

Studies in zebrafish identify a conserved axis that mediates metabolic stress–induced pancreatic islet inflammation.
Abstract
Islet inflammation is an important etiopathology of type 2 diabetes; however, the underlying mechanisms are not well defined. Using complementary experimental models, we discovered RIPK3-dependent IL1B induction in β cells as an instigator of islet inflammation. In cultured β cells, ER stress activated RIPK3, leading to NF-kB–mediated proinflammatory gene expression. In a zebrafish muscle insulin resistance model, overnutrition caused islet inflammation, β cell dysfunction, and loss in an ER stress–, ripk3-, and il1b-dependent manner. In mouse islets, high-fat diet triggered the IL1B expression in β cells before macrophage recruitment in vivo, and RIPK3 inhibition suppressed palmitate-induced β cell dysfunction and Il1b expression in vitro. Furthermore, in human islets grafted in hyperglycemic mice, a marked increase in ER stress, RIPK3, and NF-kB activation in β cells were accompanied with murine macrophage infiltration. Thus, RIPK3-mediated induction of proinflammatory mediators is a conserved, previously unrecognized β cell response to metabolic stress and a mediator of the ensuing islet inflammation.
INTRODUCTION
Type 2 diabetes (T2D) is a worldwide health concern with more than 600 million people projected to have the disease by 2045 (1). T2D is characterized by pancreatic islet dysfunction, including insufficient insulin secretion, hyperglucagonemia, and insulin resistance at multiple sites (2). Impaired insulin secretion is the major determinant of whether hyperglycemia develops in response to insulin resistance, with most T2D-associated genetic loci predicted to affect islet function (3). Multiple molecular mechanisms have been proposed for the β cell dysfunction and loss in T2D, including islet inflammation, endoplasmic reticulum (ER) stress, oxidative stress, amyloid deposition, β cell dedifferentiation, and glucose- and lipid-induced changes (2). Supporting a role of intraislet inflammation is the increased number of islet macrophages found both in T2D pancreatic samples and in animal models (4–6).
The origin and molecular mechanisms responsible for islet inflammation in T2D are not well defined. The islet inflammation is primarily driven by the interleukin-1 (IL-1) system resulting from nuclear factor κB (NF-κB) and/or NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome activation, leading to increased leukocytes, primarily macrophages (7). The chronic low-grade inflammatory state associated with T2D may contribute to the initiation of islet inflammation (8), but NF-κB and/or inflammasome can also be activated directly by saturated fatty acids or ER stress in cultured islets (9–13). Although IL1B has been detected in β cell lines (12, 13) and in β cells exposed to a diabetic milieu (14), whether it is expressed in β cells before macrophage recruitment is unknown. Furthermore, because of the low abundance (14), whether IL1B is sufficient to trigger islet inflammation remains an unanswered question.
To investigate the molecular events involved in islet inflammation and β cell loss in T2D, we used a zebrafish model of muscle insulin resistance (zMIR) (15), mouse β cell line, mouse islets in vitro and in vivo, and human islet transplants. We identified an essential role for receptor-interacting protein kinase 3 (Ripk3), a central mediator of several inflammatory pathways in other tissues, in overnutrition-induced β cell loss in zMIR fish. We discovered a critical role of ER stress–activated RIPK3 in proinflammatory gene expression in the mouse β cell line. We demonstrated that Ripk3-dependent il1b induction in β cells is sufficient to recruit macrophages to the islet in zMIR. We confirmed that mice on a high-fat diet (HFD) for 1 week had an increased IL1B in β cells and that inhibition of RIPK3 prevented palmitate-induced Il1b induction and β cell dysfunction in primary mouse islets. Furthermore, we showed the association between islet inflammation and β cell–specific activation of ER stress, RIPK3, and NF-κB in human islets transplanted in diabetic mice. These studies revealed a conserved and previously unappreciated β cell response to metabolic stress that leads to islet inflammation. Modulation of RIPK3 may be a mechanism to reduce β cell loss in T2D.
RESULTS
Overnutrition leads to β cell loss and dysfunction in zMIR fish
Using a transgenic zMIR (fig. S1A) (15), we investigated the β cell response to multiple days of overnutrition (Fig. 1A). Fish were challenged with overnutrition starting at 6 days postfertilization (dpf) (16). In response to overnutrition, zMIR and control fish initially had a similar β cell compensatory response from hours 8 to 56 (Fig. 1, B, C, and C′, and fig. S1B) and a similar whole-body free glucose levels (surrogate for blood glucose) at hour 56 (Fig. 1E). Reflecting the challenge of insulin-resistant state, zMIR fish had increased insulin gene transcription at hours 8 and 56 (Fig. 1F and fig. S1C). There were also a greater number of insulin granules per β cell (Fig. 1, G and H). However, after hour 72, β cell number in the zMIR islet markedly declined (Fig. 1, B, D, and D′), compared with hour 56 (Fig. 1C), although the fish were grossly indistinguishable from the controls (fig. S1D). A more detailed time course analysis between hours 56 and 72 indicated that the decline started at hour 64 (Fig. 1I). The loss of β cells in the zMIR larvae led to an increase in whole-body free glucose level (Fig. 1E) and a decrease in insulin mRNA levels (Fig. 1F), indicating that the remaining β cells were dysfunctional and unable to compensate for the increased insulin demand from overnutrition and insulin resistance. In contrast, α cell number was unchanged in response to overnutrition (Fig. 1, J and K). These results indicate that the challenge of insulin resistance and overnutrition leads to specific dysfunction and loss of β cells.
Fig. 1. Overnutrition leads to β cell loss in zMIR fish.

(A) Schematic of multiple sessions of overnutrition in zebrafish. Each session consisted of an 8-hour overnutrition treatment as indicated by the yellow rectangle and a 16-hour treatment in nutrient-free media as indicated by the white rectangle. Time 0 is the beginning of the first overnutrition session. (B) β cell number in fish challenged with multiple sessions of overnutrition. Data represent means ± SEM (n > 20 fish per time point). ***P < 0.001; two-way ANOVA, Tukey’s multiple comparisons test. (C to D′) Images of Tg(ins:H2BmCherry)-labeled β cells in control (C and D) and zMIR larvae (C′ and D′) at hours 56 and 72, respectively. Scale bars, 10 μm. (E) Whole-body free glucose content at hour 72. Data represent means ± SEM (n = 20 fish per group). *P < 0.05; one-way ANOVA, Tukey’s multiple comparisons test. (F) Insulin mRNA expression levels assessed by qRT-PCR in fish at hours 56 and 72. Data represent means ± SEM (n > 4 fish per group). *P < 0.05; one-way ANOVA, Tukey’s multiple comparisons test. ns, not significant. (G) Representative EM images of β cells at hour 64. Scales bar, 1 μm. (H) Quantification of granules per β cell section in islets from control and zMIR fish at hour 64. Data represent means ± SEM, n = 3 independent experiments. ***P < 0.001. (I) β cell number dynamics during the 16 hours of nutrient-free media from hours 56 to 72. Data represent means ± SEM (n > 25 fish per time point). ***P < 0.001; two-way ANOVA, Tukey’s multiple comparisons test. (J) Images of Tg(gcga:EGFP)-labeled α cells in the control and zMIR larvae at hour 72. (K) α cell number at hour 72 (n > 20 fish per group).
β cell loss and dysfunction require Ripk3
To identify the molecular mechanisms underlying the β cell loss in zMIR fish, we screened a number of candidate compounds for their ability to suppress the loss. Of the three cell death inhibitors administered at hours 56 to 72, the RIPK1 inhibitor necrostatin-1 (Nec-1) and the RIPK3 inhibitor GSK’872 both prevented β cell loss (Fig. 2A). In contrast, the pan caspase inhibitor Z-VAD-FMK (zVAD) had little effect (Fig. 2A). These results suggest a role of Ripk1 and Ripk3 in the overnutrition-induced β cell loss.
Fig. 2. β cell loss requires Ripk3.
