

Abstract
As long-read sequencing technologies are becoming increasingly popular, several methods have been developed for the discovery and analysis of structural variants (SVs) from long reads. Long reads enable detection of SVs that could not be previously detected from short-read sequencing, but computational methods must adapt to unique challenges and opportunities presented by long-read sequencing. Here, we summarize over 50 long-read based methods for SV detection, genotyping and visualization, and discuss how new telomere-to-telomere genome assemblies and pangenome efforts can improve the accuracy and drive the development of SV callers in the future.
Background
Structural variants (SVs) are generally defined as large genomic alterations longer than 50bp1, 2,3, 4,5. They are prevalent in the human genome together with single-nucleotide variants (SNVs) and small insertions or deletions (indels) as a result of important biological processes such as DNA repair and replication, meiotic recombination, and retrotransposition6. Some SVs result in the removal or addition of genetic material from the genome, whereas other SVs simply cause rearrangements of the genome. Figure 1 shows several common SV types: deletions (DEL), novel insertions (INS), inter- and intra-chromosomal translocations (TLC), inversions (INV), tandem repeats (TR), and duplications (DUP) with two subtypes (tandem duplications (TAN), and inter- or intra-chromosomal interspersed duplications (INT)), as well as more complex loci that can now be found using long reads, such as nested inversion and tandem repeat expansions. Unlike SNVs and indels, SVs are considered to be the largest source of genomic variation in terms of the number of base pairs altered2, 7–9. Thus, they can have a pronounced effect on phenotypic diversity. For instance, a 900kbp inversion in 17q21.31 MAPT locus is found in European-Mediterranean populations with 20% allele frequency but is largely absent in other populations10. This locus is associated with neurological disease and is subject to positive selection. Additionally, many SVs play critical roles in conferring susceptibility to inherited diseases and cancer11–13.
Figure 1.
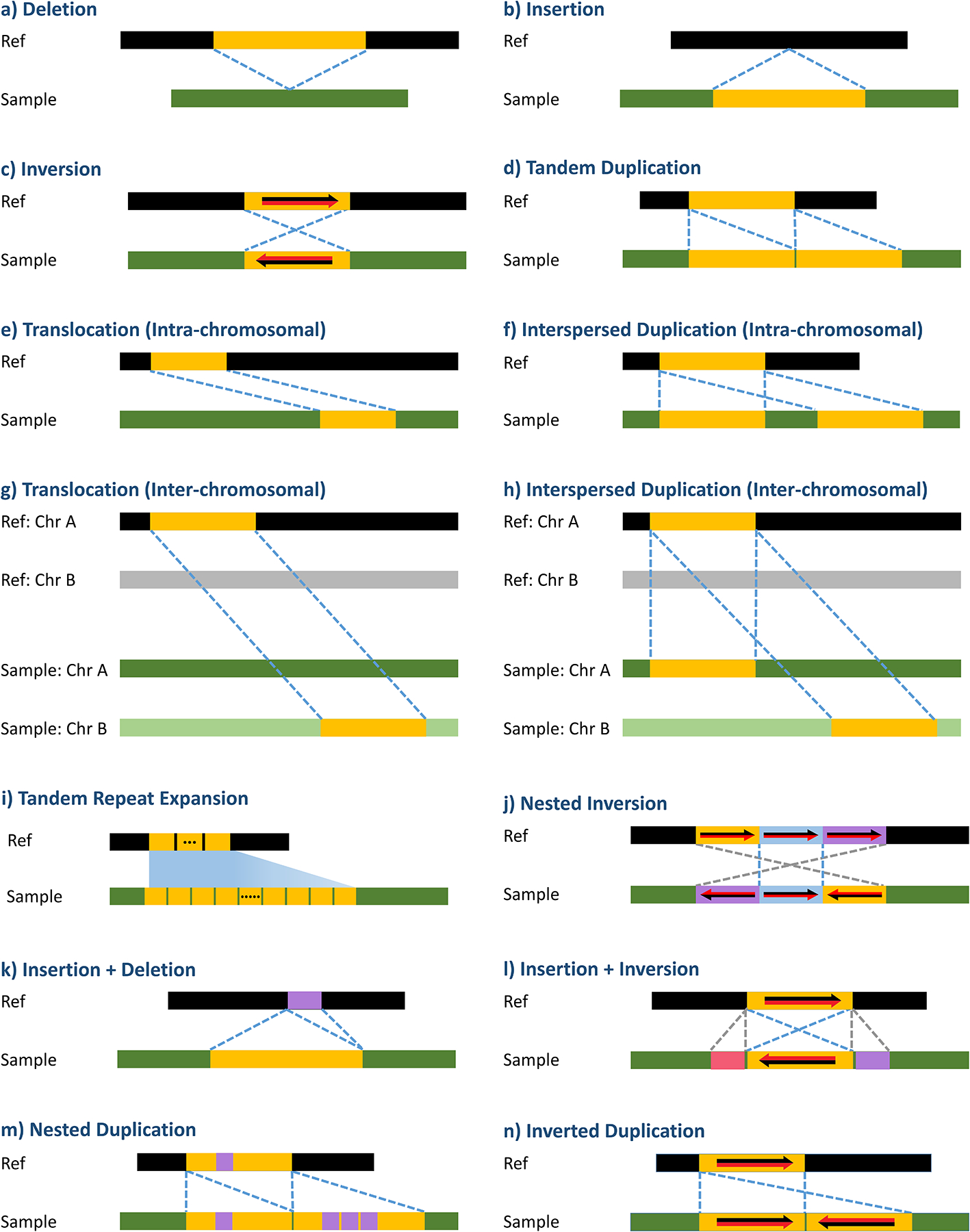
An illustration of different types of SVs. “Ref” refers to reference genome, and “Sample” refers to a query genome.
The substantial contribution of SVs to human diseases has stimulated the development of various wet-lab and computational techniques for their detection. Before next-generation sequencing (NGS) became widely available, two main approaches for SV detection existed: array-based methods14 such as oligonucleotide arrays and SNP arrays for genome wide scanning; and locus-specific assays such as quantitative polymerase chain reaction (qPCR)14, multiplex ligation-dependent probe amplification (MLPA) and NanoString15 for targeted regions. However, both methods have significant limitations. Array-based methods have limited resolution and cannot accurately detect structural variants that do not change DNA dosage, while most multiplex locus-specific assays have limited scalability and cannot be used for whole-genome study. Over the past 10 years, the deployment of high-throughput short-read sequencing—particularly paired-end sequencing—led to a surge in the development of computational methods for SV identification at a genomic scale3–5, 16–18.
Several studies have analyzed the strengths and weaknesses of these computational methods. Kosugi et al.4 evaluated 66 short-read SV callers and found most algorithms performed well only for specific SV types and size ranges. Similarly, Cameron et al.3 evaluated 10 short-read SV callers and found that assembly-based SV callers and SV callers using multiple sources of evidence (such as read depth, paired-end, and split reads) tended to perform better. They also noted the need for development of specialized algorithms for SV calling from low complexity regions with simple and tandem repeats. In particular, short-read based methods struggle with detecting longer SVs, complex SVs (such as chained fusion19 or chromothripsis20 or kilobase-scale repeat arrays21), SVs occurring in repetitive regions22, or segmental duplications, which are hotspots of chromosomal rearrangements23.
Long-read sequencing technologies, such as Oxford Nanopore Technologies (ONT)24 and Pacific Biosciences (PB or PacBio)25, provide new possibilities to tackle several challenges that cannot be resolved with short-read sequencing alone. Termed method of the year 2022 by Nature Methods26, long-read sequencing produces reads that are tens of kbp long, compared to the reads produced in short-read sequencing, which are typically 150–250bp long. Besides ONT and PacBio, several short-read sequencing systems have been adapted to produce synthetic long-reads, such as Illumina’s “complete long-read technology” (previously known as Infinity)26, st LFR27, and TELL-seq28. Long reads can span entire SVs in many cases and achieve better mappability in repetitive genomic regions. They enable the determination of long-range haplotypes, as well as the identification of small indels and SVs in complex genomic regions29, variants in coding regions for genes with many pseudogenes, and phasing of distant alleles. Finally, they allow us to distinguish highly homologous regions30.
Recent whole-genome long-read sequencing studies31–34 have made it clear that most SVs are missed by short-read sequencing or clinical microarrays but are detectable using long-read sequencing. A study published in February 2021 demonstrates that long-read and strand-specific sequencing techniques identified 107,590 SVs, and 68% are not discovered by short-read sequencing35. Thus, long-read sequencing has been successfully used in population-wide studies to improve characterization of SVs detected by short-read sequencing2. More importantly, the identification of pathogenic SVs and tandem repeat expansions, which cannot be sequenced via short-read sequencing, has proven essential in facilitating disease diagnoses and the evaluation of potential treatments30. For example, targeted long-read sequencing for patient diagnosis by Merker et al.36 were able to identify a 2184bp de novo deletion in the PRKARIA gene associated with Carney complex, a rare genetic disorder.
However, compared to short-read sequencing, the different error profiles of long-read sequencing present a different set of challenges, despite the improved sensitivity in finding SVs missed by short reads. PacBio suffers from high rate of random false insertions37 which can be partially addressed by circular consensus sequencing (CCS) to generate high-fidelity (HiFi) reads38, while Nanopore sequencing suffers from both random and systematic indel errors39, 40 which can make read alignment and SV detection more difficult (although different strategies, such as linear consensus41 or UMIs42, can be used to reduce errors). Furthermore, recent improvements of basecalling accuracy by the R10 flowcells, with two pinch points and longer barrel, allow the breakpoints to be accurately estimated, especially when the reads are assembled. Thus, considering the differences in read length, sequence type (paired-end versus single-end) and error profiles, short-read based computational methods cannot be directly used on long reads, and dozens of novel computational methods have been developed specifically for long-read sequencing to identify SVs over the past few years43–45. Nevertheless, there is a lack of a comprehensive summary of long-read based SV callers, as existing review studies have missed many recent methods developed over the last few years4, 5, 46, 47. To further improve long-read based SV analysis and detection, it is critical to comprehensively summarize how the state-of-the-art methods detect SVs and to discuss potential limitations or areas of improvements in existing methods. In addition, the recent release of a complete human genome, T2T-CHM13, will have important implications for the development of future SV callers48. In this review, we survey over 50 long-read based methods for SV/repeat discovery, genotyping, visualization and benchmarking, and their application in cancer and population-scale SV calling. We broadly classify SV callers into generalized SV detection methods (including assembly and alignment-based ones) and specialized SV detection methods. Later, we discuss the contribution of long-read sequencing towards the development of gap-less reference genomes and pan-genomes, and how these innovations will shape the future of SV calling.
