

Abstract
Vascular anomalies are malformations or tumors of the blood or lymphatic vasculature and can be life-threatening. Although molecularly targeted therapies can be life-saving, identification of the molecular etiology is often impeded by lack of accessibility to affected tissue samples, mosaicism or insufficient sequencing depth. In a cohort of 356 participants with vascular anomalies, including 104 with primary complex lymphatic anomalies (pCLAs), DNA from CD31+ cells isolated from lymphatic fluid or cell-free DNA from lymphatic fluid or plasma underwent ultra-deep sequencing thereby uncovering pathogenic somatic variants down to a variant allele fraction of 0.15%. A molecular diagnosis, including previously undescribed genetic causes, was obtained in 41% of participants with pCLAs and 72% of participants with other vascular malformations, leading to a new medical therapy for 63% (43/69) of participants and resulting in improvement in 63% (35/55) of participants on therapy. Taken together, these data support the development of liquid biopsy-based diagnostic techniques to identify previously undescribed genotype–phenotype associations and guide medical therapy in individuals with vascular anomalies.
Vascular anomalies are malformations or tumors of the vasculature that can result in significant morbidity and mortality. Vascular anomalies are caused by germline or post-zygotic pathogenic variants in genes that regulate cell growth and vascular development1–3. Complex or extensive vascular anomalies are often intractable despite interventions such as sclerotherapy, embolization or surgery. In recent years, targeted therapy has played an increasingly crucial role in the management of these diseases. Identification of the molecular causes is essential to provide efficacious therapy4–11; however, genetic profiling is often limited by difficulty in obtaining affected tissues and limited sequencing depth. The lack of this information limits options and the precision of targeted therapy, especially for pCLAs, which describe four separate but overlapping disorders of lymphatic malformations. Generalized lymphatic anomaly (GLA) is characterized by lymphatic malformations in bones and organs. Kaposiform lymphangiomatosis (KLA) is a subtype of GLA in which histology of the lesion may demonstrate spindle-shaped lymphatic endothelial cells. In Gorham–Stout disease (GSD), also known as vanishing bone disease, the lymphatic malformations lead to erosion of the cortical bone, and over time, the bone ‘vanishes’ on X-ray imaging. Central conducting lymphatic anomaly (CCLA) involves malformations of the central collecting lymphatic vessels. Together, these disorders may present with chylous effusions or ascites, bone fractures, pain and various other symptoms. Clinical diagnosis is established by medical history, physical examination, imaging studies and histology. To address these limitations, we established new techniques, including analysis of unconventional samples such as endothelial cells isolated from lymphatic fluid and cell-free DNA (cfDNA), deep coverage exome sequencing, and ultra-deep targeted sequencing using unique molecular identifiers (UMIs), in the molecular profiling of these diseases. We report results from the analysis of 356 participants with complex vascular anomalies. Appropriate determination of the causative pathogenic variants can identify previously unrecognized genotype-phenotype associations and guide medical management to improve therapeutic precision in these complex medical conditions.
Results
Study cohort and participant demographics
Based on the International Society for the Study of Vascular Anomalies (ISSVA) classification for vascular anomalies, the cohort included three major disease categories: vascular malformations (n = 326), vascular tumors (n = 17) and unclassified vascular anomalies (n = 13). The study cases with vascular malformations were further divided into capillary malformation (including nonsyndromic capillary malformation, (diffuse) capillary malformation with overgrowth, reticulate capillary malformation, cutis marmorata telangiectatica congenita (CMTC), hereditary hemorrhagic telangiectasia (HHT) and capillary malformation of capillary malformation-arteriovenous malformation (CM-AVM); n = 11; Fig. 1c), common lymphatic malformations (n = 36; Fig. 1c), GLA (n = 16; Fig. 1c), KLA (n = 15; Fig. 1c), GSD (n = 4; Fig. 1c), CCLA (n = 54; Fig. 1c), CCLA with lymphatic malformation (n = 3; Fig. 1c), primary lymphedema (n = 6; Fig. 1c), CCLA with primary lymphedema (n = 12; Fig. 1c), other lymphatic conduction disorders (including plastic bronchitis, and protein-losing enteropathy resulting from elevated central venous pressure and/or elevated lymphatic production and inflammation secondary to congenital heart defect or Fontan procedure; n = 88; Fig. 1c), venous malformations (including common venous malformations, glomuvenous malformation (GVM), and blue rubber bleb nevus syndrome (BRBNS); n = 48; Fig. 1c), sporadic arteriovenous malformation (AVM, n = 12; Fig. 1c), combined vascular malformations (n = 5; Fig. 1c) and overgrowth syndromes associated with vascular malformation (including Klippel-Trenaunay syndrome (KTS), Parkes Weber syndrome, megalencephaly-capillary malformation syndrome (MCAP), congenital lipomatous overgrowth, vascular malformations, epidermal nevi, scoliosis/skeletal and spinal syndrome (CLOVES) and generally PROS; n = 16; Fig. 1c). Unclassified vascular anomalies included angiokeratoma, PTEN hamartoma syndrome, fibroadipose vascular anomaly (FAVA) or multifocal lymphangioendotheliomatosis with thrombocytopenia (MLT), as well as two cases without recognizable phenotype after expert panel review and discussion (n = 13; Fig. 1c). Vascular tumors included one each of infantile hemangioma, tufted angioma, angiosarcoma, spindle-cell hemangioma, Dabska tumor, angiofibroma, lipoblastoma, epithelioid hemangioendothelioma, benign vascular tumor (BRAF) and benign lipomatous tumor, three with congenital hemangioma and four with kaposiform hemangioendothelioma (n = 17; Fig. 1c).
Fig. 1 |. Clinical overview.
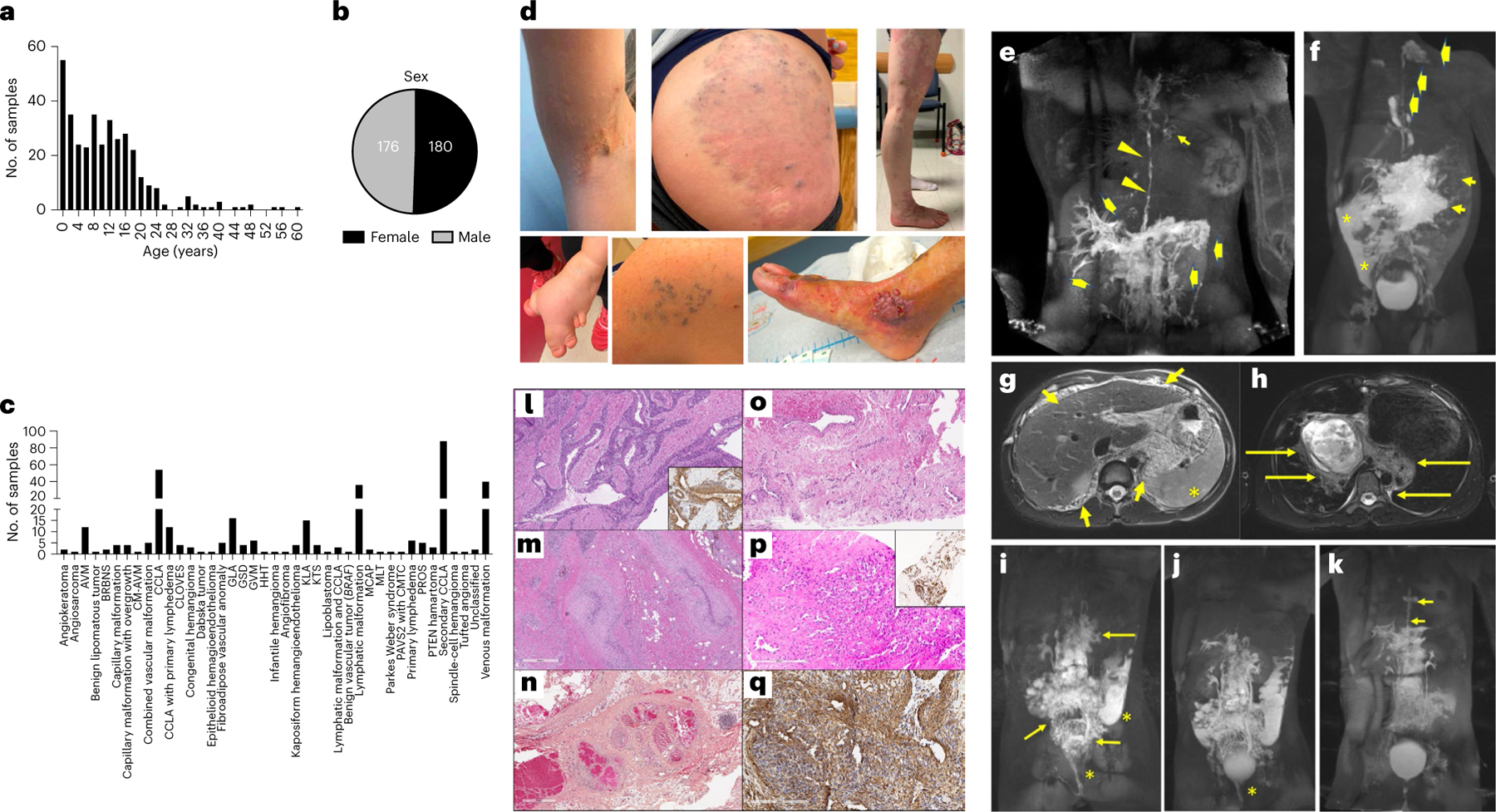
a,b, Age and sex of participants. c, Number of samples per ISSVA diagnosis. d, Example images of different participants that show microcystic lymphatic malformation, venous malformation and KTS (top panel, left to right); and syn-macrodactyly in a participant with CLOVES, GVM, and AVM (bottom panel, left to right). e, MR image of a participant with MAP2K1-CCLA (CLA135). Maximum intensity projection (MIP) after inguinal lymph node injection showing dilated and irregular thoracic duct (arrowheads), mediastinal lymphatic perfusion (short arrow) and retrograde flow in the liver and intestinal lymphatics (thick arrows). f, MR image of a participant with BRAF-GLA (CVA119). MIP from inguinal lymph node injection showing opacification of abdominal lymphatic malformation (short arrows), perfusion of cervical lymphatic malformation by a left-sided thoracic duct (thick arrows) and peritoneal lymphatic leak (asterisks). g,h, MR images of a 12-year-old male participant with PIK3CA-KLA and massive portal vein thrombosis (CVA52). Diffuse microcystic lymphatic malformation in the abdomen (short arrows) and cysts in the spleen (asterisk) (g) and lymphatic malformation involving the posterior mediastinum and hila (long arrows) (h). i,j, MR images of a participant with PIK3CA-CCLA (CVA65). MIP after injection of mesenteric (i) and liver (j) lymphatic vessels showing perfusion of the confluent abdominopelvic lymphatic malformation (long arrows) and leak of contrast into the sigmoid colon and vagina (asterisk). k, For CVA65 (same participant as in i,j), DCMRL images obtained 3 months after initiation of alpelisib and pelvic lymphovenous anastomosis showing reduced perfusion of pelvic lymphatics, resolved leak and dilated and tortuous thoracic duct (short arrows). l, Clinical histology showing dilated, tortuous venous channels with thickened smooth muscle coats containing multiple layers of perivascular cells similar to GVM due to a mosaic PDGFRB variant. Smooth muscle actin (inset) stains the smooth muscle and perivascular component (CVA172). m, Clinical histology showing tortuous and thickened or fibrotic compressed venous channels within a fibrous stroma containing fat, lymphocytes and hemosiderin due to a mosaic TEK variant (CVA19). n, Clinical histology showing a mixed vascular lesion containing dilated venous channels with ‘mulberry’ arrangement, thickened arterioles as well as nerve and abundant mature adipose tissue, consistent with PTEN hamartoma of soft tissue (CVA223). o, Clinical histology showing combined venous-capillary malformation due to a mosaic RASA1 (CVA05). p,q, Clinical histology of an unclassified vascular lesion with slit-like CD31+ (p, inset) endothelial cells and foci of surrounding cells positive for smooth muscle actin due to a mosaic GNAQ (CVA02) (q). For l–q, clinical histology was performed once with adequate positive and negative controls.
