

Abstract
Lymph node stromal cells (LNSC) are an often overlooked component of the immune system, but play a crucial role in maintaining tissue homeostasis and orchestrating immune responses. Our understanding of the functions these cells serve in the context of bacterial infections remains limited. We previously showed that Listeria monocytogenes, a facultative intracellular foodborne bacterial pathogen, must replicate within an as-yet-unidentified cell type in the mesenteric lymph node (MLN) to spread systemically. Here, we show that L. monocytogenes could invade, escape from the vacuole, replicate exponentially, and induce a type I IFN response in the cytosol of two LNSC populations infected in vitro, fibroblastic reticular cells (FRC) and blood endothelial cells (BEC). Infected FRC and BEC also produced a significant chemokine and pro-inflammatory cytokine response after in vitro infection. Flow cytometric analysis confirmed that GFP+ L. monocytogenes were associated with a small percentage of MLN stromal cells in vivo following foodborne infection of mice. Using fluorescent microscopy, we showed that these cell-associated bacteria were intracellular L. monocytogenes and the number of infected FRC and BEC changed over the course of a three-day infection in mice. Ex vivo culturing of these infected LNSC populations revealed viable, replicating bacteria that grew on agar plates. These results highlight the unexplored potential of FRC and BEC to serve as suitable growth niches for L. monocytogenes during foodborne infection and to contribute to the pro-inflammatory environment within the MLN that promotes clearance of listeriosis.
Summary Sentence
Fibroblastic reticular cells are readily infected during foodborne listeriosis; blood endothelial cells are less efficiently invaded but support exponential growth of Listeria monocytogenes.
1. Introduction
Lymph node stromal cells (LNSC) are non-hematopoietic structural cells responsible for compartmentalizing the lymph node and facilitating homeostasis by regulating immune cell migration and interactions within the node. Four subsets of CD45neg LNSC can be defined using expression of cell surface markers gp38 (podoplanin) and CD31 (platelet endothelial cell adhesion molecule): gp38posCD31neg fibroblastic reticular cells (FRC), gp38posCD31pos lymphoid endothelial cells (LEC), gp38negCD31pos blood endothelial cells (BEC), and gp38negCD31neg double negative cells (DNC) [1, 2]. FRC are found in multiple compartments within the lymph node and can be further divided into nine subsets that vary in functionality [2, 3]. For example, marginal FRC that line the cortical area are thought to give rise to follicular dendritic cells and promote B cell follicle formation during infection [4, 5]. In contrast, T cell zone FRC support naïve T cell homeostasis and facilitate interactions between T cells and dendritic cells [6]. LEC compose the floor and ceilings of lymphatic vessels, subcapsular sinuses, and medullary sinuses and function to attract and then guide antigen-bearing immune cells entering the node [7, 8]. Additionally, naïve lymphocytes enter the lymph node through high endothelial venules, which are specialized areas of blood vessels formed, in part, by BEC that generate a chemokine gradient and express a variety of adhesion molecules [9–11]. Little is known about the DNC, a potentially heterogenous population of non-hematopoietic cells currently defined only by the lack of cell surface molecules associated with well characterized LNSC.
Listeria monocytogenes, a facultative intracellular foodborne bacterial pathogen, readily overcomes the MLN barrier and spreads systemically, resulting in life-threatening systemic infections in susceptible individuals. The ability of L. monocytogenes to invade a variety of mammalian cells is thought to be its primary virulence strategy. Once internalized, L. monocytogenes readily escape from phagocytic or endocytic vacuoles and replicate in the cytosol [12, 13]. Within hours after vacuolar escape, L. monocytogenes polymerize host actin and use actin-based motility to spread into adjacent cells without encountering the extracellular milieu [14]. We previously found that this intracellular life cycle was essential for L. monocytogenes egress from the MLN following foodborne infection; however, the key cell types in the MLN required for intracellular growth and systemic spread remain unknown [15].
