with Bradford 1X Dye Reagent (Biorad, Cat#5000205) in a 1:50 ratio and monitored in a 96‐well plate on a plate reader. Samples were aliquoted using cell lysis buffer with protease and phosphatase inhibitors and 4X Laemmli protein sample buffer (Biorad, Cat#1610747). Samples were warmed for 10 minutes at 75 °C and separated using a 4% to 15% precast protein gel (Biorad, Cat#4561083) at 100 V for 1 hour. Gels were transferred onto PVDF membrane (Biorad, Cat#1620177) for 3 hours at 900 mA at 4 °C. PVDF membranes were blocked with T20 blocking buffer (Thermo, Cat#37543) for 1 hour at room temperature (RT) and probed with primary antibodies FLNC (Sigma, Cat#HPA006135, 1:1000, ABClonal Cat#13018, 1:1000), BAG3 (Proteintech, Cat#10599‐1‐AP, 1:1000), HSPB8 (small heat shock protein B8; Proteintech, Cat#15287‐1‐AP, 1:1000), HSP70 (heat shock protein 70; Proteintech, Cat#10995‐1‐AP, 1:1000), p62/SQSTM1 (Proteintech, Cat#18420‐1‐AP, 1:1000), LC3 (Sigma, Cat#L8918, 1:1000), and α‐sarcomeric actin (Sigma, Cat#A2172, 1:1000) in T20 blocking buffer o/n at 4 °C. Immunoblots were washed with 1X TBS‐Tween and secondary antibodies goat antimouse (Jackson ImmunoResearch, Cat#115‐035‐003, 1:2500) and goat antirabbit (Jackson ImmunoResearch, Cat#111‐035‐003, 1:2500) conjugated to horseradish peroxidase in T20 blocking buffer for 1 hour at RT. Immunoblots were imaged using PICO (Thermo, Cat#34580) and Femto (Thermo, Cat#34096) chemiluminescent substrates on the iBright 1500 (Invitrogen). Protein loading was measured using the Memcode reversible protein stain kit (Thermo, Cat#24585). The National Institutes of Health Image J Fiji plug‐in was used to quantify blots.
For immunofluorescence microscopy, iPSC‐CMs (3 × 105 cells) were replated on Matrigel‐coated 12 mm Micro coverglass slips (Electron Microscopy Sciences, Cat#72231‐01) in a 24‐well plate. At days 19 to 25, coverglass slips were washed with 1X PBS and fixed with 2% paraformaldehyde. Glass coverslips were washed with 1X PBS, permeabilized with 0.2% Triton, and blocked with 5% BSA for 1 hour at RT. Coverslips were incubated with primary antibodies FLNC (ABClonal, Cat#A13018, 1:200) and alpha‐actinin (Sigma, Cat#A7811, 1:1000) in 5% BSA overnight at 4 °C. Coverslips were washed with 0.1% Tween and secondary antibodies goat antirabbit IgG (immunoglobulin G; Invitrogen, Cat#A11012, 1:1000) and donkey antimouse IgG (Invitrogen, Cat#A21202, 1) were added at 1:1000 in 5% BSA for 1 hour at RT. Coverslips were washed and mounted onto glass slides with Prolong Gold (Thermo, Cat#P36930) and imaged (Zeiss, Axio Imager M2).
Proteotoxic Stress With Bortezomib
iPSC‐CMs were replated onto 12‐well plates at 2 × 106 cells/well and treated at day 30 with proteasome inhibitor, bortezomib (Cell Signaling, 2204S) at varying concentrations or 0.01% DMSO for 24 hours. Cells were collected and protein was isolated and aliquoted as described. Protein expression was measured using immunoblotting.
Multielectrode Array Measurements
iPSC‐CMs were replated onto a 96‐well multielectrode array plate (Nanion Technologies, Cat#201003). Media change was completed 1 hour before the treatment plate was read on the CardioExcyte 96 platform (Nanion Technologies). iPSC‐CMs were paced at 1 Hz, 20 ms burst length and 10% intensity at a sweep duration of 30 seconds with a repetition interval of 10 minutes. iPSC‐CMs were treated with bortezomib or DMSO when stabilized after ~10 sweeps by adding to existing media in a 1:4 dilution for final concentration of 0.1 μM or 0.01%, respectively. Quality control and data analysis was performed using DataControl software for the CardioExcyte Nanion.
Engineered Heart Tissue Generation
Engineered heart tissue (EHTs) were generated as described across 2 posts. 30 , 31 Contractility index was determined using both strength and rate of contraction using postdeformation of beating EHTs. EHTs were exposed to either 0.1 μM bortezomib or 0.01% DMSO on day 20 for 24 hours, and the contractility index was determined at 24 hours. Images were acquired and scored blinded to genotype and treatment. EHTs were scored separately for both strength and rate of contraction on a 0 to 5 scale across a minimum of 4 videos per condition. Scores were summed for strength and rate across each genotype/treatment group. A score of 0 in either category represented no contraction and a score of 5 in either category represented strong or fast contraction, respectively.
Statistical Analysis
Data were analyzed using GraphPad Prism and specific statistical tests are indicated in text and figure legends. When 2 or more groups were analyzed, ANOVA was implemented across groups with testing for multiple comparisons, specific tests are described based on distribution of data; unpaired t tests were used to compare across 2 groups. Experimental outcomes were described as means±SEM, significance was assessed as P<0.05.
Ethical Approval
All participants provided informed consent for cell donation and access to medical record information under the approval of the Northwestern University Institutional Review Board. Probands underwent clinical next generation‐based sequencing as part of standard of care. Additional family members underwent cascade screening when available. All subjects were of European or European/Ashkenazi Jewish ancestry.
Results
Cardiomyopathy and Arrhythmias With FLNC Mutations
A 40‐year‐old woman presented with cough and shortness of breath 8 weeks after giving birth to her fifth child. She was found to have a dilated left ventricle with severely globally reduced function (Figure 1 and Table, Family A, III‐1). Cardiac magnetic resonance imaging reported a left ventricular (LV) end diastolic volume index=170 mL/m2 and LV ejection fraction of 20%. There was a midmyocardial stripe of late gadolinium enhancement in the midbasal interventricular septum extending into the midbasal inferior wall. Laboratory values were significant for normal troponin and markedly elevated NT‐proBNP (N‐terminal pro‐B‐type natriuretic peptide; >4000 pg/mL). Cardiac telemetry showed nonsustained ventricular tachycardia (NSVT) (Figure 1), so she was discharged from the hospital with an external defibrillator vest while she recovered from presumed peripartum cardiomyopathy. Over the next several months, she had limited improvement in LV ejection fraction despite up titration of guideline‐directed medical therapy, and she continued to have NSVT.
Figure 1. Clinical findings in a woman with dilated cardiomyopathy and mild, nonprogressive skeletal myopathy.
