

Abstract
Background
The only clinically approved drug that reduces doxorubicin cardiotoxicity is dexrazoxane, but its application is limited due to the risk of secondary malignancies. So, exploring alternative effective molecules to attenuate its cardiotoxicity is crucial. Colchicine is a safe and well‐tolerated drug that helps reduce the production of reactive oxygen species. High doses of colchicine have been reported to block the fusion of autophagosomes and lysosomes in cancer cells. However, the impact of colchicine on the autophagy activity within cardiomyocytes remains inadequately elucidated. Recent studies have highlighted the beneficial effects of colchicine on patients with pericarditis, postprocedural atrial fibrillation, and coronary artery disease. It remains ambiguous how colchicine regulates autophagic flux in doxorubicin‐induced heart failure.
Methods and Results
Doxorubicin was administered to establish models of heart failure both in vivo and in vitro. Prior studies have reported that doxorubicin impeded the breakdown of autophagic vacuoles, resulting in damaged mitochondria and the accumulation of reactive oxygen species. Following the administration of a low dose of colchicine (0.1 mg/kg, daily), significant improvements were observed in heart function (left ventricular ejection fraction: doxorubicin group versus treatment group=43.75%±3.614% versus 57.07%±2.968%, P=0.0373). In terms of mechanism, a low dose of colchicine facilitated the degradation of autolysosomes, thereby mitigating doxorubicin‐induced cardiotoxicity.
Conclusions
Our research has shown that a low dose of colchicine is pivotal in restoring the autophagy activity, thereby attenuating the cardiotoxicity induced by doxorubicin. Consequently, colchicine emerges as a promising therapeutic candidate to improve doxorubicin cardiotoxicity.
Keywords: autophagy, colchicine, doxorubicin cardiotoxicity, heart failure
Subject Categories: Cardiotoxicity, Heart Failure, Cardiomyopathy, Cardio-Oncology
Nonstandard Abbreviations and Acronyms
- AVs
autophagic vacuoles
- hiPSC‐CMs
human‐induced pluripotent stem cell‐derived cardiomyocytes
- ROS
reactive oxygen species
Research Perspective.
What Is New?
This study demonstrated that a low dose of colchicine promoted autolysosome degradation, clearing the damaged mitochondria and reducing reactive oxygen species accumulation induced by doxorubicin.
Inhibition of autolysosome degradation by chloroquine abolished the colchicine‐mediated improvement in doxorubicin cardiotoxicity.
What Question Should Be Addressed Next?
Our results suggest the potential clinical application of low‐dose colchicine for patients receiving chemotherapy, and we highlight the importance of monitoring blood drug concentrations when administering colchicine.
Cardiovascular disease is a leading cause of death worldwide, with heart failure emerging as a prevalent complication across various cardiovascular diseases, 1 culminating in a 5‐year annual mortality of approximately 50%. 2 Doxorubicin is a conventional chemotherapeutic drug that has been used for treating malignancies. However, the notable cardiotoxic effects of doxorubicin significantly limit its application and can lead to life‐threatening adverse drug reactions. As a result, many patients receiving chemotherapy develop heart failure due to doxorubicin exposure. Doxorubicin treatment has damaging effects on cardiomyocytes, primarily manifesting in (1) the excessive accumulation of reactive oxygen species (ROS), (2) DNA damage, and (3) the destruction of mitochondria. 3 Dexrazoxane stands as the only pharmacological drug clinically approved for preventing doxorubicin cardiotoxicity. 4 , 5 Unfortunately, the use of this drug is restricted due to the associated hazards of secondary malignancies, making it urgent to find alternative treatments to eliminate the cardiotoxicity.
Colchicine is recognized as an anti‐inflammatory medication that has been identified as a therapeutic target for familial Mediterranean fever, gouty arthritis, and Behçet disease. 6 By inhibiting ROS and inflammation, low doses of colchicine can effectively treat cardiovascular diseases such as pericarditis, postprocedural atrial fibrillation, and coronary artery disease. 7 , 8 , 9 , 10 , 11 Recently, colchicine has been shown to be beneficial in mice with heart failure or heart failure with preserved ejection fraction due to its anti‐inflammatory effect. 12 , 13 This underscores the potential of colchicine as a crucial supplementary therapeutic target for cardiovascular diseases in the future. 10 However, the mechanisms through which colchicine ameliorates heart failure have not been thoroughly investigated, leaving uncertainties regarding the potential involvement of other mechanisms in this process.
Autophagy is an evolutionarily conserved process of intracellular protein degradation and recycling that plays a critical role at different developmental stages of heart failure. 14 , 15 The actin cytoskeleton is involved in the formation, delivery, and maturation processes of autophagy. 16 As an important component of the actin cytoskeleton, microtubules are dose‐dependently depolymerized by colchicine. This action blocks autolysosome degradation in cancer cells, 17 skeletal muscle cells, 18 and liver cells under high‐dose administration. 19 However, the impact of colchicine on cardiomyocytes, specifically in the context of doxorubicin‐induced heart failure, remains largely unknown.
Therefore, we aimed to investigate how doxorubicin‐induced cardiotoxicity was influenced with or without colchicine treatment, both in vivo and in vitro. Furthermore, we assessed the autophagy activity under varying concentrations of colchicine and sought to determine the efficacy of colchicine in attenuating doxorubicin‐induced heart failure in preclinical models of heart failure.
METHODS
The data supporting the results of this study are available from the corresponding author upon reasonable request. We used the Animal Research: Reporting of In Vivo Experiments reporting guidelines. 20
Ethics Statement
All human‐induced pluripotent stem cell (hiPSC) research was permitted by the First Affiliated Hospital of Zhengzhou University ethics committee under protocol number 2018‐KY‐38. Informed consent was obtained from the volunteers beforehand.
All animal protocols and procedures were approved by the Institute Animal Care and Use Committee of the Academy of Medical Sciences at Zhengzhou University (ZZU‐SBRC2020060105) as well as Zhengzhou Weisa Biotechnology Co., Ltd. (V3A02021000001).
Animal Experiments
The 5‐week‐old Syrian hamsters used in this study were acquired from Beijing Vital River Laboratory Animal Technology Co., Ltd. The standard chow was purchased from XieTong Organism Co., Ltd. (Jiangsu, China).
Syrian hamsters were housed in a facility with restricted access (HH‐A‐5II, SZhouhuang), with 3 hamsters per cage, where they were exposed to a 14/10‐hour light/dark cycle. They were given 1 week to adapt to environmental changes before conducting the formal experiment. To assess the influence of colchicine on normal hamsters, relevant reports were consulted. 12 Hamsters were orally administered phosphate‐buffered saline (PBS) and varying doses of colchicine from 5 to 16 weeks of age daily. Echocardiographic analysis was conducted at 16 weeks of age to evaluate their heart performance.
Doxorubicin was dissolved in PBS and used to induce heart failure in male hamsters at a dose of 1.5 mg/kg in accordance with our preexperiment. At 6 weeks of age, male wild‐type hamsters were grouped using a random number table and were administered doxorubicin or PBS through intraperitoneal injection once a week for 6 weeks. Heart function was evaluated using echocardiographic analysis at 16 weeks. Colchicine or saline were administered orally to the hamsters at least 1 week before the first doxorubicin injection. Each group of Syrian hamsters underwent survival analysis and were euthanized at 16 weeks of age. The order of gastric lavage treatments and cardiac ultrasound examinations was randomized, and the data were analyzed by a blinded observer. If a golden hamster lost 20% of its weight within a week, the experiment on this animal was terminated as a humanitarian action.
hiPSC‐Cardiomyocytes Generation
Initially, peripheral venous blood was collected from a heart donor, and subsequently, peripheral blood mononuclear cells were separated from the blood through density gradient centrifugation. The peripheral blood mononuclear cells were cultured using StemProTM‐34 SFM medium (Thermo Fisher), supplemented with stem cell factor (SCF) (100 ng/mL, Thermo Fisher), FLT3 (fms‐like tyrosine kinase 3; 100 ng/mL, Thermo Fisher), IL‐3 (interleukin‐3; 20 ng/mL, Cell Signaling), and IL‐6 (20 ng/mL, Cell Signaling). Peripheral blood mononuclear cells were transduced with the CytoTune‐iPS 2.0 Sendai Reprogramming Kit (Thermo Fisher, A16517). After 24 hours (Day 1), the cells were cultured with fresh StemPro‐34 SFM for 2 days. Subsequently, they were transferred onto a 6‐well plate coated with Matrigel (BD Biosciences, 354 277) and cultured for an additional 24 hours. The medium (StemPro‐34 SFM) was changed twice a day. On Day 10, the medium was replaced with mTeSR1 (Stem Cell, 85 850) and refreshed daily. On Days 18 to 21, we manually collected stem cell‐like clones with the aid of microscopic observation and transferred them onto a 48‐well plate coated with Matrigel. The stem cells were cultured in PBS with 0.5 mmol/L EDTA from Thermo Fisher Scientific, without MgCl2 or CaCl2.