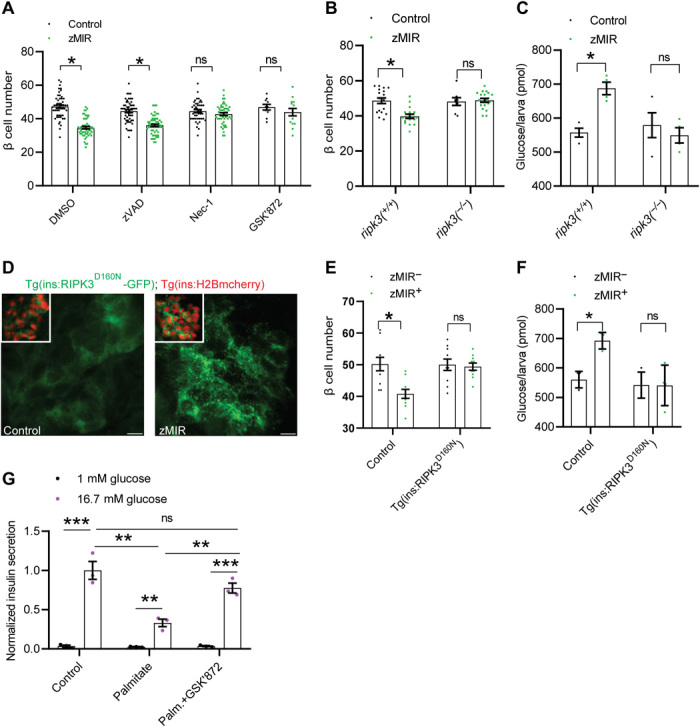
(A) β cell–protective effect of a RIPK1 inhibitor (Nec-1) and a RIPK3 inhibitor (GSK’872). Data represent means ± SEM (n > 15 per group). *P < 0.05; one-way ANOVA, Tukey’s multiple comparisons test. DMSO, dimethyl sulfoxide. (B) Requirement of ripk3 for the β cell loss. Data represent means ± SEM (n > 10 per group). *P < 0.05; one-way ANOVA, Tukey’s multiple comparisons test. (C) Maintenance of whole-body glucose content in ripk3−/−, zMIR larvae after three sessions of overnutrition. Data represent means ± SEM (n = 4 per group). *P < 0.05; one-way ANOVA, Tukey’s multiple comparisons test. (D) Punctate distribution of mutant RIPK3 in zMIR but not in non-zMIR β cells at hour 64. Scale bars, 5 μm. (E) Protective effect of β cell–specific inhibition of RIPK3 on β cells. Data represent means ± SEM (n > 25 per group). *P < 0.05; one-way ANOVA, Tukey’s multiple comparisons test. (F) Effect of β cell–specific inhibition of RIPK3 on whole-body free glucose content. Data represent means ± SEM (n = 4 per group). *P < 0.05; one-way ANOVA, Tukey’s multiple comparisons test. (G) GSK’872 prevents palmitate-impaired insulin secretion. Average insulin secretion per islet at 1 mM and 16.7 mM glucose from mouse islets treated with or without 0.5 mM palmitate in the presence or absence of 3 μM GSK’872. Data represent means ± SEM (n = 3 per group). **P ≤ 0.01 and ***P ≤ 0.001; two-way ANOVA, Tukey’s multiple comparisons test.
RIPK3 and its upstream activator RIPK1 play a critical role in inflammation and necroptosis (17, 18). To validate the pharmacological data genetically, we generated germline loss-of-function mutations of ripk1 and ripk3 using CRISPR-Cas9 (fig. S2, A to C). Loss of either Ripk1 or Ripk3 function did not lead to an overt phenotype, which is similar to the findings in Ripk3−/− mice (19) and inconsistent with the neonatal lethal phenotype of Ripk1−/− mice (20). The lack of developmental defects in ripk1−/− may be due to the maternal supply of ripk1 mRNA or its protein product, or other species differences. Nevertheless, in neither ripk3−/−; zMIR fish nor ripk1−/−; zMIR fish, overnutrition caused β cell loss (Fig. 2B and fig. S2D). Furthermore, ripk3−/−; zMIR fish also maintained normal whole-body free glucose content (Fig. 2C), consistent with normal β cell number and function. The results confirm the pharmacological results and indicate that both ripk1 and ripk3 are required for overnutrition-induced β cell dysfunction and loss in zMIR fish.
To determine whether Ripk1 and Ripk3 act cell autonomously in β cells, we expressed a dominant negative form of RIPK3 or RIPK1 to inhibit the activation of the pathway (21). As expected, the transgenes were expressed specifically in β cells (fig. S2, E and F). The enhanced green fluorescent protein (EGFP) tag allowed visualization of subcellular localization of the mutant RIPK3 or RIPK1. The distribution of mutant RIPK3, but not mutant RIPK1, in zMIR fish at the time of β cell loss was punctate (Fig. 2D and fig. S2G). The punctate distribution is a known feature of activated RIPK3, as it forms amyloid fibers (22–24). This suggests that the endogenous Ripk3 was activated and forms nonfunctional polymers with the mutant RIPK3. The punctate RIPK3 distribution was not observed at earlier stages in the zMIR fish (fig. S2C), suggesting a link between RIPK3 activation and β cell dysfunction and loss. These transgenes prevented β cell dysfunction and loss in zMIR fish, leading to normal whole-body free glucose level (Fig. 2, D to F, and fig. S2, H and I). To determine whether this RIPK3 function is conserved in mammals, we tested the effect of the RIPK3 inhibition with GSK’872 in the mouse primary islets cultured in the presence of palmitate, which impairs the glucose-induced insulin secretion (25). In this model, GSK’872 exerted a significant protection of the β cell function (Fig. 2G). These results support a cell-intrinsic role of Ripk1 and Ripk3 in β cell dysfunction and loss.
ER stress activation of RIPK3 contributes to inflammation in mammalian β cells
Overnutrition is known to cause ER stress and oxidative stress in mammalian β cells (26). To extend the zebrafish findings to mammalian β cells and to test whether these cellular stresses activate RIPK3, we examined mouse insulinoma-derived MIN6 cells (27) cultured in conditions known to cause oxidative stress or ER stress. Conditions that cause ER stress, including treatment with thapsigargin or high glucose plus palmitate (GP), increased phospho-RIPK3 (p-RIPK3) protein levels (Fig. 3A). In contrast, treatment with H2O2, high glucose, or palmitate alone did not increase the p-RIPK3 levels (Fig. 3A). As expected, conditions leading to RIPK3 activation also strongly induced the expression of ER stress markers (fig. S3A). In contrast, conditions that did not activate RIPK3 caused little or no increase in these makers (fig. S3A). Cells treated with thapsigargin or GP had extensive RIPK3 puncta compared with control cells (Fig. 3B), also indicative of RIPK3 activation (22–24). Cells in the non-RIPK3 activation conditions showed negligible RIPK3 aggregation (fig. S3B). Nec-1 and GSK’872, both inhibitors of the Ripk pathway, significantly decreased the levels of p-RIPK3 (Fig. 3C) and RIPK3 puncta (Fig. 3D).
Fig. 3. ER stress activation of RIPK3 contributes to inflammation in mammalian β cells.