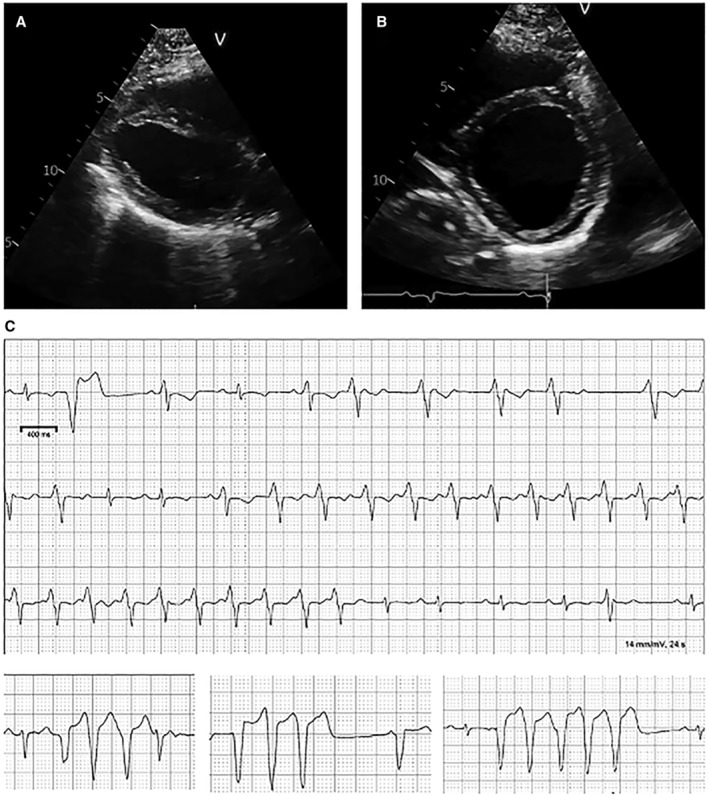
This individual presented 8 weeks postpartum with progressive heart failure symptoms and markedly reduced LV function. Her LV was markedly dilated (LV internal dimension in diastole, 6.1 cm with LV ejection fraction of 30%). (A) Parasternal long axis view. (B) Short axis view. (C) NSVT was present on telemetric monitoring. The top tracing represents a continuous strip; in the second row of the tracing, an idioventricular tachycardia was seen which then accelerates into ventricular tachycardia in the third row. Atrioventricular dissociation is present consistent with ventricular tachycardia. LV indicates left ventricular; and NSVT, nonsustained ventricular tachycardia.
Table 1.
Summary of Genetic and Clinical Findings
| Pedigree (person) | Heterozygous FLNC variants | Number | AF | Clinical information |
|---|---|---|---|---|
| A, III‐1* , † |
c.1948C>T, p.Arg650X/ c.970‐4A>G |
rs532143625 |
NA 0.005% |
DCM, NSVT |
| A, II‐5 | c.970‐4A>G | 0.005% | NSVT | |
| B, III‐1* | c.970‐4A>G | rs532143625 | 0.005% | DCM, NSVT |
| B, III‐2 | c.970‐4A>G | 0.005% | DCM, NSVT | |
| B, II‐5 | Inferred from proband | 0.005% | DCM, NSVT, cardiac transplant | |
| X, III‐1* | c.8143_8144delGT, p. Val2715fs87X | NA | ||
| Y, II‐5* | c.7371delT, p. Glu2458SerfsX71 | NA | ||
| Y, III‐4 | Inferred from proband | NA | Sudden cardiac death | |
| Z, II‐1* | c.318C>G, Phe106Leu | rs886037829 | 0.005% | LGMD, atrial fibrillation, hypertrophy |
| Z, II‐2 | c.318C>G, Phe106Leu | 0.005% | LGMD | |
| Z, II‐3 | NA | Aborted cardiac arrest |
AF indicates allele frequency gnomAD; DCM, dilated cardiomyopathy; LGMD, limb‐girdle muscular dystrophy; NA, not found in gnomAD; and NSVT, nonsustained ventricular tachycardia.
Proband.
The two genotypes for A, III‐1 (compound heterozygote) are shown.
She had hypotonia at birth, delayed walking, scoliosis requiring surgery, and internally rotated tibia and metatarus adductus of the feet. Because of these findings, she had a muscle biopsy at age 8, which was read as lipid storage myopathy (images unavailable). At the time of her adult presentation with heart failure, she was fully ambulatory and participated regularly in exercise activities with minimal to no weakness. Her history included 5 successful pregnancies, including the most recent uneventful delivery. Clinical genetic testing with a 168‐gene cardiomyopathy and arrhythmia panel identified 2 FLNC variants in exon 5 (c.970‐4A>G) and exon 12 (c.1948C>T, p.Arg650X) (Figure 2). The premature truncation p.Arg650X is not found in population databases. The exon 5 variant is present in 0.005% of the gnomAD cohort and was shown to disrupt splicing and cause a range of arrhythmogenic cardiomyopathy in 4 unrelated families characterized from multiple centers. 32 Her mother was found be heterozygous for c.970‐4A>G but did not carry p.Arg650X, and at age 70, she had a long history of ventricular ectopy without syncope and normal LV function (Figure 2 and Table, Family A, II‐5). Her children had either the p.Arg650X or c.970‐4A>G, confirming the compound heterozygous status, and other family members were not available for testing.
Figure 2. Summary of FLNC variants and pedigrees in this study.
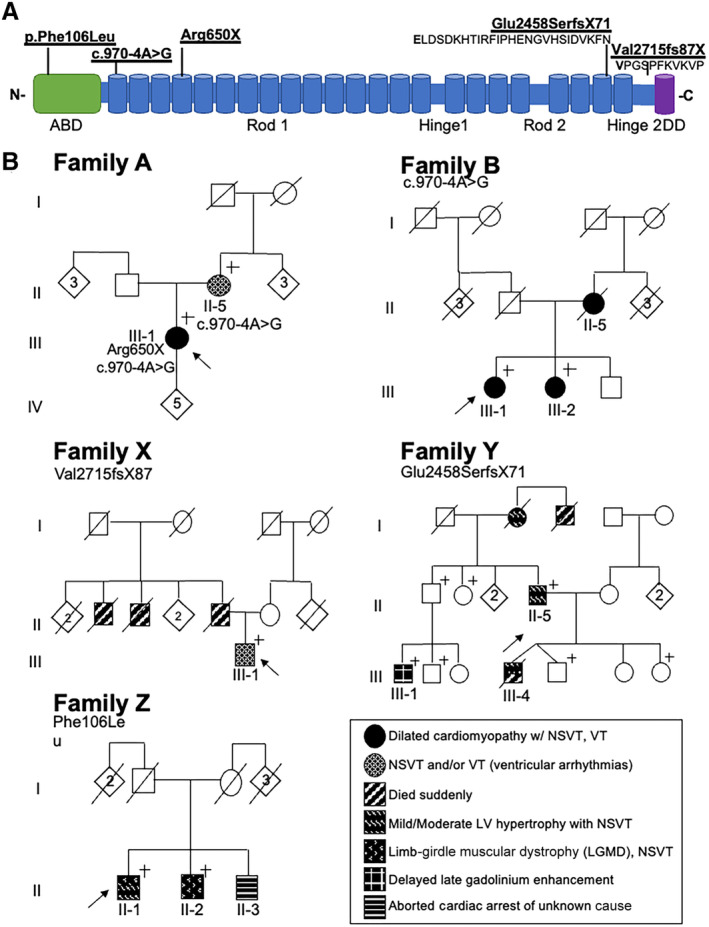
(A) Schematic of the filamin C protein and the position of the FLNC variants in the pedigrees. (B) Pedigrees of the families. The proband in family A carries 2 different FLNC variants in trans, p.Arg650X, and c.970‐4A>G, making her a compound heterozygote. The remaining families have a single FLNC variant in the heterozygous state. Family B has 1 of the 2 FLNC alleles (c.970‐4A>G) seen in the proband in Family A, but these 2 families are not related. Family X and Y have premature truncations in FLNC (p.Glu2458fsX71, p.Val2515fsX87), whereas family Z has a missense FLNC variant (p.Phe106Leu). ABD indicates actin binding domain; DD, dimerization domain; LV, left ventricular; and NSVT, nonsustained ventricular tachycardia.+ denotes that individual carries familial FLNC variant; circle denotes female, square denotes male; rhombus, live birth, sex unavailable, number indicates number of individuals; line indicates deceased.
