

Abstract
Background
The purpose of this study was to investigate a therapeutic approach targeting the inflammatory response and consequent remodeling from ischemic myocardial injury.
Methods and Results
Coronary thrombus aspirates were collected from patients at the time of ST‐segment–elevation myocardial infarction and subjected to array‐based proteome analysis. Clinically indistinguishable at myocardial infarction (MI), patients were stratified into vulnerable and resilient on the basis of 1‐year left ventricular ejection fraction and death. Network analysis from coronary aspirates revealed prioritization of tumor necrosis factor‐α signaling in patients with worse clinical outcomes. Infliximab, a tumor necrosis factor‐α inhibitor, was infused intravenously at reperfusion in a porcine MI model to assess whether infliximab‐mediated immune modulation impacts post‐MI injury. At 3 days after MI (n=7), infliximab infusion increased proregenerative M2 macrophages in the myocardial border zone as quantified by immunofluorescence (24.1%±23.3% in infliximab versus 9.29%±8.7% in sham; P<0.01). Concomitantly, immunoassays of coronary sinus samples quantified lower troponin I levels (41.72±7.34 pg/mL versus 58.11±10.75 pg/mL; P<0.05) and secreted protein analysis revealed upregulation of injury‐modifying interleukin‐2, ‐4, ‐10, ‐12, and ‐18 cytokines in the infliximab‐treated cohort. At 4 weeks (n=12), infliximab treatment resulted in significant protective influence, improving left ventricular ejection fraction (53.9%±5.4% versus 36.2%±5.3%; P<0.001) and reducing scar size (8.31%±10.9% versus 17.41%±12.5%; P<0.05).
Conclusions
Profiling of coronary thrombus aspirates in patients with ST‐segment–elevation MI revealed highest association for tumor necrosis factor‐α in injury risk. Infliximab‐mediated immune modulation offers an actionable pathway to alter MI‐induced inflammatory response, preserving contractility and limiting adverse structural remodeling.
Keywords: immune modulation, infliximab, myocardial infarction, porcine model, TNFα inhibition
Subject Categories: Animal Models of Human Disease, Inflammation, Ischemia, Myocardial Regeneration
Nonstandard Abbreviations and Acronyms
- ACC
American College of Cardiology
- AHA
American Heart Association
- CS
coronary sinus
- cTNI
cardiactroponin I
- IACUC
institutional animal care and use committee
- IC
intracoronary
- IV
intravenous
- LAD
left anterior descending
- LCx
leftcircumflex
- LVEF
left ventricular ejection fraction
- MACE
major adversecardiac events
- MI
myocardial infarction
- NYHA
New York Heart Association
- PCI
percutaneous coronary intervention
- RCA
right coronary artery
- STEMI
ST‐elevation myocardial infarction
Research Perspective.
What Is New?
Differential proteomic profiling of coronary aspirates from patients presenting with ST‐segment–elevation myocardial infarction prioritized tumor necrosis factor‐α as a molecular risk for irreversible myocardial injury.
Infliximab‐mediated immune modulation subsequently limited injury in a porcine model of myocardial infarction, with preservation of left ventricular function and reduced scar burden.
What Question Should Be Addressed Next?
Integration of infliximab infusion, without disruption of door‐to‐balloon time algorithms, may provide a future strategy to limit injury and promote myocardial recovery at the time of ST‐segment–elevation myocardial infarction.
Despite advances in acute coronary syndrome management, achieved by shortening ischemic time, device‐based interventions and novel pharmacotherapy, ST‐segment–elevation myocardial infarction (STEMI) continues to drive in‐hospital death and long‐term heart failure globally. 1 , 2 Parameters such as age, left ventricular ejection fraction (LVEF), cardiovascular risk factors, renal function, and cardiac enzyme levels are often combined within algorithms designed to forecast the risk of major adverse cardiac events following STEMI. 3 , 4 , 5 A recognized contributor to myocardial damage following ST‐segment–elevation myocardial infarction (MI) is inflammation. 6 , 7 , 8 , 9 Defining the interlink of molecular mediators with clinical outcome would provide new targets to reduce injury.
Available biomarkers identify injury with high sensitivity and accuracy, yet fall short in distinguishing systemic from myocardial inflammation, offering limited prognostic insight. 8 , 10 Notably, biomarkers derived from systemic serological samples are influenced by multiple organ systems, confounding active processes within the injured myocardium, especially under hemodynamic compromise. 6 , 7 , 11 , 12 , 13 Direct sampling of the coronary bed, which supplies infarcted myocardial tissue, would thus be advantageous, as it may best inform on factors intimately involved in modulating scar formation and remodeling. 14 , 15 , 16
Ischemic injury in the heart leads to an innate inflammatory response triggering the recruitment of neutrophils and macrophages whose intrinsic role is to clear dead tissue and orchestrate resolution of injury. 14 , 15 , 17 , 18 , 19 Macrophages are critical mediators of the immune response, fostering tissue recovery and repair in the heart. 20 , 21 , 22 , 23 Recruited to the site of injury, macrophages are reactive to the local molecular terrain shaped by the initial presence of inflammatory cells and cytokines that determine their cellular phenotype and function. 24 , 25 , 26 , 27 Polarization to the M1 phenotype promotes inflammation in the area of injury, while M2 macrophages initiate repair and augment regeneration. 17 , 19 , 28 Cytokine cues such as interferon‐γ and tumor necrosis factor‐α (TNFα) polarize toward M1, while interleukin‐4 and interleukin‐10 drive polarization toward the M2 phenotype. 29 , 30 , 31 If left unchecked, a protracted inflammatory response can lead to impaired tissue healing and worsening of cardiac function. 14 Given the centrality of the immune system in myocardial injury, decoding local proinjurious cytokine signaling at the time of STEMI may prove useful for understanding the fate of the myocardium after infarction.
Here, coronary thrombus aspirates secured at the time of percutaneous coronary intervention (PCI) from patients with STEMI were assessed to map the local proteomic landscape in the peri‐infarction setting. Obtained from patients who were clinically indistinguishable at the time of presentation, 1‐year clinical follow‐up segregated proteomic data from vulnerable and resilient groups on the basis of adverse cardiac events. Differential proteomic profile of coronary aspirates prioritized TNFα as a molecular risk for irreversible myocardial injury. This prompted investigation of infliximab, a TNFα inhibitor, in a porcine model of MI.
METHODS
STEMI Patient Study Design
This study was performed under institutional review board approvals and with informed consent. The data that support the findings of this study may be available from the corresponding author upon reasonable request. Patients between the ages of 40 and 82 years old (n=25) underwent coronary thrombus aspiration before percutaneous coronary intervention (PCI) for STEMI due to an occlusive lesion in the left anterior descending (LAD), left circumflex, or right coronary artery at the Cardiovascular Center Aalst and at Mayo Clinic, Rochester. Thrombectomy was performed only when deemed to be an appropriate step in management before PCI. A mechanical aspiration thrombectomy device (eg, CAT Rx) was used for sample collection.
Presenting demographics and data needed to calculate the Mayo Clinic Risk Model were collected. This model has been previously described and is used to predict risk of major adverse cardiac events (death, Q‐wave MI, stroke, and need for emergency coronary artery bypass graft) following PCI. Calculation is determined by the addition of integer scores for the presence of 8 possible risk factors. 3 , 4 , 32 The 5 patient/procedural characteristics included are age, congestive heart failure, New York Heart Association functional class ≥III, urgent/emergent PCI, chronic renal disease, and preprocedural cardiogenic shock. The remaining variables consist of angiographic characteristics and include left main stenosis ≥70%, multivessel disease (>70% in >1 major epicardial coronary artery or in their major branches), and presence of thrombus in any lesion. 32
All patients were managed according to American College of Cardiology/American Heart Association practice guidelines. 33 Patients were followed up regularly at the Aalst Cardiovascular Center or at Mayo Clinic, Rochester, Minnesota, both regarded as centers of excellence for cardiovascular disease.
Human STEMI Coronary Aspirate
Fresh coronary aspirates (consisting of thrombus and coronary blood) were stored in EDTA tubes and centrifuged at 4 °C. The plasma supernatant was collected, treated with a protease inhibitor, split into working aliquots, flash frozen, and stored at –80 °C within 120 minutes of coronary sampling. Coronary aspirates were included in this study if the coronary intervention was without complication and Thrombolysis in Myocardial Infarction grade 3 flow was restored. Aspirates were processed for plasma extraction, subjected to proteomic assessment with the generation of a heatmap plotting plasma protein expression profile for each patient (Figure 1A and 1B).
Figure 1. Coronary thrombus aspiration during STEMI provides the serological resource for high‐throughput proteomic assessment.
