

Abstract
Forkhead box P3 (FOXP3) is the master fate-determining transcription factor in regulatory T (Treg) cells and is essential for their development, function, and homeostasis. Mutations in FOXP3 cause immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome, and aberrant expression of FOXP3 has been implicated in other diseases such as multiple sclerosis and cancer. We previously demonstrated that pre-mRNA splicing of FOXP3 RNAs is highly sensitive to levels of DExD-box polypeptide 39B (DDX39B), and here we investigate the mechanism of this sensitivity. FOXP3 introns have cytidine (C)-rich/uridine (U)-poor polypyrimidine (py) tracts that are responsible for their inefficient splicing and confer sensitivity to DDX39B. We show that there is a deficiency in the assembly of commitment complexes (CCs) on FOXP3 introns, which is consistent with the lower affinity of U2AF2 for C-rich/U-poor py tracts. Our data indicate an even stronger effect on the conversion of CCs to pre-spliceosomes. We propose that this is due to an altered conformation that U2AF2 adopts when it binds to C-rich/U-poor py tracts and that this conformation has a lower affinity for DDX39B. As a consequence, CCs assembled on FOXP3 introns are defective in recruiting DDX39B, and this leads to the inefficient assembly of pre-spliceosome complexes.
Keywords: pre-mRNA splicing, DDX39B, FOXP3, autoimmunity, IPEX syndrome, multiple sclerosis
INTRODUCTION
DExD-box polypeptide 39B (DDX39B), an ortholog of Sub2 in Saccharomyces cerevisiae, is an ATP-dependent RNA helicase with various roles in mRNA processing, including mRNA nucleocytoplasmic export (Luo et al. 2001; Sträßer et al. 2002; Masuda et al. 2005; Yamazaki et al. 2010) and pre-mRNA splicing (Shen et al. 2007, 2008). DDX39B was initially discovered by immunologists and referred to as HLA-B associated transcript 1 (BAT1) due to the genomic location of BAT1 in the major histocompatibility complex (MHC) class III region (Spies et al. 1989). Early studies demonstrated an anti-inflammatory role for DDX39B (Allcock et al. 2001), and additional observations suggest genetic associations of variants in DDX39B with autoimmune diseases such as type I diabetes, rheumatoid arthritis, and atopic dermatitis (Todd et al. 2007; Quiñones-Lombraña et al. 2008; Paternoster et al. 2012). RNA biologists independently identified DDX39B and named it U2 auxiliary factor 65-associated protein 56 kDa (UAP56), due to its association with the splicing factor U2AF65 (now known as U2AF2) (Fleckner et al. 1997). Further studies showed that DDX39B plays critical roles in the early steps of spliceosome formation (Shen et al. 2007, 2008).
The spliceosome is a multimegadalton ribonucleoprotein complex that facilitates pre-mRNA splicing, a process by which introns are removed and exons are ligated to form mature RNAs. Hundreds of factors are required for the proper formation and function of the spliceosome, including five small nuclear ribonucleoprotein complexes (snRNPs): U1, U2, U4, U5, and U6. These snRNPs are composed of uridine-rich small nuclear RNAs (U snRNAs) and their associated proteins (Lerner and Steitz 1979; Lerner et al. 1980; Kastner et al. 2019). Additional proteins or trans-acting factors not associated with snRNPs are also required and are generally referred to as splicing factors (SFs) (Gozani et al. 1994; Kohtz et al. 1994). Sequence motifs within the pre-mRNA interact, directly or indirectly, with these SFs. These motifs include the 5′ splice site (SS), branch point sequence (BPS), polypyrimidine (py) tract, and the 3′SS. The earliest steps in the assembly of splicing complexes involve the binding of the U1 snRNP to the 5′SS and binding of splicing factor 1 (SF1) and the U2 auxiliary factor (U2AF) heterodimer to the BPS and the py tract/3′SS, respectively (Mount et al. 1983; Reed and Maniatis 1985; Zamore et al. 1992; Berglund et al. 1998). These interactions, which do not require ATP, form the early (E) or commitment complex (CC) (Michaud and Reed 1991; Jamison et al. 1992). U2AF2, the larger component of the U2AF heterodimer, recruits DDX39B in an ATP-dependent fashion to displace SF1 from the BPS (Fleckner et al. 1997; Kistler and Guthrie 2001; Zhang and Green 2001) and, together with another DExD-box helicase, DDX46, allows the U2 snRNP to bind, forming the pre-spliceosome (Zhang et al. 2021). Additionally, DDX39B enhances the stability of the pre-spliceosome by interacting with U1 snRNA at the 5′SS (Martelly et al. 2021). It is important to note that the helicase activity of DDX39B is not required for pre-spliceosome formation (Shen et al. 2008). The pre-spliceosome seeds the formation of the spliceosome, in which the tri-snRNP, composed of the U4, U6, and U5 snRNPs, interacts with the pre-mRNA. For proper interaction of U4 and U6 snRNPs with the intron, the RNA duplex they form must be unwound by the helicase activity of DDX39B (Shen et al. 2008) and another helicase called SNRNP200 (also known as BRR2) (Absmeier et al. 2016). The spliceosome becomes activated upon the dissociation of U1 and U4 snRNPs from the complex, enabling two transesterification reactions to occur (for review, see Will and Lührmann 2011). Ultimately, these reactions produce a free intron, which initially is in the form of a lariat, and a spliced product with ligated exons. Thus, DDX39B acts in at least two distinct steps in spliceosome assembly: formation of the pre-spliceosome and formation of the spliceosome.
Given the importance of DDX39B in both splicing and immunoregulation, our group sought to link these two roles. First, we demonstrated that DDX39B activates the inclusion of exon 6 of interleukin-7 receptor (IL7R) (Galarza-Muñoz et al. 2017), which is critical for T cell homeostasis. Exon 6 of IL7R encodes for the transmembrane domain of the protein, therefore its inclusion leads to the production of membrane-bound IL7R, whereas its exclusion leads to production of a soluble form of IL7R (sIL7R), which has been associated with increased risk of an autoimmune disease called multiple sclerosis (MS) (Lundström et al. 2013). Furthermore, we showed genetic epistasis between a single-nucleotide polymorphism (SNP) in the 5′ untranslated region (UTR) of DDX39B, rs2523506, and an SNP in exon 6 of IL7R, rs6897932. The population homozygous for the risk allele of both SNPs has an approximately threefold increased risk of developing MS than those homozygous for the non-risk allele of both SNPs (Galarza-Muñoz et al. 2017). Recently, we showed that DDX39B is essential for proper expression and function of Forkhead box P3 (FOXP3) (Hirano et al. 2023), which is critical for proper development, function, and homeostasis of regulatory T (Treg) cells (Georgiev et al. 2019). We demonstrated that efficient removal of FOXP3 introns was sensitive to DDX39B levels, and specifically required its ATP-binding and ATPase activity but not its helicase activity (Hirano et al. 2023). These findings suggest that DDX39B promotes FOXP3 splicing during the transition of CC to pre-spliceosome complex assembly. Furthermore, nine of 11 FOXP3 introns have cytidine (C)-rich/uridine (U)-poor py tracts, and therefore the effect of DDX39B on FOXP3 splicing is amplified in total transcript production (Hirano et al. 2023).
Building on our recently published work, our aim here is to elucidate the mechanistic basis underlying the exquisite dependence of FOXP3 introns on DDX39B. We show that there is a deficiency in the assembly of CCs on FOXP3 introns, which is consistent with the lower affinity of U2AF2 for C-rich/U-poor py tracts. Our data indicate an even stronger effect on the conversion of CCs to pre-spliceosomes. We propose that this is due to an altered conformation that U2AF2 adopts when it binds to C-rich/U-poor py tracts and that this conformation has a lower affinity for DDX39B. Consequently, CCs assembled on FOXP3 introns are defective in recruiting DDX39B and ultimately lead to the inefficient assembly of pre-spliceosome complexes. Thus, FOXP3 splicing, expression, and function are all diminished, potentially impacting Treg cell function and having implications in autoimmune disorders.
RESULTS
C-rich/U-poor polypyrimidine tracts of FOXP3 introns are responsible for their inefficient splicing
To study the mechanism underpinning the inefficient splicing of FOXP3 introns (Hirano et al. 2023), we aimed to recapitulate this effect in vitro using two different sets of splicing substrates. The first set of substrates were based on PIP7A, a construct that has been shown to splice efficiently in an in vitro system (Fig. 1A; Gil et al. 1991). We replaced its py tract sequence (20 nt upstream of the 3′SS) with that of FOXP3 intron 11 (PIP7A-F3i11wt) or with a U-rich mutant, which we name PIP7A-F3i11pU (Fig. 1A). We incubated these uniformly radiolabeled splicing substrates with HeLa nuclear extracts in the absence or presence of ATP and creatine phosphate (CP) for 1 h. RNA from these in vitro splicing (IVS) reactions were isolated and loaded onto a 15% denaturing gel to visualize the splicing intermediates. The percentage (%) spliced (also referred to as splicing efficiency) in the presence of ATP/CP for each substrate was quantified in three independent experiments, using three different HeLa nuclear extract preparations. The splicing efficiency of the PIP7A splicing was 45%, for PIP7A-F3i11pU it was 47%, but for PIP7A-F3i11wt it was 8% (Fig. 1B). Furthermore, we conducted a time course of IVS for PIP7A-F3i11wt and PIP7A-F3i11pU to determine their splicing kinetics. At 15 min of incubation with nuclear extract and ATP/CP, we did not observe splicing products with either substrate (Fig. 1C; Supplemental Fig. S1); however, after 30 min, PIP7A-F3i11pU spliced with an efficiency of 34%, while PIP7A-F3i11wt remained unspliced (Fig. 1C). By 45 min, PIP7A-F3i11wt began to splice, albeit much less efficiently than PIP7A-F3i11pU. The splicing of both substrates continued to increase at 60 and 90 min; however, at both time points, PIP7A-F3i11wt splicing was approximately two times less efficient than the splicing of PIP7A-F3i11pU (Fig. 1C).