A second proband (Family B) was found with heterozygous FLNC c.970‐4A>G and dilated cardiomyopathy (DCM; Figure 2 and Table, Family B, III‐1). This proband had DCM with a LV ejection fraction of 35%, left bundle‐branch block, and NSVT in her 50s. Her sister was diagnosed with DCM at age 47 (Figure 2 and Table 1, Family B, III‐2) with an LV ejection fraction of 8%; both sisters were confirmed to carry heterozygous FLNC c.970‐4A>G. The proband's mother, before her death, was diagnosed with DCM and underwent cardiac transplant in her early 60s (Figure 2 and Table, Family B, II‐5). Family A and Family B both carry FLNC c.970‐4A>G, but these 2 families derive from different parts of the United States and share no known relationship. This variant has been linked to cardiomyopathy and arrhythmias in independent reports 1 , 8 , 32 and is interpreted as pathogenic in ClinVar (Accession Number SCV002587791.1). The c.970‐4A>G variant activates a cryptic splice site, introducing a 3bp insertion creating a premature termination codon documented in patient‐derived cells. 32 iPSC‐CMs with the c.970‐4A>G variant display spontaneous irregular action potentials, recapitulating the arrhythmogenic propensity seen in patients. 32
The proband in Family X was heterozygous for the FLNC frameshift variant (c.8143_8144delGT, p.Val2715fs87X) (Figure 2 and Table, Family X III‐1). This patient was evaluated because of chest pain and intermittent episodes of dizziness at age 52. On telemetric monitoring he had NSVT, and treadmill stress testing identified runs of VT. Cardiac magnetic resonance imaging showed normal LV cavity size and function with an LV ejection fraction of 66%, mild concentric hypertrophy, and evidence of midmyocardial enhancement highly suspicious of infiltrative cardiomyopathy. His father died suddenly at age 69, and 2 of the proband's uncles died suddenly in their 40s.
The proband in Family Y (II‐5) came to medical attention when his son suffered sudden cardiac death at age 17 (III‐4), and his autopsy identified hypertrophic cardiomyopathy with a septum thickness of 2.0 cm. Whole genome sequencing on the proband identified heterozygous FLNC c.7371delT, p.Glu2458Serfs71X. This proband had mild hypertrophic cardiomyopathy and NSVT, and an implantable cardioverter‐defibrillator was implanted (Figure 2 and Table, Family Y, II‐5). The proband's mother had a diagnosis of hypertrophic cardiomyopathy at age 65 and died suddenly at age 93, and a maternal great uncle died suddenly at age 16 without a clear cause. One relative (Family Y, III‐1) was found with extensive delayed enhancement on cardiac magnetic resonance imaging. Site‐specific testing of this person (Family Y, III‐1) in a clinical genetic laboratory identified the same FLNC variant (FLNC c.7371delT, p.Glu2458Serfs71X), which was interpreted by the testing laboratory as pathogenic.
Family Z had 2 brothers who developed muscle weakness in their 30s. The proband was heterozygous for a FLNC variant of uncertain significance (c.318C>G, p.Phe106Leu). His brother had a similar presentation and carried the same FLNC variant of uncertain significance (Figure 2 and Table, Family Z, II‐1, II‐2). The proband had a history of atrial fibrillation, and cardiac magnetic resonance imaging showed mild basal anterior wall hypertrophy with a maximal wall thickness of 1.5 cm with focal increased signal of delayed enhancement in the midanteroseptal wall. A summary of the pedigrees is shown in Figure 2 with genetic and clinical findings presented in Table. These findings underscore the association of FLNC with variable cardiomyopathy, arrhythmia, and limb‐girdle muscular dystrophy subtypes.
Filamin C Is Markedly Reduced in iPSC‐CMs From the Individual With Biallelic FLNC Pathogenic Variants
iPSC‐CMs were generated from probands representing the different FLNC pathogenic variants. Quantitative reverse transcription polymerase chain reaction using primers to both the 5′ and 3′ portions of the FLNC transcript showed no consistent difference in the FLNC iPSC‐CM lines compared with healthy control iPSC‐CMs, suggesting that nonsense mediated decay was not present (Figure S2). Using immunoblotting with an antibody to the filamin C carboxy‐terminus, we evaluated filamin C protein production in iPSC‐CMs from healthy control lines and lines carrying FLNC variants p.Arg650X,c.970‐4A>G, p.Glu2458SerfsX71 (referred to as p.Glu2458fs), p.Val2715fs87X (referred to p.Val2715fs), and p.Phe106Leu (Figure 3A, Figure S3). Compared with healthy control iPSC‐CMs, the amount of full‐length filamin C protein was reduced in the 3 lines with FLNC truncation variants (p.Arg650X, p.Glu2458fs, p.Val2715fs) but not in the missense line p.Phe106Leu (Figure 3B). The heterozygous FLNC iPSC‐CMs retained some expression of filamin C, as expected. The smaller 30 kDa band in the p.Val2715fs lane was present in the healthy control iPSC‐CMs, so the smaller band is unrelated to a specific FLNC mutation (Figure S3). An antibody generated to amino acids 2160‐2340 was also tested and detected relatively normal amounts of filamin C in the heterozygous iPSC‐CMs and reduced but detectable full length filamin C in the IPSC‐CMs with Arg650X/c.970‐4G>A (Figure S4). The differences in these antibodies may relate to differences in sensitivity or specificity, since the Arg650X/c.970‐4G>A FLNC line is not expected to produce filamin C, consistent with immunoblots in Figure 3B and Figure S3. Both antibodies concur that filamin C protein was most reduced in p.Arg650X iPSC‐CMs, consistent with 2 FLNC variants in trans as a compound heterozygote (c.970‐4A>G), and the reduction in this line is consistent with the more severe phenotype in this patient.
Figure 3. Filamin C protein expression and localization in iPSC‐CMs from patients carrying truncating variants.
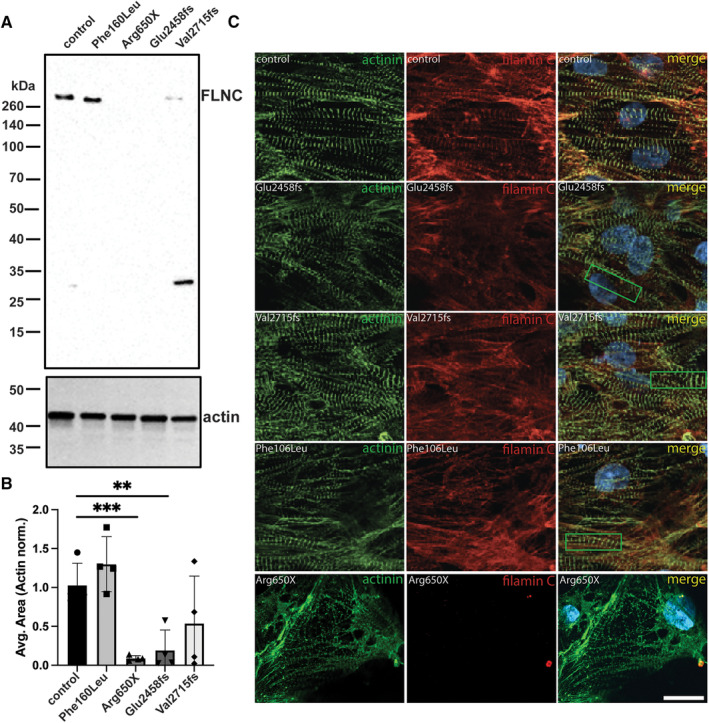
(A) Immunoblotting of iPSC‐CMs derived from FLNC missense p.Phe106Leu and FLNC truncations, p.Arg650X, p.Glu2458fs, and p.Val2715fs. (B) Quantification of immunoblots from 4 different differentiations is shown. Data are shown as mean±SEM with significance determined using a 1‐way ANOVA, Dunnett's multiple comparisons test was used to determine significance. **P=0.017, ***P=0.0074. (C) Immunofluorescence microscopy showed reduced filamin C in patient‐derived iPSC‐CMs. The left column shows α‐actinin (green) and demonstrates sarcomeres in each iPSC‐CM line. The appearance of the sarcomeres was similar between the truncation variants and the missense line, with all FLNC variants demonstrating some degree of Z band disruption (examples in green boxes in merged image). The middle column shows filamin C staining highlighting sarcomere‐associated filamin C in the healthy control and in the patient‐cell lines (red channel). Scale bar, 20 μm. iPSC‐CM indicates induced pluripotent stem cell‐cardiomyocytes.