(A) Western blot analysis of the p-RIPK3 levels in MIN6 cells treated with different stress conditions. Data represent means ± SEM (n = 3 per group). ***P < 0.001; one-way ANOVA, Tukey’s multiple comparisons test. (B) Immunofluorescence analysis of RIPK3 distribution in MIN6 cells treated with different stress conditions. F-actin stain outlines individual cells. DNA stain labels nuclei. Scale bars, 10 μm. (C) Effects of Nec-1 and GSK’872 on thapsigargin- and GP-induced increase in p-RIPK3. Data represent means ± SEM (n = 3 per group). *P < 0.05, **P < 0.01, and ***P < 0.001; one-way ANOVA, Tukey’s multiple comparisons test. (D) Effects of Nec-1 and GSK’872 on the thapsigargin- and GP-induced increase in RIPK3 polymerization. Scale bars, 10 μm. (E) Effects of 4-PBA and TUDCA on the thapsigargin- and GP-induced increase in p-RIPK3. Data represent means ± SEM (n = 3 per group). **P < 0.01; one-way ANOVA, Tukey’s multiple comparisons test. (F) Effects of 4-PBA and TUDCA on the thapsigargin- and GP-induced increase in RIPK3 polymerization. Scale bars, 10 μm. (G) Effect of RIPK3 inhibition on the expression of proinflammatory genes in MIN6 cells. Bars labeled with a different letter are significantly different (P < 0.05), while those sharing a letter are not significantly different (P > 0.05). (H) Western blot analysis of p-p65 levels in MIN6 cells treated with different stress conditions. Data represent means ± SEM (n = 3 per group). ***P < 0.001; one-way ANOVA, Tukey’s multiple comparisons test. (I) Effects of Nec-1 and GSK’872 on the thapsigargin- and GP-induced increase in p-p65. Data represent *P < 0.05 and means ± SEM (n = 3 per group). **P < 0.01; one-way ANOVA, Tukey’s multiple comparisons test. (J) Effects of 4-PBA and TUDCA on the thapsigargin- and GP-induced increase in p-p65. Data represent means ± SEM (n = 3 per group). *P < 0.05 and **P < 0.01; one-way ANOVA, Tukey’s multiple comparisons test.
To determine whether ER stress is necessary for RIPK3 activation by conditions of metabolic stress, we tested the effect of pharmacological relief of ER stress. Both 4-phenylbutyric acid (4-PBA) and tauroursodeoxycholic acid (TUDCA) decreased the p-RIPK3 levels (Fig. 3E) and reduced the RIPK3 puncta (Fig. 3F). Therefore, ER stress is an upstream and necessary step for metabolic stress–induced RIPK3 activation.
ER stress is known to up-regulate genes linked to oxidative stress and inflammation in β cells (28, 29). To determine whether activated RIPK3 modulates the transcriptional response to ER stress, we examined expression of proinflammatory and prooxidant genes (30). Transcripts of il1b, inhba, cxcl12, cxcl11, and ccl2, all genes encoding proinflammatory products, were significantly increased in cells treated with thapsigargin or GP. RIPK3 inhibitor GSK’872 significantly dampened this up-regulation (Fig. 3G). ER stress conditions also significantly increased the expression of prooxidant genes (steap4, ptgst2, and nos2). However, GSK’872 had no effect (fig. S3C).
NF-κB is the major regulator of proinflammatory genes. We therefore investigated whether RIPK3 activation contributes to NF-κB activation. The canonical pathway of NF-κB activation involves Ser536 phosphorylation and nuclear translocation of p65, leading to enhanced transcriptional activity (31–33). The levels of Ser536-phosphorylated p65 were increased in MIN6 cells cultured in conditions causing ER stress (Fig. 3H). Accordingly, thapsigargin or GP treatment caused nuclear translocation of p65 in both MIN6 cells and rat insulinoma-derived INS-1 832/13 cells (fig. S3, D and E). The increase induced by GP, but not thapsigargin, was blunted by Nec-1 and GSK’872 (Fig. 3I and fig. S3D) as well as by 4-BPA and TUDCA (Fig. 3J and fig. S3D). This may be due to the additional effects induced by thapsigargin that are independent of the RIPK3 pathway. These data indicate that the RIPK3 activity contributes to GP-induced NF-κB activation in β cells. Together, these data indicate that ER stress in β cells activates RIPK3, and this contributes to inflammation.
To determine whether ER stress occurs in the zMIR islet, we compared the expression levels of ER stress markers in isolated islets from zMIR and controls at hour 64. There was a significant increase in dnaj3, atf4, edem, and ddit3 expression in the zMIR islets (fig. S4A). The ER stress in zMIR β cells was further supported by electron microscopy, showing that the ER lumen of the zMIR fish at hour 64 was dilated (fig. S4, B and B′). Consistent with the hypothesis that ER stress acts upstream of RIPK3, we found that induction of ER stress markers still occurred in the islet of Tg(ins:RIPK3D160N-EGFP, LR); zMIR fish (fig. S4, C and D). To determine whether ER stress is necessary for overnutrition-dependent islet inflammation, β cell dysfunction, and death in zMIR, we treated fish with ER stress inhibitor TUDCA during the second and third sessions of overnutrition. The treatment prevented the loss of β cells in zMIR larvae (fig. S4E). These results provide in vivo support for the connection of persistent ER stress to overnutrition-induced β cell loss.
RIPK3-dependent il1b induction is essential for β cell loss in zMIR fish
To determine whether overnutrition also induces the expression of inflammatory cytokines in zMIR islets, we examined the islet expression of known several candidate inflammatory cytokines and chemokines. At hour 64, expression of il8a and il1b was increased in the zMIR islets (Fig. 4A). These changes were islet specific and not found in whole animals, indicating that the islet expression changes were not due to systemic inflammation (fig. S5A). These changes in islet expression were not found at hour 56 (Fig. 4B). To determine which islet cell types were responsible for the increased il8a and il1b expression, we used RNAscope to demonstrate that the il1b expression was increased in β cells of zMIR fish, while the il8a increase occurred in an unidentified nonendocrine cell type at the islet periphery (Fig. 4C). Although il1b mRNA was occasionally detected outside of the islet, it seemed to be secondary to the β cell expression, possibly from dead β cells or responding nonislet cells, as it depended on RIPK3 function in β cells (see below). To determine whether overnutrition also induces the IL1b expression in mammalian β cells, we examined islets from mice that had been on an HFD for 1 week that have normal fasting glycemia (34) and should have no macrophage expansion in the islet (5). We detected an increase in the IL1B in β cells (Fig. 4, D and D′). There was also an increased IL1B in some non–β cells, possibly α cells based on the location. To determine whether the Ripk3 kinase activity in β cells is required for the il1b induction, we examined the il8a and il1b expression levels using RNAscope in Tg(ins:RIPK3D160N-EGFP, LR); zMIR (Fig. 4E) and Tg(ins:RIPK1K45A-EGFP, LR); zMIR fish (fig. S5E). The induction of both il1b and il8a was lost. This was further supported by the quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis (Fig. 4, F and G). To test whether the RIPK3 also functions similarly in the mammalian islet, we used palmitate-treated ER primary islets (35). Palmitate increased the expression of Il1b and markers of ER stress. GSK’872 suppressed the Il1b induction but not the ER stress markers (Fig. 4H). The results indicate that the RIPK3 kinase activity–dependent il1b induction in β cells is a conserved early response to metabolic stress.
Fig. 4. RIPK3-dependent il1b induction is essential for β cell loss in zMIR fish.