FIGURE 1.

C-rich/U-poor polypyrimidine tract of FOXP3 intron 11 weakens the splicing efficiency of a robust splicing substrate. (A) Schematic diagram of PIP7A-based splicing substrates. The sequence of all three constructs remains the same except the PIP7A py tract sequence (20 nt upstream of the 3′SS) is replaced with that of FOXP3 intron 11 (PIP7A-F3i11wt) or a U-rich mutant (PIP7A-F3i11pU) (created with BioRender). (B) IVS of PIP7A-based splicing substrates. The first lane is the ladder (L) with its respective sizes on the left. Depicted on the gel are splicing intermediates from top to bottom: (*) represents a by-product of T7 RNA synthesis; unspliced substrate; intron lariat intermediate; (**) represents a by-product of T7 RNA synthesis; spliced product. Quantification of % spliced is shown on the right and calculated as (spliced/[spliced + unspliced]) × 100. Each bar represents the average ± SD of three independent experiments using three different nuclear extract preparations; (*) P < 0.05, Student's t-test. (C) IVS time course of PIP7A-F3i11wt and PIP7A-F3i11pU substrates. Depicted on the gel are splicing intermediates from top to bottom: intron lariat intermediate; intron lariat; unspliced substrate; and spliced product.
The second set of substrates used for IVS was derived from the FOXP3 sequence. These RNAs encompassed the last 10 nt of FOXP3 exon 11, followed by intron 11 with either the wt or U-rich mutant py tract sequence and the first 20 nt of exon 12, and referred to as F3i11wt-IVS and F3i11pU-IVS, respectively. The splicing efficiency of these RNAs was visualized via end point RT-PCR after 90 min of splicing incubation. In the presence of ATP/CP, there was almost no detectable splicing of F3i11wt-IVS, while splicing of F3i11pU-IVS was robust with three different nuclear extracts (Supplemental Fig. S2). Taken together, these data demonstrate that the IVS system recapitulates what we have previously shown in vivo, which is that the C-rich/U-poor py tract of FOXP3 intron 11 hinders its ability to splice efficiently. Furthermore, the in vitro results support the conclusion that the effect of the py tract was directly on splicing and not an indirect effect on transport or stability (Hirano et al. 2023).
Inefficient assembly of commitment complexes on FOXP3 intron 11
We recently demonstrated that complementation of FOXP3 intron splicing in DDX39B-depleted cells requires expression of DDX39B with wild-type (wt) ATP-binding and ATPase activity, but does not require helicase activity (Hirano et al. 2023). This suggests that the DDX39B-dependence of FOXP3 introns is likely due to a barrier in pre-spliceosome formation, as opposed to a barrier in later steps in spliceosome assembly. To determine how DDX39B facilitates the assembly of early splicing complexes on FOXP3 introns, we generated RNAs similar to the F3i11-IVS substrates but with a tobramycin aptamer at the end of exon 12 (F3i11wt-tobra and F3i11pU-tobra, respectively, in Fig. 2A). We first assessed whether these tobramycin aptamer-containing constructs recapitulated sensitivity to DDX39B. To do so, we transfected these constructs in HeLa cells treated with a nonsilencing control (siNSC) or a DDX39B-targeted (siDDX39B) siRNA (Fig. 2B), and then measured % spliced of FOXP3 intron 11 via end point RT-PCR (Fig. 2C). The splicing efficiency of F3i11wt-tobra was 19% and decreased to 11% upon DDX39B depletion (Fig. 2C; P < 0.0001, Student's t-test). On the contrary, F3i11pU-tobra was fully spliced in cells treated with either siNSC or siDDX39B and thus resulted in 100% splicing efficiency (Fig. 2C). Together these data recapitulate our previous observations in vivo with other FOXP3 intron 11 splicing reporters (Hirano et al. 2023) and validate the use of these tobramycin aptamer-containing RNAs to study why FOXP3 introns are inefficiently spliced and very sensitive to DDX39B expression.
FIGURE 2.

Splicing of a FOXP3 intron 11 tobramycin aptamer construct is sensitive to DDX39B. (A) Schematic diagram of FOXP3 intron 11-tobramycin constructs, which encompass the last 10 nt of exon 11, intron 11 wt (F3i11wt-tobra) or U-rich mutant (F3i11pU-tobra), first 20 nt of exon 12, followed by the tobramycin aptamer sequence. Highlighted in pink are nucleotide changes made to F3i11wt-tobra to create a stretch of 13 consecutive Us in py tract of F3i11pU-tobra. Black arrows represent the location of primers used for end point PCR to quantify splicing efficiency (created with BioRender). (B) Representative western blot of siRNA-mediated depletion of DDX39B (siDDX39B) compared to siNSC. β-Actin was used as a loading control. (C) End point PCR analysis of FOXP3 intron splicing from the F3i11wt-tobra and F3i11pU-tobra constructs in HeLa cells treated with siNSC or siDDX39B. Quantification of % splicing of F3i11wt-tobra (calculated as (spliced/[spliced + unspliced]) × 100) represents the average ± SD of nine biological replicates from three independent experiments; (****) P < 0.0001, Student's t-test.
To identify the differences in splicing complex formation on F3i11wt-tobra and F3i11pU-tobra RNAs, we incubated them with HeLa nuclear extracts for 30 min ± ATP/CP and then added tobramycin to elute the RNA and its interacting partners (Hartmuth et al. 2004). We analyzed the eluates using mass spectrometry to determine the protein abundance (refer to Materials and Methods; Supplemental Fig. S3) for each of four conditions: (i) F3i11wt-tobra without ATP/CP; (ii) F3i11pU-tobra without ATP/CP; (iii) F3i11wt-tobra with ATP/CP; and (iv) F3i11pU-tobra with ATP/CP. Each condition had four replicates that were derived from two independent experiments.
The initial splicing complex that assembles on pre-mRNA is the CC (also known as E complex) (Fig. 3A), and its formation does not require ATP. To assess the differences in CC formation on F3i11wt-tobra and F3i11pU-tobra, we compared the abundance of CC protein components that interacted with these two RNAs. U1 snRNP components, U1A, U1C, and U1-70K, were ∼50% lower in eluates from F3i11wt-tobra compared to F3i11pU-tobra (Fig. 3B; Supplemental Fig. S4). The abundance of proteins that bind the 3′ end of the intron, SF1, U2AF1, and U2AF2, were approximately three- to fourfold lower in F3i11wt-tobra eluates compared to those from F3i11pU-tobra (Fig. 3C; Supplemental Fig. S4).
FIGURE 3.

Inefficient assembly of CCs on FOXP3 intron 11. (A) Diagram of a CC assembly on pre-mRNA. The splice site motifs are recognized by the U1 snRNP, SF1, and U2AF heterodimers, respectively, forming the CC (created with BioRender). (B) Abundance of U1 snRNP components on F3i11wt-tobra and F3i11pU-tobra (see Materials and Methods for the calculation of abundance). (C) Abundance of SF1 and U2AF heterodimers on F3i11wt-tobra and F3i11pU-tobra. (D) Abundance of U2 snRNP components on F3i11wt-tobra and F3i11pU-tobra. In B–E, all incubations were done in the absence of added ATP, and each bar graph is the median ± interquartile range of four replicates from two independent experiments; (*) P < 0.05, Mann–Whitney test.
The formation of CC does not require DExD/H-box helicases, therefore it was not surprising to observe very low levels of DDX46 and DDX39B in eluates from either substrate (Fig. 3D). It is important to note that DDX39B has a 90% identical paralog named DDX39A, which we observe binding to both constructs (Fig. 3D). Although the role of DDX39A in spliceosome formation is not well understood, data from our group suggest that it does play a role in splicing (Banerjee et al. 2024).
Additionally, the formation of CC does not require the binding of U2 snRNP; however, our group and others have shown U2 snRNP binding to pre-mRNA substrates in the absence of ATP, and the U2 snRNA base pairs with the branch point region (Jamison and Garcia-Blanco 1992; Valcarcel et al. 1996; Das et al. 2000) and in some cases, these complexes can be chased to splice (Perriman and Ares 2000). That said, the binding of U2 snRNP components in the absence of ATP/CP was 50% less on F3i11wt-tobra compared to F3i11pU-tobra (Fig. 3E; Supplemental Fig. S4). Together these data illustrate that CC, as measured primarily by the interactions with U1 snRNP, SF1 and U2AF heterodimer, assembles on FOXP3 introns less efficiently (∼50%) than on variants with U-rich py tracts.
