

SUMMARY
p53 was discovered 45 years ago as a SV40 large T antigen binding protein, coded by the most frequently mutated TP53 gene in human cancers. As a transcription factor, p53 is tightly regulated by a rich network of post-translational modifications to execute its diverse functions in tumor suppression. Although early studies established p53-mediated cell-cycle arrest, apoptosis, and senescence as the classic barriers in cancer development, a growing number of new functions of p53 have been discovered and the scope of p53-mediated anti-tumor activity is largely expanded. Here, we review the complexity of different layers of p53 regulation, and the recent advance of the p53 pathway in metabolism, ferroptosis, immunity, and others that contribute to tumor suppression. We also discuss the challenge regarding how to activate p53 function specifically effective in inhibiting tumor growth without harming normal homeostasis for cancer therapy.
Keywords: p53, tumor suppression, p63, p73, MDM2, MDMX, apoptosis, cell-cycle arrest, senescence, genome stability, metabolism, ferroptosis, stem cell dynamics, cell competition, metastasis, immunity, p53 mutation, targeting p53, cancer treatment
INTRODUCTION
Discovered in 1979, the p53 protein, encoded by the tumor protein p53 (TP53, or p53) gene, has captivated the attention of both the cancer research community and the pharmaceutical industry, positioning it the most extensively studied gene.1 Over the past 45 years of research, p53 has consistently yielded both surprise and excitement, albeit accompanied by persistent confusions. Numerous efforts have been dedicated to understanding p53. In return for this, we now have a better picture of this gene (Figure 1). However, there are still certain aspects of p53 that remain unclear. In this review, we first introduce the basic information about p53 and its regulatory mechanisms. We then summarize the functions of p53 and its roles in both normal physiological conditions and various pathological disorders, particularly cancers. Finally, we discuss the therapeutic applications of targeting p53 and the unaddressed issues in the p53 field. Due to space constraints, many original research papers and important reviews on p53 could not be cited.
Figure 1. Timeline of research in the p53 field over the past 45 years.
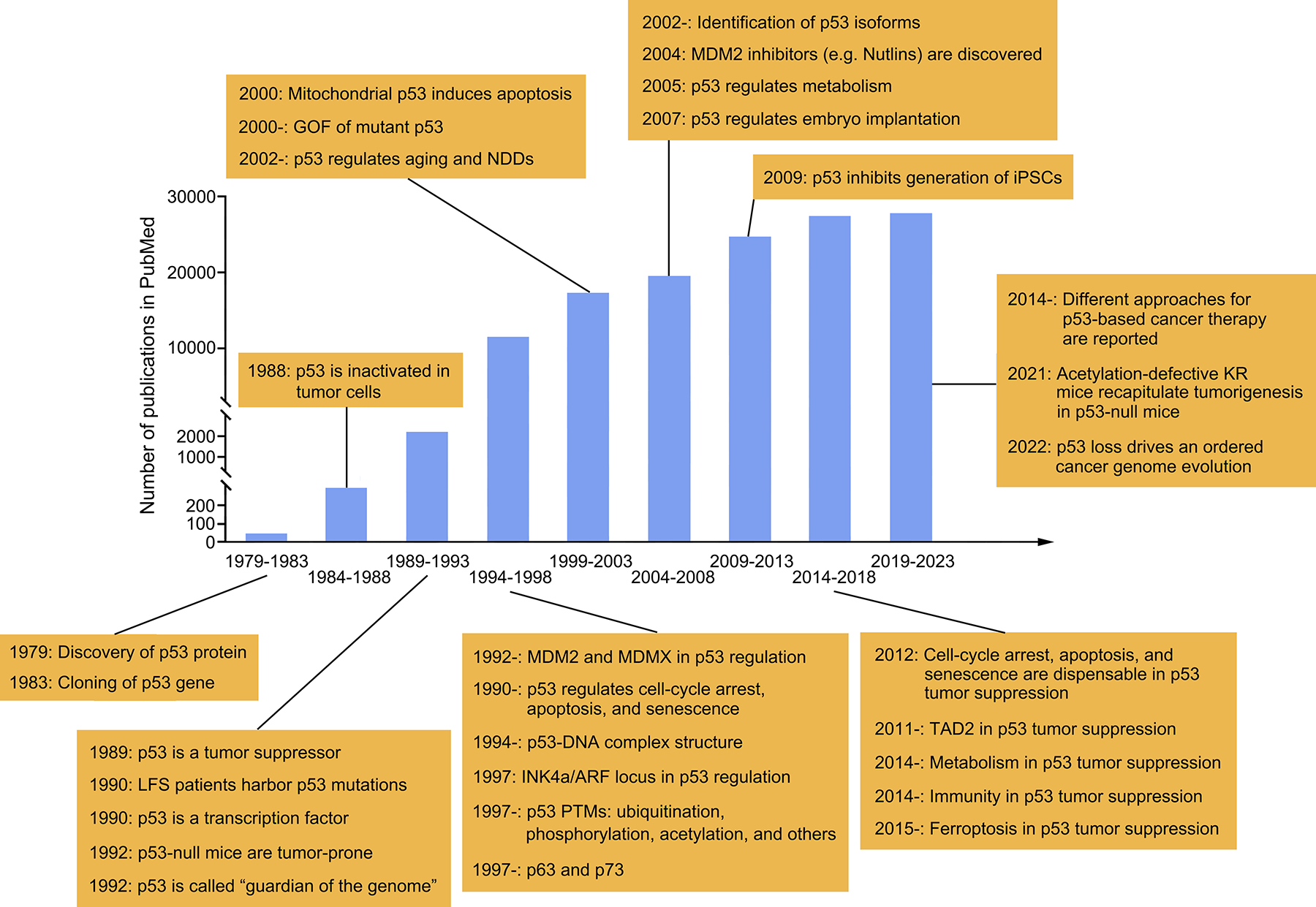
This figure shows the number of publications recorded in PubMed every five years since 1979, along with key discoveries about p53. Due to space constraints, many excellent studies cannot be included here. LFS, Li-Fraumeni syndrome; PTM, post-translational modification; GOF, gain-of-function; NDD, neurodegenerative disease; iPSC, induced pluripotent stem cell; TAD2, transactivation domain 2; KR, lysine-to-arginine mutation.
DISCOVERY, EVOLUTION, AND STRUCTURE OF p53
The study of p53 has experienced several pivotal twists and turns over the past decades, mirroring the tortuous development of oncology, characterized by significant paradigm shifts.2–5 In 1979, the p53 protein was independently discovered by several laboratories during research on cells transformed by simian virus 40 (SV40) or through other methods.6–9 Initially, p53 was believed to be an oncogene involved in cell transformation. However, in 1989, a series of papers revealed that wild-type (WT) p53 is, in fact, a tumor suppressor (Figure 1).2–5,10–14
The human p53 gene family comprises three members p53, p63, and p73. This family originated at least 800 million years ago and followed by gene duplication and structural diversification.15,16 An intriguing discovery is that the p53 family gene is present in some unicellular eukaryotes, such as the choanoflagellate Monosiga brevicollis, suggesting its important role in the evolution of multicellular organisms. The p53 family gene in unicellular eukaryotes is believed to maintain genome stability in response to various stresses. The p53 gene emerged in the earliest vertebrates and has since been evolutionarily conserved.15,16 The presence of p53 family in lower organisms is remarkable, given their minimal risk of cancer. This raises a question about the primitive role of p53 family in evolution. It is possible that this family was initially involved in maintaining the integrity of germline cells, with its well-known tumor suppression capability emerging much later.
The p53 protein primarily functions as a transcription factor (TF), although TF-independent activity has also been implicated (Figure 2).15,17,18 The full-length p53 protein (FLp53) in human comprises 393 amino acids, which are organized into five different domains: the N-terminal transactivation domain (TAD), the proline-rich domain (PRD), the central DNA-binding domain (DBD), the tetramerization domain (TD), and the C-terminal regulatory domain (CTD). The TAD of p53 is divided into two subdomains TAD1 and TAD2.15,19 In unstressed cells, p53 protein adopts a mixture of monomeric, dimeric, and tetrameric states, with dimer predominating.20 Upon diverse types of stress signals (including DNA damage, oncogene activation, ribosomal stress, telomere erosion, nutrient deprivation, and hypoxia), the majority of p53 proteins rapidly assemble into a functional tetramer (a dimer of dimers) via its TD. By using the DBD, this tetramer recognizes the p53 binding sites located at either the promoters or enhancers of target genes to modulate transcription. Unlike the well-folded DBD, whose structure has been solved, the TAD and CTD of p53 are intrinsically disordered, facilitating their interaction with cofactors for transcription mediation.15 These two domains are also the major regions to undergo post-translational modifications (PTMs).21 The PRD also contributes to the activity of p53.22 p53 is recruited to DNA via a specific response element (RE) composed by two decameric repeats: RRRCWWGYYY (R, purine; W, A or T; and Y, pyrimidine).23 p53 directly regulates the transcription of more than 300 target genes. In view of the indirect targets, p53 is believed to mediate the expression of several thousands of genes.24 Majority of the reported targets are protein-coding genes, but p53 also regulates various non-coding RNAs.25
Figure 2. Regulation of p53.
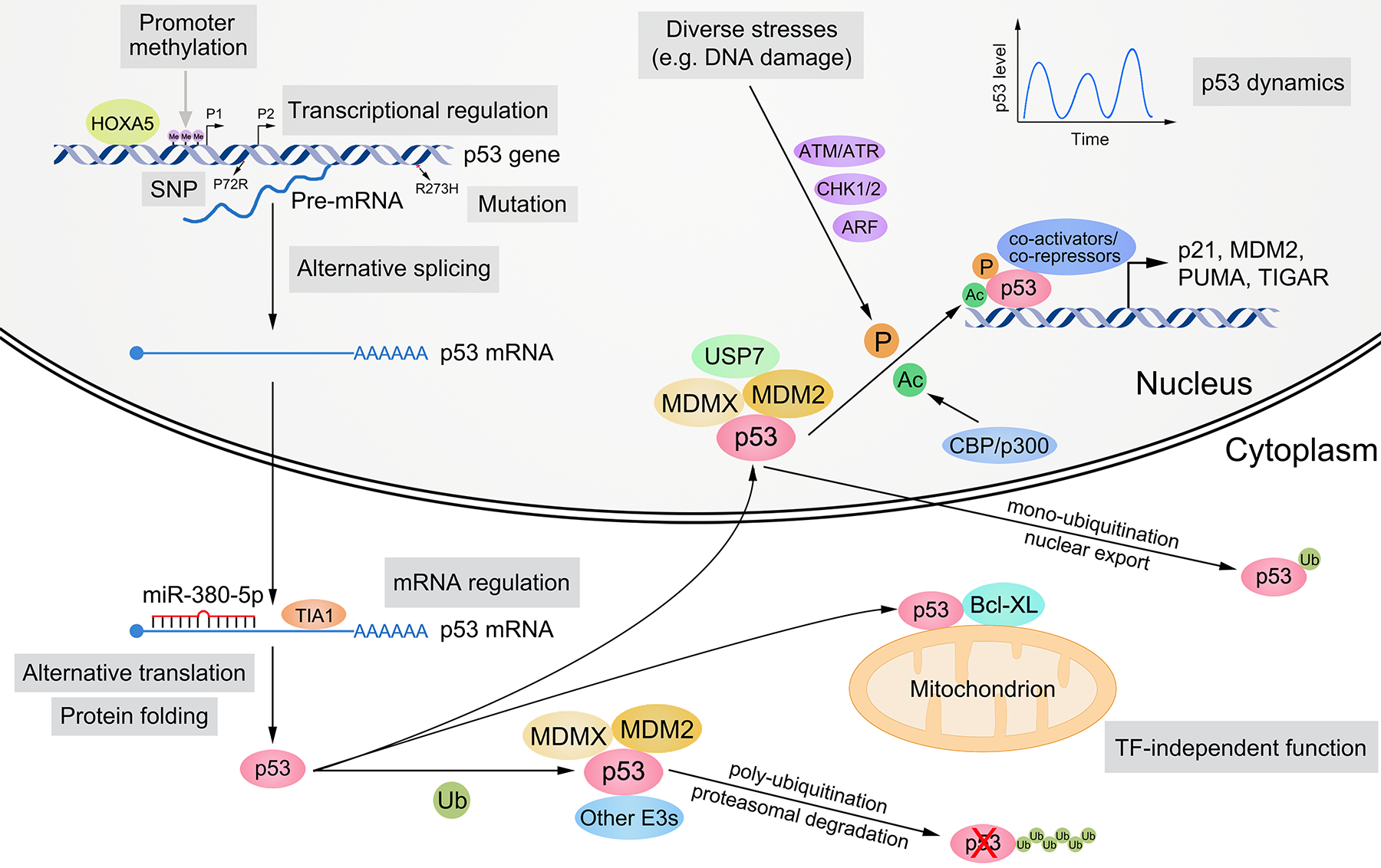
The expression and activity of p53 are controlled by multilayered regulation at the DNA, RNA, and protein levels. At the DNA level, SNPs (e.g. P72R) and mutations (e.g. R273H) may occur in the p53 gene. p53 possesses two promoters, which can be methylated and silenced. The transcription of p53 gene is activated or suppressed by various TFs (e.g. HOXA5). At the RNA level, the cellular localization, stability, and translation of p53 mRNA are modulated by RNA-binding proteins (e.g. TIA1) and ncRNAs (e.g. miR-380–5p). p53 pre-mRNA and mRNA can undergo alternative splicing and alternative translation, respectively. At the protein level, p53 folding, stability, cellular localization, DNA binding, transactivation ability, and target selection are primarily mediated by post-translational modifications (e.g. ubiquitination, phosphorylation, and acetylation) and cofactors (e.g. MDM2, MDMX, and CBP). Diverse stress signals (e.g. DNA damage) can activate p53, and its activity as a TF is highly dynamic. p53 also exhibits TF-independent function in cytoplasm (e.g. promoting apoptosis via interacting with Bcl-XL). P1 and P2, promoter 1 and 2; SNP, single nucleotide polymorphism; E3, E3 ubiquitin ligase; TF, transcription factor; Me, methylation; Ub, ubiquitination; P, phosphorylation; Ac, acetylation.
REGULATION OF p53: PTM IS THE KEY
To accurately execute its multifaceted functions, the expression and activity of p53 are subject to elaborate and multilayered regulation at the protein, DNA, and RNA levels (Figure 2).23,26
Regulation of p53 at the protein level
The p53 protein can undergo many types of PTMs, including ubiquitination, phosphorylation, acetylation, methylation, SUMOylation, NEDDylation, O-GlcNAcylation, ADP-ribosylation, UFMylation, hydroxylation, β-hydroxybutyrylation, sulfation, and isoLG adduction.21,27–29 Different stress signals determine the site and type of PTMs. Many PTMs of p53 are reversible. The overall effect of PTMs on p53 include altering its protein level, cellular localization, cofactor recruitment, target selectivity, and even protein aggregation.21,29,30 Among these, ubiquitination, phosphorylation, and acetylation are the most common and influential in affecting p53 activity.
Ubiquitination typically occurs at the C-terminal lysine residues of p53. Mouse double minute 2 homolog (MDM2) is the most well-known regulator of p53, ubiquitinating p53 to maintain low protein levels in unstressed cells or to export nuclear p53 to the cytoplasm.21 Many stimuli and regulators activate p53 through alleviating repression by MDM2.21 For instance, the p14ARF protein can stabilize p53 via inhibiting MDM2-mediated degradation of p53. Convincing evidence supporting the importance of MDM2-mediated p53 inhibition comes from mouse models, where the MDM2 deficiency-caused embryonic lethality can be rescued by deleting p53.31,32 Interestingly, MDM2 is itself a target gene of p53.33 The MDM2-p53 feedback loop forms the core in p53-associated pathways. MDMX (or MDM4), a family member of MDM2, although lacking E3 ubiquitin ligase activity itself, can form a heterodimer with MDM2 to enhance the degradation of p53. Other E3 ubiquitin ligases that can degrade p53 include ARF-BP1/HUWE1, COP1, CHIP, and Pirh2.21
Serine and threonine phosphorylation sites span across the p53 protein. The phosphorylation of p53 by ATM represents the earliest mechanism demonstrating how p53 responds to DNA damage.34,35 Phosphorylation at S15, T18, and S20 disrupts the binding and inhibition of p53 by MDM2, while enhancing interaction with transcription cofactors such as CBP. As a result, p53-mediated transcription is activated to induce cell-cycle arrest and apoptosis.36–38 Severe DNA damage further phosphorylates p53 at S46, strengthening apoptosis.39
Acetylation of several lysine residues in the DBD is critical for p53’s ability to activate key targets responsible for cell-cycle arrest, apoptosis, senescence, ferroptosis, and mTOR inhibition in a promoter-specific manner. The impact of p53 acetylation in tumor suppression is best illustrated by a series of acetylation-defective knock-in mouse models.40–46 For example, although the p53–3KR mutant, retaining its DNA binding activity, fails to activate the major targets such as p21 and PUMA critical for cell-cycle arrest, apoptosis, and senescence, the p53–3KR mutant mice are not tumor prone.41 However, further elimination of its ability to regulate ferroptosis and the mTOR pathway by p53–4KR and p53–5KR mutants recapitulates the loss of its tumor suppressor function as observed in p53-null mice.43 Notably, the role of acetylation in the CTD is complexed by the fact that the same lysine residues are also modified by methylation, ubiquitination, SUMOylation, and NEDDylation, in addition to acetylation. Thus, the CTD acetylation-defective mutant (p53–6KR and p53–7KR) mice fail to show dramatic impact in tumor suppression as these mutants eliminate both positive and negative effects on p53 function by different types of PTMs.45,46 Indeed, the acetylation–mimicking (p53-KQ) mutant mice show substantial p53 activation in transcription and tumor suppression without increasing p53 protein levels, underscoring the role of the CTD acetylation in vivo.42,44
Taken together, a gamut of the various PTMs cooperatively orchestrate the activity of p53. A serious caveat is that, albeit many PTMs are proven crucial for regulating p53 in vitro, the in vivo functions might be more complex.21 Knock-in mouse models serve as valuable tools for elucidating the functions of specific PTMs in modulating p53 activity.