iPSC‐CMs were evaluated for protein localization using immunofluorescence microscopy, comparing it to healthy control iPSC‐CMs. Localization of α‐actinin was used to evaluate sarcomere organization (green imaging in Figure 3C), with sarcomeres visualized in each of the patient‐derived iPSC‐CM lines. In healthy control human iPSC‐CMs, filamin C colocalized with α‐actinin (merged images in Figure 3C) and appeared qualitatively less in patient‐derived iPSC‐CMs, despite sarcomere structures being present (Figure 3C, red channel). This phenomena was previously reported in 2 other FLNC truncating variants. 33 In addition, Z‐line disorganization was observed in iPSC‐CM lines with p.Glu2458fs, p.Val2715fs, and p.Phe106Leu (Figure 3C, green boxes). The p.Arg650X line had no obvious filamin C (red) (Figure 3C, bottom row of images), consistent with the immunoblotting data.
Defective Response to Proteotoxic Stress in the Absence of Filamin C
As an actin crosslinking protein, filamin C is a regulator of cellular stress and injury. 34 After exercise or with experimentally induced cell injury, filamin C is enriched at sites of injury in skeletal myofibers and in cardiomyocytes, where it participates in the repair response including sarcomere rebuilding. 20 , 35 Under conditions of mechanical stress, filamin C is upregulated to manage injury and repair responses in skeletal muscle and cardiomyocytes. 36 , 37 In skeletal muscle, unfolded or misfolded filamin C is disposed of by chaperone‐assisted selective autophagy, which engages BAG3 and heat shock proteins to target unfolded proteins to lysosomes for degradation. 19 , 21 To evaluate the role of filamin C in iPSC‐CMs, we generated FLNC null iPSCs from a healthy control cell line using CRISPR‐Cas9 using guide RNAs targeting an exon encoding the actin binding domain (ABD) of filamin C. This line is referred to as FLNC ABD−/−, and this approach provides an isogenic control (ABD, Figure 4A and Figure S5). FLNC ABD−/− iPSCs carried 2 different alleles that disrupted the reading frame, and no specific off‐target mutations were detected (Figures S6 and S7). FLNC ABD−/− iPSC‐CMs had reduced mRNA expression, and filamin C protein expression was undetectable (Figures S8 and S9).
Figure 4. Impaired response to proteotoxic stress in human iPSC‐CMs lacking filamin C (FLNC ABD−/−).
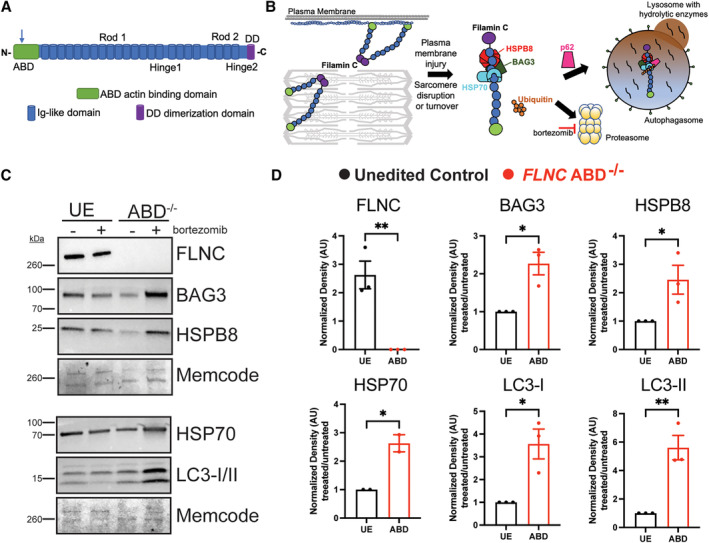
(A) A healthy control human iPSC line was gene edited in the ABD to generate FLNC ABD−/− cells (blue arrow marks position of deletion). (B) Filamin C is found at the Z disk and plasma membrane. Misfolded or unfolded filamin C protein is bound by chaperones like BAG3 and heat shock proteins and then disposed through proteasomal degradation or autophagy. 19 (C) An unedited, isogenic control and FLNC ABD−/− iPSCs (ABD) were differentiated to cardiomyocytes and then treated with the proteasome inhibitor bortezomib (0.1 μm) for 24 h. In the absence of filamin C, bortezomib exposure results in an increase in chaperone proteins and autophagy markers in iPSC‐CMs. D) Compared with the unedited, isogenic control, FLNC ABD−/− iPSC‐CMs displayed an increase in the chaperone proteins BAG3, HSP70 and HSPB8 when exposed to bortezomib. FLNC ABD−/− iPSC‐CMs also had an increase in LC3‐1 and LC3‐II after bortezomib compared with unedited control iPSC‐CMs. Expression levels for FLNC are shown as fold change compared with unedited control. Expression levels for all other proteins are normalized to bortezomib response in the unedited control cells. **P=0.006 FLNC, *0.013 BAG3, *0.032 HSP70, *0.045 HSPB8, *0.017 LC3‐I and **0.006 LC3‐II. The normalized data were analyzed with an unpaired t‐test and is shown as mean±SEM. ABD indicates actin binding domain; DD, dimerization domain; iPSC‐CM, induced pluripotent stem cell‐cardiomyocytes; and UE, unedited control.
BAG3 and heat shock proteins are chaperones that sequester and concentrate misfolded proteins, which is an important function for mechanically stressed cardiomyocytes and skeletal myofibers 19 , 24 , 38 , 39 (Figure 4B). Additionally, abnormal lysosomal accumulation was previously described as a feature of FLNC disruption in iPSC‐CMs. 40 Therefore, we assessed expression of proteins after proteasome inhibition using bortezomib, an inhibitor of the 26S proteasome. We treated both isogenic control and FLNC ABD−/− iPSC‐CMs with bortezomib (0.1 μm) for 24 hours (Figure 4C and Figure 4D, Figures S10 and S11). Under these conditions, this low dose of bortezomib is expected to partially perturb the proteasome, whereas higher doses elicited toxicity. Treatment of FLNC ABD−/− iPSC‐CMs with low‐dose bortezomib (0.1 μm) showed an increase in the chaperone proteins BAG3, HSPB8 (small heat shock protein B8), and HSP70 (heat shock protein 70) compared with isogenic unedited control iPSC‐CMs. Bortezomib also stimulated an increase in the autophagy markers LC3‐I and LC3‐II (Figure 4D) compared with the isogenic control. We interpret the increase of chaperones and autophagy markers as consistent with the role of filamin C as a scaffold to help concentrate misfolded proteins for disposal. In the absence or reduction of filamin C, cardiomyocytes as less able to tolerate perturbations in protein turnover, including during cardiomyocyte hypertrophy or regression.