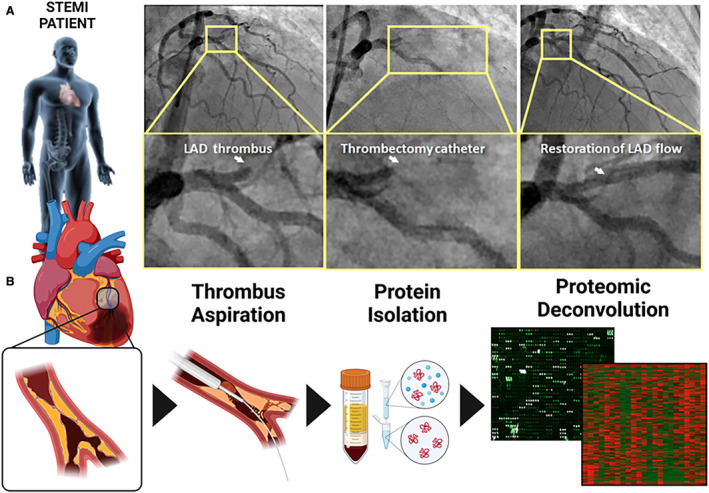
A, Angiographic depiction of an occlusive thrombus in the left anterior descending artery (left panel) and subsequent insertion of thrombectomy catheter with clot aspiration/restoration of blood flow (middle and right panels). B, Clots extracted via a coronary aspiration catheter (left 3 panels) were subjected to high‐throughput proteomic assessment to uncover the protein content of coronary blood at the time of reperfusion in STEMI. LAD indicates left anterior descending artery; and STEMI, ST‐segment–elevation myocardial infarction.
After 1‐year follow‐up, coronary aspirate samples from those with a significant drop in LVEF on routine echocardiogram following MI or cardiac death during this preset time frame were categorized as vulnerable (n=11), while samples from those who did not were categorized as resilient (n=14). Patients in the vulnerable and resilient cohorts received the same standard of care.
Proteomic Evaluation
Coronary aspirate samples stored at –80 °C were thawed and prepared for ELISA‐based antibody array analysis using L‐Series Human L507 Array Membrane Kit (Raybiotech, Peachtree Corners, Georgia). Proteomic analysis was performed according to the manufacturer's instructions. The complete antibody array target list is outlined in Figure S1. Dry slides were analyzed using Axon GenePix Pro 6 software (Molecular Devices, San Jose, CA) for quantification per manufacturer's instructions.
Complex Biological Network Analysis for Patient‐Derived Samples
Differentially expressed array proteins were submitted for network analysis using Ingenuity Pathways Knowledge Base (Ingenuity Systems, Redwood City, CA) to identify associated functional subnetworks. These were merged into a composite network in Ingenuity Pathways Knowledge Base and depicted using the network visualization program Cytoscape 3.8.2, with network topology characterized using Network Analyzer. 34 Computed properties included node degree, degree distribution, and clustering coefficient distribution. 35 , 36 , 37 , 38 Ingenuity Pathways Knowledge Base also prioritized overrepresented molecular and physiological functions and canonical pathways associated with the resolved interaction network, with P values calculated using Fisher's exact test.
Porcine MI Model
Porcine experimental protocols were approved by the institutional animal care and use committee. Animal Research: Reporting of In Vivo Experiments reporting guidelines were used. 39 Domestic Yorkshire Cross pigs of both sexes aged 4 to 6 months and weighing 30 to 40 kg were used for the study. Pigs were housed together on site. Animals received aspirin (20 mg/kg daily), clopidogrel (75 mg daily), and amiodarone (800 mg twice daily) starting 3 days before surgery. Immediately before the infarction procedure, amiodarone (300‐mg bolus), heparin (5000–10 000 IU titrated to activated clotting time) and lidocaine (50 mg IV bolus) were administered. Procedures were carried out on site where animals are housed. Animals were anesthetized with an intramuscular injection of Telazol/Xylazine and maintained under isoflurane inhalation at 1% to 3% for the duration of the LAD‐STEMI procedure. To this end, pigs were placed in the supine position, with ECG and hemodynamic monitoring performed throughout the procedure. Transthoracic echocardiography imaging was obtained before the procedure. Percutaneous access to the femoral artery and vein was performed under ultrasound guidance using a modified Seldinger technique. A 0.035‐inch guidewire was passed through the needle to secure luminal access, and a 6 to 8Fr introducer was secured into place. An 8Fr AcuNav intracardiac echocardiography probe (Siemens Medical Solutions, Malvern, PA) was positioned in the right atrium of the heart for a baseline evaluation of the heart and intraprocedural imaging of infarct progression. A 6Fr coronary guide catheter was advanced to the coronary arteries, Omnipaque™ (GE Healthcare, Chicago, IL) to visualize LAD coronary anatomy. A 0.014‐inch guidewire was advanced into the distal portion of the LAD and an angioplasty balloon catheter (3–4 mm diameter) was advanced distal to the first diagonal branch of the LAD. The balloon was then inflated and maintained above nominal rating to completely obstruct blood flow for 90 minutes. After the 90 minutes, the balloon was deflated, and the myocardium was allowed to reperfuse (Figure 2A and 2B and Figure S2). The first diagonal branch of the LAD is selected as left ventricular perfusion is highly similar across pigs past this landmark. 40 In addition, the aim is to create a sizable infarct while maintaining animal survival.
Figure 2. Angiography of the porcine heart MI model.
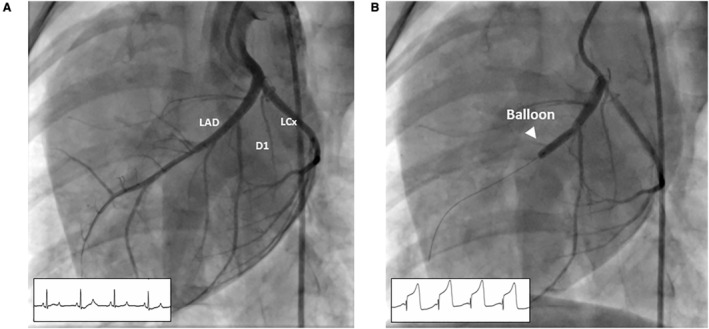
A, Preinfarction angiogram. Distribution of coronary artery blood flow along the anterior portion of the left ventricle is determined for proper balloon placement in the mid‐LAD. B, The angioplasty balloon is inflated to completely occlude the coronary artery as documented by repeat angiography and ST deviation on ECG monitoring. D1 indicates first diagonal branch; LAD, left anterior descending artery; and LCX, left circumflex artery.
Experimental Protocol
Based on the TNFα centrality of the network from proteomic analysis of patients with STEMI, we decided to test if a singular pharmacologic intervention with the TNFα inhibitor infliximab could minimize myocardial injury and potentiate recovery in the porcine MI model. Comparison of human and porcine TNFα sequencing resulted in 87% identity overlap (Figure S3, Video S1). 41 , 42 To understand the early inflammatory response post‐MI, we analyzed samples 3 days after injury with subsequent animal euthanasia. Seven pigs were divided into 2 subgroups: group I (n=4) underwent the infarct procedure described above and received a single administration of infliximab at 5 mg/kg IV immediately upon reperfusion and group II (n=3) underwent the infarct procedure with a sham infusion of saline. Randomization to infliximab or sham infusion was performed by the veterinary technician. The operator was blinded to randomization and analysis. Infliximab dosing was chosen on the basis of the therapeutic regimen in humans. Intracardiac echocardiography, hemodynamics and blood samples were taken at baseline and 3 days after MI (Figure 3A).
Figure 3. Porcine MI model and experimental protocol timeline.
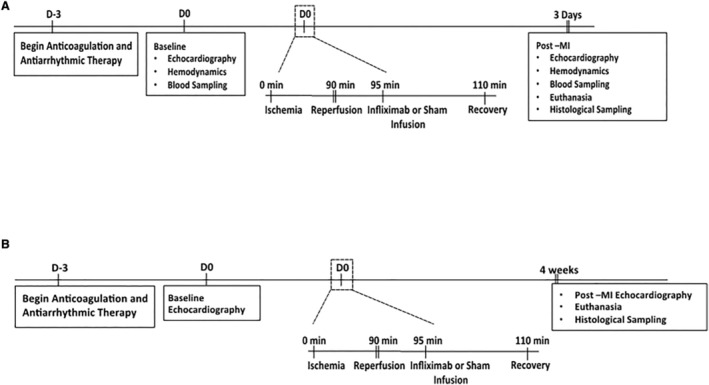
A, Timeline for assessment of initial response to injury 3 days after MI in pigs receiving infliximab (n=4) vs control infusion (m=3). B, Timeline for assessment of late response to injury 4 weeks post‐MI in pigs receiving infliximab (n=5) vs control infusion (n=7). MI indicates myocardial infarction.
To determine the infliximab‐mediated molecular and cellular adjustment impact of long‐term myocardial recovery, myocardial function and scar size were quantified 4‐weeks after infarction. Twelve pigs were divided into 2 subgroups: group I (n=5) underwent the infarct procedure and received a single administration of infliximab intravenous or via intracoronary injection at 5 mg/kg immediately upon reperfusion and group II (n=7) underwent the infarct procedure with no infliximab administration. Once again, the operator was blinded to randomization, and the veterinary technician administered the infliximab or sham infusion. Intravenous administration was chosen as the initial method for infliximab administration, and intracoronary injection used in subsequent procedures in 2 animals due to ease of administration and comparable bioavailability. Intracardiac echocardiography was performed at baseline and at 4 weeks following MI (Figure 3B).
All pigs were operated on consecutively by the same operationalists. Four pigs underwent infarction but died due to procedural complications and were therefore not included in the study analysis. One other pig was excluded due to refractory ventricular fibrillation during MI requiring multiple shocks and pharmacologic interventions, despite surviving for 30 days. Exclusion was because continuous 90‐minute occlusion was not performed.