Genetic study strategy
A tiered testing strategy was implemented using multiple sample types to detect somatic variants with various variant allele frequencies (VAFs, Fig. 2a–c). When a biopsy of the affected lesion was not recommended because of risk to the patient, chylous fluid for cfDNA isolation and endothelial cell enrichment, and blood for cfDNA isolation were collected to provide alternative sample types (Fig. 2a,c). In individuals with vascular anomalies that are probably due to somatic mosaicism (for example, common venous malformation and PIK3CA-related overgrowth spectrum (PROS)), we performed deep exome sequencing (~400×) when tissue (frozen or formalin-fixed paraffin-embedded (FFPE) skin or other lesional biopsy) was available. We aimed to detect somatic variants with a VAF of > 2%. For nine randomly selected individuals with negative results from deep exome sequencing, we pursued deeper exome sequencing (~1,500×) to increase our capacity to detect variants with lower VAF (< 2%). As amplicon-based gene panels are widely used in cancer diagnostics, we designed a panel of 35 candidate disease genes (referred to as the amplicon panel) and aimed for a VAF of < 2%. Low VAF is often confounded by sequencing errors that generally occur at a rate of ~1% per base on Illumina platforms, as well as DNA polymerase errors during PCR enrichment12. To correct these errors, the addition of UMIs, as barcodes, to sequencing reads has been shown to substantially improve the accuracy of detecting lower VAFs by reconstructing consensus sequences with error correction13. We then designed a broader vascular anomaly panel with 49 genes with UMIs (referred to henceforth as the UMI panel). UMI panel sequencing reached an average raw read depth of ~86,570×, which enabled us to collapse the data to an average effective depth of ~1,892× and to confidently call somatic variants with a VAF of > 0.2%. As germline RASopathies are the largest known molecular contributor to CCLA, we performed routine exome sequencing (~76×) on genomic DNA (gDNA) isolated from blood or saliva from participants with complex lymphatic anomalies (CLAs) including GLA, KLA, GSD and CCLA4,14.
Fig. 2 |. Methods and technical aspects of the study.
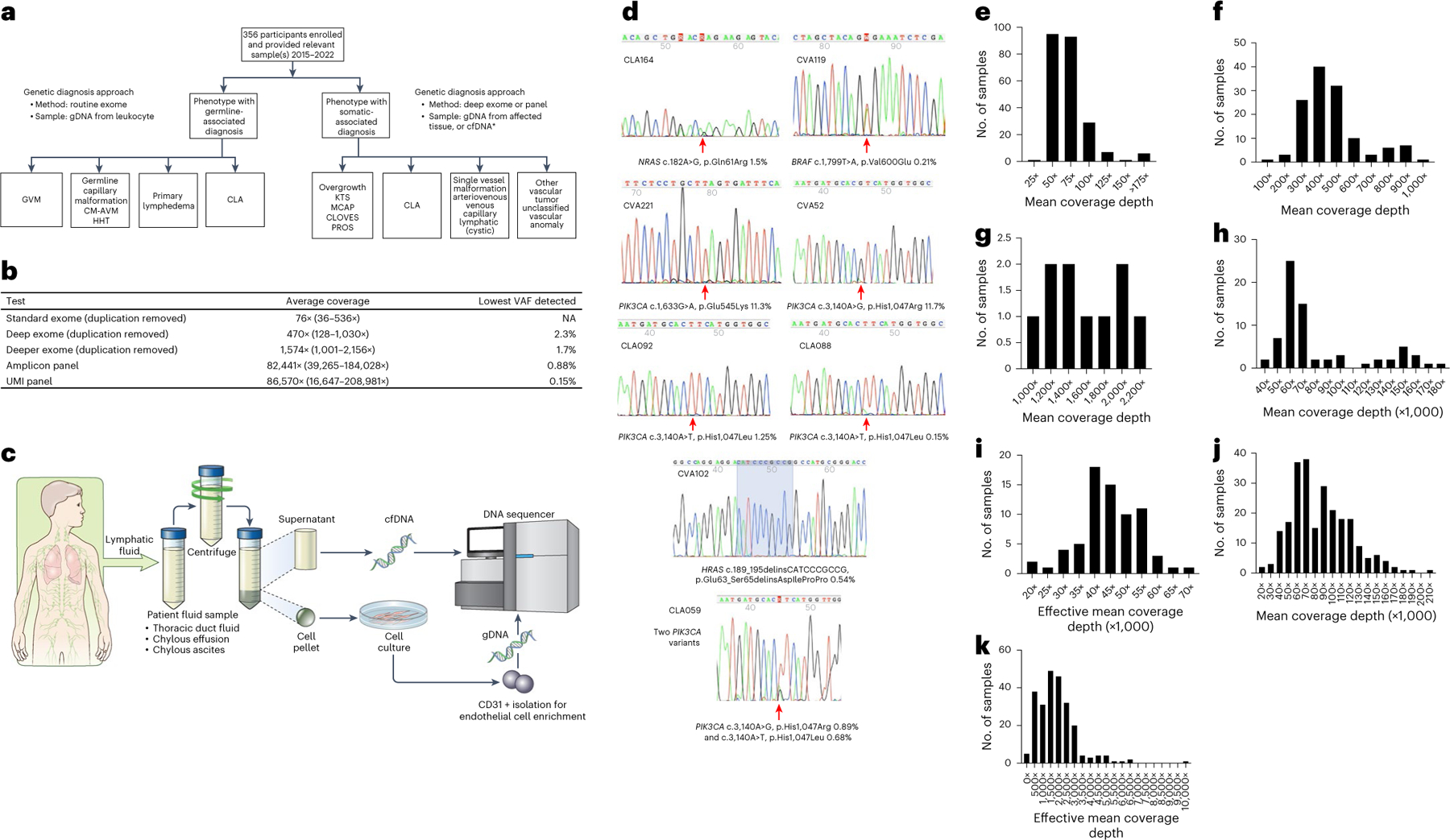
a, Genetics study workflow. CLA includes CCLA, KLA, GLA, GSD and secondary CCLA. *Affected tissue samples included fresh, frozen or FFPE tissue, cell pellets from lymphatic fluid samples, endothelial cell enrichment from lymphatic fluid samples and cfDNA isolated from either plasma or lymphatic fluid. b, Average coverage depth and lowest VAF detected by different sequencing strategies. c, Lymphatic fluid analysis procedure. Thoracic duct fluid, chylous effusion or chylous ascites were collected as part of clinical care. An aliquot was provided to the research laboratory. The sample was centrifuged. Supernatant was used for cfDNA isolation. The cell pellet was dissociated, plated and grown under conditions that favor endothelial cell growth. If growth occurred, CD31 magnetic bead isolation was performed. Genomic DNA was isolated from this material. Both were used as samples for panel sequencing. d, Representative Sanger traces from BDA qPCR assays confirming the somatic variants identified (red arrows). e–k, Distribution of coverage of genomic sequencing. Mean coverage depth of routine exome sequencing was 76× with interquartile range (IQR) of 25 (e). Mean coverage depth of deep exome sequencing was 470× with IQR of 162 (f). Mean coverage depth of deeper exome sequencing was 1,574× with IQR of 726 (g). Mean coverage of amplicon ultra-deep panel sequencing was 82,441× with IQR of 37,290 (h). Effective mean coverage of amplicon ultra-deep panel sequencing was 44,941× with IQR of 11,383 (i). Mean coverage of UMI ultra-deep panel sequencing was 86,570× with IQR of 46,325 (j). Effective mean coverage of UMI ultra-deep panel sequencing after error correction was 1,892× with IQR of 1,346 (k).
Molecular diagnostic yield
In total, 356 unrelated participants with 41 vascular anomalies diagnosed by a multidisciplinary expert team were included in this study (Fig. 1a–c). The cohort was primarily pediatric (less than 18 years old) (Fig. 1a), and sexes were represented equally (Fig. 1b). A wide range of phenotypes were included (Fig. 1c,d).
Overall, 156 of 356 unrelated participants (43.8%) received a molecular diagnosis or strong candidate diagnosis through our staged testing study (Supplementary Table 1). Specifically, we divided the cohort into cases with CLAs, vascular malformations (including common lymphatic and venous malformations and primary lymphedema), unclassified vascular anomalies and vascular tumors (Fig. 3a). With advances in the imaging of the lymphatic system and lymphatic intervention, our group now recognizes that a disrupted lymphatic system in lymphatic conduction disorders associated with congenital heart disease may be secondary to elevated central venous pressure and/or elevated lymphatic production15–17. Therefore, CLAs were subdivided into primary (n = 104) and secondary lymphatic conduction anomaly (n = 88). Excluding these 88 cases, 146 of the remaining 268 probands (54.5%) had variants that were pathogenic or likely pathogenic (herein referred to as P/LP variants), and an additional six (2.2%) harbored a variant of unknown significance (VUS) (Supplementary Table 1). Overall, the molecular diagnostic yield was 41.3% (43/104) for pCLA and 4.5% (4/88) for secondary lymphatic conduction anomaly (Fig. 3a and Supplementary Table 1). The diagnosis rate for the remainder of the cohort was 66.5% (109/164). Specifically, vascular malformations (including primary lymphedema), unclassified vascular anomalies and vascular tumors had diagnosis rates of 71.6% (96/134), 61.5% (8/13) and 29.4% (5/17), respectively (Fig. 3a and Supplementary Table 1). All germline variants and non-hotspot somatic variants with a VAF of > 15% were validated through routine Sanger sequencing. Other somatic variants were validated using a blocker displacement amplification (BDA) quantitative PCR (qPCR) and Sanger assay (Fig. 2d and Supplementary Table 1). Two individuals for whom samples contained insufficient DNA for validation had recurrent pathogenic somatic variants in PIK3CA and NRAS, which explained their disease course (Supplementary Table 1). Thirty-four individuals had germline variants only, and 118 had somatic variants only. Interestingly, three individuals had both germline and somatic variants (dual diagnoses) (Table 1 and Supplementary Table 1).
