

Abstract
Directed evolution facilitates enzyme engineering via iterative rounds of mutagenesis. Despite the wide applications of high-throughput screening, building “smart libraries” to effectively identify beneficial variants remains a major challenge in the community. Here, we developed a new computational directed evolution protocol based on EnzyHTP, a software we have previously reported to automate enzyme modeling. To enhance the throughput efficiency, we implemented an adaptive resource allocation strategy that dynamically allocates different types of computing resources (e.g., GPU/CPU) based on the specific need of an enzyme modeling sub-task in the workflow. We implemented the strategy as a Python library and tested the library using fluoroacetate dehalogenase as a model enzyme. The results show that comparing to fixed resource allocation where both CPU and GPU are on-call for use during the entire workflow, applying adaptive resource allocation can save 87% CPU hours and 14% GPU hours. Furthermore, we constructed a computational directed evolution protocol under the framework of adaptive resource allocation. The workflow was tested against two rounds of mutational screening in the directed evolution experiments of Kemp eliminase (KE07) with a total of 184 mutants. Using folding stability and electrostatic stabilization energy as computational readout, we identified all four experimentally-observed target variants. Enabled by the workflow, the entire computation task (i.e., 18.4 μs MD and 18,400 QM single point calculations) completes in three days of wall clock time using ~30 GPUs and ~1000 CPUs.
Graphical Abstract
1. Introduction
Directed evolution has been widely employed to expand and enhance enzymes’ catalytic capability via iterative rounds of screening and selecting of enzyme variants.1, 2 Despite significant achievements in enzyme engineering, directed evolution faces big challenges in screening because of the enormously large combinatorial mutational space. Specifically, due to unknown sequence-function relationship, each round of screening may take substantial cost of time and resource. The outcome from the screening is also uncertain. Although high-throughput screening techniques have been used to facilitate directed evolution,3 designing readout for the function of enzyme mutants is highly case dependent. To accelerate the process of mutational screening, computational strategies have been employed to mechanistically infer beneficial mutations,4–8 identify hotspots,9–11 reveal structure/sequence-function relationships12–15, and build data-driven models for predictive biocatalysis.16 These foundational efforts significantly broaden the scope of which functional enzyme variants can be understood, predicted and designed, but how to build a general-purpose virtual screening platform for biocatalyst discovery remains an open question.
Previously, our lab developed EnzyHTP17 as a high-throughput enzyme modeling tool to automate the entire life cycle of enzyme modeling, including structural preparation, mutagenesis, molecular dynamic (MD), and quantum mechanics (QM) calculations. Enabled by EnzyHTP, here we further developed a computational directed evolution protocol that enables hundreds of mutants to be virtually screened based on their stability and capability of stabilizing the breaking bond in the rate-limiting transition state. Notably, prior to our work, the Kamerlin lab has pioneered the development of software, CADEE,18 for semi-automatic screening of enzyme variants. CADEE specializes in free energy calculations using empirical valence-bond (EVB), which is different from the current work that emphasizes the construction of a generic computational enzyme engineering platform.
Central to the new development and implementation is an adaptive resource allocation strategy that accelerates the high-throughput computation via dynamically requesting and distributing computing nodes for each sub-task in the high-throughput modeling workflow. Here, resource allocation refers to the process of managing different types of computing resources (e.g., CPU or GPU) for different tasks (e.g., QM or MD simulations). In high-performance computing (HPC) clusters, large amount of computing resources can be wasted if a fixed number of CPU and GPU nodes are requested based on the resource need of the most computationally demanding sub-tasks. Besides resource waste, the fixed allocation scheme can also significantly delay the completion of the entire workflow.
The strategy of adaptive resource allocation was implemented as a Python application programming interface, known as adaptive resource manager (ARMer). Based on EnzyHTP,17 we benchmarked the resource and time consumption of ARMer against fixed resource allocation scheme in the task of modeling fluoroacetate dehalogenase (FAcD)19–23. Furthermore, we constructed a computational directed evolution protocol to help screen for rate-enhancing enzyme mutants. We tested the workflow for two rounds of directed evolution screening of Kemp eliminase variants. Enabled by adaptive resource allocation, the computational directed evolution protocol offers a tool for accelerating the process of screening and selecting beneficial enzyme variants for biocatalytic uses.
2. Design and Implementation
2a. The computational directed evolution protocol.
We constructed the computational directed evolution protocol using modular Python functions in EnzyHTP (Figure 1, also see scripts Supporting Information.zip). The protocol was designed to consist of four steps. First, the workflow converts a list of user-provided starting variants into structural models using the structural preparation module. The file format of these structural models is compatible with the downstream molecular simulations. Second, for each starting variant, the workflow randomly generates a certain number of mutants (defined by the user) using the mutation engine to form the mutant library for the subsequent computational screening. Third, the workflow computes the thermostability score for all mutants in a high-throughput fashion using cartesian_ddg in Rosetta.24, 25 This interface has recently been added in EnzyHTP (see details in the Supporting Information, Text S5). The workflow filters out 80% of the mutants that are predicted to be thermally unstable relative to the wild type (i.e., positive thermostability score) in this step. Fourth, the workflow calculates electrostatic stabilization energy, Gelec,12, 26, 27 for mutants that passed the thermostability filter via performing one set of 100 ns MD simulation in AMBER28 (detailed in Text S7, Supporting Information) and QM single-point calculations (i.e., PBE1PBE/def2-SVP) in Gaussian 1629 using 100 snapshots evenly extracted from the MD trajectory. Specifically, the Gelec is derived from the dot product between the reacting bond dipole (by wavefunction analysis30) and the electric field strength of the enzyme (calculate by Coulomb’s law with MM charges, See the Supporting Information, Text S8 for details). Finally, the mutants are ranked based on the values of activity index (i.e., Gelec). Notably, completing the four steps concludes one round of computational directed evolution. A certain number of mutants (defined by the user) can be used as starting variants to initiate a new round of directed evolution. Notably, to systematically test the computational throughput capability of the protocol, we modified the fourth step by conducting the MD and QM simulations for all mutants including those that are determined to be thermally unstable in the third step. In practice, to achieve resource efficiency, we recommend users conduct these molecular simulations solely on stable mutants.
Figure 1.
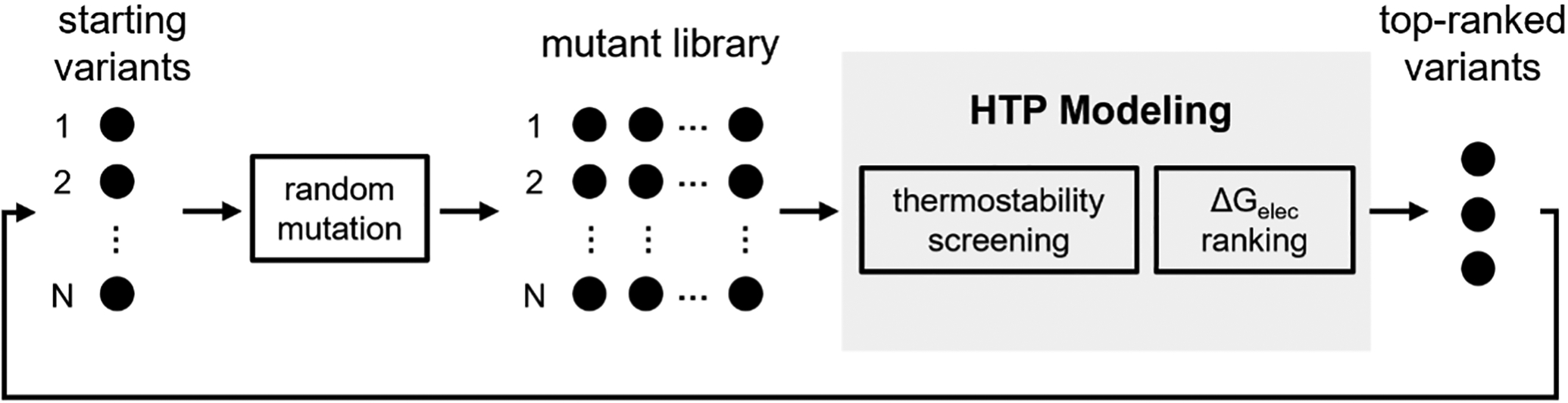
The design architecture of computational directed evolution protocol. Each black dot represents an enzyme variant.