Echocardiography
Echocardiography was conducted using a VisualSonics Vevo 2100 ultrasound system (VisualSonics, Inc., Toronto, Ontario, Canada). The hamsters were lightly anesthetized with spontaneous respiration using isoflurane (working concentration 2%–2.5%), and the air velocity was set between 500 and 700 mL/min. Chemical hair remover was applied to shave the chest surface of the hamsters to optimize the visibility of their hearts; subsequently, we used clean water for cleaning to alleviate the discomfort of the golden hamsters. Two‐dimensional and M‐mode echocardiography were used to perform parasternal long‐axis scans. The average was reported for at least 3 to 5 cardiac cycles per hamster at the level of the papillary muscle. The molecular treatment conditions of each hamster were blinded to the sonographers and investigators during image acquisition and analysis.
Histochemical and Immunohistochemical Analyses
Paraffin‐embedded sections were fixed for a minimum of 48 hours and cut at 5 μm for hematoxylin and eosin and Masson's trichrome staining. Both staining methods were performed following the manufacturer's instructions. Hematoxylin and eosin staining (Servicebio, G1120) of left ventricular myocardial tissue sections was examined under a light microscope to analyze changes in the morphology of cardiomyocytes and myofilaments. To analyze fibrosis accumulation, Masson's trichrome‐stained sections of the left ventricular myocardium were examined using image analysis software (ImageJ, National Institutes of Health). Five fields were randomly selected from each recorded image, and the final fibrosis ratio was calculated by averaging the data of these selections. The ratio for each sample was calculated by dividing the blue‐stained fibrosis area by the total recorded image area.
Real‐Time Quantitative Reverse Transcription Polymerase Chain Reaction
To extract RNA from frozen tissues or hiPSC‐carCMs, the Qiagen RNeasy Plus Mini Kit (#74134) was used in accordance with the manufacturer's instructions. Next, cDNA was obtained for real‐time quantitative reverse transcription polymerase chain reaction by reverse transcribing 500 ng of RNA using the TAKARA PrimeScript RT reagent Kit (#RR037A). ChamQ Universal SYBR qPCR Master Mix (Vazyme, Q711) was then used for mRNA expression. Primers involved in this study are listed in Table.
Table 1.
The Primer Sequences for qRT‐PCR
| GAPDH (hamster) | forward 5′‐GACATCAAGAAGGTGGTGAAGCA‐3′ |
| reverse 5′‐CATCAAAGGTGGAAGAGTGGGA‐3′ | |
| ANP (hamster) | forward 5′‐TACAGTGCAGTGTCCAACACAG‐3′ |
| reverse 5′‐TGCTTCATCATTCTGCTCACTC‐3′ | |
| BNP (hamster) | forward 5′‐TCCCAGCCAGTCTCCGGAACAA‐3′ |
| reverse 5′‐TCTTGGTCCTTCAGGAGCTGCC‐3′ | |
| GAPDH (human) | forward 5′‐GTCTCCTCTGACTTCAACAGCG −3′ |
| reverse 5′‐ACCACCCTGTTGCTGTAGCCAA −3′ | |
| ANP (human) | forward 5′‐ACAATGCCGTGTCCAACGCAGA −3′ |
| reverse 5′‐CTTCATTCGGCTCACTGAGCAC‐3′ | |
| BNP (human) | forward 5′‐TCTGGCTGCTTTGGGAGGAAGA‐3′ |
| reverse 5′‐CCTTGTGGAATCAGAAGCAGGTG‐3′ |
ANP indicates atrial natriuretic peptide, BNP, brain natriuretic peptide; and qRT‐PCR, quantitative reverse transcription polymerase chain reaction.
Electron Microscopy
After euthanizing the hamsters and immediately harvesting their hearts, we cut 1 mm3 of the left ventricular myocardium. We collected and centrifuged the hiPSC‐cardiomyocytes (hiPSC‐CMs) before putting them into fresh transmission electron microscopy (TEM) fixative (Servicebio, G1102) for preservation at 4 °C. The tissue was fixed using 0.1 mol/L phosphate buffer (pH 7.4) with 1% OsO4 at 25 °C for 2 hours and subsequently washed with 0.1 mol/L phosphate buffer (pH 7.4) 3 times for 15 minutes each. The samples underwent dehydration at 25 °C in various ethanol concentrations (30%, 50%, 70%, 80%, 95%, 100%, 100%), each for 20 minutes, followed by 2 rounds of immersion in acetone for 15 minutes. Following resin infiltration and embedding, the samples were incubated overnight at 37 °C. The samples were polymerized at 65 °C for 48 hours and then sliced into 60 to 80 nm sections using an ultra‐microtome. The samples were stained with 2.6% lead citrate (Ted Pella Inc.) for 8 minutes while avoiding CO2. They were then washed with ultra‐pure water and left to dry. Ultimately, images were captured using a TEM (Hitachi, HT7800).
Western Blotting
Heart tissues or hiPSC‐CMs were homogenized using RIPA lysis buffer (Cowin, CW2333) that contains both protease and phosphatase inhibitors (Cowin, CW2200, CW2383). Protein concentration was determined using a BCA Protein Assay Kit (US EVERBRIGHT, Suzhou, China, B6169). The proteins were separated through SDS‐PAGE (4–20%) and then transferred onto PVDF membranes (Millipore, MA). To block the membrane, 5% skimmed milk was used for 2 hours at 25 °C. The primary antibodies were left to incubate overnight at 4 °C, followed by a 2‐hour incubation with the secondary antibody at room temperature. The intensity values of the protein levels were normalized to GAPDH. The antibody information can be obtained in Data S1.
Bulk RNA‐Sequencing
RNA‐seq was performed by Shanghai OE Biotech Co., Ltd. Briefly, total RNA was extracted from hiPSC‐CMs using the mirVana miRNA Isolation Kit (Ambion‐1561). Briefly, RNA integrity was assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Using the TruSeq Stranded mRNA Sample Prep Kit (Illumina, RS‐122‐2101), a total of 4 μg RNA was used to build library construction. Next, the libraries were sequenced using the Illumina HiSeq X Ten sequencing platform. This generated 150‐bp paired‐end reads as raw data. The initial data were purified to obtain high‐quality, clean data. The genes that met the ±2‐fold change and corrected‐P value <0.05 were analyzed for gene ontology and Kyoto Encyclopedia of Genes and Genomes pathway analyses. In‐depth transcriptional information was obtained based on the human genome (NCBI_GRCh38.p13). The raw data were deposited in the Gene Expression Omnibus database under accession number GSE247345.
Statistical Analysis
Statistical testing was analyzed using GraphPad Prism 8 as described in each figure legend. Data from at least 3 independent experiments are presented as mean±SD. One‐way ANOVA followed by a Tukey multiple comparisons test was used to determine the differences between groups. The differences in the Kaplan–Meier survival curve were determined using the Mantel–Cox test. Sample sizes are presented in every figure legend. P<0.05 was considered statistically significant.
RESULTS
Colchicine Is Tolerable and Doxorubicin Induces Heart Failure in Syrian Hamsters
Given colchicine's dose‐dependent manner and the potential to reverse most side effects upon ceasing or reducing colchicine intake, we intended to first ascertain the safety and tolerability of colchicine in vivo. Male hamsters were grouped using a random number table. Saline or colchicine was orally administered at varying doses (0.1 mg/kg and 0.5 mg/kg daily) to male wild‐type hamsters (n=5/group) over an 11‐week period (Figure S1A). The oral administration of colchicine was well tolerated, with no adverse effects observed on heart performance, as assessed through echocardiographic analysis (Figure S1C through S1E). Also, the group treated with colchicine did not show any significant differences in heart morphology compared with the control group, as indicated by similar heart size and heart weight relative to body weight (Figure S1F through S1H). Mild histological changes appeared across the 3 doxorubicin‐treated groups, as evidenced in the hematoxylin and eosin and Masson staining analyses (refer to Figure S1I and S1J). Among hamsters, no death in either saline or colchicine treatment groups (Figure S1B) was observed, and no significant changes were observed in the heart failure biomarkers ANP (atrial natriuretic peptide) and BNP (B‐type natriuretic peptide) (data not shown). These findings together indicate that oral administration of colchicine at 0.1 and 0.5 mg/kg daily for 11 weeks is safe and well tolerated in Syrian hamsters.
We then investigated the cardiotoxicity induced by doxorubicin in vivo. To determine the optimal concentration of doxorubicin‐induced heart failure for Syrian hamsters, we administered varying doses of the drug (Figure S2A) and demonstrated that the optimal concentration would be 1.5 mg/kg once a week for 6 consecutive weeks (Figure S2B through S2I). Briefly, doxorubicin‐treated Syrian hamsters exhibited heart morphology atrophy (Figure S2B through S2D), a finding consistent with previous reports in mice. 21 Besides, the reduced left ventricular ejection fraction and fractional shortening indicated that the doxorubicin group developed heart dysfunction (Figure S2E through S2G). At the same time, Masson staining confirmed that the doxorubicin group developed significant fibrosis deposition in the left ventricles (Figure S2H and S2I). The levels of ANP and BNP were significantly elevated after doxorubicin administration (Figure S3E and S3F). The administration of doxorubicin at 1.5 mg/kg (once weekly) for 6 consecutive weeks also resulted in a mortality rate of approximately 50% (Figure S3G).