Prolonged Extracellular Field Potential in FLNC Mutant iPSC‐CMs Is Enhanced Under Proteasomal Inhibition
Although the cardiomyopathic expression of FLNC truncation variants is variable, a major clinical complication in FLNC cardiomyopathic mutations is ventricular arrhythmia. 2 , 8 , 9 To assess arrhythmic potential of iPSC‐CMs, we measured extracellular field potential as a proxy for action potential duration in paced iPSC‐CMs. For these studies, we tested isogenic control iPSC‐CMs and FLNC ABD−/− iPSC‐CMs. We also included the p.Arg650X/c.970‐4A>G patient‐derived iPSC‐CMs (referred to as p.Arg650X for simplicity) because this line had the greatest reduction in filamin C. At baseline, extracellular field potential was prolonged in both FLNC ABD−/− and p.Arg650X iPSC‐CMs compared with the unedited control iPSC‐CM line (compare y axes in Figure S12). These findings are consistent with prolonged action potential duration. When treated with 0.1 μm bortezomib, extracellular field potential remained normal in the isogenic control iPSC‐CMs. However, with this dose of bortezomib, the extracellular field potential remained prolonged in FLNC ABD−/− iPSC‐CMs and further prolonged in p.Arg650X iPSC‐CMs, consistent with a stress‐induced substrate for arrhythmia risk (Figure 5A). We also generated EHTs from isogenic control and FLNC ABD−/− iPSC‐CMs because this format can be more readily used to assess function given that EHTs display greater cardiomyocyte and sarcomere alignment compared with iPSC‐CMs in 2 dimensional cell culture (Figure 5B). We measured the contractility of FLNC ABD−/− and isogenic control EHTs blinded to genotype before and after 24 hours treatment with 0.1 μm bortezomib. At baseline, the contractility index of FLNC ABD−/− EHTs was significantly lower compared with isogenic controls (P=0.026), consistent with reduced function. Isogenic control EHTs had a 1.8‐fold reduction in their contractility index after bortezomib, whereas FLNC ABD−/− EHTs had a 3‐fold reduction in contractility after bortezomib (Figure 5C). The observation that FLNC ABD−/− EHTs had greater impairment after partial proteasome inhibition highlight the necessity of filamin C for normal proteostasis.
Figure 5. Bortezomib‐induced proteotoxic stress prolonged field potential duration and reduced contractility in cultured FLNC iPSC‐CMs and in FLNC ABD−/− engineered heart tissues.
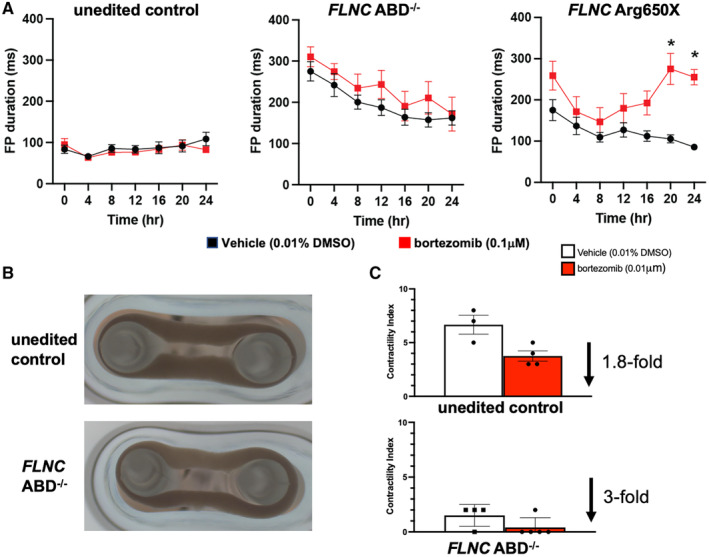
Extracellular field potential duration was monitored on the Nanion CardioExcyte‐96 platform. (A) Time course of field potential duration with bortezomib exposure (repeated measures ANOVA, Bonferroni correction P<0.001 at 20 h, and P<0.001 at 24 h) (red lines) compared with vehicle (black lines) (n=16 wells per time point, per each genotype). (B) Representative images of EHTs containing the isogenic, unedited control (top) and FLNC ABD−/− (bottom) iPSC‐CMs. (C) Contractility index was determined for EHTs before and after bortezomib treatment. Isogenic, unedited controls exhibited a 1.8‐fold reduction in contractility after bortezomib whereas FLNC ABD−/− EHTs had a 3‐fold reduction in contractility. These data demonstrate that proteotoxic stress creates a substrate for arrhythmia that can be monitored in cultured iPSC‐CMs and EHTs. Data are shown as ±SEM. ABD indicates actin binding domain; DD, dimerization domain; EHT, engineered heart tissues; FP, field potential; iPSC‐CM, induced pluripotent stem cell‐cardiomyocytes; and UE, unedited control.
Discussion
Variable Cardiomyopathy Expression With FLNC Mutations
Heterozygous truncating FLNC mutations have been associated with hypertrophic, dilated, and arrhythmogenic cardiomyopathy. 1 , 3 Among the probands reported here, we also observed multiple forms of cardiomyopathies including hypertrophic cardiomyopathy and DCM. FLNC p.Phe106Leu associated with a mild myopathic process affecting both cardiac and skeletal muscle. This variant was previously described in 2 siblings with early‐onset, lethal cardiomyopathy with FLNC compound heterozygous variants, consistent with its role in filaminopathy. 41 We previously reported one of the first cases of sudden cardiac death linked to FLNC in a young woman carrying c.3791‐1G>C, which was subsequently identified in the Ashkenazi population. 42 , 43 The FLNC c.3791‐1G>C variant disrupts a splicing acceptor at the end of intron 21 and was associated with reduced filamin C transcript expression in fibroblasts. Genetic assessment of targeted populations with arrhythmias and cardiomyopathies identified an enrichment of FLNC mutations, especially truncating variants. 2 , 3 , 8 , 9 , 44 On a population cohort level, a genotype “first” approach, where genetic findings are then correlated with electronic health record reports, also confirmed that FLNC truncations increase risk of cardiac disease. A survey of 171 948 subjects in a medical biobank found FLNC loss of function (truncation) variants associated with an increased risk of ventricular arrhythmia and dysfunction. Biobank participants with FLNC truncation variants had significantly increased odds of DCM, LV dysfunction, supraventricular tachycardia, and arrhythmia. 45 Because of findings like these and other reports, 46 the American College of Medical Genetics and Genomics recently recommended reporting pathogenic FLNC variants incidentally identified through genome sequencing citing the high penetrance and elevated risk for cardiomyopathy and sudden death. In these studies, the penetrance of FLNC variants was relatively low, supporting the idea that additional stressors or second “hits” likely contribute to disease manifestation. Our studies identify cell stimuli that further stress protein turnover may contribute to that risk.