Formation of the pre-spliceosome complex is stalled on FOXP3 introns
The transition from commitment to pre-spliceosome complex formation depends on ATP hydrolysis and requires DDX46, DDX39B, and perhaps DDX39A (Fig. 4A). To determine differences in pre-spliceosome assembly on F3i11wt-tobra and F3i11pU-tobra, we compared the abundance of proteins involved in pre-spliceosome complex (also known as A complex) assembly on both constructs in the presence of ATP/CP. The abundance of U1 snRNP components—U1A, U1C, and U1-70K were less on F3i11wt-tobra compared to its F3i11pU-tobra counterpart (Fig. 4B; Supplemental Fig. S4; P < 0.05, Mann–Whitney test for U1A and U1-70K but did not reach significance for U1C [P = 0.0571]). Furthermore, the abundance of proteins that bind the 3′ end of the intron, SF1, U2AF1, and U2AF2, were all dramatically lower on F3i11wt-tobra compared to F3i11pU-tobra (Fig. 4C; Supplemental Fig. S4). The binding of DDX46 was reduced on F3i11wt-tobra compared to F3i11pU-tobra, although this was not statistically significant, due to an outlier (Fig. 4D). There was significantly less abundance of DDX39A (P < 0.05, Mann–Whitney test) on F3i11wt-tobra compared to F3i11pU-tobra but no significant difference in the abundance of DDX39B between both constructs (Fig. 4D).
FIGURE 4.

Inefficient pre-spliceosome formation on FOXP3 intron 11. (A) Diagram of a pre-spliceosome complex. In the presence of ATP and two DExD-box helicases, DDX46 and DDX39B (DDX39A), SF1 is displaced from the BPS, allowing for U2 snRNP to bind and form the pre-spliceosome (created with BioRender). (B) Abundance of U1 snRNP components on F3i11wt-tobra and F3i11pU-tobra. (C) Abundance of SF1 and U2AF heterodimers on F3i11wt-tobra and F3i11pU-tobra. (D) Abundance of U2 snRNP components on F3i11wt-tobra and F3i11pU-tobra. In B–E, all incubations are in the presence of added ATP, and each bar graph is the median ± interquartile range of four replicates from two independent experiments; (*) P < 0.05, Mann–Whitney test.
The levels of U2 snRNP proteins found in F3i11wt-tobra eluates were ∼25% those in F3i11pU-tobra eluates (Fig. 4E; Supplemental Fig. S4). These data, along with splicing complex analysis via native gel electrophoresis that indicate a deficiency in pre-spliceosome formation on F3i11wt-tobra (Supplemental Fig. S5), demonstrate that the C-rich/U-poor py tracts of FOXP3 introns impede their ability to efficiently convert CCs to pre-spliceosome complexes. Furthermore, the difference in pre-spliceosome assembly is more pronounced than the differences observed in CC formation between F3i11wt-tobra and F3i11pU-tobra.
5′SS and py tract are necessary for recruitment of DDX39B to FOXP3 introns
Despite the marked difference (>20 times) in U2AF2 abundance between F3i11wt-tobra and F3i11pU-tobra (Fig. 4C), it was surprising to observe no statistically significant difference in the abundance of DDX39B on the two RNAs. Given the transient interactions of DDX39B with the spliceosome, we hypothesized that the reason behind this paradox could be attributed to the differences in the substrate splicing kinetics. To test this, we conducted the tobramycin RAC experiment and measured the abundance of U2AF2 and DDX39B on F3i11wt-tobra and F3i11pU-tobra over time (Fig. 5A; Supplemental Fig. S6A). At the earliest time we could measure, which we label 0 min, there was approximately threefold more binding of U2AF2 to F3i11pU-tobra than F3i11wt-tobra (Fig. 5A; Supplemental Fig. S6A). From 0 to 15 min of incubation under splicing conditions, U2AF2 abundance decreased on F3i11wt-tobra, while U2AF2 increased by approximately threefold on F3i11pU-tobra (Fig. 5A; Supplemental Fig. S6A). At 30 min of incubation, U2AF2 abundance decreased on both constructs but to a greater extent (∼90%) on F3i11wt-tobra (Fig. 5A; Supplemental Fig. S6A). At 0 min, there was little to no binding of DDX39B to either construct (Fig. 5A; Supplemental Fig. S6A). From 0 to 15 min, there was an increase of DDX39B abundance on both constructs, albeit a more robust increase on the F3i11pU-tobra construct (Fig. 5A; Supplemental Fig. S6A). At 30 min of incubation, the binding of DDX39B modestly decreased on F3i11wt-tobra while it decreased by approximately threefold on F3i11pU-tobra (Fig. 5A; Supplemental Fig. S6A). These data show that the most pronounced differences in U2AF2 and DDX39B binding between the two constructs occur at 15 min of IVS incubation. When considered alongside Figure 1C, these data suggest that differences in the splicing kinetics of the two substrates can explain why there is no difference in DDX39B but a dramatic difference in U2AF2 binding at 30 min.
FIGURE 5.

Recruitment of DDX39B to FOXP3 intron 11 requires its 5′SS and py tract. Western blot analyses of U2AF2 and DDX39B binding to (A) full-length constructs F3i11wt-tobra and F3i11pU-tobra, or (B) hemi-intron constructs F3i11wt3′end-tobra and F3i11pU3′end-tobra, after 0, 15, and 30 min of IVS incubation. Each panel represents triplicate experiments with full-length constructs or duplicate experiments with hemi-intron constructs using different nuclear extract preparations.
To further understand the influence of the py tract on its interactions with U2AF2 and DDX39B, we created hemi-intron versions of the F3i11-tobra constructs in which we deleted the 10 nt of FOXP3 exon 11 and the first 119 nt of intron 11 and refer to these RNAs as F3i11wt3′end-tobra and F3i11pU3′end-tobra (Supplemental Fig. S6B). Because these hemi-intron RNAs have no 5′SS, there should be very little binding of the U1 snRNP (as measured with U1-70K), and indeed this is what we observe (Fig. 5B; Supplemental Fig. S6C). The binding of U2AF2 to these hemi-intron RNAs is the same as observed for their full-length counterparts (Fig. 5B; Supplemental Fig. S6C). The binding of DDX39B to these substrates was ATP-dependent as observed with the full-length constructs (Fig. 5B; Supplemental Fig. S6C). Nevertheless, DDX39B bound both hemi-intron substrates equally in contrast to what was observed with the full-length substrates (Fig. 5B; Supplemental Fig. S6C). These data suggest that both the 5′SS and py tract of FOXP3 intron 11 are required for proper recruitment of DDX39B to the intron and are consistent with known ATP-dependent interactions between DDX39B and both the U1 snRNP and U2AF2 (Fleckner et al. 1997; Martelly et al. 2021).
Splicing of FOXP3 introns is sensitive to U2AF2-mediated recruitment of DDX39B
To further examine the relationship of U2AF2 and DDX39B on the splicing of FOXP3 introns, we sought to explore the importance of py tract U residues in determining how sensitive an intron is to DDX39B levels. The requirement for U residues is not evenly distributed among the length of the py tract and those in the distal half of the py tract relative to the 3′SS are most critical for U2AF2 binding (Glasser et al. 2022). This is due to the differential discrimination of the RNA recognition motifs (RRMs) of U2AF2 for non-U residues. RRM1 binds the proximal half of the py tract relative to the 3′SS, while RRM2 binds the distal half (Sickmier et al. 2006). While RRM1 binding is only modestly affected by Cs, RRM2 is more sensitive to the presence of Cs (Jenkins et al. 2013; Agrawal et al. 2014). We made a series of splicing reporters in which we mutated the distal half (distU) or proximal half (proxU) of the FOXP3 intron 11 py tract to a stretch of five consecutive U residues or both halves of the py tract with two stretches of 5 Us (polyU.v2) (Fig. 6A). The polyU.v2 splicing reporter is very similar to our previously tested polyU splicing substrate. HeLa cells were treated with either siNSC or siDDX39B siRNAs (Fig. 6B) and then transfected with the splicing reporters. As we have previously shown, depletion of DDX39B led to a significant decrease (P < 0.0001; one-way ANOVA) in splicing efficiency of FOXP3 intron 11 wt reporter (F3i11_wt) (Fig. 6C). Mutating the distal half of the py tract (F3i11_distU) increased the overall splicing efficiency to 86% and abrogated DDX39B sensitivity (Fig. 6C). On the contrary, mutating the proximal half (F3i11_proxU) increased the overall splicing efficiency of the reporter, but did not abrogate DDX39B sensitivity (Fig. 6C). Lastly, mutating both halves of the py tract (F3i11_polyU.v2) increased the splicing efficiency to 88% and abrogated DDX39B sensitivity (Fig. 6C). Together these data imply that the distal half of the py tract confers DDX39B sensitivity and is consistent with the fact that RRM2 of U2AF2 is more sensitive to the presence of Cs compared to RRM1 (Jenkins et al. 2013; Agrawal et al. 2014).
FIGURE 6.

Cytidines in the distal half of the py tract of FOXP3 intron 11 mediate its sensitivity to DDX39B. (A) Schematic diagram of FOXP3 intron 11 splicing reporters. The sequence of FOXP3 intron 11 was inserted into the open reading frame of Renilla luciferase. The sequence of all constructs remains the same except for the py tract sequence (20 nt upstream of the 3′SS). Highlighted in pink are nucleotide changes made to Rluc-F3i11_wt to create a stretch of five Us in the distal half of the py tract (Rluc-F3i11_distU), proximal half (Rluc-F3i11_proxU), or in both halves of the py tract (Rluc-F3i11_polyU.v2). Black arrows represent the location of primers used for end point PCR to quantify splicing efficiency (created with BioRender). (B) Quantification of siRNA-mediated depletion of DDX39B (siDDX39B) compared to siNSC. Quantification represents the average ± SD of nine biological replicates from three independent experiments; (****) P < 0.0001, Student's t-test. (C) End point PCR analysis of FOXP3 intron splicing from the FOXP3 intron 11 py tract mutant splicing reporters in HeLa cells treated with siNSC or siDDX39B. Quantification represents the average ± SD of nine biological replicates from three independent experiments; (****) P < 0.0001, one-way ANOVA.