Doxorubicin Induces Microtubule Dysfunction and Blocked Autolysosome Degradation
To assess the impact of doxorubicin on autophagy activity in cardiomyocytes, we conducted Western blot analysis on the hearts of doxorubicin‐treated hamsters. Doxorubicin induced a significant increase in the expression of both SQSTM1 (also known as P62) and LC3II (Figure S2J through S2L). P62, an autophagy cargo receptor, usually binds to ubiquitin‐tagged components before they undergo degradation via autophagy. During the formation of autophagosomes, LC3II is recruited to autophagosomal membranes (phagophore) and subsequently conversed into LC3I in autolysosome. 22 By conducting TEM analysis to evaluate the autophagic vacuoles (AVs), we observed increases in AVs accumulation in the doxorubicin‐treated hearts (Figure S2N and S2O). These results implied that doxorubicin functioned by either blocking the autolysosome degradation or enhancing the formation of autophagosomes. After analysis of the expression of ATG5, an important biomarker of autophagosome formation, we found that ATG5 expression was significantly decreased (Figure S2M). Thus, doxorubicin impaired autophagic activity by reducing autophagosome formation and notably blocking autolysosome degradation in Syrian hamster hearts.
We also evaluated the alteration of doxorubicin‐induced autophagy in vitro. hiPSC‐CMs recapitulated the complex cellular physiology of human cardiomyocytes quite well, showing many advantages over other in vitro models. 23 When comparing PBS‐treated hiPSC‐CMs with doxorubicin‐treated (1 mmol/L, 48 hour) ones, significant increases in P62 and LC3II accumulation were observed (Figure 1A, 1D, and 1E) along with a reduction in ATG5 expression (Figure 1F). These observations were similar to the results in vivo (Figure S2J through S2L). Among other autophagy proteins, mTOR (mammalian target of rapamycin) negatively regulates autophagy by activating phosphoSer757‐ULK1 (p‐ULK1757), thereby preventing autophagy formation. 24 In our study, doxorubicin administration resulted in significant increases in expression of p‐ULK1757and phosphoSer2448‐mTOR (p‐mTOR2448) (Figure 1B and 1C), indicating the decline in autophagosome formation. Collectively, the enhanced LC3II expression level was due to blocked autolysosome degradation.
Figure 1. Doxorubicin induces impaired autophagy in hiPSC‐CMs.
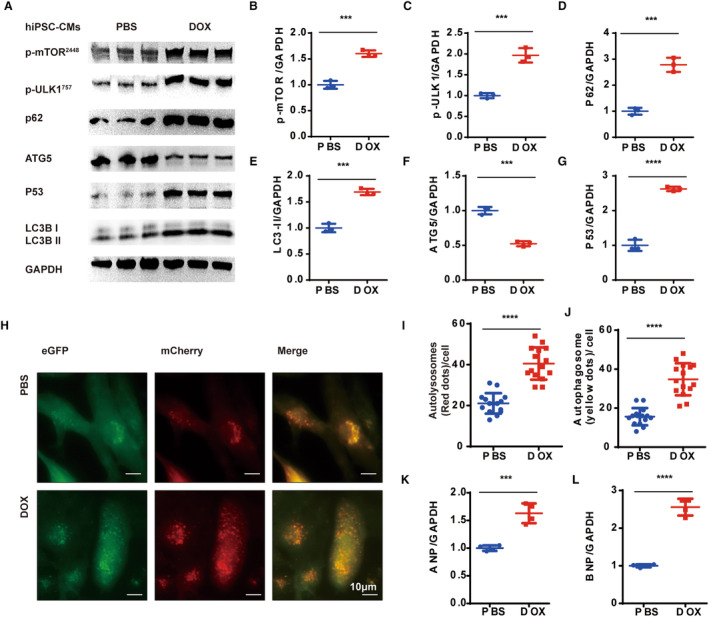
A, Representative WB analysis. B through G, Quantification of the expression of p‐mTOR, p‐ULK1, P62, LC3BII, ATG5, and P53 (n=3 per group). H, Representative images of mCherry‐eGFP‐LC3 puncta in PBS‐ and doxorubicin‐treated hiPSC‐CMs, LC3‐positive autophagosomes (merged yellow dots) and autolysosomes (red dots). I through J, Measurements of the number of LC3‐positive autophagosomes and autolysosomes per cell (n=15 per group). K and L, Quantitative PCR analysis of ANP and BNP mRNA expression (n=4 per group). All data are represented as means±SD, 2‐tailed Student t test, P>0.05, nonsignificant (ns), *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. ANP indicates atrial natriuretic peptide, BNP, brain natriuretic peptide; eGFP, enhanced green fluorescent protein; hiPSC‐CM, human‐induced pluripotent stem cell‐derived cardiomyocyte; PCR, polymerase chain reaction; and WB, Western blot.
Subsequently, we used an adenovirus vector expressing tandem mCherry‐eGPF‐LC3 (mCherry–enhanced green fluorescent protein–LC3B) to monitor the autophagic flux in vitro. The eGFP fluorescence was quenched in an acidic lysosomal environment, enabling the labeling of autophagosomes in yellow and autolysosomes in red. 25 After 1 h of glucose deprivation, both the yellow spots (autophagosomes) and red spots (autolysosomes) were obviously accumulated in the doxorubicin‐treated group (Figure 1H through 1J). Previous studies have shown that doxorubicin is capable of blocking autolysosome degradation in mice hearts. 26 We further proved that doxorubicin inhibits autolysosome degradation to suppress autophagosome formation, leading to the impairment in autophagy activity both in vivo and in vitro.
Blocking autophagy activities promotes the accumulation of damaged mitochondria, resulting in ROS overload and DNA damage. 27 To evaluate its influence on cardiomyocytes, we performed a TEM analysis. The doxorubicin‐treated hearts exhibit significant mitochondrial fragmentation and cristae defects (Figure S3C and S3D). Immunohistochemistry analysis of dihydroethidium on the left ventricles of hamsters showed significant ROS deposition in the left ventricles of doxorubicin‐treated ones (Figure S3A and S3B). Furthermore, there was a significant increase in the expression level of P53 (cellular tumor antigen P53, an important regulator responding to DNA damage and mitochondrial apoptosis) in the hiPSC‐CMs of the doxorubicin‐treated group (Figure 1A and 1G). 28 , 29 Therefore, doxorubicin blocked autophagic flux and resulted in mitochondrial damage and ROS overload.
To elucidate the mechanisms underlying doxorubicin‐induced cardiotoxicity in cardiomyocytes, we conducted transcriptome analysis on hiPSC‐CMs with and without doxorubicin administration (Figure 2A). Kyoto Encyclopedia of Genes and Genome analysis revealed that the P53 pathway was damaged (Figure 2C). Gene ontology analysis showed that the differentially expressed genes were most associated with the microtubule network (Figure 2B). The α‐tubulin immunocytochemistry staining revealed that doxorubicin treatment induced a denser microtubule network (Figure 2D). Western blot analysis confirmed this observation (Figure 2E through 2G). Previous studies have well reported the ability of the microtubule network to regulate autophagy activity, so we worked on a well‐known microtubule depolymerizer, colchicine, to elucidate how it influences autophagy in cardiomyocytes. 30 , 31
Figure 2. Doxorubicin damages the microtubule network in hiPSC‐CMs.