Pregnancy as a Risk With FLNC Pathogenic Variants
The index case presented here developed symptomatic heart failure and ventricular arrhythmias in the peripartum interval. During her evaluation, she was found to have biallelic FLNC variants, both of which disrupted filamin C expression. FLNC pathogenic variants have been described in peripartum cardiomyopathy. 47 During pregnancy, in response to increased cardiac demand, the heart grows, and in the postpartum interval this cardiac growth recedes. 48 Therefore, it is possible that filamin C is especially critical during windows of sarcomere growth or reduction. TTN gene truncations also manifest as peripartum cardiomyopathy. Both titin and filamin C are key proteins that regulate sarcomerogenesis and formation of intercalated disks, which may be especially subject to turnover with the cardiac adaptations during late pregnancy. 49 , 50 Altered proteostasis has been previously linked to atrial fibrillation, where it has been suggested to modulate intercellular communication and potentially be a target for therapeutic intervention, 51 , 52 , 53 and aspects of these same mechanisms may also be critical for ventricular arrhythmias.
Roles of Filamin C in Cardiomyocytes
In skeletal muscle, resistance exercise is associated with an increase in chaperone‐assisted selective autophagy components including filamin C. 36 When mouse hearts are subjected to transaortic construction or isoproterenol stress, filamin C is upregulated. 37 Acute injury to myotubes is associated with recruitment of Flnc mRNA and filamin C protein to the site of injury where it plays an essential role in injury repair. 35 , 54 Similarly, injury to iPSC‐CMs is also associated with recruitment of filamin C to the injury site. 20 Filamin C may serve as a scaffold onto which small and larger heat shock proteins assemble to manage excess or misfolded proteins at the sites of injury. 55 A recent study on FLNC‐related myofibrillar myopathy suggests that distinct FLNC truncations may differentially alter protein quality control. 56 Filamin C has been linked to cellular repair and protein homeostasis, and our study suggests impaired proteostasis may contribute to arrhythmia risk because even mild proteotoxic stress was sufficient to further prolong action potential in the absence of filamin C. We hypothesize improper removal of misfolded proteins creates a cellular milieu that impedes conduction unevenly across the myocardium, which increases irregular action potential firing and transmission.
A recent study using FLNC iPSC‐CMs identified an accumulation of lysosomal proteins. 40 Homozygous FLNC null lines showed reduced Z band and select sarcomere proteins, whereas heterozygous loss of FLNC resulted in an accumulation of lysosomal proteins. The heterozygous FLNC iPSC‐CMs not only had increased lysosomal content but also displayed increased autophagic flux and depletion of autophagy proteins, which could reflect a compensation for impaired proteasomal degradation and disposal of proteins through the proteasome pathways.
Conclusions
Our findings are consistent with the increase in autophagic flux based on the observed increase of LC3I/II under proteasome inhibition in IPSC‐CMs with markedly reduced filamin C. In a separate line of experiments, gene expression profiling identified excess PDGFRA (platelet derived growth factor A) signaling in FLNC null iPSC‐CMs, and the authors demonstrated that crenolanib, a receptor tyrosine kinase inhibitor with preference for mutant receptor tyrosine kinases was effective at improving contractile dysfunction in FLNC mutant iPSC‐CMs. 33 How these signaling pathways may intersect with enhanced proteasomal sensitivity and autophagy is not known, and additional investigation may help further identify how FLNC disruption leads to cardiomyopathy with enhanced arrhythmia.
Sources of Funding
This work was supported by NIH R01HL128075, NIH R00HL141698, NIH K08HL163392, American Heart Association SFRN, and Leducq Foundation. The funders played no role in the study design or interpretation.
Disclosures
Elizabeth M. McNally is a consultant for Amgen, AstraZeneca, Cytokinetics, PepGen, Pfizer, and Tenaya Therapeutics and is a founder of Ikaika Therapeutics. Alfred L. George, Jr received research funding from Praxis Precision Medicines, Tevard Biosciences, and Neurocrine Biosciences. These activities are unrelated to the content of this manuscript. The remaining authors have no disclosure to report.
Supporting information
Tables S1–S3
Figures S1–S12
This article was sent to Shu‐Fen Wung, PhD, RN, ACNP‐BC, FAAN, Guest Editor, for review by expert referees, editorial decision, and final disposition.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.123.030467
For Sources of Funding and Disclosures, see page 13.
References
- 1. Verdonschot JAJ, Vanhoutte EK, Claes GRF, Helderman‐van den Enden A, Hoeijmakers JGJ, Hellebrekers D, de Haan A, Christiaans I, Lekanne Deprez RH, Boen HM, et al. A mutation update for the FLNC gene in myopathies and cardiomyopathies. Hum Mutat. 2020;41:1091–1111. doi: 10.1002/humu.24004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ortiz‐Genga MF, Cuenca S, Dal Ferro M, Zorio E, Salgado‐Aranda R, Climent V, Padrón‐Barthe L, Duro‐Aguado I, Jiménez‐Jáimez J, Hidalgo‐Olivares VM, et al. Truncating FLNC mutations are associated with high‐risk dilated and Arrhythmogenic cardiomyopathies. J Am Coll Cardiol. 2016;68:2440–2451. doi: 10.1016/j.jacc.2016.09.927 [DOI] [PubMed] [Google Scholar]
- 3. Begay RL, Graw SL, Sinagra G, Asimaki A, Rowland TJ, Slavov DB, Gowan K, Jones KL, Brun F, Merlo M, et al. Filamin C truncation mutations are associated with arrhythmogenic dilated cardiomyopathy and changes in the cell–cell adhesion structures. JACC Clin Electrophysiol. 2018;4:504–514. doi: 10.1016/j.jacep.2017.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Begay RL, Tharp CA, Martin A, Graw SL, Sinagra G, Miani D, Sweet ME, Slavov DB, Stafford N, Zeller MJ, et al. FLNC gene splice mutations cause dilated cardiomyopathy. JACC Basic Transl Sci. 2016;1:344–359. doi: 10.1016/j.jacbts.2016.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Selcen D, Ohno K, Engel AG. Myofibrillar myopathy: clinical, morphological and genetic studies in 63 patients. Brain. 2004;127:439–451. doi: 10.1093/brain/awh052 [DOI] [PubMed] [Google Scholar]
- 6. Selcen D, Engel AG. Myofibrillar myopathies. Handb Clin Neurol. 2011;101:143–154. doi: 10.1016/b978-0-08-045031-5.00011-6 [DOI] [PubMed] [Google Scholar]
- 7. Celeghin R, Cipriani A, Bariani R, Bueno Marinas M, Cason M, Bevilacqua M, de Gaspari M, Rizzo S, Rigato I, da Pozzo S, et al. Filamin‐C variant‐associated cardiomyopathy: a pooled analysis of individual patient data to evaluate the clinical profile and risk of sudden cardiac death. Heart Rhythm. 2022;19:235–243. doi: 10.1016/j.hrthm.2021.09.029 [DOI] [PubMed] [Google Scholar]