Porcine Plasma Protein Analysis
Great cardiac vein (LAD territory) sampling was performed at baseline and 3 days after MI to assess response to injury. The coronary sinus of each heart was engaged using an Agilis NxT Steerable Introducer (St. Jude Medical, St. Paul, MN). A 6Fr multipurpose guide catheter was placed within the coronary sinus and positioned at the ostium of the great cardiac vein for subsequent blood collections (Cordis, Milpitas, CA) (Figure 4). Blood was collected, placed into EDTA tubes and centrifuged to collect plasma for analysis. Cardiac troponin I was tested using a sandwich‐based ELISA specific for porcine troponin (LS‐F23639; LifeSpan Biosciences, Inc., Seattle, WA) according to manufacturer's instructions. The assay detects cardiac troponin I levels as low as 31.25 pg/mL with a range of detection from 31.25 pg/mL to 2000 pg/mL.
Figure 4. Fluoroscopic image demonstrating coronary sinus sampling from the great cardiac vein (LAD infarct territory).
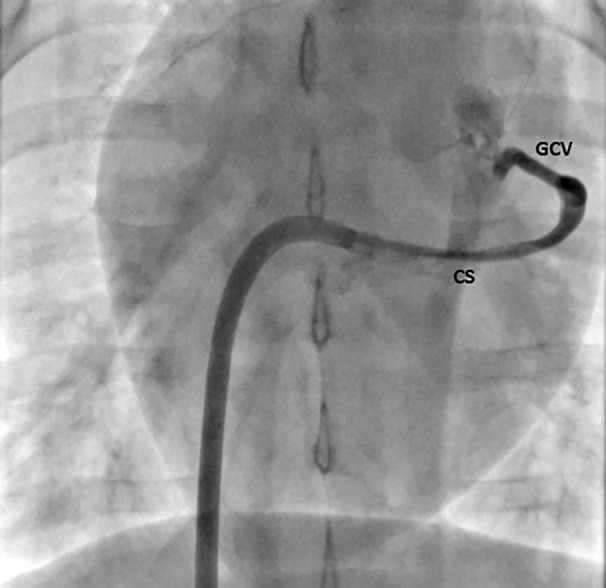
CS indicates coronary sinus; GCV, great cardiac vein; and LAD, left anterior descending artery.
Plasma cytokines were measured using a MILLIPLEX MAP Porcine Cytokine and Chemokine Magnetic Bead Panel (EMD Millipore, Billerica, MA). Plasma samples were analyzed for expression levels of 1000 proteins using RayBiotech label‐based human Antibody Array 1000 (RayBiotech, Peachtree Corners, GA) per the manufacturer's instructions. Briefly, the glass slide arrays were blocked and incubated with biotin‐labeled samples. After extensive washing, the array slides were incubated with a streptavidin‐conjugated fluorescent dye. The fluorescent signals were then visualized using a GenePix 4000B microarray Scanner system (Molecular Devices, Sunnyvale, CA) using the green channel. Protein array data were assessed and analyzed using the R programming platform (http://www.r‐project.org/). Data quality was assessed on the basis of data distribution (density curve shown in Figure S4 and correlation matrix in Figure S5A and S5B). Principal component analysis and unsupervised hierarchical clustering were performed on the entire protein expression profile of all 7 samples (Figure S5C and S5D). With heteroscedasticity (unequal variations among proteins) of data, Welch's 2‐sample t test was applied for differential analysis to identify protein markers that differentiate treated and control samples. Significant differential expressed markers were selected on the basis of P values <0.05 and fold change >2.0 (Figure S6A through S6G). Protein interaction network and pathway enrichment analyses were applied on the selected differential biomarkers. Data integration and visualization strategy were applied to reveal the core functions and identify network hubs and potential important proteins, which are in charge of crosstalk among those pathways. 43 This was achieved using R programming software with customized R codes.
Echocardiography Analysis
Left ventricular structure and function were evaluated by intracardiac echocardiography (Acuson SC 2000, AcuNav 8F; Siemens, Mountain View, CA) and compared between animals at 4 weeks after myocardial infarction with (group I) or without (group II) infliximab therapy. LVEF (%) was calculated as [(EDV‐ESV)/EDV]×100, where EDV is end‐diastolic volume, and ESV is end‐systolic volume.
Histological Preparation and Scar Size Calculation
After the 3‐day or 4‐week follow‐up, pigs were euthanized, and hearts were perfused and fixed with 4% paraformaldehyde through the aortic root. Hearts were then cut into 1‐cm cross‐sections and left in 4% paraformaldehyde overnight. Whole left ventricular sections were prepared from 4‐week follow‐up animals, paraffin embedded, and stained with Masson's trichrome. Specimens from 3‐day postinfarct were paraffin embedded and stained for M2 macrophages; mature pan‐macrophage marker [25F9] (Abcam, ab15689, 1:200) and M2 marker CD163 (Abcam, ab87099, 1 g/mL). Scar size and total myocardial area were measured in all cross‐sections. Scar size is reported as total percentage collagen area. Cross‐sectional short‐axis images were digitally superimposed and given a 5‐mm digital width in Zeiss Zen software (Zeiss Microscopy, Jena, Germany). Digitized images were rendered as a z‐stack, and a movie was generated from manipulation of the z‐stacked image (Videos S2 and S3).
Statistical Analysis
Patient clinical data are described as mean±SD and compared between groups with Student's 2‐sample t‐test. Patient demographic data were analyzed by ANOVA. Cytokine concentrations are presented as median±interquartile range and analyzed by nonparametric statistics Mann–Whitney U test. Differences were considered significant with P<0.05. Descriptive statistics for animal data are presented as mean±SD. Echocardiographic data were analyzed by nonparametric statistics Kruskal–Wallis test. Statistical analyses were performed in GraphPad Prism version 10.0.1 (GraphPad Software, Inc., San Diego, CA).
RESULTS
Vulnerable Versus Resilient Patient Cohort Following STEMI
Patients presenting with STEMI and treated with thrombectomy were indistinguishable in demographics (Table), cardiovascular risk factors (Table), hemodynamic parameters (Figure 5A), Mayo Clinic Risk Model of major adverse cardiac events (Figure 5B), and Global Registry of Acute Coronary Events score (Figure 5C) at the time of recruitment. There was also no significant difference in cardiac troponin T or C‐reactive protein serum levels at the time of MI. At the 1‐year follow‐up preset time point, coronary aspirates from patients suffering from major cardiac events were labeled as “vulnerable,” while those obtained from event‐free counterparts were designated “resilient.” Patient survival was 55% (6/11 patients) versus 100% (14/14 patients) in the vulnerable and resilient cohorts, respectively. At 1‐year follow‐up, the surviving vulnerable cohort demonstrated an LVEF reduction of 24.9% (±5.9%) compared with a 0.9% (±1.9%; P<0.01) drop in the resilient group (Figure 5D).
Table .
Baseline Characteristics of Patients Presenting With STEMI
| Baseline characteristics | Total patient cohort (N=25) | Resilient cohort (n=14) | Vulnerable cohort (n=11) | P value |
|---|---|---|---|---|
| Age, y | 65±10 | 62±12 | 68±8 | 0.27 |
| Women, % | 44 | 43 | 45 | 0.99 |
| Total ischemic time, h | 6.7±4.1 | 6.6±4.4 | 6.9±3.7 | 0.85 |
| STEMI of LAD, % | 64 | 57 | 73 | 0.74 |
| Troponin T, ng/mL | 8.2±7.5 | 6.3±6.4 | 10.6±8.4 | 0.52 |
| C‐reactive protein, mg/L | 23.0±46.5 | 29.6±58.3 | 15.7±30.2 | 0.99 |
| WBC, cells/μL | 13 042±5077 | 12 856±5254 | 13 279±5087 | 0.99 |
| Past family history CAD, % | 6 | 5 | 1 | 0.27 |
| Smoking, % | 7 | 5 | 2 | 0.62 |
| Diabetes, % | 4 | 2 | 2 | 0.97 |
| Hypercholesterolemia, % | 12 | 7 | 5 | 0.97 |
| BMI, kg/m2 | 26.4±4.5 | 26.8±5.9 | 26.0±3.2 | 0.95 |
| Statin, % | 16 | 9 | 7 | 0.79 |
| β Blocker, % | 13 | 8 | 5 | 0.99 |
| Ca2+ channel blocker, % | 1 | 0 | 1 | 0.38 |
| ACEi, % | 15 | 9 | 6 | 0.99 |
| ARB, % | 2 | 2 | 0 | 0.35 |
| Aspirin, % | 15 | 9 | 6 | 0.99 |
| NYHA score, median | 1 | 1 | 1 | 0.89 |
ACEi indicates angiotensin‐converting enzyme inhibitor; ARB, angiotensin receptor blocker; BMI, body mass index; CAD, coronary artery disease; LAD, left anterior descending artery; NYHA, New York Heart Association; STEMI, ST‐segment–elevation myocardial infarction; and WBC, white blood cell count.
Figure 5. Patients presenting with STEMI and treated with thrombectomy at Cardiovascular Center Aalast and Mayo Clinic, Rochester.