Fig. 3 |. Molecular yield and findings.
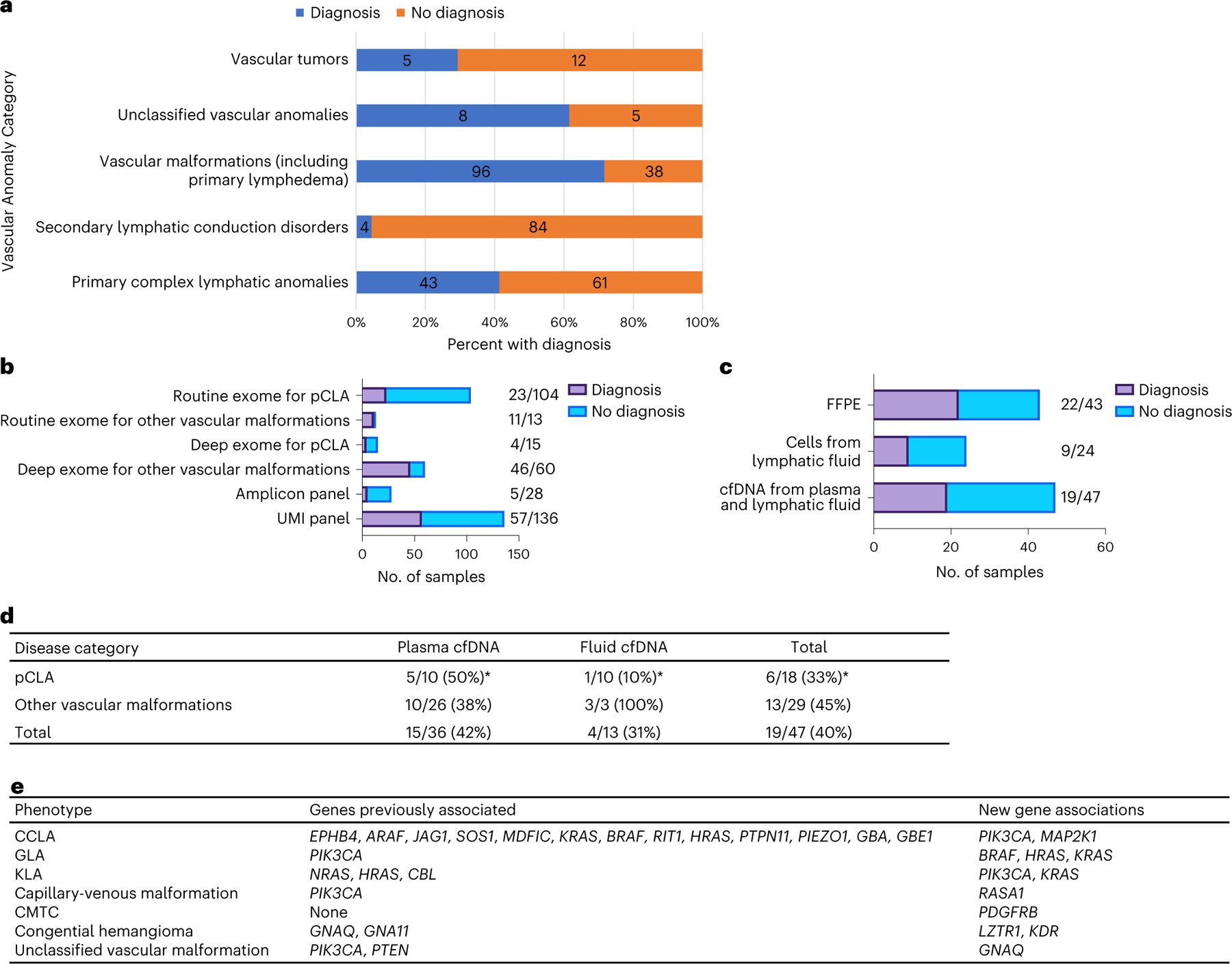
a–c, Molecular yield by diagnosis (a), methodology (b) and sample type (c). d, Further breakdown of cfDNA diagnostics for pCLA and other vascular malformations. *Two individuals were negative on both plasma and fluid cfDNA. e, New genotype–phenotype associations found in the study.
Table 1 |.
Dual diagnoses and revised clinical diagnoses based on molecular findings
| ID | Initial phenotype | Expected genetic diagnosis | Identified genetic |
|---|---|---|---|
| CVA28 | Macrocephaly, autism spectrum disorder, hamartoma | PTEN | PIK3CA (somatic), ANKRD11 (KBG syndrome) |
| CVA15 | Venous malformation, supravalvuar aortic stenosis, pulmonic stenosis, hypertrophic cardiomyopathy | TEK | TEK (somatic), RIT1 (Noonan syndrome) |
| CVA25 | NF1 and venous malformation | NF1 (with NF1-related vascular malformation) | NF1 (germline), PIK3CA (somatic) |
| CVA53 | Venous malformation by physical exam imaging, lymphatic malformation by pathology | PIK3CA | TEK (somatic) |
| CVA227 | Multiple cutaneous and mucosal venous malformations | TEK (germline) | GLMN (germline) |
| CVA172 | CMTC, GVM on pathology | GLMN | PDGFRB (somatic) |
| CVA19 | FAVA | PIK3CA | TEK (somatic) |
| CVA103 | FAVA | PIK3CA | PTEN (somatic) |
The first three cases demonstrate the utility of deep exome sequencing to identify a somatic cause, as well as the germline cause for the additional medical issues.
Diagnostic yield by procedures and sample types
Routine exome sequencing in blood or saliva.
Previous studies demonstrated germline causes of CCLA; therefore, we conducted routine exome sequencing (average 76×; Fig. 2b,e) with blood or saliva gDNA from participants (n = 104) diagnosed with pCLA by a multidisciplinary team regardless of availability of tissue samples4,10,14,18–20. Exome sequencing analysis provided a definitive diagnosis defined as a P/LP variant in 17 out of 104 (16.3%) participants and a provisional diagnosis defined as a VUS in 6 out of 104 (5.8%) (Fig. 3b and Supplementary Table 1). We also performed routine exome sequencing for those phenotypes (that is, GVM, primary lymphedema, CM-AVM, HHT and Parkes Weber syndrome) that are typically caused by germline variants (n = 13; Fig. 3b and Supplementary Table 2) and identified P/LP variants in 10 out of 13 participants and VUS in 1 out of 13 participants.
Deep exome sequencing and ultra-deep panel sequencing.
Deep exome sequencing, mainly paired with tissue biopsies (average 470×; Fig. 2b,f), detected a P/LP variant in 4 out of 15 participants with pCLAs, 46 out of 60 participants with vascular malformations (Fig. 3b) and 5 out of 11 participants with vascular tumors, providing a diagnostic yield of 64.0% (55/86) with a VAF ranging from 2.3% to 32% (Supplementary Table 3). Deeper exome sequencing (average 1,574×; Fig. 2b,g) further revealed two participants with P/LP variants with lower VAF (Supplementary Table 1). Amplicon panel sequencing (average 82,441×; Fig. 2b,h,i) identified P/LP somatic variants in 5 out of 28 participants. Finally, the UMI panel (average 86,570×; Fig. 2b,j,k) revealed 57 P/LP somatic variants (57/136) with even lower VAF, e.g. <1% (Fig. 3b and Supplementary Table 4).
FFPE blocks and slides.
Although fresh affected tissue is the ideal sample for molecular testing, biopsy may not be feasible owing to risk of bleeding or persistent effusions. Therefore, we assessed whether gDNA isolated from FFPE from a previous biopsy or surgical excision would increase molecular diagnostic yield. In 43 participants with FFPE as the only tissue sample, we made the molecular diagnosis in 22 participants (51.2%; 8 venous malformation, 7 lymphatic malformation, 2 PROS, 2 GLA, 1 KLA, AVM, MCAP, PTEN hamartoma syndrome and capillary malformation with overgrowth each; Fig. 3c and Supplementary Table 5). FFPE yielded a VAF ranging from 0.52% to 31.5% (median 6.25%) in 22 participants, suggesting that FFPE is useful in resolving genetic etiology, including low VAF, while a sensitive sequencing and validation approach is available.
Body fluid.
Previous work suggests that the causative variants may be present in endothelial cells21. We also examined gDNA isolated from endothelial cells from chylous fluid that was drained as a routine part of clinical care for pCLAs. Deep exome and/or panel sequencing with enriched endothelial cells from chylous fluid from 24 participants with pCLAs identified pathogenic variants in nine participants with VAF ranging from 0.21% to 44% (Fig. 3c and Supplementary Table 6).
While cfDNA diagnostics has gained prominence in cancer and prenatal genetic testing, its utility in vascular anomalies has also emerged11,22–27. Combining UMI panel and ultra-deep sequencing, we evaluated samples from 47 participants with heterogeneous diagnoses with cfDNA isolated from chylous or cystic lymphatic fluid supernatant or plasma. Molecular diagnosis was made in 15 plasma cfDNA and four chylous lymphatic fluid cfDNA samples (Fig. 3c,d and Supplementary Table 7).
Expanded genetic landscape in pCLA
Among the 104 participants enrolled with pCLA (29% of the cohort), we uncovered several previously undescribed genotype–phenotype associations (Fig. 3e). A 32-year-old female participant (CLA135) with capillary malformation, right-sided lateralized overgrowth, chylous pericardial effusion and CCLA (Fig. 1e) was found to have an LP variant, c.158T>G, p.F53C, in MAP2K1 (Table 2) from amplicon-based and UMI-based panels on cfDNA isolated from thoracic duct fluid (VAF of 9.8% and 12.4%, respectively) and gDNA isolated from leftover cells after cfDNA was spun down (VAF of 0.66% and 0.83%, respectively). Four participants with CCLA had somatic variants in PIK3CA, including one participant (CLA059) who had pleural effusions, pericardial effusion, ascites, constrictive pericarditis, lower extremity edema, protein-losing enteropathy and anasarca. Amplicon-based and UMI-based panel sequencing on CD31+ cells enriched from pleural fluid collected at the time of lymphangiogram identified two hotspot pathogenic somatic variants in PIK3CA (c.3140A>G, p.H1047R, VAF = 0.89%; and c.3140A>T, p.H1047L, VAF = 0.68%) (Table 2). Four participants with GLA were confirmed to have a pathogenic variant, and all had a very low VAF ranging from 0.21% to 1.3% (Table 2). We identified the BRAF hotspot variant, c.1799T>A, p.V600E, with a VAF of 0.21% using UMI panel sequencing on gDNA isolated directly from a pleural fluid sample in a 30-month-old female (CVA119) with GLA, protein-losing enteropathy and expressive language delay (Fig. 1f). In a 40-year-old participant (CVA09) with GLA characterized by lymphatic malformation of the pelvis, cutaneous microcysts, chylous effusions and chylous ascites requiring frequent paracenteses and thoracenteses, we discovered a pathogenic somatic variant in HRAS, c.191_217dup, p.M72_R73insHSAMRDQYM, with a VAF of 0.51–0.63% from UMI panel sequencing from cfDNA (Table 2). NRAS, HRAS and CBL are previously reported causes of KLA9,28,29. We identified four unreported KLA cases with the recurrent NRAS variant (c.182A>G, p.Q61R) and one KLA (CVA102) with a novel HRAS variant (c.189_195delinsCATCCCGCCG, p.Glu63_Ser65delinsAspIleProPro) (Table 2). In two participants with atypical KLA, we identified somatic PIK3CA hotspot variants (CVA221 and CVA52; Table 2, Fig. 1g,h and Supplementary Table 1). In a third participant (CLA201), using ultra-deep UMI panel sequencing, a KRAS hotspot variant, c.35G>A, p.G12D, was identified with a VAF of 0.55% and 0.39% from cfDNA isolated from blood and fluid, respectively.