DISCUSSION
We have previously demonstrated that C-rich/U-poor py tracts of FOXP3 introns cripple their ability to splice efficiently. This is consistent with work we did 30 years ago when our group showed that replacing eight consecutive Us with 13 Cs in the py tract of a splicing substrate abrogated its ability to splice and form splicing complexes in vitro (Roscigno et al. 1993). Furthermore, these C-rich/U-poor py tracts of FOXP3 introns confer sensitivity to DDX39B expression. To further understand the mechanism underlying this effect, we developed an IVS system that recapitulated what we previously reported (Figs. 1 and 2; Supplemental Figs. S1 and S2). Furthermore, our data imply that FOXP3 introns are spliced inefficiently due to a deficiency of both commitment and pre-spliceosome complex formation. All proteins that assemble on pre-mRNA to form CC were decreased, with the majority being decreased significantly, on F3i11wt-tobra compared to its U-rich counterpart, F3i11pU-tobra (Fig. 3). Given that the only difference between the two constructs is the py tract sequence, it was interesting to observe ∼50% less binding of U1 snRNP components to the F3i11wt-tobra construct (Fig. 3B). These data imply that binding of the U1 snRNP components to the 5′SS depends not only on the 5′SS sequence but also on the py tract sequence and its interactions with other trans-acting proteins (Wu and Maniatis 1993; Abovich and Rosbash 1997).
Considering that FOXP3 introns have C-rich/U-poor py tracts and that these py tracts serve as poor substrates for U2AF2 binding (Sickmier et al. 2006; Glasser et al. 2022), it was not surprising that we observed significantly less U2AF2 binding to F3i11wt-tobra compared to F3i11pU-tobra (Fig. 3C). U2AF2 directly interacts with SF1, which binds the BPS, and U2AF1, which binds the 3′SS, so it was also not surprising that we detected less binding of both proteins on F3i11wt-tobra compared to F3i11pU-tobra (Fig. 3C). Our group and others have shown ATP-independent binding of U2 snRNP components to pre-mRNA during CC assembly (Jamison and Garcia-Blanco 1992; Gozani et al. 1998), which is what we observed on both constructs but significantly less on F3i11wt-tobra (Fig. 3E). Furthermore, it has been shown that some ATP-independent U2 snRNP complexes could be chased to splice (Perriman and Ares 2000). When ATP/CP was added to this IVS system to visualize pre-spliceosome formation, the difference in abundance of SF1, U2AF heterodimer, and components of the U2 snRNP between F3i11wt-tobra and F3i11pU-tobra was compounded (Fig. 4C,E). It appears that the abundance of those proteins did not increase in the presence of ATP/CP on F3i11wt-tobra but drastically increased on F3i11pU-tobra (Figs. 3 and 4). These data suggest that the initial binding of U2AF2 on F3i11pU-tobra drives the splicing reaction forward, while its binding on F3i11wt-tobra is suboptimal and perhaps forms “functionally dead/weak” CC, thereby hindering the ability of proper pre-spliceosome assembly on FOXP3 introns.
Formation of the pre-spliceosome complex is ATP-dependent and is mediated by DExD-box RNA helicases, including DDX46, DDX39B, and possibly DDX39A (Fig. 4A). Although the difference in DDX46 abundance was not statistically different between the two constructs, there was ∼50% less binding to F3i11wt-tobra compared to F3i11pU-tobra. This is in line with the fact that DDX46 interacts with the U2 snRNP to facilitate its binding and stabilization to the BPS (Liang and Cheng 2015; Zhang et al. 2021). On the contrary, the abundance of the two DDX39 paralogs on the two constructs is quite interesting. As previously mentioned, the role of DDX39A in splicing has not been specified, as many of the studies on splicing focus on its 90% identical paralog, DDX39B. Nonetheless, our data demonstrated that DDX39A depletion does not phenocopy the decrease in splicing of FOXP3 introns observed upon DDX39B depletion. However, overexpressing DDX39A in DDX39B-depleted cells can rescue the splicing of FOXP3 introns (Banerjee et al. 2024). Moreover, here we show that DDX39A binds to both constructs in the absence of ATP/CP, and its abundance is increased on both constructs in the presence of ATP/CP, albeit significantly more on F3i11pU-tobra (Fig. 4D). Together these data suggest that DDX39A responds to ATP/CP addition similar to its paralog DDX39B and DDX46, and can possibly substitute for DDX39B in pre-mRNA splicing.
The recruitment of DDX39B to the intron is mediated by the binding of U2AF2 to the py tract and allows for SF1 to be displaced from the BPS to enable U2 snRNP binding (Fleckner et al. 1997; Kistler and Guthrie 2001). The binding of U2AF2 to the py tract depends on its strength, specifically RRM2 recognition of a U-rich distal half mediates a conformational change that allows RRM1 to bind the proximal half of the py tract (Mackereth et al. 2011). Because DDX39B interacts with U2AF2 through a region that includes the RRM1 (Fleckner et al. 1997), proper binding of U2AF2 to the py tract is essential for DDX39B recruitment and binding to the intron. In fact, this is consistent with our observation that U2AF2 and DDX39B bound F3i11pU-tobra better than F3i11wt-tobra (Fig. 5A; Supplemental Fig. S6A). Nevertheless, when we disrupt the 5′SS in hemi-intron RNAs, we do not observe more DDX39B binding to the polyU py tract (F3i11pU3′end-tobra) than F3i11wt3′end-tobra (Fig. 5B; Supplemental Fig. S6C). These data indicate that concurrent binding of the U1 snRNP to the 5′SS and U2AF2 to the py tract are critical for proper recruitment of DDX39B to FOXP3 introns. Given that both the 5′SS and py tracts of FOXP3 introns are weak (Hirano et al. 2023), simultaneous binding of the U1 snRNP and U2AF2 to the introns is infrequent, which is unfavorable in recruiting DDX39B and can explain the DDX39B-dependence of FOXP3 introns.
Nine of eleven FOXP3 introns have C-rich/U-poor py tracts and are conserved in vertebrates (Hirano et al. 2023). Due to their weak py tracts characterized by these C-rich/U-poor sequences, any aberrations to SFs that mediate their proper splicing can lead to an increase of retained FOXP3 introns. We propose a model in which suboptimal binding of U2AF2 to py tracts of FOXP3 introns, primarily due to the poor ability of U2AF2 RRM2 to accommodate C residues, leads to a twofold decrease of CC assembly compared to what is observed in introns with U-rich py tracts (Fig. 7A). More importantly, the transition from CC to pre-spliceosome complex formation on FOXP3 introns is approximately three times less efficient than in introns with U-rich py tracts (Fig. 7A). We attribute this to the fact that U2AF2 conformation is altered when its RRM2 interacts with C residues. Our proteomic data suggest that pre-spliceosome complexes formed on introns with U-rich py tracts progress into spliceosome and activated spliceosome complexes (represented with * in Fig. 7A). These data suggest that the quality of U2AF2 binding to py tracts influences the transition from commitment to pre-spliceosome complex formation through its ability to recruit DDX39B. When DDX39B is reduced (by ∼50%, denoted by * in Fig. 7B), as in individuals homozygous for the MS risk allele, A, of SNP rs2523506 in its 5′UTR (Galarza-Muñoz et al. 2017), the transition from CC to pre-spliceosome is drastically reduced (Fig. 7B). The result is intron retention, decreased FOXP3 protein, and impaired immune function.
FIGURE 7.
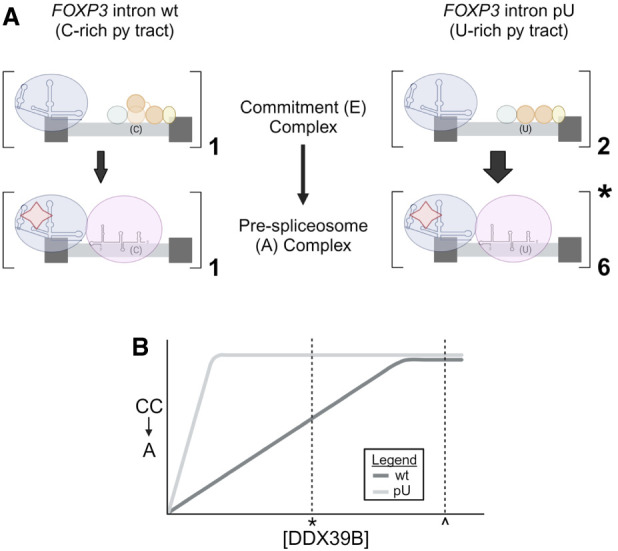
Inefficient transition from commitment to pre-spliceosome complex assembly on FOXP3 introns is dependent on DDX39B levels. (A) Comparison of commitment and pre-spliceosome complex assembly on FOXP3 intron wt and FOXP3 intron pU. Dark gray boxes represent 5′ and 3′ exons that are intercepted by an intron (light gray) with a C-rich (C) or U-rich (U) py tract. (Key: U1 snRNP = blue circle and RNA structure, SF1 = green circle, RRM1 and RRM2 of U2AF2 = orange circles, U2AF1 = yellow circle, U2 snRNP = purple circle and RNA structure, DDX39B = red star; created with BioRender.) (B) Representative graph depicting the dependency of FOXP3 intron wt compared to FOXP3 intron pU on DDX39B levels to transition from CC to pre-spliceosome (A) complex. Normal expression of DDX39B is represented by ^, and decreased expression (∼50%) of DDX39B is represented by * (created with BioRender).