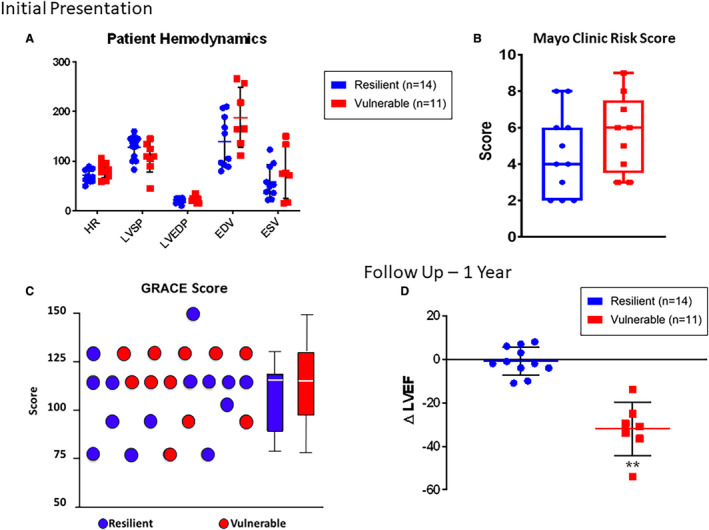
A, Patients evaluated at the time of STEMI were found to be indistinguishable with respect to direct hemodynamic measurements. B, Patients were furthermore indistinguishable with regard to Mayo Clinic risk score for prediction of major adverse cardiac events following PCI as well as with (C) the GRACE (Global Registry of Acute Coronary Events) Score. D, At 1‐year follow‐up, patients were distinguished as vulnerable vs resilient on the basis of survival or ≥20% drop in LVEF following MI. The surviving vulnerable cohort demonstrated an LVEF reduction of 24.9% (±5.9%) compared with a 0.9% (±1.9%; P<0.01) drop in the resilient group. Data are reported as mean±SD. Asterisks indicate statistical significance. EDV indicates end‐diastolic volume; ESV, end‐systolic volume; HR, heart rate; LVEF, left ventricular ejection fraction; LVEDP, left ventricular end‐diastolic pressure; LVSP, left ventricular systolic pressure; MACE, major adverse cardiac event; PCI, percutaneous coronary intervention; and STEMI, ST‐segment–elevation myocardial infarction.
Differential Proteome Profile in Vulnerable Versus Resilient Coronary Aspirates
Quantitative protein array identified, of 1080 proteins, 19 significantly distinct proteins in thrombus aspirates from vulnerable versus resilient groups (2‐fold difference, P<0.05). Upon systems analysis, these 19 discriminatory factors clustered into a scale‐free network composed of 65 nodes linked by 417 pairwise connections (Figure 6A). Network topology displayed nonstochastic architecture with hierarchical tendencies (Figure 6B). The biological network prioritized TNFα centrality, with 51 connections (Figure 6B). Evaluation for overrepresented molecular and physiological functions revealed prioritization of hematological, immunological, and cardiovascular functions (Figure 6C). Canonical pathway assessment indicated overrepresented calcium regulation, retinoic acid signaling, and endothelial inflammation (Figure 6D).
Figure 6. Proteomic dissection of bio‐banked coronary aspirates from patients with STEMI segregated on the basis of long‐term clinical outcomes (vulnerable versus resilient).
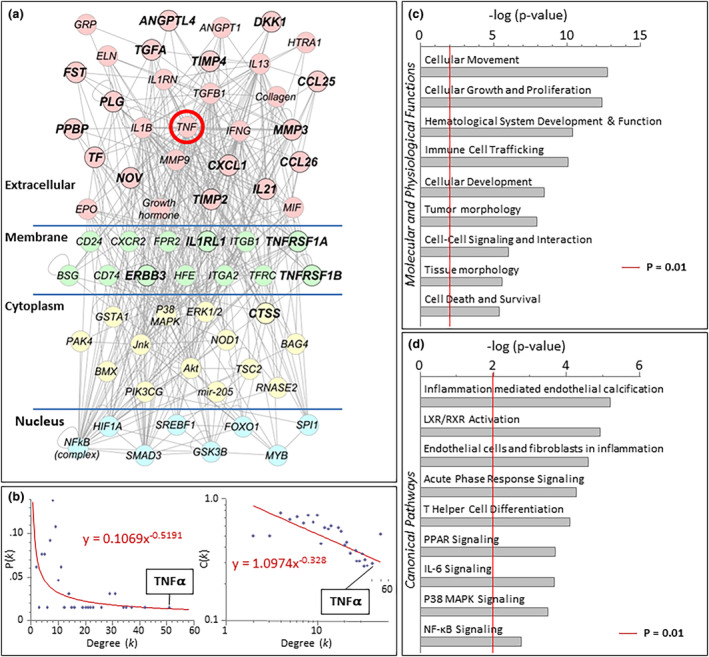
Analysis was performed with Ingenuity Pathways Knowledge Base (Ingenuity Systems, Redwood City, California). A, A TNF⍺‐centric network mediating worse outcomes was revealed. B, Following ingenuity pathway analysis, network topology ranked TNF⍺ as the most highly connected factor. Degree (k) is the number of connections that a node possesses, which can be plotted against either the proportion of total nodes with a specific degree [P(k)] to derive the degree distribution of the network or plotted against the clustering coefficient for nodes with degree k [C(k)] to derive the clustering coefficient for the network. The first graph [k vs P(k)] indicates whether a network possesses scale‐free topology, while the second graph [k vs C(k)] indicates whether the network possesses a hierarchical architecture. Cytoscape's Network Analyzer tool was used to derive the degree of distribution and clustering coefficient. C, Evaluation of overrepresented molecular and physiologic functions revealed cellular movement, cellular growth and proliferation, and hematological system development and function as the most highly overrepresented pathways (D) and inflammation of endothelium as the most highly ranked canonical pathway. Significance of P<0.01 is demonstrated by the red line in panels C and D. IL‐6 indicates interleukin‐6; LXR, liver X receptor; MAPK, mitogen‐activated protein kinase; NF‐κB, nuclear factor kappa‐light‐chain enhancer of activated B cells; PPAR, peroxisome proliferator‐activated receptor; RXR, retinoid X receptor; STEMI, ST‐segment–elevation myocardial infarction; and TNFα, tumor necrosis factor‐α.
Early Phase Porcine Infarction and Biological Impact of Infliximab
Luminex‐array cytokine profiling of samples from the great cardiac vein (LAD territory venous drainage) at 3 days after MI revealed that infliximab induced significant alteration from baseline values in interleukin‐2 (56.4 versus 126.7%; P<0.05), interleukin‐4 (53.2 versus 208.7%; P<0.05), interleukin‐10 (42.5 versus 111.1%; P<0.05), interleukin‐12 (59.8 versus 81.0%, P<0.05), and interleukin‐18 (72.8 versus 97.2%; P<0.01) within the myocardial microenvironment between MI controls and treated animals, respectively (Figure 7A through 7J). Despite infliximab therapy, no difference in TNFα protein concentration in plasma could be measured at day 3 as a percentage of baseline (Figure 7F; P=NS). Cardiac‐specific troponin I at day 3 was significantly different (41.72±7.34 pg/mL versus 58.11±10.75 pg/mL; P<0.05) (Figure 7K). Beyond cytokine and troponin quantitation, to evaluate the biological impact of infliximab following MI, histological profiling was performed documenting immune cell infiltration in the infarct border (Figure 8A). Immunofluorescence staining analysis of the infarct border zone showed that infliximab treatment increased the percentage of CD163+ M2 macrophages (24.1%±23.3% for infliximab versus 9.29%±8.7% for sham; P<0.01) (Figure 8B and 8C). Analysis of intracardiac echocardiography 3 days after MI was not performed due to missingness of data from difficult image views.
Figure 7. Infliximab‐mediated immune modulation alters the cytokine and cellular profile of the infarct bed.
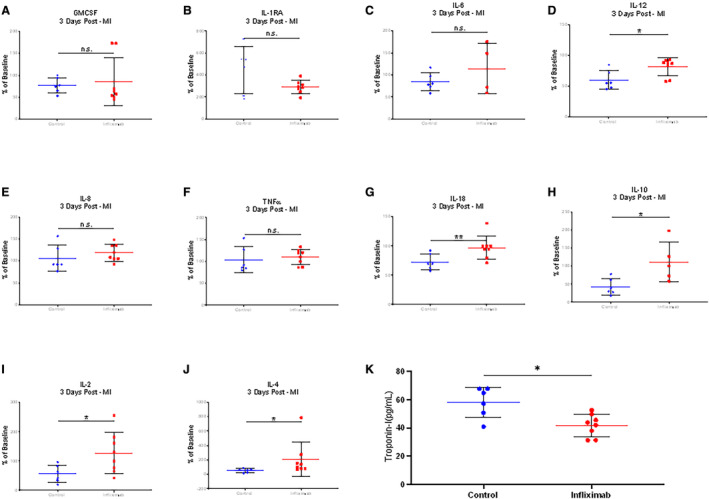
Blood was collected from the ostium of the great cardiac vein 3 days after myocardial infarction. A through J, Cytokine profiling was distinguished with respect to interleukin‐2, ‐4, ‐10, ‐12, and ‐18 activity in infliximab‐treated (n=4) vs untreated pigs (n=3). K, Quantitation of cardiac troponin I levels was lower in infliximab‐treated (n=4) vs untreated (n=3) animals (41.72±7.34 pg/mL vs 58.11±10.75 pg/mL; P<0.05). Asterisks indicate statistical significance. GM‐CSF indicates granulocyte‐macrophage colony‐stimulating factor; IL, interleukin; MI, myocardial infarction; and TNFα, tumor necrosis factor‐α.