Generalized SV Detection
Generalized SV detection methods from long-read datasets can call several common SV types in an entire genome. This contrasts with specialized SV detection methods, which target a specific SV type or a specific genomic region. Computational methods for generalized SV detection usually have two components: 1) alignment against a reference genome and 2) generation of consensus calls from individual reads. Based on how these two components are implemented, there are two types of generalized SV detection methods: alignment of 1) raw reads and 2) assembled reads. Alignment of a query sequence, whether a long-read or a contig, can contain concrete evidence for an SV, typically referred to as an “SV signature”. Examples of such SV signatures are gaps within alignments, clipped bases, and split alignments. Alignment-based SV detection methods directly use alignment information and extract SV signatures from each long-read and then combine evidence across overlapping reads to inform a consensus SV call. Assembly-based methods, on the other hand, generate a consensus sequence or contig from long reads using de novo or reference-guided assembly, and extract SV signatures from the alignment of a contig against a reference genome. The general framework of SV detection using long-read sequencing is illustrated in Figure 2 and a short summary of each method is described in Table 1.
Figure 2.
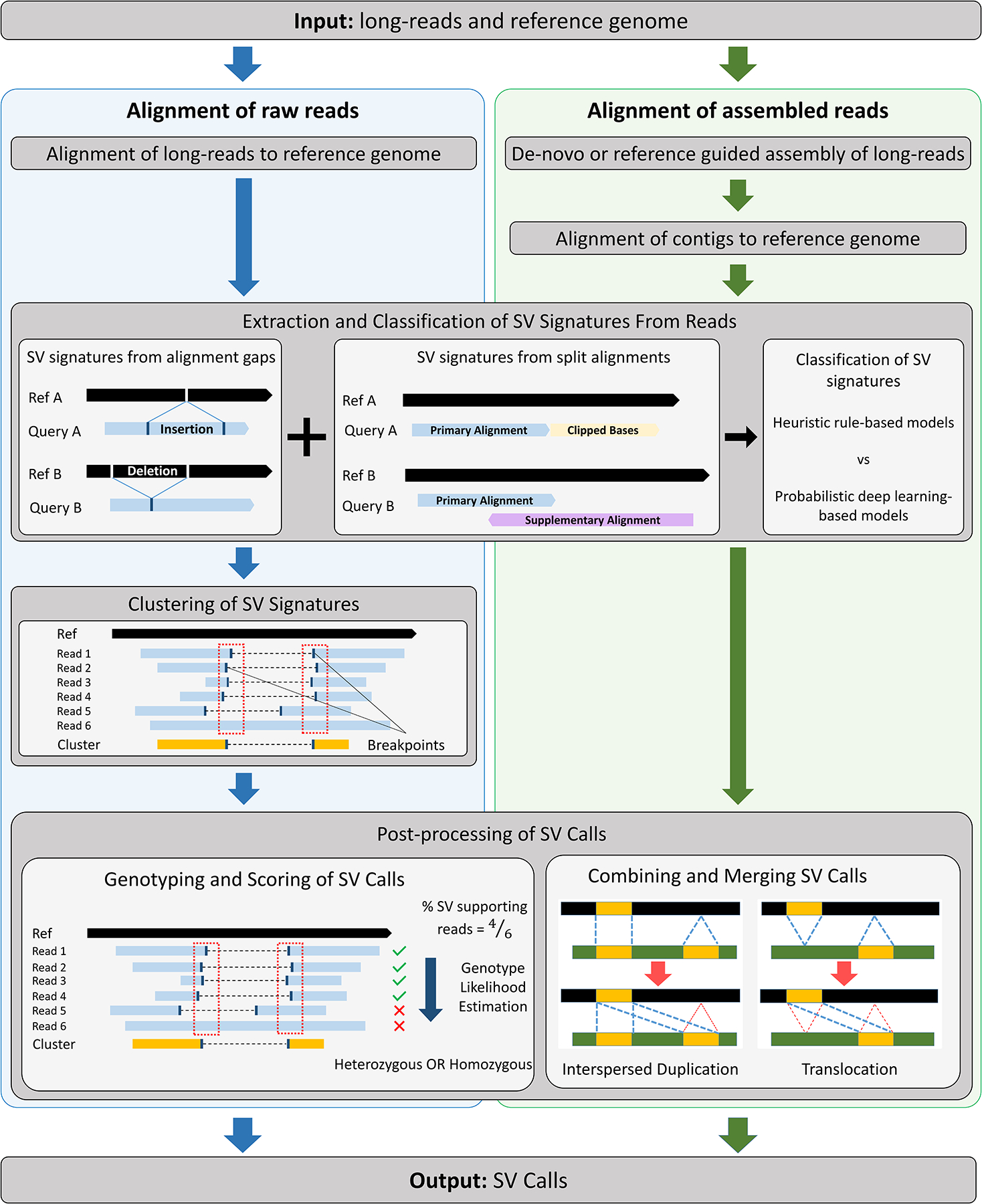
The general framework of SV detection using long-read sequencing. Alignment and assembly-based methods are described in the left and right columns respectively. Alignment and assembly-based methods use alignments of query sequences, which can be long reads or contigs respectively, to extract SV signatures that show evidence of an SV. SV Signatures can be extracted from alignment gaps, split alignments or clipped bases and these signatures are classified into various SV types. Alignment-based methods cluster SV signatures from multiple reads to give a consensus SV call. For assembly-based methods, contigs can be assembled through de-novo assembly for the whole genome or locally assembled with help of reference sequence. SV callers employ various post-processing steps such as assignment of quality scores to SV calls, genotyping and merging of simple SV calls into complex SVs.
Table 1.
The list of existing computational SV detection methods using long reads. Various methods are highlighted according to different sub-categories below. Assembly-based: de novo assembly (blue), and local assembly (orange). Alignment-based: general SV detection (grey), complex SV detection in targeted regions (yellow), and specific SV class detection (green); SV detection with combined strategy (pink). Repeat expansion: detection on long-read sequences (cyan), and detection on raw Nanopore signals (grey).
| Tool | Platform | Comments | ||
| Assembly-based SV detection | De-novo assembly | PAV35 | PB, ONT | Generates phased SV callsets for haplotype-resolved assemblies from contig alignments against a reference genome. |
| Dip-call141 | PB | Detects large insertions and deletions from haplotype-resolved genome assemblies. | ||
| SVIM-asm77 | PB, ONT | Detects SV from diploid assemblies by pairing similar SVs from opposite haplotypes. | ||
| SyRI79 | PB, ONT | Detects SVs, as well as small variants inside rearranged regions between two genome assemblies. | ||
| Smartie-SV87 | - | Aligns contigs assembled from any type of sequencing against a reference genome. | ||
| Assemblytics78 | - | Can detect SVs, repeat expansions and contractions from contigs. | ||
| Reference guided local assembly | PhasedSV90 | PB | Creates haplotype partitioned local assemblies and supports trio assembly for accurate SV detection. | |
| MsPAC91 | PB, ONT | Uses HMM on multiple sequence alignment of haplotype partitioned local assemblies. | ||
| PBSV75 | PB | Uses local multiple sequence realignment to detect SVs. | ||
| SVDSS92 | PB | Performs local assembly of sample-specific substrings into larger superstrings which are clustered and then used for SV detection. | ||
| Alignment-based SV detection | Rule-based | cuteSV49 | PB, ONT | Uses a heuristic method to detect and genotype SVs. |
| NanoSV55 | PB, ONT | Uses a random forest to filter false positive SVs. | ||
| SVIM52 | PB, ONT | Uses a custom distance metric and graphs to cluster SVs and detects both tandem and interspersed duplications. | ||
| Sniffles59 | PB, ONT | Can detect complex nested SVs and estimate parameters from data set and uses NGMLR aligner. | ||
| Sniffles260 | PB, ONT | Supports somatic and population level SV calling. | ||
| SENSV54 | ONT | Uses a novel SV-aware aligner to refine breakpoints, especially for detecting long SVs (>100kbp) using low coverage ONT reads. | ||
| PBHoney56 | PB | Uses characteristics and error profile of PB sequencing. | ||
| NanoVar51 | PB, ONT | Optimized for SV detection from low-depth sequencing. | ||
| Duet50 | ONT | Incorporates SNP signatures to enable phased SV detection and genotyping. | ||
| SKSV57 | PB | Generates improved read alignment profiles for SV calling and genotyping. | ||
| DeBreak58 | PB, ONT | Identifies SVs via a density-based clustering of SV candidates obtained from alignments and uses de novo assembly detect large SVs spanning multiple reads. | ||
| Picky53 | PB, ONT | Uses a greedy seed-and-extend algorithm to improve alignment and can detect tandem duplications. | ||
| Deep learning-based | SVision69 | PB, ONT | a deep learning approach to resolve simple and complex structural variants. | |
| BreakNet67 | PB | Predicts deletions via a CNN-LSTM deep learning model trained with feature matrices from read alignments pileup. | ||
| MAMnet68 | PB, ONT | Predicts insertions and deletions via a CNN-LSTM deep learning model trained with variant signature matrices constructed from read alignment pileups. | ||
| Ensemble Methods | NextSV71 | PB | Ensemble of cuteSV2 and Sniffles2 used with minimap2 and NGMLR aligners. | |
| combiSV74 | Combines results from six SV callers into a single call set with increased recall and precision. | |||
| Specialized SV detection | Complex SVs | SVision69 | PB, ONT | Resolves complex SVs using a CNN trained on read alignment features encoded in image format. |
| CORGi94 | PB, ONT | Detects and visualizes complex genomic rearrangements in a local region. | ||
| TSD95 | PB | Detects and visualizes complex SVs in targeted PB deep-sequencing. | ||
| Miscellaneous SV subtypes | rCANID96 | PB, ONT | Novel element insertion detection. | |
| rMETL97 | PB, ONT | Mobile element insertion or deletion detection. | ||
| npInv98 | PB, ONT | Non-allelic homologous recombination inversion detection. | ||
| Repeat Expansion Detection | Sequence-based | RepeatHMM101 | PB, ONT | Repeat detection from long reads using HMM. |
| Tandem-genotypes105 | PB, ONT | Repeat detection from long reads using copy number histogram analysis. | ||
| PacmonsTR102 | PB | Repeat detection from long reads using pairHMM. | ||
| Straglr106 | PB, ONT | Scans the genome for large insertions and generates a list of coordinates and motifs which are used to genotype tandem repeats. | ||
| adVNTR103 | PB | Uses trained HMMs to genotype target variable number tandem repeats obtained with specific sequencing technologies. | ||
| RepLong107 | PB | Repeat detection from long reads using network modularity optimization. | ||
| NanoRepeat104 | PB, ONT | Repeat detection from long reads using Gaussian mixture models. | ||
| Signal-based | STRique108 | ONT | Repeat detection using Nanopore raw signals and HMM. | |
| NanoSatellite109 | ONT | Uses squiggle-based algorithm on Nanopore raw signals. | ||
| DeepRepeat110 | ONT | Repeat detection using deep learning on Nanopore signals. | ||
| SV Genotyping | Sniffles59 | PB, ONT | Computes the fraction of supporting reads for each variant against the reference and then uses allele frequency to predict genotype. | |
| cuteSV49 | PB, ONT | Genotypes are predicted by computing the maximum likelihood of each zygosity as a function of supporting reads. | ||
| cuteSV2142 | PB, ONT | Regenotyping SVs through an accurate force-calling method. | ||
| Samplot119 | PB, ONT | Generates images with read depth and alignment information for SVs, and uses a trained ResNet-like model to predicts deletion genotypes based on these images. | ||
| svviz2120 | PB, ONT | Displays the number of supporting reads assigned to each allele, which can be used to estimate zygosity. | ||
| SVJedi121 | PB, ONT | Generates representative allele sequences for each SV and then aligns reads to these sequences to estimate allele frequencies for genotyping. | ||
| VaPoR122 | PB | Scores SV predictions by analyzing the k-mer recurrence and estimates genotype likelihood by fitting a Gaussian mixture model to the score distribution. | ||
| LRCaller118 | ONT | Alignment features are used to genotype each SV directly from long reads. | ||
| TT-Mars123 | PB | Genotypes SVs by matching their local regions to haplotype-resolved assemblies. | ||
| Somatic SV detection | Sniffles260 | PB, ONT | Increases sensitivity for low-frequency SVs and additional filtering and preprocessing steps to enable non-germline SV calling. | |
| DeBreak58 | PB, ONT | Detects non-germline SVs with clustered breakpoints in cancer genomes. | ||
| Nanomonsv115 | PB, ONT | Detects somatic SVs from paired tumor and matched control long-read sequencing data. | ||