At the protein level, recruitment of cofactors is another important parameter influencing p53 activity. p53 protein can bind to a plethora of interacting partners, including both activators and repressors, which profoundly affect its folding, stability, cellular localization, DNA binding, transactivation ability, and target selection.23,47 For example, a group of chaperone proteins regulate p53’s folding and stability.48–50 MDM2 and MDMX bind the TAD of p53 to restrict its transactivation activity, independently of MDM2’s role as an E3 ubiquitin ligase.51 Several transcription regulators such as PBRM1, SET, and Dicer are able to interact with p53 in an acetylation-dependent manner.40 The components of the m6A methyltransferase complex, METTL3 and RBM15, interact with p53 to selectively modify the mRNAs of a group of p53 target genes.52,53
The local chromatin structure and epigenetic state also play a crucial role in dictating p53’s DNA binding and effective transcriptional induction. In glioblastoma, BRD8 maintains a repressive chromatin state by retaining histone variant H2AZ at p53 target loci, thus blocking p53 recruitment.54 TRIM24 simultaneously binds p53 and unmethylated H3K4, impeding chromatin opening by p53.55 p53 can cooperate with other locally bound TFs to establish an accessible chromatin state and boost transcription.56
Regulation of p53 at the DNA and RNA levels
The modulation at the DNA and RNA levels substantially influences the protein level of p53.26 p53 gene possesses two promoters, which leads to alternative transcriptional initiation.57 The promoter region of the p53 gene can undergo DNA methylation and histone methylation, thereby impacting its transcription.58,59 Multiple TFs control the transcription of the p53 gene.26 The pre-mRNA of p53 may undergo alternative splicing.60 Besides, the stability, localization, and translation of p53 mRNA are all tightly regulated.61–63
p53 activation is not a simple all-or-none mode, but a dynamic process. The heterogeneity of cells, the characteristic of stresses, the diversified regulatory factors above, and the stability of target genes together determine the dynamics of p53 activity.23,64,65
VARIANTS, ISOFORMS, AND FAMILY MEMBERS OF p53
Besides the diverse regulatory means acting on WT FLp53, the field is further complicated by the presence of p53 variants (including single nucleotide polymorphisms (SNPs) and mutants), isoforms, and other family members (Figure 2).
p53 SNPs: twins are different
The P72R polymorphism is the most extensively studied SNP of p53. The P72 variant has a weaker ability to induce apoptosis compared to the R72 variant,66 and thus is associated with a longer lifespan but a higher cancer mortality than the latter.67,68 However, there is also report that people bearing the P72 variant are less susceptible to HPV-associated carcinogenesis.69 Mice carrying the R72 variant of p53 show a higher propensity for metabolic dysfunction,70 yet they also exhibit increased rates of embryo implantation in females71. There are also several ethnic-specific SNPs in the p53 gene that have implications for disease susceptibility and treatment.72
p53 mutants: guardian has a dark side
Beyond SNPs, the p53 gene undergoes other types of genetic variation, such as deletions, and more commonly, mutations. p53 is among the most frequently mutated genes in cancers, with over half of all cancers possessing a mutated p53 allele. Missense mutation is the predominant mutation type of p53. There are six notable hotspots for missense mutations: R175, G245, R248, R249, R273, and R282, that all locate in the DBD, accounting for nearly 30% of all missense mutations of p53. Other sites with relatively higher incidences of missense mutations include H179 and Y220.15,73,74 All missense mutations affect the thermal stability of p53 to varying degrees. Generally, missense mutations can be classified into contact mutations, which maintain the overall conformation of p53 but impair DNA binding (e.g., R248W and R273H), and structural/conformational mutations, which significantly alter the conformation and stability of the DBD (e.g., R175H, G245S, and R249S), all disrupting p53’s TF activity. R196, R213, R306, and R342 are major sites for nonsense mutations.15,73,74 While most cancer patients acquire p53 mutations in their somatic cells, a notable exception is seen in patients with Li-Fraumeni syndrome (LFS), who carry a germline mutant p53 and have up to a 90% lifetime risk of developing one or more cancers and 50% of them get cancer before 30 years old.75,76 Notably, normal somatic cells may also harbor p53 mutations and other oncogenic mutations,77,78 raising the question of whether these cells are predisposed to oncogenesis under certain conditions.
The functional outcome of mutant p53 can be attributed to three different effects: (1) Loss-of-function (LOF): mutant p53 loses the activity of WT p53. For example, p53 mutants are often impaired in their ability to induce cell-cycle arrest and apoptosis.79 (2) Dominant-negative effect (DNE): mutant p53 interferes with the function of any remaining WT p53. In human pluripotent stem cells, mutant p53 confers an advantage in accelerating self-renewal.80 However, this DNE in normal tissues may not be as common as in cancer cells, as the levels of mutant p53 in normal cells are often kept low, similar to WT p53. (3) Gain-of-function (GOF): mutant p53 acquires additional activities not observed by loss of WT p53, typically through interaction with specific cofactors. The GOF of mutant p53 is largely attributed to its accumulation to high levels in cancer cells. For instance, mutant p53 can co-aggregate with WT p53 and other tumor suppressors (such as p63 and p73).81,82 The p53 hotspot mutants gain anti-ferroptosis activity to promote tumor growth.83,84 By modulating the expression of pro-metastatic targets, the p53 mutants are able to enhance the metastatic potential of cancer cells in different mouse models.83,85–88 Depsite the overwhelming evidence indicating the importance of the GOF of p53 mutants,89 one recent study showed that the LOF but not the GOF of p53 mutants is more critical under different experimental settings90. Nevertheless, categorizing p53 mutations into only one of these three effects is over-simplistic and whether the GOF or the LOF plays a more dominant role in vivo is likely context-dependent. It is clear that the overall outcome induced by p53 mutations results from the combination of all the three effects described above.
p53 isoforms: one root, many branches
Beyond FLp53, the p53 gene can produce other protein isoforms, resulting from promoter selection, alternative splicing, alternative translation, and even post-translational cleavage of FLp53.57 Attributable to combinations of four distinct N-termini (ATG1, Δ40, Δ133, and Δ160) and three different C-termini (α, β, and γ), there are twelve canonical isoforms of the p53 protein. The expression of these isoforms depends on the isoform type, developmental stage, tissue type, and is also subject to various regulatory mechanisms. These isoforms may possess unique activities, with the N-terminal isoforms being better investigated than the C-terminal ones. Particularly, they are often dysregulated in different cancers.57 For example, Δ40p53 suppresses tumor cell growth,91 while Δ160p53 can cooperate with mutant p53 to facilitate tumorigenesis92.
p63 and p73: siblings stand together
p53 shares high structural similarity with p63 and p73, especially in the DBD. Yet they also show differences in structure.15 These structural features result in both shared and distinct functions among the three proteins. Both p63 and p73 are able to regulate a number of well-known p53 targets,93 and exhibit tumor suppression activity94. Compared to p53, p63 and p73 have more roles in germ cell protection, fertility maintenance, and development regulation.16,95 For instance, p63 knockout (KO) mice exhibit defects in limb, craniofacial, and epithelial development, while p73 KO mice display neurological abnormalities.95
FUNCTIONS OF p53: DIVERSITY AND COMPLEXITY
p53 regulates a vast range of functions, constituting the complex p53 network (Figure 3).96
Figure 3. Functions and physiopathological roles of p53.
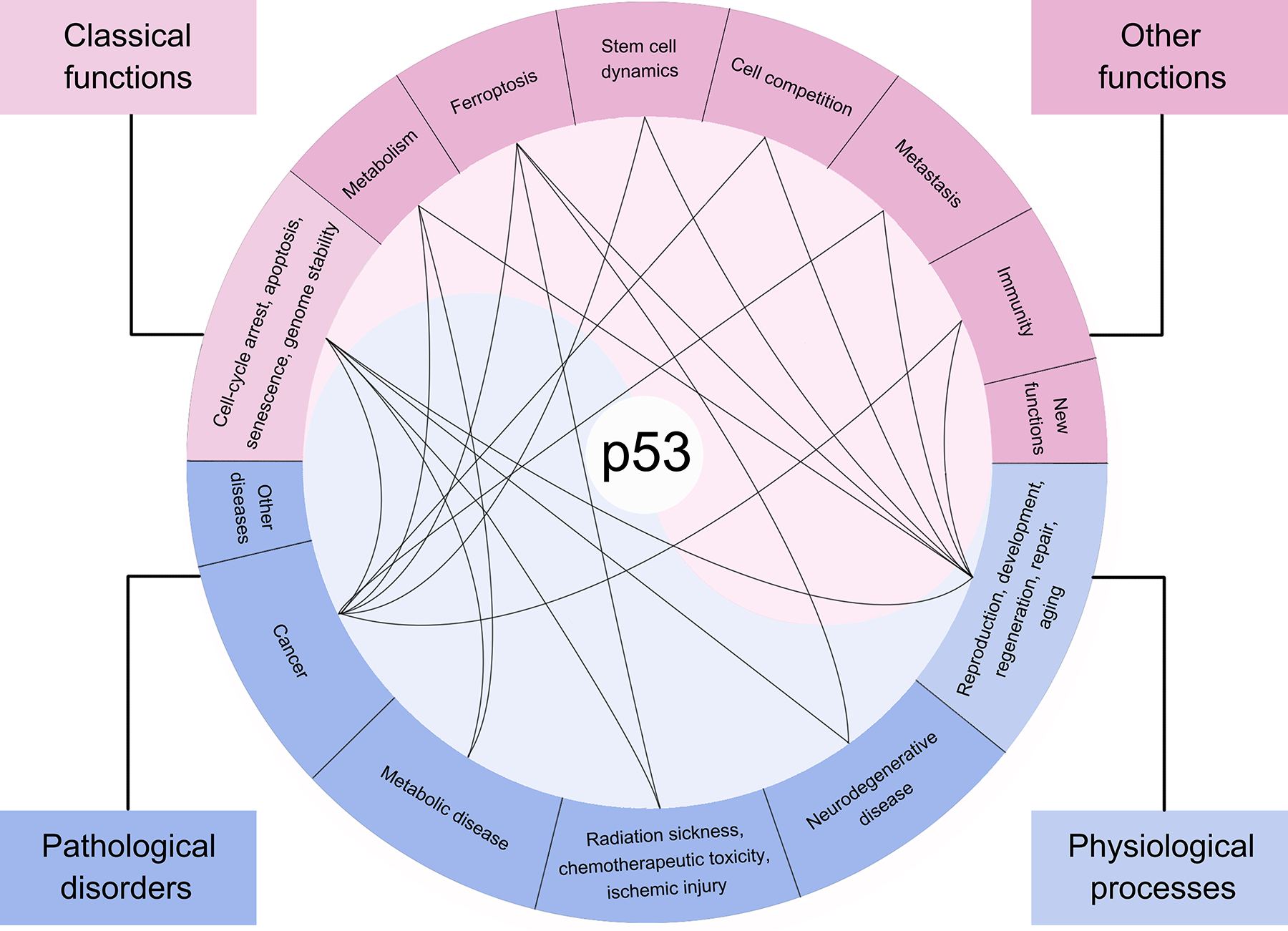
p53 exhibits diverse and complex functions: classical functions (including inducing cell-cycle arrest, apoptosis, and senescence, and maintaining genome stability) and other functions (such as mediating metabolism, ferroptosis, stem cell dynamics, cell competition, metastasis, and immunity). Due to its wide array of functions, p53 plays a crucial role in numerous physiological processes (e.g., reproduction, development, regeneration, repair, and aging) and pathological disorders (like neurodegenerative disease, radiation sickness, chemotherapeutic toxicity, ischemic injury, metabolic disease, and cancer). The black curves illustrate how specific functions of p53 contribute to its role in linked physiological or pathological processes.
Cell-cycle arrest, apoptosis, senescence, and genome stability
The inductions of cell-cycle arrest, apoptosis, and senescence are among the earliest discovered functions of p53.2 Various stress signals can induce p53 to exert these functions, with DNA damage being the most potent trigger. Upon DNA damage, p53 is stabilized and activated to arrest the cell cycle, providing cells with a time window and adequate material and energy spared from cell cycle progression to repair damaged DNA. If the damage is too severe to repair, apoptosis and senescence will be elicited by p53 to eliminate the compromised cells. Notably, the outcome of p53 activation is also determined by the type of cell and DNA damage. These three activities are well accepted as the major barriers to prevent tumorigenesis.
On the other hand, failure to eliminate the damage cells leads to genomic instability. The loss of p53, including loss of heterozygosity (LOH) and biallelic inactivation or deletion, promotes genomic instability and drives the evolution of the tumor cell genome.97–99 p53 is well known as “guardian of the genome”,100 thus a highly relevant activity of p53 is to directly promote DNA damage repair. Indeed, a number of p53 targets have been identified to contribute to the DNA repair process.101,102 Nevertheless, it remains unclear whether p53-mediated activation of those DNA repair-related targets is sufficient to suppress tumorigenesis independent of other p53 activities.
Metabolism and ferroptosis
p53 is a master regulator in modulating metabolism of glucose, lipids, amino acids, nucleotides, iron, and redox processes. It also regulates autophagy and has broad crosstalks with key metabolic regulators such as AMPK, AKT, and mTOR.103 This function of p53 links it to several metabolic disorders, especially cancers. In general, p53 represses anabolic processes (such as de novo lipogenesis and nucleotide synthesis), while promotes catabolisms (including oxidative phosphorylation, lipolysis, and fatty acid oxidation). The enhanced glycolysis generates multiple molecular materials for biosynthesis in cancer cells and thus is inhibited by p53, as well.103 These activities of p53 counteract the rapid proliferation requirements of cancer cells, thus leading to tumor suppression.
However, p53 manifests bidirectional roles in many metabolic processes. This paradoxical activity lies in the context-dependent nature of p53 function.96,104 The role of p53 in ROS control provides a good example. When there is a low intensity of ROS (indicating mild, transient, tolerable, and mitigable stresses), p53 plays an anti-oxidative role to reduce it to safeguard the cell from damage (pro-survival). On the contrary, when ROS levels are excessively high and potentially cause uncontrollable damage (representing severe, prolonged, detrimental, and unrelievable stresses), p53 further intensifies the ROS, leading to cell death and limiting harm in the damaged cells, thus protecting nearby undamaged cells (pro-death).47,103,105
Ferroptosis is an iron-dependent form of regulated cell death that occurs upon excess levels of lipid peroxidation, tightly linked with metabolic pathways. Several metabolic targets of p53 including SLC7A11, VKORC1L1, GLS2, and PLTP are directly involved in modulating ferroptosis.106–111 Similar to apoptosis during the DNA damage response, ferroptosis is able to eliminate the severe damaged cells during metabolic stress.112 By using acetylation-defective p53 mouse models, p53-mediated ferroptosis is implicated as an important arsenal in tumor suppression41,43 and interestingly, the defect in p53-mediated ferroptosis caused by an African-specific p53 SNP impairs its tumor suppressive function.113 Unlike apoptosis, initiation of canonical ferroptosis often relies on the treatment of cells with ferroptosis inducers, such as GPX4 inhibitors. It remains unclear whether ferroptosis-dependent tumor suppression requires ferroptosis inducers in vivo. Notably, a recent study showed that PHLDA2-mediated phosphatidic acid peroxidation triggers a non-canonical ferroptotic response in the absence of common ferroptosis inducers.114 Since p53 is able to promote both canonical and non-canonical ferroptotic processes, it will be interesting to examine which ferroptotic pathway plays a more dominant role in p53-mediated tumor suppression.
Stem cell dynamics and cell competition
Stem cells share many similarities with cancer cells, including sustained proliferative ability, reprogrammed metabolism, and the core transcription network. Therefore, it is not surprising that p53 restricts the cell stemness and modulates cell fate in various types of stem cells.115,116 In embryonic stem cells (ESCs), p53 inhibits genes that maintain stemness while activating those related to differentiation.117,118 In diverse types of adult stem cells (ASCs), p53 represses self-renewal, promotes depletion, maintains quiescence, or stimulates the differentiation.116 The ability of p53 to limit stemness is critical to its tumor suppression function, as it impedes the formation of cancer stem cells.119,120 Specific differentiation route guided by p53 contributes to tumor suppression in lung cancer.121 Loss or mutation of p53 may cause dedifferentiation, cell reprogramming, and increased cellular plasticity in cancers.122,123 The course of generating induced pluripotent stem cells (iPSCs) resembles dedifferentiation and cell transformation. p53 serves as a major brake in this process, and silencing p53 greatly improves the efficiency of iPSC generation.124,125
Cell competition is vital in development, tissue injury repair, tumor evolution, and metastasis. Generally, as p53 inhibits anabolism and proliferation while promoting cell death, which are not beneficial for cell to outcompete neighboring cells, a high level of p53 activity often marks a “loser” cell state in cell competition.126 However, there is a study that reports p53 activity is required for supercompetitor cells to eliminate nearby normal cells in Drosophila.127 The regulation of cell competition by p53 has significant physiological implications.128–131 Cells harboring mutant p53 may undergo clonal expansion, potentially driving tumor initiation and evolution.80,132,133 However, these p53 mutant cells are not always retained, as they may experience necroptosis by competing with nearby normal cells, or be outcompeted by cells with mutations in other genes that confer higher fitness.134,135
Metastasis
p53 suppresses metastasis at its multiple stages and in both cell-autonomous and non-cell-autonomous manners.136 In tumor cells, p53 restricts their mobility and epithelial-mesenchymal transition (EMT) process.136 Metastatic cancer cells in circulatory system may undergo anoikis and ferroptosis, both of which are promoted by p53 to prevent cancer cell migration to new sites.106,136 At each step of metastatic spread, cancer cells adopt specialized metabolic programs to meet their energy and biomolecular requirements, which can be counteracted by p53.103,137 On the other hand, p53 shapes a tumor microenvironment (TME) that is unfavorable for metastasis. For example, p53 restrains the angiogenesis and lymphangiogenesis, blocking the main metastatic routes via blood and lymphatic systems.138,139 It also maintains the integrity of extracellular matrix and enhances tumor cell adhesion to it, limiting tumor cell movement.140 Moreover, p53 hinders pro-metastatic inflammation.141
Immunity
Another essential function of p53 is to regulate immune response. p53 functions in both innate and adaptive immunity through multiple mechanisms.142,143 Both p53 in tumor cells and non-tumor cells synergize to construct a tumor-suppressive immune network. In tumor cells, p53 indirectly represses PD-L1 expression by upregulating miR-34, sensitizing tumor cells to anti-tumor immune response and immunotherapy.144 p53 activates cGAS-STING pathway to induce anti-tumor activity.145 In a mouse liver carcinoma model, restoring p53 expression induces tumor cell senescence, triggering the release of inflammatory cytokines and eliciting an innate immune response to eliminate tumor cells.146
In hepatic stellate cells, p53-induced senescence also exhibits a tumor-suppressive effect in liver cancer, establishing a senescence-associated secretory phenotype (SASP) that bolsters M1 macrophage polarization to maintain a tumor-inhibitory TME.147,148 In a subtype of murine myeloid precursor cells, p53 drives their differentiation into Ly6c+CD103+ monocytic antigen-presenting cells, enhancing anti-tumor immunity.149
Loss of p53 in tumor cells or TME cells significantly reverses the tumor-suppressive immune microenvironment to an immunosuppressive condition, promoting the immune tolerance or escape of tumor cells, or establish an inflammatory environment conducive to tumor metastasis.150,151 Mutant p53 may stimulate tumor cell immune evasion,143,152 and intriguingly, p53 mutants themselves can generate neoantigens that may be novel immunotherapeutic targets.153–155
p53 participates in autoimmune responses and immune defenses against various pathogens as well.142,156 Noteworthily, not all immunity-related activities of p53 promote immune cell function or are beneficial to health. p53 may inhibit the proliferation and function in certain T cell subtypes.157,158 For instance, p53 suppresses antigen-non-specific CD4+ T cell proliferation, a process that can be abolished by T cell receptor (TCR) signaling.158 Some viruses rely on p53 activity for cell-cycle arrest and replication.156
Rethinking the multitudinous functions of p53
p53 is such a powerful regulator with an array of diverse and complex functions that summarizing its roles in just a few words is challenging. These functions are intricately orchestrated by this single protein to achieve a unified biological purpose. Simplified expressions are often used to describe the working model of p53, such as “guardian of the genome”,100 “protector or killer”,47 and “pro-survival or pro-death”159. The working mode of p53 originates from the protection of germ cells by the ancestral p53 gene in lower organisms.160 Human p53 plays a similar protective role in somatic cells. In the majority of species expressing p53 or p53-like gene, this gene is essentially a stress-responder, but not merely a tumor suppressor. Therefore, p53 could also be called the “guardian of the cell”.21 The tumor-suppressive role of p53 may be considered coincidental, as some of its guardian functions are inherently antagonistic to many cancer cell characteristics. Thus, p53’s normal activity incidentally represses tumor initiation and development.
p53 IN PHYSIOLOGY AND PATHOLOGY: GUARDIAN IS AN ALL-ROUNDER
Due to its myriad functions, p53 is involved in a multitude of biological processes in normal physiology. However, both normal and dysregulated p53 activity also contribute to various disorders (Figure 3).