2b. Adaptive resource manager (ARMer).
To properly manage resource allocations for computational screening tasks, the adaptive resource allocation strategy employs a “workflow script” that runs a single-CPU thread to manage sub-tasks for the entire high-throughput workflow. Using commands implemented in the ARMer Python library, the workflow script configures, submits, and monitors new jobs in HPC clusters that pertain to the actual need of computing resources in a sub-task of the workflow. This is in sharp contrast to the fixed resource allocation scheme where maximal computing resources are requested.
The ARMer Python library consists of two classes: the Job class and HPC class (Supporting Information, Figure S1). The Job class (called ClusterJob in the code) defines properties and functions that are associated with job configuration, submission, and dynamic monitoring of job completion (Figure 2). The HPC class (subclasses of ClusterInterface in the code) supports the Job class with properties and functions to mediate shell input/output in user’s local HPC where ARMer is deployed. This “plug-in” interface design allows easy support for any new HPC. Similar to subprocess.run in the Python standard library, ARMer library enables the “workflow script” to run shell commands on other computational nodes – these commands are wrapped in the job scripts in the HPC clusters (see more discussion in the Supporting Information, Figure S1 and Text S1).
Figure 2.
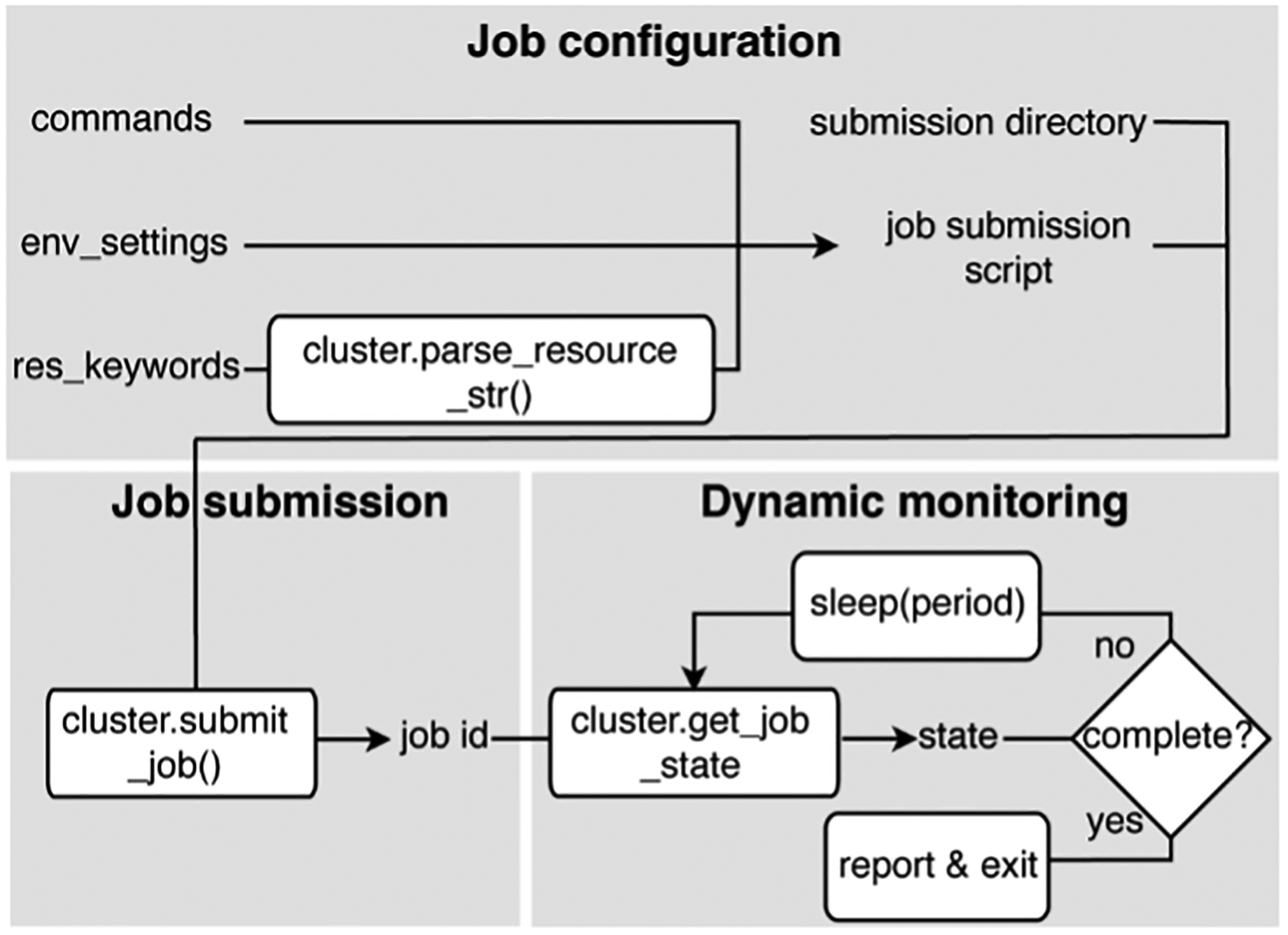
Variables and functions for job configuration, submission, and dynamic monitoring defined in the Job class.
With the job object instantiated, a job script for the required task can be generated (Supporting Information, Figure S3) and then submitted by the submit() method (Figure 2 and Supporting Information, Figure S2). Notably, the format of the job script, the submission commands, and other HPC-dependent information are obtained from the HPC class object that is instantiated and passed to the cluster argument described above. Once the job has been submitted, a job ID is added to the object by the function. By tracing the job ID, the “workflow script” can monitor the status of a job object in the queue, and mediate the status by killing, holding, or releasing the job (Figure 2). Notably, the capability of dynamically monitoring the job completion status is vital to high-throughput modeling workflow. This is because the workflow involves multiple different types of simulation sub-tasks that must be sequentially operated.
Two methods have been implemented to achieve dynamic monitoring – they are: wait_to_end()and wait_to_array_end() method. The wait_to_end() method checks the status of a job in the job queue with a certain period of time (i.e., every 30 seconds) and exits upon the detection of messages that indicate job completion, error, or cancellation. The wait_to_array_end() method takes multiple job objects and submits them in one job array. Similarly, the method monitors the status of all jobs in the array regularly, and dynamically append new jobs to the array up to the maximal capacity (i.e., array size).
Implementation of ARMer in EnzyHTP enables an if_cluster_job option for all computationally insensitive functions (like MD, QM and free energy calculation). With this option turned on, the computational directed evolution protocol described in the Section 2a can achieve adaptive resource allocation.
3. Results and Discussion
3a. Test of adaptive resource allocation using fluoroacetate dehalogenase.
We employed fluoroacetate dehalogenase (FAcD)19–23 as a model system and conducted a high-throughput workflow of enzyme simulations using EnzyHTP.17 The workflow consists of four sequential sub-tasks, namely, 1) mutant structure construction, 2) 100 ns MD simulation, 3) 100 QM cluster calculation, and 4) post-analysis. (See details in Supporting Information Text S2 Notably, the model system and workflow have been applied in our previous work to test the throughput capability of EnzyHTP.17 However, unlike the previous workflow that samples 100 variants with minimalist resource cost, the current workflow only tests one enzyme variant (i.e, K83D, Supporting Information, Figure S4) with MD and QM simulations set up to meet resource demand in actual computational research (Supporting Information.zip).
We benchmarked the resource and time consumption of the workflow for two strategies: fixed resource allocation versus adaptive resource allocation, on our local HPC at Vanderbilt, i.e., advanced computing center for research and education (ACCRE). Using fixed resource allocation, all computing resources (i.e., 1 GPU and 8 CPU) involved in the workflow are requested by a single submission script in one shot. Figure 3 shows the simulation type, time cost (i.e., vertical axis), and resource demand (i.e., GPU in orange and CPU in blue) associated with each sub-task in the workflow. Both CPU and GPU are requested for the entire workflow (Figure 3). However, resource waste is observed. Specifically, in the ~14 hours of MD simulation, only one GPU is used but the CPUs are primarily in idle mode (time-waste: ~8×14 = ~112 CPU hours); in the 3 hours of QM calculations, CPUs are used but the GPU is not (time-waste: 3 GPU hours). In the minimization and post-analysis sub-tasks, CPU and GPU are also not exploited, albeit the resource cost is trivial. Apparently, with fixed resource allocation, the collective resource waste is significant.