MATERIALS AND METHODS
Cell culture
HeLa cells were purchased from ATCC (CCL-2) and grown at 37°C in Dulbecco's modified Eagle medium (DMEM) (Genesee Scientific) with 10% fetal bovine serum (FBS) (Genessee Scientific) and 1% (v/v) penicillin–streptomycin (Thermo Fisher Scientific). Cells were tested for mycoplasma contamination using MycoAlert Mycoplasma Detection Kit (Lonza).
Plasmids
PIP7A plasmid was previously described (Gil et al. 1991). To construct PIP7A-F3i11wt and PIP7A-F3i11pU plasmids, synthesized DNA fragments were purchased from Integrated DNA Technologies and subcloned into PIP7A at KpnI and HindIII restriction enzyme sites. To construct tobramycin aptamer-containing plasmids, F3i11wt-tobra, F3i11pU-tobra, F3i11wt3′end-tobra, and F3i11pU3′end-tobra, synthesized DNA fragments were purchased from Integrated DNA Technologies and subcloned into pcDNA5 plasmid at HindIII and NotI restriction enzyme sites. FOXP3 intron 11 wt splicing reporter plasmid was previously described (Hirano et al. 2023). To construct FOXP3 intron 11 py tract mutant splicing reporters, synthesized DNA fragments were purchased from Twist Bioscience and subcloned into pcDNA3.1 plasmid at KpnI and NotI restriction enzyme sites. All constructs were confirmed by Sanger sequencing (Azenta).
In vitro splicing
Uniformly [α-32P]-UTP radiolabeled RNAs were transcribed with T7 RNA polymerase (New England Biolabs) from PIP7A-based templates that were linearized with HindIII. The labeling reaction consisted of 1× T7 reaction buffer, 2 µL T7 RNA polymerase mix, 1 µg of DNA template and final concentrations of 1 mM ATP, 1 mM CTP, 0.2 mM GTP, 4 µM UTP, 2 µM [α-32P]-UTP (BLU507X250UC, Revvity), 0.8 mM m7G cap analog, and 5 mM DTT (all reagents from New England Biolabs unless noted). Reactions were incubated at 37°C for 6 h. Two microliters of DNase I was then added and reactions were incubated at 37°C for an additional 15 min. RNAs were then isolated using TRIzol LS (Invitrogen), following the manufacturer's protocol.
Splicing reactions were performed as described previously (Jamison et al. 1992). For each 25 µL splicing reaction, 5 × 105 cpm of [α-32P]-UTP radiolabeled RNA was incubated at 30°C with a splicing mixture composed of final concentrations of 33% (v/v) nuclear extract (prepared following methods described by Dignam et al. 1983 and Lee et al. 1988), 62 mM KCl, 2 mM MgCl2, 1 mM ATP, and 5 mM CP. Reactions were immediately placed on ice and the volumes were brought up to 75 µL with nuclease-free water (Ambion). To stop the splicing reactions, 75 µL of high salt buffer (10 mM Tris-HCl [pH 7.5], 50 mM EDTA, 7 M urea, 0.1 M LiCl, 0.5% SDS) was added. To identify splicing intermediates, RNA was extracted using TRIzol LS (Invitrogen) following the manufacturer's protocol, and then subjected to electrophoresis on a 15% denaturing TBE–urea gel (SequaGel) at 15 W for 4 h. The gel was then fixed in a solution of 10% methanol and 10% acetic acid for 30 min. The gel was visualized by exposing it to a phosphorimaging screen for 2 h at −80°C, and the image was developed using a phosphorimager (Typhoon FLA and Typhoon Trio+). Splicing efficiency was measured by densitometry analysis in ImageJ and quantified as (spliced/[spliced + unspliced]) × 100.
To identify splicing complexes formed on F3i11-IVS substrates, uniformly [α-32P]-UTP radiolabeled RNAs were transcribed with T7 RNA polymerase (New England Biolabs) from templates PCR amplified from F3i11wt-tobra and F3i11pU-tobra plasmids using forward primer 5′-TGACCTCCATAGAAGACACC-3′ and reverse primer 5′-CTATACTAAGCCGGATCCCC-3′. The labeling reaction and RNA isolation methods were described above. These RNAs were then incubated at 30°C with splicing mixtures for a designated amount of time. Following incubation, heparin was added to a final concentration of 0.5 mg/mL and the reaction was incubated at 30°C for an additional 5 min. The reactions were then subjected to native gel (4% polyacrylamide/50 mM Tris-glycine) (Konarska and Sharp 1987) electrophoresis at 200 V for 3 h, then transferred to Whatman paper and dried at 80°C for 1 h. The dried gel was exposed to a phosphorimaging screen for 8–14 h and developed using a phosphorimager (Typhoon Trio+).
To visualize the splicing efficiency of F3i11-IVS substrates, RNAs were transcribed from DNA templates prepared as above. The in vitro transcription reaction consisted of 1× T7 reaction buffer, 2 µL T7 RNA polymerase mix, 1 µg of DNA template and final concentrations of 10 mM ATP, CTP, and UTP, 2 mM GTP, 8 mM m7G cap analog, and 5 mM DTT (all reagents from New England Biolabs). Reactions were incubated at 37°C overnight. Two microliters of DNase I was then added and reactions were incubated at 37°C for an additional 15 min. RNAs were then isolated using TRIzol LS (Invitrogen), following the manufacturer's protocol. These RNAs were then incubated with splicing mixtures in the absence or presence of ATP/CP at 30°C for 90 min. Reactions were immediately placed on ice, the volumes were brought up to 75 µL with nuclease-free water (Ambion), and the splicing reactions were stopped with the addition of 75 µL of the high salt buffer. RNAs were then extracted using an RNA Clean & Concentrator kit (Zymo Research), following the manufacturer's protocol. Reverse transcription was performed on total RNA using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific), following the manufacturer's protocol. To visualize the splicing efficiency of these substrates, end point PCR (2X Taq Master Mix, New England Biolabs) was conducted with forward primer 5′-CCCTATAGTGCACCTGGAAG-3′ and reverse primer 5′-CTATACTAAGCCGGATCCCC-3′. The thermocycling conditions were 95°C for 30 sec, followed by 45 cycles of 95°C for 30 sec, 58°C for 30 sec, 68°C for 30 sec, and final extension at 68°C for 5 min. PCR products were detected by electrophoresis on a 6% TBE gel (Invitrogen), stained with 1× SYBR Gold Nucleic Acid Gel Stain (Thermo Fisher Scientific) for 10 min, and then visualized with a UV transilluminator.
FOXP3 splicing reporter assays
A total of 5 × 104 HeLa cells were seeded in 24-well plates and reverse transfected at a final concentration of 10 nM with a siNSC (all-stars nontargeting control, Qiagen) or a DDX39B-targeting (siDDX39B: Hs_BAT1_13, Qiagen) siRNA using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific). After 24 h, 500 ng of tobramycin aptamer-containing constructs (Fig. 2) or 500 ng of FOXP3 intron 11 py tract mutant constructs (Fig. 6) were transfected using Lipofectamine 3000 (Thermo Fisher Scientific), and the media was changed 6 h later. At 24 h post plasmid transfection, total RNA was isolated and protein was harvested.
RNA was isolated using a Direct-zol RNA kit with on-column DNase I treatment (Zymo Research), following the manufacturer's protocol. Reverse transcription was performed with 500 ng of RNA using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific), following the manufacturer's protocol. To quantify the splicing efficiency of the tobramycin aptamer-containing constructs, end point PCR (2X Taq Master Mix, New England Biolabs) was conducted with forward primer 5′-CCCTATAGTGCACCTGGAAG-3′ and reverse primer 5′-CTATACTAAGCCGGATCCCC-3′. The thermocycling conditions were 95°C for 30 sec, followed by 35 cycles of 95°C for 30 sec, 58°C for 30 sec, 68°C for 30 sec, and final extension at 68°C for 5 min. To measure the splicing efficiency of the FOXP3 intron 11 py tract mutant constructs, end point PCR was performed using forward primer 5′-TTGGTATGGGCAAATCAGGC-3′ and reverse primer 5′-TGGACGACGAACTTCACCTT-3′. The thermocycling conditions were 95°C for 30 sec, followed by 18 cycles of 95°C for 30 sec, 59°C for 30 sec, 68°C for 30 sec, and final extension at 68°C for 5 min. PCR products were detected by electrophoresis on a 6% TBE gel (Invitrogen) and stained with 1× SYBR Gold Nucleic Acid Gel Stain (Thermo Fisher Scientific) for 10 min then visualized with a UV transilluminator. Splicing efficiency was measured by densitometry analysis in ImageJ and quantified as (spliced/[spliced + unspliced]) × 100.