Reproduction, development, regeneration, repair, and aging
p53 predominantly influences maternal reproduction.161,162 Mechanistically, p53 transactivates leukemia inhibitory factor (LIF), a key cytokine for implantation, in the uterus. Lower LIF levels in p53-null female mice cause impaired implantation.162 In women under 35 with infertility, the P72 variant of p53, which has a weaker transactivating effect on LIF, is overrepresented, suggesting a positive correlation between p53 activity and implantation rate.71
p53’s regulation of development is rooted in its pivotal roles in the cell cycle, cell death, stem cell dynamics, and cell competition.116 p53 maintains genomic integrity in stem cells of embryo, promote differentiation, and permit normal development.116 The activity of p53 in early development must be kept in check, as evidenced by that deregulated p53 (loss, hyperactivation, and mutation) is linked to a variety of developmental defects in mice and humans.163,164 p53 also functions in distinct types of ASCs to maintains proper tissue hierarchy by protecting their integrity, holding them in dormancy, fine-tuning differentiation route, preventing unlimited proliferation, unordered differentiation, and dedifferentiation, plus suppressing tumorigenesis.116 p53 also plays a role in tissue regeneration.165 A recent study reported that p53 facilitates alveolar regeneration by regulating AT1 differentiation.121 As the wound healing course has many similarities with metastasis, p53 is supposed to impede it.166
Theoretically, p53’s activity has both pro-aging and anti-aging effects. On one hand, p53-mediated stress (particularly the DNA damage, a key driver of aging) response supports cell survival, removes damaged cells, protects tissue integrity, and maintains organismal homeostasis, potentially delaying aging. However, excessive and persistent DNA damage response by p53 has the opposite effect on aging.167 On the other hand, stem cell depletion by p53 accelerates aging process,38,168 yet there are counter-examples.169,170 Furthermore, functions of p53 in modulating senescence, metabolism, immune activity, and its interplay with sirtuins are also related to aging mediation.171,172 The relationship between p53 activity, tumor suppression, and aging has been investigated in different mouse models and little consensus has reached, which may stem from the highly context-dependent nature of p53’s function.170,173–177 Insights into the relationship between p53, cancers, and aging may be gleaned from studying animals other than mice.178 A notable example is the elephant, which has twenty copies of the p53 gene and exhibits a low tumor incidence along with a long lifespan.179 More efforts are required to dig out the role of p53 in aging, hoping to develop interventions that could better balance tumor suppression with aging, or possibly achieve longevity with a reduced incidence of tumors.
Neurodegenerative disease (NDD)
p53’s role in aging regulation is closely related to its function in various NDDs.180–183 The p53 protein level is often elevated in these disorders. Primarily, p53 contributes to NDD pathology by inducing apoptosis. Recently, ferroptosis has been recognized as playing a significant role in NDDs.184 As a master regulator of ferroptosis,106 it is logical to speculate that p53 participates in NDD progression via regulating ferroptosis. Additionally, p53 protein aggregation is implicated in Alzheimer’s disease.185 When developing drugs targeting p53 pathway for cancer treatment or slowing aging in elder people, the impact of p53 on NDDs should be considered.
Radiation sickness, chemotherapeutic toxicity, and ischemic injury
Exposure to radiation and genotoxic reagents, whether accidental or as part of cancer treatment, activates p53-dependent apoptosis and ferroptosis, causing pathologies in various organs.106,186 Particularly, this mechanism underlies major side effects in tumor radiotherapy and chemotherapy.187,188 Upon genotoxic stresses, the presence of p53 is not always harmful. The response of p53 to radiation is tissue-specific; while it stimulates radiation-related cell death in hematopoietic system, hair follicle, and spinal cord, it offers a radioprotective effect on gastrointestinal tract.186,189 Similarly, apoptosis and ferroptosis promoted by p53 lead to ischemic injuries in organs such as brain (e.g. stroke), kidney (e.g. kidney transplantation), and heart (e.g. myocardial infarction).106,186
Metabolic disease
p53 plays a complicated role in diverse metabolic diseases, including obesity, diabetes, alcoholic and non-alcoholic fatty liver diseases (AFLD and NAFLD), and cardiovascular diseases.103,190,191 Its multifaceted activities, particularly in metabolic regulation, affect these disorders in different cells, tissues, and organs, such as pancreatic β-cell, liver, muscle, and adipose tissue. Contradictory results are often reported about functions of p53 in these diseases. For example, while p53 in skeletal muscle progenitor cells and agouti-related peptide neurons protects against obesity,192,193 the liver R72 variant of p53, with enhanced transactivation ability, promotes fat accumulation and NAFLD.70 Besides, in endothelial cells p53 exacerbates dietary obesity-related metabolic abnormalities.194 Hence, when discussing the function of p53 in systemic metabolism and associated metabolic diseases, it must be put into a specific setting. Care must be taken when targeting p53 to treat certain metabolic diseases to avoid disrupting other metabolic processes and minimize the risk of predisposing recipient cells to cancer.
Cancer
As previously mentioned, p53 possesses an arsenal of functions to combat all the hallmarks of cancer,195 which may be lost or reversed due to its mutation, deletion, or repression in cancer cells (Figure 4).
Figure 4. p53 and cancer hallmarks.
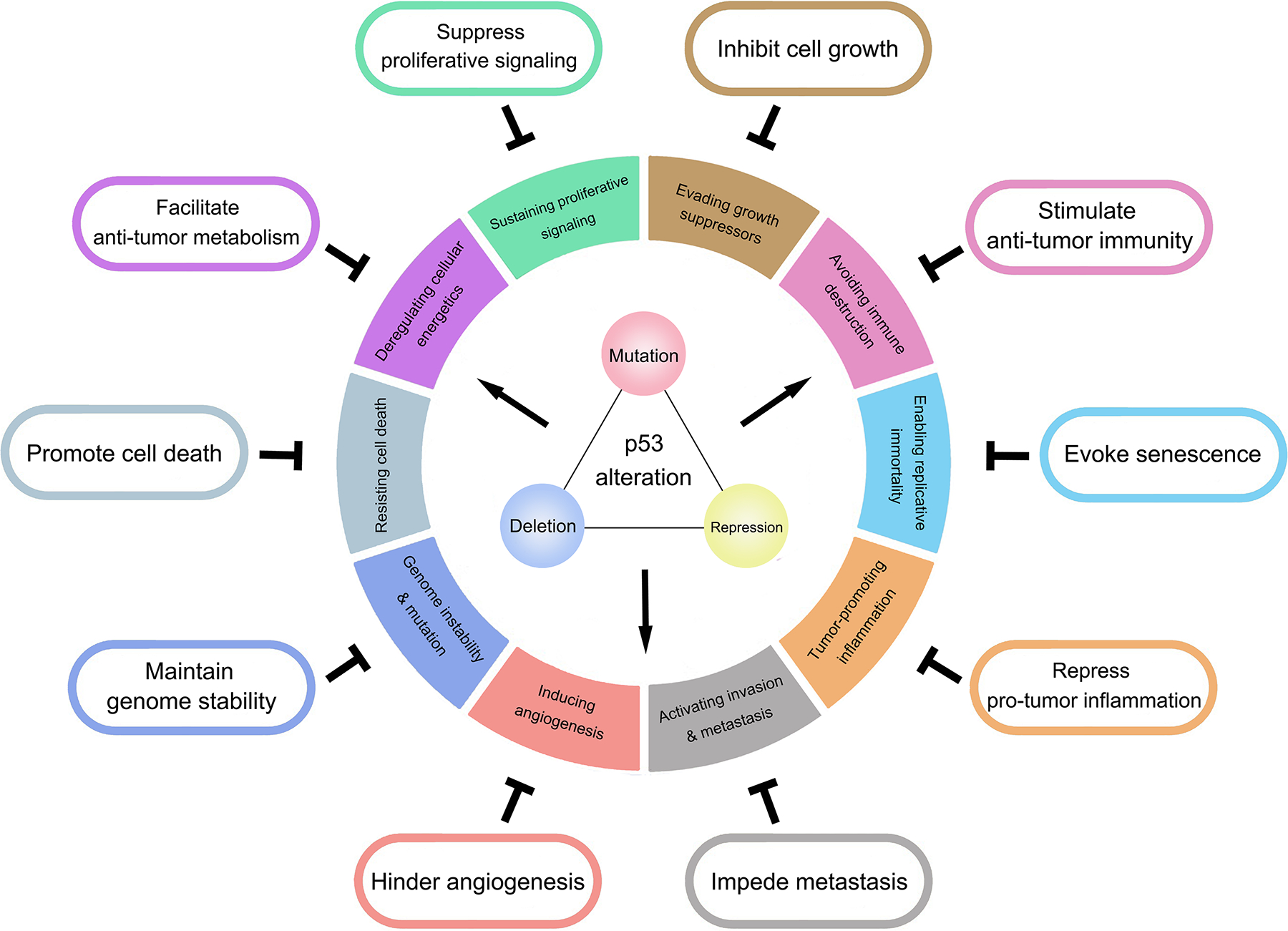
Activity of WT p53 antagonizes all the hallmarks of cancer, as depicted in the surrounding ovals. In contrast, alterations in p53, including repression, mutation, and deletion, promote these hallmarks. It is partially adapted from reference195.
While a mutation in one p53 allele may be sufficient for cellular transformation when the other WT p53 allele remains intact,196,197 LOH of p53 gene is often observed. The mechanisms behind p53 mutations are not entirely clear. Some may result from environmental and chemical carcinogens such as ultraviolet radiation, aflatoxin, and tobacco smoke, which leave characteristic fingerprints on the p53 gene.73,198 The frequency and spectrum of p53 mutations are dictated by factors such as gender, tissue type, and both cell-autonomous and non-cell-autonomous mechanisms in tumor evolution.199–203 In addition, the timing of p53 mutations varies among different tumors.198 Even within the same tumor type, different p53 mutants may not lead to the same phenotype.204 As introduced before, mutant p53 promotes tumor development via LOF, DNE, and GOF, which are well-elucidated in mouse models.79,196,197 While extensive focus has been placed on p53 hotspot mutations, other mutations should not be overlooked. For instance, missense mutations disrupting p53 oligomerization can lead to tumorigenesis.205,206 Missense mutations in the PRD of p53 may potentially impact its tumor-suppressive functions.207 Serum antibodies against mutant p53, immunohistochemistry of mutant p53, and DNA fragments of mutant p53 in tumor cells or body fluids and feces can be used as diagnostic and prognostic biomarkers in tumor patients.198,208–211 p53 mutations also act as predictors for the efficacy of certain tumor treatments and are useful for patient stratification.212
The p53 gene is located on the short arm of chromosome 17 (17p13.1), a region frequently deleted in tumors. p53 loss promotes tumor growth and metastasis in many different ways.97,98,123,151,213 p53 deletion is often linked with the loss of nearby genes, such as POLR2A and EIF5A, which also contributes to tumorigenesis and creates therapeutic vulnerabilities by targeting the co-deleted genes.214,215
As p53 mutation and deletion have significant impact on carcinogenesis, mutating or deleting p53 have been effectively applied to generate various mouse tumor models.196,197,216,217 A tightly relevant question is, among the LOF, DNE, and GOF of p53 alteration, which is the predominant mechanism underpinning p53-related tumor initiation and progression? While GOF of p53 mutants has garnered plenty of evidence supporting its tumorigenic role,218 there are also studies suggesting that LOF and DNE contribute more to cell transformation than GOF90,201,219. The moderate LOH rate (40–60%) in LFS patients has implications for this question.220 More well-controlled and tumor type-specific studies are warranted to clarify this issue. The answer is likely highly context-dependent.
A notable percentage of tumors retain two intact WT p53 alleles. These tumors may use different methods to attenuate p53 activity and its associated pathway. A common way is to enhance inhibition of p53 protein level, nuclear import, promoter recruitment, and transactivation activity by upregulating its negative regulators.54,63,221 The amplification of the MDM2 gene in sarcoma and glioblastoma is a notable example.221,222 Besides amplification, the T309G SNP of MDM2 and deletion of p14ARF both enhance repression of WT p53 by MDM2 in tumors.221,223,224 Many oncogenic viruses (such as SV40, EBV, and KHSV) transform cells partially by inactivating p53.221 Sometimes, WT p53 is misfolded into a pseudo-mutant conformation due to multiple mechanisms.74 In addition to inhibiting p53 directly, disruption of p53 downstream effectors also impairs the tumor-suppressive effect of WT p53.225
Although p53 is a well-known tumor suppressor, sporadic reports indicate that under certain circumstances, WT p53 activity may facilitate tumorigenesis and tumor survival. This can occur in the precancerous stage226 and during tumor growth227–230. Activation of WT p53 may also impair the efficacy of tumor treatment, promote resistance to anti-tumor drugs, and cause tumor relapse.231–234 These cases stem from the hijacking of one or more functions of p53 by precancerous or tumor cells to survive and develop. Correspondingly, some bona fide p53 target genes, such as MDM2 and TIGAR,235,236 can function as oncogenes. Hence, p53 should not be seen as a stereotypical tumor suppressor, but as a context-dependent guardian of the cell.
The list of disorders influenced by p53 extends to some other diseases. Taken together, beyond its most famous function in cancer, p53 is a highly health-relevant gene in both normal physiology and pathology (Figure 3). Health is achieved by maintaining the body’s homeostasis, which is a dynamic and balanced process. As a major stress-responder, p53 guards cellular homeostasis against various stresses. However, its activity can have both positive and negative effects on health and should be kept in equilibrium. Both insufficient and excessive p53 activities can disrupt a healthy state. Hence, the double-edged power of p53 should be seriously and carefully considered in health promotion and disease treatment strategies targeting p53.
TARGETING p53 FOR DISEASE TREATMENT: ALL ROADS LEAD TO HEALTH
Targeting p53 in cancer
Its “tumor-suppressive TF” nature makes p53 seemingly undruggable. Nevertheless, a multitude of methods have been developed to enhance or recover the WT function of p53 depending on its status in cancer cells (whether repressed, mutated, or lost) (Figure 5). For more information, readers are referred to recent reviews.74,237
Figure 5. Targeting p53 in cancer.
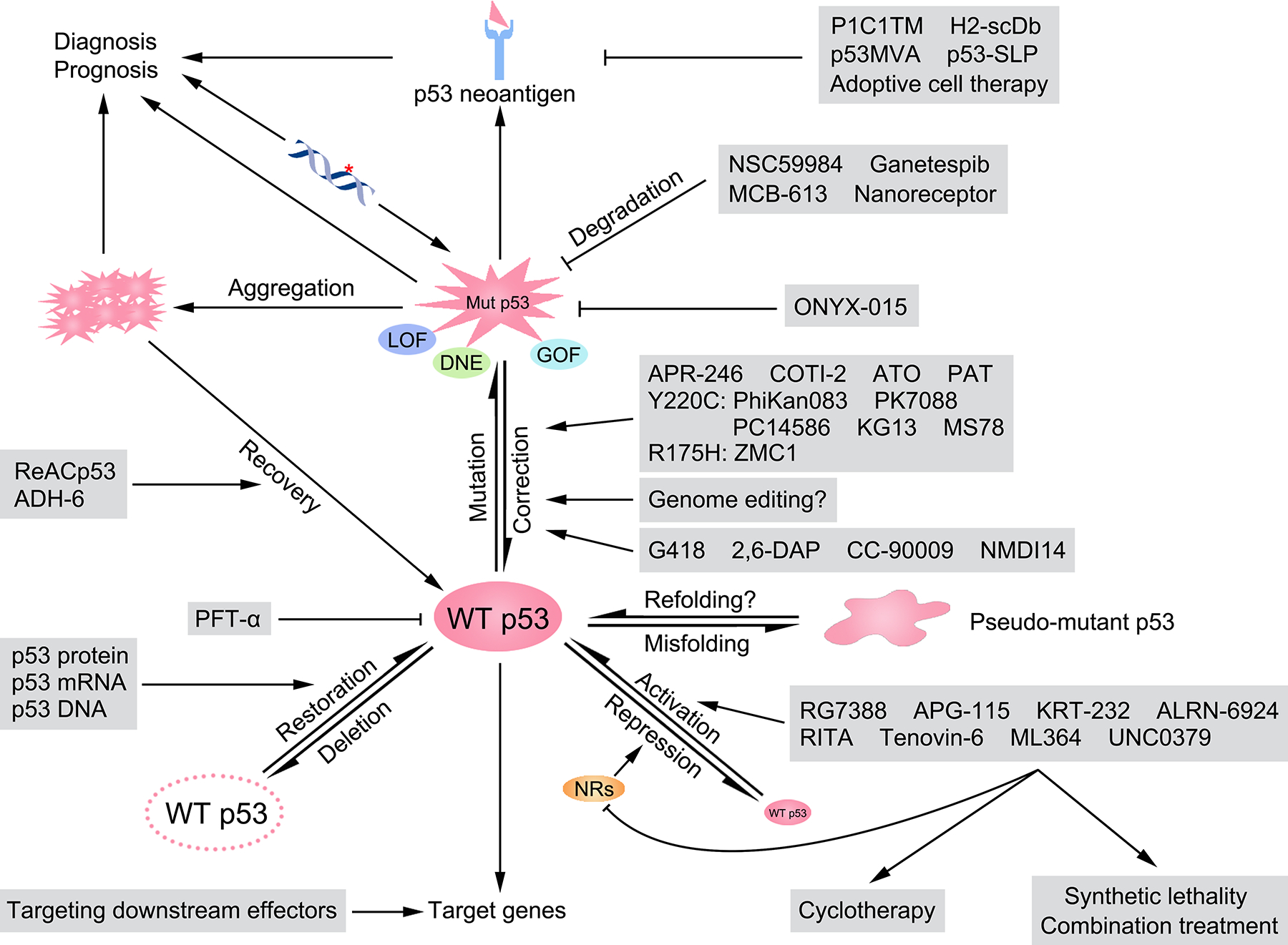
Various methods have been developed to target p53 for tumor treatment. In tumors retaining WT p53, RG7388, APG-115, KRT-232, and ALRN-6924 are used to disrupt the PPIs between p53 and MDM2 or MDMX, while RITA, tenovin-6, ML364, and UNC0379 target other negative regulators of p53. Activation of p53 can be utilized in cyclotherapy to protect normal cells, or in combination with other treatments for synergistic tumor eradication. WT p53 can be misfolded into a pseudo-mutant conformation, which may be reversed with appropriate drugs. Downstream targets of p53 are also potential therapeutic targets to partially reactivate the p53 signaling pathway. In tumors containing p53 missense mutations, APR-246, COTI-2, ATO, and PAT are capable of restoring the WT conformation of many p53 mutants. Specific agents such as PhiKan083, PK7088, PC14586, KG13, and MS78 target the p53 Y220C mutation, while ZMC1 is used for the p53 R175H mutant. Additionally, genome editing may be useful in correcting p53 gene mutations. NSC59984, ganetespib, MCB-613, and nanoreceptors are able to degrade mutant p53. ONYX-015, an oncolytic virus, specifically kills tumor cells with p53 mutations. Agents like ReACp53 and ADH-6 resolve the aggregation of mutant p53, partially restoring WT p53 functions. Antibodies like P1C1TM and H2-scDb, which recognize neoantigens derived from mutant p53, mediate tumor cell elimination by immune cells. p53MVA and p53-SLP are p53 vaccines used in immunotherapy. Mutant p53 neoantigens are also useful for developing adoptive cell therapies. Mutant p53 DNA fragments and proteins (including their aggregates) can be utilized for tumor diagnosis and prognosis. In tumors with p53 nonsense mutations, G418, 2,6-DAP, CC-90009, and NMDI14 can induce the readthrough of p53 mutant mRNAs, or inhibiting NMD. In p53-null tumors, delivery of p53 protein, mRNA, and DNA may restore p53 expression and eliminate tumor cells. LOF, loss-of-function; DNE, dominant-negative effect; GOF, gain-of-function; NR, negative regulator of p53.