Figure 3.
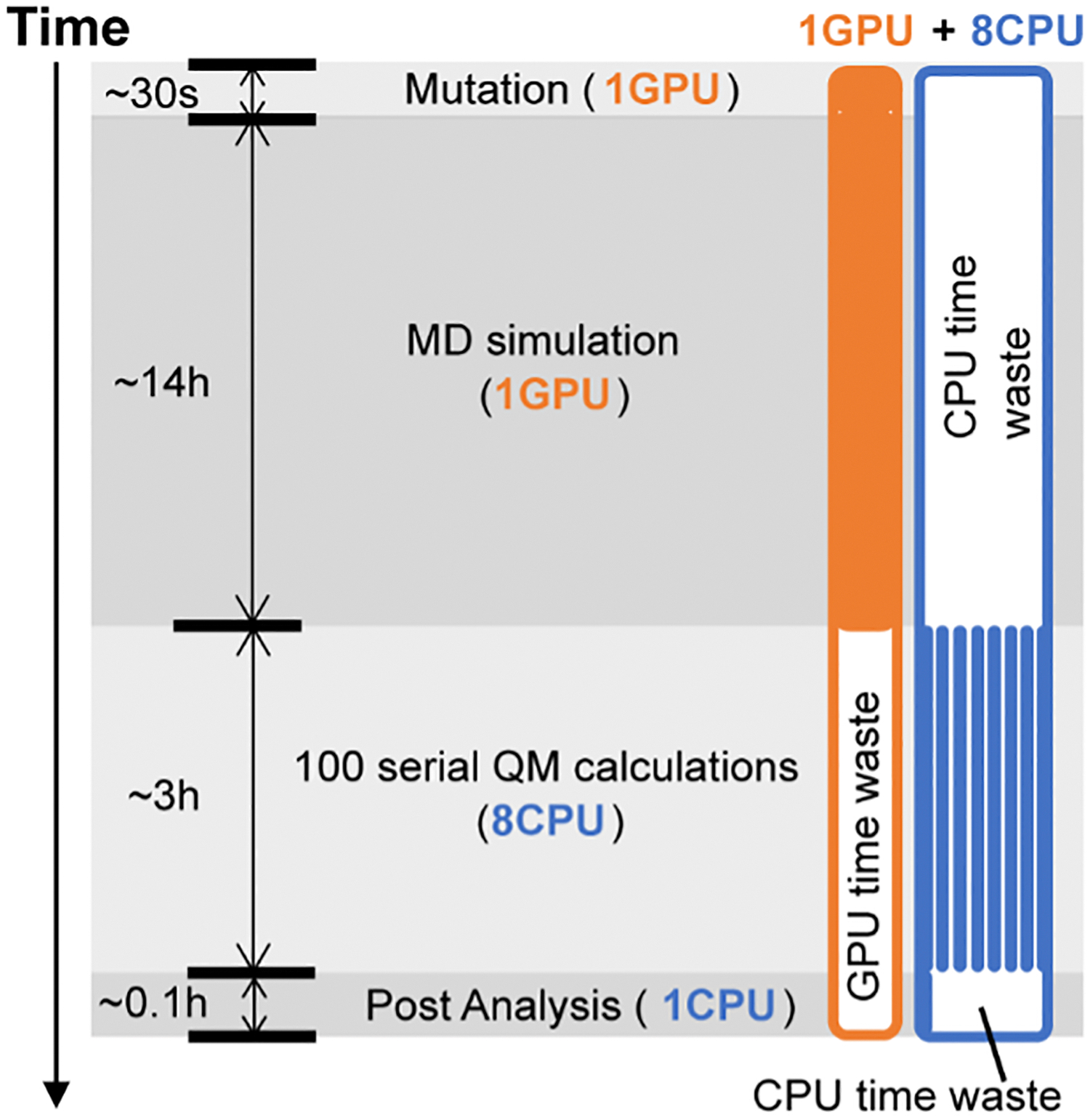
The high-throughput workflow of FAcD modeling using fixed resource allocation strategy, in which 1 GPU (in orange) and 8 CPUs (in blue) are requested in the beginning of the workflow. The type of modeling sub-tasks, time cost, and resource consumption are noted on the Figure.
Using adaptive resource allocation, a 96-hour wall-clock time, single CPU job is submitted that operates a Python “workflow script” to allocate resources for sequential sub-tasks involved in the workflow (Figure 4). The “workflow script” manages the sub-tasks using commands implemented in the ARMer library (detailed in the Design and Implementation section). Compared to fixed allocation strategy that directly executes sub-tasks using the allocated CPU or GPU, this workflow script configures resource-demanding sub-tasks (i.e., need >1 CPU or ≥1 GPU) in a new job script and then submits the job to the queue (i.e., setting if_cluster_job = ‘True’ in the code).
Figure 4.
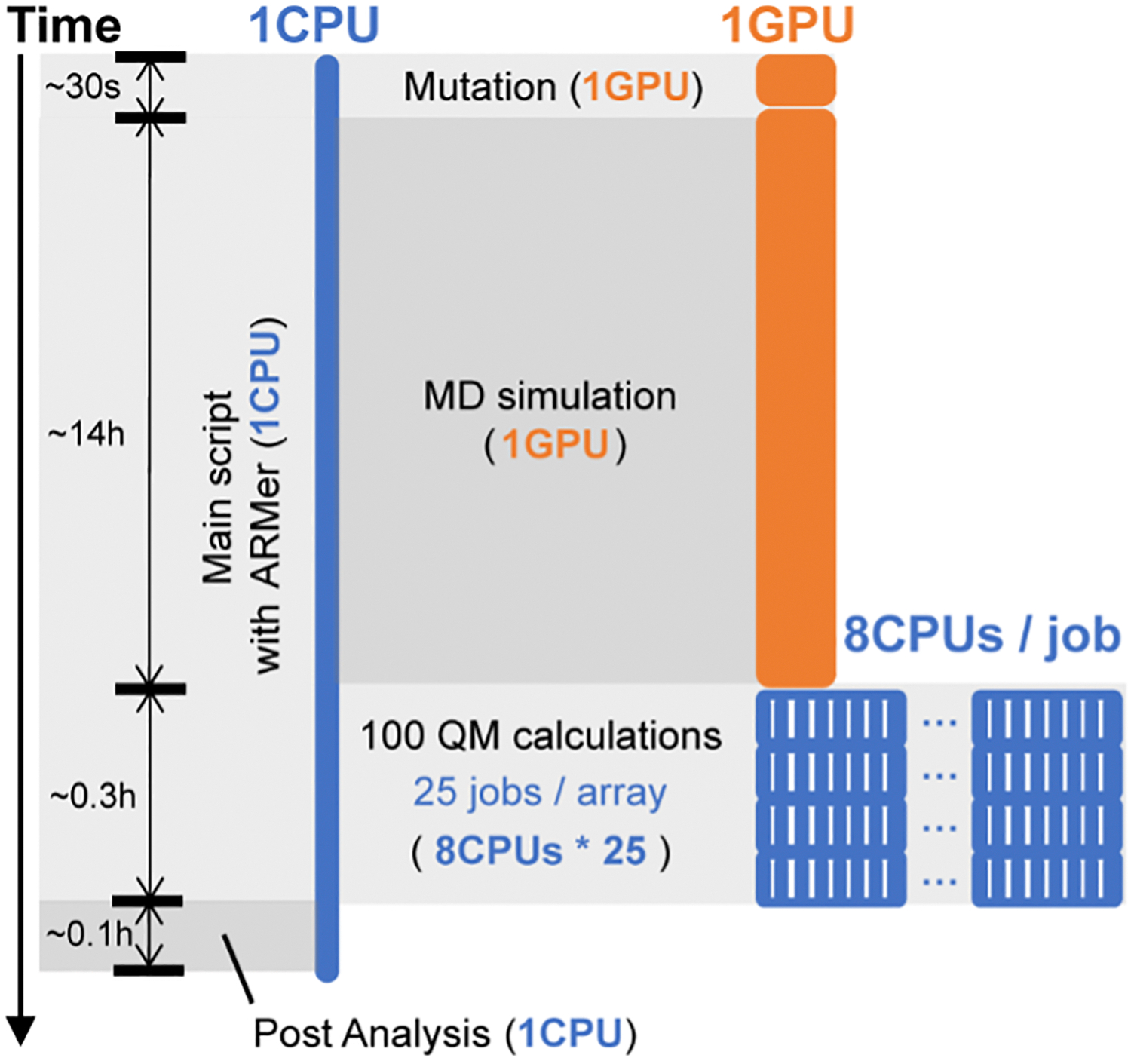
The high-throughput workflow of FAcD modeling using adaptive resource allocation strategy, in which a “workflow script” the runs on a single-CPU thread operates the modeling sub-tasks (i.e., mutation, MD, and QM) by configuring, submitting, and monitoring new job scripts. The MD job requests 1 GPU (in orange) and each QM job 8 CPUs (in blue). To submit and run individual QM calculations in parallel, a job array with a size of 25 is employed. The type of modeling sub-tasks, time cost, and resource consumption are noted on the Figure.