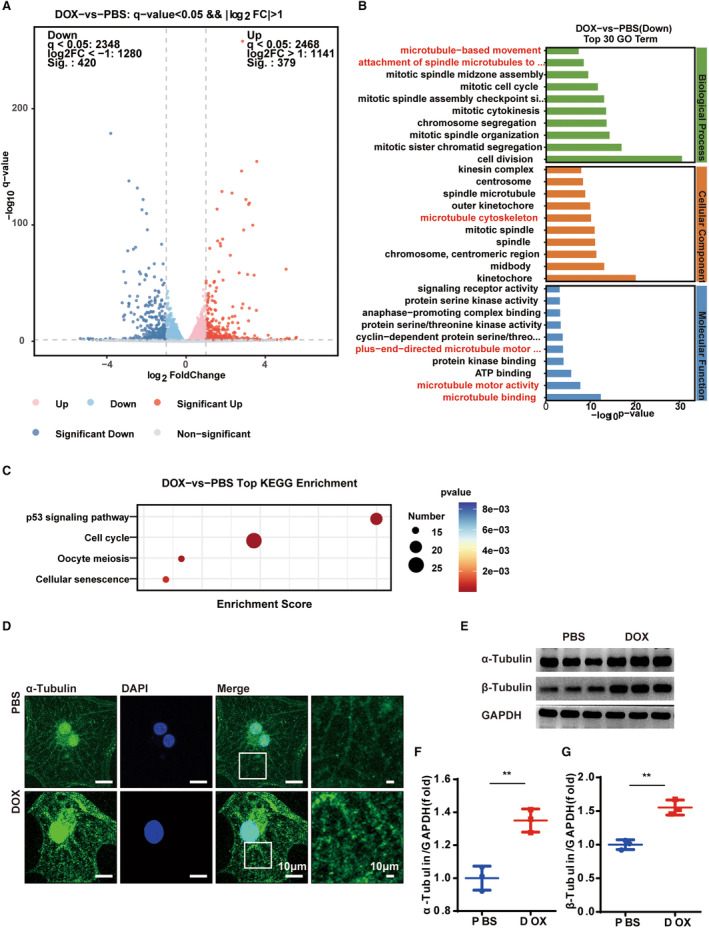
A, Volcano plot of the differentially expressed genes. B and C, GO and KEGG enrichment analysis of the PBS‐ and doxorubicin‐treated hiPSC‐CMs. N=3 per group. D, Representative images of the α‐tubulin immunocytochemistry staining for labeling the microtubule network. Similar results were found in more than 3 different repeats for each group. E, Representative WB analysis of α‐ and β‐tubulin. F and G, Quantitative analysis of α‐ and β‐tubulin (n=3 per group), means±SD, 2‐tailed Student t test, P>0.05, nonsignificant (ns), *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. GO indicates gene ontology; hiPSC‐CM, human‐induced pluripotent stem cell‐derived cardiomyocyte; KEGG, Kyoto Encyclopedia of Genes and Genomes; and WB, Western blot.
Low Dose of Colchicine Promotes the Autolysosome Degradation in hiPSc‐CMs
To determine the optimal concentration of colchicine used, we treated the hiPSc‐CMs with colchicine at different concentrations (0, 1, 2.5, 5, 10, and 20 nmol/L) for 48 hours (Figure 3A). The low‐concentration dosage induced a decrease in LC3BII expression level while giving a high dosage at 20 nmol/L resulted in an increase in the LC3BII expression (Figure 3B). Among the low dosages, 1 to 10 nmol/L showed a relatively stable level of p62 expression, which increased significantly at a dosage of 20 nmol/L (Figure 3C). The decreased expression of LC3BII would be attributed to decreased autophagosome formation or accelerated autolysosome degradation. To address this, chloroquine or vehicle was introduced to the PBS or colchicine‐treated hiPSc‐CMs to inhibit the degradation of LC3BII. The dosage of 5 nmol/L colchicine resulted in a decreased expression of LC3BII (vehicle‐treated groups). However, under chloroquine administration (10 μmol/L, 2 hour), the expression of LC3BII remained unchanged between the PBS or colchicine‐treated groups (Figure 3D). Collectively, the low‐dose administration of colchicine actually promoted autolysosome degradation but did not inhibit autophagosome formation.
Figure 3. Colchicine bidirectionally regulates autophagy in hiPSC‐CMs.
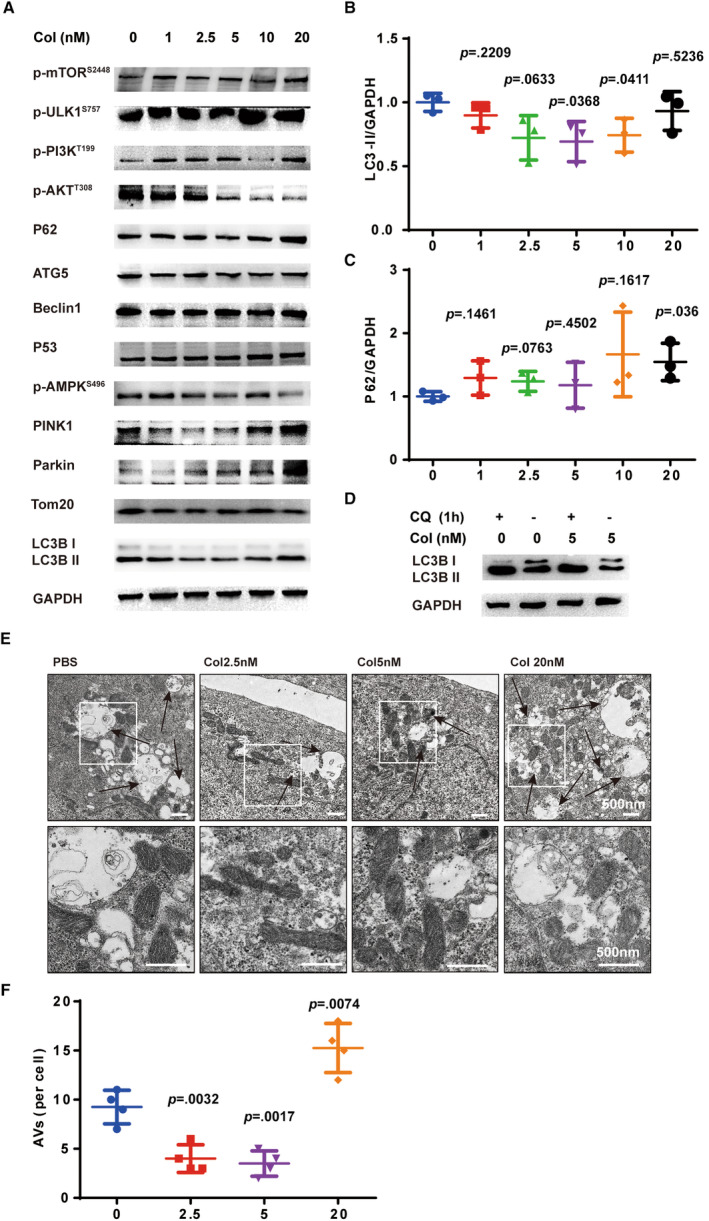
A, Representative WB analysis of a panel of autophagy associated with proteins. B and C, Quantification of the expression of P62 and LC3B‐II (n=3 per group). D, Representative WB analysis of LC3B‐II treated with +/− colchicine (5 nmol/L, 48 h), +/− chloroquine (10 μmol/L, 1 h). Similar results were found in more than 3 different repeats for each group. E, Representative TEM images for labeling AVs and mitochondria under different doses of colchicine. F, Measurements of AVs per cell (3 random fields per sample, n=4 per group). Means±SD, 2‐tailed Student t test comparing with the colchicine (0 nmol/L group). AVs indicates autophagic vacuoles; hiPSC‐CM, human‐induced pluripotent stem cell‐derived cardiomyocyte; TEM, transmission electron microscopy; and WB, Western blot.
To further explain these observations, we measured other proteins associated with autophagy. The expression levels of ATG5 and beclin1, another important protein participating in autophagosome formation, were relatively stable under colchicine treatment. Meanwhile, a mild influence on p‐AMPK496, p‐mTOR2448, and p‐ULK1757 expression was observed (Figure 3A). These results indicated that using low‐dose colchicine produced a gentle influence on the autophagosome formation process. For the PI3K/AKT pathway, an increased level of p‐PI3K199 expression but a decreased level of p‐AKT308 expression were observed, indicating the presence of other mechanisms of cross‐talking. The colchicine also resulted in a dose‐dependent increase in the expression level of P53. The mitophagy process under colchicine administration was also evaluated. Results revealed that the expression of PINK1 was decreased within 1 to 5 nmol/L of colchicine treatment but increased at 20 nmol/L. Parkin was increased dose‐dependently by colchicine and Tom20 remains relatively stable (Figure 3A). In a word, the autophagy formation activity were mildly influenced within 1 to 10 nmol/L of colchicine. Next, TEM analysis was performed to evaluate the autophagy activity under different dosages of colchicine. Under the glucose deprivation process (1 hour), the use of 2.5 nmol/L or 5 nmol/L colchicine accelerated the autolysosome degradation as evidenced by the decreased AVs. Administering 20 nmol/L of colchicine, on the other hand, resulted in a significant enhancement in AV accumulation (Figure 3E and 3F). Therefore, low‐dose colchicine was potentially an autolysosome degradation accelerator for the doxorubicin‐induced blocking of autophagic flux. Meanwhile, the blocking of autolysosome degradation by 20 nmol/L colchicine suggests that a high dosage may not be an appropriate therapeutic concentration for doxorubicin cardiotoxicity.
Given that colchicine inhibited microtubule depolymerization in a dose‐dependent manner, α‐tubulin immunofluorescence staining was conducted on hiPSC‐CMs. At a concentration of 5 nmol/L (48 hour), a reduction in microtubule density was observed without apparent damage to the microtubule network (Figure S4). Therefore, we proved that doxorubicin impeded autolysosome degradation, induced damage to mitochondria, and led to an overload of ROS. Meanwhile, at a low concentration (1–10 nmol/L), colchicine had a positive effect on the promotion of autolysosome degradation.