In cancer cells retaining WT p53, an intuitive idea is to abrogate p53’s inhibition. This can be achieved by reducing the protein level (using siRNA, proteolysis-targeting chimera (PROTAC), or molecular glue) or activity of negative regulators of p53. However, these methods often lack specificity, as many p53 inhibitory factors have additional targets.238 Hence, lots of efforts have been made to identify molecules that specifically disrupt the protein-protein interactions (PPIs) between p53 and its negative regulators. MDM2 is the major target for elevating WT p53’s protein level and activity. Nutlin, the first small molecule to interfere with MDM2’s binding and degradation of p53 was proposed in 2004.239 Based on it, several derivatives including RG7112 and a more potent RG7388 (idasanutlin) have been developed and tested in clinical trials.74,237,240 However, in the phase 3 MDM2 antagonist Idasanutlin in Relapsed or Refractory acute myeloid leukemia for Overall Survival (MIRROS) trial, although an encouraging overall response rate was observed in cytarabine plus idasanutlin group over cytarabine plus placebo group, the primary endpoint (overall survival) was not reached.241 What’s more, idasanutlin treatment may cause hematological and gastrointestinal toxicities, highlighting the potential side effects of p53-activating drugs.240 Other small molecular inhibitors of the MDM2-p53 interaction include APG-115242 and AMG 232 (KRT-232)243, both undergoing clinical trials. ALRN-6924, a stapled α-helical peptide, simultaneously relieves the inhibition of MDM2 and MDMX on p53.244 Other negative regulators of p53, such as USP7,245 HPV E6,246 SIRT1/2,247 VPRBP,248 and SETD8,249 are promising targets for p53 activation. Given that PTMs are crucial for regulating p53 activity, targeting these cofactors holds attractive potential. Again, how to specifically affect the PTM status of p53 is a big challenge. Cofactors adjusting WT p53’s protein folding, cellular localization, DNA recruitment, and activity dynamics should be considered for targeting as well. Other aspects related to p53 expression and activity, including alternative splicing and translation, mRNA stability, and SNPs, can be targeted, too.
In cancer cells with p53 missense mutations, primary efforts are focused on restoring WT p53 activity. The existence of pseudo-mutant p53,74 second-site reversion of mutant p53,250 and temperature-sensitive mutant p53251 imply that the conformations of WT and mutant p53 might be interchangeable. This possibility opens the door to restoring the tumor-suppressive conformation of mutant p53 using specially designed small molecules (correctors). CP-31398, discovered in 1999, was the first compound found to enable mutant p53 to activate transcription and suppress tumor growth.252 After that, many more correctors are identified, for instance PRIMA-1.253 The degradation metabolite of PRIMA-1, methylene quinuclidinone (MQ), covalently binds to the thiol group of cysteine in mutant p53, restoring the WT conformation. Nevertheless, it also alters the redox state of the cell independently of p53 by reacting with other proteins. Its derivatives PRIMA-1MET (APR-246, or eprenetapopt) and APR-548 are undergoing multiple clinical trials.237 A thiosemicarbazone drug COTI-2, is also in a clinical trial.254 Arsenic trioxide (ATO), a drug to treat acute promyelocytic leukemia, has been found to rescue many p53 mutants, with varying efficiencies depending on the solvent accessibility and temperature sensitivity of the mutants.255,256 The antiparasitic drug potassium antimony tartrate (PAT) also rescues temperature-sensitive p53 mutants, like p53-V272M.257 These correctors above are responsible for rescuing multiple p53 mutants, but their broad-spectrum nature may limit efficiency for specific mutations. Targeted correctors for particular mutations would be more suitable for personalized treatment. For example, PhiKan083,258 PK7088,259 PC14586,260 and KG13261 are designed to correct the p53-Y220C mutant, because of its special conformation. Additionally, MS78, an acetylation targeting chimera (AceTAC), promotes the K382 acetylation of p53-Y220C, thereby specially rescuing its tumor suppressive activity.262 For a more common p53-R175H mutant, the thiosemicarbazone drug ZMC1 (NSC319726) is discovered, facilitating zinc binding and forcing the mutant into a WT conformation.263 The aggregation of mutant p53 can be relieved by ReACp53264 and ADH-6265. The dissociated mutant p53 partly recovers tumor-suppressive activity. Some GOF aspects of mutant p53 are targetable, too. NSC59984, for example, promotes mutant p53 degradation,266 releasing sequestered p73 to inhibit tumor growth. Other drugs, such as ganetespib,267 MCB-613,268 and biomimetic nanoreceptor,269 also promote mutant p53 degradation.
To treat nonsense mutations of p53, chemicals inducing translational readthrough or inhibiting nonsense-mediated mRNA decay (NMD), such as G418, 2,6-DAP, CC-90009, and NMDI14 can be utilized.74,237 For p53-mutated cancer cells, an alternative approach is oncolytic virus therapy. ONYX-015, a modified adenovirus, replicates only in p53-inactivated cells, thus selectively targeting cancer cells with p53 mutations while sparing normal cells.270
To treat p53-null tumors, one approach is to introduce p53 protein or use gene therapy to deliver p53 mRNA or DNA into tumor cells, thereby reinstating the expression of WT p53 protein.271–273 Advances in adeno-associated virus (AAV) and nanoparticle techniques may enhance the development and application of p53 gene therapy. The potential of CRISPR-Cas9 base editing technology for correcting p53 mutations in tumors is intriguing.
Targeting p53 can involve diverse cell types in the TME to achieve a tumor-suppressive outcome. For example, p53 activation induces the expression of endogenous retroviruses, which can potentiate immunotherapy.274 As previously introduced, mutant p53 can generate neoantigens targetable by immunotherapy.153–155 The engineered antibody P1C1TM, for instance, can distinguish between different p53-derived peptide-MHCs on WT and p53 mutant cells, mediating cytotoxicity or serving as an antibody-drug conjugate (ADC) to specifically eliminate p53 mutated tumor cells.154 The bispecific antibody H2-scDb, which bridges cancer cells presenting a p53-R175H neoantigen with T cells, effectively facilitates the destruction of cancer cells by T cells.153 Identifying mutant p53-derived neoantigens can also enhance adoptive cell therapies (such as CAR-T and TCR-T)155 and the development of p53 vaccines275,276. The success of mRNA vaccine technology encourages further exploration in the p53 vaccine direction. Targeting p53 in immune cells and tumor stroma cells (e.g. cancer-associated fibroblasts) could enhance anti-tumor effects as well.147,277,278
The status of p53 can significantly impact the sensitivity to certain cancer treatments.279,280 Often, p53-targeted therapies are combined with other treatments for two primary purposes: to enhance the efficacy of activating or restoring p53,281 and to exploit synthetic lethal effects created by simultaneously activating p53 and intervening in other pathways282–284. For instance, in acute myeloid leukemia, the activation of p53 coupled with Bcl-2 inhibition helps overcome resistance to individual treatments and stimulates apoptosis in cancer cells.282
Beyond directly targeting the p53 protein, key downstream components, especially those effectors critical for p53-mediated tumor suppression, can also be targeted to partially reactivate the p53 signaling pathway. This approach, while potentially less effective than activating WT p53, may be more feasible and serve as a complement to p53-targeting strategies.122,225,285 Besides the therapeutic and prophylactic applications, the diagnostic and prognostic values of p53 should not be overlooked either.198,208–212,286
Although there never lacks ideas to target p53, only a minority of them can enter clinical trial, not to mention the successful approval into clinical application. There are several obstacles to overcome before a p53-targeted drug can truly benefit the cancer patients. Firstly, the absorption, distribution, metabolism, and excretion (ADME) of the drug need to be optimized. Secondly, the efficiency of the p53-targeted therapy is influenced by various factors. Although p53 has potent tumor-suppressive capabilities, its activation does not always guarantee efficient tumor cell eradication.287,288 Strategies such as patient stratification and combination treatments may enhance efficacy. Thirdly, the low specificity of some drugs may result in off-target effects. Fourthly, the side effects caused by the on-target or off-target drug toxicity are among the major safety concerns. This is particularly evident in normal cells when activating WT p53 by blocking MDM2-p53 interaction. Given that the induction of cell-cycle arrest, apoptosis, and senescence is dispensable in p53-triggered tumor suppression,289 developing methods to retain p53’s anti-tumor effect without harming normal cells is a realistic goal. Another concern arises from the possibility that in normal cells harboring p53 mutations, MDM2 inhibitors could potentially activate the oncogenic function of these p53 mutants by stabilizing them. This consideration warrants serious attention in clinical practice. Specific drug delivery to tumor cells could reduce toxicity. It’s worth noting, as previously discussed, that p53-mediated cell death contributes significantly to the side effects experienced during tumor radiotherapy and chemotherapy.187,188 Consequently, suppressing p53 activity might be beneficial in reducing these treatment-related side effects.290 Interestingly, if controlled well, the mild activation of p53 could be employed in cyclotherapy, serving to protect normal cells.291 Fifthly, p53-targeted therapy poses the risk of driving tumor evolution, and selection of new mutations of p53 or alterations in the p53 pathway, leading to treatment resistance and tumor relapse.74,292,293 Appropriate combination treatments may help to mitigate this risk. Sixthly, p53 rarely promotes tumor survival, progression, drug resistance, and relapse, as mentioned before. In such scenarios, careful consideration is necessary to determine whether activating p53 is advisable. With progress in fields like artificial intelligence,294 structural biology, and multi-omics, these problems will be eventually addressed.
Targeting p53 in other physiological and pathological settings
The exploration of targeting p53 in normal physiological processes and non-cancerous diseases is not as extensive as in cancers. However, given p53’s broad impact, strategically modulating its activity could potentially enhance health by improving normal physiological functions and alleviating p53-associated disorders. For example, activating p53 in the uterus might increase the success rate of embryo implantation, while inhibiting p53 might prevent NDDs and reduce ischemic organ injury. In the context of metabolic disorders and aging, the role of p53 remains debatable and warrants further investigation. To suppress p53 activity, various methods can be employed, such as using p53 inhibitors,186,290 disrupting the PPIs between p53 and its activators,295 or augmenting the function of negative regulators of p53. These approaches essentially mirror those used for activating p53 but in reverse. A word of caution is that suppressing p53 might inadvertently increase the risk of tumorigenesis. Strategies like short-term intervention and cell type-specific delivery of p53-inhibitory drugs could decrease this risk. Looking ahead, it is full of hope that targeting p53 can improve health outcomes beyond just cancer treatment.
INTRIGUING QUESTIONS IN THE p53 FIELD: OPEN QUESTIONS, BRIGHT FUTURE
Great progress has been made over the past 45 years in p53 research. Basic regulations, functions, and therapeutic potentials of p53 have been elucidated. Nevertheless, there are still some fundamental questions awaiting to answer.
Firstly, what is the function of basal p53? In resting cell, both the expression and activity of p53 protein are kept low, until various stimuli stabilize and activate it. However, obvious stress is not always required for p53 to carry out some of its functions, such as regulating stem cell dynamics, cell competition, ROS level, and immune activity.105,142,296 So how does basal p53 work in unstressed cells? Does the basal activity of p53 contribute to tumor suppression? Basal p53 can bind to the promoters or enhancers of its target genes, establishing a primed state for rapid responses to stresses.23 Importantly, it also maintains a specific expression profile of target genes.297 Particularly, basal p53 is responsible for the baseline level of some tumor suppressor genes like PTEN.298 There may be differences in the target gene lists between basal and activated p53. Evidence also suggests that robust stabilization,44 full transactivation ability,297 and tetrameric conformation205 of p53 are not absolutely necessary for its tumor suppression effect. Therefore, leveraging the basal activity of p53 may help minimize side effects on normal cells and reduce the risk of selecting p53 mutations resulting from its activation. Studying the function of basal p53 requires an appropriate model. While knocking out p53 in a tumor cell line might provide some insights, transferring cells to tissue culture can be a p53-inducing stress. p53 levels in cultured cells may not reflect a truly unstressed basal state, which presents a caveat when interpreting in vitro results. More importantly, cultured cells lack the diverse environmental cues present in vivo and thus cannot fully simulate the actual tumorigenic process. Moreover, the phenotype observed in p53 KO mice should not be solely attributed to the loss of basal p53 activity, as the functions of stress-activated p53 are eliminated, too.
Secondly, what is the contribution of p53’s repressive target genes to its function? p53’s role is not limited to activating transcription. It has also been found to repress the expression of many genes,299 with the notable examples such as SLC7A11,107 NANOG,117 and VKORC1L1109. In most cases, p53 suppresses gene expression indirectly, either through transcriptional or post-transcriptional mechanisms. It competes with other TFs for DNA binding,120,300 activates negative regulators of gene expression,301–304 and suppresses the expression of transcription activators.305 Otherwise, direct repression of transcription by p53, through binding to special REs,117,119,306,307 interfering with enhancer function,118,305 and remolding chromatin structure305 are reported as well. Although the direct transcriptional repression by p53 is a subject of ongoing debate308 and requires further investigation, it is clear that hundreds of genes are downregulated upon p53 activation or upregulated in its absence. Future studies are expected to shed more light on whether and how p53 functions as a direct transcription repressor, and the roles these repressive target genes play within the p53 network.
Thirdly, are genetically engineered mouse models (GEMMs) sufficiently effective for p53 research? While GEMMs have significantly advanced our understanding of p53,289,309 they also present inherent limitations. The most notable is the species gap between humans and mice, as many human p53 targets are not shared by mouse p53,310 not to mention the differences in immune system or other biological systems. The knowledge about p53 got from in vitro studies is often tested in mouse models. Nonetheless, most reproducible results in mice could not be directly translated to applications in humans. Currently, humanized mouse models311,312 and human organoids313 serve as useful supplements to GEMMs. However, these alternatives have their limitations as well. Consequently, there is a pressing need to develop more practical and human physiopathologically relevant systems for p53 research.
Fourthly, what are the underpinning mechanisms and biological relevance of the context-dependent activity of p53? A reiterated feature of p53 activity in above sections is its context-dependency. The target profile of p53 exhibits significant organ specificity and spatiotemporal variations.314 Heterogeneity in p53 expression is also evident within a single tumor.315 Even within the same cell, the target selectivity and functional outcome of p53 activation are determined by a variety of variables.23 In addition, gender can dictate p53-regulated human behavior.316 When interpreting experimental results related to p53, placing them within a specific context is essential.104 Further effort is still warranted for a deeper understanding of p53 activity and the development of context-specific treatment regimens that target p53.
Fifthly, is targeting p53 truly a practical approach for treating cancers? The differential alterations of p53 in cancers and the extensive variety of p53 missense mutations complicate the design of specific drugs for each type of p53 alteration. The context-dependency of p53 activity adds another layer of complexity to this issue. However, targeting p53 as a treatment for cancers remains an enticing approach to pursue. Considering the frequent side effects, are there other elements in the p53 pathway that could serve as safer targets than MDM2, without compromising anti-tumor efficacy? Is it possible to achieve a “two-drugs-cure-all” goal by activating or inhibiting p53 to treat all p53-associated diseases, and improve overall health, particularly in extending longevity? This ambition may seem lofty, but it is certainly worth exploring.
Likewise, several other important questions remain unanswered. New targets of p53 are continuously being identified. A notable example is zinc finger matrin-type 3 (ZMAT3), which plays a critical role in p53-dependent tumor suppression.317–320 What are the unidentified targets and functions of p53? What targets or functions are indispensable for p53 to mediate tumor suppression? What is the functional diversity and evolutionary significance of p53’s two TADs? How do they cooperate in executing p53’s tumor-suppressive function? Some progress has been made in this area,19,297 but it has not been fully addressed. Are the non-cell-autonomous activities of p53 viable targets for cancer treatment? Under what circumstances does p53 shift to support tumor development? Can we leverage p63 and p73 to synergize with p53 targeting in the treatment of cancers or other diseases? The list of the questions could be longer. New questions will emerge in the future. It is on the way to find answers to these questions, our understanding of p53 is refreshed and deepened.