In the MD simulation sub-task, the workflow script configures shell commands that run AMBER28 simulations in a job script along with the GPU request and environment settings. The workflow script then submits the MD job and regularly monitors the completion status of the job. After confirming the completion of MD, the workflow script will continue operating the QM calculation sub-task in the workflow. Since the 100 QM calculations are independent, the workflow script can submit multiple QM jobs (8 CPU each) simultaneously to the job array so that they can run in parallel up to the size limit of job array (i.e., 25 jobs) in local HPC cluster (Figure 4). New jobs will be submitted once the “workflow script” detects open slots on the array (see discussion of parallel computing using Python subprocess module, Supporting Information Text S3). With an array size of 25 jobs, one would expect an ideal time acceleration by a factor of 25 given the ideal condition of no job queueing time (i.e., ~2.4 hours as compared to 60 hours with all job running serially). Overall, with adaptive resource allocation, the resource waste can be minimized.
We compared the computational cost (Supporting Information, Figure S5) and wall clock time (Supporting Information, Figure S6) for different sub-tasks of the workflow using fixed resource allocation (in red) versus adaptive resource allocation (in green) on ACCRE. In contrast, fixed resource allocation involves 14% resource waste in GPU-based MD simulation and 87% in CPU-based QM calculations, while adaptive resource allocation makes full use of these computing resources in every sub-task of the workflow (Supporting Information, Figure S5). For wall clock time, both strategies differ most significantly in the QM calculation sub-task (Supporting Information, Figure S6). Enabled by parallel submission of individual QM jobs to a job array (i.e., 25 jobs per array), adaptive resource allocation strategy completes 100 QM calculations by 0.35 hours. With fixed resource allocation (i.e., 8 CPUs), the calculations are completed by 2.8 hours. With larger QM system size and higher theory level, the QM resource consumption will be more significant. As such, the waste of resource and time for fixed resource allocation is expected to inflate.
With new jobs generated, submitted, and monitored on the fly by adaptive resource allocation strategy, the resource waste observed by fixed resource allocation is no long expected because resources are requested and distributed based on the need of individual sub-task in the workflow. We should note that one potential caveat of adaptive resource allocation strategy is the time spent for queueing. In our test, the total amount of time spent for job queueing is about 12 minutes, which is relatively trivial. On extremely crowded HPC, we assume job queueing time to be much longer. The fixed resource allocation scheme suffers less from the queueing, but the overall cost of time and resource is substantial.
Besides being resource-efficient, adaptive resource allocation scheme also prevents conflict of software environment setting. Under the adaptive resource allocation scheme, the workflow script can customize specific environment setting based on the requirement of an individual job. In contrast, the fixed resource allocation scheme will have to load all environmental variables in the beginning of the submission job, which can cause conflict for different software as the workflow proceeds.
3b. Test of the computational directed evolution protocol.
We employed Kemp eliminase KE07 as the model enzyme (originally designed by Röthlisberger et al.31) to test the computational directed evolution protocol for its ability to predict experimentally-observed rate-enhancing KE07 mutants in two rounds of directed evolution experiments. KE07 was selected for three reasons. First, the crystal structure of KE07 has been determined (PDB code: 2RKX31), which provides a reliable structural template to generate structural models for mutants. Second, the lead variants associated with each round of KE07 screening in directed evolution have been reported.32 This provides reference data to examine the predictive capability of the computational protocol. Third, KE07 is known to involve a general acid-base mechanism – the substrate 5-nitrobenzisoxazole is deprotonated by a nearby carboxylate (side chain of Glu or Asp) to form 2-hydroxy-5-nitrobenzonitrile via one single transition state.31 The relatively simple mechanistic picture reduces the complexity of having to consider the variation of the rate-limiting step upon mutation. A detailed protocol for structural preparation of KE07-substrate complex is described in the Supporting Information, Text S6.
We selected two out of seven continuous rounds of directed evolution (i.e., round 5 and 6) that leads to the identification of KE0731. Both rounds use error-prone PCR for mutagenesis. Experimentally, round 5 starts from six KE07 variants from the previous round and conducts error-prone PCR with 1±1 mutations per mutant. Twelve active variants (only two were reported, Figure 5) from round 5 were filtered into round 6 for error-prone PCR with 3±1 mutations per mutant. This results in twenty active variants (only two were reported, Figure 5) that go into round 7.
Figure 5.
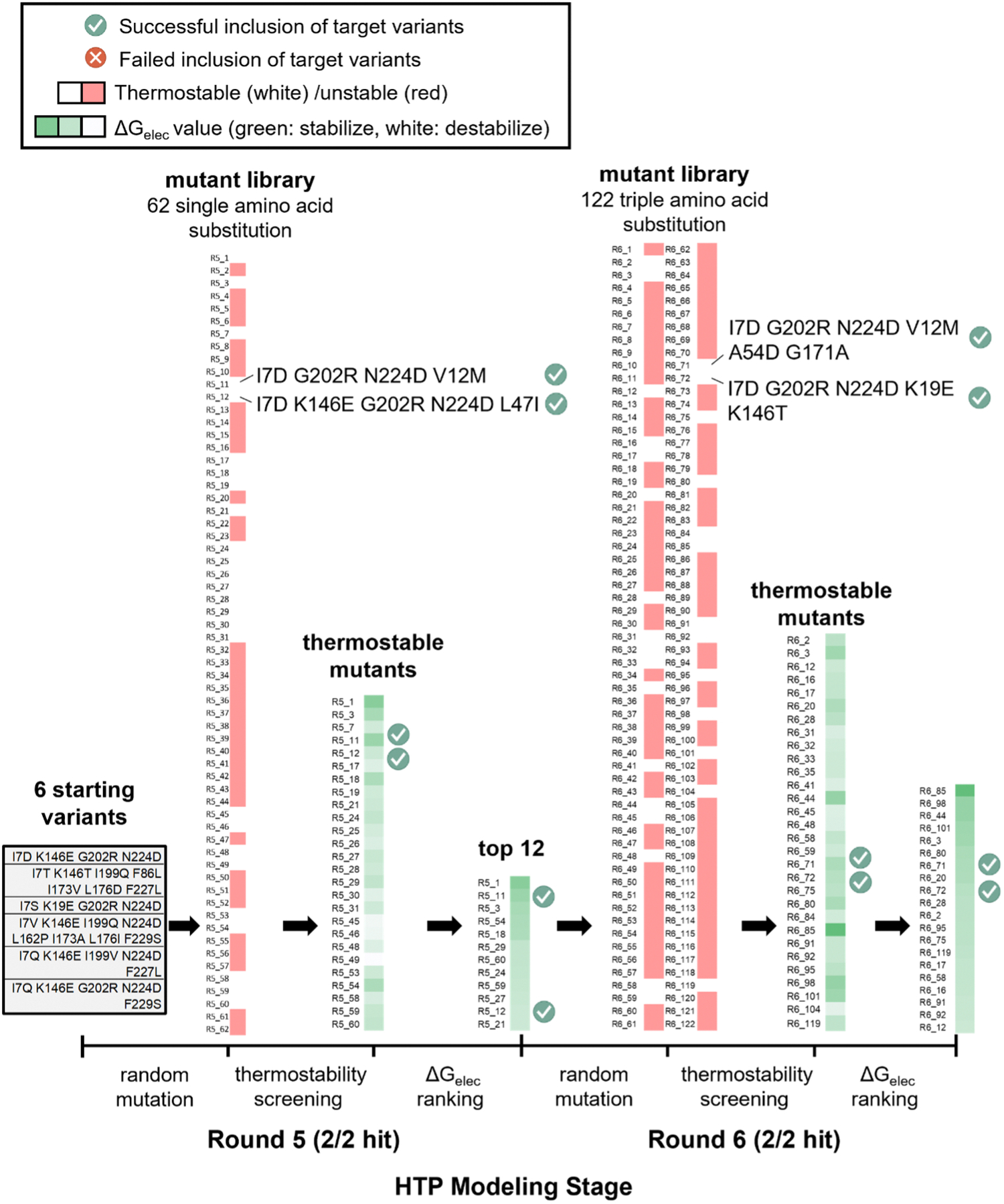
The computational protocol for two rounds of directed evolution. Round 5 and 6 out of the seven rounds of experimental directed evolution of Kemp eliminase are used in the test. Specific stages of the workflow are shown on the bottom axis from left to right. The results of each stage are listed above the axis. Specific information about each mutant is listed in the Supporting Information, Table S1. The modeling results of thermostability and electrostatic stabilization energy (i.e., Gelec) are represented as color-coded blocks next to the mutants. For the thermostability score, the thermally stable and unstable mutants are shown in white and red, respectively. For Gelec, a color gradient scheme from darker green to white is applied, where darker green refers to stronger electric field with greater capability of stabilizing the bond cleavage. The same color scale is applied for both R5 and R6. “Green check” or “red cross” represents the successful or failed inclusion of an experimentally-observed target mutant in a screening stage. The 6 starting variants are derived from the experimental work of Röthlisberger et al.31
Based on the experimental protocol, we set up a computational workflow of directed evolution, aiming to test whether we are able to distinguish the experimentally-observed target variants from a bunch of randomly-generated mutations through the in silico screening protocol (Figure 5). Specifically, for round 5, the workflow uses an identical set of variants as the experimental directed evolution protocol (i.e., six starting variants), builds a library of sixty mutants with single amino acid substitution through generating ten random mutants for each starting variant, computes thermostability score and electrostatic stabilization energy for each mutant by MD and QM simulations, and selects twelve top-ranked mutants with stable fold and strong electric field to promote deprotonation in the rate-limiting transition state (Figure 5). These twelve mutants (same number as the experimental setup) are used as starting variants to initiate the round 6. For round 6, random triple mutations are generated with ten mutants for each starting variant to form a library of 120 mutants. This round of screening results in twenty top-ranked mutants (same number as the experimental setup). For each round, we manually added the two experimentally observed target variants in the mutant library (i.e., 62 and 122 mutants in total for round 5 and 6, respectively) and test whether these two mutants can be filtered in as the top-ranked mutants based on the computational scoring of protein thermostability and electrostatic stabilization energy (i.e., Gelec). By statistics, the chance of observing the target variants among a group of randomly selected mutants is 3.5% for round 5, 2.5% for round 6, and 0.091% for both round 5 and round 6 (Supporting Information, Text S10), which presents a challenging test to the predictive capability of the computational directed evolution protocol.