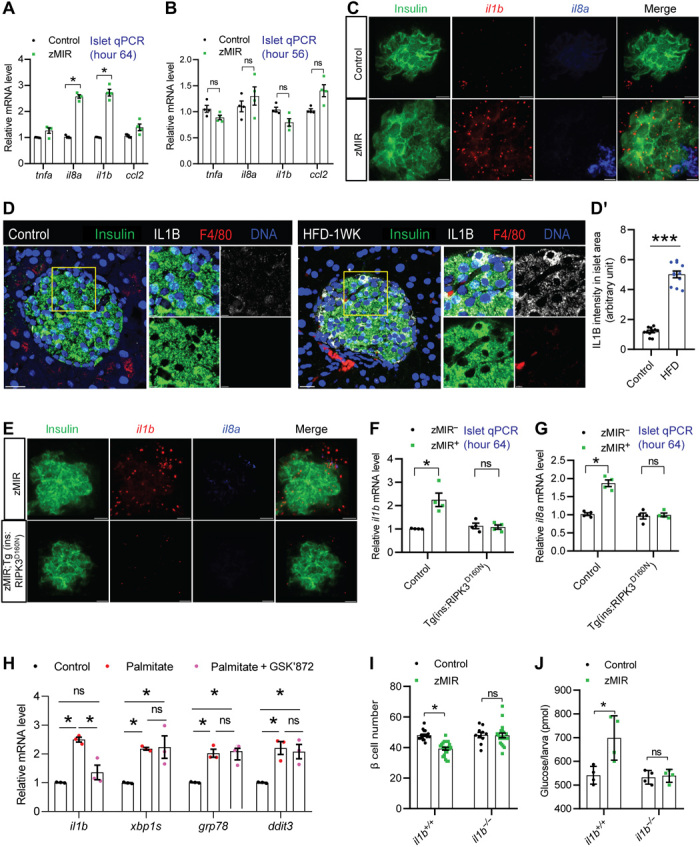
(A) qRT-PCR analysis of candidate macrophage attractants in the islets at hour 64. Data represent means ± SEM (n = 4 per group). *P < 0.05; one-way ANOVA, Tukey’s multiple comparisons test. (B) qRT-PCR analysis of candidate macrophage attractants in the islets at hour 56. Data represent means ± SEM (n = 4 per group). *P < 0.05; one-way ANOVA, Tukey’s multiple comparisons test. (C) RNAscope analysis of insulin, il1b, and il8a at hour 64 in the control and zMIR fish. Scale bars, 10 μm. (D) Representative IL1B and F4/80 immunofluorescence images of control and 1-week HFD mouse pancreas sections (D′) and quantification of IL1B signal intensity in islet area. Scale bars, 20 μm. Inset scale bar, 5 μm. Data represent means ± SEM (n = 3 per group). ***P ≤ 0.001; one-way ANOVA, Tukey’s multiple comparisons test. (E) RNAscope analysis of insulin, il1b, and il8a at hour 64 in the zMIR and zMIR; Tg(ins: RIPK3D160N-GFP) fish. Scale bars, 10 μm. (F and G) Islet il1b expression and il8a by qRT-PCR. Data represent means ± SEM (n = 4 per group). *P < 0.05; one-way ANOVA, Tukey’s multiple comparisons test. (H) qRT-PCR analysis of mouse islets treated with the control medium or medium containing palmitate (0.5 mM) in the presence or absence of 3 μM GSK for 48 hours. *P < 0.05. (I) Requirement of il1b for β cell loss. Data represent means ± SEM (n > 9 per group). *P < 0.05; one-way ANOVA, Tukey’s multiple comparisons test. (J) Preservation of whole-body free glucose content in the il1b−/− zMIR larvae after three sessions of overnutrition. Data represent means ± SEM (n = 4 per group). *P < 0.05; one-way ANOVA, Tukey’s multiple comparisons test.
To determine whether IL1B is required for β cell dysfunction and loss, we inactivated il1b by generating frame-shift mutations using CRIPSR-Cas9 (fig. S5, C and D). Loss of il1b function in zMIR fish prevented β cell loss (Fig. 4I) and normalized the whole-body free glucose content (Fig. 4J). Thus, the β cell–specific induction of il1b as a result of overnutrition and insulin resistance is necessary for β cell dysfunction and loss.
il1b induction in β cells is necessary for recruiting macrophages to the islet
A hallmark of islet inflammation is an increase in innate immune cells. The two predominant innate immune cells in the zebrafish are macrophages that can be identified using Tg(mpeg1:EGFP) (36) and neutrophils that can be identified using Tg(mpo:EGFP) (37). We performed live imaging in intact fish from hours 64 to 70 at 30-min intervals. In control animals, we found that macrophages did not contact the islet (Fig. 5A). In zMIR fish, however, macrophages were found to be in close contact with the islet (Fig. 5B) coincident with il1b induction and β cell loss. The contact is transient, as macrophages usually leave the islet within 1 hour (Fig. 5C). Higher-resolution images confirmed the live-imaging results (Fig. 5, D, E, and E′). In addition, macrophage processes were observed to have surrounded the zMIR β cells (Fig. 5, D, E, and E′). In contrast to the macrophages, the live-imaging analysis indicated that neutrophils did not contact the islet either in the control (fig. S6A) or zMIR animals (fig. S6B).
Fig. 5. il1b induction is necessary for recruiting macrophage to the islet.

(A and B) Representative images from live imaging. Live control (A) or zMIR (B) fish were imaged for macrophages (green) and β cells (red) every 30 min starting at hour 64. (C to E) Representative images from the fixed control larvae (C and C′) and zMIR larvae (D to E′). A single confocal slice (C, D, and E) and the corresponding projection (C′, D′, and E′) of the islet are shown. The inset in (E) shows a macrophage surrounding a β cell. Scale bars, 10 μm. (F and F′) Requirement of Ripk3 for macrophage recruitment as shown by representative images (F) and distance between macrophage and nearest β cell (F′). Scale bars, 10 μm. Data represent means ± SEM (n = 10 per group). *P < 0.05; one-way ANOVA, Tukey’s multiple comparisons test. Mø stands for macrophage. (G and G′) Requirement of il1b for macrophage infiltration as shown by representative images (G) and by macrophage–to–β cell distance (G′) at hour 66. Scale bars, 10 μm. Data represent means ± SEM (n = 10 per group). ***P < 0.001; one-way ANOVA, Tukey’s multiple comparisons test.
To determine whether macrophage recruitment requires ripk1, ripk3, and il1b, we determined the macrophage β cell distance at hour 66. Loss of ripk1, ripk3 (Fig. 5, F and F′), or il1b (Fig. 5, G and G′, and fig. S6C) prevented macrophage recruitment. These results suggest that the Ripk3-dependent Il1b induction in β cells is necessary for recruiting macrophages to the islet.
ER stress–RIPK3 inflammation axis is conserved in human β cells in vivo
To determine whether the ER stress–RIPK3 inflammation axis is present in mammalian β cells in vivo, human islets were transplanted under the kidney capsule of NOD.Cg-PrkdcscidIl2rgtm1Wjl/Sz (NSG); Tg(Ins2-HBEGF)6832)Ugfm/Sz (NSG-DTR) mice, a model that allows prolonged human islet graft survival and also allows rapid and complete ablation of mouse β cells by diphtheria toxin (DT) (Fig. 6A and table S1) (38). After the human islet engraftment, mice were treated with DT and quickly became hyperglycemic, and mouse β cells were markedly depleted (fig. S7, A and B) (38). The human islet grafts were analyzed for the expression of GRP78 (an ER stress marker), RIPK3, phospho-p65, and F4/80 (a murine macrophage marker). In the grafts from hyperglycemic mice, both the GRP78 signal and RIPK3 puncta were markedly increased in the human β cells, indicating ER stress and RIPK3 activation, respectively (Fig. 6, B to C*). RIPK3 activation was not detected in α cells (Fig. 6, B and B′), again indicating a β cell–specific mechanism. An increase in phospho-p65 staining in β cells was also found in the grafts from DT-treated mice (Fig. 6, D′ and D*, arrows), suggesting activation of proinflammatory signaling. Of note, a phospho-p65 signal was also found in non–β cells (Fig. 6D′, arrowheads), possibly macrophages. A basal GRP78 expression, RIPK3 puncta, and phospho-p65 staining were found in some β cells in grafts from phosphate-buffered saline (PBS)–treated mice (Fig. 6, B, C, and E). This may result from the higher basal blood glucose in mice than in humans. When the signals of insulin, GRP78, and phospho-p65 were quantified in individual β cells from DT-treated mice, there was a strong inverse relationship between insulin and either marker, suggesting that ER stress and inflammation are associated with β cell dysfunction (fig. S7C). Although mouse macrophages were found in the grafts regardless of treatment, there was a significant increase in macrophage density in the grafts from the DT-treated animals (Fig. 6E*). In addition, RIPK3 was activated rarely in the macrophages in grafts from the PBS-treated mice, but frequently in those from the DT-treated mice (Fig. 6, E and E*). RIPK3-activated macrophages were often in close vicinity to β cells with RIPK3 activation (Fig. 6E′, arrows), suggesting cross-talk between the two cell types.