CONCLUSIONS
The past 45 years of p53 research represent a remarkable journey. As discussed above, countless discoveries result in a much better understanding of the complexity of p53 functions under different physiological settings. The diverse tumor suppression mechanisms including both classical activities and a growing number of other p53 functions raise a more interesting issue about which pathway is more critical for suppressing tumor growth in different types of human cancers. Moreover, although p53 is well accepted as a tumor suppressor, not all p53-induced activities are necessary for tumor suppression. For example, p53-mediated pro-survival activity seems at odds with its growth-suppressive function in tumors; however, this activity is potentially important for normal cell homeostasis, allowing normal cell survival during stress responses. The challenge remains how to activate p53 function for cancer therapy. The difficulties associated with translating the MDM2-targeting approach into clinical application raises a serious issue about how to kill cancer cells without harming normal tissues. It remains unclear whether the toxicity induced by MDM2 inhibitors (particularly in the bone marrow) is unique for the MDM2 pathway or for p53 activation in general. If the former is the case, targeting different pathways for p53 activation should be seriously considered.248 Notably, in contrast to the classic activities such as apoptosis, many p53 targets involved in metabolism, ferroptosis, and immunity do not directly cause severe harm to cell viability.321 It will be interesting to examine whether the specific activation of those pathways effectively suppresses tumor growth without causing severe toxicity.
ACKNOWLEDGEMENTS
We greatly appreciate Dr. Arnold J. Levine for critical comments and helpful suggestions on the manuscript. This work was supported by the National Cancer Institute of the National Institutes of Health under Award R35CA253059, RO1CA258390, and RO1CA254970 (to W.G.). Owing to space limitations, we sincerely apologize for not citing all the important publications of p53 in this review. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
DECLARATION OF INTERESTS
O. Tavana is currently an employee of AstraZeneca and has stock ownership in AstraZeneca. All other authors declare that they have no conflict of interest.
REFERENCES
- 1.Dolgin E (2017). The most popular genes in the human genome. Nature 551, 427–431. [DOI] [PubMed] [Google Scholar]
- 2.Levine AJ & Oren M (2009). The first 30 years of p53: growing ever more complex. Nat Rev Cancer 9, 749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hainaut P & Wiman KG (2009). 30 years and a long way into p53 research. Lancet Oncol 10, 913–919. [DOI] [PubMed] [Google Scholar]
- 4.Soussi T (2010). The history of p53. A perfect example of the drawbacks of scientific paradigms. EMBO Rep 11, 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barnoud T, Indeglia A & Murphy ME (2021). Shifting the paradigms for tumor suppression: lessons from the p53 field. Oncogene 40, 4281–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lane DP & Crawford LV (1979). T-Antigen Is Bound to a Host Protein in Sv40-Transformed Cells. Nature 278, 261–263. [DOI] [PubMed] [Google Scholar]
- 7.Linzer DIH & Levine AJ (1979). Characterization of a 54k Dalton Cellular Sv40 Tumor-Antigen Present in Sv40-Transformed Cells and Uninfected Embryonal Carcinoma-Cells. Cell 17, 43–52. [DOI] [PubMed] [Google Scholar]
- 8.Kress M, May E, Cassingena R & May P (1979). Simian-Virus 40-Transformed Cells Express New Species of Proteins Precipitable by Anti-Simian Virus-40 Tumor Serum. J Virol 31, 472–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deleo AB, Jay G, Appella E, Dubois GC, Law LW & Old LJ (1979). Detection of a Transformation-Related Antigen in Chemically-Induced Sarcomas and Other Transformed-Cells of the Mouse. P Natl Acad Sci USA 76, 2420–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eliyahu D, Michalovitz D, Eliyahu S, Pinhasikimhi O & Oren M (1989). Wild-Type P53 Can Inhibit Oncogene-Mediated Focus Formation. P Natl Acad Sci USA 86, 8763–8767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finlay CA, Hinds PW & Levine AJ (1989). The P53 Proto-Oncogene Can Act as a Suppressor of Transformation. Cell 57, 1083–1093. [DOI] [PubMed] [Google Scholar]
- 12.Baker SJ, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM, Vantuinen P, Ledbetter DH, Barker DF, Nakamura Y, et al. (1989). Chromosome-17 Deletions and P53 Gene-Mutations in Colorectal Carcinomas. Science 244, 217–221. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi T, Nau MM, Chiba I, Birrer MJ, Rosenberg RK, Vinocour M, Levitt M, Pass H, Gazdar AF & Minna JD (1989). P53 - a Frequent Target for Genetic Abnormalities in Lung-Cancer. Science 246, 491–494. [DOI] [PubMed] [Google Scholar]
- 14.Nigro JM, Baker SJ, Preisinger AC, Jessup JM, Hostetter R, Cleary K, Bigner SH, Davidson N, Baylin S, Devilee P, et al. (1989). Mutations in the P53 Gene Occur in Diverse Human-Tumor Types. Nature 342, 705–708. [DOI] [PubMed] [Google Scholar]
- 15.Joerger AC & Fersht AR (2016). The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu Rev Biochem 85, 375–404. [DOI] [PubMed] [Google Scholar]
- 16.Levine AJ (2020). p53: 800 million years of evolution and 40 years of discovery. Nat Rev Cancer 20, 471–480. [DOI] [PubMed] [Google Scholar]
- 17.Green DR & Kroemer G (2009). Cytoplasmic functions of the tumour suppressor p53. Nature 458, 1127–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S & Miyazono K (2009). Modulation of microRNA processing by p53. Nature 460, 529–U111. [DOI] [PubMed] [Google Scholar]
- 19.Raj N & Attardi LD (2017). The Transactivation Domains of the p53 Protein. Csh Perspect Med 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gaglia G, Guan YH, Shah JV & Lahav G (2013). Activation and control of p53 tetramerization in individual living cells. P Natl Acad Sci USA 110, 15497–15501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu YQ, Tavana O & Gu W (2019). p53 modifications: exquisite decorations of the powerful guardian. J Mol Cell Biol 11, 564–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walker KK & Levine AJ (1996). Identification of a novel p53 functional domain that is necessary for efficient growth suppression. P Natl Acad Sci USA 93, 15335–15340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hafner A, Bulyk ML, Jambhekar A & Lahav G (2019). The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Bio 20, 199–210. [DOI] [PubMed] [Google Scholar]
- 24.Fischer M (2017). Census and evaluation of p53 target genes. Oncogene 36, 3943–3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen S, Thorne RF, Zhang XD, Wu M & Liu LX (2021). Non-coding RNAs, guardians of the p53 galaxy. Semin Cancer Biol 75, 72–83. [DOI] [PubMed] [Google Scholar]
- 26.Hollstein M & Hainaut P (2010). Massively regulated genes: the example of TP53. J Pathol 220, 164–173. [DOI] [PubMed] [Google Scholar]
- 27.Liu J, Guan D, Dong MG, Yang JJ, Wei HB, Liang Q, Song LZ, Xu L, Bai JJ, Liu C, et al. (2020). UFMylation maintains tumour suppressor p53 stability by antagonizing its ubiquitination. Nat Cell Biol 22, 1056–1063. [DOI] [PubMed] [Google Scholar]
- 28.Xu PF, Xi Y, Wang PC, Luka ZM, Xu MS, Tung HC, Wang JY, Ren SR, Feng DC, Gao B, et al. (2022). Inhibition of p53 Sulfoconjugation Prevents Oxidative Hepatotoxicity and Acute Liver Failure. Gastroenterology 162, 1226–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gokulan RC, Paulrasu K, Azfar J, El-Rifai W, Que JW, Boutaud OG, Ban YG, Gao Z, Buitrago MG, Dikalov SI, et al. (2023). Protein adduction causes non-mutational inhibition of p53 tumor suppressor. Cell Rep 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X, Li CF, Zhang L, Wu CY, Han LX, Jin GX, Rezaeian AH, Han F, Liu CF, Xu C, et al. (2016). TRAF6 Restricts p53 Mitochondrial Translocation, Apoptosis, and Tumor Suppression. Mol Cell 64, 803–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luna RMD, Wagner DS & Lozano G (1995). Rescue of Early Embryonic Lethality in Mdm2-Deficient Mice by Deletion of P53. Nature 378, 203–206. [DOI] [PubMed] [Google Scholar]
- 32.Jones SN, Roe AE, Donehower LA & Bradley A (1995). Rescue of Embryonic Lethality in Mdm2-Deficient Mice by Absence of P53. Nature 378, 206–208. [DOI] [PubMed] [Google Scholar]
- 33.Barak Y, Juven T, Haffner R & Oren M (1993). Mdm2 Expression Is Induced by Wild Type-P53 Activity. Embo J 12, 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB & Siliciano JD (1998). Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 281, 1677–1679. [DOI] [PubMed] [Google Scholar]
- 35.Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, et al. (1998). Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281, 1674–1677. [DOI] [PubMed] [Google Scholar]
- 36.Shieh SY, Ikeda M, Taya Y & Prives C (1997). DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91, 325–334. [DOI] [PubMed] [Google Scholar]
- 37.Chao C, Herr D, Chun J & Xu Y (2006). Ser18 and 23 phosphorylation is required for p53-dependent apoptosis and tumor suppression. Embo J 25, 2615–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu DP, Ou LD, Clemenson GD, Chao C, Lutske ME, Zambetti GP, Gage FH & Xu Y (2010). Puma is required for p53-induced depletion of adult stem cells. Nat Cell Biol 12, 993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y, et al. (2000). p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell 102, 849–862. [DOI] [PubMed] [Google Scholar]
- 40.Xia Z, Kon N, Gu AP, Tavana O & Gu W (2022). Deciphering the acetylation code of p53 in transcription regulation and tumor suppression. Oncogene 41, 3039–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li TY, Kon N, Jiang L, Tan MJ, Ludwig T, Zhao YM, Baer R & Gu W (2012). Tumor Suppression in the Absence of p53-Mediated Cell-Cycle Arrest, Apoptosis, and Senescence. Cell 149, 1269–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang DL, Kon N, Lasso G, Jiang L, Leng WC, Zhu WG, Qin J, Honig B & Gu W (2016). Acetylation-regulated interaction between p53 and SET reveals a widespread regulatory mode. Nature 538, 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kon N, Ou Y, Wang SJ, Li H, Rustgi AK & Gu W (2021). mTOR inhibition acts as an unexpected checkpoint in p53-mediated tumor suppression. Gene Dev 35, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kon N, Churchill M, Li H, Mukherjee S, Manfredi JJ & Gu W (2021). Robust p53 Stabilization Is Dispensable for Its Activation and Tumor Suppressor Function. Cancer Research 81, 935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krummel KA, Lee CJ, Toledo F & Wahl GM (2005). The C-terminal lysines fine-tune P53 stress responses in a mouse model but are not required for stability control or transactivation. Proc Natl Acad Sci U S A 102, 10188–10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feng LJ, Lin TX, Uranishi H, Gu W & Xu Y (2005). Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Mol Cell Biol 25, 5389–5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vousden KH & Prives C (2009). Blinded by the Light: The Growing Complexity of p53. Cell 137, 413–431. [DOI] [PubMed] [Google Scholar]
- 48.Boysen M, Kityk R & Mayer MP (2019). Hsp70-and Hsp90-Mediated Regulation of the Conformation of p53 DNA Binding Domain and p53 Cancer Variants. Mol Cell 74, 831–843. [DOI] [PubMed] [Google Scholar]
- 49.Dahiya V, Agam G, Lawatscheck J, Rutz DA, Lamb DC & Buchner J (2019). Coordinated Conformational Processing of the Tumor Suppressor Protein p53 by the Hsp70 and Hsp90 Chaperone Machineries. Mol Cell 74, 816–830. [DOI] [PubMed] [Google Scholar]
- 50.Trinidad AG, Muller PAJ, Cuellar J, Klejnot M, Nobis M, Valpuesta JM & Vousden KH (2013). Interaction of p53 with the CCT Complex Promotes Protein Folding and Wild-Type p53 Activity. Mol Cell 50, 805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kruse JP & Gu W (2009). Modes of p53 Regulation. Cell 137, 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raj N, Wang MX, Seoane JA, Zhao RL, Kaiser AM, Moonie NA, Demeter J, Boutelle AM, Kerr CH, Mulligan AS, et al. (2022). The Mettl3 epitranscriptomic writer amplifies p53 stress responses. Mol Cell 82, 2370–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang J, Wei J, Sun R, Sheng H, Yin K, Pan Y, Jimenez R, Chen S, Cui XL, Zou Z, et al. (2023). A lncRNA from the FTO locus acts as a suppressor of the m(6)A writer complex and p53 tumor suppression signaling. Mol Cell 83, 2692–2708 e2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun XQ, Klingbeil O, Lu B, Wu CZ, Ballon C, Ouyang M, Wu XL, Jin Y, Hwangbo Y, Huang YH, et al. (2023). BRD8 maintains glioblastoma by epigenetic reprogramming of the p53 network. Nature 613, 195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Isbel L, Iskar M, Durdu S, Weiss J, Grand RS, Hietter-Pfeiffer E, Kozicka Z, Michael AK, Burger L, Thomä NH, et al. (2023). Readout of histone methylation by Trim24 locally restricts chromatin opening by p53. Nat Struct Mol Biol 30, 948–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Catizone AN, Uzunbas GK, Celadova P, Kuang S, Bose D & Sammons MA (2020). Locally acting transcription factors regulate p53-dependent cis-regulatory element activity. Nucleic Acids Res 48, 4195–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao LQ & Sanyal S (2022). p53 Isoforms as Cancer Biomarkers and Therapeutic Targets. Cancers 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hodge DR, Peng B, Cherry JC, Hurt EM, Fox SD, Kelley JA, Munroe DJ & Farrar WL (2005). Interleukin 6 supports the maintenance of p53 tumor suppressor gene promoter methylation. Cancer Res 65, 4673–4682. [DOI] [PubMed] [Google Scholar]
- 59.Macedo-Silva C, Miranda-Goncalves V, Tavares NT, Barros-Silva D, Lencart J, Lobo J, Oliveira A, Correia MP, Altucci L & Jeronimo C (2023). Epigenetic regulation of TP53 is involved in prostate cancer radioresistance and DNA damage response signaling. Signal Transduct Target Ther 8, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang Y, Horikawa I, Ajiro M, Robles AI, Fujita K, Mondal AM, Stauffer JK, Zheng ZM & Harris CC (2013). Downregulation of splicing factor SRSF3 induces p53β, an alternatively spliced isoform of p53 that promotes cellular senescence. Oncogene 32, 2792–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aschtgen MS, Fragkoulis K, Sanz G, Normark S, Selivanova G, Henriques-Normark B & Peuget S (2022). Enterobacteria impair host p53 tumor suppressor activity through mRNA destabilization. Oncogene 41, 2173–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Díaz-Muñoz MD, Kiselev VY, Le Novère N, Curk T, Ule J & Turner M (2017). Tia1 dependent regulation of mRNA subcellular location and translation controls p53 expression in B cells. Nat Commun 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Swarbrick A, Woods SL, Shaw A, Balakrishnan A, Phua Y, Nguyen A, Chanthery Y, Lim L, Ashton LJ, Judson RL, et al. (2010). miR-380–5p represses p53 to control cellular survival and is associated with poor outcome in MYCN-amplified neuroblastoma. Nat Med 16, 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stewart-Ornstein J, Iwamoto Y, Miller MA, Prytyskach MA, Ferretti S, Holzer P, Kallen J, Furet P, Jambhekar A, Forrester WC, et al. (2021). p53 dynamics vary between tissues and are linked with radiation sensitivity. Nat Commun 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paek AL, Liu JC, Loewer A, Forrester WC & Lahav G (2016). Cell-to-Cell Variation in p53 Dynamics Leads to Fractional Killing. Cell 165, 631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dumont P, Leu JIJ, Della Pietra AC, George DL & Murphy M (2003). The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet 33, 357–365. [DOI] [PubMed] [Google Scholar]
- 67.Zhao Y, Wu L, Yue X, Zhang C, Wang J, Li J, Sun X, Zhu Y, Feng Z & Hu W (2018). A polymorphism in the tumor suppressor p53 affects aging and longevity in mouse models. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Heemst D, Mooijaart SP, Beekman M, Schreuder J, de Craen AJ, Brandt BW, Slagboom PE, Westendorp RG & Long Life study, g. (2005). Variation in the human TP53 gene affects old age survival and cancer mortality. Exp Gerontol 40, 11–15. [DOI] [PubMed] [Google Scholar]
- 69.Storey A, Thomas M, Kalita A, Harwood C, Gardiol D, Mantovani F, Breuer J, Leigh IM, Matlashewski G & Banks L (1998). Role of a p53 polymorphism in the development of human papillomavirus-associated cancer. Nature 393, 229–234. [DOI] [PubMed] [Google Scholar]
- 70.Kung CP, Leu JIJ, Basu S, Khaku S, Anokye-Danso F, Liu Q, George DL, Ahima RS & Murphy ME (2016). The P72R Polymorphism of p53 Predisposes to Obesity and Metabolic Dysfunction. Cell Rep 14, 2413–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kang HJ, Feng Z, Sun Y, Atwal G, Murphy ME, Rebbeck TR, Rosenwaks Z, Levine AJ & Hu WW (2009). Single-nucleotide polymorphisms in the p53 pathway regulate fertility in humans. P Natl Acad Sci USA 106, 9761–9766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fischer NW, Ma YHV & Gariépy J (2023). Emerging insights into ethnic-specific TP53 germline variants. Jnci-J Natl Cancer I 115, 1145–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hainaut P & Pfeifer GP (2016). Somatic TP53 Mutations in the Era of Genome Sequencing. Cold Spring Harb Perspect Med 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tuval A, Strandgren C, Heldin A, Palomar-Siles M & Wiman KG (2023). Pharmacological reactivation of p53 in the era of precision anticancer medicine. Nat Rev Clin Oncol. [DOI] [PubMed] [Google Scholar]
- 75.Malkin D, Li FP, Strong LC, Fraumeni JF Jr., Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA, et al. (1990). Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250, 1233–1238. [DOI] [PubMed] [Google Scholar]
- 76.Srivastava S, Zou ZQ, Pirollo K, Blattner W & Chang EH (1990). Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature 348, 747–749. [DOI] [PubMed] [Google Scholar]
- 77.Krimmel JD, Schmitt MW, Harrell MI, Agnew KJ, Kennedy SR, Emond MJ, Loeb LA, Swisher EM & Risques RA (2016). Ultra-deep sequencing detects ovarian cancer cells in peritoneal fluid and reveals somatic TP53 mutations in noncancerous tissues. Proc Natl Acad Sci U S A 113, 6005–6010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee-Six H, Olafsson S, Ellis P, Osborne RJ, Sanders MA, Moore L, Georgakopoulos N, Torrente F, Noorani A, Goddard M, et al. (2019). The landscape of somatic mutation in normal colorectal epithelial cells. Nature 574, 532–537. [DOI] [PubMed] [Google Scholar]