Figure 5 shows the outcome of each screening stage of the computational directed evolution protocol. In the mutant library of round 5 (i.e., 62 mutants), the thermostability scores vary from −6.4 to 56.3 Rosetta energy unit (REU) with a standard deviation of 17.3 REU; 16 mutants are predicted to be more stable than the wild-type (i.e., negative thermostability score) and 46 less stable (i.e., positive thermostability score). For screening, we defined the “stable” mutants (labeled in white, Figure 5) to consist of all mutants that are more stable than the KE07 wild-type (i.e., 16 mutants) plus 20% of the top-ranked mutants with a positive thermostability score (i.e., 10 mutants). We allowed for a slight magnitude of stability dropping because the loss of stability might be a potential trade-off for catalytic enhancement. Notably, the empirical cutoff value used here was defined prior to the computation and used consistently for both round 5 and round 6. Among the 26 stable mutants, the Gelec values vary from 0.5 to 3.1 kcal/mol with a standard deviation of 0.6 kcal/mol; the 12 mutants with top-ranked Gelec values were sent for screening in round 6. Throughout round 5, both experimentally-observed target mutants are found in the final list.
In the mutant library of round 6 (i.e., 122 mutants), the thermostability scores vary from −12.5 REU to 68.3 REU with a standard deviation of 13.9 REU. Using the same screening strategy as described for round 5, the workflow identifies 34 stable mutants. Among the stable mutants, Gelec values vary from −0.4 kcal/mol to 2.5 kcal/mol with a standard deviation of 0.6 kcal/mol. Based on the ranking of Gelec values, we selected the top 20 mutants, in which both target mutants are included.
Using the computational directed evolution protocol, all four experimentally observed beneficial mutations were identified two continuous rounds of mutagenesis. The hit rate is significantly higher than that derived from random sampling. (The chance is 0.091%) Accelerated by ARMer, the entire workflow finishes in 3 days with ~30 GPUs and ~1,000 CPUs (See hardware specifications in Supporting Information, Text S9). This corresponds to a total of 18.4 μs MD and 18,400 QM single point calculations, which includes Gelec calculation of all mutants. Under the same technical configuration, if MD and QM calculations are conducted only on thermostable mutants, the computational workflow is projected to complete in one single day.
3c. Limitation and future development of the computational directed evolution protocol.
The computational directed evolution protocol, which was demonstrated in the enzyme engineering of Kemp Eliminase KE07, offers a groundwork for in silico construction of physics-inspired smart libraries for protein engineering. Below, we discuss the limitation of this proof-of-concept protocol, along with future plans and ongoing efforts towards its refinement.
1. The mutation engine.
The current algorithm for mutant structure construction involves the integration of tLEaP and MM minimization as described in the EnzyHTP paper.17 This method relies on molecular dynamics equilibration to fix artifacts introduced in the model construction step and to sample different sidechain conformations (rotamers). In rare situations, the approach has been found to fail when the generated conformations involve unphysical interactions that cannot be resolved by molecular dynamics equilibration, such as a structure in which the lysine side chain penetrate through the aromatic ring of tryptophan.17 To develop a more robust mutation engine for the next generation of EnzyHTP, we are in the process of testing different strategies for mutant structure construction, including sidechain rotamer library,33, 34 Monte Carlo Markov Chain (MCMC) search,24, 25, 35 and deep learning-based methods.36, 37
2. The algorithm for constructing “smart” mutation library.
To mimic error-prone PCR, the current protocol employs the strategy of random single amino acid substitution to generate KE07 mutants. However, in real-life applications, the random mutagenesis algorithm is ineffective in identifying beneficial mutations due to the large mutational landscape for protein engineering. In addition, the high computational cost of classical modeling and quantum chemistry simulation impedes the ability of computational screening to match the scale of experimental ultra-throughput screening (i.e., >106 per week). As such, an urgent need is to develop new algorithms for constructing smart mutation libraries. Instead of searching for beneficial mutations on a broader sequence space, a more reasonable approach would be to use scientific hypotheses to guide the search of mutation hotspots that involve a higher likelihood for functional enhancement. The guiding hypotheses could be the optimization of electric field strength and distribution, the active site configuration and dynamics, the mediation of global protein flexibility, and so on. Furthermore, developing machine learning models that can dynamically formulate specific guiding hypotheses for enzyme engineering based on initial molecular simulation data is an active area of focus in our ongoing refinement of the computational directed evolution protocol.
3. The MD sampling scheme.
The current conformational sampling scheme adopts one set of 100 ns MD run for each mutant. The time length used here is longer than the MD simulations performed in the previous study of Kiss et al. (i.e., 20 ns).38 As such, we calculated the Gelec values of mutants using only the first 50 ns out of each 100 ns simulation (Supporting Information, Table S2). We tested the correlation of the rankings of Gelec values before and after the trajectory truncation. The Spearman correlation coefficient of both rankings is 0.89 and 0.89 for R5 and R6 mutants, respectively. This shows that conformational sampling with a shorter MD run works reasonably well in selecting beneficial mutants for the Kemp eliminase system. This also helps save substantial amount of GPU resources during the computational screening protocol. An ongoing implementation in our group involves the replacement of single 100 ns MD run by multiple replicas of shorter MD runs. This strategy has been shown to increase the efficiency of conformational sampling for computing functional descriptors as screening readouts.27, 39, 40 Nonetheless, the successful use of shorter MD sampling critically depends on the assumption that the system doesn’t undergo significant conformational changes. While this might be true for enzyme mutants with minimal structural differences from the wild type, it may not be the case for mutants that exhibit local misfolding or activate various conformational states.