Ly6Chi monocytes are the predominant cell type associated with L. monocytogenes in the MLN three days post-infection; however, primary Ly6Chi monocytes sort-purified from uninfected animals did not support intracellular growth of L. monocytogenes [16]. Likewise, L. monocytogenes could invade primary conventional dendritic cells sort-purified from naïve mice but survived intracellularly for only a few hours [17]. These results suggested that the critical intracellular growth niches in the MLN were likely to be a minor population of cells associated with L. monocytogenes. In the present study, we assessed the relative efficiency of L. monocytogenes invasion, survival, and intracellular replication in various stromal cell subsets. Using both transformed cell lines and primary cells isolated from the MLN, we show that at least two stromal subsets (FRC and BEC) can support exponential intracellular growth of L. monocytogenes and mount a significant inflammatory cytokine/chemokine response during a L. monocytogenes infection.
2. Materials and Methods
2.1. Bacteria
The mouse-adapted [18] derivatives of L. monocytogenes EGDe used in this study (SD2000, parental InlAm-expressing; SD2710, constitutively expresses GFP; and SD2001, control strain with empty vector pPL2 integrated) were described previously [19]. L. monocytogenes strains were cultured in Brain Heart Infusion (BHI) broth (Difco) at either 30°C (for in vivo infection of mice) or 37°C (for in vitro assays) until either mid-log phase or early stationary phase and then aliquots were prepared, stored at −80°C, and thawed prior to use as described [20].
2.2. Cell culture
Murine SVEC4–10 cells were obtained from the American Type Culture Collection (CRL-2181) and used only at low (< 13) passage. The cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM; cat. # 11960–051, Life Technologies) supplemented with GlutaMAX (Life Technologies Cat. # 35050–061), 10% Fetal Bovine Serum (FBS; cat. #100–106, GeminiBio), and penicillin/streptomycin in 100 mm dishes (Corning) and incubated at 37°C in 7% CO2. The day before infection, SVEC4–10 cells were seeded at 2.5 × 105 cells/well in 6-well dishes (Corning) or at 1 × 105 cells/well onto round glass coverslips (12 mm diameter) in 24-well dishes (Corning) in media lacking antibiotics. L2 fibroblasts were maintained in DMEM supplemented with 2.5 mM L-glutamine, 10% FBS, and penicillin/streptomycin in 100-mm dishes at 37°C in 7% CO2. For plaque assays, L2 fibroblasts were seeded at 2.5 × 105 cells/well in 6-well dishes.
2.3. Intracellular growth assay
SVEC4–10 cells were seeded at 1 × 105/well in 24-well dishes (Corning). Primary stromal cells were seeded at 3 × 104cells/well in half area 96-well flat bottom dishes (Corning, cat. #3696). Aliquots of L. monocytogenes were thawed, incubated shaking at 37°C in BHI broth for 1.5 h, washed once in PBS (Gibco cat. #14190–144), and then used to infect cells at a multiplicity of infection (MOI) of 10. Plates were centrifuged for 5 min at 300 x g to synchronize infection. Extracellular bacteria were removed 1 h later by washing cells 3 times with pre-warmed (37°C) PBS and then suspended in media containing 10 μg/ml gentamicin for at least 20 min. At each timepoint, cells were washed once and either sterile water was added to the wells with vigorous pipetting up and down or coverslips were harvested and placed in sterile H2O and then vortexed. To ensure cell survival was uniform among each stromal subset throughout the assay, we evaluated viability using trypan blue staining at each timepoint. Following cell lysis, serial dilutions were prepared and plated on BHI agar. For analysis of invasion efficiency, the percentage of each inoculum internalized at 1 hour post-infection was calculated by dividing the number of CFU recovered at each time point by the total number of CFU added to each well (% invasion).