- 8. Gigli M, Stolfo D, Graw SL, Merlo M, Gregorio C, Nee Chen S, Dal Ferro M, Paldino MA, de Angelis G, Brun F, et al. Phenotypic expression, natural history, and risk stratification of cardiomyopathy caused by filamin C truncating variants. Circulation. 2021;144:1600–1611. doi: 10.1161/circulationaha.121.053521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Akhtar MM, Lorenzini M, Pavlou M, Ochoa JP, O'Mahony C, Restrepo‐Cordoba MA, Segura‐Rodriguez D, Bermúdez‐Jiménez F, Molina P, Cuenca S, et al. Association of left ventricular systolic dysfunction among carriers of truncating variants in filamin C with frequent ventricular arrhythmia and end‐stage heart failure. JAMA Cardiol. 2021;6:891–901. doi: 10.1001/jamacardio.2021.1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kley RA, Leber Y, Schrank B, Zhuge H, Orfanos Z, Kostan J, Onipe A, Sellung D, Güttsches AK, Eggers B, et al. FLNC‐associated myofibrillar myopathy: new clinical, functional, and proteomic data. Neurol Genet. 2021;7:e590. doi: 10.1212/nxg.0000000000000590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kley RA, Maerkens A, Leber Y, Theis V, Schreiner A, van der Ven PF, Uszkoreit J, Stephan C, Eulitz S, Euler N, et al. A combined laser microdissection and mass spectrometry approach reveals new disease relevant proteins accumulating in aggregates of filaminopathy patients. Mol Cell Proteomics. 2013;12:215–227. doi: 10.1074/mcp.M112.023176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liao WC, Juo LY, Shih YL, Chen YH, Yan YT. HSPB7 prevents cardiac conduction system defect through maintaining intercalated disc integrity. PLoS Genet. 2017;13:e1006984. doi: 10.1371/journal.pgen.1006984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van der Ven PF, Obermann WM, Lemke B, Gautel M, Weber K, Fürst DO. Characterization of muscle filamin isoforms suggests a possible role of gamma‐filamin/ABP‐L in sarcomeric Z‐disc formation. Cell Motil Cytoskeleton. 2000;45:149–162. doi: [DOI] [PubMed] [Google Scholar]
- 14. Kong SW, Hu YW, Ho JW, Ikeda S, Polster S, John R, Hall JL, Bisping E, Pieske B, dos Remedios CG, et al. Heart failure‐associated changes in RNA splicing of sarcomere genes. Circ Cardiovasc Genet. 2010;3:138–146. doi: 10.1161/circgenetics.109.904698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vorgerd M, van der Ven PF, Bruchertseifer V, Löwe T, Kley RA, Schröder R, Lochmüller H, Himmel M, Koehler K, Fürst DO, et al. A mutation in the dimerization domain of filamin c causes a novel type of autosomal dominant myofibrillar myopathy. Am J Hum Genet. 2005;77:297–304. doi: 10.1086/431959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kley RA, Serdaroglu‐Oflazer P, Leber Y, Odgerel Z, van der Ven PF, Olivé M, Ferrer I, Onipe A, Mihaylov M, Bilbao JM, et al. Pathophysiology of protein aggregation and extended phenotyping in filaminopathy. Brain. 2012;135:2642–2660. doi: 10.1093/brain/aws200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chevessier F, Schuld J, Orfanos Z, Plank AC, Wolf L, Maerkens A, Unger A, Schlötzer‐Schrehardt U, Kley RA, von Hörsten S, et al. Myofibrillar instability exacerbated by acute exercise in filaminopathy. Hum Mol Genet. 2015;24:7207–7220. doi: 10.1093/hmg/ddv421 [DOI] [PubMed] [Google Scholar]
- 18. Dalkilic I, Schienda J, Thompson TG, Kunkel LM. Loss of FilaminC (FLNc) results in severe defects in myogenesis and myotube structure. Mol Cell Biol. 2006;26:6522–6534. doi: 10.1128/mcb.00243-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Collier MP, Benesch JLP. Small heat‐shock proteins and their role in mechanical stress. Cell Stress Chaperones. 2020;25:601–613. doi: 10.1007/s12192-020-01095-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leber Y, Ruparelia AA, Kirfel G, van der Ven PF, Hoffmann B, Merkel R, Bryson‐Richardson RJ, Fürst DO. Filamin C is a highly dynamic protein associated with fast repair of myofibrillar microdamage. Hum Mol Genet. 2016;25:2776–2788. doi: 10.1093/hmg/ddw135 [DOI] [PubMed] [Google Scholar]
- 21. Arndt V, Dick N, Tawo R, Dreiseidler M, Wenzel D, Hesse M, Fürst DO, Saftig P, Saint R, Fleischmann BK, et al. Chaperone‐assisted selective autophagy is essential for muscle maintenance. Curr Biol. 2010;20:143–148. doi: 10.1016/j.cub.2009.11.022 [DOI] [PubMed] [Google Scholar]
- 22. Tedesco B, Vendredy L, Timmerman V, Poletti A. The chaperone‐assisted selective autophagy complex dynamics and dysfunctions. Autophagy. 2023;19:1619–1641. doi: 10.1080/15548627.2022.2160564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Höhfeld J, Benzing T, Bloch W, Fürst DO, Gehlert S, Hesse M, Hoffmann B, Hoppe T, Huesgen PF, Köhn M, et al. Maintaining proteostasis under mechanical stress. EMBO Rep. 2021;22:e52507. doi: 10.15252/embr.202152507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fang X, Bogomolovas J, Wu T, Zhang W, Liu C, Veevers J, Stroud MJ, Zhang Z, Ma X, Mu Y, et al. Loss‐of‐function mutations in co‐chaperone BAG3 destabilize small HSPs and cause cardiomyopathy. J Clin Invest. 2017;127:3189–3200. doi: 10.1172/jci94310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ohiri JC. Modeling Filamin C Mutations Causing Cardiomyopathy . 2023. Dissertation Northwestern University. Accessed March 24, 2024. https://nufia.library.northwestern.edu/concern/generic_works/pc289j492?locale=en.
- 26. Kim EY, Page P, Dellefave‐Castillo LM, McNally EM, Wyatt EJ. Direct reprogramming of urine‐derived cells with inducible MyoD for modeling human muscle disease. Skelet Muscle. 2016;6:32. doi: 10.1186/s13395-016-0103-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim EY, Barefield DY, Vo AH, Gacita AM, Schuster EJ, Wyatt EJ, Davis JL, Dong B, Sun C, Page P, et al. Distinct pathological signatures in human cellular models of myotonic dystrophy subtypes. JCI Insight. 2019;4:e122686. doi: 10.1172/jci.insight.122686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, Lan F, Diecke S, Huber B, Mordwinkin NM, et al. Chemically defined generation of human cardiomyocytes. Nat Methods. 2014;11:855–860. doi: 10.1038/nmeth.2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Concordet JP, Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018;46:W242–W245. doi: 10.1093/nar/gky354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gacita AM, Fullenkamp DE, Ohiri J, Pottinger T, Puckelwartz MJ, Nobrega MA, McNally EM. Genetic variation in enhancers modifies cardiomyopathy gene expression and progression. Circulation. 2021;143:1302–1316. doi: 10.1161/circulationaha.120.050432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tiburcy M, Hudson JE, Balfanz P, Schlick S, Meyer T, Chang Liao ML, Levent E, Raad F, Zeidler S, Wingender E, et al. Defined engineered human myocardium with advanced maturation for applications in heart failure modeling and repair. Circulation. 2017;135:1832–1847. doi: 10.1161/circulationaha.116.024145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. O'Neill MJ, Chen SN, Rumping L, Johnson R, van Slegtenhorst M, Glazer AM, Yang T, Solus JF, Laudeman J, Mitchell DW, et al. Multicenter clinical and functional evidence reclassifies a recurrent noncanonical filamin C splice‐altering variant. Heart Rhythm. 2023;20:1158–1166. doi: 10.1016/j.hrthm.2023.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen SN, Lam CK, Wan YW, Gao S, Malak OA, Zhao SR, Lombardi R, Ambardekar AV, Bristow MR, Cleveland J, et al. Activation of PDGFRA signaling contributes to filamin C‐related arrhythmogenic cardiomyopathy. Sci Adv. 2022;8:eabk0052. doi: 10.1126/sciadv.abk0052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kley RA, van der Ven PF, Olivé M, Höhfeld J, Goldfarb LG, Fürst DO, Vorgerd M. Impairment of protein degradation in myofibrillar myopathy caused by FLNC/filamin C mutations. Autophagy. 2013;9:422–423. doi: 10.4161/auto.22921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roman W, Pinheiro H, Pimentel MR, Segalés J, Oliveira LM, García‐Domínguez E, Gómez‐Cabrera MC, Serrano AL, Gomes ER, Muñoz‐Cánoves P. Muscle repair after physiological damage relies on nuclear migration for cellular reconstruction. Science. 2021;374:355–359. doi: 10.1126/science.abe5620 [DOI] [PubMed] [Google Scholar]