- 79.Kim MP & Lozano G (2018). Mutant p53 partners in crime. Cell Death Differ 25, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Merkle FT, Ghosh S, Kamitaki N, Mitchell J, Avior Y, Mello C, Kashin S, Mekhoubad S, Ilic D, Charlton M, et al. (2017). Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature 545, 229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li JZ, Guo M, Chen L, Chen ZC, Fu Y & Chen YH (2022). p53 amyloid aggregation in cancer: function, mechanism, and therapy. Exp Hematol Oncol 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xu J, Reumers J, Couceiro JR, De Smet F, Gallardo R, Rudyak S, Cornelis A, Rozenski J, Zwolinska A, Marine JC, et al. (2011). Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat Chem Biol 7, 285–295. [DOI] [PubMed] [Google Scholar]
- 83.Su Z, Kon N, Yi J, Zhao H, Zhang W, Tang Q, Li H, Kobayashi H, Li Z, Duan S, et al. (2023). Specific regulation of BACH1 by the hotspot mutant p53(R175H) reveals a distinct gain-of-function mechanism. Nat Cancer 4, 564–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dibra D, Xiong S, Moyer SM, El-Naggar AK, Qi Y, Su X, Kong EK, Korkut A & Lozano G (2024). Mutant p53 protects triple-negative breast adenocarcinomas from ferroptosis in vivo. Sci Adv 10, eadk1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Weissmueller S, Manchado E, Saborowski M, Morris J.P.t., Wagenblast E, Davis CA, Moon SH, Pfister NT, Tschaharganeh DF, Kitzing T, et al. (2014). Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell 157, 382–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Efe G, Dunbar KJ, Sugiura K, Cunningham K, Carcamo S, Karaiskos S, Tang Q, Cruz-Acuna R, Resnick-Silverman L, Peura J, et al. (2023). p53 Gain-of-Function Mutation Induces Metastasis via BRD4-Dependent CSF-1 Expression. Cancer Discov 13, 2632–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, Getlik M, Barsyte-Lovejoy D, Al-awar R, Katona BW, Shilatifard A, et al. (2015). Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 525, 206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schulz-Heddergott R, Stark N, Edmunds SJ, Li J, Conradi LC, Bohnenberger H, Ceteci F, Greten FR, Dobbelstein M & Moll UM (2018). Therapeutic Ablation of Gain-of-Function Mutant p53 in Colorectal Cancer Inhibits Stat3-Mediated Tumor Growth and Invasion. Cancer Cell 34, 298–314 e297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tang QS, Su ZY, Gu W & Rustgi AK (2020). Mutant p53 on the Path to Metastasis. Trends Cancer 6, 62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang Z, Burigotto M, Ghetti S, Vaillant F, Tan T, Capaldo BD, Palmieri M, Hirokawa Y, Tai L, Simpson DS, et al. (2023). Loss-of-function but not gain-of-function properties of mutant TP53 are critical for the proliferation, survival and metastasis of a broad range of cancer cells. Cancer Discov. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ota A, Nakao H, Sawada Y, Karnan S, Wahiduzzaman M, Inoue T, Kobayashi Y, Yamamoto T, Ishii N, Ohashi T, et al. (2017). Delta40p53alpha suppresses tumor cell proliferation and induces cellular senescence in hepatocellular carcinoma cells. J Cell Sci 130, 614–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Candeias MM, Hagiwara M & Matsuda M (2016). Cancer-specific mutations in p53 induce the translation of Delta160p53 promoting tumorigenesis. EMBO Rep 17, 1542–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Berkers CR, Maddocks OD, Cheung EC, Mor I & Vousden KH (2013). Metabolic regulation by p53 family members. Cell Metab 18, 617–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Flores ER, Sengupta S, Miller JB, Newman JJ, Bronson R, Crowley D, Yang A, McKeon F & Jacks T (2005). Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 7, 363–373. [DOI] [PubMed] [Google Scholar]
- 95.Dotsch V, Bernassola F, Coutandin D, Candi E & Melino G (2010). p63 and p73, the ancestors of p53. Cold Spring Harb Perspect Biol 2, a004887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang MX & Attardi LD (2022). A Balancing Act: p53 Activity from Tumor Suppression to Pathology and Therapeutic Implications. Annu Rev Pathol-Mech 17, 205–226. [DOI] [PubMed] [Google Scholar]
- 97.Baslan T, Morris JP, Zhao Z, Reyes J, Ho YJ, Tsanov KM, Bermeo J, Tian S, Zhang S, Askan G, et al. (2022). Ordered and deterministic cancer genome evolution after p53 loss. Nature 608, 795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dudgeon C, Chan C, Kang WF, Sun Y, Emerson R, Robins H & Levine AJ (2014). The evolution of thymic lymphomas in p53 knockout mice. Gene Dev 28, 2613–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu G, Parant JM, Lang G, Chau P, Chavez-Reyes A, El-Naggar AK, Multani A, Chang S & Lozano G (2004). Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet 36, 63–68. [DOI] [PubMed] [Google Scholar]
- 100.Lane DP (1992). Cancer. p53, guardian of the genome. Nature 358, 15–16. [DOI] [PubMed] [Google Scholar]
- 101.Mello SS & Attardi LD (2018). Deciphering p53 signaling in tumor suppression. Curr Opin Cell Biol 51, 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Panatta E, Butera A, Mammarella E, Pitolli C, Mauriello A, Leist M, Knight RA, Melino G & Amelio I (2022). Metabolic regulation by p53 prevents R-loop-associated genomic instability. Cell Rep 41. [DOI] [PubMed] [Google Scholar]
- 103.Liu Y & Gu W (2022). The complexity of p53-mediated metabolic regulation in tumor suppression. Semin Cancer Biol 85, 4–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kastenhuber ER & Lowe SW (2017). Putting p53 in Context. Cell 170, 1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kruiswijk F, Labuschagne CF & Vousden KH (2015). p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol 16, 393–405. [DOI] [PubMed] [Google Scholar]
- 106.Liu YQ & Gu W (2022). p53 in ferroptosis regulation: the new weapon for the old guardian. Cell Death Differ 29, 895–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jiang L, Kon N, Li TY, Wang SJ, Su T, Hibshoosh H, Baer R & Gu W (2015). Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chu B, Kon N, Chen DL, Li TY, Liu T, Jiang L, Song SJ, Tavana O & Gu W (2019). ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol 21, 579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yang X, Wang Z, Zandkarimi F, Liu YQ, Duan SF, Li ZM, Kon N, Zhang ZG, Jiang XJ, Stockwell BR, et al. (2023). Regulation of VKORC1L1 is critical for p53-mediated tumor suppression through vitamin K metabolism. Cell Metabolism 35, 1474–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Suzuki S, Venkatesh D, Kanda H, Nakayama A, Hosokawa H, Lee E, Miki T, Stockwell BR, Yokote K, Tanaka T, et al. (2022). GLS2 Is a Tumor Suppressor and a Regulator of Ferroptosis in Hepatocellular Carcinoma. Cancer Res 82, 3209–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gnanapradeepan K, Indeglia A, Stieg DC, Clarke N, Shao C, Dougherty JF, Murali N & Murphy ME (2022). PLTP is a p53 target gene with roles in cancer growth suppression and ferroptosis. J Biol Chem 298, 102637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chen D, Chu B, Yang X, Liu Z, Jin Y, Kon N, Rabadan R, Jiang X, Stockwell BR & Gu W (2021). iPLA2β-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat Commun 12, 3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, Scott JP, Cai KQ, Campbell MR, Porter DK, et al. (2016). An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev 30, 918–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yang X, Wang Z, Samovich SN, Kapralov AA, Amoscato AA, Tyurin VA, Dar HH, Li Z, Duan S, Kon N, et al. (2024). PHLDA2-mediated phosphatidic acid peroxidation triggers a distinct ferroptotic response during tumor suppression. Cell Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Olivos DJ & Mayo LD (2016). Emerging Non-Canonical Functions and Regulation by p53: p53 and Stemness. Int J Mol Sci 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Koifman G, Aloni-Grinstein R & Rotter V (2019). p53 balances between tissue hierarchy and anarchy. J Mol Cell Biol 11, 553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lin T, Chao C, Saito S, Mazur SJ, Murphy ME, Appella E & Xu Y (2005). p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol 7, 165–171. [DOI] [PubMed] [Google Scholar]
- 118.Li M, He Y, Dubois W, Wu X, Shi J & Huang J (2012). Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Mol Cell 46, 30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Godar S, Ince TA, Bell GW, Feldser D, Donaher JL, Bergh J, Liu A, Miu K, Watnick RS, Reinhardt F, et al. (2008). Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell 134, 62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tschaharganeh DF, Xue W, Calvisi DF, Evert M, Michurina TV, Dow LE, Banito A, Katz SF, Kastenhuber ER, Weissmueller S, et al. (2014). p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell 158, 579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kaiser AM, Gatto A, Hanson KJ, Zhao RL, Raj N, Ozawa MG, Seoane JA, Bieging-Rolett KT, Wang M, Li I, et al. (2023). p53 governs an AT1 differentiation programme in lung cancer suppression. Nature 619, 851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Morris J.P.t., Yashinskie JJ, Koche R, Chandwani R, Tian S, Chen CC, Baslan T, Marinkovic ZS, Sanchez-Rivera FJ, Leach SD, et al. (2019). α-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 573, 595–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Amit M, Takahashi H, Dragomir MP, Lindemann A, Gleber-Netto FO, Pickering CR, Anfossi S, Osman AA, Cai Y, Wang R, et al. (2020). Loss of p53 drives neuron reprogramming in head and neck cancer. Nature 578, 449–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Krizhanovsky V & Lowe SW (2009). Stem cells: The promises and perils of p53. Nature 460, 1085–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhao Y, Yin X, Qin H, Zhu F, Liu H, Yang W, Zhang Q, Xiang C, Hou P, Song Z, et al. (2008). Two supporting factors greatly improve the efficiency of human iPSC generation. Cell Stem Cell 3, 475–479. [DOI] [PubMed] [Google Scholar]
- 126.Wagstaff L, Goschorska M, Kozyrska K, Duclos G, Kucinski I, Chessel A, Hampton-O’Neil L, Bradshaw CR, Allen GE, Rawlins EL, et al. (2016). Mechanical cell competition kills cells via induction of lethal p53 levels. Nat Commun 7, 11373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.de la Cova C, Senoo-Matsuda N, Ziosi M, Wu DC, Bellosta P, Quinzii CM & Johnston LA (2014). Supercompetitor status of Drosophila Myc cells requires p53 as a fitness sensor to reprogram metabolism and promote viability. Cell Metab 19, 470–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sun XL, Chen ZH, Guo X, Wang J, Ge M, Wong SZH, Wang T, Li S, Yao M, Johnston LA, et al. (2023). Stem cell competition driven by the Axin2-p53 axis controls brain size during murine development. Dev Cell 58, 744–759 e711. [DOI] [PubMed] [Google Scholar]
- 129.Dejosez M, Ura H, Brandt VL & Zwaka TP (2013). Safeguards for Cell Cooperation in Mouse Embryogenesis Shown by Genome-Wide Cheater Screen. Science 341, 1511–1514. [DOI] [PubMed] [Google Scholar]
- 130.Lima A, Lubatti G, Burgstaller J, Hu D, Green AP, Di Gregorio A, Zawadzki T, Pernaute B, Mahammadov E, Perez-Montero S, et al. (2021). Cell competition acts as a purifying selection to eliminate cells with mitochondrial defects during early mouse development. Nat Metab 3, 1091–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kozyrska K, Pilia G, Vishwakarma M, Wagstaff L, Goschorska M, Cirillo S, Mohamad S, Gallacher K, Salas REC & Piddini E (2022). p53 directs leader cell behavior, migration, and clearance during epithelial repair. Science 375, eabl8876. [DOI] [PubMed] [Google Scholar]
- 132.Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, Cagan A, Murai K, Mahbubani K, Stratton MR, et al. (2018). Somatic mutant clones colonize the human esophagus with age. Science 362, 911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Colom B, Alcolea MP, Piedrafita G, Hall MWJ, Wabik A, Dentro SC, Fowler JC, Herms A, King C, Ong SH, et al. (2020). Spatial competition shapes the dynamic mutational landscape of normal esophageal epithelium. Nat Genet 52, 604–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Watanabe H, Ishibashi K, Mano H, Kitamoto S, Sato N, Hoshiba K, Kato M, Matsuzawa F, Takeuchi Y, Shirai T, et al. (2018). Mutant p53-Expressing Cells Undergo Necroptosis via Cell Competition with the Neighboring Normal Epithelial Cells. Cell Rep 23, 3721–3729. [DOI] [PubMed] [Google Scholar]
- 135.Murai K, Skrupskelyte G, Piedrafita G, Hall M, Kostiou V, Ong SH, Nagy T, Cagan A, Goulding D, Klein AM, et al. (2018). Epidermal Tissue Adapts to Restrain Progenitors Carrying Clonal p53 Mutations. Cell Stem Cell 23, 687–699 e688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Powell E, Piwnica-Worms D & Piwnica-Worms H (2014). Contribution of p53 to metastasis. Cancer Discov 4, 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bergers G & Fendt SM (2021). The metabolism of cancer cells during metastasis. Nat Rev Cancer 21, 162–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ruddell A, Kelly-Spratt KS, Furuya M, Parghi SS & Kemp CJ (2008). p19/Arf and p53 suppress sentinel lymph node lymphangiogenesis and carcinoma metastasis. Oncogene 27, 3145–3155. [DOI] [PubMed] [Google Scholar]
- 139.Teodoro JG, Evans SK & Green MR (2007). Inhibition of tumor angiogenesis by p53: a new role for the guardian of the genome. J Mol Med-Jmm 85, 1175–1186. [DOI] [PubMed] [Google Scholar]
- 140.Zou ZQ, Gao CL, Nagaich AK, Connell T, Saito S, Moul JW, Seth P, Appella E & Srivastava S (2000). p53 regulates the expression of the tumor suppressor gene maspin. J Biol Chem 275, 6051–6054. [DOI] [PubMed] [Google Scholar]
- 141.Schwitalla S, Ziegler PK, Horst D, Becker V, Kerle I, Begus-Nahrmann Y, Lechel A, Rudolph KL, Langer R, Slotta-Huspenina J, et al. (2013). Loss of p53 in Enterocytes Generates an Inflammatory Microenvironment Enabling Invasion and Lymph Node Metastasis of Carcinogen-Induced Colorectal Tumors. Cancer Cell 23, 93–106. [DOI] [PubMed] [Google Scholar]
- 142.Muñoz-Fontela C, Mandinova A, Aaronson SA & Lee SW (2016). Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat Rev Immunol 16, 741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Blagih J, Buck MD & Vousden KH (2020). p53, cancer and the immune response. Journal of Cell Science 133. [DOI] [PubMed] [Google Scholar]
- 144.Cortez MA, Ivan C, Valdecanas D, Wang XH, Peltier HJ, Ye YP, Araujo L, Carbone DP, Shilo K, Giri DK, et al. (2016). PDL1 Regulation by p53 via miR-34. Jnci-J Natl Cancer I 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ghosh M, Saha S, Li JY, Montrose DC & Martinez LA (2023). p53 engages the cGAS/STING cytosolic DNA sensing pathway for tumor suppression. Mol Cell 83, 266–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C & Lowe SW (2007). Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V, et al. (2013). Non-Cell-Autonomous Tumor Suppression by p53. Cell 153, 449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L & Lowe SW (2008). Senescence of activated stellate cells limits liver fibrosis. Cell 134, 657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sharma MD, Rodriguez PC, Koehn BH, Baban B, Cui Y, Guo G, Shimoda M, Pacholczyk R, Shi H, Lee EJ, et al. (2018). Activation of p53 in Immature Myeloid Precursor Cells Controls Differentiation into Ly6c(+)CD103(+) Monocytic Antigen-Presenting Cells in Tumors. Immunity 48, 91–106 e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bezzi M, Seitzer N, Ishikawa T, Reschke M, Chen M, Wang GC, Mitchell C, Ng C, Katon J, Lunardi A, et al. (2018). Diverse genetic-driven immune landscapes dictate tumor progression through distinct mechanisms. Nat Med 24, 165–175. [DOI] [PubMed] [Google Scholar]
- 151.Zhu MR, Kim J, Deng Q, Ricciuti B, Alessi JV, Eglenen-Polat B, Bender ME, Huang HC, Kowash RR, Cuevas I, et al. (2023). Loss of p53 and mutational heterogeneity drives immune resistance in an autochthonous mouse lung cancer model with high tumor mutational burden. Cancer Cell 41, 1731–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ghosh M, Saha S, Bettke J, Nagar R, Parrales A, Iwakuma T, van der Velden AWM & Martinez LA (2021). Mutant p53 suppresses innate immune signaling to promote tumorigenesis. Cancer Cell 39, 494–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hsiue EH, Wright KM, Douglass J, Hwang MS, Mog BJ, Pearlman AH, Paul S, DiNapoli SR, Konig MF, Wang Q, et al. (2021). Targeting a neoantigen derived from a common TP53 mutation. Science 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Low L, Goh A, Koh J, Lim S & Wang CI (2019). Targeting mutant p53-expressing tumours with a T cell receptor-like antibody specific for a wild-type antigen. Nat Commun 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kim SP, Vale NR, Zacharakis N, Krishna S, Yu Z, Gasmi B, Gartner JJ, Sindiri S, Malekzadeh P, Deniger DC, et al. (2022). Adoptive Cellular Therapy with Autologous Tumor-Infiltrating Lymphocytes and T-cell Receptor-Engineered T Cells Targeting Common p53 Neoantigens in Human Solid Tumors. Cancer Immunol Res 10, 932–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Aloni-Grinstein R, Charni-Natan M, Solomon H & Rotter V (2018). p53 and the Viral Connection: Back into the Future. Cancers 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Concepcion AR, Wagner LE, Zhu JJ, Tao AY, Yang J, Khodadadi-Jamayran A, Wang YH, Liu MH, Rose RE, Jones DR, et al. (2022). The volume-regulated anion channel LRRC8C suppresses T cell function by regulating cyclic dinucleotide transport and STING-p53 signaling. Nat Immunol 23, 287–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Watanabe M, Moon KD, Vacchio MS, Hathcock KS & Hodes RJ (2014). Downmodulation of tumor suppressor p53 by T cell receptor signaling is critical for antigen-specific CD4(+) T cell responses. Immunity 40, 681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Liu Y, Leslie PL & Zhang YP (2021). Life and Death Decision-Making by p53 and Implications for Cancer Immunotherapy. Trends Cancer 7, 226–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Levine AJ (2019). The many faces of p53: something for everyone. J Mol Cell Biol 11, 524–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Levine AJ, Tomasini R, McKeon FD, Mak TW & Melino G (2011). The p53 family: guardians of maternal reproduction. Nat Rev Mol Cell Bio 12, 259–265. [DOI] [PubMed] [Google Scholar]