| SHARC116 | ONT | Uses low coverage long-read sequencing to detect SVs in cancer genomes. A random forest model trained on SV features filters false positive SV calls. | ||
| CAMPHOR117 | ONT | Detects somatic SVs by comparing SVs identified from tumor samples against those from matched control samples. | ||
Alignment-based methods
Alignment-based SV callers include software tools such as cuteSV49, Duet50, NanoVar51, SVIM52, Picky53, SENSV54, NanoSV55, PBHoney56, SKSV57, DeBreak58, Sniffles59 and its improved successor Sniffles260. These tools employ a similar three-step framework: 1) align long reads against a reference genome, 2) determine SV signatures from the alignment of each read and classify them into SV types, and 3) use a clustering method to group similar SV signatures from various reads and get their consensus to reliably identify SVs. Since alignment can be orders of magnitude faster than assembly for large genomes, alignment-based SV calling methods tend to be faster than assembly-based methods.
Alignment of long reads to reference genome.
Alignment-based SV callers usually take an alignment file (in BAM format) generated by an aligner as input and infer SV signatures directly from the alignments. Therefore, SV calling accuracy is greatly affected by the choice of alignment algorithm, as well as the parameter settings of the aligners. To mitigate this issue, some SV callers either use specialized aligners or carry out alignment themselves using a well-known aligner with specialized parameters. For example, Sniffles59 works best with alignments generated by its companion aligner NGMLR59, designed specifically to facilitate SV detection using a convex gap-score model which consolidates several smaller insertions/deletions into large gaps in alignment. On the other hand, Picky53 and SENSV54 use the LAST61 and minimap262 aligner respectively to carry out an initial round of alignment, and then improve these alignments with an additional algorithm. SENSV uses its own SV-aware and gap-tolerant aligner, SV-DP, to refine breakpoints of large SVs, whereas Picky uses a greedy seed-and-extend algorithm to generate an improved alignment profile for each read. In a similar fashion, SKSV57 generates a pseudo-alignment profile using a custom dynamic programming algorithm that exploits the low error rate of HiFi reads to skip base level operations in alignment to achieve fast alignment and SV signature detection.
Classification and clustering of SV signatures.
SV signatures are generally identified by scanning read alignments for alignment gaps, clipped bases, or split alignment. However, some SV callers incorporate additional types of evidence. For instance, SENSV54 compares sequencing depth of the query genome against a panel-of-normals (PON) reference dataset consisting of ONT reads on 24 human genomes that do not share SVs. In this context, genomic intervals with abnormal depth in the query genome are considered to contain SVs. PBHoney56, on the other hand, identifies discordant regions with a high rate of small variants relative to adjacent genomic regions based on the hypothesis that a mis-aligned SV sequence will lead to more alignment mismatches.
After identifying SV signatures, most SV callers classify them using heuristic rule-based methods—a series of if-else conditional statements—specific to each SV class based upon criteria such as the length of gaps in alignments, differences in mapping orientations of segments, or the presence of more than one breakpoint. However, each read supporting a putative SV can have a different breakpoint or signature due to the existence of repetitive genomic regions and relatively high error rates of long reads (except when protocols such as PacBio HiFi are used). Therefore, it is critical to identify which SV signatures across the reads arise from the same SV and combine these independent predictions of SV sequences and breakpoints to get a reliable SV call. This is carried out by clustering algorithms, typically agglomerative clustering, which also often use rule-based methods to merge SV signatures based on factors such as breakpoint proximity and length of SV. A more flexible approach is taken by DeBreak58 which uses a density-based clustering that automatically adjusts clustering parameters for different SV types and sizes.
There are two main challenges that alignment-based methods need to address. First, a robust SV classifier needs to have an exhaustive list of cases in which the same SV can be represented by various combinations of SV signatures. Second, it is possible to overfit these classification and clustering rules to certain long-read error profiles or alignment artifacts of certain aligners. Many SV callers have hard-coded thresholds in their classification and clustering rules for important metrics such as maximum distance allowed between breakpoints for merging SV clusters. As a result, any improvements in read length and sequencing accuracy, modifications in algorithms of existing aligners, or development of new aligners can render many such ad-hoc rules obsolete or create new types of SV signatures which may not be accounted for. For instance, longer read length leads to fewer reads being split into multiple alignments, and as a result a tandem duplication might appear as a simple insertion instead of a pair of overlapping split alignments. Several published SV callers, such as cuteSV49 and Sniffles59, are under active development and are improving upon the SV signature detection, classification, and clustering methods of previous SV callers by adapting to the changing landscape of long-read sequencing technology.
Application of deep-learning to SV detection from long reads.
Over the past several years, deep learning-based methods have been successfully applied to detect small variants63–66 from long reads. Recently, a few deep-learning based methods been developed to tackle long-read SV calling using model-based inference to classify SVs as opposed to rule-based inference. Compared with rule-based methods, deep learning offers the ability to learn complex abstractions from labeled datasets without expert guidance. For instance, BreakNet67 and MAMnet68 generate a feature matrix for each 200bp subregion of the genome by extracting various alignment features such as read depth or number of deletions and apply a convolutional neural network (CNN) to this feature matrix. Then, a recurrent neural network analyzes CNN output of a series of contiguous subregions to predict the probability of an SV in each subregion. Afterwards, a clustering approach is used to define breakpoints more precisely from individual read alignments. A different approach is used by SVision69, another deep learning-based SV caller that can detect all types of SVs from long-read sequencing and is especially optimized for complex structural variants. SVision converts the alignment profiles of each read against the reference genome into an image that is fed to a CNN which assigns a probability score for various SV classes occurring in different segments of the image. After that, a graph-based approach is used to characterize complex SV events. A combination of both approaches has been applied by the SV caller Cue70 which creates a feature image with detailed alignment statistics of all reads in a large genomic interval and uses CNN to identify SV types and breakpoints directly on the image without the need of breakpoint clustering. We expect additional deep learning approaches that directly take alignments and genomic context features as input may be developed in the coming years, which can complement existing alignment-based and assembly-based SV callers.
Post-processing of SV calls.
Post processing of variant calls to assign confidence scores, determine complex SV types, and filter false positive SV calls is an important component of alignment-based SV callers. Several SV callers, such as SVIM52 and DeBreak58, carry out an additional step to combine or reclassify different SV calls, especially insertions, to identify different types of duplications and their sources of origin. Most tools discard any SV signature cluster with low read support, and an additional genotyping step using Bayesian likelihood estimation on the number of reads supporting different alleles can determine zygosity and genotype likelihood. For example, NanoVar51 and NanoSV55 use a neural network and random forest respectively, to assign confidence scores to SV calls using features such as: number of alignment mismatches near a breakpoint, number and fraction of reads supporting a breakpoint versus reference allele, and amount of deviation in breakpoints and SV lengths across supporting reads. SENSV54 takes a more thorough approach to validate an SV candidate. It constructs a local alternative reference sequence by inserting the SV into the reference sequence according to the predicted breakpoints, and a final SV call is made if the reads overlapping the SV have better alignment against the alternative allele than the reference genome. Lastly, incorporation of long-range haplotype information provided by long reads has become a staple of small variant detection, but most alignment-based SV callers ignore this crucial piece of information. Recently, Duet50—an SV calling framework for ONT sequencing data—extended cuteSV49 to allow for SNV-assisted SV calling and phasing. It filters out false SVs based on low haplotype confidence of supporting reads and shows improved performance in low coverage samples.
Improving SV calling by combining several tools.