Table 2 |.
Expanded genetic landscape in vascular anomalies
| Participant ID | Genotype and VAF | Solved approach | Sample type | Distinguishing clinical features | Current diagnosisa |
|---|---|---|---|---|---|
| CLA062 | KRAS c.35G>A, p.G12D, 1.4% | Amplicon panel | gDNA fluid | Conduction abnormalities on MRL | CCLA |
| CLA063 | KRAS c.G35T, p.G12V, 0.77% | UMI panel | gDNA fluid | Conduction abnormalities on MRL | CCLA |
| CLA149 | KRAS c.35G>A, p.G12D, 44% | Amplicon panel | gDNA fluid | Conduction abnormalities on MRL | CCLA |
| CLA163 | KRAS c.436G>A, p.A146T, 3.9% | Deep exome | gDNA tissue | Conduction abnormalities on MRL | CCLA |
| CLA088 | PIK3CA c.A3140T, p.H1047L, 0.15% | UMI panel | gDNA fluid | Conduction abnormalities on MRL | CCLA |
| CLA092 | PIK3CA c.3140A>T. p.H1047L, 1.25% | UMI panel | gDNA fluid | Conduction abnormalities on MRL | CCLA |
| CVA65 | PIK3CA c.3129G>A, p.M1043I, 5.6% | Deep exome | gDNA tissue | Cystic abdomino-pelvic lymphatic malformation with conduction abnormalities on MRL | Features of GLA and CCLA |
| CLA059 | PIK3CA c.3140A>G, p.H1047R, 0.88%; and c.3140A>T, p.H1047L, 0.68% | Amplicon panel | gDNA fluid | Conduction abnormalities on MRL | CCLA |
| CLA060 | BRAF c.T1799A, p.V600E, 0.91% | UMI panel | gDNA tissue | History of excised abdominal lymphatic malformation; conduction abnormalities on MRL | CCLA |
| CLA135 | MAP2K1 c.158T>G, p.F53C, 0.8% | UMI panel | gDNA fluid | CCLA, capillary malformation, overgrowth, pericardial effusion | CCLA |
| CVA231 | KRAS c.167_211dup, p.Q70_Y71ins15, 1.22% | UMI panel | gDNA tissue | Mediastinal lymphatic malformation; bone, liver, splenic lesions | GLA |
| CVA211 | PIK3CA c.263G>A, p.R88Q, 1.3% | UMI panel | gDNA FFPE | Circular bony lesions, spleen, cortex intact | GLA |
| CVA119 | BRAF c.1799T>A, p.V600E, 0.21% | UMI panel | gDNA fluid | Abdominal lymphatic malformation with conduction abnormalities | Features of GLA and CCLA |
| CVA09 | HRAS c.191_217dup, p.M72_R73insHSAMRDQYM, 0.51% | UMI panel | cfDNA fluid | Pelvic lymphatic malformation, conduction abnormalities on MRL | Features of GLA and CCLA |
| CLA164 | NRAS c.182A>G, p.Q61R, 1.48% | UMI panel | cfDNA plasma | Diffuse-appearing lesions on imaging (sternum, vertebrae, femurs, tibias) | KLA |
| CVA14 | NRAS c.182A>G, p.Q61R, 0.54% | UMI panel | cfDNA plasma | Diffuse-appearing lesions on imaging (thoracolumbar vertebrae, rib, femurs, mediastinal edema); elevated D-dimer | KLA |
| CVA177 | NRAS c.182A>G, p.Q61R, 5.1% | UMI panel | gDNA FFPE | Diffuse-appearing lesions on imaging (thoracolumbar vertebrae), mediastinal LM | KLA |
| CVA102 | HRAS c.189_195delinsCATCCCGCCG, p.E63_S65delinsDIPP, 0.54% | UMI panel | gDNA tissue | Diffuse appearing lesions on imaging | KLA |
| CVA221 | PIK3CA c.1633G>A, p.E545K, 11.3% | Deep exome | gDNA tissue | Spindle cells (primary or secondary); diffuse enhancement on imaging typical for KLA | KLA |
| CVA52 | PIK3CA c.3140A>G, p.H1047R, 0.41% | UMI panel | cfDNA plasma | Mediastinal lymphatic malformation; skull, liver, splenic lesions with diffuse appearance | KLA |
| CLA201 | KRAS c.35G>A, p.G12D, 0.55% | UMI panel | cfDNA plasma | KLA | |
| CVA02 | GNAQ c.626A>T, p.Q209L, 6% | Deep exome | gDNA tissue | Subcutaneous lesion noted at 10 years old | Unclassified vascular malformation |
| CVA05 | RASA1 c.1422del, p.(Asn474Lysfs*9), 8%; and c.475_476del, p.(Leu159Glyfs*20) 12.2% | Deep exome | gDNA tissue | Histopathology (Fig. 1o) | Capillary-venous malformation |
| CVA172 | PDGFRB c.1998C>A, p.N666K, 32% | Deep exome | gDNA tissue | CMTC, histopathology (Fig. 1l) | PAVS2 with CMTC |
| CVA03 | LZTR1 c.848G>A, p.R283Q, 4% | Deep exome | gDNA tissue | GLUT1 negative | Congenital hemangioma |
| CVA260 | KDR c.2341_2342insAAGACTTCTGGCTAC, p.L780_L781insQDFWL, 18.1% | Deep exome | gDNA tissue | GLUT1 negative | Congenital hemangioma |
Expanded genetic landscape for other vascular anomalies
In total, we identified 27 variants not described in vascular anomalies in 16 genes and 12 previously undescribed genotype–phenotype pairs in 15 participants (Supplementary Table 1). PIK3CA variants were most prevalent, observed in 61 individuals with blood and lymphatic malformations (that is, venous malformation, lymphatic malformation, CLOVES, MCAP, FAVA, KTS, combined capillary-lymphatic malformation, CCLA and KLA; Supplementary Table 8). Previously undescribed genotype–phenotype associations include LZTR1 (c.848G>A, p.R283Q) and KDR (c.2341_2342insAAGACTTCTGGCTAC, p.L780_L781insQDFWL) with congenital hemangioma (Fig. 3e and Table 2); unclassified vascular malformation with a recurrent somatic GNAQ variant (c.626A>T, p.Q209L; CVA02; Fig. 1p,q, Fig. 3e and Table 2); a combined capillary-venous malformation (CVA05) with two somatic RASA1 variants confirmed with immunostaining on repeat samples (Fig. 1o and Table 2) and CMTC (CVA172) with a somatic pathogenic variant, c.1998C>A, p.N666K, in PDGFRB (Fig. 1l and Table 2). Although a previous article reported a somatic pathogenic variant in GNA11 as a cause for CMTC, in our opinion the photographs of the dermatological findings in those patients are more consistent with a reticulated capillary malformation30. Additionally, we confirmed prior associations with somatic PTEN variants in PTEN hamartomas31–33 (Fig. 1n) and HRAS with AVMs34.
Functional characterization of RASopathy variants
To confirm the pathogenic effects of the identified variants and inform the therapeutic options as we previously described4,9–11, we functionally characterized a few variants in either cellular assays or in the zebrafish model. We demonstrated that BRAF and RAF1 variants increased p-ERK and enhanced sprouting capacity relative to wild type, and that biochemistry and morphology reversal could be achieved by MEK inhibition (Extended Data Fig. 1a–d). Comparably, expression of KRAS-A146T in zebrafish resulted in thoracic duct dilatation and tissue expansion (Extended Data Fig. 1e–g).
Impact of genetic testing on diagnosis and medical care
The genetic findings impacted medical care by providing an updated diagnosis or a dual molecular diagnosis, or by directing medication administration. In eight cases, the molecular genetics finding led to an updated diagnosis and dual diagnoses (Table 1), including a clinically diagnosed FAVA found to have a recurrent pathogenic somatic TEK variant and thus reclassified as a venous malformation (Fig. 1m), suggesting that deep exome sequencing (~400×) is an efficient method to evaluate both somatic and germline variants in patients with syndromic features.
In several cases, the medication was changed based on the finding. Three of these cases were previously reported4,9,10. Over 40% (69/156) of the participants who received a genetic diagnosis in our study were on a medical therapy or planned to initiate medical therapy (Fig. 4a). Genetic testing results supported the medication choice for 36% (25/69) of participants already on medical therapy, led to a change or a plan to change therapy in 25% (17/69) and led to an initiation or plan to initiate therapy in 38% (26/69) (Fig. 4b). Interestingly, the genetic finding did not support the medical therapy in one participant (CVA231), but this individual had improved on the medication, so this was continued, suggesting that further research is needed to investigate the crosstalk between signaling pathways in vascular malformations (Fig. 4b,c). The disease improved for the majority of participants on medical therapy (35/55 or 63%). Three participants had mixed response (5%), nine participants had stable disease (16%) and three participants had worsening of their disease on medical therapy (5%) (Fig. 4c). In five participants, it was too early or no information was available to assess disease response.
Fig. 4 |. Medical impact of molecular testing.