Colchicine Alleviated Doxorubicin‐Induced Autophagy Blockade
In the subsequent phase of our study, we investigated whether colchicine could reverse the autophagy dysfunction induced by doxorubicin in cardiomyocytes. Our results showed that 2.5 nmol/L colchicine significantly decreased doxorubicin‐induced P62 accumulation. However, P62 levels gradually increased at 5 and 10 nmol/L (Figure 4A and 4B). The elevation of LC3BII induced by doxorubicin was significantly reduced at 5 nmol/L of colchicine, a response not clearly observed at 2.5 nmol/L and 10 nmol/L (Figure 4E). Therefore, we postulated that the administration of colchicine at a low dose could mitigate the doxorubicin‐induced inhibition of autolysosome degradation. To validate our findings, hiPSc‐CMs were collected for TEM analysis after 1 hour of glucose deprivation. We observed a notable reduction in both doxorubicin‐induced AV accumulation and damaged mitochondria with the additional administration of 5 nmol/L colchicine (Figure 4F through 4H). Using adenovirus vector‐mCherry‐eGFP‐LC3 vectors, a significant reduction was evident in the accumulation of yellow and red spots (autophagosomes and autolysosomes, respectively) induced by doxorubicin under glucose deprivation (1 hour) with the additional administration of 5 nmol/L colchicine (Figure 4I through 4K). Collectively, these findings illustrated the efficacy of low‐dose colchicine administration in alleviating doxorubicin‐induced autolysosome degradation. In hamster hearts, administering 0.1 mg/kg colchicine daily significantly attenuated the doxorubicin‐induced elevation in LC3BII levels (Figure S5A and S5B), and the accumulation of AVs was notably diminished as well (Figure S5C and S5D). These observations suggested that colchicine accelerated autolysosome degradation process in vitro and in vivo.
Figure 4. Colchicine attenuates the doxorubicin induced autophagy blocking in hiPSC‐CMs.
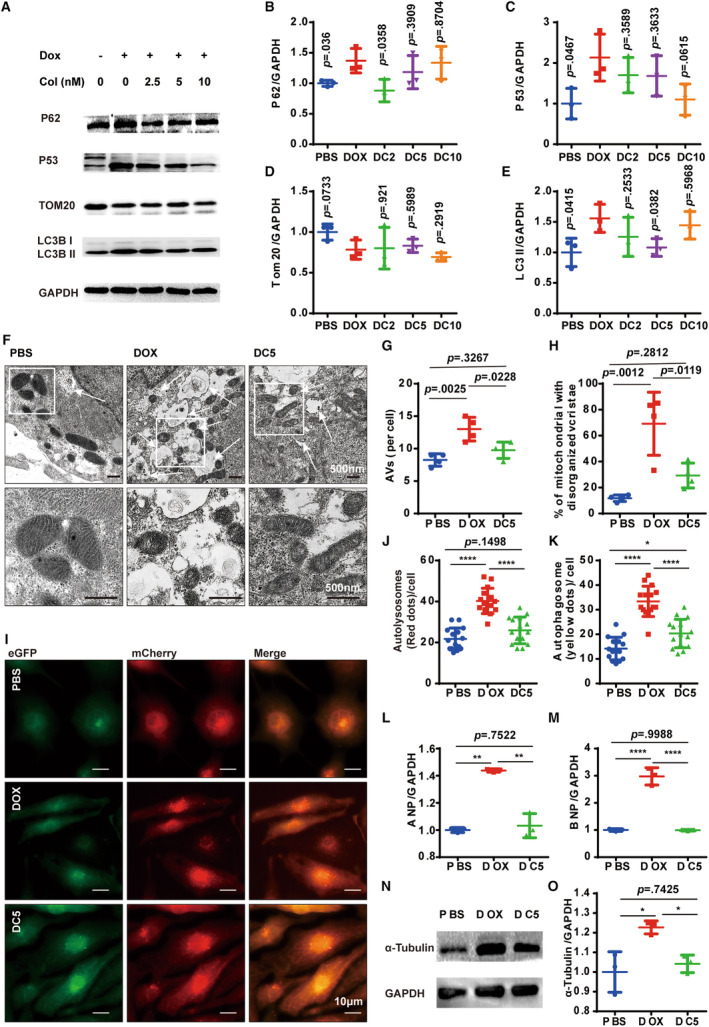
A, Representative WB analysis of a panel of autophagy associated with proteins. B through E, Quantification of the expression of P62, P53, Tom20, and LC3B‐II (n=3 per group). F through H, Representative TEM image and quantification analysis of AVs and mitochondria in different groups (3 random fields per sample, n=4 per group). I, Representative images of mCherry‐eGFP‐LC3 puncta in different groups. J and K, Measurements of the number of LC3‐positive autophagosomes and autolysosomes per cell (n=15 per group). L and M, Quantitative PCR analysis of ANP and BNP mRNA expression (n=4 per group). For B through E, 2‐tailed Student t test comparing with the doxorubicin group; N and O. Representative WB analysis of α‐tubulin (n=3 per group). G through O, 1‐way ANOVA (Tukey post test). P>0.05, values are presented, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. AVs indicates autophagic vacuoles; DC, doxorubicin+different concentrations of colchicine (nmol/L); eGFP, enhanced green fluorescent protein; hiPSC‐CM, human‐induced pluripotent stem cell‐derived cardiomyocyte; PCR, polymerase chain reaction; TEM, transmission electron microscopy; and WB, Western blot.
Next, we aimed to evaluate the potential benefits of colchicine‐induced autophagy rebalancing for cellular homeostasis. Tom20, a mitochondrial outer membrane protein, displayed minor changes, suggesting that accelerated autolysosome degradation did not lead to mitochondrial deficiency (Figure 4D). However, the presence of damaged mitochondria was significantly decreased following colchicine (5 nmol/L) administration both in vitro (Figure 4F and 4H) and in vivo (Figure S5E and S5F). Furthermore, the levels of ROS exhibited a notable reduction following colchicine administration in doxorubicin‐treated hearts (Figure S5G and S5H). To explore whether colchicine mitigated doxorubicin‐induced DNA damage in hiPSc‐CMs, P53 expression was evaluated. We observed a slight decrease in P53 expression in doxorubicin‐treated hiPSc‐CMs upon the additional administration of colchicine (Figure 4C).
The mechanism by which low‐dose colchicine accelerates autolysosome degradation remains unclear. Our findings show that doxorubicin triggered significant damage to the microtubule system (Figure 2B), leading to an increase in microtubule density (Figure 2D through 2F). The microtubule lattice is known to regulate autophagy activity. We observed that the elevated microtubule density caused by doxorubicin was significantly reduced with low‐dose colchicine treatment (Figure 4N and 4O, Figure S4). This modulation of microtubule density by colchicine could be a potential mechanism of autophagy rebalancing in this context.
Colchicine Attenuated Doxorubicin‐Induced Heart Failure
We further investigated whether administering low‐dose colchicine could alleviate heart failure symptoms (Figure 5A). Notably, the daily administration of 0.1 mg/kg colchicine significantly mitigated both the heart morphologic changes and the loss of body weight induced by doxorubicin, whereas the effectiveness was less pronounced with the 0.5 mg/kg dosage (Figure 5B through 5D). In addition, the administration of 0.1 mg/kg colchicine improved heart fibrosis deposition and myocyte fiber disorder (Figure 5E and 5F) in the hearts of doxorubicin‐treated Syrian hamsters. Also, the improvement in left ventricular ejection fraction and fractional shortening indicated the efficacy of colchicine in restoring normal heart functions in the 0.1 mg/kg but not in the 0.5 mg/kg group (Figure 5G through 5I, Table S1). Additionally, colchicine enhanced the levels of ANP and BNP in the 0.1 mg/kg group, although this effect was significantly diminished in the 0.5 mg/kg group (Figure 5K and 5L). However, despite these improvements, 0.1 mg/kg colchicine only slightly increased survival rates, which could be attributed to the limited sample size (Figure 5J). These results underscore the importance of selecting an appropriate working dose of colchicine to achieve an optimal treatment effect.
Figure 5. Low‐dose colchicine alleviates heart failure in Syrian hamsters.