Fig. 6. The ER stress–RIPK3 inflammation axis is conserved in human β cells in vivo.

(A) Experimental design. (B to B*) Representative RIPK3 immunofluorescence images of human islet grafts from the PBS-treated (B) and DT-treated mice (B′) and (B*) and quantification of the percentage of β cells with the RIPK3 activation (B*). Data represent means ± SEM (n = 4 per group). ***P < 0.001; one-way ANOVA, Tukey’s multiple comparisons test. Scale bars, 20 μm. Inset scale bar, 5 μm. (C to C*) Representative RIPK3 and GRP78 immunofluorescence images of human islet grafts from the PBS-treated (C) and DT-treated mice (C′) and quantification of the percentage of β cells with the RIPK3 activation (C*). Data represent means ± SEM (n = 4 per group). ***P < 0.001; one-way ANOVA, Tukey’s multiple comparisons test. (D to D*) Representative GRP78 and p-p65 immunofluorescence images of human islet grafts from the PBS-treated (D) and DT-treated mice (D′) and quantification of the percentage of β cells with strong p-p65 signal (D*). Data represent means ± SEM (n = 4 per group). ***P < 0.001; one-way ANOVA, Tukey’s multiple comparisons test. (E to E*) Representative RIPK3 and F4/80 immunofluorescence images of human islet grafts from the PBS-treated (E) and DT-treated mice (E′) and quantification of the total and RIPK3-positive macrophages (E*). Data represent means ± SEM (n = 4 per group). ***P < 0.001; one-way ANOVA, Tukey’s multiple comparisons test. The transplants and human islets were part of a prior report (38), and sections of the graft were analyzed in the current study.
Together, these data demonstrate a conserved β cell response to metabolic stress, where the resultant ER stress activates RIPK3, leading to induction of inflammatory genes and macrophage recruitment and/or activation.
DISCUSSION
In T2D, the initial islet response to insulin resistance involves an increase in β cell number and insulin secretion (compensation) (39) followed by an eventual reduction in β cell number and insulin secretion (decompensation) leading to hyperglycemia (40, 41). Here, we present a model combining skeletal muscle insulin resistance and overnutrition that displays an accelerated progression from β cell compensation to decompensation. Using live imaging and testing candidate compounds, we uncovered a key role for Ripk3-dependent β cell expression of proinflammatory mediators and the consequent macrophage recruitment as critical steps to β cell dysfunction and death. We found that RIPK3 activation requires ER stress and contributes to NF-κB activation and the ensuing induction of proinflammatory genes in cultured mammalian β cells. We also observed similar β cell responses in human islet grafts exposed to a hyperglycemic environment in vivo. Therefore, we have identified Ripk3-dependent β cell expression of proinflammatory mediators as a conserved response to metabolic stress. The results suggest that β cell–derived proinflammatory mediators are sufficient to induce islet inflammation and contribute to T2D pathogenesis.
By screening candidate inhibitors of β cell loss in the zMIR fish, we identified Ripk3 and its upstream kinase Ripk1 and validated their role genetically. RIPK3 is a well-known modulator of innate immunity independent of its function in necroptosis (42–44). However, its immunomodulatory function has mainly been characterized in macrophage and dendritic cells. RIPK3 contains a RIP homotypic interaction motif (RHIM) and is activated by other RHIM-containing proteins, such as RIPK1. Once activated, RIPK3 self-polymerizes via the RHIM (22–24), which phosphorylates and activates its substrate mixed lineage kinase domain-like (MLKL), causing NF-κB activation, membrane rupture, and programmed necrosis (necroptosis) (17, 18). The release of damage-associated molecular patterns (DAMPs) after membrane rupture can trigger inflammation (45).
RIPK3 can also cause inflammation independent of MLKL by activating NF-κB and NLRP3 inflammasome in certain cell types and contexts (46). In the human embryonic kidney 293 and Phoenix-A cells, RIPK3 overexpression can result in NF-κB activation (46). Our study demonstrates a proinflammatory role of RIPK3 in β cells. We found that overnutrition-induced il1b induction in zMIR β cells depends on Ripk3 function. This may be through inflammasomes since RIPK3 can also activate the NLRP3 inflammasome, thereby promoting Il1b maturation. However, the zebrafish has a very different NLR repertoire from mammals and does not have an NLRP3 ortholog (47). Whether and how Ripk3 regulates Il1b maturation in zebrafish is an open question and will be a subject of future research.
We demonstrated an essential role of il1b induction in macrophage infiltration in the zMIR islet. This agrees with a recent study showing macrophage infiltration in zebrafish with transgenic il1b expression in β cells (48). Although IL1b transcripts and protein have been detected in β cells from T2D donors (14, 49) and from T2D animals but not controls (14), whether it is expressed before the development of hyperglycemia is unknown. Our study shows an increased Il1b expression in β cells before macrophage recruitment both in the zebrafish and mice under metabolic stress. il1b expression in β cells is low, as it could not be detected by conventional in situ hybridization in zebrafish islets and required the use of a more sensitive RNAscope. Likewise, recent bulk RNAseq and single-cell sequencing of diabetic islets did not identify an enrichment of proinflammatory genes in β cells of T2D donors (50–52), likely indicating their low abundance. Nevertheless, even low levels of il1b seem to be sufficient for initiating macrophage recruitment in zMIR fish. Although our results support the critical role of Il1b in β cell dysfunction and loss, we cannot conclude whether Il1b from β cells or macrophage is directly responsible for these effects, as il1b was absent in both cell types in the il1b−/− fish. Future studies will use cell type–specific il1b inactivation or rescue to determine the role of macrophage-derived Il1b in β cell dysfunction and loss.
Nutrient overload is known to cause both ER stress and oxidative stress in β cells (26). We discovered that ER stress, but not oxidative stress, activates RIPK3 in cultured mouse β cells. RIPK3 activity also contributes to NF-κB activation and the expression of proinflammatory cytokines and chemokines. We further validated the in vitro findings in in vivo settings in zMIR fish and human islet grafts in diabetic mice. Although the in vivo results from human β cells are correlative, when combined with the zebrafish data, they suggest that inflammation from the ER stress activation of RIPK3 is a conserved β cell response to metabolic stress. It is likely that RIPK3 activation requires prolonged or unresolvable ER stress, since even a single exposure to overnutrition is known to cause mild ER stress in wild-type zebrafish (53), and suppression of ER stress just during the second and third sessions of overnutrition was sufficient to prevent β cell loss. Several other mechanisms have been discovered that mediate metabolic stress–induced β cell expression of proinflammatory mediators. Our study adds another layer to the mechanisms that tie stress to inflammation. For example, saturated fatty acids, which are increased in prediabetes and diabetes, induce β cell expression of proinflammatory mediators by directly activating Toll-like receptor 2 (TLR2) and TLR4 (10). ER stress can induce β cell expression of proinflammatory mediators by thioredoxin interacting protein (TXNIP)–mediated activation of NLRP3 inflammasome (12, 13). Here, we identified an additional mechanism, and future studies will focus on dissecting the pathway from ER stress to activation of RIPK3 as well as the role and mechanisms of macrophages in β cell loss and dysfunction.
While we used three complimentary models and they all support the findings, they may not fully represent the etiology of T2D onset in humans. First, all the zebrafish studies were carried out in 6- to 9-day-old fish, while T2D is usually an adult-onset condition. Nevertheless, pediatric T2D is increasing in prevalence (54), probably due to increasing prevalence of severe obesity (55). Second, β cell loss in the zMIR model is rapid. Although such a model facilitates mechanistic investigations, a similar model has not been reported in mammals. Third, while MIN6 cells were used to identify the role of ER stress in RIPK3 activation, cultured cells may not completely recapitulate β cells in their natural setting. Last, even though we extended our finding to human islets in vivo, these islets were transplanted in immunodeficient mice and may differ from β cells in their natural environment. However, these types of models are necessary to reveal the potential molecular mechanisms in human disease pathology that cannot be longitudinally studied otherwise.