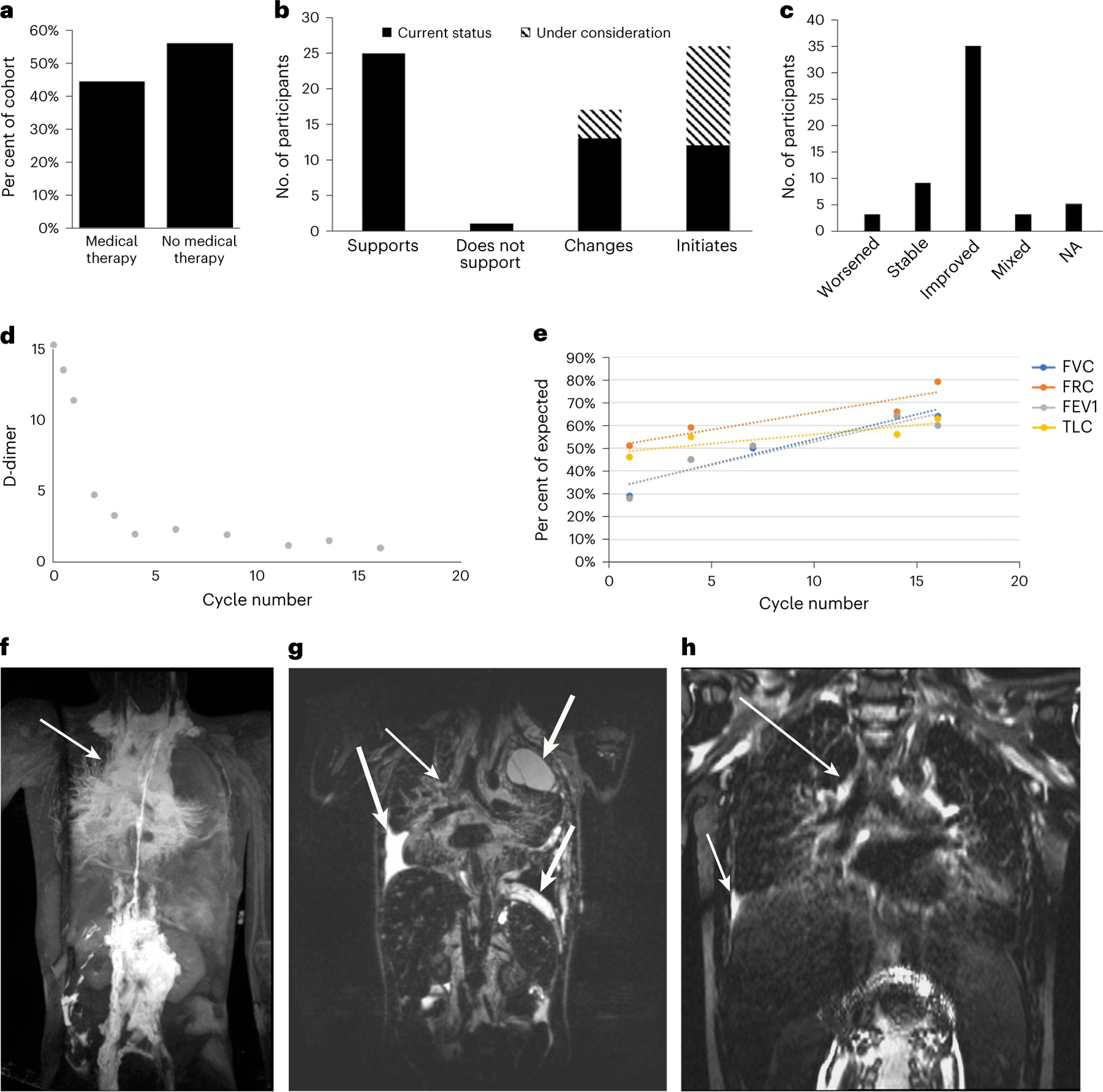
a, Of the participants in the study, 44% (69) were already on medical therapy or planned to initiate medical therapy, and 56% (87) were not on medical therapy. b, Distribution of the impact of the molecular finding on medical therapy. The molecular finding supported the chosen therapy in 25 participants and did not support the medication in 1 participant. The molecular diagnosis led to a change in therapy in 13 participants and 4 additional participants planned to change therapy based on the finding. Twelve participants commenced therapy based on the genetic diagnosis and 14 more are planning to initiate therapy. c, Response to therapy. Three participants had worsening of disease, 9 participants had stable disease, 35 participants had improvement in disease and 3 participants had a mixed response to disease. For five participants, it was too early to assess disease response or information was not available (not applicable, NA). d, D-dimer decreased from 37 mg L−1 FEU at the start (off graph) to below 5 mg L−1 FEU within two cycles (CVA14). e, Pulmonary function tests show an increase in forced vital capacity (FVC), forced residual capacity (FRC), forced expiratory volume (FEV1) and total lung capacity (TLC). f–h, MR images before and after trametinib treatment. DCMRL (f) and T2 space (g) demonstrate significant mediastinal perfusion (f, thin arrow) and thickening (g, thin arrow) as well as pleural effusions (g, thick arrow). These findings are significantly reduced post treatment (h, arrows).
A noteworthy case was identified in a 9-year-old female (CVA65) who presented with a history of protein-losing enteropathy for 3 years and chylorrhea from the rectum and vagina, as well as severe abdominal pain from extensive abdominal-pelvic lymphatic malformation. Sirolimus (target level of 10–13 ng ml−1) led to a response in leakage but was discontinued due to adverse effects. Dynamic contrast-enhanced magnetic resonance lymphangiography (DCMRL) by mesenteric and liver injection showed channels leaking into the sigmoid colon and the vaginal vault (Fig. 1i,j). She then underwent laparotomy for glue embolization of pelvic lymphatic conglomerates and a lymphovenous anastomosis of a large retroperitoneal lymphatic channel to the right gonadal vein. Sigmoidoscopy with lymphatic dye injection was performed to guide the intervention and perform a rectal biopsy. Deep exome sequencing of gDNA isolated from rectal wall tissue biopsy identified a PI3KCA pathogenic variant c.3129G>A, p.M1043I with a VAF of 5.6%. This led to the initiation of alpelesib at 100 mg daily, which was well-tolerated and led to improvement on DCMRL (Fig. 1k).
Another noteworthy case was identified in a 9-year-old male (CVA14) with KLA characterized by chylothorax with spindled endothelial cells on cytology, sclerotic vertebral lesions and mediastinal lymphatic enhancement on DCMRL, and elevated D-dimer. Sirolimus treatment alone resulted in incremental improvement. cfDNA isolated from plasma collected prior to the start of trametinib therapy identified a somatic, hotspot, pathogenic variant in NRAS (c.182A>G, p.Q61R, VAF = 0.54%). Given his worsening performance status, poor quality of life, disease refractory to standard therapy and suspicion of activation of the Ras–MAPK pathway, his treatment was changed to trametinib, a MEK1 and MEK2 inhibitor. After 1 month of trametinib therapy, his D-dimer levels decreased from 37 mg l−1 fibrinogen equivalent unit (FEU) to 13 mg l−1 FEU, and by two cycles were below 5 mg l−1 FEU (Fig. 4d). His pulmonary function tests have improved due to resolution of pleural effusions and reduction of mediastinal involvement (Fig. 4e–h).
A third noteworthy case was identified in a previously healthy 20-year-old male (CVA221) with KLA characterized by a soft tissue lesion with multiple small enhancing vessels and calcifications involving the mesentery, retroperitoneum, presacral area, some areas of the pelvis, and extending into the mediastinum; cystic-appearing lesions in the pelvic bones, spine and spleen; elevated angiopoietin-2 (4,519 pg ml−1, normal range 1,434–4,141 pg ml−1) and pathology of the soft tissue lesions demonstrating occasional areas of spindled-to-round cells and small D2–40 and PROX1 positive lymphatic channels. He started sirolimus, resulting in resolution of pleural effusions, anemia, thrombocytopenia and abdominal pain; and improvement in fatigue, hepatosplenomegaly and D-dimer from a maximum of 116 μg ml−1 to < 0.2 μg ml−1. However, his improvement stalled on sirolimus. Deep exome sequencing (249×) on gDNA isolated from an abdominal mass biopsy identified a somatic pathogenic variant in PIK3CA (c.1633G>A, p.E545K, VAF = 11.3%), which was confirmed on follow-up clinical testing. Given the molecular diagnosis, his treatment was switched to alpelisib and he has continued to improve on a dose of 125 mg daily.
Discussion
Identification of the molecular cause for complex vascular anomalies is essential given the recent advances in therapy. Therefore, we used a tiered testing strategy incorporating deep exome sequencing, ultra-deep gene panels and an affordable blocker-based qPCR validation assay to comprehensively identify both germline and somatic variants down to a VAF of 0.15%. In total, we identified 24 participants with P/LP variants with a VAF of ≤ 1%, which would be challenging to detect by conventional methods. Indeed, in one participant with CCLA (CVA119), we identified and validated a BRAF variant (VAF = 0.21%) that was not reported in the clinical testing because it was below the reportable limit.
Another aim of our study was to evaluate the use of multiple sample types (including lymphatic fluid, cyst fluid, lymph node biopsy, fresh lesion tissue, frozen tissue, FFPE and plasma or fluid cfDNA) in genetic testing, which allowed us to provide a molecular diagnosis to 41% of participants with pCLA and 72% of those with other vascular malformations. Using deep panels with cfDNA as the sample generated VAFs ranging from 0.20% to 10.62% (median 0.51%), at or below the range of VAFs reported in previous studies22–25,27. Notably, we have now expanded the use of cfDNA to chylous fluid in patients with pCLAs, although future studies are needed to compare the efficacy of plasma to that of chylous fluid in pCLAs. Although we were able to detect these low VAFs, it will be important to develop more sensitive methods that could be used to track VAF and measure therapeutic response. This low VAF may not represent the real allele fraction in participants owing to the unconventional sample type, but the variant itself could inform proper molecular-guided therapy.
Importantly, sequencing of gDNA isolated from CD31+ cells that were enriched from lymphatic fluid identified variants with high VAF, validating our enrichment strategy. For example, in one CCLA participant (CLA149), an amplicon panel on gDNA isolated from enriched endothelial cells revealed a hotspot KRAS variant (c.35G>A, p.G12D, VAF = 44%), suggesting that culture may enrich cell populations carrying the variant or that the variant is mainly carried in the endothelial cells. This indicated that development of lymphatic fluid-based molecular diagnostics coupled with the sensitive sequencing method is likely to be highly efficacious for resolving pCLA cases.
Of note, this is the largest prospective molecular study of individuals with pCLAs (n = 104). Prior to our study, it was widely accepted that GLA was caused by somatic variants in PIK3CA35, and that KLA was caused by a specific recurrent somatic variant in NRAS29. We identified pathogenic somatic variants as novel causes in different CLAs, including PIK3CA in CCLA and KLA, HRAS and BRAF in GLA, KRAS in KLA and MAP2K1 as a candidate gene for CCLA. We also confirmed BRAF as a cause of CCLA and HRAS as a cause of KLA14,28. Interestingly, participants CVA119 and CVA09 presented with multiple lymphatic malformations and conduction abnormalities on DCMRL. Although conduction abnormalities may be seen in GLA, these participants with variants in genes typically seen in CCLA may have a new phenotype and disease entity that encompasses both GLA and CCLA. This hypothesis is interesting to consider given the demonstration of novel genotypes associated with multiple CLA phenotypes.
Although we recently discovered KRAS variants in CCLA14,36, we report here two additional CCLA cases, reinforcing the association. Furthermore, we confirmed two KRAS variants (c.35G>A, p.G12D, 0.55% and c.167_211dup, p.(Gln70_Tyr71ins15), 1.22%) in one KLA and GLA case each. Considering that KRAS was recently studied in GSD37,38, it appears that KRAS variants can cause all CLA types (that is, CCLA, GLA, KLA and GSD) in addition to capillary malformations and AVMs39. Given these findings, further translational work is needed to evaluate how pathogenic variants in the same gene can cause different pCLA phenotypes.