Since the performance of alignment-based methods is significantly impacted by the choice of aligners and SV callers, better performance can be achieved by combining several tools46, 71. Zhou et al.46 compared performances of NanoSV55 and Sniffles59 when used with three different aligners: minimap262, NGMLR59 and GraphMap72. Their analysis shows significant changes in recall, especially for insertions, when the same SV caller works with different aligners. Moreover, different combinations of SV callers and aligners each had many unique calls, even when an SV caller or aligner is kept the same. This complicates the choice of SV callers or aligners since some SVs might only be detected by a certain combination of aligner and SV caller. One way to overcome this problem is by leveraging ensemble methods such as the latest version of NextSV71, which runs cuteSV49 and Sniffles59 with the two aligners minimap262 and NGMLR59 and merges their SV calls. Similarly, SURVIVOR73 and combiSV74 can merge several user-provided SV call sets to generate a consensus. Whereas SURVIVOR can merge SV calls from any tool, combiSV works specifically with the outputs of six well-established long-read SV callers (cuteSV, Sniffles, SVIM, NanoSV, NanoVar and PBSV75) and merges their SV calls by exploiting tool-specific strengths and weaknesses. A different approach is taken by Vulcan76, a long-read alignment framework that combines two distinct alignment gap scoring models from minimap262 and NGMLR59. It uses minimap2 for fast alignment of reads against a reference, identifies sub-optimally aligned reads based on edit distance, and then realigns them using NGMLR’s more sensitive gap penalty algorithm. Their evaluation on the HG002 benchmark dataset shows improvement in overall F1-score compared to using minimap2 or NGMLR alone, with significant improvements in detecting duplications.
Assembly-based methods
Assembly-based methods for SV calling first assemble contigs from individual sequencing reads and then map contigs against a reference genome to discover SVs through indels and split alignments. There are usually two strategies to assemble contigs: de-novo assembly and reference-guided local assembly. In de-novo assembly methods, contigs are constructed from all reads in a genome sequencing dataset without any prior knowledge of a reference genome. It involves a computationally intensive step of calculating read overlap among millions of reads. In contrast, reference-guided local assembly methods map reads to a reference genome first, and then reads aligned to a small genomic region of interest are selected. Then, a multiple sequence aligner or a de-novo assembly tool can be used to create contigs from this much smaller group of reads. In either case, SV callers align contigs or consensus sequences back to the reference genome to detect SVs.
SV calling from de-novo assemblies.
De-novo assembly-based SV callers typically rely on an external assembly tool to generate contigs from sequencing reads. These SV callers take user generated alignments of the contigs and usually recommend which aligner and parameter settings work best with the SV caller. Since assembled contigs can be treated as a particularly long sequencing read (with longer read length and higher per-base accuracy), some alignment-based SV callers have been modified to call SVs from diploid assemblies. For instance, SVIM-asm77 uses essentially the same SV signature detection and classification heuristic as SVIM52 but without the complicated process of getting consensus from multiple discordant reads. Similarly, cuteSV49 and SVision69, both alignment-based SV callers, have added support for SV calling from assembled contigs by using parameter settings suitable for low depth input. PAV35, Assemblytics78 and SyRI79, on the other hand, are tools developed specifically for detecting SVs from de-novo assemblies. Before applying the typical strategy for detecting SV signatures from alignment of diploid assemblies, PAV improves contig alignments by trimming redundant mappings, which improves breakpoint resolution of large tandem duplications and large repeat-mediated deletions. Similarly, both Assemblytics78 and SyRI79 anchor assembled contigs to a reference genome by identifying uniquely mappable segments of contigs, which is analogous to PAV’s alignment trimming strategy. While Assemblytics simply discards contig alignments without a unique anchor, SyRI identifies all anchoring regions between the assembled genome and reference genome and regards regions between these anchors as SV hotspots. However, SyRI requires chromosome-level assemblies as input, which can be difficult to generate using long-read assembly alone; it has been estimated that constructing chromosome-scale assemblies requires at least 30-fold coverage of reads longer than 100kbp80.
A major advantage of calling SVs from de-novo assemblies is the identification of large-scale genomic alterations and novel insertions (>100kbp) and better resolution of large repetitive loci. This is especially helpful for PacBio HiFi methods, where reads are typically shorter than 20kbp. Compared to long reads, assembled contigs have higher base level accuracy and better mappability in genomic regions containing large repeats, which improves precision of SV breakpoint inference due to error-correction of bases from read consensus, and contigs can be easily analyzed using SV visualization tools. Moreover, highly rearranged structure of some genomic regions can confound read alignment, but assembling reads into contigs before alignment can minimize the reference bias in such regions. Although long-read de-novo assembly tools81 require intensive computational resources compared to long-read alignment tools, the runtime has decreased significantly over the past few years, making them an increasingly attractive option. For instance, Shafin et al.80 report that Shasta, a fast assembly tool, can assemble the human genome from ONT reads in 5.5 hours using 128 CPUs and 1Tb of memory, whereas hifiasm82 can assemble the human genome in 10 hours using 48 CPUs. A recent comparison study by Lin et. al. found that the assembly-based approach produces higher consistency SV calls from long reads, reaching higher recall and precision across SVs of varying complexity83. The assembly approach was also able to detect 4,625 additional SVs, many of which were unresolvable via read alignments likely due to SV signature ambiguity in the clustering step. Nevertheless, while assembly-based strategies achieve higher performance with >20-fold coverage long-read data, the alignment-based strategy can achieve 90% recall with only 5-fold coverage even in complex regions83, 84, which is consistent with previous findings59, 85. Thus, while assembly-based methods require sufficient sequencing coverage, alignment-based methods may be preferable for clinical applications where obtaining high-coverage long-read data is difficult83.
Improving SV calling using chromosome-scale and haplotype-resolved assemblies.
Several long-read assemblers, such as Canu86, collapse homologous alleles into a haploid assembly. Additionally, early tools for SV calling from assemblies, such as Smartie-SV87 and Assemblytics, assumed a haploid genome, leading to low sensitivity for heterozygous variants. Therefore, polyploid assemblies are crucial for accurate SV detection and several strategies have been developed to tackle them. For diploid genomes, these methods fall into two categories: 1) generating dual assemblies in which contigs consist of mixed haplotypes from each parent, and 2) generating fully haplotype-resolved assemblies in which each contig comes from one parental haplotype. Initial attempts at creating diploid assemblies required multiple steps including 1) generating of an initial assembly graph; 2) phasing heterozygous SNPs and identifying the haplotype of each read; 3) generating a haplotype-resolved assembly88. However, the emergence of HiFi reads has led to the development of fast and easy-to-use assembly tools such as hifiasm89 and HiCanu59, that can compete with alignment tools in terms of simplicity. While a single long-read sequencing sample is sufficient for generating dual assemblies, such assemblies are prone to haplotype switching and can have limited utility in analyzing the interplay of distant alleles, genotype imputation, and the study of recombination or evolutionary patterns. On the other hand, generating haplotype-resolved assemblies requires either parental sequencing data for trio-binning82 or orthogonal sequencing such as Hi-C82 or Strand-seq60 for scaffolding, which can be costly. A recent study by Cheng et al.89 has shown that haplotype-resolved assemblies created by hifiasm using 30-fold PacBio HiFi reads with parental HiFi reads or Hi-C sequencing yield higher quality assemblies than ONT. Despite the increased cost, haplotype-resolved assembly strategies in conjunction with chromosome-scale assembly methods hold tremendous potential in revolutionizing SV calling.
SV calling from reference-guided local-assemblies.
Some limitations of de-novo assemblies can be addressed by using reference-guided local-assembly based SV callers such as PhasedSV90, MsPAC91, PBSV75 and SVDSS92. These types of methods are much faster than de-novo assembly for a whole genome. Additionally, using a reference as a guide avoids some problems associated with de-novo assemblies such as assembly collapse around segmental duplications. PhasedSV90 and MsPAC91 use SNV calls from read alignment to phase the reads. Then they divide the reference genome into small regular intervals and use a de-novo assembly tool to locally assemble contigs from each haploid set of reads in each interval. However, carrying out local assembly for the whole genome can still be very inefficient. Therefore, some SV callers are more selective. For instance, PBSV75, designed for PacBio reads, extracts only reads with SV signatures and generates consensus sequences around potential SV breakpoints using multiple sequence alignment. Similarly, DeBreak58 carries out local de-novo assembly only for regions with large numbers of clippings to identify large insertions. Unfortunately, local assembly-based methods have lagged behind other SV calling methods in terms of development and ease of use, showing a significant need for improvement in this area, especially for ONT reads. SVDSS92, a recently developed tool for HiFi reads, demonstrates the strengths of local assembly-based SV calling approaches, especially for multi-allelic SVs. It identifies sequences unique to a sample with regard to the reference and extends them until they include anchoring sites for potential SV breakpoints92. These sample-specific sequences are clustered into haplotypes and then assembled by locally applying the Partial-Order Alignment algorithm92, 93. However, currently SVDSS supports the detection of insertions and deletions only.
Specialized SV Detection
Complex SV detection in targeted regions.
While most genomic rearrangements resemble the common SV types shown in Figure 1, some rearrangements are complex and cannot be classified into known SV categories. Therefore, it can be helpful to elucidate how different segments of a complex SV relate to each other and various parts of the reference genome. This can be challenging for generalized SV detection methods that are optimized for fast genome-wide variant detection of common SV types. However, often a gene or genomic region of interest is known a priori, especially in clinical or diagnostic settings, due to the presence of phenotypic evidence. In such cases, specialized SV callers designed for SV detection in small targeted regions, such as CORGi94 and TSD95, or generalized SV detection tool SVision (in graph mode) can employ an extensive algorithm to resolve arbitrarily complex rearrangements that would otherwise be computationally infeasible for whole genome SV calling. For instance, to generate an alignment profile or correct order of split alignments for a read, CORGi and TSD use dynamic programming algorithms, whereas Picky53, a generalized SV caller, uses a faster greedy seed-and-extend algorithm which can often lead to suboptimal solutions. These tools characterize a complex SV by breaking it down as a sequence of basic SVs or rearrangements relative to the reference genome and create a plot depicting the alterations for visual analysis.
SV detection for a specific SV type.
Some other SV callers are designed to detect a specific type of SV, especially novel insertions. For example, rCANID96 (read Clustering and Assembly-based Novel Insertion Detection) is designed to detect novel insertions based on the idea that large novel insertions can result in reads that are completely unmapped or are partially mapped with large clippings – such reads are usually ignored by alignment-based SV callers. rCANID assembles contigs from such reads and extends the contigs using fully aligned reads that overlap them. This allows rCANID to anchor insertion-containing contigs to reference genome via alignment, and inserted sequences are checked for any matches in the reference genome to determine novel insertions. Similarly, rMETL97 is designed to detect mobile element insertions or deletions. It aligns reads supporting SV breakpoints to sequences of known mobile elements to determine if the inserted or deleted sequence is a mobile element. By default, rMETL uses Alu, L1 and SVA sequences obtained from previous studies, but users can input their own library of mobile element sequences. Another computational tool, npInv98 (nanopore Inversion) is designed to detect inversions and places a special emphasis on differentiating between inversions mediated by non-allelic homologous recombination and inversions mediated by non-homologous end joining.