In summary, we identified a conserved β cell inflammatory response to metabolic stress. The study identified a role of ER stress–activated RIPK3 in proinflammatory cytokine expression in β cells. Our study also demonstrated, at least in zebrafish, that the relatively low il1b induction is enough to cause islet inflammation as indicated by macrophage recruitment. Future studies should delineate the mechanistic link between ER stress and RIPK3 activation, determine why only a fraction of β cells are lost, and clarify the role of Il1b and macrophages in β cell dysfunction and loss. Such studies may provide new targets for drug development with the goal of preserving β cell function and preventing β cell loss.
MATERIALS AND METHODS
Animals
Zebrafish
Zebrafish were raised in an aquatic habitat system on a 14/10-hour light/dark cycle. Embryos were raised at 28.5°C in an incubator on a 14/10-hour light/dark cycle. All animals used for imaging and/or counting β cells carry Tg(ins:H2B-mCherry) that marks all β cells with mCherry (16). Other published transgenic and mutant strains used for this study are listed in table S2. Transgenic lines expressing dominant-negative human RIPK1 and human RIPK3 (21) in β cells were generated using the Tol2 System (56). Mutations in il1b, ripk3,and ripk1 were generated using CRISPR-Cas9 as described previously (57) using recombinant Cas9 (PNA Bio). Except for ripk1, two knockout lines in each gene were characterized for an initial cross-validation before selecting one line for all the experiments (figs. S3, A to C, and S5, C to D).
Isolation and culture of mouse pancreatic islets
The animals were handled in compliance with guidelines approved by the Vanderbilt University Institutional Animal Care and Use Committee protocols (protocol no. M1600063-01). All of the mice used in these studies were 18-week-old age-matched C57BL/6J males. Mouse pancreata were digested with collagenase P (Roche, Basel, Switzerland), and islets were isolated via density gradient centrifugation as previously described (58, 59). The islets were allowed to recover for 24 hours after isolation in RPMI 1640 supplemented with 15% fetal bovine serum, glucose (11 mM), and bovine serum albumin (0.5 mg/ml) at 37°C in 5% CO2. Islets were then treated with control medium or medium containing palmitate (0.5 mM) in the presence or absence of glycogen synthase kinase (GSK) (3 μM) for 48 hours. Islets were used either for qPCR or for insulin secretion measurements. The insulin secretion measurements from static incubations were conducted as previously described (60). Insulin concentrations were determined using Insulin Rodent Chemiluminescence enzyme-linked immunosorbent assay (ALPCO, Salem, NH, USA) under the conditions indicated in the figure legends.
Rodent β cell culture
Mouse MIN6 cells and rat INS-1 832/13 cells were cultured in standard conditions (27, 61). Cells were seeded either in a 6- or 12-well plate. After 24 hours, the cells were cultured with metabolic stress and control media for 24 hours and harvested for RNA extraction, immunostaining, or Western blot.
Human islets
Functionally validated human islets [2000 islet equivalents (IEQ)] from Integrated Islet Distribution Program (http://iidp.coh.org/) were transplanted into the kidney capsule of 18-week-old male NSG-DTR mice as described (38, 62). Characteristics of the islets are summarized in table S1. After 2 weeks of engraftment, the mice were injected with 5 ng of DT or PBS and monitored for 4 weeks. The human islet graft and pancreas from the recipient mouse were collected as previously reported (38).
Overnutrition and compound treatment
Overnutrition is achieved by culturing larvae in 5% chicken egg yolk solution for 8 hours as described (16, 63). For multiple days of overnutrition, larvae were rinsed and kept in nutrient-free 0.3× Danieau buffer for 16 hours before the next session of overnutrition treatment. The following compounds were used with their concentration in parenthesis: Z-VAD-FMK (200 μM), TUDCA (100 μM), Nec-1, (20 μM), and GSK’872 (5 μM).
In situ hybridization
Zebrafish larvae were fixed at hour 64 and embedded into O.C.T. Islet-containing frozen sections (14 μm) were selected for in situ hybridization using the RNAscope 2.0 High-Definition kit [Advanced Cell Diagnostics (ACD), Hayward, California, USA] according to the manufacturer’s instructions. Briefly, slides were thawed at room temperature for 10 min before baking at 60°C for 45 min. The sections were then postfixed in prechilled 4% paraformaldehyde (PFA) for 15 min at 4°C, washed in three changes of PBS for 5 min each before dehydration through 50, 70, 100%, and 100% ethanol for 5 min each. After pretreatment, slides were performed with target retrieval reagents in the steamer at 95°C for 5 min. Probes hybridization and signal amplification were performed according to the manufacturer’s instructions. The signal for each probe was revealed by Opal 520, Opal 570, and Opal 690 (Akoya Biosciences) according to the ACD manual. After the final wash with wash buffer reagents from ACD, slides were mounted with ProLong Diamond (Thermo Fisher Scientific), and images were acquired on a Zeiss LSM780. Images were analyzed by Imaris (Oxford Instruments).
Immunofluorescence
Cultured rodent β cells on coverslips were treated with experimental conditions for 24 hours. The cells were rinsed with ice-cold PBS and fixed with 4% PFA for 10 min at room temperature followed by permeabilization in 0.1% sodium citrate plus 0.1% Triton X-100. The cells were then incubated with RIPK3 antibody (1:100) for 2 hours at room temperature. The cells were then washed with cold PBS three times for 3 min each and incubated with Alexa Fluor 488–labeled anti-rabbit immunoglobulin G secondary antibody (1:1000) (Invitrogen) at room temperature for 1 hour. After three washes with cold PBS, cells were stained with Hoechst 33342 (0.5 μg/ml) and 1.65 μM phalloidin. The coverslips were mounted on slides with ProLong Diamond mounting medium (Thermo Fisher Scientific). Images were obtained from a Zeiss LSM880 using 63× oil lens.
Immunofluorescence of mouse pancreas and human islet graft sections was performed as described (38). Briefly, recovered human islet grafts and mouse pancreas were fixed in 4% PFA in PBS and cryopreserved. Five-micrometer frozen sections were cut and stained. Primary antibodies used were guinea pig anti-human insulin (1:500) (DAKO, A0564), mouse anti-glucagon (1:500) (Abcam, ab10988), rabbit anti-human RIPK3 (1:200) (Aviva Systems Biology), mouse anti-human Grp78 (1:100) (Invitrogen), rat anti-mouse F4/80 [CI:A3-1] (1:100) (Abcam, ab6640), and rabbit anti-IL1B (1:100; Novus, NB600-633). Images were obtained using a Zeiss LSM880.
Statistics
For multiple group comparisons, a one-way analysis of variance (ANOVA) test was performed, and a post hoc Tukey’s multiple comparisons test was used. A P value less than 0.05 was considered statistically significant. Values represent means ± SEM. Analyses were done using GraphPad Prism.
Study approval
All human islet studies were approved by the Vanderbilt Institutional Review Board. All animal studies were approved by the Vanderbilt Institutional Animal Care and Use Committee.