Figure 8. Histological profiling of the infarct border zone in early phase myocardial infarction.
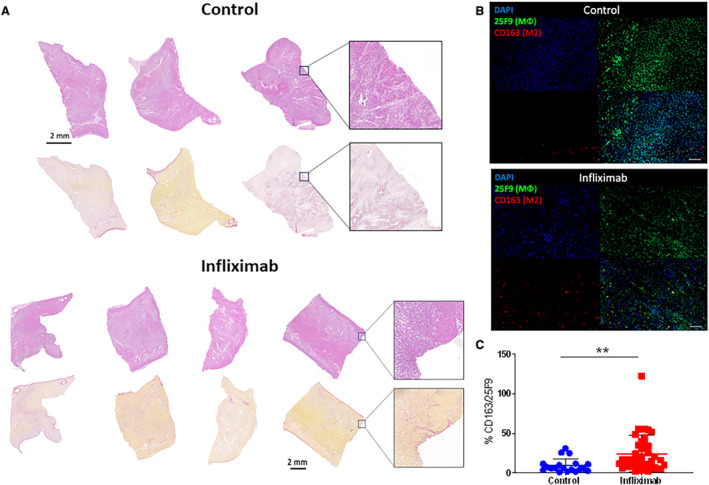
A, Hematoxylin and eosin and Sirius Red staining of the infarction border of control (n=3) and infliximab‐treated (n=4) cohorts reveals a similar pattern of injury at day 3. B and C, Immunofluorescence staining analysis of the infarct border zone showing percentage of M2 macrophages (CD163) compared with total mature macrophages (25F9). Immunofluorescence visualization of macrophage populations with 25F9 (green: total population) vs CD163 (red: M2 population) revealed a significant upregulation of M2 cells following infliximab treatment. Scale bar: 100 μm. Data reported as mean±SD. Asterisks indicates statistical significance.
Infliximab‐Mediated Proteomic Alteration of the Infarcted Microenvironment
Proteomics profiling of coronary sinus plasma collected 3 days following MI was performed in treated and untreated porcine cohorts. Analysis of data distribution curves (Figure S4), correlation (Figure S5A and S5B), unsupervised hierarchical clustering (Figure S5C), and principal component analysis (Figure S5D) revealed consistently altered protein expression profiles between infliximab treated and control samples. Differential analysis using Welch's 2‐sample t test identified 323 proteins with a P value of <0.05 and fold change >2 (Figure 9; Figures S6 through S8). Gene interaction networks (Figure 9A through 9E) revealed hub genes with more than 20 interaction partners: POMC, PLG, SERPINA1, CCR8, CASR, VEGFD, ITGB3, TIMP3, LEFTY2, CCL5, IGF1, CCR3, TGFB3, PDGFB, PF4, PPBP, EGF, SPARC, KNG1, TGFB2, and THBS1. Those genes served as hubs in interaction networks and are vital for signaling transduction. Gene function pathway enrichment analysis on those differential proteins highlighted that they are involved in important functions including inflammatory response, cell–cell signaling, positive regulation of cell proliferation, monocyte chemotaxis, cellular response to tumor necrosis factor, neutrophil chemotaxis, cellular response to interleukin‐1, lymphocyte chemotaxis, and negative regulation of angiogenesis (Figures 7 and 9; Tables S1 and S2). The crosstalk between identified pathways encompassed many C‐C chemokine ligand (CCL) family genes (Figure 9B through 9D). Overall, histological and proteomics profiling day 3 after MI, stratified by infliximab exposure, revealed a polarization toward M2 macrophages (Figure 8) and deconvoluted a microenvironment immunotitrated away from inflammation and toward a proregenerative state.
Figure 9. Integrated function and network analysis from coronary sinus samples 3 days after porcine myocardial infarction reveal that differentially expressed proteins are involved in inflammatory response, chemotaxis, and cell signaling.
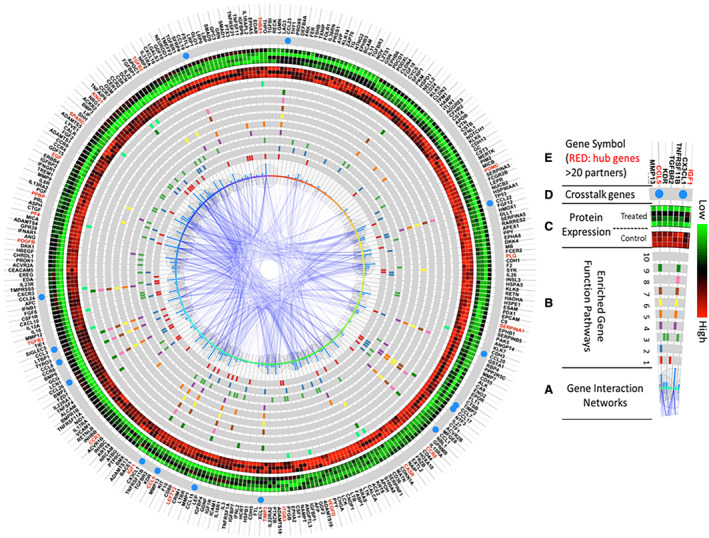
The enriched pathway analysis was generated using R programming software, and the top 10 most significant were selected. A, The center of this figure represents gene interaction networks. The larger the bubbles or higher the blue bars, the more interacting network partners connect to a given protein. B, Genes involved in the top 10 most significantly enriched function pathways are highlighted in each gray circle. The various colors represent genes in different pathways. From the innermost to outermost circles, the pathways consist of: (1) inflammatory response; (2) cell–cell signaling; (3) positive regulation of cell proliferation; (4) monocyte chemotaxis; (5) cellular response to tumor necrosis factor; (6) neutrophil chemotaxis; (7) cellular response to interleukin‐1; (8) lymphocyte chemotaxis; (9) positive regulation of inflammatory response; (10) negative regulation of angiogenesis. C, Protein expression levels of the control cohort (n=3; inner circle—red predominant) and infliximab‐treated cohort (n=4; outer circle: green predominant). Green represents higher expression, and red represents lower expression. D, Genes present in >5 of the top 10 enriched pathways, represented by a blue circle, serve as crosstalk between pathways. (E) Official gene symbols. Proteins with >20 interacting partners are labeled in red, and proteins with <20 interacting partners are labeled in black.
Late‐Phase Porcine MI
Echocardiographic evaluation revealed an increase in anterior wall thickness in diastole (7.07±0.91 mm for postinfarction infliximab treated (n=3) and 3.73±1.22 mm for postinfarction sham; P<0.05; Figure 10A). LVEF decreased immediately following MI (26.0±13.7% versus 35.3±5.3%) compared with baseline (62.7±8.1% versus 62.0±6.5%), in infliximab‐treated versus untreated pigs, respectively, with preservation of LVEF at 4 weeks (53.9%±5.4% versus 36.2%±5.3%; P<0.001) (Figure 10B). Echocardiography data were not available for 1 pig in the late‐phase control cohort due to difficult image views. Histological staining of whole porcine left ventricular cross‐sections with Masson's trichrome revealed a significantly diminished scar burden and minimization of compensatory hypertrophy in the contralateral wall in the infliximab‐treated group. Quantification of scar area on histology revealed a significant decrease in scar size (8.31%±10.9% for infliximab versus 17.41%±12.5% for sham; P<0.05) (Figure 10C and 10D, Figure S9, Videos S2 and S3). Infliximab was tolerated in all pigs treated with no adverse effects.
Figure 10. Late‐phase porcine myocardial infarction.
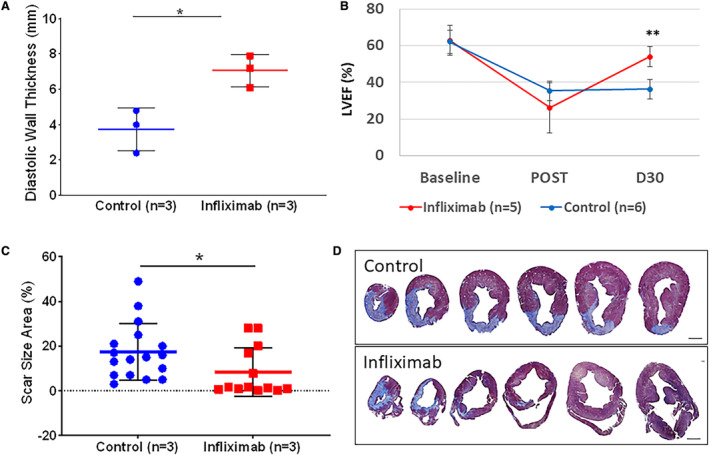
A, Apical anteroseptal wall thickness was obtained from intracardiac echocardiography. Evaluation revealed an increase in anterior wall thickness in diastole (7.07±0.91 mm for postinfarction infliximab treated [n=3] and 3.73±1.22 mm for control [n=3]; P<0.05) 4 weeks after MI. B, LVEF was obtained from intracardiac echocardiography at baseline (62.7±8.1 vs 62.0±6.5%) and immediately after MI (26.0±13.7 vs 35.3±5.3%), in infliximab‐treated (n=5) and control (n=6) cohorts. Preservation of LVEF at 30 days after MI was observed in the infliximab cohort (53.9±5.4 vs 36.2±5.3%; P<0.001). C, Systemic infliximab treatment (n=3) resulted in reduced scar size and diminished hypertrophic adverse remodeling in the left ventricle compared with untreated infarcted hearts (n=3) 4 weeks after MI (8.31±10.9% for infliximab vs 17.41±12.5% for control; P<0.05). Data points depict scar size area measurements from histology sections. D, Representative histological cross‐sections of pig hearts 4 weeks after infarction stained with Masson's trichrome to visualize collagen (blue) and myocardium (red/purple). Analysis performed with nonparametric statistical Kruskal–Wallis test. Asterisks indicate statistical significance. Scale bar represents 10 mm. LVEF indicates left ventricular ejection fraction; and MI, myocardial infarction.