In summary, we report a comprehensive sequencing and validation approach by resolving the molecular diagnosis for 156 of 356 unrelated participants (43.8%) with vascular anomalies. Deep exome sequencing is an efficacious strategy, especially for dual diagnoses, when high quality tissue samples are available. Smaller gene panels based on UMIs provide sufficient sequencing depth and present a robust way to identify the molecular etiology of vascular anomalies, from DNA isolated from FFPE or cfDNA from multiple sources. Importantly, use of cfDNA from plasma and chylous fluid provides a promising alternative, less invasive sampling strategy that will improve outcomes for patients. Such a comprehensive approach may be considered for clinical laboratories to establish molecular diagnoses for vascular anomalies, especially pCLA. Our findings enabled us to identify previously undescribed genotype–phenotype associations, inform decision making for currently available therapies, and may help provide a knowledge base for the design and implementation of future clinical trials along with the emergence of multiple inhibitors in both PI3K–mTOR and Ras–MAPK pathways.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41591-023-02364-x.
Methods
Study cohort
In total, a cohort of 356 unrelated participants or legally authorized representatives or parents of minors provided consent to join a research study approved by the Institutional Review Board of The Children’s Hospital of Philadelphia (CHOP) (protocols #06-004886 and #16-013278) over 7 years (April 2015–September 2022). Written informed consent for publication of clinical photographs was obtained separately at CHOP directly from participants or from legally authorized representatives or parents of minors. We enrolled participants of both sexes, and this was determined from the medical record. Sex was not an inclusion criterion for the study. None of the participants received any compensation. Participants were included if they had a vascular anomaly, including vascular malformation, CLA or vascular tumor. Participants with vascular malformations generally due to somatic mosaicism were included if they had an affected tissue sample (including fresh lesion biopsy, frozen tissue, FFPE block, or unstained slides from previous biopsy or surgical excision) or a plasma cfDNA sample collected.
After review of history, physical exam, pathology, imaging and clinical genetic studies, a multidisciplinary panel of expert clinicians (hematologists, oncologists, geneticist, radiologists, cardiologists, dermatologist, pathologists, pulmonologists and plastic surgeons) from the CHOP Comprehensive Vascular Anomalies Frontier Program provided a diagnosis using the ISSVA 2018 classification3. Two participants (CVA02 and CVA116) did not have a recognizable phenotype after expert panel review; therefore, they were categorized as unclassified vascular malformations.
Seventeen participants (13 with P/LP variants and four with VUS; Supplementary Table 1) were previously described by our group, including one clinical research study4,9,10,14. Inclusion of these participants enabled us to better evaluate the diagnostic utility of different sequencing methods and sample types.
Impact of genetic testing
Participants with a P/LP variant identified by genetic testing were evaluated for the impact of the genetic result on their medical care in two ways: whether it redefined clinical diagnosis and whether it guided the type of medical therapy recommended by the team. Genetic testing was defined as impacting diagnosis if the molecular result led to a change in diagnosis or provided an additional diagnosis (compared to the diagnosis after clinical evaluation). In our multidisciplinary clinic, participants often receive an updated diagnosis after initial clinical evaluation. In other centres without VA experience, genetic testing may have a greater impact in providing a precise diagnosis.
Molecular diagnosis can affect medical therapy in many ways. Sirolimus is a first-line medical therapy for several conditions and may be prescribed based on phenotype alone (without genotype). However, recent research led to the approval of alpelisib for PIK3CA-related conditions, and small case studies demonstrate the efficacy of trametinib, a MEK inhibitor, for CLAs. Therefore, we defined the impact of molecular diagnosis on therapy in four ways. The first is that the molecular diagnosis supports the current therapy (‘supports’). The second is that the molecular diagnosis leads to a change in therapy or a change is planned based on the results (‘changes’). The third way is that the molecular diagnosis leads to the initiation of medical therapy (‘initiates’). The final way is that the molecular diagnosis does not support the medical therapy.
For participants on medical therapy, responses were classified as ‘worsened’ if their primary disease showed progression, ‘stable’ if there was no change in status, ‘mixed’ if there was improvement in one domain but worsening in others or ‘improved’ if there was general improvement in at least one or more characteristic of the primary disease. Some participants were excluded from analysis if medication was recently initiated and it was too early to assess response or if this information was unavailable from the records.
Acquired lymphatic conduction disorder
Some, but not all individuals with congenital heart disease and related cardiac surgery will develop lymphatic conduction disorders17. Therefore, we reasoned that acquired lymphatic conduction disorders may be caused by somatic mosaicism. We enrolled 88 such participants with unknown pathophysiology, and 29 fluid DNA or cfDNA samples underwent either deep exome or panel sequencing. As part of the cohort of 356 participants, written informed consent for these participants was similarly obtained directly from participants or from legally authorized representatives or parents of minors. In addition to germline variants that underlie structural defects, such as a DNAH5 nonsense variant, compound heterozygous variants in CFTR and NF1 truncating variant, one pathogenic somatic variant, c.547C>T, p.R183C, with a VAF of 3.2%, in GNA11 was identified in one participant with CCLA and Down syndrome. The participant (CLA185) had a history of atrioventricular septal defect and tetralogy of Fallot status post repair (partial ASD closure, atrioventricular canal repair, pulmonary artery reconstruction, ligation and division of the patent ductus arteriosus, repair of tetralogy of Fallot with non-transannular patch and pulmonary valvotomy), pulmonary hypertension on sildenafil, high-risk B-cell acute lymphoblastic leukemia in remission, bilateral chylous effusions and reticulated capillary malformation. DCMRL demonstrated the presence of a small, attenuated thoracic duct above the level of the mid-mediastinum with connection to the left venous angle and diffuse retrograde hepatic and mesenteric flow with dispersion of contrast material within the lower mediastinum, bilateral pleural spaces, left pulmonary hilum, bowel, perihepatic and perisplenic tissues with resultant bilateral chylothorax, trace ascites and protein-losing enteropathy. Genomic study for the remaining 84 participants was unrevealing, including negative routine exome sequencing for all 84 probands, deep exome sequencing for seven participants when either tissue or fluid sample was available and ultra-deep panel sequencing for 29 participants with tissue, fluid or cfDNA, demonstrating that a somatic variant as the cause was uncommon (1/30). This is in agreement with previous work that there can be secondary causes of conduction disorders16.
Endothelial cell enrichment from fluid samples
Tissue culture dishes (10 cm, Falcon) were coated with 54 mg of fibronectin (Millipore, 1 mg cm−2) in 5 ml of 0.1 M sodium carbonate (Sigma), pH 9.4 for 20 min at 37 °C. After removal of the fibronectin solution, the plates were washed with PBS, and the PBS was removed. Cells were collected from lymphatic fluid samples by centrifugation (200×g, 10 min), resuspended in complete endothelial growth media (ECGM-MV2, Promocell) supplemented with 20% fetal bovine serum (FBS, VWR) and plated on one fibronectin coated plate. The following day, the medium was replaced with fresh ECGM-MV2 or 20% FBS. Media were replaced weekly. Depending on the amount of cell growth, cells were either processed for gDNA isolation or were processed using anti-CD31 magnetic Dynabeads (Thermo Fisher) as follows. For each sample, 20 ml of Dynabeads were washed twice with 0.1% bovine serum albumin (BSA) in PBS, using a DynaMag to isolate the beads. Cells were collected using the endothelial cell detach kit (Promocell) and pelleted by centrifugation (200×g, 10 min). Cells were resuspended in 0.5 ml of 0.1% BSA and were mixed with the washed beads. The cell–bead mixture was incubated on a rotator at 4 °C for 20 min. The volume was increased to 1 ml with 0.1% BSA, and the tube was placed on the DynaMag for 2 min. Unbound cells were removed. The tube was removed from the magnet, the cells were resuspended in 1 ml of 0.1% BSA, and the tube was placed back on the magnet for 2 min. Unbound material was removed and collected, and this wash procedure was repeated for a total of five washes. After washing, the beads were resuspended in 10 ml of ECGM-MV2 supplemented with 10% FBS and plated onto 10-cm tissue culture plates pre-coated with 0.5 mg/cm2 fibronectin as above. The unbound cells collected from the washes were pelleted and resuspended in 10 ml of ECGM-MV2 supplemented with 10% FBS and plated onto another series of 10-cm tissue culture plates pre-coated with 0.5 mg cm−2 fibronectin. To enrich for cells with pathogenic variants that may lend a growth factor advantage, isolated cells were cultured, with weekly media replacement, until proliferation was no longer observed. gDNA was then isolated (DNeasy Blood and Tissue kit, Qiagen).
Sequencing
All next-generation sequencing was performed on an Illumina NovaSeq in the Center for Applied Genomics at the Children’s Hospital of Philadelphia.
(Deep) exome sequencing and analysis.
Exome sequencing was performed using a Twist Human Core Exome Capture Kit (TWIST Bioscience). For DNA isolated from blood, approximately 50 million paired-end 101-bp reads were generated with a median insert size of ~250 bp mainly for germline variant calling. We performed routine exome sequencing from gDNA from blood or saliva for those who had pCLAs, GVM, primary lymphedema, CM-AVM syndrome, HHT and Parkes Weber syndrome, given that these phenotypes are typically caused by germline variants.
To identify both germline and somatic variants, we performed deep exome sequencing on DNA isolated from endothelial cell enrichment, tissue biopsy and archived FFPE. We constructed three libraries to increase fragment complexity for deep exome sequencing (~400×). Data were analyzed using BWA-mem v0.7.12 for alignment40 and Picard v1.97 for PCR duplication removal. The resulting BAM file was fed to GATK-Queue v2.6.5 for germline variant calling and GATK-Mutect2 v4.1.4.0 for somatic variant calling41. ANNOVAR42 and SnpEff43 were then used to functionally annotate the variants and collect minor allele frequency (MAF) data from the 1000 Genomes Project, ESP6500SI, ExAC, gnomAD and Kaviar. Subsequent variant filtration and prioritization were based on MAF in either population dataset or function annotation such as non-synonymous, exonic, splicing-altering and frameshift. Subsequent gene prioritization was performed on the basis of deleterious prediction and biological relevance by referring to the Online Mendelian Inheritance in Man (OMIM) database and Human Gene Mutation Database (HGMD).
Amplicon-based gene panel with ultra-deep targeted sequencing and analysis.