Whole-cell lysates were harvested in 1× RIPA buffer (Cell Signaling Technologies) supplemented with 1× protease inhibitor cocktail (Cell Signaling Technologies). Lysates were centrifuged at 13,000g for 15 min at 4°C, and protein concentration was quantified by BCA (Thermo Fisher Scientific). For western blot analysis, 15 µg of total protein prepared in 1× LDS sample buffer (GenScript) supplemented with 5% 2-mercaptoethanol was incubated at 70°C for 10 min and loaded on 4%–12% precast PAGE gels (GenScript), transferred to nitrocellulose membranes (Bio-Rad), and blotted with antibodies against DDX39B (1:5000; ab18106, Abcam) and β-actin (1:2000; sc-47778, Santa Cruz) as a loading control.
Tobramycin RNA affinity chromatography
Tobramycin RNA affinity chromatography was carried out as described by Hartmuth et al. (2004) and analyzed by western blot and mass spectrometry. RNAs were transcribed with T7 RNA polymerase from tobramycin aptamer-containing templates linearized with NotI. Reactions consisted of 1 µg of DNA template and final concentrations of 1× T7 reaction buffer, 10 mM each of ATP, GTP, UTP, and CTP, 5 mM DTT, and 2 µL T7 RNA polymerase mix. Reactions were incubated at 37°C for 16 h. Two microliters of DNase I was then added to the reaction and incubated at 37°C for 15 min before RNAs were isolated using TRIzol LS (Invitrogen), following the manufacturer's protocol.
Tobramycin-coupled Sepharose matrix was prepared following the protocol in Hartmuth et al. (2004). Briefly, 1 mL of NHS-activated Sepharose (Cytiva) was washed four times with 4.5 mL of 1 mM HCl then incubated with 1 mL of 5 mM tobramycin in coupling buffer (0.2M NaHCO3, 0.5M NaCl, pH 8.3 [NaOH]) at 4°C overnight. The tobramycin matrix was then collected by centrifugation at 250g for 5 min and then incubated with 8 mL of blocking buffer A (0.2 M NaHCO3, 0.1 M NaCl, 1 M ethanolamine, pH 8.0 [HCl]) for 2 h at room temperature with head-over-tail rotation. The matrix was then collected by centrifugation at 250g for 5 min and washed three times with 10 mL of PBS and stored in 2 mL of PBS containing 0.02% NaN3 at 4°C.
To prepare tobramycin matrix-bound RNA, a packed tobramycin matrix volume of 15 µL was blocked in 250 µL of blocking buffer B (20 mM Tris-HCl [pH 8.1 and 21°C], 1 mM CaCl2, 1 mM MgCl2, 300 mM KCl, 0.1 mg/mL tRNA, 0.5 mg/mL BSA, 0.01% NP-40, 0.2 mM DTT) at 4°C overnight with head-over-tail rotation. The tobramycin matrix was then collected by centrifugation at 250g for 5 min and then incubated with RNAs. To prepare RNAs, 60 pmol of RNA was added to 400 µL of binding buffer (20 mM Tris-HCl [pH 8.1 and 21°C], 1 mM CaCl2, 1 mM MgCl2, 145 mM KCl, 0.1 mg/mL tRNA, 0.2 mM DTT), and the mixture was heated at 95°C for 5 min. The RNA-binding buffer mixture was then cooled to room temperature for 15–30 min before being added to the preblocked tobramycin matrix. This RNA and tobramycin matrix mixture was then incubated at 4°C for 1–1.5 h with head-over-tail rotation. The tobramycin matrix-bound RNA was then collected by centrifugation at 250g for 5 min and then washed three times with 1 mL of RNA washing buffer (20 mM Tris-HCl [pH 8.1 and 21°C], 1 mM CaCl2, 1 mM MgCl2, 145 mM KCl, 0.1% NP-40, 0.2 mM DTT). The RNAs bound the tobramycin matrix at an efficiency of 55%–58%, which is similar to Hartmuth et al. (2004).
To identify proteins bound to the RNAs, tobramycin matrix-bound RNA was incubated at 30°C with head-over-tail rotation with the IVS mixture consisting of final concentrations of 33% (v/v) precleared nuclear extract, 62 mM KCl, 2 mM MgCl2, 1 mM ATP, and 5 mM CP. ATP and CP were omitted from the splicing mixture in designated −ATP samples. After incubation under splicing conditions for a designated amount of time, the tobramycin matrix-bound RNA was centrifuged at 250g for 5 min at 4°C and washed three times with 500 µL of W150 buffer (20 mM Tris-HCl [pH 8.1 at 21°C], 1 mM CaCl2, 1 mM MgCl2, 150 mM KCl, 0.1% NP-40, 0.2 mM DTT). To elute the RNA and interacting proteins, 125 µL of elution buffer (20 mM Tris-HCl [pH 8.1 at 21°C], 1 mM CaCl2, 3 mM MgCl2, 145 mM KCl, 5 mM tobramycin, 0.2 mM DTT) was added to the matrix and incubated at 4°C for 10 min with head-over-tail rotation. The eluates were collected by centrifugation at 250g for 5 min at 4°C and stored at −20°C until further analysis. It is important to note that 85%–90% of the tobramycin matrix-bound RNA was released upon elution, which is similar to Hartmuth et al. (2004).
For western blot analysis, 20 µL of eluates prepared in 1× LDS sample buffer (GenScript) supplemented with 5% 2-mercaptoethanol were incubated at 70°C for 10 min and loaded on 4%–12% precast PAGE gels (GenScript), transferred to nitrocellulose membranes (Bio-Rad), and blotted with antibodies against DDX39B (1:5000; ab18106, Abcam), U2AF2 (1:2000; U4758, Sigma), U1-70K (1:2000; 05-1588, EMD Millipore) and SF3B1 (1:1000; 14434S, Cell Signaling Technologies).
Mass spectrometry analysis
For mass spectrometry analysis, eluates were sent to the University of Texas Medical Branch Mass Spectrometry Facility. Samples were prepared according to standard proteomic sample prep workflow, which includes reduction and alkylation of cysteines and S-trap-based trypsin digestion. Briefly, 1 µL of benzonase (70664-10KUN, MilliporeSigma) was added to 25 µL of the eluate and heated at 37°C for 30 min. Twenty-five microliters of 10% SDS, 100 mM tetraethylammonium bicarbonate (TEAB, pH 7.1) and 1 µL of 0.25M Tris(2-carboxyethyl)phosphine (TCEP) (77720, Thermo Fisher Scientific) was added and the mixture was heated to 65°C for 30 min. The sample was then cooled to room temperature and 1 μL of 0.5M iodoacetamide was added and allowed to react for 30 min in the dark. Then, 2.7 μL of 12% phosphoric acid was then added to the protein solution followed by 165 μL of S-Trap buffer solution (90% Methanol, 100 mM TEAB final; pH 7.1). The resulting solution was administered to an S-Trap spin column (Protifi) and passed through the column using a bench-top centrifuge at 4000g for 30 sec. The spin column was then washed two times with 150 μL of delipidation wash (50% MTBE/50% MeOH) and centrifuged at 1200 rpm for 1 min. The spin column was then washed two times with 150 μL of S-Trap buffer solution and centrifuged at 1200 rpm for 1 min. Trypsin (V5280, Promega) was then added to the protein mixture in a ratio of 1:25 in 50 mM TEAB, pH 8, and incubated at 37°C overnight. Peptides were eluted with 75 µL of 50% acetonitrile (ACN), 0.2% formic acid followed by 75 µL of 80% ACN, 0.2% formic acid, and centrifuged at 1200 rpm for 1 min. The combined peptide solution was then dried in a speed vacuum at room temperature for 1.5 h and resuspended in 2% ACN, 0.1% formic acid, and 97.9% water. A total of 20 µL of each sample was added in an autosampler vial.
Peptide mixtures were analyzed by nanoflow liquid chromatography-tandem mass spectrometry (nanoLC–MS/MS) using a nanoLC chromatography system (UltiMate 3000 RSLCnano, Dionex), coupled online to a Thermo Orbitrap Eclipse mass spectrometer (Thermo Fisher Scientific) through a nanospray ion source. A direct injection method is used onto an analytical column; Aurora (75 µm × 25 cm, 1.6 µm) from (ionopticks). After equilibrating the column in 98% solvent A (0.1% formic acid in water) and 2% solvent B (0.1% formic acid in ACN), the samples (2 µL in solvent A) were injected (300 nL/min) by gradient elution onto the C18 column as follows: isocratic at 2% B, 0–10 min; 2%–27% 10–98 min, 27%–45% B, 98–102 min; 45%–90% B, 102–103 min; isocratic at 90% B, 103–104 min; 90%–15%, 104–106 min; 15%–90% 106–108 min; isocratic for 2 min; 90%–2%, 110–112 min; and isocratic at 2% B, till 120 min. All LC–MS/MS data were acquired using an Orbitrap Eclipse in positive ion mode using a top speed data-dependent acquisition (DDA) method with a 3-sec cycle time. The survey scans (m/z 375–1500) were acquired in the Orbitrap at 50,000 resolution (at m/z = 400) in profile mode, with a maximum injection time of 100 msec and an AGC target of 400,000 ions. The S-lens RF level was set to 30. Isolation was performed in the quadrupole with a 1.6-Da isolation window, and HCD MS/MS acquisition was performed in profile mode using the orbitrap at a resolution of 7500 using the following settings: parent threshold = 5000; collision energy = 30%; using the default settings. Monoisotopic precursor selection (MIPS) and charge state filtering were on, with charge states 2–6 included. Dynamic exclusion was used to remove selected precursor ions, with a ±10 ppm mass tolerance, for 60 sec after the acquisition of one MS/MS spectrum.