DISCUSSION
In this study, coronary aspirates at time of STEMI, with long‐term clinical follow‐up, allowed deconvolution of pathways that precipitate heart failure following MI and identified an association with TNFα. This discovery prompted leveraging of existing pharmacologic modalities to diminish the extent of myocardial injury in the setting of ischemic insult. Accordingly, infliximab, an established TNFα inhibitor, was found to have a cardioprotective impact in a porcine model of acute myocardial infarction. Systemic infusion of infliximab, at the time of reperfusion, readjusted the myocardial infarct bed by stimulating M2 polarization of macrophages, known to drive regeneration following injury. 20 , 21 Cytokine and proteomic profiling of blood samples 3 days after injury from the venous territory of infarction revealed a shift in the immune landscape away from inflammation and toward repair. Further documentation of benefit was attained 30 days following MI, with echocardiographic and histological evidence for infliximab‐mediated preservation of myocardial structure and function. This study offers a pharmacologic approach to preserve heart function by modifying the interface between the immune system and myocardium following infarction. Although several works have highlighted a duality in the immune system from injurious to regenerative, limited evidence has existed for a clinically translatable approach to alter the immune landscape in setting of STEMI. 15 , 16 Infliximab was here repurposed as a single intravenous regimen to acutely limit significant myocardial injury.
In acute myocardial infarction, the historical supposition has been that ischemia and ischemia–reperfusion injury are master regulators of long‐term outcomes. 44 The centrality that these factors play has recently been challenged as reduction of ischemic time, beyond a threshold, no longer appears to influence morbidity and death. 1 Furthermore, a large body of preclinical and basic science evidence has pointed to the immune system as having a key role in the evolution of injury resolution. 14 , 15 , 16 , 17 , 44 Following myocardial infarction, resident cardiac macrophages are diminished as a large influx of immature monocytes and neutrophils clear necrotic debris. 14 , 45 Proinflammatory cytokines such as TNFα, interleukin‐1β, and‐interleukin‐6 are increased early after infarction. Excessive inflammation either early or in a chronic unrestrained fashion can result in significant and irreversible consequences on myocardial structure and function. 14 , 30 , 31 Timely resolution of the inflammatory response is therefore critical in prevention of adverse remodeling, with recruitment of reparative macrophages and release of inhibitory mediators such as TGF‐β, interleukin‐10, and interleukin‐4. 14 , 30 , 31 , 45 In the present study, administration of a single dose of infliximab at the time of reperfusion altered the cellular profile of the infarct bed, with significantly increased activity of inhibitory cytokines, and a resultant cardioprotective effect. Notably, despite infliximab administration, no difference in TNFα protein concentration in plasma could be measured at day 3 as a percentage of baseline. This phenomenon has been reported previously, given that TNFα is antibody bound and not removed from circulation. 46 , 47
Therefore, it appears that the immune system, with pharmacologic restraint of inflammation, may provide regenerative influence following injury. 26 , 48 , 49 This notion not only challenges the paradigm that the myocardium has no regenerative reserve but also challenges the assumption that regeneration is perpetuated only by endogenous cells within the heart. 50 Injury models in zebrafish have suggested a role for endocardial and epicardial regeneration following the initial inflammatory response to injury, by creating a scaffold by which to support the myocardium in the regenerative process. 51 , 52 Mounting evidence now points to immunomodulatory monocytes as well, and a shift toward M2 macrophages, as critical to creating a microenvironment in which regeneration following injury can ensue. 16 , 29 , 45 , 53
Previous preclinical studies have shown increased expression of TNFα on single‐cell sequencing following MI in murine models, in tandem with the impact of targeted immune cell modulation on infarct size. 54 , 55 Recently, there has been increased interest in the use of TNFα inhibition in mitigation of myocardial injury. A study by Zheng et al 56 investigated the use of both a novel TNFα inhibitor and infliximab, administered both intravenously and via local injection to infarcted myocardium, 3 days after injury in a rat model of MI via suture ligation. At ≈1 month following infarction, preservation of cardiac function and reduced scar size were noted in the treated cohort compared with untreated controls. Local intramyocardial injection proved more cardioprotective compared with intravenous infusion, which may have important clinical implications for patients presenting with acute MI requiring emergency coronary artery bypass grafting. Regardless, systemic treatment with infliximab appears to be cardioprotective in preclinical models of MI. Infliximab has also been successfully used in other porcine models of TNFα‐driven disease, strengthening the supposition for cross‐species reactivity with this medication. 57
In the present study, evaluation of late‐phase porcine MI revealed an increase in anterior diastolic wall thickness, preservation of LVEF, and reduced scar size in infliximab‐treated pigs compared with sham infusion. Review of scar pattern revealed the greatest improvement at the base, with persistence of apical injury in infliximab‐treated pigs. This perhaps indicates lesser anti‐inflammatory effect from infliximab in the absence of robust collateral circulation and anoxia‐driven mechanism of injury at the apex. Blood sampling from the great cardiac vein 3 days after injury revealed significantly lower cardiac‐specific troponin I in infliximab‐treated pigs compared with control. Reported values for cardiac‐specific troponin I in porcine models vary considerably in the literature depending on study methods, sample collection, and assay used. One study with a similar model of MI in mini pigs reported a cardiac troponin I of 4.6 pg/mL in control pigs with no MI and 388 pg/mL 24 hours after MI in (untreated) pigs. 58 Another study assessing cardiac biomarkers in a porcine LAD stenosis model reported peak troponin concentrations at 18 hours following injury. 59 The troponin levels in the present study were collected on day 3 following MI, and therefore do not represent peak concentrations.
This study is strengthened by the nature of the porcine model, given known similarities in coronary anatomy and cardiac pathophysiology between pigs and humans. 40 In addition, our ischemia–reperfusion approach mimics that of the clinical presentation for MI, and therapeutic delivery at the time of reperfusion is easily achievable. Of note, the bioavailability of infliximab would not be altered with intracoronary delivery versus intravenous, and the duration of infusion would require prolonged intracoronary access, imposing undue risk to the patient. Endomyocardial delivery in the setting of STEMI is also not viable due to the higher proclivity of the myocardium to rupture with manipulation.
The work presented herein performed proteomics analysis of coronary aspirate samples taken from patients during the treatment of STEMI. High‐throughput profiling of these samples segregated on the basis of 1‐year clinical follow‐up revealed that the acute proinflammatory and antiangiogenic response to injury was associated with poorer long‐term outcomes following MI. The 19 differentially regulated proteins in vulnerable versus protected cohorts point to significant alterations in immune cell chemotaxis, including eosinophils, neutrophils, macrophages, and T‐cell behavior. Following infliximab, coronary sinus sampling notably corrected these programs toward the protected phenotype with particular impact notable on eotaxin and neutrophil activating peptide‐2 (NAP2) influencing cell chemotaxis, transforming growth factor influencing T‐cell behavior, interleukin‐1–mediated alteration of macrophage behavior, and induction of proangiogenic events. This altered expression profile in infliximab‐treated cohorts, served to preemptively activate the regenerative program at the time of injury to prevent cellular apoptosis. In this way, contractility was preserved, and the heart set along a trajectory away from adverse remodeling. Indeed, the early post‐MI death of the 5 vulnerable cohort patients may also have an association with increased TNFα concentration and subsequent disruption of calcium regulation/risk of cardiac arrhythmia. Use of existing immunomodulatory medication (such as infliximab) may provide a strategy, beyond door‐to‐balloon times, to limit injury and promote myocardial recovery at the time of STEMI. The ability to integrate infliximab infusion without disruption of door‐to‐balloon time algorithms make this approach potentially feasible. The data presented herein serve as proof of concept warranting further preclinical exploration and clinical testing of this paradigm.