A PCR-based amplicon custom 35-gene panel (AKT1, AKT2, AKT3, ARAF, BRAF, CBL, EPHB4, FLT4, GNA11, GNA14, GNAQ, HRAS, ITGA9, KDR, KRAS, LZTR1, MAP2K1, MTOR, NF1, NRAS, PIK3CA, PTEN, PTPN11, RAF1, RASA1, RIT1, SHOC2, SMARCA4, SOS1, SPRED1, TEK, TP53, TSC1, TSC2 and VEGFC) was designed with the Illumina AmpliSeq platform v7.454, with a total targeted region of 96 kb. Libraries were generated with the Illumina AmpliSeq Library PLUS and were sequenced to an average coverage depth of ~82,000× (Fig. 2b,h) with paired-end 151-bp reads. Variant calling was performed by using Illumina’s DNA amplicon workflow, which incorporates BWA40 and Pisces44. The resulted BAM files had an effective mean coverage depth of 44,941× (Fig. 2i). The resulted VCF files were subsequently annotated similarly by using ANNOVAR and SnpEff, and the candidate variant was prioritized as mentioned above.
UMI-based gene panel with ultra-deep targeted sequencing and analysis.
A capture-based custom 49-gene panel (AKT1, AKT2, AKT3, ARAF, BRAF, CBL, CCM2, CDH5, EPHB4, FLT4, FOXC2, GATA2, GLMN, GNA11, GNA14, GNAQ, HRAS, IDH1, IDH2, ITGA9, KDR, KRAS, KRIT1, LZTR1, MAP2K1, MAP2K2, MAP3K3, MTOR, NF1, NF2, NRAS, PIK3CA, PROX1, PTEN, PTPN11, PTPN14, RAF1, RASA1, RIT1, RRAS, SHOC2, SOS1, SOS2, SPRED1, TEK, TP53, TSC1, TSC2 and VEGFC) with UMIs was designed with IDT, and libraries were constructed with an IDT xGen Prism DNA Library Prep Kit and Wash Kit following the manufacturer’s instruction. Libraries were sequenced to an average coverage depth of ~87,000× (Fig. 2b) with a targeted region of 233 kb with paired-end 101-bp reads. Read alignment, deduplication, error correction based on UMI information and variant calling were performed using BWA-mem v0.7.12, Picard v2.26.0, fgbio v1.3.0 and VarDict v1.8.2, respectively. Essentially, all reads with the same start–stop position in alignment and UMI could be grouped, filtered and collapsed as a single read family, with the most likely sequence being retained. Then, after error correction, all read family sequences could be used for variant calling to greatly reduce false positive rates. The effective mean coverage depth after error correction is provided in Fig. 2k. The resulting VCF files were subsequently annotated similarly by using ANNOVAR and SnpEff, and the candidate variant was prioritized as mentioned above.
Rationale of the tiered sequencing approach.
Our first-line testing approach with deep exome sequencing was cost-effective. Out of 86 (64%) sequenced samples (excluding acquired lymphatic anomalies), 55 were positive for somatic variants with VAFs ranging from 2.3% to 44%, the majority (77%) being a relatively low VAF (≤ 10%; Supplementary Table 1). However, VAF can be less than 2%. To test whether deep exome sequencing with additional sequencing depth would increase our capacity to discover somatic variants with a lower VAF (< 2%), we selected nine individuals with negative findings and one positive control and made additional exome libraries to increase the fragment complexity. Ten samples reached an average sequencing depth of 1,574× (ranging from 1,001× to 2,156×; Fig. 2g) with 94.0% and 60.6% of targeted regions covered at least 500× and 1,000×, respectively. Two previously negative lymphatic malformation cases were returned positive with a VAF of 1.7% and 2.0%, as well as the positive control with common venous malformations (Supplementary Table 1). These variants were subsequently validated by BDA qPCR and Sanger assay. This suggested that deeper exome sequencing could identify low VAF (< 2%). However, due to the substantially increased sequencing cost, we decided to apply the targeted gene panel for the unresolved cases. Among them, six samples had enough DNA for an ultra-deep UMI-based gene panel, which was performed with an average raw depth of 85,252× and an average effective (after error correction) depth of 1,368×. Ultimately, three additional cases (CCLA, venous malformation and PTEN hamartoma) were resolved with a range of VAF (BRAF, c.1799T>A, p.V600E, 0.21%; TEK, c.2740C>T, p.L914F, 1.3%; and PTEN, c.264_265insCCTTTTGAAG, p.D92Afs*3, 15%, respectively) (Supplementary Table 1). Interim analysis demonstrated that the UMI panel was more efficacious than deep exome sequencing on gDNA isolated from FFPE. Therefore, we then used a UMI panel for FFPE samples, as FFPE always yielded a relatively low amount of DNA, which was insufficient for both approaches. This result led us to pursue a UMI-based panel for all cases that were not resolved by deep exome sequencing. Indeed, superior coverage of ultra-deep panel sequencing led to multiple molecular diagnoses with the majority of the VAF being less than 1% (Supplementary Table 1). Generally, clinical diagnostic laboratories set a VAF criterion of 1% or higher, which is limited by some intrinsic factors in the testing such as ~1% error rate in next-generation sequencing platforms and amplification error during PCR enrichment. The UMI-based approach provided an opportunity to correct such errors by utilizing the 8-bp UMI on both ends of sequencing reads.
Variant confirmation.
The BDA qPCR and Sanger assay, described elsewhere45, was used to validate somatic variants with relatively low VAF. The qPCR and Sanger assay, including primer and blocker (Supplementary Table 9), was purchased from NuProbe or IDT. Each tested sample (that is, tissue DNA, cfDNA, or peripheral blood gDNA) was mixed with blocker and without blocker in two separated tubes for a qPCR run. PCR reactions were then performed in a final volume of 10 ml using the ViiA7 real-time PCR system (Thermo Scientific) with incubation at 95 °C for 3 min, followed by 45 cycles of 95 °C for 10 s and 60 °C for 30 s. Subsequently, Sanger sequencing of the amplified products of the tube with blocker was used to visualize the somatic variant, and the tube without blocker acted as control.
Cellular characterization of variants in primary human dermal lymphatic endothelial cells.
Primary human dermal lymphatic endothelial cells acquired from Promocell (catalog number C-12216; from juvenile foreskin) were retrovirally transduced with wild type or different variants and cultured for 48–72 h. Transduced cells were plated in a 96-well plate (20,000 cells per well) in the presence of carrier (0.1% DMSO) or 300 nM trametinib and cultured for 24 h. Cells were lysed, lysates were cleared by centrifugation and analyzed by SDS–PAGE and western blotting with pERK (Cell Signaling Technology no. 4376) and GAPDH (Santa Cruz Biotechnology no. sc-47724). Data were quantified, and the p-ERK signals were normalized to the GAPDH signals. These ratios were further normalized to the untreated wild type ratio and plotted. For sprouting assays, transduced cells were cultured for 24 h under conditions that promote spheroid formation. Spheroids were embedded in 2% collagen and cultured for a further 24 h. Spheroids were imaged with Z-stacks on an EVOS XL microscope, images were flattened, and sprouts were counted and measured for length using the NeuronJ plugin for FIJI. Cumulative sprout length per sphere was calculated by summing all of the sprout lengths on each individual sphere, and the number of sprouts per sphere was counted.
Zebrafish transgene expression.
All procedures using zebrafish were approved by the Institutional Animal Care and Use Committee of CHOP (IAC 001154) and the Animal Care and Use Committee of NICHD (ASP 22-032) in accordance with the Guide for the Care and Use of Laboratory Animals (NRC 2011). Human KRAS cDNAs were cloned into the p3E multisite gateway vector p3E. Expression constructs were assembled using a Tol2 backbone and the multisite Gateway cloning system46. The resulting constructs have the lymphovenous mrc1a promoter, followed by mCherry and then KRAS in the same reading frame. To express transgenes, vectors (25 pg) were co-injected with transposase messenger RNA (25 pg) into tg(mrc1a:GFP) fish. The zebrafish mrc1a promoter drives expression in the vein and lymphatic endothelium. Larvae were mounted in low-melting agarose, and multiple Z-stack images were taken with a Zeiss LSM710 confocal microscope using a ×20 lens. Confocal Z-stacks of images were superimposed into a maximum intensity projection using Zeiss Zen software. To quantify edema, transgene vector was co-injected with transposase messenger RNA into wild type zebrafish of the EK strain at the one-cell stage, and larvae were screened for edema at 5 days post fertilization. Zebrafish between 0 and 7 days post fertilization were sex neutral, so there was no bias towards male or female animals in the study. Tg(mrc1a:GFP)y251 zebrafish were a gift from the Weinstein Lab and in the casper background47.
Extended Data
Extended Data Fig. 1 |. Functional characterization of BRAF-F486S, RAF1-T145P, and KRAS-A146T.
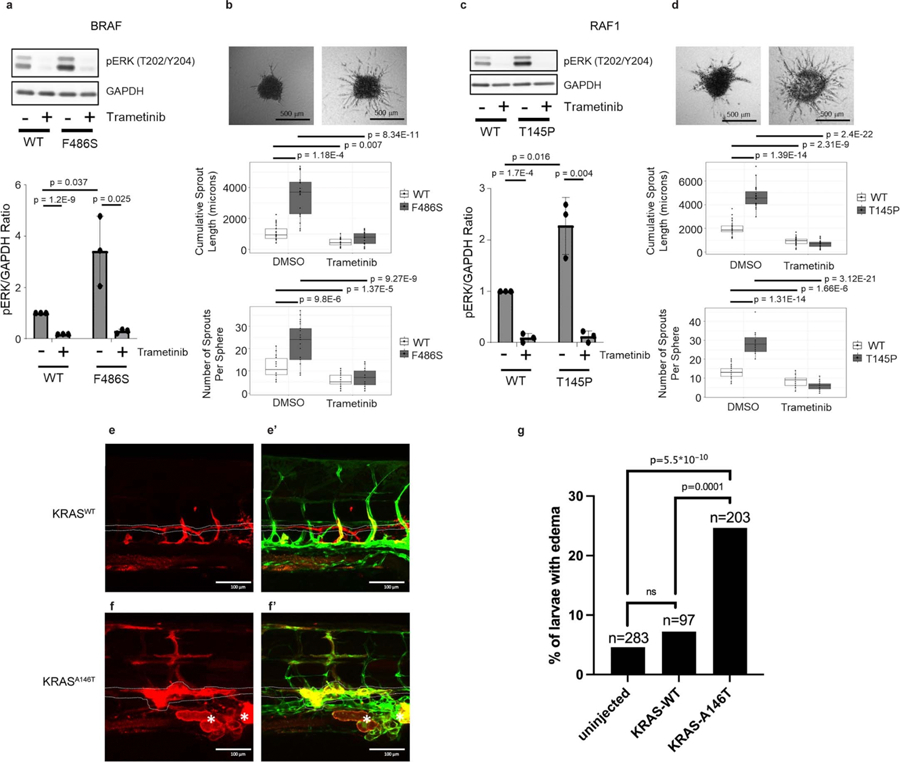
Transduction of BRAF-F486S (a) or RAF1-T145P (c) significantly increased the level of p-ERKs and Trametinib treatment led to a significant reduction of p-ERKs. Three-dimensional lymphatic spheroid sprouting assay showed elevated sprouting activity in HDLECs expressing BRAF-F486S (b) or RAF1-T145P (d) compared to its WT as measured by both cumulative sprout length and number of sprouts. Trametinib treatment led to a significant reduction of both cumulative sprout length and number of sprouts. For a-d, three independent experiments were performed. P values were calculated using 2-sided t-tests and included in each panel (degree of freedom was included in the Source Data for Extended Data Fig. 1), and corrected for multiple testing using the FDR (Benjamini and Hochberg) method. Bar graphs in panels a and c represent mean fold change in the ratio of pERK to GAPDH, normalized to the untreated wild type. Error bars represent standard deviations. In the box and whisker plots in panels b and d, the line in the middle of the box represents the medians, tops and bottoms of the boxes represent the 25th and 75th quartiles respectively, and the whiskers extend to 1.5 times the interquartile range beyond the 25th and 75th quartiles. All the data points that are summarized by the boxplots are superimposed as dots on the plots; minima and maxima can be determined by the highest and lowest dots. e, e’, Expression of KRASWT had no impact on lymphatic vessel morphology in trunk. f, f’, Expression of KRASA146T resulted in lymphatic tissue expansion (asterisks) and dilation of the thoracic duct (dotted line). Red: Expression of transgene in trunk of zebrafish at 5dpf, Green: mrc1a:GFF labeling lymphatic vessels. g, Quantitation of WT EK larvae that were assayed for pericardial edema at 5 dpf. Injected embryos were screened for transgenic mCherry expression in endothelial cells prior to quantitation so that only transgenic expressing embryos were counted. The p-values in the graph were calculated via Fisher’s Exact tests (two-sided) between samples as indicated, followed by Bonferroni correction. Indicated n are the total number of larvae assayed per condition.