Tandem mass spectra were extracted and charge state deconvoluted using Proteome Discoverer 2.5 (Thermo Fisher Scientific). Deisotoping was not performed. All MS/MS spectra were searched against a Uniprot Human database using Sequest. Searches were performed with a parent ion tolerance of 5 ppm and a fragment ion tolerance of 0.60 Da. Trypsin was specified as the enzyme, allowing for two missed cleavages. Fixed modification of carbamidomethyl (C) and variable modifications of oxidation (M) and deamidation were specified in Sequest. The processing nodes were Spectrum Files RC, Spectrum Selector, Sequest HT, Percolator, and also included Minora Feature Detector. The consensus nodes were MSF Files, Feature Mapper, Precursor Ions Quantifier, and also included PSM Grouper, Peptide Validator, Peptide and Protein Filter, and Protein Annotation. To quantify peptide abundance, the intensities of the top three peptides for each protein were averaged and referred to as raw protein abundance values. For peptides that had missing values, low abundance resampling was conducted to replace the missing values with random values sampled from the lower 5% of detected values. The raw protein abundance values for each protein were then normalized to the sample with the highest total peptide amount and referred to as normalized protein abundance values.
We elected to represent the median ± interquartile range of the raw, as opposed to normalized, protein abundance values for each of the four conditions (Figs. 3 and 4). Our rationale for this is that these RNAs are being incubated with nuclear extract and splicing conditions in order to specifically identify differences in their splicing complex formation. Therefore, the raw protein abundance values in the eluates are a representation of the biological differences between splicing complexes being formed on F3i11wt-tobra and F3i11pU-tobra. Furthermore, Supplemental Figure S3 displays the log10 transformed raw protein abundance values of all proteins in each replicate of each condition (−ATP:F3i11wt-tobra = light blue, −ATP:F3i11pU-tobra = light orange, +ATP:F3i11wt-tobra = blue, +ATP:F3i11pU-tobra = orange) and indicates that the differences in total abundance is not statistically significant (Mann–Whitney test) between F3i11wt-tobra and F3i11pU-tobra in both minus and plus ATP conditions.
Data analysis and statistics
Bar graphs in Figures 1 and 2 represent the mean ± SD and the statistical tests performed were Student's t-test. Bar graphs in Figures 3 and 4 and Supplemental Figures S3 and S4 represent the median ± interquartile range and the statistical tests conducted were Mann–Whitney test. The bar graphs in Figure 6 represent the mean ± SD and the statistical tests performed were Student's t-test for panel B and one-way ANOVA for panel C. Asterisks represent levels of statistical significance: (*) P < 0.05, (**) P < 0.01, (***) P < 0.001 and (****) P < 0.0001. All statistical analyses were conducted using GraphPad Prism 9.4.1 software.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
C.K.N. acknowledges support from the Human Pathophysiology and Translational Medicine program at the University of Texas Medical Branch. W.K.R. acknowledges support from the Cancer Prevention Research Institute of Texas (Grant: RP190682). M.A.G.-B. acknowledges support from the National Institutes of Health (Grant: P01 AI150585) and Startup Funds from the University of Virginia School of Medicine. We thank members of the Garcia-Blanco laboratory, especially Dr. Shefali Banerjee for her input and discussions and Debbie Kennedy for reviewing the manuscript. We thank Dr. W. Sam Fagg at the University of Texas Medical Branch for his insights during the work and feedback on the manuscript. We also thank Dr. Clara Kielkopf and Mary Pulvino from the University of Rochester for their input and expertise on the U2AF heterodimer.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079933.123.
Freely available online through the RNA Open Access option.
MEET THE FIRST AUTHOR
Chloe Nagasawa.

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Chloe Nagasawa is the first author of this paper, “Inefficient recruitment of DDX39B impedes pre-spliceosome assembly on FOXP3 introns.” Chloe is a PhD candidate at the University of Texas Medical Branch in Dr. Mariano Garcia-Blanco's lab, which recently moved to the University of Virginia. The focus of her research is to understand how alterations to essential splicing factors affect spliceosome formation and influence the development of autoimmunity.
What are the major results described in your paper and how do they impact this branch of the field?
Here we describe the splicing mechanism of FOXP3 introns, which have C-rich/U-poor polypyrimidine (py) tracts and determine why their splicing is sensitive to an essential splicing factor called DDX39B. We show that inefficient binding of U2AF2 to the C-rich/U-poor py tracts of FOXP3 introns leads to poor recruitment of DDX39B, which ultimately contributes to a deficiency in pre-spliceosome formation. The information presented in this manuscript can help explain why introns with C-rich/U-poor py tracts are spliced poorly.
What led you to study RNA or this aspect of RNA science?
I began studying the mechanism of RNA splicing during my master's program at Kansas City University under the mentorship of Dr. Douglas Bittel. Since then, I have maintained an interest in understanding the intricacies of splicing and how aberrant splicing influences disease pathogenesis.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
Given the dramatic difference of U2AF2 binding between the FOXP3 intron 11 wt and polyU mutant RNAs, it was surprising to see no significant difference in DDX39B binding (Fig. 4D). This result prompted us to conduct time course experiments (Fig. 5; Supplemental Fig. S6), which revealed that the binding of both U2AF2 and DDX39B to the two RNAs is dramatically different at early time points.
If you were able to give one piece of advice to your younger self, what would that be?
One piece of advice I would give my younger self is to appreciate the grind.
REFERENCES
- Abovich N, Rosbash M. 1997. Cross-intron bridging interactions in the yeast commitment complex are conserved in mammals. Cell 89: 403–412. 10.1016/S0092-8674(00)80221-4 [DOI] [PubMed] [Google Scholar]
- Absmeier E, Santos KF, Wahl MC. 2016. Functions and regulation of the Brr2 RNA helicase during splicing. Cell Cycle 15: 3362–3377. 10.1080/15384101.2016.1249549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal AA, McLaughlin KJ, Jenkins JL, Kielkopf CL. 2014. Structure-guided U2AF65 variant improves recognition and splicing of a defective pre-mRNA. Proc Natl Acad Sci 111: 17420–17425. 10.1073/pnas.1412743111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allcock RJN, Williams JH, Price P. 2001. The central MHC gene, BAT1, may encode a protein that down-regulates cytokine production. Genes Cells 6: 487–494. 10.1046/j.1365-2443.2001.00435.x [DOI] [PubMed] [Google Scholar]
- Banerjee S, Nagasawa CK, Widen SG, Garcia-Blanco MA. 2024. Parsing the roles of DExD-box proteins DDX39A and DDX39B in alternative RNA splicing. Nucl Acids Res (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund JA, Abovich N, Rosbash M. 1998. A cooperative interaction between U2AF65 and mBBP/SF1 facilitates branchpoint region recognition. Genes Dev 12: 858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Zhou Z, Reed R. 2000. Functional association of U2 snRNP with the ATP-independent spliceosomal complex E. Mol Cell 5: 779–787. 10.1016/S1097-2765(00)80318-4 [DOI] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RG. 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res 11: 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleckner J, Zhang M, Valcárcel J, Green MR. 1997. U2AF65 recruits a novel human DEAD box protein required for the U2 snRNP-branchpoint interaction. Genes Dev 11: 1864–1872. 10.1101/gad.11.14.1864 [DOI] [PubMed] [Google Scholar]
- Galarza-Muñoz G, Briggs FBS, Evsyukova I, Schott-Lerner G, Kennedy EM, Nyanhete T, Wang L, Bergamaschi L, Widen SG, Tomaras GD, et al. 2017. Human epistatic interaction controls IL7R splicing and increases multiple sclerosis risk. Cell 169: 72–84.e13. 10.1016/j.cell.2017.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiev P, Charbonnier L-M, Chatila TA. 2019. Regulatory T cells: the many faces of Foxp3. J Clin Immunol 39: 623–640. 10.1007/s10875-019-00684-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil A, Sharp PA, Jamison SF, Garcia-Blanco MA. 1991. Characterization of cDNAs encoding the polypyrimidine tract-binding protein. Genes Dev 5: 1224–1236. 10.1101/gad.5.7.1224 [DOI] [PubMed] [Google Scholar]