Limitations
While there is considerable consistency in coronary anatomy across pigs, there are inherent challenges in creation of an MI with the goal of maintaining survival and limiting life‐threatening arrhythmia. As a result, some variation in the optimal location for balloon inflation within the LAD and infarction size between animals is to be anticipated. The location of the first diagonal branch is used to help maintain consistency and to create a sizable infarction while preserving the animal. In addition, reported values for cardiac troponin I in pigs vary considerably in the literature. Cardiac troponin I levels in the present study were obtained at day 3 after MI and therefore do not represent peak concentration. This was a procedural limitation imposed by our institutional animal care and use committee to afford animals adequate recovery time following MI. Given the consistency in TNFα concentration as measured on ELISA in infliximab‐treated and untreated cohorts, it is not possible to definitively confirm attenuation of TNFα activity as the mechanism for the results attained in the study. Furthermore, the missingness of a diversity of macrophage antibodies in the porcine model precluded validation of macrophage polarization toward the M2 state beyond CD163/25F9. Infliximab was tested in the porcine MI model given the association with TNFα seen in vulnerable patient coronary aspirates and resulted in preservation of myocardial structure and function. This conclusion is strengthened by other preclinical studies demonstrating and documenting the role of TNFα in acute MI. 54 , 55 , 56 While an 87% overlap exists in sequence identities between human and porcine TNFα, there remain some inherent discrepancies. Importantly, the binding region of infliximab carries significant overlap following ChimeraX superposition. Infliximab has successfully been used in other porcine models of TNFα‐driven disease, supporting cross‐species reactivity. The data presented in this study are associative but compelling for a positive response to infliximab‐mediated immune modulation. A need remains for further studies detailing and confirming the precise mechanisms of action.
Sources of Funding
This work was supported by the Van Cleve Cardiac Regenerative Medicine Program, the Marriott Family Foundation, and the National Institutes of Health (R01 HL134664; T32 HL07111).
Disclosures
Drs Livia, Behfar, Terzic, and Stalboerger and Mayo Clinic have financial interest in Rion Inc., which is unrelated to this manuscript. The remaining authors have no disclosures to report.
Supporting information
Tables S1–S2
Figures S1–S9
Videos S1
Video S1
Videos SS3
This manuscript was sent to Hani Jneid, MD, Associate Editor, for review by expert referees, editorial decision, and final disposition.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.123.032172
For Sources of Funding and Disclosures, see page 15.
References
- 1. Menees DS, Peterson ED, Wang Y, Curtis JP, Messenger JC, Rumsfeld JS, Gurm HS. Door‐to‐balloon time and mortality among patients undergoing primary PCI. N Engl J Med. 2013;369:901–909. doi: 10.1056/NEJMoa1208200 [DOI] [PubMed] [Google Scholar]
- 2. Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, et al. Heart disease and stroke statistics—2017 update: a report from the American Heart Association. Circulation. 2017;135:e146–e603. doi: 10.1161/CIR.0000000000000485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Singh M, Rihal CS, Lennon RJ, Garratt KN, Holmes DR Jr. Comparison of Mayo Clinic risk score and American College of Cardiology/American Heart Association lesion classification in the prediction of adverse cardiovascular outcome following percutaneous coronary interventions. J Am Coll Cardiol. 2004;44:357–361. doi: 10.1016/j.jacc.2004.03.059 [DOI] [PubMed] [Google Scholar]
- 4. Singh M, Gersh BJ, Li S, Rumsfeld JS, Spertus JA, O'Brien SM, Suri RM, Peterson ED. Mayo Clinic risk score for percutaneous coronary intervention predicts in‐hospital mortality in patients undergoing coronary artery bypass graft surgery. Circulation. 2008;117:356–362. doi: 10.1161/CIRCULATIONAHA.107.711523 [DOI] [PubMed] [Google Scholar]
- 5. Lewis EF, Moye LA, Rouleau JL, Sacks FM, Arnold JM, Warnica JW, Flaker GC, Braunwald E, Pfeffer MA. Predictors of late development of heart failure in stable survivors of myocardial infarction: the CARE study. J Am Coll Cardiol. 2003;42:1446–1453. doi: 10.1016/S0735-1097(03)01057-X [DOI] [PubMed] [Google Scholar]
- 6. Donoghue ML, Morrow DA, Cannon CP, Jarolim P, Desai NR, Sherwood MW, Murphy SA, Gerszten RE, Sabatine MS. Multimarker risk stratification in patients with acute myocardial infarction. J Am Heart Assoc. 2016;5:e002586. doi: 10.1161/JAHA.115.002586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vasan RS. Biomarkers of cardiovascular disease. Circulation. 2006;113:2335–2362. doi: 10.1161/CIRCULATIONAHA.104.482570 [DOI] [PubMed] [Google Scholar]
- 8. Wang TJ, Gona P, Larson MG, Tofler GH, Levy D, Newton‐Cheh C, Jacques PF, Rifai N, Selhub J, Robins SJ, et al. Multiple biomarkers for the prediction of first major cardiovascular events and death. N Engl J Med. 2006;355:2631–2639. doi: 10.1056/NEJMoa055373 [DOI] [PubMed] [Google Scholar]
- 9. Chia S, Senatore F, Raffel OC, Lee H, Wackers FJT, Jang I‐K. Utility of cardiac biomarkers in predicting infarct size, left ventricular function, and clinical outcome after primary percutaneous coronary intervention for ST‐segment elevation myocardial infarction. J Am Coll Cardiol Intv. 2008;1:415–423. doi: 10.1016/j.jcin.2008.04.010 [DOI] [PubMed] [Google Scholar]
- 10. Zethelius B, Berglund L, Sundstrom J, Ingelsson E, Basu S, Larsson A, Venge P, Arnlov J. Use of multiple biomarkers to improve the prediction of death from cardiovascular causes. N Engl J Med. 2008;358:2107–2116. doi: 10.1056/NEJMoa0707064 [DOI] [PubMed] [Google Scholar]
- 11. Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. 2000;101:2981–2988. doi: 10.1161/01.CIR.101.25.2981 [DOI] [PubMed] [Google Scholar]
- 12. Chapman RE, Spinale FG. Extracellular protease activation and unraveling of the myocardial interstitium: critical steps toward clinical applications. Am J Physiol Heart Circ Physiol. 2004;286:H1–h10. doi: 10.1152/ajpheart.00609.2003 [DOI] [PubMed] [Google Scholar]
- 13. Fertin M, Dubois E, Belliard A, Amouyel P, Pinet F, Bauters C. Usefulness of circulating biomarkers for the prediction of left ventricular remodeling after myocardial infarction. Am J Cardiol. 2012;110:277–283. doi: 10.1016/j.amjcard.2012.02.069 [DOI] [PubMed] [Google Scholar]
- 14. Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159–173. doi: 10.1161/CIRCRESAHA.111.243162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mann DL. The emerging role of innate immunity in the heart and vascular system: for whom the cell tolls. Circ Res. 2011;108:1133–1145. doi: 10.1161/CIRCRESAHA.110.226936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Forbes SJ, Rosenthal N. Preparing the ground for tissue regeneration: from mechanism to therapy. Nat Med. 2014;20:857–869. doi: 10.1038/nm.3653 [DOI] [PubMed] [Google Scholar]
- 17. Mortensen RM. Immune cell modulation of cardiac remodeling. Circulation. 2012;125:1597–1600. doi: 10.1161/CIRCULATIONAHA.112.097832 [DOI] [PubMed] [Google Scholar]
- 18. Epelman S, Liu PP, Mann DL. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat Rev Immunol. 2015;15:117–129. doi: 10.1038/nri3800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J, Yamamoto T, Takeshima A, Shinmura K, Shen W, et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol. 2013;62:24–35. doi: 10.1016/j.yjmcc.2013.04.023 [DOI] [PubMed] [Google Scholar]
- 20. Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D, Dieli F, Ghisletti S, Natoli G, De Baetselier P, et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc Natl Acad Sci U S A. 2009;106:14978–14983. doi: 10.1073/pnas.0809784106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ben‐Mordechai T, Holbova R, Landa‐Rouben N, Harel‐Adar T, Feinberg MS, Abd Elrahman I, Blum G, Epstein FH, Silman Z, Cohen S, et al. Macrophage subpopulations are essential for infarct repair with and without stem cell therapy. J Am Coll Cardiol. 2013;62:1890–1901. doi: 10.1016/j.jacc.2013.07.057 [DOI] [PubMed] [Google Scholar]
- 22. Gombozhapova A, Rogovskaya Y, Shurupov V, Rebenkova M, Kzhyshkowska J, Popov SV, Karpov RS, Ryabov V. Macrophage activation and polarization in post‐infarction cardiac remodeling. J Biomed Sci. 2017;24:13. doi: 10.1186/s12929-017-0322-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel‐Duby R, Sadek HA, Olson EN. Macrophages are required for neonatal heart regeneration. J Clin Invest. 2014;124:1382–1392. doi: 10.1172/JCI72181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fujiu K, Wang J, Nagai R. Cardioprotective function of cardiac macrophages. Cardiovasc Res. 2014;102:232–239. doi: 10.1093/cvr/cvu059 [DOI] [PubMed] [Google Scholar]