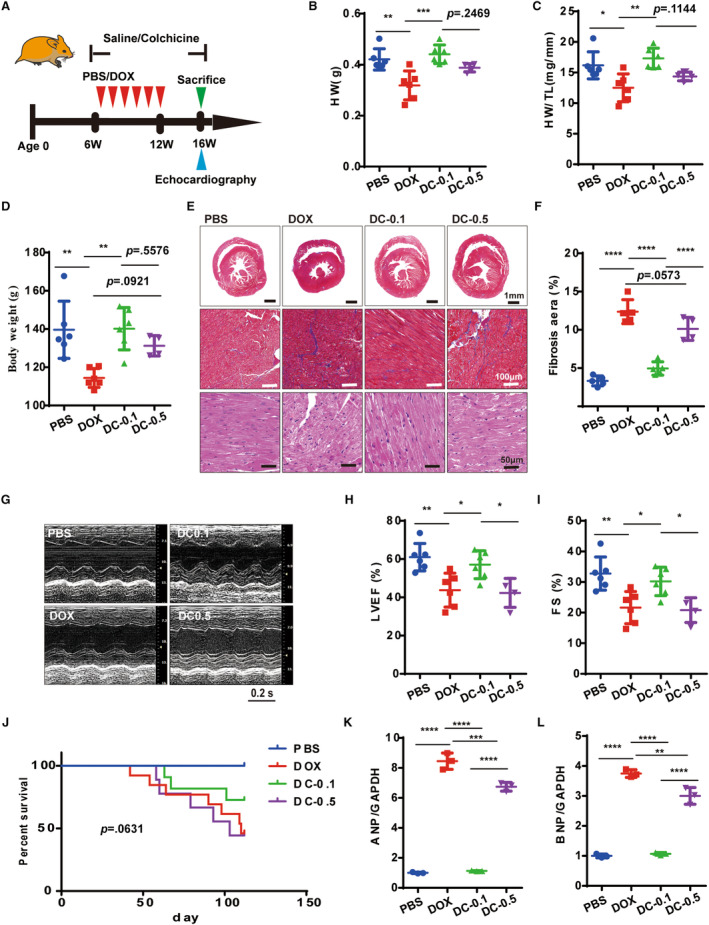
A, Experimental protocol illustrating the in vivo therapeutic effects of colchicine in alleviating heart failure. B and C, Measurements of HW and ratios of HW/TL (n=4–6 per group). D, Measurements of body weight (n=4–6 per group). E, Representative Masson's trichrome and hematoxylin and eosin staining of heart sections from different groups. Similar results were found in more than 3 different hamsters for each group. F, Measurements of fibrosis (n=4–6 per group). G through I, Representative M‐mode echocardiographic images and LVEF, FS measurements (n=4–6 per group). J, Kaplan–Meier survival analysis of hamsters in different groups (n=8–13 per group), means±SD, P value was determined by log‐rank (Mantel–Cox) test. K through L, Quantitative PCR analysis of ANP and BNP mRNA expression (n=3 per group). For these data, means±SD, 1‐way ANOVA (Tukey post test). P>0.05, values are presented, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. ANP indicates atrial natriuretic peptide, BNP, brain natriuretic peptide; DC‐0.1 and DC‐0.5, hamsters treated with doxorubicin and colchicine 0.1 and 0.5 mg/kg, respectively; FS, fractional shortening; HW/TL, heart weight to tibial length weight; LVEF, left ventricular ejection fraction; and PCR, polymerase chain reaction.
To explore whether enhanced autophagy activity could alleviate heart failure characteristics in vitro, 5 nmol/L of colchicine was applied to doxorubicin‐treated hiPSc‐CMs. The ANP and BNP expressions were significantly reduced (Figure 4L and 4M). Also, both damaged mitochondria (Figure 4F and 4H, Figure S5E and S5F) and elevated ROS levels (Figure S5G and S5H) showed improvement with colchicine treatment, both in vitro and in vivo. Therefore, administering colchicine could improve heart function and promote cellular homeostasis.
Inhibition of Autolysosome Degradation In Vitro Attenuates the Advantageous Effects of Colchicine in Heart Failure
To investigate the role of autolysosome degradation in the colchicine‐dependent effects on doxorubicin cardiotoxicity, doxorubicin‐treated hiPSc‐CMs were coexposed to colchicine (5 nmol/L) and chloroquine (2 μmol/L) for 48 hours. Western blot analysis revealed that chloroquine effectively inhibited the colchicine‐induced LC3BII degradation in the hiPSc‐CMs, indicating the successful blockade of autolysosome degradation (Figure 6A and 6B). Using the adenovirus vector‐mCherry‐eGFP‐LC3 vectors, we also confirmed that chloroquine nullified the effects of colchicine in doxorubicin‐treated hiPSc‐CMs (Figure 6C through 6E). Additionally, α‐actinin immunostaining on hiPSc‐CMs revealed that chloroquine also abolished the sarcomeric improvement induced by colchicine (Figure 6F). The ANP and BNP expressions were both increased in the chloroquine‐treated hiPSc‐CMs (Figure 6G and 6H). The colchicine‐dependent benefits for damaged mitochondria (Figure 6I) were also abolished by the additional use of chloroquine. We also assessed changes in ROS levels with the addition of chloroquine. Colchicine (5 nmol/L) effectively reduced ROS overload in doxorubicin‐treated hiPSc‐CMs, but this protective effect was suppressed with the use of chloroquine (Figure 6J and 6K). These results indicated that inhibiting autolysosome degradation by chloroquine abrogated the cardioprotective effects of colchicine during the progression of doxorubicin cardiotoxicity in vitro.
Figure 6. Blockade of autophagic flux abrogates the beneficial effects of colchicine on hiPSC‐CMs.
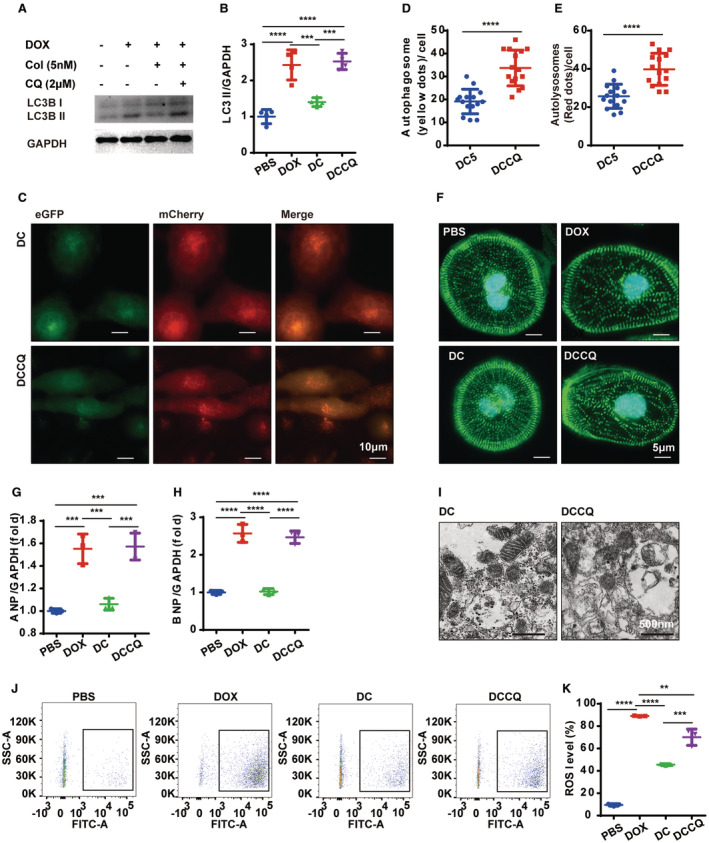
A and B, Representative WB image and analysis of LC3B‐II (n=3 per group). C, Representative images of mCherry‐eGFP‐LC3 puncta in different groups. D through E, Measurements of the number of LC3‐positive autophagosomes and autolysosomes per cell (n=15 per group). F, Representative images of the α‐actinin immunocytochemistry staining for labeling the sarcomere network in different groups, Similar results were found in more than 3 different repeats for each group. G and H. Quantitative PCR analysis of ANP and BNP mRNA expression (n=3 per group). I, Representative TEM analysis of mitochondria in different groups (3 random fields per sample, n=4 per group). J and K, Representative ROS levels and analysis by flow cytometry (n=3 per group). P>0.05, nonsignificant (ns), *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. ANP indicates atrial natriuretic peptide, BNP, brain natriuretic peptide; DCCQ, doxorubicin+colchicine+chloroquine; eGFP, enhanced green fluorescent protein; hiPSC‐CM, human‐induced pluripotent stem cell‐derived cardiomyocyte; PCR, polymerase chain reaction; ROS, reactive oxygen species; TEM, transmission electron microscopy; and WB, Western blot.
DISCUSSION
In our study, we demonstrated that administering colchicine at a low dose prevented the exacerbation of doxorubicin‐induced cardiotoxicity and myocardial damage. This was achieved by restoring the balance of autophagy activity in both Syrian hamsters and hiPSc‐CMs models. The doxorubicin‐induced cardiotoxicity negatively affected autophagy formation, hindering the degradation of autolysosomes. This impairment in autophagic processes subsequently led to mitochondrial damage and ROS overload and ultimately resulted in heart failure. On the other hand, low‐dose colchicine significantly boosted autolysosome degradation in doxorubicin‐affected cardiomyocytes, mitigated cellular homeostasis disruption, and ultimately prevented heart failure in vivo and in vitro. What is more, the lysosome function inhibitor chloroquine nullified the cardioprotective effects mediated by colchicine, underscoring the crucial downstream mechanism of promoting autolysosome degradation.