2.4. Microscopy
For differential “in/out” staining of L. monocytogenes, cells were spun onto chamber slides (SPL Life Sciences cat. # 30108) for 6 min at 600 rpm using a Cytospin, washed three times with cold buffer (Ca2+/Mg2+-free HBSS/1% FBS/1 mM EDTA) and then incubated for 20 min on ice with polyclonal anti-Listeria antibodies (BD Difco cat. # 223021) that were diluted 1:100 in PBS containing 3% bovine serum albumin (BSA; ThermoFisher # A11037). Following incubation, the cells were washed with cold buffer and then incubated with goat anti-rabbit IgG–Texas Red diluted 1:200 in PBS + BSA for 20 min on ice. Air-dried slides were fixed with formalin for 10 min at 4°C, washed with PBS, and mounted under coverslips with ProLong Diamond antifade (Molecular Probes). For F-actin staining, fixed cells were washed three times with PBS then permeabilized using Tris-buffered saline (TBS; MP Biomedicals cat. #819623) supplemented with 1% BSA and 0.1% Triton X-100 (Sigma) for 30 min at room temperature. Permeabilized cells were incubated with goat anti-rabbit IgG-Texas Red-X phalloidin (ThermoFisher) diluted 1:100 in TBS+BSA for 30 min at room temperature. The cells were washed eight times in TBS-TX (TBS supplemented with 0.1% Triton X-100) followed by eight washes with TBS alone. Cells were visualized using either an Invitrogen EVOS M5000 cell imager or a Nikon A1R Confocal System with a 100X oil immersion objective and analyzed with Nikon NIS Elements AR software (version 4.50).
2.5. Plaque Assay
Plaque assays were performed as previously described [21]. Briefly, cells were grown to confluency overnight in a 6-well plate at 37°C in 7% CO2. An overnight culture of L. monocytogenes was prepared, back-diluted the following day, and grown to mid-log phase. SVEC4–10 cells or L2 fibroblasts were washed with warm (37°C) PBS and infected with mid-log phase L. monocytogenes at various dilutions; plates were centrifuged for 5 min at 300 x g to synchronize infection. One hour later, cells were washed three times with warm PBS, and suspended in medium containing 0.7% agarose and 10 μg/ml gentamicin. Following a four day incubation at 37°C/7% CO2, each well was overlayed with medium containing 0.7% agarose and 0.5% neutral red and then incubated for 6–8 hours at 37°C/7% CO2. Plaques were photographed after incubation and measured with a digital ruler (Adobe Photoshop).
2.6. Mice
BALBcBy/J mice were either purchased from The Jackson Laboratory (Bar Harbor, ME) or bred in a specific pathogen-free (SPF) facility at The University of Kentucky. For foodborne infections, four-week old female mice were adapted to an ASBL-2 room with a 14-hour light cycle (7 PM to 9 AM) for at least two weeks and then used in experiments when they were between 6 to 10 weeks old. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Kentucky.
2.7. Enzymatic digestion of MLN
Mesenteric lymph nodes that drain the small intestine (sMLN) and colon (cMLN) were aseptically harvested as described [22]. Where the use of peripheral lymph nodes (PLN) was specified, axillary, brachial and inguinal nodes were harvested. Lymph nodes were pierced using a sterile 25G needle and placed in 2 mL of freshly made media consisting of RPMI 1640 supplemented with 0.6 mg/mL Dispase II (0.9 U/mg; Roche), 0.2 mg/mL Collagenase P (1.9 U/mg; Roche), and 0.1 mg DNase I (120 U/mL; Worthington) and digested as previously described [23].
2.8. Flow cytometry
Single-cell suspensions were incubated with fluorescently conjugated monoclonal antibodies (Biolegend) specific for the following cell surface markers: CD16/32 (Fc Block; clone 93), CD45 (clone 30-F11), CD31 (clone 390), and podoplanin (clone PMab-1). Cells were fixed with 10% neutral buffered formalin (VWR cat. #16004–128) prior to analysis. Flow cytometry data were acquired using a FACSymphony (BD Biosciences) and analyzed using FlowJo software (Version 10). The percentage of Listeria-associated (GFP+) cells in each population was determined by using cells from mice infected with L. monocytogenes SD2001 as a negative gating control as described previously [16, 17, 19].