- 162.Hu WW, Feng ZH, Teresky AK & Levine AJ (2007). p53 regulates maternal reproduction through LIF. Nature 450, 721–U728. [DOI] [PubMed] [Google Scholar]
- 163.Bowen ME & Attardi LD (2019). The role of p53 in developmental syndromes. J Mol Cell Biol 11, 200–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Jones NC, Lynn ML, Gaudenz K, Sakai D, Aoto K, Rey JP, Glynn EF, Ellington L, Du C, Dixon J, et al. (2008). Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat Med 14, 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Charni M, Aloni-Grinstein R, Molchadsky A & Rotter V (2017). p53 on the crossroad between regeneration and cancer. Cell Death Differ 24, 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Qin Y, Wu K, Zhang Z, Pan RL, Lin ZQ, Zhang WY, Huang SS, Dai JJ, Huang R, Gong SQ, et al. (2022). NLRC3 deficiency promotes cutaneous wound healing due to the inhibition of p53 signaling. Bba-Mol Basis Dis 1868. [DOI] [PubMed] [Google Scholar]
- 167.Ou HL & Schumacher B (2018). DNA damage responses and p53 in the aging process. Blood 131, 488–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Rossi DJ, Jamieson CHM & Weissman IL (2008). Stems cells and the pathways to aging and cancer. Cell 132, 681–696. [DOI] [PubMed] [Google Scholar]
- 169.Liu L, Charville GW, Cheung TH, Yoo B, Santos PJ, Schroeder M & Rando TA (2018). Impaired Notch Signaling Leads to a Decrease in p53 Activity and Mitotic Catastrophe in Aged Muscle Stem Cells. Cell Stem Cell 23, 544–556 e544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Begus-Nahrmann Y, Lechel A, Obenauf AC, Nalapareddy K, Peit E, Hoffmann E, Schlaudraff F, Liss B, Schirmacher P, Kestler H, et al. (2009). p53 deletion impairs clearance of chromosomal-instable stem cells in aging telomere-dysfunctional mice. Nat Genet 41, 1138–1143. [DOI] [PubMed] [Google Scholar]
- 171.Lopez-Otin C, Blasco MA, Partridge L, Serrano M & Kroemer G (2013). The hallmarks of aging. Cell 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ong ALC & Ramasamy TS (2018). Role of Sirtuin1-p53 regulatory axis in aging, cancer and cellular reprogramming. Ageing Res Rev 43, 64–80. [DOI] [PubMed] [Google Scholar]
- 173.Maier B, Gluba W, Bernier B, Turner T, Mohammad K, Guise T, Sutherland A, Thorner M & Scrable H (2004). Modulation of mammalian life span by the short isoform of p53. Genes Dev 18, 306–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu XB, Soron G, Cooper B, Brayton C, et al. (2002). p53 mutant mice that display early ageing-associated phenotypes. Nature 415, 45–53. [DOI] [PubMed] [Google Scholar]
- 175.Ferbeyre G & Lowe SW (2002). Ageing - The price of tumour suppression? Nature 415, 26–27. [DOI] [PubMed] [Google Scholar]
- 176.Garcia-Cao I, Garcia-Cao M, Martin-Caballero J, Criado LM, Klatt P, Flores JM, Weill JC, Blasco MA & Serrano M (2002). “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. Embo J 21, 6225–6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Viña J, Matheu A, Maraver A, Klatt P, Flores I, Garcia-Cao I, Borras C, Flores JM, Blasco MA & Serrano M (2007). Delayed aging through damage protection by the Arf/p53 pathway:: Importance of antioxidant defence. Free Radical Res 41, S62–S62. [Google Scholar]
- 178.Bartas M, Brazda V, Volna A, Cerven J, Pecinka P & Zawacka-Pankau JE (2021). The Changes in the p53 Protein across the Animal Kingdom Point to Its Involvement in Longevity. Int J Mol Sci 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Abegglen LM, Caulin AF, Chan A, Lee K, Robinson R, Campbell MS, Kiso WK, Schmitt DL, Waddell PJ, Bhaskara S, et al. (2015). Potential Mechanisms for Cancer Resistance in Elephants and Comparative Cellular Response to DNA Damage in Humans. JAMA 314, 1850–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Checler F & Alves da Costa C (2014). p53 in neurodegenerative diseases and brain cancers. Pharmacol Ther 142, 99–113. [DOI] [PubMed] [Google Scholar]
- 181.Szybinska A & Lesniak W (2017). P53 Dysfunction in Neurodegenerative Diseases - The Cause or Effect of Pathological Changes? Aging Dis 8, 506–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Maor-Nof M, Shipony Z, Lopez-Gonzalez R, Nakayama L, Zhang YJ, Couthouis J, Blum JA, Castruita PA, Linares GR, Ruan K, et al. (2021). p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR). Cell 184, 689–708 e620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Mansky RH, Greguske EA, Yu D, Zarate N, Intihar TA, Tsai W, Brown TG, Thayer MN, Kumar K & Gomez-Pastor R (2023). Tumor suppressor p53 regulates heat shock factor 1 protein degradation in Huntington’s disease. Cell Rep 42, 112198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Reichert CO, de Freitas FA, Sampaio-Silva J, Rokita-Rosa L, Barros PL, Levy D & Bydlowski SP (2020). Ferroptosis Mechanisms Involved in Neurodegenerative Diseases. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Farmer KM, Ghag G, Puangmalai N, Montalbano M, Bhatt N & Kayed R (2020). P53 aggregation, interactions with tau, and impaired DNA damage response in Alzheimer’s disease. Acta Neuropathol Com 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Gudkov AV & Komarova EA (2010). Pathologies associated with the p53 response. Cold Spring Harb Perspect Biol 2, a001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Lowe SW, Schmitt EM, Smith SW, Osborne BA & Jacks T (1993). P53 Is Required for Radiation-Induced Apoptosis in Mouse Thymocytes. Nature 362, 847–849. [DOI] [PubMed] [Google Scholar]
- 188.Lowe SW, Ruley HE, Jacks T & Housman DE (1993). P53-Dependent Apoptosis Modulates the Cytotoxicity of Anticancer Agents. Cell 74, 957–967. [DOI] [PubMed] [Google Scholar]
- 189.Komarova EA, Kondratov RV, Wang K, Christov K, Golovkina TV, Goldblum JR & Gudkov AV (2004). Dual effect of p53 on radiation sensitivity in vivo: p53 promotes hematopoietic injury, but protects from gastro-intestinal syndrome in mice. Oncogene 23, 3265–3271. [DOI] [PubMed] [Google Scholar]
- 190.Lacroix M, Riscal R, Arena G, Linares LK & Le Cam L (2020). Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Mol Metab 33, 2–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Men HB, Cai H, Cheng QL, Zhou WQ, Wang X, Huang S, Zheng Y & Cai L (2021). The regulatory roles of p53 in cardiovascular health and disease. Cell Mol Life Sci 78, 2001–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Molchadsky A, Ezra O, Amendola PG, Krantz D, Kogan-Sakin I, Buganim Y, Rivlin N, Goldfinger N, Folgiero V, Falcioni R, et al. (2013). p53 is required for brown adipogenic differentiation and has a protective role against diet-induced obesity. Cell Death Differ 20, 774–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Quiñones M, Al-Massadi O, Folgueira C, Bremser S, Gallego R, Torres-Leal L, Haddad-Tóvolli R, García-Caceres C, Hernandez-Bautista R, Lam BYH, et al. (2018). p53 in AgRP neurons is required for protection against diet-induced obesity via JNK1. Nat Commun 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Yokoyama M, Okada S, Nakagomi A, Moriya J, Shimizu I, Nojima A, Yoshida Y, Ichimiya H, Kamimura N, Kobayashi Y, et al. (2014). Inhibition of Endothelial p53 Improves Metabolic Abnormalities Related to Dietary Obesity. Cell Rep 7, 1691–1703. [DOI] [PubMed] [Google Scholar]
- 195.Hanahan D & Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 196.Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, et al. (2004). Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 119, 861–872. [DOI] [PubMed] [Google Scholar]
- 197.Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D & Jacks T (2004). Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119, 847–860. [DOI] [PubMed] [Google Scholar]
- 198.Rivlin N, Brosh R, Oren M & Rotter V (2011). Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2, 466–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Haupt S, Caramia F, Herschtal A, Soussi T, Lozano G, Chen H, Liang H, Speed TP & Haupt Y (2019). Identification of cancer sex-disparity in the functional integrity of p53 and its X chromosome network. Nat Commun 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Rodriguez-Meira A, Norfo R, Wen S, Chedeville AL, Rahman H, O’Sullivan J, Wang G, Louka E, Kretzschmar WW, Paterson A, et al. (2023). Single-cell multi-omics identifies chronic inflammation as a driver of TP53-mutant leukemic evolution. Nat Genet 55, 1531–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Giacomelli AO, Yang X, Lintner RE, McFarland JM, Duby M, Kim J, Howard TP, Takeda DY, Ly SH, Kim E, et al. (2018). Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet 50, 1381–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Hoyos D, Zappasodi R, Schulze I, Sethna Z, de Andrade KC, Bajorin DF, Bandlamundi C, Callahan MK, Funt SA, Hadrup SR, et al. (2022). Fundamental immune-oncogenicity trade-offs define driver mutation fitness. Nature 606, 172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS, Lamprecht TL, Shen D, Hundal J, Fulton RS, et al. (2015). Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 518, 552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Xiong S, Chachad D, Zhang Y, Gencel-Augusto J, Sirito M, Pant V, Yang P, Sun C, Chau G, Qi Y, et al. (2022). Differential Gain-of-Function Activity of Three p53 Hotspot Mutants In Vivo. Cancer Res 82, 1926–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Gencel-Augusto J, Su XP, Qi Y, Whitley EM, Pant V, Xiong SB, Shah VT, Lin JRM, Perez E, Fiorotto ML, et al. (2023). Dimeric p53 Mutant Elicits Unique Tumor-Suppressive Activities through an Altered Metabolic Program. Cancer Discov 13, 1230–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Choe JH, Kawase T, Xu A, Guzman A, Obradovic AZ, Low-Calle AM, Alaghebandan B, Raghavan A, Long KT, Hwang PM, et al. (2023). Li-Fraumeni Syndrome-Associated Dimer-Forming Mutant p53 Promotes Transactivation-Independent Mitochondrial Cell Death. Cancer Discov 13, 1250–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hoyos D, Greenbaum B & Levine AJ (2022). The genotypes and phenotypes of missense mutations in the proline domain of the p53 protein. Cell Death Differ 29, 938–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Lubin R, Zalcman G, Bouchet L, Tredaniel J, Legros Y, Cazals D, Hirsch A & Soussi T (1995). Serum P53 Antibodies as Early Markers of Lung-Cancer. Nat Med 1, 701–702. [DOI] [PubMed] [Google Scholar]
- 209.Lobello C, Tichy B, Bystry V, Radova L, Filip D, Mraz M, Montes-Mojarro IA, Prokoph N, Larose H, Liang HC, et al. (2021). STAT3 and TP53 mutations associate with poor prognosis in anaplastic large cell lymphoma. Leukemia 35, 1500–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Singh N, Piskorz AM, Bosse T, Jimenez-Linan M, Rous B, Brenton JD, Gilks CB & Kobel M (2020). p53 immunohistochemistry is an accurate surrogate for TP53 mutational analysis in endometrial carcinoma biopsies. J Pathol 250, 336–345. [DOI] [PubMed] [Google Scholar]
- 211.Wong D, Luo P, Oldfield LE, Gong H, Brunga L, Rabinowicz R, Subasri V, Chan C, Downs T, Farncombe KM, et al. (2024). Early Cancer Detection in Li-Fraumeni Syndrome with Cell-Free DNA. Cancer Discov 14, 104–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Sinn M, Sinn BV, Treue D, Keilholz U, Damm F, Schmuck R, Lohneis P, Klauschen F, Striefler JK, Bahra M, et al. (2020). TP53 Mutations Predict Sensitivity to Adjuvant Gemcitabine in Patients with Pancreatic Ductal Adenocarcinoma: Next-Generation Sequencing Results from the CONKO-001 Trial. Clin Cancer Res 26, 3732–3739. [DOI] [PubMed] [Google Scholar]
- 213.Sethi NS, Kikuchi O, Duronio GN, Stachler MD, McFarland JM, Ferrer-Luna R, Zhang YX, Bao CY, Bronson R, Patil D, et al. (2020). Early TP53 alterations engage environmental exposures to promote gastric premalignancy in an integrative mouse model. Nat Genet 52, 219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Liu YH, Zhang XN, Han C, Wan GH, Huang XX, Ivan C, Jiang DH, Rodriguez-Aguayo C, Lopez-Berestein G, Rao PH, et al. (2015). TP53 loss creates therapeutic vulnerability in colorectal cancer. Nature 520, 697–U286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Liu Y, Chen C, Xu Z, Scuoppo C, Rillahan CD, Gao J, Spitzer B, Bosbach B, Kastenhuber ER, Baslan T, et al. (2016). Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature 531, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kucherlapati MH (2023). Mouse models in colon cancer, inferences, and implications. Iscience 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S & Tuveson DA (2005). Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483. [DOI] [PubMed] [Google Scholar]
- 218.Muller PAJ & Vousden KH (2013). p53 mutations in cancer. Nat Cell Biol 15, 2–8. [DOI] [PubMed] [Google Scholar]
- 219.Boettcher S, Miller PG, Sharma R, McConkey M, Leventhal M, Krivtsov AV, Giacomelli AO, Wong W, Kim J, Chao S, et al. (2019). A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 365, 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Varley JM, Evans DGR & Birch JM (1997). Li-Fraumeni syndrome - A molecular and clinical review. Brit J Cancer 76, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wasylishen AR & Lozano G (2016). Attenuating the p53 Pathway in Human Cancers: Many Means to the Same End. Cold Spring Harb Perspect Med 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Oliner JD, Kinzler KW, Meltzer PS, George DL & Vogelstein B (1992). Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 358, 80–83. [DOI] [PubMed] [Google Scholar]
- 223.Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, Bargonetti J, Bartel F, Taubert H, Wuerl P, et al. (2004). A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell 119, 591–602. [DOI] [PubMed] [Google Scholar]
- 224.Zhang Y, Xiong Y & Yarbrough WG (1998). ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 92, 725–734. [DOI] [PubMed] [Google Scholar]
- 225.Marney CB, Anderson ES, Adnan M, Peng KL, Hu Y, Weinhold N & Schmitt AM (2021). p53-intact cancers escape tumor suppression through loss of long noncoding RNA. Cell Rep 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Zeng J, Hills SA, Ozono E & Diffley JFX (2023). Cyclin E-induced replicative stress drives p53-dependent whole-genome duplication. Cell 186, 528–542 e514. [DOI] [PubMed] [Google Scholar]
- 227.Maddocks OD, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E & Vousden KH (2013). Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493, 542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Tajan M, Hock AK, Blagih J, Robertson NA, Labuschagne CF, Kruiswijk F, Humpton TJ, Adams PD & Vousden KH (2018). A Role for p53 in the Adaptation to Glutamine Starvation through the Expression of SLC1A3. Cell Metabolism 28, 721–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Kim J, Yu LL, Chen WC, Xu YX, Wu M, Todorova D, Tang QS, Feng BB, Jiang L, He JJ, et al. (2019). Wild-Type p53 Promotes Cancer Metabolic Switch by Inducing PUMA-Dependent Suppression of Oxidative Phosphorylation. Cancer Cell 35, 191–203. [DOI] [PubMed] [Google Scholar]
- 230.Makino Y, Hikita H, Fukumoto K, Sung JH, Sakano Y, Murai K, Sakane S, Kodama T, Sakamori R, Kondo J, et al. (2022). Constitutive Activation of the Tumor Suppressor p53 in Hepatocytes Paradoxically Promotes Non-Cell Autonomous Liver Carcinogenesis. Cancer Research 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Cho YH, Ro EJ, Yoon JS, Mizutani T, Kang DW, Park JC, Kim TI, Clevers H & Choi KY (2020). 5-FU promotes stemness of colorectal cancer via p53-mediated WNT/β-catenin pathway activation. Nat Commun 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Sole L, Lobo-Jarne T, Alvarez-Villanueva D, Alonso-Maranon J, Guillen Y, Guix M, Sangrador I, Rozalen C, Vert A, Barbachano A, et al. (2022). p53 wild-type colorectal cancer cells that express a fetal gene signature are associated with metastasis and poor prognosis. Nat Commun 13, 2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Jackson JG, Pant V, Li Q, Chang LL, Quintas-Cardama A, Garza D, Tavana O, Yang P, Manshouri T, Li Y, et al. (2012). p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell 21, 793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Webster MR, Fane ME, Alicea GM, Basu S, Kossenkov AV, Marino GE, Douglass SM, Kaur A, Ecker BL, Gnanapradeepan K, et al. (2020). Paradoxical Role for Wild-Type p53 in Driving Therapy Resistance in Melanoma. Mol Cell 77, 681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E & Vousden KH (2006). TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126, 107–120. [DOI] [PubMed] [Google Scholar]
- 236.Cheung EC, DeNicola GM, Nixon C, Blyth K, Labuschagne CF, Tuveson DA & Vousden KH (2020). Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell 37, 168–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Hassin O & Oren M (2023). Drugging p53 in cancer: one protein, many targets. Nature Reviews Drug Discovery 22, 127–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Adams CM, Mitra R, Xiao YC, Michener P, Palazzo J, Chao AL, Gour J, Cassel J, Salvino JM & Eischen CM (2023). Targeted MDM2 Degradation Reveals a New Vulnerability for p53-Inactivated Triple-Negative Breast Cancer. Cancer Discov 13, 1210–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. (2004). In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848. [DOI] [PubMed] [Google Scholar]
- 240.Yee K, Papayannidis C, Vey N, Dickinson MJ, Kelly KR, Assouline S, Kasner M, Seiter K, Drummond MW, Yoon SS, et al. (2021). Murine double minute 2 inhibition alone or with cytarabine in acute myeloid leukemia: Results from an idasanutlin phase 1/1b study small star, filled. Leuk Res 100, 106489. [DOI] [PubMed] [Google Scholar]
- 241.Konopleva MY, Röllig C, Cavenagh J, Deeren D, Girshova L, Krauter J, Martinelli G, Montesinos P, Schäfer JA, Ottmann O, et al. (2022). Idasanutlin plus cytarabine in relapsed or refractory acute myeloid leukemia: results of the MIRROS trial. Blood Adv 6, 4147–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Aguilar A, Lu J, Liu L, Du D, Bernard D, McEachern D, Przybranowski S, Li X, Luo R, Wen B, et al. (2017). Discovery of 4-((3’R,4’S,5’R)-6”-Chloro-4’-(3-chloro-2-fluorophenyl)-1’-ethyl-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamido)bicyclo[2.2.2]octane-1-carboxylic Acid (AA-115/APG-115): A Potent and Orally Active Murine Double Minute 2 (MDM2) Inhibitor in Clinical Development. J Med Chem 60, 2819–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Sun D, Li Z, Rew Y, Gribble M, Bartberger MD, Beck HP, Canon J, Chen A, Chen X, Chow D, et al. (2014). Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J Med Chem 57, 1454–1472. [DOI] [PubMed] [Google Scholar]