4. The mutant structure and substrate-binding configurations.
In the current protocol, mutant structures are directly predicted from the reference structure. This process uses the mutation engine in EnzyHTP that relies on AMBER tLEaP to generate and optimize the predicted structure. The reference structure is obtained from the KE07 WT crystal structure (PDB code: 2RKX) with the substrate docked in the active site. To investigate the reliability of this approach, we predicted mutant structures for two KE mutants, I7D and N224D, and compared these structures with a mutant crystal structure that contains both mutations (PDB ID: 3IIO).32 Notably, both mutations have been reported to change local hydrogen bonding interactions in the active site.32 The goal is to investigate whether the hydrogen bonding patterns observed in the mutant crystal structure can be reflected in our protocol. We found that the predicted mutant structures of I7D and N224D using the mutation generation engine involve a different active site hydrogen bonding pattern from the crystal structure. Specifically, the predicted structure of I7D shows a hydrogen bonding interaction between K222 and E101, which is different from the mutant crystal structure where the hydrogen bond forms between K222 and D7. The predicted structure of N224D does not involve a hydrogen bond between D224 and H201 which presents in the mutant crystal structure (Supporting Information, Figure S7A). These results show that the mutation generation engine fails to predict the active site hydrogen bond network of a mutant when its pattern differs from that of the wild-type reference structure. However, for both I7D and N224D mutants, despite using the predicted mutant structure as a starting point, the MD equilibration reproduces the hydrogen bonding network configuration observed from the mutant crystal structure (Supporting Information, Figure S7B). This result shows that the mutation-equilibration approach has the capability of informing the impact of mutation on the active-site conformational states for the mutant structures. The change the H-bonding network in the active site caused by I7D and N224D mutations are reproduced through the MD equilibration.
A complex scenario in mutant structure involves the emergence of multiple active site configurations that bind to the substrate differently. Hong et al. discovered that some mutations of KE07 introduced in rounds 5 and 6 create two more substrate binding configurations, namely configuration B and C (with respect to the native state, configuration A). Configurations B and C are believed to contribute to the enhancement of the catalytic efficiency.41 These configurations mainly differ by the conformations of W50 sidechain that is in pi-stacking with the substrate (Supporting Information, Figure S8A). In this work, all generated mutant structures bind to the substrate only in the form of configuration A. To investigate whether configurations B and C can be sampled through 100 ns MD, we collected representative snapshots along the MD trajectories for four experimentally observed mutants and all top-ranking mutants identified in the protocol, and then performed clustering analysis using the RMSD values of the active site residues, including E101, W50, Y128, and the substrate (the snapshots can be found under representative_structures/, Supporting Information.zip). The results show that only configuration A is sampled in the MD trajectories, and configurations B and C are not observed (Supporting Information, Figure S8B and Figure S9). This is likely caused by the fact that the distance between the substrate and E101 is constrained in the MD simulation, making it energetically difficult to flip the W50 sidechain and sample alternative configurations. This highlights the importance of considering alternative substrate binding configurations in the design protocol. An ongoing development in our lab is to implement a reactive conformation docking algorithm in EnzyHTP, which is expected to enable high-throughput docking between different mutant conformers and the substrate.
5. The scoring function.
The current protocol only employs Gelec as the score function for reactivity assessment. The Gelec value of a mutant reflects the stabilization of enzyme interior electric field on a reacting dipole, which presumably accounts for the contribution of an enzyme mutant in decreasing the active energy or enthalpy of the chemical reaction (See details in Supporting Information, Text S8). However, the value does not inform the impact of mutation on activation entropy – a major factor that affects the catalytic efficiency. For example, in the KE07 system, R6_72 involves a weaker electrostatic stabilization effect than R5_11 (i.e., a higher Gelec value by 0.65 kcal/mol), which is qualitatively consistent with the experimental observation that R6_72 has a higher activation energy by about 2 kcal/mol.41 However, R5_11 was observed to involve a greater pre-exponential factor than R6_72. The favorable activation entropy in R5_11 overrides the enthalpic difference and leads to a lower free activation barrier than R6_72. This example highlights the importance of accounting for entropic effects in reactivity assessment. An ongoing effort in our lab aims to design a scoring function that reflects the impact of mutation on activation entropy. We believe that incorporating the entropic scoring in the protocol will further improve the computational accuracy and reliability.
6. The reaction mechanism.
The model enzyme, Kemp eliminase, has a well-established mechanism that involves one single rate-limiting step, i.e., substrate C–H deprotonation. As such, the directed evolution workflow tested here only focused on the engineering of enzyme interior electric field to stabilize the deprotonation step through mutagenesis. In real-life applications, a mutation may shift the rate-limiting step or even change the reaction mechanism entirely. For example, hydrolases typically involve a competition between the nucleophilic step and the covalent intermediate dissociation step.42 De novo designed Diels–Alderases involve the dilemma between transition state stabilization versus product inhibition, same as the artificial enzymes derived from catalytic antibody.43 Consequently, the future development of computational directed evolution workflow needs to simultaneously consider the impact of mutation on multiple rate-limiting steps. The key challenge is how to build a computationally efficient score function that evaluates activation barriers of multiple elementary steps. Electronic structure calculations provide a promising strategy because of its capability of locating transition states and predicting free energy barriers, but these methods fall short in computational efficiency when used for virtual screening. In addition, the selection of QM theories also introduce uncertainty in the prediction. To address the challenge, new scoring functions should be designed that augment first principle modeling with machine learning models trained from experimental kinetics data.
7. The generality of the protocol.
Though only demonstrated for the mutational screening of KE07, the computational directed evolution protocol is generalizable to other enzyme targets, because the mutation generation engine, molecular modeling engine, and functional assessment module are independent from the specific application. Compared to experimental screening that needs to associate enzyme activity to cell growth, color change, or spectroscopic readout, the computational protocol is facile to adapt. However, we should note that the scoring functions used for functional assessment might be subject to change for different enzymes. Specifically, although electrostatic stabilization energy used for KE07 is a general descriptor correlated with enzyme activity, locating the specific reacting bond dipole, along which Gelec value should be computed, can become a non-trivial question for reactions involving rearrangement of multiple bonds. In addition, entropic effects can substantially influence substrate binding and chemical activation. For engineering industrial enzymes (e.g., amylases, lipases, etc.), one also needs to implement descriptors that represent the impact of mutations on optimal catalytic temperature and solubility in nonaqueous solutions. Consequently, developing a comprehensive set of scoring functions that predict desired enzyme properties in silico, is pivotal to advancing towards a more automated computational enzyme engineering process.
4. Conclusion
We designed a computational directed evolution protocol based on EnzyHTP. Through iterative rounds of screening, the workflow can generate random mutations and identify mutants with enhanced catalytic functions. To accelerate the throughput capability, we developed a Python library, ARMer, to adaptively allocate resources for computational sub-tasks in a high-throughput molecular modeling workflow in HPC clusters. The ARMer Python library consists of two classes: the Job class that defines variables and functions for job configuration, submission, and monitoring; the HPC class that supports the Job class to mediate external input/output in HPC clusters and users can easily customize a subclass for their local HPC clusters (Supporting Information, Text S1). Using commands implemented in ARMer, a “workflow script” can run on a single CPU thread to generate, submit, and monitor new jobs that call external software to execute sub-tasks.
To test the efficiency, we employed ARMer to manage the sub-tasks in the enzyme modeling workflow for FAcD using EnzyHTP. The sub-tasks include enzyme mutant structure construction, 100 ns MD simulation, 100 QM calculations, and electronic structure analysis. We compared the consumption of time and resource between two allocation strategies: fixed resource allocation versus adaptive resource allocation. Fixed resource allocation involves significant resource waste: in the 14 hours of MD simulation, only one GPU is used but the CPUs are primarily in idle mode (time-waste: 8x14 = 112 CPU hours); in the 3 hours of QM calculations, CPUs are used but the GPU is not (time-waste: 3 GPU hours). In contrast, the adaptive resource allocation makes full use of both computing resources in the corresponding sub-tasks of the workflow. Moreover, ARMer allows parallel submission of multiple smaller computational jobs in a job array and provides customized environment settings for each software used in the workflow.