Acknowledgments
We thank members of the groups of W.C., A.C.P., and D.A.J. and the Vanderbilt β cell community for the helpful discussions. Tg(mpeg1:EGFP) fish were a gift from S. Leach and M. Levesque, irf8 mutants were provided by W. Talbot, and Tg(mpeg1:NTR-mcherry) fish were from S. Wu. Human islets were from IIDP. MIN-6 cells were from R. Stein. INS-1 832/13 cells were from C. Newgard. Some mouse pancreatic sections were provided by M. Gannon. We thank J. Kang for the support in the bioinformatics analysis. Funding: This study is supported by DK117147 (to W.C. and A.C.P.), DK109407 (to W.C.), DK-097392 (to D.A.J.), and DK-115620 (to D.A.J.). Confocal imaging and image analysis were performed in part through the use of the Vanderbilt Cell Imaging Shared Resource. The human islet transplantation research was performed using resources and/or funding provided by the National Institute of Diabetes and Digestive and Kidney Diseases–supported HIRN (RRID:SCR_014393; DK104211, DK108120, and DK112232), DK106755, DK72473, DK89572, and DK20593, grants from the JDRF, and the Department of Veterans Affairs (BX000666). Human pancreatic islets were provided by the NIDDK-funded Integrated Islet Distribution Program at the City of Hope (NIH grant no. 2UC4 DK098085). Author contributions: Conceptualization: W.C., L.A.M., and B.Y. Funding acquisition: W.C., A.C.P., and D.A.J. Investigation: B.Y., L.A.M., K.E.Z., L.Y., Z.T., and L.Z. Methodology: L.A.M., L.Y., and K.E.Z. Resources: D.A.J., C.D., and A.C.P. Supervision: W.C. Validation: B.Y. Visualization: B.Y. and L.A.M. Writing, original draft: B.Y., L.A.M., A.C.P., and W.C. Writing, review and editing: C.D. and D.A.J. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eabd7272/DC1
REFERENCES AND NOTES
- 1.Cho N. H., Shaw J. E., Karuranga S., Huang Y., da Rocha Fernandes J. D., Ohlrogge A. W., Malanda B., IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 138, 271–281 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Halban P. A., Polonsky K. S., Bowden D. W., Hawkins M. A., Ling C., Mather K. J., Powers A. C., Rhodes C. J., Sussel L., Weir G. C., β-cell failure in type 2 diabetes: Postulated mechanisms and prospects for prevention and treatment. J. Clin. Endocrinol. Metab. 99, 1983–1992 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petersen M. C., Shulman G. I., Mechanisms of insulin action and insulin resistance. Physiol. Rev. 98, 2133–2223 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lytrivi M., Igoillo-Esteve M., Cnop M., Inflammatory stress in islet β-cells: Therapeutic implications for type 2 diabetes? Curr. Opin. Pharmacol. 43, 40–45 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Ying W., Lee Y. S., Dong Y., Seidman J. S., Yang M., Isaac R., Seo J. B., Yang B.-H., Wollam J., Riopel M., McNelis J., Glass C. K., Olefsky J. M., Fu W., Expansion of Islet-resident macrophages leads to inflammation affecting β cell proliferation and function in obesity. Cell Metab. 29, 457–474.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eguchi K., Nagai R., Islet inflammation in type 2 diabetes and physiology. J. Clin. Invest. 127, 14–23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Böni-Schnetzler M., Donath M. Y., Increased IL-1β activation, the culprit not only for defective insulin secretion but also for insulin resistance? Cell Res. 21, 995–997 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Imai Y., Dobrian A. D., Morris M. A., Nadler J. L., Islet inflammation: A unifying target for diabetes treatment? Trends Endocrinol. Metab. 24, 351–360 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Böni-Schnetzler M., Boller S., Debray S., Bouzakri K., Meier D. T., Prazak R., Kerr-Conte J., Pattou F., Ehses J. A., Schuit F. C., Donath M. Y., Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin-1 receptor I. Endocrinology 150, 5218–5229 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Eguchi K., Manabe I., Oishi-Tanaka Y., Ohsugi M., Kono N., Ogata F., Yagi N., Ohto U., Kimoto M., Miyake K., Tobe K., Arai H., Kadowaki T., Nagai R., Saturated fatty acid and TLR signaling link β cell dysfunction and islet inflammation. Cell Metab. 15, 518–533 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Hotamisligil G. S., Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lerner A. G., Upton J.-P., Praveen P. V. K., Ghosh R., Nakagawa Y., Igbaria A., Shen S., Nguyen V., Backes B. J., Heiman M., Heintz N., Greengard P., Hui S., Tang Q., Trusina A., Oakes S. A., Papa F. R., IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 16, 250–264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oslowski C. M., Hara T., O’Sullivan-Murphy B., Kanekura K., Lu S., Hara M., Ishigaki S., Zhu L. J., Hayashi E., Hui S. T., Greiner D., Kaufman R. J., Bortell R., Urano F., Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab. 16, 265–273 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maedler K., Sergeev P., Ris F., Oberholzer J., Joller-Jemelka H. I., Spinas G. A., Kaiser N., Halban P. A., Donath M. Y., Glucose-induced β cell production of IL-1β contributes to glucotoxicity in human pancreatic islets. J. Clin. Invest. 110, 851–860 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maddison L. A., Joest K. E., Kammeyer R. M., Chen W., Skeletal muscle insulin resistance in zebrafish induces alterations in β-cell number and glucose tolerance in an age and diet dependent manner. Am. J. Physiol. Endocrinol. Metab. 308, E662–E669 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maddison L. A., Chen W., Nutrient excess stimulates β-cell neogenesis in zebrafish. Diabetes 61, 2517–2524 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pasparakis M., Vandenabeele P., Necroptosis and its role in inflammation. Nature 517, 311–320 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Shan B., Pan H., Najafov A., Yuan J., Necroptosis in development and diseases. Genes Dev. 32, 327–340 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moriwaki K., Chan F. K.-M., RIP3: A molecular switch for necrosis and inflammation. Genes Dev. 27, 1640–1649 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelliher M. A., Grimm S., Ishida Y., Kuo F., Stanger B. Z., Leder P., The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity 8, 297–303 (1998). [DOI] [PubMed] [Google Scholar]
- 21.Cho Y. S., Challa S., Moquin D., Genga R., Ray T. D., Guildford M., Chan F. K.-M., Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun L., Wang H., Wang Z., He S., Chen S., Liao D., Wang L., Yan J., Liu W., Lei X., Wang X., Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Mompeán M., Li W., Li J., Laage S., Siemer A. B., Bozkurt G., Wu H., McDermott A. E., The structure of the necrosome RIPK1-RIPK3 core, a human hetero-amyloid signaling complex. Cell 173, 1244–1253.e10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li J., Quade T. M., Siemer A. B., Napetschnig J., Moriwaki K., Hsiao Y.-S., Damko E., Moquin D., Walz T., Dermott A. M., Chan F. K.-M., Wu H., The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150, 339–350 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joseph J. W., Koshkin V., Zhang C.-Y., Wang J., Lowell B. B., Chan C. B., Wheeler M. B., Uncoupling protein 2 knockout mice have enhanced insulin secretory capacity after a high-fat diet. Diabetes 51, 3211–3219 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Qiu H., Schlegel V., Impact of nutrient overload on metabolic homeostasis. Nutr. Rev. 76, 693–707 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Miyazaki J.-I., Araki K., Yamato E., Ikegami H., Asano T., Shibasaki Y., Oka Y., Yamamura K.-I., Establishment of a pancreatic β cell line that retains glucose-inducible insulin secretion: Special reference to expression of glucose transporter isoforms. Endocrinology 127, 126–132 (1990). [DOI] [PubMed] [Google Scholar]