Repeat expansion detection
Repeat expansions are a special type of SV where inserted sequences are tandemly repeated DNA motifs that can be 2–6bp long (short tandem repeats, STR), or longer than 6bp (variable number tandem repeats, VNTR). Whereas normal STR alleles usually have a comparatively smaller number of repeats, pathogenic STR alleles can be expanded by tens or hundreds of repeat copies, and they have been found to cause several genetic disorders.99 For instance, Huntington’s disease is caused by expansion of the CAG motif in the HTT gene from 6–35 copies in normal individuals to ≥ 36 copies in patients.100 In such cases, determining the exact copy number of a repeat motif can be extremely important, but generalized SV callers are not designed to estimate SV lengths with such precision. As a result, several tools dedicated to STR expansion detection using long reads have been developed, which fall broadly into two categories: repeat expansion detection via 1) nucleotide sequences in long reads or 2) ionic signals from Nanopore sequencing. These tools typically produce a distribution of repeat count estimates across all reads in a targeted region instead of a mean SV length prediction.
Repeat expansion detection from long-read nucleotide sequences.
Methods in this category can be applied to both PacBio and Oxford Nanopore sequencing. A common strategy is to carefully realign sequences up-stream and down-stream of the repeat region on a read to accurately determine its boundaries. Next, repeat count of the read is determined by analyzing the intervening tandem repeat sequence, and a repeat count distribution is generated over all reads overlapping the repeat locus. To calculate repeat counts, some tools use a probabilistic model, such as Hidden Markov Models used by RepeatHMM101, PacmonsTR102and adVNTR103, or a heuristic algorithm in the case of NanoRepeat104, Tandem-genotypes105 and Straglr106. For example, NanoRepeat aligns a read against a series of alternative reference sequences containing an increasing number of repeat units. Repeat count in the read is determined by the repeat count of the alternative reference that provides the best alignment. A Gaussian mixture model can be fit to the repeat count distribution to determine bi-allelic repeat counts and genotype. A different approach is used by RepLong107 which can detect repetitive regions in a genome without the use of a reference genome. It uses pairwise mapping of long reads with each other to identify boundaries of repeat regions and can be useful for the detection of novel repeat regions which are missing from prior knowledge or reference genome annotations. However, RepLong only detects repetitive regions and does not provide any information on copy number changes.
Repeat expansion detection from Oxford Nanopore ionic signals.
Ionic signals generated from the passage of nucleotides through a pore in Oxford Nanopore sequencing can also be used to detect repeat expansions. Basecalling, the process of translating signals into a nucleotide sequence, is evolving and improving at a rapid pace, but per-base accuracy (in the absence of consensus calling from multiple rounds of sequencing) is still lower than short-read sequencing, especially for repetitive sequences. Errors in basecalling can prevent accurate repeat counts by interrupting motifs in a tandem repeat region and sequence-based methods discussed earlier are unable to correct such errors. However, this can be circumvented by analyzing the underlying ionic signal. Several tools such as STRique108, NanoSatellite109 and DeepRepeat110 have been developed that process the raw signals directly and infer repeat counts based on the repetitive nature of the signals corresponding to tandem repeats. STRique108 and NanoSatellite109 both compare Nanopore signals of long reads containing repeats against a simulated Nanopore signal for the corresponding target reference sequence, whereas DeepRepeat used a deep-learning model and leverages self-similarity of signals associated with repeat units in a long-read without aligning the signal to the reference signal.
Somatic SV calling
Somatic mutations play an important role in the initiation and progression of various types of cancers. Somatic SVs can activate oncogenes, disrupt tumor suppressor genes, create/abolish non-coding gene regulatory sequences, and generate gene fusions. Accurate detection of somatic SVs can aid in cancer diagnosis, the development of therapeutic drugs and treatment, and the detection of driver genes.111 Somatic SV calling refers to detecting SVs from a patient’s tumor tissue sample and determining which variants are somatic versus germline. This often involves calling SVs from a normal tissue sample and comparing them with tumor SV calls. However, tumor samples show a high degree of mosaicism, contain low allele frequency variants and show complex rearrangement patterns involving several breakpoints112, such as chromoplexy and chromothripsis, which can be difficult to resolve using short reads. Moreover, most SV callers are designed for germline variants and evaluated on cell-line samples, and they often lack sensitivity for detecting low frequency somatic SVs. This highlights the need to develop specialized SV calling methods for somatic SVs, and such methods fall into two categories depending upon whether they detect somatic SVs from tumor sample only, or from comparison of normal and tumor samples.
Several studies have used long-read sequencing to identify somatic SVs in cancer cells58, 113–115 and shown that alignment-based SV callers are better suited than assembly-based methods for this task. A proof-of-principal study by Euskirchen et al.114 demonstrated that deep amplicon sequencing with Nanopore MinION can allow SV detection (using NanoSV55), copy number profiling, SNV detection, and methylation profiling for same-day diagnosis of brain tumors at very low cost. Some long-read SV callers, such as Sniffles260 and DeBreak58, have special modes for somatic variant calling from tumor samples aimed at improving recall of low allele frequency variants. For instance, the non-germline mode of Sniffles2 decreases the minimum read support requirement for SV detection and disables read coverage-based filtering to improve recall, while imposing a strict filter on read alignment quality to remove sequencing and alignment artifacts. As a result, Sniffles2 can accurately detect somatic SVs with allele frequency as low as 7% on samples with 70X coverage by ONT.60 Another SV caller Nanomonsv115 creates a consensus sequence for putative somatic SVs from a tumor sample with flanking reference sequences and filters out any SV candidate as germline, if reads from the control sample can be aligned against the SV sequence accurately. In general, somatic SV calling from tumor-only sample requires deep coverage of high accuracy reads to distinguish somatic SVs from sequencing errors. This can be accomplished using PacBio HiFi sequencing or the newly developed Oxford Nanopore kit14 (R10.4.1) flowcells in combination with targeted amplicon sequencing or CRISPR-based capture to increase sequencing depth.
Methods that compare reads or SV calls from tumor and normal tissue samples can allow more precise differentiation of somatic SVs from germline SVs without the need for deep sequencing. Several long-read tools have been developed for this purpose using well-known SV callers. For instance, SHARC116 is a pipeline that uses NanoSV55 to call SVs from the tumor sample only but implements several SV filtering steps such as random forest classification to filter germline SVs. It further compares filtered SV calls against a user-provided list of germline SV calls (which could be from a database or normal tissue from the patient) and prioritizes remaining SVs based on likelihood of being somatic. In a similar manner, CAMPHOR117 separately calls SVs from both tumor and control samples and distinguishes between them using breakpoint proximity. However, in a clinical setting, it is often difficult to acquire sufficient samples for both normal and tumor tissues which complicates the adoption of long-read sequencing for somatic SV calling.
Population-scale SV calling
Short-read sequencing can be used to carry out whole genome sequencing with high coverage at relatively low cost, which makes it well suited for large-scale cohort studies. Long-read sequencing studies have been limited to smaller population cohorts due to the higher costs. However, the landscape is changing due to the continuously decreasing cost of Nanopore and PacBio sequencing, especially with the wider adoption of the Nanopore PromethION sequencer. Recent developments have shown that even low coverage long-read samples can reliably detect SVs using alignment-based SV calling.83, 85 However, false positives from the high sequencing error of long reads can be significantly amplified in a large cohort SV callset, thus requiring extensive variant filtering and validation. In the largest population-scale SV detection study to date using long-read sequencing, Beyter et al.118 used Nanopore PromethION sequencing datasets of 3,622 Icelanders with a median coverage of 17.2x to create a catalog of 133,886 SVs. Their pipeline uses Sniffles to detect SVs in individuals, refines breakpoints using short read data, and validates them by comparing raw Nanopore read signal against alternative and reference allele sequences. SV calls from individuals are merged using graph-based clustering to remove redundant but slightly different representations of the same SVs across samples. The merged SV set is genotyped using both long and short reads and imputed into long-range phased haplotypes of 166,281 Icelanders. A final high-confidence SV set is created by filtering SVs according to imputation accuracy. Their analysis shows that population-scale SV discovery and genotyping from long-read sequencing data are more reliable and accurate than from short-read data, especially in tandem repeat regions. A comprehensive review of population scale SV discovery from long-read sequencing approaches can be found here85.
SV Genotyping, Visualization and Benchmark Evaluation Tools
Tools for SV genotyping from long reads.
SV genotyping and validation refers to the task of determining whether a known SV is present in a query genome. Typically, these are high-confidence SVs whose breakpoints and sequences are known with high accuracy through population-wide studies. This is a much simpler task than the SV calling or discovery procedures described earlier. Therefore, specialized algorithms developed for genotyping can produce more accurate results. A common approach is to create an alternative reference sequence that contains the SV at known breakpoints and examine read alignments against both the normal reference and alternative reference sequences. Although a few SV callers (Sniffles59and cuteSV49) and SV visualization tools (Samplot119 and svviz2120) allow SV genotyping, several tools have been developed specifically for SV genotyping from long reads (SVJedi121, VaPoR122, and LRCaller118) as well as for genotyping from haplotype-resolved assemblies (TT-Mars123). A survey and evaluation of state-of-the art long-read genotyping tools can be found here124.
Tools for SV visualization from long reads.
Several computational tools are available for visualization and manual inspection of SV calls in the context of long-read sequencing data. Unlike generic alignment viewers such as IGV125, they have additional functionalities to facilitate more detailed examination of SVs. For example, svviz2120 is a read viewer for validating putative structural variants using both short and long reads. It takes a candidate SV call and BAM files as inputs, searches the BAM files for reads relevant to the SV and realigns them against putative variant alleles as well as the reference allele. Alignments are plotted in a genome browser format, showing each allele and its supporting reads for visual validation of breakpoints and genotype. New Genome Browser (NGB)126 is a web client-server tool with an interface like IGV, but it has been developed with specific features for visualization of SVs and their supporting reads. Ribbon127 is a genome visualization tool for viewing alignments of reads and assembled contigs against a reference genome. It can take additional VCF or BED files of SVs for visual validation. Ribbon can show complex alignment profiles of reads and display translocations across chromosomes. Figure 3 shows a Ribbon plot for ONT and PacBio reads of HG002 overlapping a 36,400bp inversion at chr10:47023408 identified by cuteSV, Sniffles, and SVIM. Thus, SV visualization tools like Ribbon allow for convenient manual validation of SVs independent of SV calling tools.