- 244.Carvajal LA, Ben Neriah D, Senecal A, Benard L, Thiruthuvanathan V, Yatsenko T, Narayanagari SR, Wheat JC, Todorova TI, Mitchell K, et al. (2018). Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci Transl Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Tavana O & Gu W (2017). Modulation of the p53/MDM2 interplay by HAUSP inhibitors. J Mol Cell Biol 9, 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Zhao CY, Szekely L, Bao W & Selivanova G (2010). Rescue of p53 function by small-molecule RITA in cervical carcinoma by blocking E6-mediated degradation. Cancer Res 70, 3372–3381. [DOI] [PubMed] [Google Scholar]
- 247.Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, McCarthy A, Appleyard V, Murray KE, Baker L, et al. (2008). Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 13, 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Yi J, Tavana O, Li H, Wang D, Baer RJ & Gu W (2023). Targeting USP2 regulation of VPRBP-mediated degradation of p53 and PD-L1 for cancer therapy. Nat Commun 14, 1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Veschi V, Liu ZH, Voss TC, Ozbun L, Gryder B, Yan CH, Hu Y, Ma AQ, Jin J, Mazur SJ, et al. (2017). Epigenetic siRNA and Chemical Screens Identify SETD8 Inhibition as a Therapeutic Strategy for p53 Activation in High-Risk Neuroblastoma. Cancer Cell 31, 50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Wieczorek AM, Waterman JLF, Waterman MJF & Halazonetis TD (1996). Structure-based rescue of common tumor-derived p53 mutants. Nat Med 2, 1143–1146. [DOI] [PubMed] [Google Scholar]
- 251.Michalovitz D, Halevy O & Oren M (1990). Conditional Inhibition of Transformation and of Cell-Proliferation by a Temperature-Sensitive Mutant of P53. Cell 62, 671–680. [DOI] [PubMed] [Google Scholar]
- 252.Foster BA, Coffey HA, Morin MJ & Rastinejad F (1999). Pharmacological rescue of mutant p53 conformation and function. Science 286, 2507–2510. [DOI] [PubMed] [Google Scholar]
- 253.Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, Bergman J, Wiman KG & Selivanova G (2002). Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med 8, 282–288. [DOI] [PubMed] [Google Scholar]
- 254.Westin SN, Nieves-Neira W, Lynam C, Salim KY, Silva AD, Ho RT, Mills GB, Coleman RL, Janku F & Matei D (2018). Safety and early efficacy signals for COTI-2, an orally available small molecule targeting p53, in a phase I trial of recurrent gynecologic cancer. Cancer Research 78. [Google Scholar]
- 255.Chen S, Wu JL, Liang Y, Tang YG, Song HX, Wu LL, Xing YF, Yan N, Li YT, Wang ZY, et al. (2021). Arsenic Trioxide Rescues Structural p53 Mutations through a Cryptic Allosteric Site. Cancer Cell 39, 225–239. [DOI] [PubMed] [Google Scholar]
- 256.Song HX, Wu JL, Tang YG, Dai YT, Xiang XR, Li Y, Wu LL, Wu JQ, Liang Y, Xing YF, et al. (2023). Diverse rescue potencies of p53 mutations to ATO are predetermined by intrinsic mutational properties. Sci Transl Med 15. [DOI] [PubMed] [Google Scholar]
- 257.Tang YG, Song HX, Wang ZY, Xiao SJ, Xiang XR, Zhan HE, Wu LL, Wu JL, Xing YF, Tan Y, et al. (2022). Repurposing antiparasitic antimonials to noncovalently rescue temperature-sensitive p53 mutations. Cell Rep 39. [DOI] [PubMed] [Google Scholar]
- 258.Boeckler FM, Joerger AC, Jaggi G, Rutherford TJ, Veprintsev DB & Fersht AR (2008). Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc Natl Acad Sci U S A 105, 10360–10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Liu X, Wilcken R, Joerger AC, Chuckowree IS, Amin J, Spencer J & Fersht AR (2013). Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res 41, 6034–6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Dumbrava EE, Johnson ML, Tolcher AW, Shapiro G, Thompson JA, El-Khoueiry AB, Vandross AL, Kummar S, Parikh AR, Munster PN, et al. (2022). First-in-human study of PC14586, a small molecule structural corrector of Y220C mutant p53, in patients with advanced solid tumors harboring a Y220C mutation. Journal of Clinical Oncology 40. [Google Scholar]
- 261.Guiley KZ & Shokat KM (2023). A Small Molecule Reacts with the p53 Somatic Mutant Y220C to Rescue Wild-type Thermal Stability. Cancer Discov 13, 56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Kabir M, Sun N, Hu XP, Martin TC, Yi JJ, Zhong Y, Xiong Y, Kaniskan HÜ, Gu W, Parsons R, et al. (2023). Acetylation Targeting Chimera Enables Acetylation of the Tumor Suppressor p53. J Am Chem Soc 145, 14932–14944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Yu X, Vazquez A, Levine AJ & Carpizo DR (2012). Allele-Specific p53 Mutant Reactivation. Cancer Cell 21, 614–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Soragni A, Janzen DM, Johnson LM, Lindgren AG, Nguyen ATQ, Tiourin E, Soriaga AB, Lu J, Jiang L, Faull KF, et al. (2016). A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell 29, 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Palanikumar L, Karpauskaite L, Al-Sayegh M, Chehade I, Alam M, Hassan S, Maity D, Ali L, Kalmouni M, Hunashal Y, et al. (2021). Protein mimetic amyloid inhibitor potently abrogates cancer-associated mutant p53 aggregation and restores tumor suppressor function. Nat Commun 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Zhang SL, Zhou LL, Hong B, van den Heuvel APJ, Prabhu VV, Warfel NA, Kline CLB, Dicker DT, Kopelovich L & El-Deiry WS (2015). Small-Molecule NSC59984 Restores p53 Pathway Signaling and Antitumor Effects against Colorectal Cancer via p73 Activation and Degradation of Mutant p53. Cancer Research 75, 3842–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Alexandrova EM, Yallowitz AR, Li D, Xu S, Schulz R, Proia DA, Lozano G, Dobbelstein M & Moll UM (2015). Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 523, 352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Padmanabhan A, Candelaria N, Wong KK, Nikolai BC, Lonard DM, O’Malley BW & Richards JS (2018). USP15-dependent lysosomal pathway controls p53-R175H turnover in ovarian cancer cells. Nat Commun 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Huang XW, Cao ZY, Qian JY, Ding T, Wu YX, Zhang H, Zhong SQ, Wang XL, Ren XG, Zhang W, et al. (2024). Nanoreceptors promote mutant p53 protein degradation by mimicking selective autophagy receptors. Nat Nanotechnol. [DOI] [PubMed] [Google Scholar]
- 270.Heise C, SampsonJohannes A, Williams A, McCormick F, VonHoff DD & Kirn DH (1997). ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat Med 3, 639–645. [DOI] [PubMed] [Google Scholar]
- 271.Yang ZF, Ka-Li Sun J, Lee MM & Chan MK (2022). Restoration of p53 activity via intracellular protein delivery sensitizes triple negative breast cancer to anti-PD-1 immunotherapy. J Immunother Cancer 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Kong N, Tao W, Ling X, Wang J, Xiao Y, Shi S, Ji X, Shajii A, Gan ST, Kim NY, et al. (2019). Synthetic mRNA nanoparticle-mediated restoration of p53 tumor suppressor sensitizes p53-deficient cancers to mTOR inhibition. Sci Transl Med 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Pirollo KF, Nemunaitis J, Leung PK, Nunan R, Adams J & Chang EH (2016). Safety and Efficacy in Advanced Solid Tumors of a Targeted Nanocomplex Carrying the p53 Gene Used in Combination with Docetaxel: A Phase 1b Study. Mol Ther 24, 1697–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Zhou X, Singh M, Sanz Santos G, Guerlavais V, Carvajal LA, Aivado M, Zhan Y, Oliveira MMS, Westerberg LS, Annis DA, et al. (2021). Pharmacologic Activation of p53 Triggers Viral Mimicry Response Thereby Abolishing Tumor Immune Evasion and Promoting Antitumor Immunity. Cancer Discov 11, 3090–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Speetjens FM, Kuppen PJ, Welters MJ, Essahsah F, Voet van den Brink AM, Lantrua MG, Valentijn AR, Oostendorp J, Fathers LM, Nijman HW, et al. (2009). Induction of p53-specific immunity by a p53 synthetic long peptide vaccine in patients treated for metastatic colorectal cancer. Clin Cancer Res 15, 1086–1095. [DOI] [PubMed] [Google Scholar]
- 276.Hardwick NR, Carroll M, Kaltcheva T, Qian D, Lim D, Leong L, Chu P, Kim J, Chao J, Fakih M, et al. (2014). p53MVA therapy in patients with refractory gastrointestinal malignancies elevates p53-specific CD8+ T-cell responses. Clin Cancer Res 20, 4459–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Procopio MG, Laszlo C, Al Labban D, Kim DE, Bordignon P, Jo SH, Goruppi S, Menietti E, Ostano P, Ala U, et al. (2015). Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat Cell Biol 17, 1193–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Arandkar S, Furth N, Elisha Y, Nataraj NB, van der Kuip H, Yarden Y, Aulitzky W, Ulitsky I, Geiger B & Oren M (2018). Altered p53 functionality in cancer-associated fibroblasts contributes to their cancer-supporting features. P Natl Acad Sci USA 115, 6410–6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, et al. (2013). p53 status determines the role of autophagy in pancreatic tumour development. Nature 504, 296–300. [DOI] [PubMed] [Google Scholar]
- 280.Emerling BM, Hurov JB, Poulogiannis G, Tsukazawa KS, Choo-Wing R, Wulf GM, Bell EL, Shim HS, Lamia KA, Rameh LE, et al. (2013). Depletion of a Putatively Druggable Class of Phosphatidylinositol Kinases Inhibits Growth of p53-Null Tumors. Cell 155, 844–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Hoe KK, Verma CS & Lane DP (2014). Drugging the p53 pathway: understanding the route to clinical efficacy. Nature Reviews Drug Discovery 13, 217–236. [DOI] [PubMed] [Google Scholar]
- 282.Pan R, Ruvolo V, Mu H, Leverson JD, Nichols G, Reed JC, Konopleva M & Andreeff M (2017). Synthetic Lethality of Combined Bcl-2 Inhibition and p53 Activation in AML: Mechanisms and Superior Antileukemic Efficacy. Cancer Cell 32, 748–760 e746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Sullivan KD, Padilla-Just N, Henry RE, Porter CC, Kim J, Tentler JJ, Eckhardt SG, Tan AC, DeGregori J & Espinosa JM (2012). ATM and MET kinases are synthetic lethal with nongenotoxic activation of p53. Nat Chem Biol 8, 646–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Abraham SA, Hopcroft LEM, Carrick E, Drotar ME, Dunn K, Williamson AJK, Korfi K, Baquero P, Park LE, Scott MT, et al. (2016). Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nature 534, 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Moon SH, Huang CH, Houlihan SL, Regunath K, Freed-Pastor WA, Morris J.P.t., Tschaharganeh DF, Kastenhuber ER, Barsotti AM, Culp-Hill R, et al. (2019). p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 176, 564–580 e519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Heinzl N, Maritschnegg E, Koziel K, Schilhart-Wallisch C, Heinze G, Yang WL, Bast RC, Sehouli J, Braicu EI, Vergote I, et al. (2023). Amyloid-like p53 as prognostic biomarker in serous ovarian cancer-a study of the OVCAD consortium. Oncogene 42, 2473–2484. [DOI] [PubMed] [Google Scholar]
- 287.Feldser DM, Kostova KK, Winslow MM, Taylor SE, Cashman C, Whittaker CA, Sanchez-Rivera FJ, Resnick R, Bronson R, Hemann MT, et al. (2010). Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature 468, 572–U249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Junttila MR, Karnezis AN, Garcia D, Madriles F, Kortlever RM, Rostker F, Swigart LB, Pham DM, Seo Y, Evan GI, et al. (2010). Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature 468, 567–U244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Kaiser AM & Attardi LD (2018). Deconstructing networks of p53-mediated tumor suppression. Cell Death Differ 25, 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV & Gudkov AV (1999). A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 285, 1733–1737. [DOI] [PubMed] [Google Scholar]
- 291.Rao B, Lain S & Thompson AM (2013). p53-Based cyclotherapy: exploiting the ‘guardian of the genome’ to protect normal cells from cytotoxic therapy. Br J Cancer 109, 2954–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Martins CP, Brown-Swigart L & Evan GI (2006). Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 127, 1323–1334. [DOI] [PubMed] [Google Scholar]
- 293.Jung J, Lee JS, Dickson MA, Schwartz GK, Le Cesne A, Varga A, Bahleda R, Wagner AJ, Choy E, de Jonge MJ, et al. (2016). TP53 mutations emerge with HDM2 inhibitor SAR405838 treatment in de-differentiated liposarcoma. Nat Commun 7, 12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Bhinder B, Gilvary C, Madhukar NS & Elemento O (2021). Artificial Intelligence in Cancer Research and Precision Medicine. Cancer Discov 11, 900–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Allen BL, Quach K, Jones T, Levandowski CB, Ebmeier CC, Rubin JD, Read T, Dowell RD, Schepartz A & Taatjes DJ (2022). Suppression of p53 response by targeting p53-Mediator binding with a stapled peptide. Cell Rep 39, 110630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Vousden KH & Ryan KM (2009). p53 and metabolism. Nat Rev Cancer 9, 691–700. [DOI] [PubMed] [Google Scholar]
- 297.Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Broz DK, Basak S, Park EJ, McLaughlin ME, et al. (2011). Distinct p53 Transcriptional Programs Dictate Acute DNA-Damage Responses and Tumor Suppression. Cell 145, 571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Pappas K, Xu J, Zairis S, Resnick-Silverman L, Abate F, Steinbach N, Ozturk S, Saal LH, Su T, Cheung P, et al. (2017). p53 Maintains Baseline Expression of Multiple Tumor Suppressor Genes. Mol Cancer Res 15, 1051–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Böhlig L & Rother K (2011). One Function-Multiple Mechanisms: The Manifold Activities of p53 as a Transcriptional Repressor. J Biomed Biotechnol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Li B & Lee MY (2001). Transcriptional regulation of the human DNA polymerase delta catalytic subunit gene POLD1 by p53 tumor suppressor and Sp1. J Biol Chem 276, 29729–29739. [DOI] [PubMed] [Google Scholar]
- 301.Uxa S, Bernhart SH, Mages CFS, Fischer M, Kohler R, Hoffmann S, Stadler PF, Engeland K & Muller GA (2019). DREAM and RB cooperate to induce gene repression and cell-cycle arrest in response to p53 activation. Nucleic Acids Res 47, 9087–9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Imbriano C, Gnesutta N & Mantovani R (2012). The NF-Y/p53 liaison: well beyond repression. Biochim Biophys Acta 1825, 131–139. [DOI] [PubMed] [Google Scholar]
- 303.Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, et al. (2007). Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 26, 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M, et al. (2010). A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 142, 409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Porter JR, Fisher BE, Baranello L, Liu JC, Kambach DM, Nie ZQ, Koh WS, Luo J, Stommel JM, Levens D, et al. (2017). Global Inhibition with Specific Activation: How p53 and MYC Redistribute the Transcriptome in the DNA Double-Strand Break Response. Mol Cell 67, 1013–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Hoffman WH, Biade S, Zilfou JT, Chen J & Murphy M (2002). Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem 277, 3247–3257. [DOI] [PubMed] [Google Scholar]
- 307.Amson R, Pece S, Lespagnol A, Vyas R, Mazzarol G, Tosoni D, Colaluca I, Viale G, Rodrigues-Ferreira S, Wynendaele J, et al. (2012). Reciprocal repression between P53 and TCTP. Nat Med 18, 91–99. [DOI] [PubMed] [Google Scholar]
- 308.Fischer M, Steiner L & Engeland K (2014). The transcription factor p53: not a repressor, solely an activator. Cell Cycle 13, 3037–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Donehower LA & Lozano G (2009). 20 years studying p53 functions in genetically engineered mice. Nat Rev Cancer 9, 831–841. [DOI] [PubMed] [Google Scholar]
- 310.Fischer M (2021). Mice Are Not Humans: The Case of p53. Trends Cancer 7, 12–14. [DOI] [PubMed] [Google Scholar]
- 311.Walsh NC, Kenney LL, Jangalwe S, Aryee KE, Greiner DL, Brehm MA & Shultz LD (2017). Humanized Mouse Models of Clinical Disease. Annu Rev Pathol 12, 187–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Day CP, Merlino G & Van Dyke T (2015). Preclinical mouse cancer models: a maze of opportunities and challenges. Cell 163, 39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Hofer M & Lutolf MP (2021). Engineering organoids. Nat Rev Mater 6, 402–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Pant V, Sun C & Lozano G (2023). Tissue specificity and spatio-temporal dynamics of the p53 transcriptional program. Cell Death Differ 30, 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Xue YZ, San Luis B & Lane DP (2019). Intratumour heterogeneity of p53 expression; causes and consequences. Journal of Pathology 249, 274–285. [DOI] [PubMed] [Google Scholar]
- 316.Parikh S, Parikh R, Michael K, Bikovski L, Barnabas G, Mardamshina M, Hemi R, Manich P, Goldstein N, Malcov-Brog H, et al. (2022). Food-seeking behavior is triggered by skin ultraviolet exposure in males. Nat Metab 4, 883–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Bieging-Rolett KT, Kaiser AM, Morgens DW, Boutelle AM, Seoane JA, Van Nostrand EL, Zhu C, Houlihan SL, Mello SS, Yee BA, et al. (2020). Zmat3 Is a Key Splicing Regulator in the p53 Tumor Suppression Program. Mol Cell 80, 452–469 e459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Janic A, Valente LJ, Wakefield MJ, Di Stefano L, Milla L, Wilcox S, Yang HY, Tai L, Vandenberg CJ, Kueh AJ, et al. (2018). DNA repair processes are critical mediators of p53-dependent tumor suppression. Nat Med 24, 947–953. [DOI] [PubMed] [Google Scholar]
- 319.Brennan MS, Brinkmann K, Romero Sola G, Healey G, Gibson L, Gangoda L, Potts MA, Lieschke E, Wilcox S, Strasser A, et al. (2024). Combined absence of TRP53 target genes ZMAT3, PUMA and p21 cause a high incidence of cancer in mice. Cell Death Differ 31, 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Muys BR, Anastasakis DG, Claypool D, Pongor L, Li XL, Grammatikakis I, Liu M, Wang X, Prasanth KV, Aladjem MI, et al. (2021). The p53-induced RNA-binding protein ZMAT3 is a splicing regulator that inhibits the splicing of oncogenic CD44 variants in colorectal carcinoma. Genes Dev 35, 102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Levine AJ (2024). Improving T cell killing and understanding senescence: Possible roles for TP53 in cancer immunotherapy. Proc Natl Acad Sci U S A 121, e2402533121. [DOI] [PMC free article] [PubMed] [Google Scholar]