- 28.Chaudhari N., Talwar P., Parimisetty A., Lefebvre d’Hellencourt C., Ravanan P., A molecular web: Endoplasmic reticulum stress, inflammation, and oxidative stress. Front. Cell. Neurosci. 8, 213 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Juliana C. A., Yang J., Cannon C. E., Good A. L., Haemmerle M. W., Stoffers D. A., A PDX1-ATF transcriptional complex governs β cell survival during stress. Mol. Metab. 17, 39–48 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Good A. L., Cannon C. E., Haemmerle M. W., Yang J., Stanescu D. E., Doliba N. M., Birnbaum M. J., Stoffers D. A., JUND regulates pancreatic β cell survival during metabolic stress. Mol. Metab. 25, 95–106 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen L.-F., Williams S. A., Mu Y., Nakano H., Duerr J. M., Buckbinder L., Greene W. C., NF-κB RelA phosphorylation regulates rela acetylation. Mol. Cell. Biol. 25, 7966–7975 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buss H., Dörrie A., Schmitz M. L., Hoffmann E., Resch K., Kracht M., Constitutive and interleukin-1-inducible phosphorylation of p65 NF-κB at serine 536 is mediated by multiple protein kinases including IκB kinase (IKK)-α, IKKβ, IKKε, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J. Biol. Chem. 279, 55633–55643 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Ghosh S., May M. J., Kopp E. B., NF-κB and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 16, 225–260 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Mosser R. E., Maulis M. F., Moullé V. S., Dunn J. C., Carboneau B. A., Arasi K., Pappan K., Poitout V., Gannon M., High-fat diet-induced β-cell proliferation occurs prior to insulin resistance in C57Bl/6J male mice. Am. J. Physiol. Endocrinol. Metab. 308, E573–E582 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Igoillo-Esteve M., Marselli L., Cunha D. A., Ladrière L., Ortis F., Grieco F. A., Dotta F., Weir G. C., Marchetti P., Eizirik D. L., Cnop M., Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia 53, 1395–1405 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Ellett F., Pase L., Hayman J. W., Andrianopoulos A., Lieschke G. J., mpeg1 promoter transgenes direct macrophage-lineage expression in zebrafish. Blood 117, e49–e56 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Renshaw S. A., Loynes C. A., Trushell D. M. I., Stone E., Ingham P. W., Whyte M. K. B., A transgenic zebrafish model of neutrophilic inflammation. Blood 108, 3976–3978 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Dai C. H., Kayton N. S., Shostak A., Poffenberger G., Cyphert H. A., Aramandla R., Thompson C., Papagiannis I. G., Emfinger C., Shiota M., Stafford J. M., Greiner D. L., Herrera P. L., Shultz L. D., Stein R., Powers A. C., Stress-impaired transcription factor expression and insulin secretion in transplanted human islets. J. Clin. Investig. 126, 1857–1870 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bergman M., Pathophysiology of prediabetes and treatment implications for the prevention of type 2 diabetes mellitus. Endocrine 43, 504–513 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Hanley S. C., Austin E., Assouline-Thomas B., Kapeluto J., Blaichman J., Moosavi M., Petropavlovskaia M., Rosenberg L., β-cell mass dynamics and islet cell plasticity in human type 2 diabetes. Endocrinology 151, 1462–1472 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Rahier J., Guiot Y., Goebbels R. M., Sempoux C., Henquin J. C., Pancreatic β-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab. 10, 32–42 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Peltzer N., Walczak H., Cell death and inflammation – A vital but dangerous liaison. Trends Immunol. 40, 387–402 (2019). [DOI] [PubMed] [Google Scholar]
- 43.He S., Wang X., RIP kinases as modulators of inflammation and immunity. Nat. Immunol. 19, 912–922 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Shlomovitz I., Zargrian S., Gerlic M., Mechanisms of RIPK3-induced inflammation. Immunol. Cell Biol. 95, 166–172 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Kaczmarek A., Vandenabeele P., Krysko D. V., Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity 38, 209–223 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Moriwaki K., Chan F. K.-M., Necroptosis-independent signaling by the RIP kinases in inflammation. Cell. Mol. Life Sci. 73, 2325–2334 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meunier E., Broz P., Evolutionary convergence and divergence in NLR function and structure. Trends Immunol. 38, 744–757 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Delgadillo-Silva L. F., Tsakmaki A., Akhtar N., Franklin Z. J., Konantz J., Bewick G. A., Ninov N., Modelling pancreatic β-cell inflammation in zebrafish identifies the natural product wedelolactone for human islet protection. Dis. Model. Mech. 12, dmm036004 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Böni-Schnetzler M., Thorne J., Parnaud G., Marselli L., Ehses J. A., Kerr-Conte J., Pattou F., Halban P. A., Weir G. C., Donath M. Y., Increased interleukin (IL)-1β messenger ribonucleic acid expression in β-cells of individuals with type 2 diabetes and regulation of IL-1β in human islets by glucose and autostimulation. J. Clin. Endocrinol. Metab. 93, 4065–4074 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Segerstolpe Å., Palasantza A., Eliasson P., Andersson E.-M., Andréasson A.-C., Sun X., Picelli S., Sabirsh A., Clausen M., Bjursell M. K., Smith D. M., Kasper M., Ämmälä C., Sandberg R., Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab. 24, 593–607 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xin Y., Kim J., Okamoto H., Ni M., Wei Y., Adler C., Murphy A. J., Yancopoulos G. D., Lin C., Gromada J., RNA sequencing of single human islet cells reveals type 2 diabetes genes. Cell Metab. 24, 608–615 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Dorrell C., Schug J., Lin C. F., Canaday P. S., Fox A. J., Smirnova O., Bonnah R., Streeter P. R., Stoeckert C. J. Jr., Kaestner K. H., Grompe M., Transcriptomes of the major human pancreatic cell types. Diabetologia 54, 2832–2844 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li M., Page-McCaw P., Chen W., FGF1 mediates overnutrition-induced compensatory β-cell differentiation. Diabetes 65, 96–109 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mayer-Davis E. J., Lawrence J. M., Dabelea D., Divers J., Isom S., Dolan L., Imperatore G., Linder B., Marcovina S., Pettitt D. J., Pihoker C., Saydah S., Wagenknecht L.; SEARCH for Diabetes in Youth Study , Incidence trends of type 1 and type 2 diabetes among youths, 2002-2012. N. Engl. J. Med. 376, 1419–1429 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Skinner A. C., Skelton J. A., Prevalence and trends in obesity and severe obesity among children in the United States, 1999-2012. JAMA Pediatr. 168, 561–566 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Kawakami K., Takeda H., Kawakami N., Kobayashi M., Matsuda N., Mishina M., A transposon-mediated gene trap approach identifies developmentally regulated genes in zebrafish. Dev. Cell 7, 133–144 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Yin L., Maddison L. A., Li M., Kara N., LaFave M. C., Varshney G. K., Burgess S. M., Patton J. G., Chen W., Multiplex conditional mutagenesis using transgenic expression of Cas9 and sgRNAs. Genetics 200, 431–441 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dickerson M. T., Bogart A. M., Altman M. K., Milian S. C., Jordan K. L., Dadi P. K., Jacobson D. A., Cytokine-mediated changes in K+ channel activity promotes an adaptive Ca2+ response that sustains β-cell insulin secretion during inflammation. Sci. Rep. 8, 1158 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vierra N. C., Dadi P. K., Jeong I., Dickerson M., Powell D. R., Jacobson D. A., Type 2 diabetes-associated K+ channel TALK-1 modulates β-cell electrical excitability, second-phase insulin secretion, and glucose homeostasis. Diabetes 64, 3818–3828 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dadi P. K., Luo B., Vierra N. C., Jacobson D. A., TASK-1 potassium channels limit pancreatic α-cell calcium influx and glucagon secretion. Mol. Endocrinol. 29, 777–787 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hohmeier H. E., Mulder H., Chen G., Henkel-Rieger R., Prentki M., Newgard C. B., Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 49, 424–430 (2000). [DOI] [PubMed] [Google Scholar]
- 62.Shultz L. D., Lyons B. L., Burzenski L. M., Gott B., Chen X., Chaleff S., Kotb M., Gillies S. D., King M., Mangada J., Greiner D. L., Handgretinger R., Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2Rγnull mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 174, 6477–6489 (2005). [DOI] [PubMed] [Google Scholar]
- 63.Li M., Maddison L. A., Page-McCaw P., Wenbiao C., Overnutrition induces β-cell differentiation through prolonged activation of β-cells in zebrafish larvae. Am. J. Physiol. Endocrinol. Metab. 306, E799–E807 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zecchin E., Filippi A., Biemar F., Tiso N., Pauls S., Ellertsdottir E., Gnügge L., Bortolussi M., Driever W., Argenton F., Distinct delta and jagged genes control sequential segregation of pancreatic cell types from precursor pools in zebrafish. Dev. Biol. 301, 192–204 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eabd7272/DC1