Figure 3.
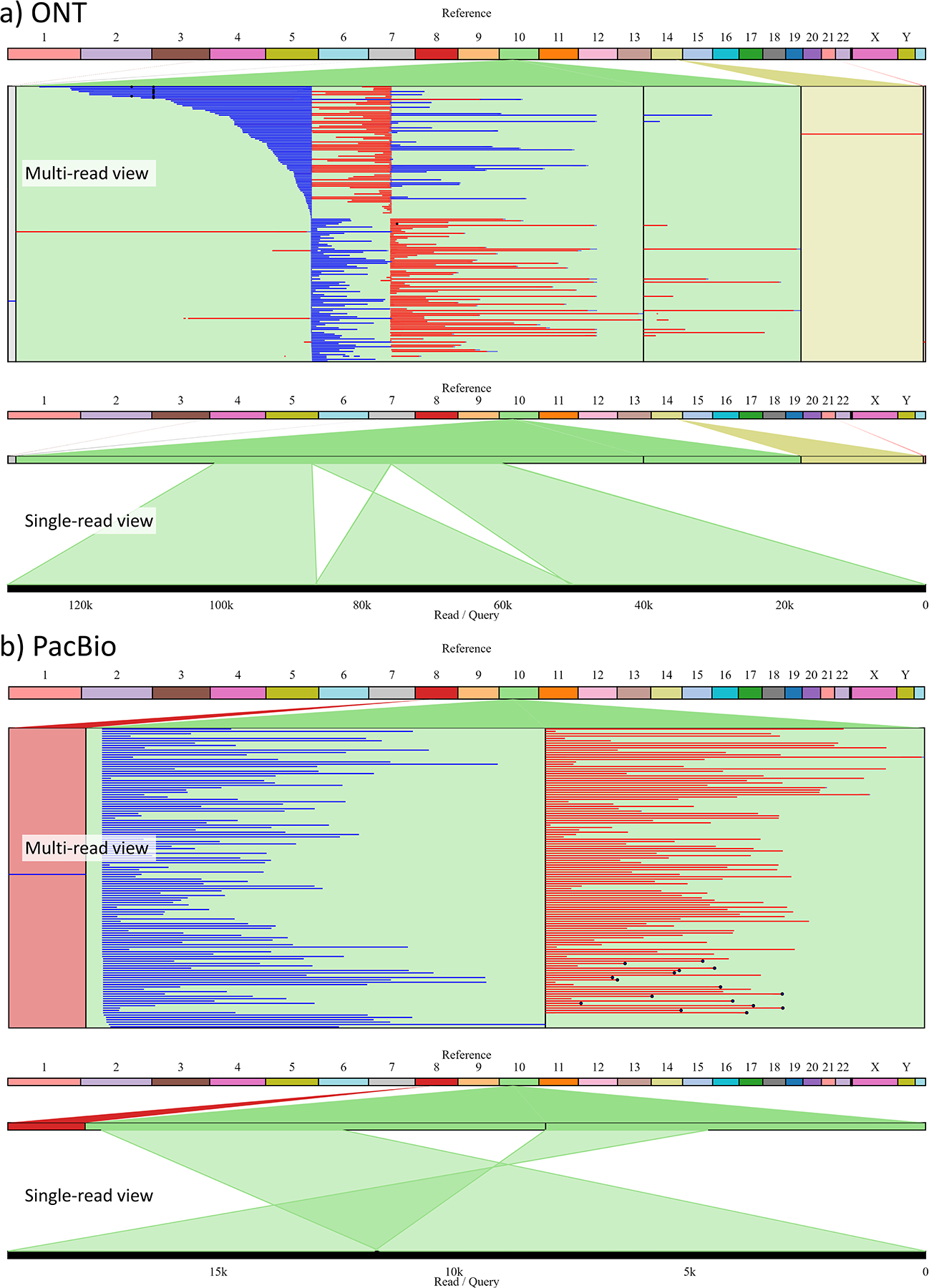
Ribbon plot of a HG002 ONT and PacBio reads for a 36,400bp inversion at chr10:4,7023,408 (GRCh37) identified by cuteSV, Sniffles and SVIM. a) and b) show ONT and PacBio reads of HG002 respectively. Multi-read view shows all reads overlapping the inversion, with split alignments of each read in a single row. Blue and red colors represent mapping orientation of split alignments. Single-read view shows split alignments of a single read in detail. Several ONT reads span the entire inversion and cover several tens kbp of flanking region.
SV benchmark evaluation tools.
A few computational tools have been developed to compare two different SV call sets and can be used to evaluate SV calling performance when a ground truth set is available. One such tool, SURVIVOR73, provides three functional SV modules: SV simulation, SV operation by merging/comparing SV calls within a sample or among population, and SV evaluation regardless of variant caller or sequencing technology. Another evaluation tool Truvari128 provides functions to annotate VCF files and compare VCF files with a consistency report between multiple VCFs. It can also produce performance metrics from comparison of predicted and benchmark SVs. One shortcoming of these popular SV evaluation tools is that they perform a pairwise comparison of SVs, which can result in the mischaracterization of complex rearrangements, especially in tandem repeat regions. As haplotype-resolved genome assemblies and phased SV ground truth sets become available, it is important to develop better evaluation tools that leverage haplotype information for SV comparison. One such example is hap-eval129, a haplotype-aware SV evaluation tool. Instead of simply comparing each pair of SVs in a putative SV call set and ground truth call set, hap-eval clusters nearby SV calls and assembles them into haplotypes based on available genotype and phasing information. The assembled haplotypes for the truth set and calls are then compared, and the best-matching haplotypes are evaluated.
Long-read based reference genomes and SV call sets
Choice of reference genome is a crucial component of SV detection, since most methods rely on alignment against a reference genome for SV detection. The first assembled draft human reference genome has served as a powerful tool for variant discovery and has undergone decades of additional refinement, with the most recent version being GRCh38130–132. Nevertheless, GRCh38 has not been without its limitations. First, even with alternative haplotypes in the assembly, it does not capture the diversity of the worldwide population: it is a mixture of ethnicities derived from the genomes of approximately 20 individuals which were predominantly European and African133. In addition, GRCh38 represents a mosaic of haplotypes from many individuals, which leads to errors from SVs occurring between haplotypes134. This highlights the importance of single haplotype genome assemblies with sequence continuity across highly repetitive and structurally variant portions of the genome134. Finally, in addition to telomeres and centromeres, more than 100 million nucleotides remain unresolved due to repetitive sequences or complex genomic regions which are difficult to reconstruct132. Recent advances in sequencing technology have produced long reads capable of spanning and closing these gaps: PacBio HiFi sequencing generates 10–20 kb length reads with 99.9% accuracy, while Oxford Nanopore’s platform can generate sequences with moderate coverage for hundreds of kilobases or even longer34. Recently, the Telomere-to-Telomere (T2T) Consortium leveraged both technologies to produce the first complete assembly of a human genome sequence for nearly all chromosomes48, 135. The T2T-CHM13 genome was derived from a single homozygous complete hydatidiform mole, and thus it represents a complete human haplotype with minor exceptions135. Although T2T-CHM13 is complete, in some cases long-read transcript sequences map better to GRCh38, suggesting that both capture different structurally variant haplotypes136. Thus, to detect and characterize variants across the full diversity of human genetic variation, ongoing efforts led by the Human Pangenome Reference Consortium are focused on compiling a collection of all common haplotypes in the human population into a database of reference genomes known as a pangenome137, 138. Moving away from reliance on a single reference for SV detection will thus require the development of tools—including alignment, haplotype representation, and variant detection tools—with the flexibility to leverage these different reference formats. This is an area of potential exploration for the future development of SV detection tools.
A major clinical application of SV detection is for genome-wide association studies (GWAS) to characterize SV types and their functional consequences. Thus, there are ongoing efforts to develop a comprehensive dataset of variants for association studies including gnomAD-SV with SVs resolved from a diverse cohort of 14,891 genomes139, and a recent study resolving SVs from 3,202 high-coverage samples, including 602 complete trios136, 140. An important limitation of these datasets is that variants were discovered using short read sequences which have decreased sensitivity in repetitive regions where SVs are often located139, 140. To improve sensitivity in detecting large SVs, Ebert et. al. used long-read sequencing data to assemble 64 highly complete and contiguous human haplotype genomes from a diverse population, and identified 107,590 SVs, of which 68% were not discovered with short-read sequencing methods35. Thus, the development of long-read SV callers capable of leveraging data from large sets of diverse human genomes will improve the discovery and characterization of SVs for association studies.
Summary
In the current survey, we reviewed the development of over 50 long-read based methods for SV discovery and analysis. We describe the shared analysis strategies and unique data processing approaches for these methods and discuss how improvements in sequencing accuracy and read length in the past few years has led to a new generation of SV callers. We also discuss how the new telomere-to-telomere genome assemblies and pangenome efforts can improve accuracy and drive the development of future SV calling strategies. Box 1 summarizes three key factors that can lead to significant improvements in the accuracy and robustness of SV detection from long-reads: advances in 1) long-read sequencing technologies, 2) long-read assembly and alignment methods, and 3) computational methods for SV detection from long-reads. In conclusion, long-read sequencing enables the detection of SVs that could not be previously detected from conventional short-read sequencing, yet there is still substantial room for improvement in computational methods to identify disease relevant SVs in the future.
Box 1. Directions for improvements in long-read SV calling.
Advances in long-read sequencing technologies
Improved basecalling accuracy and longer read length from new protocols for consensus sequencing and duplex sequencing.
Application of PCR-free Cas9-based enrichment or adaptive sampling to examine targeted regions.
Development of library preparation protocols to sequence ultra-long reads with high yield.
Development of low-cost synthetic long-read platforms (such as stLFR, TELL-Seq, Infinity).
Advances in long-read assembly and alignment methods
Development of automated assembly tools for chromosome-scale and haplotype-resolved assemblies.
Creation of additional telomere-to-telomere reference genomes.
Creation of pangenomes to catalog common haplotypes in diverse human populations.
Creation of comprehensive catalogs for specific complex regions (such as MHC and 22q11.2).
Development of new tools and standards for graph pangenome alignments, SV representation and SV calling.
Advances in computational methods for long-read SV calling
Application of deep learning in probabilistic inference-based SV detection.
Creation of ensemble methods to leverage the strengths of various SV callers and create reliable confidence measures.