Supplementary Material
Acknowledgements
We thank the patients and their families for participating in the research. We also thank the Comprehensive Vascular Anomaly Program for clinical care. The work was supported by a Children’s Hospital of Philadelphia Frontier Program Grant (D.M.A., H.H.), Children’s Hospital of Philadelphia K-Readiness Grant (S.E.S.), the National Center for Advancing Translational Sciences of the National Institutes of Health (NIH) under award number 5R21TR003331 (D.L.), a research grant from the Lymphatic Malformation Institute (D.L.), and the Eunice Kennedy Shriver National Institute of Child Health and Human Development under award number ZIA-HD009003-01 (S.E.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. We thank A. Hoofring of NIH Medical Arts for his assistance with the preparation of Fig. 2c.
Footnotes
Competing interests
H.H. and CHOP are equity holders in Nobias Therapeutics, which is developing MEK inhibitor therapy for complex lymphatic anomalies. D.M.A. is a consultant for Novartis Pharmaceuticals and Nobias. K.M.S. is a consultant for Novartis Pharmaceuticals, which makes Vijoice (alpelisib), a selective PI3Ka inhibitor. The remaining authors declare no competing interests.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41591-023-02364-x.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41591-023-02364-x.
Peer review information Nature Medicine thanks Douglas Marchuk and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling editor: Anna Maria Ranzoni, in collaboration with the Nature Medicine team.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
De-identified next-generation sequencing data for participants are available through controlled access at dbGaP with accession number phs003197.v1.p1. Data will be made available for secondary research only after investigators have obtained approval from the NIH to use the requested data for a particular project. Source data are provided with this paper.
References
- 1.Makinen T, Boon LM, Vikkula M & Alitalo K Lymphatic malformations: genetics, mechanisms and therapeutic strategies. Circ. Res 129, 136–154 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Queisser A, Seront E, Boon LM & Vikkula M Genetic basis and therapies for vascular anomalies. Circ. Res 129, 155–173 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Wassef M et al. Vascular anomalies classification: recommendations from the international society for the study of vascular anomalies. Pediatrics 136, e203–e214 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Li D et al. ARAF recurrent mutation causes central conducting lymphatic anomaly treatable with a MEK inhibitor. Nat. Med 25, 1116–1122 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Adams DM et al. Efficacy and safety of sirolimus in the treatment of complicated vascular anomalies. Pediatrics 137, e20153257 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ricci KW et al. Efficacy of systemic sirolimus in the treatment of generalized lymphatic anomaly and Gorham-Stout disease. Pediatr. Blood Cancer 66, e27614 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hammill AM et al. Sirolimus for the treatment of complicated vascular anomalies in children. Pediatr. Blood Cancer 57, 1018–1024 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Triana P et al. Sirolimus in the treatment of vascular anomalies. Eur. J. Pediatr. Surg 27, 86–90 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Foster JB et al. Kaposiform lymphangiomatosis effectively treated with MEK inhibition. EMBO Mol. Med 12, e12324 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dori Y et al. Severe lymphatic disorder resolved with MEK inhibition in a patient with Noonan syndrome and SOS1 mutation. Pediatrics 146, e20200167 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Chowers G, et al. Treatment of severe kaposiform lymphangiomatosis positive for NRAS mutation by MEK inhibition. Pediatr Res 10.1038/s41390-022-01986-0 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pfeiffer F et al. Systematic evaluation of error rates and causes in short samples in next-generation sequencing. Sci. Rep 8, 10950 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith T, Heger A & Sudbery I UMI-tools: modeling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Res 27, 491–499 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu M et al. Genetics etiologies and genotype phenotype correlations in a cohort of individuals with central conducting lymphatic anomaly. Eur. J. Hum. Genet 30, 1022–1028 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mackie AS, Veldtman GR, Thorup L, Hjortdal VE & Dori Y Plastic bronchitis and protein-losing enteropathy in the Fontan patient: evolving understanding and emerging therapies. Can. J. Cardiol 38, 988–1001 (2022). [DOI] [PubMed] [Google Scholar]
- 16.Dori Y & Smith CL Lymphatic disorders in patients with single ventricle heart disease. Front Pediatr 10, 828107 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh RM et al. Prevalence and cause of early Fontan complications: does the lymphatic circulation play a role? J. Am. Heart Assoc 9, e015318 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li D et al. Pathogenic variant in EPHB4 results in central conducting lymphatic anomaly. Hum. Mol. Genet 27, 3233–3245 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Byrne AB et al. Pathogenic variants in MDFIC cause recessive central conducting lymphatic anomaly with lymphedema. Sci. Transl. Med 14, eabm4869 (2022). [DOI] [PubMed] [Google Scholar]
- 20.Li D et al. Expanded phenotypic spectrum of JAG1-associated diseases: central conducting lymphatic anomaly with a pathogenic variant in JAG1. Clin. Genet 99, 742–743 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Konczyk DJ et al. Arteriovenous malformation MAP2K1 mutation causes local cartilage overgrowth by a cell-non autonomous mechanism. Sci. Rep 10, 4428 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zenner K et al. Cell-free DNA as a diagnostic analyte for molecular diagnosis of vascular malformations. Genet. Med 23, 123–130 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun Y et al. Cell-free DNA from plasma as a promising alternative for detection of gene mutations in patients with Maffucci syndrome. Hereditas 159, 4 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wiggins JM, Ali S & Polsky D Cell-free DNA in dermatology research. J. Invest. Dermatol 142, 1523–1528 e1521 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palmieri M et al. Cell-free DNA next-generation sequencing liquid biopsy as a new revolutionary approach for arteriovenous malformation. JVS Vasc. Sci 1, 176–180 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Biderman Waberski M et al. Urine cell-free DNA is a biomarker for nephroblastomatosis or Wilms tumor in PIK3CA-related overgrowth spectrum (PROS). Genet. Med 20, 1077–1081 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen WL et al. The utility of cerebrospinal fluid-derived cell-free DNA in molecular diagnostics for the PIK3CA-related megalencephaly-capillary malformation (MCAP) syndrome: a case report. Cold Spring Harb. Mol. Case Stud 8, a006188 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allen-Rhoades W et al. Cellular variant of kaposiform lymphangiomatosis: a report of three cases, expanding the morphologic and molecular genetic spectrum of this rare entity. Hum. Pathol 122, 72–81 (2022). [DOI] [PubMed] [Google Scholar]
- 29.Barclay SF et al. A somatic activating NRAS variant associated with kaposiform lymphangiomatosis. Genet. Med 21, 1517–1524 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schuart C et al. Cutis marmorata telangiectatica congenita being caused by postzygotic GNA11 mutations. Eur. J. Med. Genet 65, 104472 (2022). [DOI] [PubMed] [Google Scholar]
- 31.Rofes P et al. Mosaicism in PTEN-new case and comment on the literature. Eur. J. Hum. Genet 30, 641–644 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou XP et al. Germline and germline mosaic PTEN mutations associated with a Proteus-like syndrome of hemihypertrophy, lower limb asymmetry, arteriovenous malformations and lipomatosis. Hum. Mol. Genet 9, 765–768 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Caux F et al. Segmental overgrowth, lipomatosis, arteriovenous malformation and epidermal nevus (SOLAMEN) syndrome is related to mosaic PTEN nullizygosity. Eur. J. Hum. Genet 15, 767–773 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Konczyk DJ et al. Arteriovenous malformation associated with a HRAS mutation. Hum. Genet 138, 1419–1421 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodriguez-Laguna L et al. Somatic activating mutations in PIK3CA cause generalized lymphatic anomaly. J. Exp. Med 216, 407–418 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sheppard SE et al. Lymphatic disorders caused by mosaic, activating KRAS variants respond to MEK inhibition. JCI Insight 10.1172/jci.insight.155888 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Homayun-Sepehr N et al. KRAS-driven model of Gorham-Stout disease effectively treated with trametinib. JCI Insight 6, e149831 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nozawa A et al. A somatic activating KRAS variant identified in an affected lesion of a patient with Gorham-Stout disease. J. Hum. Genet 65, 995–1001 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Chang CA et al. Novel findings and expansion of phenotype in a mosaic RASopathy caused by somatic KRAS variants. Am. J. Med. Genet. A 185, 2829–2845 (2021). [DOI] [PubMed] [Google Scholar]
- 40.Li H & Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DePristo MA et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43, 491–498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang K, Li M & Hakonarson H ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cingolani P et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dunn T et al. Pisces: an accurate and versatile variant caller for somatic and germline next-generation sequencing data. Bioinformatics 35, 1579–1581 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu LR, Chen SX, Wu Y, Patel AA & Zhang DY Multiplexed enrichment of rare DNA variants via sequence-selective and temperature-robust amplification. Nat. Biomed. Eng 1, 714–723 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Villefranc JA, Amigo J & Lawson ND Gateway compatible vectors for analysis of gene function in the zebrafish. Dev. Dyn 236, 3077–3087 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jung HM et al. Development of the larval lymphatic system in zebrafish. Development 144, 2070–2081 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
De-identified next-generation sequencing data for participants are available through controlled access at dbGaP with accession number phs003197.v1.p1. Data will be made available for secondary research only after investigators have obtained approval from the NIH to use the requested data for a particular project. Source data are provided with this paper.