- 36. Ulbricht A, Gehlert S, Leciejewski B, Schiffer T, Bloch W, Höhfeld J. Induction and adaptation of chaperone‐assisted selective autophagy CASA in response to resistance exercise in human skeletal muscle. Autophagy. 2015;11:538–546. doi: 10.1080/15548627.2015.1017186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Collier MP, Alderson TR, de Villiers CP, Nicholls D, Gastall HY, Allison TM, Degiacomi MT, Jiang H, Mlynek G, Fürst DO, et al. HspB1 phosphorylation regulates its intramolecular dynamics and mechanosensitive molecular chaperone interaction with filamin C. Sci Adv. 2019;5:eaav8421. doi: 10.1126/sciadv.aav8421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Janin A, N'Guyen K, Habib G, Dauphin C, Chanavat V, Bouvagnet P, Eschalier R, Streichenberger N, Chevalier P, Millat G. Truncating mutations on myofibrillar myopathies causing genes as prevalent molecular explanations on patients with dilated cardiomyopathy. Clin Genet. 2017;92:616–623. doi: 10.1111/cge.13043 [DOI] [PubMed] [Google Scholar]
- 39. Klimek C, Kathage B, Wördehoff J, Höhfeld J. BAG3‐mediated proteostasis at a glance. J Cell Sci. 2017;130:2781–2788. doi: 10.1242/jcs.203679 [DOI] [PubMed] [Google Scholar]
- 40. Agarwal R, Paulo JA, Toepfer CN, Ewoldt JK, Sundaram S, Chopra A, Zhang Q, Gorham J, DePalma SR, Chen CS, et al. Filamin C cardiomyopathy variants cause protein and lysosome accumulation. Circ Res. 2021;129:751–766. doi: 10.1161/circresaha.120.317076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reinstein E, Gutierrez‐Fernandez A, Tzur S, Bormans C, Marcu S, Tayeb‐Fligelman E, Vinkler C, Raas‐Rothschild A, Irge D, Landau M, et al. Congenital dilated cardiomyopathy caused by biallelic mutations in filamin C. Eur J Hum Genet. 2016;24:1792–1796. doi: 10.1038/ejhg.2016.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Golbus JR, Puckelwartz MJ, Dellefave‐Castillo L, Fahrenbach JP, Nelakuditi V, Pesce LL, Pytel P, McNally EM. Targeted analysis of whole genome sequence data to diagnose genetic cardiomyopathy. Circ Cardiovasc Genet. 2014;7:751–759. doi: 10.1161/circgenetics.113.000578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oz S, Yonath H, Visochyk L, Ofek E, Landa N, Reznik‐Wolf H, Ortiz‐Genga M, Monserrat L, Ben‐Gal T, Goitein O, et al. Reduction in filamin C transcript is associated with arrhythmogenic cardiomyopathy in Ashkenazi Jews. Int J Cardiol. 2020;317:133–138. doi: 10.1016/j.ijcard.2020.04.005 [DOI] [PubMed] [Google Scholar]
- 44. Paldino A, Dal Ferro M, Stolfo D, Gandin I, Medo K, Graw S, Gigli M, Gagno G, Zaffalon D, Castrichini M, et al. Prognostic prediction of genotype vs phenotype in genetic cardiomyopathies. J Am Coll Cardiol. 2022;80:1981–1994. doi: 10.1016/j.jacc.2022.08.804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Carruth ED, Qureshi M, Alsaid A, Kelly MA, Calkins H, Murray B, Tichnell C, Sturm AC, Baras A, Lester Kirchner H, et al. Loss‐of‐function FLNC variants are associated with arrhythmogenic cardiomyopathy phenotypes when identified through exome sequencing of a general clinical population. Circ Genom Precis Med. 2022;15:e003645. doi: 10.1161/circgen.121.003645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nafissi NA, Abdulrahim JW, Kwee LC, Coniglio AC, Kraus WE, Piccini JP, Daubert JP, Sun AY, Shah SH. Prevalence and phenotypic burden of monogenic arrhythmias using integration of electronic health records with genetics. Circ Genom Precis Med. 2022;15:e003675. doi: 10.1161/circgen.121.003675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Goli R, Li J, Brandimarto J, Levine LD, Riis V, McAfee Q, DePalma S, Haghighi A, Seidman JG, Seidman CE, et al. Genetic and phenotypic landscape of peripartum cardiomyopathy. Circulation. 2021;143:1852–1862. doi: 10.1161/circulationaha.120.052395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yoshida K, Saucerman JJ, Holmes JW. Multiscale model of heart growth during pregnancy: integrating mechanical and hormonal signaling. Biomech Model Mechanobiol. 2022;21:1267–1283. doi: 10.1007/s10237-022-01589-y [DOI] [PubMed] [Google Scholar]
- 49. Martin TG, Kirk JA. Under construction: the dynamic assembly, maintenance, and degradation of the cardiac sarcomere. J Mol Cell Cardiol. 2020;148:89–102. doi: 10.1016/j.yjmcc.2020.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lyon RC, Lange S, Sheikh F. Breaking down protein degradation mechanisms in cardiac muscle. Trends Mol Med. 2013;19:239–249. doi: 10.1016/j.molmed.2013.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Batista‐Almeida D, Martins‐Marques T, Ribeiro‐Rodrigues T, Girao H. The role of proteostasis in the regulation of cardiac intercellular communication. Adv Exp Med Biol. 2020;1233:279–302. doi: 10.1007/978-3-030-38266-7_12 [DOI] [PubMed] [Google Scholar]
- 52. Hoogstra‐Berends F, Meijering RA, Zhang D, Heeres A, Loen L, Seerden JP, Kuipers I, Kampinga HH, Henning RH, Brundel BJ. Heat shock protein‐inducing compounds as therapeutics to restore proteostasis in atrial fibrillation. Trends Cardiovasc Med. 2012;22:62–68. doi: 10.1016/j.tcm.2012.06.013 [DOI] [PubMed] [Google Scholar]
- 53. Li N, Brundel B. Inflammasomes and proteostasis novel molecular mechanisms associated with atrial fibrillation. Circ Res. 2020;127:73–90. doi: 10.1161/circresaha.119.316364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McNally EM, Demonbreun AR. Resealing and rebuilding injured muscle. Science. 2021;374:262–263. doi: 10.1126/science.abm2240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ulbricht A, Eppler FJ, Tapia VE, van der Ven PF, Hampe N, Hersch N, Vakeel P, Stadel D, Haas A, Saftig P, et al. Cellular mechanotransduction relies on tension‐induced and chaperone‐assisted autophagy. Curr Biol. 2013;23:430–435. doi: 10.1016/j.cub.2013.01.064 [DOI] [PubMed] [Google Scholar]
- 56. Sellung D, Heil L, Daya N, Jacobsen F, Mertens‐Rill J, Zhuge H, Döring K, Piran M, Milting H, Unger A, et al. Novel filamin C myofibrillar myopathy variants cause different pathomechanisms and alterations in protein quality systems. Cells. 2023;12:1321. doi: 10.3390/cells12091321 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S3
Figures S1–S12