Creation of ensemble methods to combine diverse range of sequencing/mapping platforms and sequencing approaches (such as HiC, Pore-C, synthetic long reads)
Development of specialized SV callers for more complex loci (such as nested inversion, inverted duplication) and tandem mixed-motif repeat expansions.
Incorporation of haplotype information, epi-haplotype information and phased local assemblies in SV calling.
Development of high precision somatic SV callers for tumor samples that are sensitive to SVs with low variant allele fraction.
SV discovery and genotyping from long-read sequencing in population scale studies.
Acknowledgements
We thank several readers who worked on structural variants to provide valuable feedback on our preprint, including Anagha Gouru, Yunyun Zhou and Yu Hu. The thank the developers of the various software tools described in the manuscript to make their tool available with detailed documentations. We also thank anonymous reviewers in their insightful comments to restructure/refocus the manuscript and improve the informativeness of the manuscript. The survey is in part supported by NIH grant GM132713 and the CHOP Research Institute.
Footnotes
Ethics Declaration
The authors declare no competing interests.
References
- 1.Zook JM et al. A robust benchmark for detection of germline large deletions and insertions. Nature biotechnology (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study represents a gold standard SV benchmark for the HG002 genome containing nearly ten thousand insertions and deletions validated by several orthogonal technologies.
- 2.Sudmant PH et al. An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cameron DL, Di Stefano L & Papenfuss AT Comprehensive evaluation and characterisation of short read general-purpose structural variant calling software. Nature Communications 10, 3240 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kosugi S et al. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biology 20, 117 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahmoud M et al. Structural variant calling: the long and the short of it. Genome Biology 20, 246 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bickhart D & Liu G The challenges and importance of structural variation detection in livestock. Frontiers in Genetics 5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conrad DF et al. Origins and functional impact of copy number variation in the human genome. Nature 464, 704–712 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kidd JM et al. A Human Genome Structural Variation Sequencing Resource Reveals Insights into Mutational Mechanisms. Cell 143, 837–847 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korbel JO et al. Paired-End Mapping Reveals Extensive Structural Variation in the Human Genome. Science 318, 420–426 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]; An important study demonstrating extensive presence of SVs in human genomes using paired-end sequencing.
- 10.Sudmant PH et al. Diversity of human copy number variation and multicopy genes. Science (New York, N.Y.) 330, 641–646 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cortés-Ciriano I et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nature Genetics 52, 331–341 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rees E & Kirov G Copy number variation and neuropsychiatric illness. Curr Opin Genet Dev 68, 57–63 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stankiewicz P & Lupski JR Structural variation in the human genome and its role in disease. Annual review of medicine 61, 437–455 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Feuk L, Carson AR & Scherer SW Structural variation in the human genome. Nature Reviews Genetics 7, 85–97 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Geiss GK et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol 26, 317–325 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Ye K, Schulz MH, Long Q, Apweiler R & Ning Z Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rausch T et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Layer RM, Chiang C, Quinlan AR & Hall IM LUMPY: a probabilistic framework for structural variant discovery. Genome Biology 15, R84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan EKF et al. Optical mapping reveals a higher level of genomic architecture of chained fusions in cancer. Genome Res 28, 726–738 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kloosterman WP & Cuppen E Chromothripsis in congenital disorders and cancer: similarities and differences. Curr Opin Cell Biol 25, 341–348 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Dai Y et al. Single-molecule optical mapping enables quantitative measurement of D4Z4 repeats in facioscapulohumeral muscular dystrophy (FSHD). Journal of medical genetics 57, 109–120 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alkan C, Sajjadian S & Eichler EE Limitations of next-generation genome sequence assembly. Nat Methods 8, 61–65 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharp AJ et al. Segmental duplications and copy-number variation in the human genome. Am. J. Hum. Genet 77, 78–88 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Branton D et al. The potential and challenges of nanopore sequencing. Nature biotechnology 26, 1146–1153 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eid J et al. Real-time DNA sequencing from single polymerase molecules. Science (New York, N.Y.) 323, 133–138 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Marx V Method of the year: long-read sequencing. Nat Methods 20, 6–11 (2023). [DOI] [PubMed] [Google Scholar]
- 27.Wang O et al. Efficient and unique cobarcoding of second-generation sequencing reads from long DNA molecules enabling cost-effective and accurate sequencing, haplotyping, and de novo assembly. Genome Res 29, 798–808 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Z et al. Ultralow-input single-tube linked-read library method enables short-read second-generation sequencing systems to routinely generate highly accurate and economical long-range sequencing information. Genome Res 30, 898–909 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olson ND et al. PrecisionFDA Truth Challenge V2: Calling variants from short and long reads in difficult-to-map regions. Cell Genomics 2, 100129 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mantere T, Kersten S & Hoischen A Long-Read Sequencing Emerging in Medical Genetics. Front Genet 10, 426 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi L et al. Long-read sequencing and de novo assembly of a Chinese genome. Nature Communications 7, 12065 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parikh H et al. svclassify: a method to establish benchmark structural variant calls. BMC Genomics 17, 64 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pendleton M et al. Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nature Methods 12, 780–786 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jain M et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol 36, 338–345 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ebert P et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 372 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; A study on SV detection from haplotype resolved assemblies generated from long-reads and Strand-seq which identified three times as many SVs compared to short-reads.
- 36.Merker JD et al. Long-read genome sequencing identifies causal structural variation in a Mendelian disease. Genetics in Medicine 20, 159–163 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carneiro MO et al. Pacific biosciences sequencing technology for genotyping and variation discovery in human data. BMC Genomics 13, 375 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wenger AM et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nature biotechnology 37, 1155–1162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Menegon M et al. On site DNA barcoding by nanopore sequencing. PLOS ONE 12, e0184741 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krishnakumar R et al. Systematic and stochastic influences on the performance of the MinION nanopore sequencer across a range of nucleotide bias. Scientific reports 8, 3159 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li C et al. INC-Seq: accurate single molecule reads using nanopore sequencing. GigaScience 5, 34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karst SM et al. High-accuracy long-read amplicon sequences using unique molecular identifiers with Nanopore or PacBio sequencing. Nat Methods 18, 165–169 (2021). [DOI] [PubMed] [Google Scholar]
- 43.Aganezov S et al. Comprehensive analysis of structural variants in breast cancer genomes using single-molecule sequencing. Genome Res 30, 1258–1273 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sone J et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nature Genetics 51, 1215–1221 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Miao H et al. Long-read sequencing identified a causal structural variant in an exome-negative case and enabled preimplantation genetic diagnosis. Hereditas 155, 32 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou A, Lin T & Xing J Evaluating nanopore sequencing data processing pipelines for structural variation identification. Genome Biology 20, 237 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luan M-W, Zhang X-M, Zhu Z-B, Chen Y & Xie S-Q Evaluating Structural Variation Detection Tools for Long-Read Sequencing Datasets in Saccharomyces cerevisiae. Frontiers in Genetics 11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nurk S et al. The complete sequence of a human genome. Science (New York, N.Y.) 376, 44–53 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; The study describes the first complete human reference genome T2T-CHM13 which allows SV detection in centromeric region, telomeric region, and other complex regions.
- 49.Jiang T et al. Long-read-based human genomic structural variation detection with cuteSV. Genome Biol 21, 189 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou Y, Leung AW, Ahmed SS, Lam TW & Luo R Duet: SNP-assisted structural variant calling and phasing using Oxford nanopore sequencing. BMC bioinformatics 23, 465 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tham CY et al. NanoVar: accurate characterization of patients’ genomic structural variants using low-depth nanopore sequencing. Genome Biol 21, 56 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heller D & Vingron M SVIM: structural variant identification using mapped long reads. Bioinformatics 35, 2907–2915 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gong L et al. Picky comprehensively detects high-resolution structural variants in nanopore long reads. Nature methods 15, 455–460 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leung HCM et al. Detecting structural variations with precise breakpoints using low-depth WGS data from a single oxford nanopore MinION flowcell. Scientific reports 12, 4519 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cretu Stancu M et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat Commun 8, 1326 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.English AC, Salerno WJ & Reid JG PBHoney: identifying genomic variants via long-read discordance and interrupted mapping. BMC bioinformatics 15, 180 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu Y et al. SKSV: ultrafast structural variation detection from circular consensus sequencing reads. Bioinformatics (2021). [DOI] [PubMed] [Google Scholar]
- 58.Chen Y et al. Deciphering the exact breakpoints of structural variations using long sequencing reads with DeBreak. Nat Commun 14, 283 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sedlazeck FJ et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods 15, 461–468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes a highly accurate alignment based long-read SV caller and its companion aligner NGMLR. Sniffles is one of the earliest methods for long-read SV calling and is still widely used today.
- 60.Smolka M et al. Comprehensive Structural Variant Detection: From Mosaic to Population-Level. bioRxiv, 2022.2004.2004.487055 (2022). [Google Scholar]
- 61.Kielbasa SM, Wan R, Sato K, Horton P & Frith MC Adaptive seeds tame genomic sequence comparison. Genome Res 21, 487–493 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li H Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ahsan MU, Liu Q, Fang L & Wang K NanoCaller for accurate detection of SNPs and indels in difficult-to-map regions from long-read sequencing by haplotype-aware deep neural networks. Genome Biol 22, 261 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luo R et al. Exploring the limit of using a deep neural network on pileup data for germline variant calling. Nature Machine Intelligence 2, 220–227 (2020). [Google Scholar]
- 65.Poplin R et al. A universal SNP and small-indel variant caller using deep neural networks. Nature biotechnology 36, 983–987 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Shafin K et al. Haplotype-aware variant calling with PEPPER-Margin-DeepVariant enables high accuracy in nanopore long-reads. Nat Methods 18, 1322–1332 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luo J et al. BreakNet: detecting deletions using long reads and a deep learning approach. BMC Bioinformatics 22, 577 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ding H & Luo J MAMnet: detecting and genotyping deletions and insertions based on long reads and a deep learning approach. Brief Bioinform 23 (2022). [DOI] [PubMed] [Google Scholar]
- 69.Lin J et al. SVision: a deep learning approach to resolve complex structural variants. Nat Methods 19, 1230–1233 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; An innovative deep-learning based inference model for complex SV detection. It converts read alignment into an image that is analyzed by convolutional neural networks.