- 26. Hulsmans M, Sam F, Nahrendorf M. Monocyte and macrophage contributions to cardiac remodeling. J Mol Cell Cardiol. 2016;93:149–155. doi: 10.1016/j.yjmcc.2015.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ruparelia N, Godec J, Lee R, Chai JT, Dall'Armellina E, McAndrew D, Digby JE, Forfar JC, Prendergast BD, Kharbanda RK, et al. Acute myocardial infarction activates distinct inflammation and proliferation pathways in circulating monocytes, prior to recruitment, and identified through conserved transcriptional responses in mice and humans. Eur Heart J. 2015;36:1923–1934. doi: 10.1093/eurheartj/ehv195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and Postmyocardial infarction remodeling. Circ Res. 2004;94:1543–1553. doi: 10.1161/01.RES.0000130526.20854.fa [DOI] [PubMed] [Google Scholar]
- 29. Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121:2437–2445. doi: 10.1161/CIRCULATIONAHA.109.916346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shintani Y, Ito T, Fields L, Shiraishi M, Ichihara Y, Sato N, Podaru M, Kainuma S, Tanaka H, Suzuki K. IL‐4 as a repurposed biological drug for myocardial infarction through augmentation of reparative cardiac macrophages: proof‐of‐concept data in mice. Sci Rep. 2017;7:6877. doi: 10.1038/s41598-017-07328-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jung M, Ma Y, Iyer RP, DeLeon‐Pennell KY, Yabluchanskiy A, Garrett MR, Lindsey ML. IL‐10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation. Basic Res Cardiol. 2017;112:33. doi: 10.1007/s00395-017-0622-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Singh M, Lennon RJ, Holmes DR Jr, Bell MR, Rihal CS. Correlates of procedural complications and a simple integer risk score for percutaneous coronary intervention. J Am Coll Cardiol. 2002;387‐393:387–393. doi: 10.1016/S0735-1097(02)01980-0 [DOI] [PubMed] [Google Scholar]
- 33. Gara PT, Kushner FG, Ascheim DD, Casey DE, Chung MK, de Lemos JA, Ettinger SM, Fang JC, Fesmire FM, Franklin BA, et al. 2013 ACCF/AHA guideline for the management of ST‐elevation myocardial infarction. Circulation. 2012;61:e78–e140. doi: 10.1016/j.jacc.2012.11.019 [DOI] [PubMed] [Google Scholar]
- 34. Smoot ME, Ono K, Ruscheinski J, Wang P‐L, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Arrell DK, Niederlander NJ, Faustino RS, Behfar A, Terzic A. Cardioinductive network guiding stem cell differentiation revealed by proteomic cartography of tumor necrosis factor alpha‐primed endodermal secretome. Stem Cells. 2008;26:387–400. doi: 10.1634/stemcells.2007-0599 [DOI] [PubMed] [Google Scholar]
- 36. Behfar A, Perez‐Terzic C, Faustino RS, Arrell DK, Hodgson DM, Yamada S, Puceat M, Niederlander N, Alekseev AE, Zingman LV, et al. Cardiopoietic programming of embryonic stem cells for tumor‐free heart repair. J Exp Med. 2007;204:405–420. doi: 10.1084/jem.20061916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arrell DK, Zlatkovic J, Kane GC, Yamada S, Terzic A. ATP‐sensitive K+ channel knockout induces cardiac proteome remodeling predictive of heart disease susceptibility. J Proteome Res. 2009;8:4823–4834. doi: 10.1021/pr900561g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zlatkovic‐Lindor J, Arrell DK, Yamada S, Nelson TJ, Terzic A. ATP‐sensitive K(+) channel‐deficient dilated cardiomyopathy proteome remodeled by embryonic stem cell therapy. Stem Cells. 2010;28:1355–1367. doi: 10.1002/stem.465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U, et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLoS Biol. 2020;14:e3000410. doi: 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Weaver ME, Pantely GA, Bristow JD, Ladley HD. A quantitative study of the anatomy and distribution of coronary arteries in swine in comparison with other animals and man. Cardiovasc Res. 1986;20:907–917. doi: 10.1093/cvr/20.12.907 [DOI] [PubMed] [Google Scholar]
- 41. Liang S, Dai J, Hou S, Su L, Zhang D, Guo H, Hu S, Wang H, Rao Z, Guo Y, et al. Structural basis for treating tumor necrosis factor α (TNFα)‐associated diseases with the therapeutic antibody infliximab. J Biol Chem. 2013;288:13799–13807. doi: 10.1074/jbc.M112.433961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Meng EC, Goddard TD, Pettersen EF, Couch GS, Pearson ZJ, Morris JH, Ferrin TE. UCSF ChimeraX: tools for structure building and analysis. Protein Sci. 2023;32:e4792. doi: 10.1002/pro.4792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li X, Martinez‐Fernandez A, Hartjes KA, Kocher JP, Olson TM, Terzic A, Nelson TJ. Transcriptional atlas of cardiogenesis maps congenital heart disease interactome. Physiol Genomics. 2014;46:482–495. doi: 10.1152/physiolgenomics.00015.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hausenloy DJ, Yellon DM. Myocardial ischemia‐reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123:92–100. doi: 10.1172/JCI62874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sager HB, Kessler T, Schunkert H. Monocytes and macrophages in cardiac injury and repair. J Thorac Dis. 2017;9:S30–S35. doi: 10.21037/jtd.2016.11.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marotte H, Maslinski W, Miossec P. Circulating tumour necrosis factor‐α bioactivity in rheumatoid arthritis patients treated with infliximab: link to clinical response. Arthritis Res Ther. 2005;7:R149–R155. doi: 10.1186/ar1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Amini Kadijani A, Asadzadeh Aghdaei H, Sorrentino D, Mirzaei A, Shahrokh S, Balaii H, Nguyen VQ, Mays JL, Reza ZM. Transmembrane TNF‐alpha density, but not soluble TNF‐alpha level, is associated with primary response to infliximab in inflammatory bowel disease. Clin Transl Gastroenterol. 2017;8:e117. doi: 10.1038/ctg.2017.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Frangogiannis NG. The immune system and the remodeling infarcted heart: cell biological insights and therapeutic opportunities. J Cardiovasc Pharmacol. 2014;63:185–195. doi: 10.1097/FJC.0000000000000003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sattler S, Fairchild P, Watt FM, Rosenthal N, Harding SE. The adaptive immune response to cardiac injury—the true roadblock to effective regenerative therapies? NPJ Reg Med. 2017;2:19. doi: 10.1038/s41536-017-0022-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cahill TJ, Choudhury RP, Riley PR. Heart regeneration and repair after myocardial infarction: translational opportunities for novel therapeutics. Nat Rev Drug Discov. 2017;16:699–717. doi: 10.1038/nrd.2017.106 [DOI] [PubMed] [Google Scholar]
- 51. Guo QY, Yang JQ, Feng XX, Zhou YJ. Regeneration of the heart: from molecular mechanisms to clinical therapeutics. Mil Med Res. 2023;10:18. doi: 10.1186/s40779-023-00452-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Godwin JW, Debuque R, Salimova E, Rosenthal NA. Heart regeneration in the salamander relies on macrophage‐mediated control of fibroblast activation and the extracellular landscape. NPJ Reg Med. 2017;2:22. doi: 10.1038/s41536-017-0027-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. González‐Rosa JM, Burns CE, Burns CG. Zebrafish heart regeneration: 15 years of discoveries. Regeneration (Oxf). 2017;4:105–123. doi: 10.1002/reg2.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vafadarnejad E, Rizzo G, Krampert L, Arampatzi P, Arias‐Loza AP, Nazzal Y, Rizakou A, Knochenhauer T, Bandi SR, Nugroho VA, et al. Dynamics of cardiac neutrophil diversity in murine myocardial infarction. Circ Res. 2020;127:e232–e249. doi: 10.1161/CIRCRESAHA.120.317200 [DOI] [PubMed] [Google Scholar]
- 55. Zhuang L, Wang Y, Chen Z, Li Z, Wang Z, Jia K, Zhao J, Zhang H, Xie H, Lu L, et al. Global characteristics and dynamics of single immune cells after myocardial infarction. J Am Heart Assoc. 2022;11:e027228. doi: 10.1161/JAHA.122.027228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zheng W, Zhou T, Zhang Y, Ding J, Xie J, Wang S, Wang Z, Wang K, Shen L, Zhu Y, et al. Simplified α2‐macroglobulin as a TNF‐α inhibitor for inflammation alleviation in osteoarthritis and myocardial infarction therapy. Biomaterials. 2023;301:1–12. doi: 10.1016/j.biomaterials.2023.122247 [DOI] [PubMed] [Google Scholar]
- 57. Martínez‐Fernández de la Cámara C, Olivares‐González L, Hervás D, Salom D, Millán JM, Rodrigo R. Infliximab reduces Zaprinast‐induced retinal degeneration in cultures of porcine retina. J Neuroinflammation. 2014;172:1–14. doi: 10.1186/s12974-014-0172-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hou C, Lin C, Wu C, Tsung L, Lin M, Chen Y, Chang S, Hsu C, Liao C, Shih Y, et al. Cardiac function evaluation of bone marrow mesenchymal stromal cells intracoronary transplantation in acute myocardial infarction. Lee‐Sung Mini‐PIG Model. Cytotherapy. 2020;22:S75. doi: 10.1016/j.jcyt.2020.03.119 [DOI] [Google Scholar]
- 59. Feng YJ, Chen C, Fallon JT, Lai T, Chen L, Knibbs DR, Waters DD, Wu AH. Comparison of cardiac troponin I, creatine kinase‐MB, and myoglobin for detection of acute ischemic myocardial injury in a swine model. Am J Clin Pathol. 1998;110:70–77. doi: 10.1093/ajcp/110.1.70 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S2
Figures S1–S9
Videos S1
Video S1
Videos SS3