Enabled by ARMer and EnzyHTP, we constructed the computational directed evolution protocol and tested it against two continuous rounds of experimental directed evolution for Kemp eliminase, KE07. Using the workflow, we successfully reproduced all four experimentally observed target mutants in 3 days with a total of 18.4 μs MD and 18,400 QM single point calculations by ~30 GPUs and ~1000 CPUs. Under the same technical configuration, the computational workflow is estimated to complete in one single day if molecular simulations are conducted only on thermostable mutants. This speed allows the in-silico screening of >1000 mutants in a week. Compared to experimental ultra-throughput screening (e.g., >106 mutants per week), the computational workflow has a special advantage of informing molecular details of enzyme dynamics and electronic structures, guiding the development of scientific hypotheses or data-driven models for optimization of mutants. In addition, the functional readout is also much easier to set up in a computational workflow for reactions whose outcomes cannot be detected by colorimetry and fluorimetry or associated with cell growth. Therefore, the computational directed evolution protocol holds promise to accelerate the process of enzyme engineering.
Supplementary Material
ACKNOWLEDGMENT
This research was supported by the startup grant from Vanderbilt University and the fellowship of Vanderbilt Institute of Chemical Biology. The PI thanks the sponsorship from Rosetta Commons Seed Grant Award. This work was also supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM146982. This work used SDSC Dell Cluster with AMD Rome HDR IB at Expanse from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program, which is supported by National Science Foundation grants BIO200057.44
Footnotes
Supporting Information. Architecture of the ARMer Python library; Details about Architecture of the ARMer Python library; Example code of ARMer API.; A sample job script that performs QM calculation using Gaussian 16; Structure of FAcD K83D mutant and the active site cluster; The comparison of computing resource cost; The comparison of wall-clock time cost; The comparison between modeled sidechain conformation and the experimentally determined sidechain conformation upon the mutation of I7D and N224D; The comparison between modeled substrate binding configurations and experimentally determined binding configurations; More structures of modeled substrate binding configurations; Details of the benchmark workflow; Choice of parallelization strategy; Discrepancy of expected and actual speed up from ARMer parallelization; Details of the thermostability score calculation using cartesian_ddg; Details of structure preparation and parameterization; Details of the molecular dynamics simulation; Geometric restraints applied in the MD simulation; Details of the electrostatic stabilization energy calculation; Specifications of hardware used in this work; Statistical analysis for the chance of randomly identifying the two target mutants in the top-ranked mutants; List of mutants in the computational directed evolution workflow; List of mutants with Gelec calculated based on the first 50ns of the MD (PDF)
Input files for the computational directed evolution test: input structure; workflow python script; job submission script of the workflow script; Configuration files of the MD simulation; Representative structures of MD trajectories of all top-ranking mutants and experimentally-observed mutants (ZIP).
The authors declare no competing financial interest.
Data and Software Availability.
The code and sample input for ARMer is publically available at https://github.com/ChemBioHTP/ARMer. The input files and structures are provided as part of the SI files. EnzyHTP is available from https://github.com/ChemBioHTP/EnzyHTP/tree/develop. Workflow scripts for the computational directed evolution protocol are available from https://github.com/ChemBioHTP/EnzyHTP/blob/develop/Test_file/KE_accre/R6/KE-metrics_complete.py. AMBER 18 is available from http://ambermd.org/. Gaussian 16 is available from https://gaussian.com/.
References
- 1.Arnold FH; Volkov AA, Directed evolution of biocatalysts. Curr. Opin. Chem. Biol 1999, 3, 54–59. [DOI] [PubMed] [Google Scholar]
- 2.Wang Y; Xue P; Cao M; Yu T; Lane ST; Zhao H, Directed Evolution: Methodologies and Applications. Chem. Rev 2021. [DOI] [PubMed] [Google Scholar]
- 3.Jones KA; Snodgrass HM; Belsare K; Dickinson BC; Lewis JC, Phage-Assisted Continuous Evolution and Selection of Enzymes for Chemical Synthesis. ACS Central Science 2021, 7, 1581–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tishkov VI; Pometun AA; Stepashkina AV; Fedorchuk VV; Zarubina SA; Kargov IS; Atroshenko DL; Parshin PD; Shelomov MD; Kovalevski RP; Boiko KM; Eldarov MA; D’Oronzo E; Facheris S; Secundo F; Savin SS, Rational Design of Practically Important Enzymes. Moscow Univ. Chem. Bull. (Engl. Transl.) 2018, 73, 1–6. [Google Scholar]
- 5.Gao L; Zou Y; Liu X; Yang J; Du X; Wang J; Yu X; Fan J; Jiang M; Li Y; Houk KN; Lei X, Enzymatic control of endo- and exo-stereoselective Diels–Alder reactions with broad substrate scope. Nature Catalysis 2021, 4, 1059–1069. [Google Scholar]
- 6.Narayan ARH; Jiménez-Osés G; Liu P; Negretti S; Zhao W; Gilbert MM; Ramabhadran RO; Yang Y-F; Furan LR; Li Z; Podust LM; Montgomery J; Houk KN; Sherman DH, Enzymatic hydroxylation of an unactivated methylene C–H bond guided by molecular dynamics simulations. Nature Chemistry 2015, 7, 653–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vavra O; Damborsky J; Bednar D, Fast approximative methods for study of ligand transport and rational design of improved enzymes for biotechnologies. Biotechnol. Adv 2022, 60, 108009. [DOI] [PubMed] [Google Scholar]
- 8.Chica RA; Doucet N; Pelletier JN, Semi-rational approaches to engineering enzyme activity: combining the benefits of directed evolution and rational design. Curr. Opin. Biotechnol 2005, 16, 378–384. [DOI] [PubMed] [Google Scholar]
- 9.Bhowmick A; Sharma SC; Honma H; Head-Gordon T, The role of side chain entropy and mutual information for improving the de novo design of Kemp eliminases KE07 and KE70. Physical Chemistry Chemical Physics 2016, 18, 19386–19396. [DOI] [PubMed] [Google Scholar]
- 10.Khersonsky O; Lipsh R; Avizemer Z; Ashani Y; Goldsmith M; Leader H; Dym O; Rogotner S; Trudeau DL; Prilusky J; Amengual-Rigo P; Guallar V; Tawfik DS; Fleishman SJ, Automated Design of Efficient and Functionally Diverse Enzyme Repertoires. Mol. Cell 2018, 72, 178–186.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Romero-Rivera A; Garcia-Borràs M; Osuna S, Role of Conformational Dynamics in the Evolution of Retro-Aldolase Activity. ACS Catalysis 2017, 7, 8524–8532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fried SD; Boxer SG, Electric Fields and Enzyme Catalysis. Annu. Rev. Biochem 2017, 86, 387–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang Y; Yan B; Chen Y; Juarez RJ; Yang ZJ, Molecular Dynamics-Derived Descriptor Informs the Impact of Mutation on the Catalytic Turnover Number in Lactonase Across Substrates. The Journal of Physical Chemistry B 2022, 126, 2486–2495. [DOI] [PubMed] [Google Scholar]
- 14.Clements H; Flynn A; Nicholls B; Grosheva D; Hyster T; Sigman M, In; Chemrxiv: 2021. [Google Scholar]
- 15.Xie WJ; Warshel A, Natural Evolution Provides Strong Hints about Laboratory Evolution of Designer Enzymes. Proc. Natl. Acad. Sci. U.S.A 2022, 119, e2207904119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang Y; Ran X; Yang ZJ, Data-Driven Enzyme Engineering to Identify Function-Enhancing Enzymes. Protein Engineering, Design and Selection 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shao Q; Jiang Y; Yang ZJ, EnzyHTP: A High-Throughput Computational Platform for Enzyme Modeling. J. Chem. Inf. Model 2022, 62, 647–655. [DOI] [PubMed] [Google Scholar]