- 70.Popic V et al. A deep learning framework for structural variant discovery and genotyping. bioRxiv, 2022.2004.2030.490167 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fang L, Hu J, Wang D & Wang K NextSV: a meta-caller for structural variants from low-coverage long-read sequencing data. BMC Bioinformatics 19, 180 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sović I et al. Fast and sensitive mapping of nanopore sequencing reads with GraphMap. Nature Communications 7, 11307 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jeffares DC et al. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat Commun 8, 14061 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dierckxsens N, Li T, Vermeesch JR & Xie Z A benchmark of structural variation detection by long reads through a realistic simulated model. Genome Biol 22, 342 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Biosciences, P, Vol. 2020 (Pacific Biosciences, 2017). [Google Scholar]
- 76.Fu Y, Mahmoud M, Muraliraman VV, Sedlazeck FJ & Treangen TJ Vulcan: Improved long-read mapping and structural variant calling via dual-mode alignment. GigaScience 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Heller D & Vingron M SVIM-asm: structural variant detection from haploid and diploid genome assemblies. Bioinformatics (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nattestad M & Schatz MC Assemblytics: a web analytics tool for the detection of variants from an assembly. Bioinformatics 32, 3021–3023 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Goel M, Sun H, Jiao W-B & Schneeberger K SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biology 20, 277 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shafin K et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nature biotechnology 38, 1044–1053 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes the Shasta toolkit for fast de-novo assembly from Oxford Nanopore sequencing, which allows a 6 hour run time for assembly.
- 81.Marx V Long road to long-read assembly. Nature Methods 18, 125–129 (2021). [DOI] [PubMed] [Google Scholar]
- 82.Garg S et al. Chromosome-scale, haplotype-resolved assembly of human genomes. Nature biotechnology 39, 309–312 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes an accurate assembly tool for PacBio HiFi reads that can generate chromosome-scale and haplotype resolved assemblies using trio or HiC data.
- 83.Lin J, Jia P, Wang S & Ye K Comparison and benchmark of long-read based structural variant detection strategies. bioRxiv, 2022.2008.2009.503274 (2022). [Google Scholar]
- 84.Zhao X et al. Expectations and blind spots for structural variation detection from long-read assemblies and short-read genome sequencing technologies. The American Journal of Human Genetics 108, 919–928 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.De Coster W, Weissensteiner MH & Sedlazeck FJ Towards population-scale long-read sequencing. Nature Reviews Genetics 22, 572–587 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Koren S et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27, 722–736 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kronenberg ZN et al. High-resolution comparative analysis of great ape genomes. Science (New York, N.Y.) 360, eaar6343 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chin CS et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods 13, 1050–1054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cheng H et al. Haplotype-resolved assembly of diploid genomes without parental data. Nature biotechnology 40, 1332–1335 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chaisson MJP et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nature communications 10, 1784–1784 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; A key study that demonstrated six-fold increase in SV detection from local assembly based SV calling compared to short read sequencing.
- 91.Rodriguez OL, Ritz A, Sharp AJ & Bashir A MsPAC: a tool for haplotype-phased structural variant detection. Bioinformatics 36, 922–924 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Denti L, Khorsand P, Bonizzoni P, Hormozdiari F & Chikhi R SVDSS: structural variation discovery in hard-to-call genomic regions using sample-specific strings from accurate long reads. Nature Methods (2022). [DOI] [PubMed] [Google Scholar]
- 93.Lee C, Grasso C & Sharlow MF Multiple sequence alignment using partial order graphs. Bioinformatics 18, 452–464 (2002). [DOI] [PubMed] [Google Scholar]
- 94.Stephens Z, Wang C, Iyer RK & Kocher JP Detection and visualization of complex structural variants from long reads. BMC bioinformatics 19, 508 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Meng G et al. TSD: A Computational Tool To Study the Complex Structural Variants Using PacBio Targeted Sequencing Data. G3 (Bethesda, Md.) 9, 1371–1376 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jiang T, Fu Y, Liu B & Wang Y Long-Read Based Novel Sequence Insertion Detection With rCANID. IEEE transactions on nanobioscience 18, 343–352 (2019). [DOI] [PubMed] [Google Scholar]
- 97.Jiang T, Liu B, Li J & Wang Y rMETL: sensitive mobile element insertion detection with long read realignment. Bioinformatics 35, 3484–3486 (2019). [DOI] [PubMed] [Google Scholar]
- 98.Shao H et al. npInv: accurate detection and genotyping of inversions using long read sub-alignment. BMC bioinformatics 19, 261 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Paulson H Repeat expansion diseases. Handb Clin Neurol 147, 105–123 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bates GP et al. Huntington disease. Nat Rev Dis Primers 1, 15005 (2015). [DOI] [PubMed] [Google Scholar]
- 101.Liu Q, Zhang P, Wang D, Gu W & Wang K Interrogating the “unsequenceable” genomic trinucleotide repeat disorders by long-read sequencing. Genome Med 9, 65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ummat A & Bashir A Resolving complex tandem repeats with long reads. Bioinformatics 30, 3491–3498 (2014). [DOI] [PubMed] [Google Scholar]
- 103.Bakhtiari M, Shleizer-Burko S, Gymrek M, Bansal V & Bafna V Targeted genotyping of variable number tandem repeats with adVNTR. Genome Res 28, 1709–1719 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fang L et al. Haplotyping SNPs for allele-specific gene editing of the expanded huntingtin allele using long-read sequencing. HGG Adv 4, 100146 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mitsuhashi S et al. Tandem-genotypes: robust detection of tandem repeat expansions from long DNA reads. Genome Biol 20, 58 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chiu R, Rajan-Babu IS, Friedman JM & Birol I Straglr: discovering and genotyping tandem repeat expansions using whole genome long-read sequences. Genome Biol 22, 224 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Guo R et al. RepLong: de novo repeat identification using long read sequencing data. Bioinformatics 34, 1099–1107 (2018). [DOI] [PubMed] [Google Scholar]
- 108.Giesselmann P et al. Analysis of short tandem repeat expansions and their methylation state with nanopore sequencing. Nature biotechnology 37, 1478–1481 (2019). [DOI] [PubMed] [Google Scholar]
- 109.De Roeck A et al. NanoSatellite: accurate characterization of expanded tandem repeat length and sequence through whole genome long-read sequencing on PromethION. Genome Biology 20, 239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fang L et al. DeepRepeat: direct quantification of short tandem repeats on signal data from nanopore sequencing. Genome Biol 23, 108 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rheinbay E et al. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature 578, 102–111 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Campbell PJ et al. Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sakamoto Y, Zaha S, Suzuki Y, Seki M & Suzuki A Application of long-read sequencing to the detection of structural variants in human cancer genomes. Computational and Structural Biotechnology Journal 19, 4207–4216 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Euskirchen P et al. Same-day genomic and epigenomic diagnosis of brain tumors using real-time nanopore sequencing. Acta Neuropathologica 134, 691–703 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shiraishi Y et al. Precise characterization of somatic structural variations and mobile element insertions from paired long-read sequencing data with nanomonsv. bioRxiv, 2020.2007.2022.214262 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Valle-Inclan JE et al. Optimizing Nanopore sequencing-based detection of structural variants enables individualized circulating tumor DNA-based disease monitoring in cancer patients. Genome Medicine 13, 86 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fujimoto A et al. Whole-genome sequencing with long reads reveals complex structure and origin of structural variation in human genetic variations and somatic mutations in cancer. Genome Medicine 13, 65 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Beyter D et al. Long-read sequencing of 3,622 Icelanders provides insight into the role of structural variants in human diseases and other traits. Nature Genetics 53, 779–786 (2021). [DOI] [PubMed] [Google Scholar]; A pioneering study on SV genotyping and merging of large-scale SV call sets from long-read dataset of a large cohort of Icelandic population.
- 119.Belyeu JR et al. Samplot: a platform for structural variant visual validation and automated filtering. Genome Biol 22, 161 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Spies N, Zook JM, Salit M & Sidow A svviz: a read viewer for validating structural variants. Bioinformatics 31, 3994–3996 (2015). [DOI] [PubMed] [Google Scholar]
- 121.Lecompte L, Peterlongo P, Lavenier D & Lemaitre C SVJedi: genotyping structural variations with long reads. Bioinformatics 36, 4568–4575 (2020). [DOI] [PubMed] [Google Scholar]
- 122.Zhao X, Weber AM & Mills RE A recurrence-based approach for validating structural variation using long-read sequencing technology. GigaScience 6, 1–9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yang J & Chaisson MJP TT-Mars: structural variants assessment based on haplotype-resolved assemblies. Genome Biol 23, 110 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Duan X, Pan M & Fan S Comprehensive evaluation of structural variant genotyping methods based on long-read sequencing data. BMC Genomics 23, 324 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Robinson JT et al. Integrative genomics viewer. Nat Biotechnol 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ahdesmaki MJ et al. Prioritisation of structural variant calls in cancer genomes. PeerJ 5, e3166 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nattestad M, Aboukhalil R, Chin CS & Schatz MC Ribbon: intuitive visualization for complex genomic variation. Bioinformatics 37, 413–415 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.English AC, Menon VK, Gibbs RA, Metcalf GA & Sedlazeck FJ Truvari: refined structural variant comparison preserves allelic diversity. Genome Biology 23, 271 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sentieon (2022).
- 130.International Human Genome Sequencing, C. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004). [DOI] [PubMed] [Google Scholar]
- 131.Lander ES et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001). [DOI] [PubMed] [Google Scholar]
- 132.Schneider VA et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res 27, 849–864 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sherman RM & Salzberg SL Pan-genomics in the human genome era. Nature Reviews Genetics 21, 243–254 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Eichler EE, Clark RA & She X An assessment of the sequence gaps: unfinished business in a finished human genome. Nat Rev Genet 5, 345–354 (2004). [DOI] [PubMed] [Google Scholar]
- 135.Huddleston J et al. Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res 27, 677–685 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vollger MR et al. Segmental duplications and their variation in a complete human genome. Science (New York, N.Y.) 376, eabj6965 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Miga KH & Wang T The Need for a Human Pangenome Reference Sequence. Annu Rev Genomics Hum Genet 22, 81–102 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wang T et al. The Human Pangenome Project: a global resource to map genomic diversity. Nature 604, 437–446 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes the development of human pangenome reference from haplotype resolved assemblies to accurately represent human genomic diversity by facilitating SV discovery.
- 139.Collins RL et al. A structural variation reference for medical and population genetics. Nature 581, 444–451 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Byrska-Bishop M et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 185, 3426–3440.e3419 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Li H et al. A synthetic-diploid benchmark for accurate variant-calling evaluation. Nat Methods 15, 595–597 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cao S, Jiang T, Liu Y, Liu S & Wang Y Re-genotyping structural variants through an accurate force-calling method. bioRxiv (2022). [Google Scholar]