- Glasser E, Maji D, Biancon G, Puthenpeedikakkal AMK, Cavender CE, Tebaldi T, Jenkins JL, Mathews DH, Halene S, Kielkopf CL. 2022. Pre-mRNA splicing factor U2AF2 recognizes distinct conformations of nucleotide variants at the center of the pre-mRNA splice site signal. Nucleic Acids Res 50: 5299–5312. 10.1093/nar/gkac287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozani O, Patton Jg, Reed R. 1994. A novel set of spliceosome-associated proteins and the essential splicing factor PSF bind stably to pre-mRNA prior to catalytic step II of the splicing reaction. EMBO J 13: 3356–3367. 10.1002/j.1460-2075.1994.tb06638.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozani O, Potashkin J, Reed R. 1998. A potential role for U2AF-SAP 155 interactions in recruiting U2 snRNP to the branch site. Mol Cell Biol 18: 4752–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmuth K, Vornlocher H-P, Lührmann R. 2004. Tobramycin affinity tag purification of spliceosomes. In mRNA processing and metabolism: methods and protocols (ed. Schoenberg DR), pp. 47–64. Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- Hirano M, Galarza-Muñoz G, Nagasawa C, Schott G, Wang L, Antonia AL, Jain V, Yu X, Widen SG, Briggs FB, et al. 2023. The RNA helicase DDX39B activates FOXP3 RNA splicing to control T regulatory cell fate. Elife 12: e76927. 10.7554/eLife.76927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamison SF, Garcia-Blanco MA. 1992. An ATP-independent U2 small nuclear ribonucleoprotein particle/precursor mRNA complex requires both splice sites and the polypyrimidine tract. Proc Natl Acad Sci 89: 5482–5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamison SF, Crow A, Garcia-Blanco MA. 1992. The spliceosome assembly pathway in mammalian extracts. Mol Cell Biol 12: 4279–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins JL, Agrawal AA, Gupta A, Green MR, Kielkopf CL. 2013. U2AF65 adapts to diverse pre-mRNA splice sites through conformational selection of specific and promiscuous RNA recognition motifs. Nucleic Acids Res 41: 3859–3873. 10.1093/nar/gkt046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastner B, Will CL, Stark H, Lührmann R. 2019. Structural insights into nuclear pre-mRNA splicing in higher eukaryotes. Cold Spring Harb Perspect Biol 11: a032417. 10.1101/cshperspect.a032417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kistler AL, Guthrie C. 2001. Deletion of MUD2, the yeast homolog of U2AF65, can bypass the requirement for Sub2, an essential spliceosomal ATPase. Genes Dev 15: 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohtz JD, Jamison SF, Will CL, Zuo P, Lührmann R, Garcia-Blanco MA, Manley JL. 1994. Protein-protein interactions and 5′-splice-site recognition in mammalian mRNA precursors. Nature 368: 119–124. 10.1038/368119a0 [DOI] [PubMed] [Google Scholar]
- Konarska MM, Sharp PA. 1987. Interactions between small nuclear ribonucleoprotein particles in formation of spliceosomes. Cell 49: 763–774. 10.1016/0092-8674(87)90614-3 [DOI] [PubMed] [Google Scholar]
- Lee KAW, Bindereif A, Green MR. 1988. A small-scale procedure for preparation of nuclear extracts that support efficient transcription and pre-mRNA splicing. Gene Anal Tech 5: 22–31. 10.1016/0735-0651(88)90023-4 [DOI] [PubMed] [Google Scholar]
- Lerner MR, Steitz JA. 1979. Antibodies to small nuclear RNAs complexed with proteins are produced by patients with systemic lupus erythematosus. Proc Natl Acad Sci 76: 5495–5499. 10.1073/pnas.76.11.5495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner MR, Boyle JA, Mount SM, Wolin SL, Steitz JA. 1980. Are snRNPs involved in splicing? Nature 283: 220–224. 10.1038/283220a0 [DOI] [PubMed] [Google Scholar]
- Liang W-W, Cheng S-C. 2015. A novel mechanism for Prp5 function in prespliceosome formation and proofreading the branch site sequence. Genes Dev 29: 81–93. 10.1101/gad.253708.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundström W, Highfill S, Walsh STR, Beq S, Morse E, Kockum I, Alfredsson L, Olsson T, Hillert J, Mackall CL. 2013. Soluble IL7Rα potentiates IL-7 bioactivity and promotes autoimmunity. Proc Natl Acad Sci 110: E1761–E1770. 10.1073/pnas.1222303110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M-J, Zhou Z, Magni K, Christoforides C, Rappsilber J, Mann M, Reed R. 2001. Pre-mRNA splicing and mRNA export linked by direct interactions between UAP56 and Aly. Nature 413: 644–647. 10.1038/35098106 [DOI] [PubMed] [Google Scholar]
- Mackereth CD, Madl T, Bonnal S, Simon B, Zanier K, Gasch A, Rybin V, Valcárcel J, Sattler M. 2011. Multi-domain conformational selection underlies pre-mRNA splicing regulation by U2AF. Nature 475: 408–411. 10.1038/nature10171 [DOI] [PubMed] [Google Scholar]
- Martelly W, Fellows B, Kang P, Vashisht A, Wohlschlegel JA, Sharma S. 2021. Synergistic roles for human U1 snRNA stem-loops in pre-mRNA splicing. RNA Biol 18: 2576–2593. 10.1080/15476286.2021.1932360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda S, Das R, Cheng H, Hurt E, Dorman N, Reed R. 2005. Recruitment of the human TREX complex to mRNA during splicing. Genes Dev 19: 1512–1517. 10.1101/gad.1302205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud S, Reed R. 1991. An ATP-independent complex commits pre-mRNA to the mammalian spliceosome assembly pathway. Genes Dev 5: 2534–2546. 10.1101/gad.5.12b.2534 [DOI] [PubMed] [Google Scholar]
- Mount SM, Pettersson I, Hinterberger M, Karmas A, Steitz JA. 1983. The U1 small nuclear RNA-protein complex selectively binds a 5′ splice site in vitro. Cell 33: 509–518. 10.1016/0092-8674(83)90432-4 [DOI] [PubMed] [Google Scholar]
- Paternoster L, Standl M, Chen C-M, Ramasamy A, Bønnelykke K, Duijts L, Ferreira MA, Alves AC, Thyssen JP, Albrecht E, et al. 2012. Meta-analysis of genome-wide association studies identifies three new risk loci for atopic dermatitis. Nat Genet 44: 187–192. 10.1038/ng.1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perriman R, Ares M. 2000. ATP can be dispensable for prespliceosome formation in yeast. Genes Dev 14: 97–107. [PMC free article] [PubMed] [Google Scholar]
- Quiñones-Lombraña A, Lopez-Soto A, Ballina-Garcia FJ, Alperi-López M, Queiro-Silva R, Lopez-Vazquez A, Lopez-Larrea C, Gonzalez S. 2008. BAT1 promoter polymorphism is associated with rheumatoid arthritis susceptibility. J Rheumatol 35: 741–744. [PubMed] [Google Scholar]
- Reed R, Maniatis T. 1985. Intron sequences involved in lariat formation during pre-mRNA splicing. Cell 41: 95–105. 10.1016/0092-8674(85)90064-9 [DOI] [PubMed] [Google Scholar]
- Roscigno RF, Weiner M, Garcia-Blanco MA. 1993. A mutational analysis of the polypyrimidine tract of introns. Effects of sequence differences in pyrimidine tracts on splicing. J Biol Chem 268: 11222–11229. 10.1016/S0021-9258(18)82114-7 [DOI] [PubMed] [Google Scholar]
- Shen J, Zhang L, Zhao R. 2007. Biochemical characterization of the ATPase and helicase activity of UAP56, an essential pre-mRNA splicing and mRNA export factor. J Biol Chem 282: 22544–22550. 10.1074/jbc.M702304200 [DOI] [PubMed] [Google Scholar]
- Shen H, Zheng X, Shen J, Zhang L, Zhao R, Green MR. 2008. Distinct activities of the DExD/H-box splicing factor hUAP56 facilitate stepwise assembly of the spliceosome. Genes Dev 22: 1796–1803. 10.1101/gad.1657308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmier EA, Frato KE, Shen H, Paranawithana SR, Green MR, Kielkopf CL. 2006. Structural basis for polypyrimidine tract recognition by the essential pre-mRNA splicing factor U2AF65. Mol Cell 23: 49–59. 10.1016/j.molcel.2006.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies T, Blanck G, Bresnahan M, Sands J, Strominger JL. 1989. A new cluster of genes within the human major histocompatibility complex. Science 243: 214–217. 10.1126/science.2911734 [DOI] [PubMed] [Google Scholar]
- Sträßer K, Masuda S, Mason P, Pfannstiel J, Oppizzi M, Rodriguez-Navarro S, Rondón AG, Aguilera A, Struhl K, Reed R, et al. 2002. TREX is a conserved complex coupling transcription with messenger RNA export. Nature 417: 304–308. 10.1038/nature746 [DOI] [PubMed] [Google Scholar]
- Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F, et al. 2007. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet 39: 857–864. 10.1038/ng2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valcarcel J, Gaur RK, Singh R, Green MR. 1996. Interaction of U2AF65 RS region with pre-mRNA of branch point and promotion base pairing with U2 snRNA. Science 273: 1706–1709. [DOI] [PubMed] [Google Scholar]
- Will CL, Lührmann R. 2011. Spliceosome structure and function. Cold Spring Harb Perspect Biol 3: a003707. 10.1101/cshperspect.a003707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JY, Maniatis T. 1993. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell 75: 1061–1070. 10.1016/0092-8674(93)90316-I [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Fujiwara N, Yukinaga H, Ebisuya M, Shiki T, Kurihara T, Kioka N, Kambe T, Nagao M, Nishida E, et al. 2010. The closely related RNA helicases, UAP56 and URH49, preferentially form distinct mRNA export machineries and coordinately regulate mitotic progression. Mol Biol Cell 21: 2953–2965. 10.1091/mbc.e09-10-0913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamore PD, Patton JG, Green MR. 1992. Cloning and domain structure of the mammalian splicing factor U2AF. Nature 355: 609–614. 10.1038/355609a0 [DOI] [PubMed] [Google Scholar]
- Zhang M, Green MR. 2001. Identification and characterization of yUAP/Sub2p, a yeast homolog of the essential human pre-mRNA splicing factor hUAP56. Genes Dev 15: 30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Rigo N, Dybkov O, Fourmann J-B, Will CL, Kumar V, Urlaub H, Stark H, Lührmann R. 2021. Structural insights into how Prp5 proofreads the pre-mRNA branch site. Nature 596: 296–300. 10.1038/s41586-021-03789-5 [DOI] [PMC free article] [PubMed] [Google Scholar]