- 18.Amrein BA; Steffen-Munsberg F; Szeler I; Purg M; Kulkarni Y; Kamerlin SCL, CADEE: Computer-Aided Directed Evolution of Enzymes. IUCrJ 2017, 4, 50–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Makinen MW; Fink AL, Reactivity and Cryoenzymology of Enzymes in the Crystalline State. Annual Review of Biophysics and Bioengineering 1977, 6, 301–343. [DOI] [PubMed] [Google Scholar]
- 20.Schulz EC; Mehrabi P; Müller-Werkmeister HM; Tellkamp F; Jha A; Stuart W; Persch E; De Gasparo R; Diederich F; Pai EF; Miller RJD, The hit-and-return system enables efficient time-resolved serial synchrotron crystallography. Nat. Methods 2018, 15, 901–904. [DOI] [PubMed] [Google Scholar]
- 21.Mehrabi P; Di Pietrantonio C; Kim TH; Sljoka A; Taverner K; Ing C; Kruglyak N; Pomès R; Pai EF; Prosser RS, Substrate-Based Allosteric Regulation of a Homodimeric Enzyme. J. Am. Chem. Soc 2019, 141, 11540–11556. [DOI] [PubMed] [Google Scholar]
- 22.Kim TH; Mehrabi P; Ren Z; Sljoka A; Ing C; Bezginov A; Ye L; Pomès R; Prosser RS; Pai EF, The role of dimer asymmetry and protomer dynamics in enzyme catalysis. Science 2017, 355, eaag2355. [DOI] [PubMed] [Google Scholar]
- 23.Mehrabi P; Schulz EC; Dsouza R; Müller-Werkmeister HM; Tellkamp F; Miller RJD; Pai EF, Time-resolved crystallography reveals allosteric communication aligned with molecular breathing. Science 2019, 365, 1167–1170. [DOI] [PubMed] [Google Scholar]
- 24.Park H; Bradley P; Greisen P; Liu Y; Mulligan VK; Kim DE; Baker D; Dimaio F, Simultaneous Optimization of Biomolecular Energy Functions on Features from Small Molecules and Macromolecules. J. Chem. Theory Comput 2016, 12, 6201–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frenz B; Lewis SM; King I; DiMaio F; Park H; Song Y, Prediction of Protein Mutational Free Energy: Benchmark and Sampling Improvements Increase Classification Accuracy. Frontiers in Bioengineering and Biotechnology 2020, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fried SD; Boxer SG, Measuring Electric Fields and Noncovalent Interactions Using the Vibrational Stark Effect. Acc. Chem. Res 2015, 48, 998–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhowmick A; Sharma SC; Head-Gordon T, The Importance of the Scaffold for de Novo Enzymes: A Case Study with Kemp Eliminase. J. Am. Chem. Soc 2017, 139, 5793–5800. [DOI] [PubMed] [Google Scholar]
- 28.Case DA, Belfon HMA,K, Ben-Shalom IY, Brozell SR, Cerutti DS, Cheatham TE III, Cisneros GA, Cruzeiro VWD, Darden TA, Duke RE, Giambasu G, Gilson MK, Gohlke H, Goetz AW, Harris R, Izadi S, Izmailov SA, Jin C, Kasavajhala K, Kaymak MC, King E, Kovalenko A, Kurtzman T, Lee TS, LeGrand S, Li P, Lin C, Liu J, Luchko T, Luo R, Machado M, Man V, Manathunga M, Merz KM, Miao Y, Mikhailovskii O, Monard G, Nguyen H, O’Hearn KA, Onufriev A, Pan F, Pantano S, Qi R, Rahnamoun A, Roe DR, Roitberg A, Sagui C, Schott-Verdugo S, Shen J, Simmerling CL, Skrynnikov NR, Smith J, Swails J, Walker RC, Wang J, Wei H, Wolf RM, Wu X, Xue Y, York DM, Zhao S, and Kollman PA Amber 2021. [Google Scholar]
- 29.Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg J Williams L; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian 16 Rev. C.01, Wallingford, CT, 2016. [Google Scholar]
- 30.Lu T; Chen F, Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem 2012, 33, 580–592. [DOI] [PubMed] [Google Scholar]
- 31.Röthlisberger D; Khersonsky O; Wollacott AM; Jiang L; Dechancie J; Betker J; Gallaher JL; Althoff EA; Zanghellini A; Dym O; Albeck S; Houk KN; Tawfik DS; Baker D, Kemp elimination catalysts by computational enzyme design. Nature 2008, 453, 190–195. [DOI] [PubMed] [Google Scholar]
- 32.Khersonsky O; Röthlisberger D; Dym O; Albeck S; Jackson CJ; Baker D; Tawfik DS, Evolutionary Optimization of Computationally Designed Enzymes: Kemp Eliminases of the KE07 Series. J. Mol. Biol 2010, 396, 1025–1042. [DOI] [PubMed] [Google Scholar]
- 33.Krivov GG; Shapovalov MV; Dunbrack RL Jr., Improved prediction of protein side-chain conformations with SCWRL4. Proteins: Structure, Function, and Bioinformatics 2009, 77, 778–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schrodinger, LLC, In; 2015. [Google Scholar]
- 35.Eswar N; Webb B; Marti - Renom MA; Madhusudhan MS; Eramian D; Shen MY; Pieper U; Sali A, Comparative Protein Structure Modeling Using Modeller. Current Protocols in Bioinformatics 2006, 15, 5.6.1–5.6.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Misiura M; Shroff R; Thyer R; Kolomeisky AB, DLPacker: Deep learning for prediction of amino acid side chain conformations in proteins. Proteins: Structure, Function, and Bioinformatics 2022, 90, 1278–1290. [DOI] [PubMed] [Google Scholar]
- 37.Jumper J; Evans R; Pritzel A; Green T; Figurnov M; Ronneberger O; Tunyasuvunakool K; Bates R; Žídek A; Potapenko A; Bridgland A; Meyer C; Kohl SAA; Ballard AJ; Cowie A; Romera-Paredes B; Nikolov S; Jain R; Adler J; Back T; Petersen S; Reiman D; Clancy E; Zielinski M; Steinegger M; Pacholska M; Berghammer T; Bodenstein S; Silver D; Vinyals O; Senior AW; Kavukcuoglu K; Kohli P; Hassabis D, Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kiss G; Röthlisberger D; Baker D; Houk KN, Evaluation and ranking of enzyme designs. Protein Sci. 2010, 19, 1760–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Genheden S; Ryde U, How to obtain statistically converged MM/GBSA results. J. Comput. Chem 2009, NA-NA. [DOI] [PubMed] [Google Scholar]
- 40.Ramírez-Palacios C; Wijma HJ; Thallmair S; Marrink SJ; Janssen DB, Computational Prediction of ω-Transaminase Specificity by a Combination of Docking and Molecular Dynamics Simulations. J. Chem. Inf. Model 2021, 61, 5569–5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hong N-S; Petrović D; Lee R; Gryn’Ova G; Purg M; Saunders J; Bauer P; Carr PD; Lin C-Y; Mabbitt PD; Zhang W; Altamore T; Easton C; Coote ML; Kamerlin SCL; Jackson CJ, The evolution of multiple active site configurations in a designed enzyme. Nat. Commun 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richter F; Blomberg R; Khare SD; Kiss G; Kuzin AP; Smith AJT; Gallaher J; Pianowski Z; Helgeson RC; Grjasnow A; Xiao R; Seetharaman J; Su M; Vorobiev S; Lew S; Forouhar F; Kornhaber GJ; Hunt JF; Montelione GT; Tong L; Houk KN; Hilvert D; Baker D, Computational Design of Catalytic Dyads and Oxyanion Holes for Ester Hydrolysis. J. Am. Chem. Soc 2012, 134, 16197–16206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siegel JB; Zanghellini A; Lovick HM; Kiss G; Lambert AR; St.Clair JL; Gallaher JL; Hilvert D; Gelb MH; Stoddard BL; Houk KN; Michael FE; Baker D, Computational Design of an Enzyme Catalyst for a Stereoselective Bimolecular Diels-Alder Reaction. Science 2010, 329, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Towns J; Cockerill T; Dahan M; Foster I; Gaither K; Grimshaw A; Hazlewood V; Lathrop S; Lifka D; Peterson GD; Roskies R; Scott JR; Wilkins-Diehr N, XSEDE: Accelerating Scientific Discovery. Computing in Science & Engineering 2014, 16, 62–74. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code and sample input for ARMer is publically available at https://github.com/ChemBioHTP/ARMer. The input files and structures are provided as part of the SI files. EnzyHTP is available from https://github.com/ChemBioHTP/EnzyHTP/tree/develop. Workflow scripts for the computational directed evolution protocol are available from https://github.com/ChemBioHTP/EnzyHTP/blob/develop/Test_file/KE_accre/R6/KE-metrics_complete.py. AMBER 18 is available from http://ambermd.org/. Gaussian 16 is available from https://gaussian.com/.