Doxorubicin has been shown to impair the autophagy flux by reducing autophagy formation or inhibiting autolysosome degradation in previous studies using mice and zebrafish models. 26 , 32 Our research uses Syrian hamsters and hiPSc‐CMs models to further demonstrate that doxorubicin not only impairs autophagosome formation but also significantly blocks autolysosome degradation. The blocked autophagy flux led to the destruction of cardiomyocytes' ability to remove damaged mitochondria, resulting in an overload of ROS and heart failure. 32 The mechanism by which doxorubicin impairs autophagy activity in cardiomyocytes is still unclear. Some studies suggested it might be due to inhibited lysosome acidification 26 or decreased α‐tubulin acetylation using mouse models. 13 , 33 However, evidence is still lacking, and the mechanism needs further investigation.
In this study, RNAseq analysis conducted on doxorubicin and PBS‐treated hiPSc‐CMs revealed that doxorubicin may primarily target the microtube network. Microtubules are key cytoskeletal components of cardiomyocytes, regulating their electrical and mechanical activities. 34 Colchicine, as a microtubule disruptor, also regulates calcium homeostasis in cardiomyocytes, which is important for its antiatrial fibrillation effect. 35 , 36 , 37 Inhibition of microtubule polymerization with colchicine in both Duchenne muscular dystrophy mouse models (0.4 mg/kg per day for 4 weeks) and cardiomyocytes from patients with heart failure (10 μmol/L for 2 hours) showed significant protective effects in previous reports. 38 , 39 These findings suggested that targeting microtubules can pose a protective effect on heart failure. Previous reports demonstrated that colchicine was capable of attenuating the doxorubicin‐induced sarcomere disturbance, 13 and the disturbed cytoskeleton was able to impair the autophagy activity in different steps. 16 We proved that doxorubicin resulted in an enhanced density of microtubule, and a low dose of colchicine greatly attenuated this alteration. The collective findings from our study, along with previous research, proved that the improved autolysosome degradation induced by colchicine could be attributed to its ability to regulate microtubule dynamics.
CONCLUSIONS
In conclusion, doxorubicin‐induced cardiotoxicity led to decreased autophagy formation and especially blocked autolysosome degradation. Here, we revealed a new mechanism for how low‐dose colchicine promotes autolysosome degradation to attenuate doxorubicin‐induced cardiotoxicity through microtubule regulations. Given its low cost, established long‐term safety profile, and tolerability, low‐dose colchicine could potentially prevent doxorubicin‐induced cardiotoxicity in patients undergoing chemotherapy. Our results have also demonstrated the significance of selecting the working plasma concentration of colchicine. In the future, considerations such as body weight, renal function, and the delivery method would be essential factors to evaluate before administering colchicine.
There were also limitations in our research. As colchicine is a pleiotropic and mechanistically plausible molecule, we cannot discount the involvement of other mechanistic pathways. A prior study illustrated that colchicine, even at relatively low concentrations, can stimulate the maturation of murine bone marrow‐derived dendritic cells and cytokine production. 40 Recent research has shown that targeting the innate immune system and mitigating inflammation through colchicine can be beneficial for the cardiovascular and renal outcomes of patients with chronic kidney disease and doxorubicin‐induced cardiotoxicity. 13 , 41 Our study investigated the effect of colchicine‐induced autophagy on the regulation of doxorubicin‐induced cardiotoxicity and heart failure both in vivo and in vitro, focusing on its destabilizing effect on microtubules. Because our current findings cannot conclusively discern the individual therapeutic impact of colchicine, further exploration is necessary to pinpoint the primary mechanism definitively. Moreover, autophagy can be classified into 3 types depending on how cargos are delivered to lysosomes: macroautophagy, microautophagy, and chaperone‐mediated autophagy. Specific types, such as mitophagy, reticulophagy, ribophagy, pexophagy, lipophagy, aggrephagy, nucleophagy, and xenophagy can be inferred based on cargo selectivity. 42 Here, we mainly focused on the process of macroautophagy, where the autophagosome is transported to the lysosome, and the subsequent autolysosome formation is mediated by the cytoskeletal structure, including microtubules. Our observations revealed that doxorubicin treatment resulted in a denser microtubule network in cardiomyocytes, a condition significantly alleviated by colchicine. Because colchicine promotes autophagosome transport and autolysosome formation in our study, we anticipate that colchicine may also have beneficial effects on other types of autophagy, such as mitophagy, which will be investigated in future research.
Sources of Funding
This work was supported by Population Assessment of Influenza and Disease Activities, PANDA II (CSCF2019A04); Natural Science Foundation of the First Affiliated Hospital of Zhengzhou University for Dong; Henan Key Laboratory of Hereditary Cardiovascular Diseases.
Disclosures
None.
Supporting information
Data S1
Table S1
Figures S1–S5
Acknowledgments
We thank the animals euthanized in this study. Conceptualization: Ying Peng, Yaohe Wang, Jianzeng Dong. Methodology, formal analysis, data curation, and investigation: Ying Peng, Jianchao Zhang, Yunshu Dong, Yafei Zhai, Honglin Zheng, Mengduan Liu, Jing Zhao, Wenting Du, Yangyang Liu, Liping Sun, Xiaowei Li, Hailong Tao, Xiaoyan Zhao. Writing—original draft: Ying Peng, Yunshu Dong. Writing—review and editing: Chenglin Zhang, Deyong Long, Xin Du, Changsheng Ma, Jianzeng Dong. Supervision: Changsheng Ma, Yaohe Wang, Jianzeng Dong. Project administration: Ying Peng, Yaohe Wang, Jianzeng Dong.
This article was sent to Tochukwu M. Okwuosa, DO, Associate Editor, for review by expert referees, editorial decision, and final disposition.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.123.033700
For Sources of Funding and Disclosures, see page 15.
Contributor Information
Yaohe Wang, Email: yaohe.wang@qmul.ac.uk.
Jianzeng Dong, Email: jzdong@zzu.edu.cn.
References
- 1. Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, Boehme AK, Buxton AE, Carson AP, Commodore‐Mensah Y, et al. Heart disease and stroke Statistics‐2022 update: a report from the American Heart Association. Circulation. 2022;145:e153–e639. doi: 10.1161/CIR.0000000000001052 [DOI] [PubMed] [Google Scholar]
- 2. Braunwald E. Heart failure. JACC Heart Fail. 2013;1:1–20. doi: 10.1016/j.jchf.2012.10.002 [DOI] [PubMed] [Google Scholar]
- 3. Henriksen PA. Anthracycline cardiotoxicity: an update on mechanisms, monitoring and prevention. Heart. 2018;104:971–977. doi: 10.1136/heartjnl-2017-312103 [DOI] [PubMed] [Google Scholar]
- 4. Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6 [DOI] [PubMed] [Google Scholar]
- 5. Stansfeld A, Radia U, Goggin C, Mahalingam P, Benson C, Napolitano A, Jones RL, Rosen SD, Karavasilis V. Pharmacological strategies to reduce anthracycline‐associated cardiotoxicity in cancer patients. Expert Opin Pharmacother. 2022;23:1641–1650. doi: 10.1080/14656566.2022.2124107 [DOI] [PubMed] [Google Scholar]
- 6. Deftereos S, Giannopoulos G, Papoutsidakis N, Panagopoulou V, Kossyvakis C, Raisakis K, Cleman MW, Stefanadis C. Colchicine and the heart: pushing the envelope. J Am Coll Cardiol. 2013;62:1817–1825. doi: 10.1016/j.jacc.2013.08.726 [DOI] [PubMed] [Google Scholar]
- 7. Imazio M. Colchicine for pericarditis. Trends Cardiovasc Med. 2015;25:129–136. doi: 10.1016/j.tcm.2014.09.011 [DOI] [PubMed] [Google Scholar]
- 8. Katsanos AH, Palaiodimou L, Price C, Giannopoulos S, Lemmens R, Kosmidou M, Georgakis MK, Weimar C, Kelly PJ, Tsivgoulis G. Colchicine for stroke prevention in patients with coronary artery disease: a systematic review and meta‐analysis. Eur J Neurol. 2020;27:1035–1038. doi: 10.1111/ene.14198 [DOI] [PubMed] [Google Scholar]
- 9. Opstal TSJ, Hoogeveen RM, Fiolet ATL, Silvis MJM, The SHK, Bax WA, de Kleijn DPV, Mosterd A, Stroes ESG, Cornel JH. Colchicine attenuates inflammation beyond the inflammasome in chronic coronary artery disease: a LoDoCo2 proteomic substudy. Circulation. 2020;142:1996–1998. doi: 10.1161/CIRCULATIONAHA.120.050560 [DOI] [PubMed] [Google Scholar]
- 10. Deftereos SG, Beerkens FJ, Shah B, Giannopoulos G, Vrachatis DA, Giotaki SG, Siasos G, Nicolas J, Arnott C, Patel S, et al. Colchicine in cardiovascular disease: in‐depth review. Circulation. 2022;145:61–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li YW, Chen SX, Yang Y, Zhang ZH, Zhou WB, Huang YN, Huang ZQ, He JQ, Chen TF, Wang JF, et al. Colchicine inhibits NETs and alleviates cardiac remodeling after acute myocardial infarction. Cardiovasc Drugs Ther. 2024;38:31–41. doi: 10.1007/s10557-022-07326-y [DOI] [PubMed] [Google Scholar]
- 12. Shen S, Duan J, Hu J, Qi Y, Kang L, Wang K, Chen J, Wu X, Xu B, Gu R. Colchicine alleviates inflammation and improves diastolic dysfunction in heart failure rats with preserved ejection fraction. Eur J Pharmacol. 2022;929:175126. doi: 10.1016/j.ejphar.2022.175126 [DOI] [PubMed] [Google Scholar]
- 13. Sun X, Duan J, Gong C, Feng Y, Hu J, Gu R, Xu B. Colchicine ameliorates dilated cardiomyopathy via SIRT2‐mediated suppression of NLRP3 Inflammasome activation. J Am Heart Assoc. 2022;11:e025266. doi: 10.1161/JAHA.122.025266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kanamori H, Yoshida A, Naruse G, Endo S, Minatoguchi S, Watanabe T, Kawaguchi T, Tanaka T, Yamada Y, Takasugi N, et al. Impact of autophagy on prognosis of patients with dilated cardiomyopathy. J Am Coll Cardiol. 2022;79:789–801. doi: 10.1016/j.jacc.2021.11.059 [DOI] [PubMed] [Google Scholar]
- 15. Shao D, Kolwicz SC Jr, Wang P, Roe ND, Villet O, Nishi K, Hsu YA, Flint GV, Caudal A, Wang W, et al. Increasing fatty acid oxidation prevents high‐fat diet‐induced cardiomyopathy through regulating Parkin‐mediated mitophagy. Circulation. 2020;142:983–997. doi: 10.1161/CIRCULATIONAHA.119.043319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kast DJ, Dominguez R. The cytoskeleton‐autophagy connection. Curr Biol. 2017;27:R318–R326. doi: 10.1016/j.cub.2017.02.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Larocque K, Ovadje P, Djurdjevic S, Mehdi M, Green J, Pandey S. Novel analogue of colchicine induces selective pro‐death autophagy and necrosis in human cancer cells. PLoS One. 2014;9:e87064. doi: 10.1371/journal.pone.0087064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Buch BT, Halling JF, Ringholm S, Gudiksen A, Kjøbsted R, Olsen MA, Wojtaszewski JFP, Pilegaard H. Colchicine treatment impairs skeletal muscle mitochondrial function and insulin sensitivity in an age‐specific manner. FASEB J. 2020;34:8653–8670. doi: 10.1096/fj.201903113RR [DOI] [PubMed] [Google Scholar]
- 19. Rocha ALD, Pinto AP, Morais GPD, Marafon BB, Silva ASRD. Moderate, but not excessive, training attenuates autophagy machinery in metabolic tissues. Int J Mol Sci. 2020;21:1–21. doi: 10.3390/ijms21228416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U, et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLoS Biol. 2020;18:e3000410. doi: 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xia P, Chen J, Liu Y, Fletcher M, Jensen BC, Cheng Z. Doxorubicin induces cardiomyocyte apoptosis and atrophy through cyclin‐dependent kinase 2‐mediated activation of forkhead box O1. J Biol Chem. 2020;295:4265–4276. doi: 10.1074/jbc.RA119.011571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tanida I, Ueno T, Kominami E. LC3 and autophagy. Methods Mol Biol. 2008;445:77–88. [DOI] [PubMed] [Google Scholar]
- 23. Karakikes I, Ameen M, Termglinchan V, Wu JC. Human induced pluripotent stem cell‐derived cardiomyocytes: insights into molecular, cellular, and functional phenotypes. Circ Res. 2015;117:80–88. doi: 10.1161/CIRCRESAHA.117.305365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dos Santos NV, Saponi CF, Ryan TM, Primo FL, Greaves TL, Pereira JFB. Reversible and irreversible fluorescence activity of the enhanced green fluorescent protein in pH: insights for the development of pH‐biosensors. Int J Biol Macromol. 2020;164:3474–3484. doi: 10.1016/j.ijbiomac.2020.08.224 [DOI] [PubMed] [Google Scholar]
- 26. Li DL, Wang ZV, Ding G, Tan W, Luo X, Criollo A, Xie M, Jiang N, May H, Kyrychenko V, et al. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation. 2016;133:1668–1687. doi: 10.1161/CIRCULATIONAHA.115.017443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li A, Gao M, Liu B, Qin Y, Chen L, Liu H, Wu H, Gong G. Mitochondrial autophagy: molecular mechanisms and implications for cardiovascular disease. Cell Death Dis. 2022;13:444. doi: 10.1038/s41419-022-04906-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Boutelle AM, Attardi LD. p53 and tumor suppression: it takes a network. Trends Cell Biol. 2021;31:298–310. doi: 10.1016/j.tcb.2020.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carlsson MJ, Vollmer AS, Demuth P, Heylmann D, Reich D, Quarz C, Rasenberger B, Nikolova T, Hofmann TG, Christmann M, et al. p53 triggers mitochondrial apoptosis following DNA damage‐dependent replication stress by the hepatotoxin methyleugenol. Cell Death Dis. 2022;13:1009. doi: 10.1038/s41419-022-05446-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mackeh R, Perdiz D, Lorin S, Codogno P, Poüs C. Autophagy and microtubules—new story, old players. J Cell Sci. 2013;126:1071–1080. doi: 10.1242/jcs.115626 [DOI] [PubMed] [Google Scholar]
- 31. Monastyrska I, Rieter E, Klionsky DJ, Reggiori F. Multiple roles of the cytoskeleton in autophagy. Biol Rev Camb Philos Soc. 2009;84:431–448. doi: 10.1111/j.1469-185X.2009.00082.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Su L, Zhang J, Gomez H, Kellum JA, Peng Z. Mitochondria ROS and mitophagy in acute kidney injury. Autophagy. 2023;19:401–414. doi: 10.1080/15548627.2022.2084862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Song R, Yang Y, Lei H, Wang G, Huang Y, Xue W, Wang Y, Yao L, Zhu Y. HDAC6 inhibition protects cardiomyocytes against doxorubicin‐induced acute damage by improving α‐tubulin acetylation. J Mol Cell Cardiol. 2018;124:58–69. doi: 10.1016/j.yjmcc.2018.10.007 [DOI] [PubMed] [Google Scholar]
- 34. Caporizzo MA, Prosser BL. The microtubule cytoskeleton in cardiac mechanics and heart failure. Nat Rev Cardiol. 2022;19:364–378. doi: 10.1038/s41569-022-00692-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Caporizzo MA, Chen CY, Prosser BL. Cardiac microtubules in health and heart disease. Exp Biol Med (Maywood). 2019;244:1255–1272. doi: 10.1177/1535370219868960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kerfant BG, Vassort G, Gómez AM. Microtubule disruption by colchicine reversibly enhances calcium signaling in intact rat cardiac myocytes. Circ Res. 2001;88:E59–E65. doi: 10.1161/hh0701.090462 [DOI] [PubMed] [Google Scholar]
- 37. Lu YY, Chen YC, Kao YH, Lin YK, Yeh YH, Chen SA, Chen YJ. Colchicine modulates calcium homeostasis and electrical property of HL‐1 cells. J Cell Mol Med. 2016;20:1182–1190. doi: 10.1111/jcmm.12818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Caporizzo MA, Chen CY, Bedi K, Margulies KB, Prosser BL. Microtubules increase diastolic stiffness in failing human cardiomyocytes and myocardium. Circulation. 2020;141:902–915. doi: 10.1161/CIRCULATIONAHA.119.043930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Himelman E, Lillo MA, Nouet J, Gonzalez JP, Zhao Q, Xie LH, Li H, Liu T, Wehrens XH, Lampe PD, et al. Prevention of connexin‐43 remodeling protects against Duchenne muscular dystrophy cardiomyopathy. J Clin Invest. 2020;130:1713–1727. doi: 10.1172/JCI128190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mizumoto N, Tanaka H, Matsushima H, Vishwanath M, Takashima A. Colchicine promotes antigen cross‐presentation by murine dendritic cells. J Invest Dermatol. 2007;127:1543–1546. doi: 10.1038/sj.jid.5700699 [DOI] [PubMed] [Google Scholar]
- 41. Zoccali C, Mallamaci F. Innate immunity system in patients with cardiovascular and kidney disease. Circ Res. 2023;132:915–932. doi: 10.1161/CIRCRESAHA.122.321749 [DOI] [PubMed] [Google Scholar]
- 42. Nie T, Zhu L, Yang Q. The classification and basic processes of autophagy. Adv Exp Med Biol. 2021;1208:3–16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Table S1
Figures S1–S5