2.9. Cell Sorting
Due to the low yield of LNSC, cells pooled from three animals were used for each experiment. Prior to sorting, CD45neg cells were enriched using magnetic bead separation. MLN cells were incubated with a R-Phycoerythrin (PE)-conjugated anti-CD45 antibody for 30 minutes on ice. Cells were washed with cold buffer and then incubated with anti-PE magnetic particles (BD Biosciences cat. #557899) for 30 minutes at 4°C. CD45neg cells were enriched by placing a tube containing the stained cells against a magnet for 8 minutes and collecting the supernatant three times. The cells were then further stained and subsequently purified using an iCyt Sorter (Sony). Sorted stromal populations were suspended in Minimal Essential Media (cat. # 10370–021; Gibco) supplemented with 10% FBS. Cells were counted and viability was assessed prior to seeding in a half-area 96-well flat bottom dishes (3 × 104/well). Cells were left to recover for at least 30 minutes at 37°C in 7% CO2 prior to infection. Following the recovery period, cells were infected at an MOI of 10. In each experiment, the plated cells were used to analyze infection at two time points; no technical replicates were plated.
2.10. Multiplex Immunoassay
Supernatants were collected after each intracellular growth assay and stored at −80°C until used for subsequent Luminex assays. A custom mouse 32-plex Procartaplex kit was purchased from Life Technologies and used according to manufacturer’s recommendations. The plate was read using a Luminex 200 and data was analyzed using the ProcartaPlex analysis App (ThermoFisher).
2.11. Foodborne infection of mice
Mice were infected using the natural feeding model previously described [24, 25]. Briefly, mice were placed in cages with raised wire floors for the duration of the experiment to prevent coprophagy and food was withheld for 16–20 h prior to infection. Aliquots of L. monocytogenes were thawed in BHI at 30°C, washed once in PBS and then used to saturate 3 mm bread pieces (Kroger), followed by the addition of 2 μl of melted salted butter (Kroger). Mice were fed the L. monocytogenes-contaminated bread at the beginning of their dark cycle, and chow was returned immediately after infection.
2.12. Statistics
Statistical analyses were performed using Prism for Macintosh (version 9; GraphPad). The specific tests used for each experiment are indicated in the figure legends. P values of < 0.05 were considered significant and are indicated as follows: *, P < 0.05; **, P <0.01; ***, P < 0.001; ****, P < 0.0001.
3. Results
3.1. L. monocytogenes replicate exponentially in a stromal cell line despite inefficient invasion
To determine if L. monocytogenes could invade and replicate within stromal cells, we first used SVEC4–10, a murine blood endothelial cell line originally isolated from an axillary lymph node [26]. The cells were infected with either mid-logarithmic phase or stationary phase L. monocytogenes for one hour and then gentamicin was added to kill any extracellular bacteria. This resulted in an invasion rate of less than 0.2% for either inoculum (Fig. 1A). For comparison, non-phagocytic cells such as the human intestinal epithelial cell line Caco-2 or the murine colorectal cell line CT-26 typically internalize 1–3% of L. monocytogenes when used at a similar multiplicity of infection (Fig. 1B) [27, 28]. Thus, SVEC4–10 cells did not efficiently internalize L. monocytogenes. We did find a notable decrease in the invasion efficiency of L. monocytogenes grown at mid-log phase relative to stationary phase (Fig. 1A); this may suggest that a surface protein which promotes invasion of SVEC4–10 cells could be differentially expressed by the bacteria.
Fig. 1. L. monocytogenes replicate exponentially in SVEC4–10 cells despite inefficient invasion.
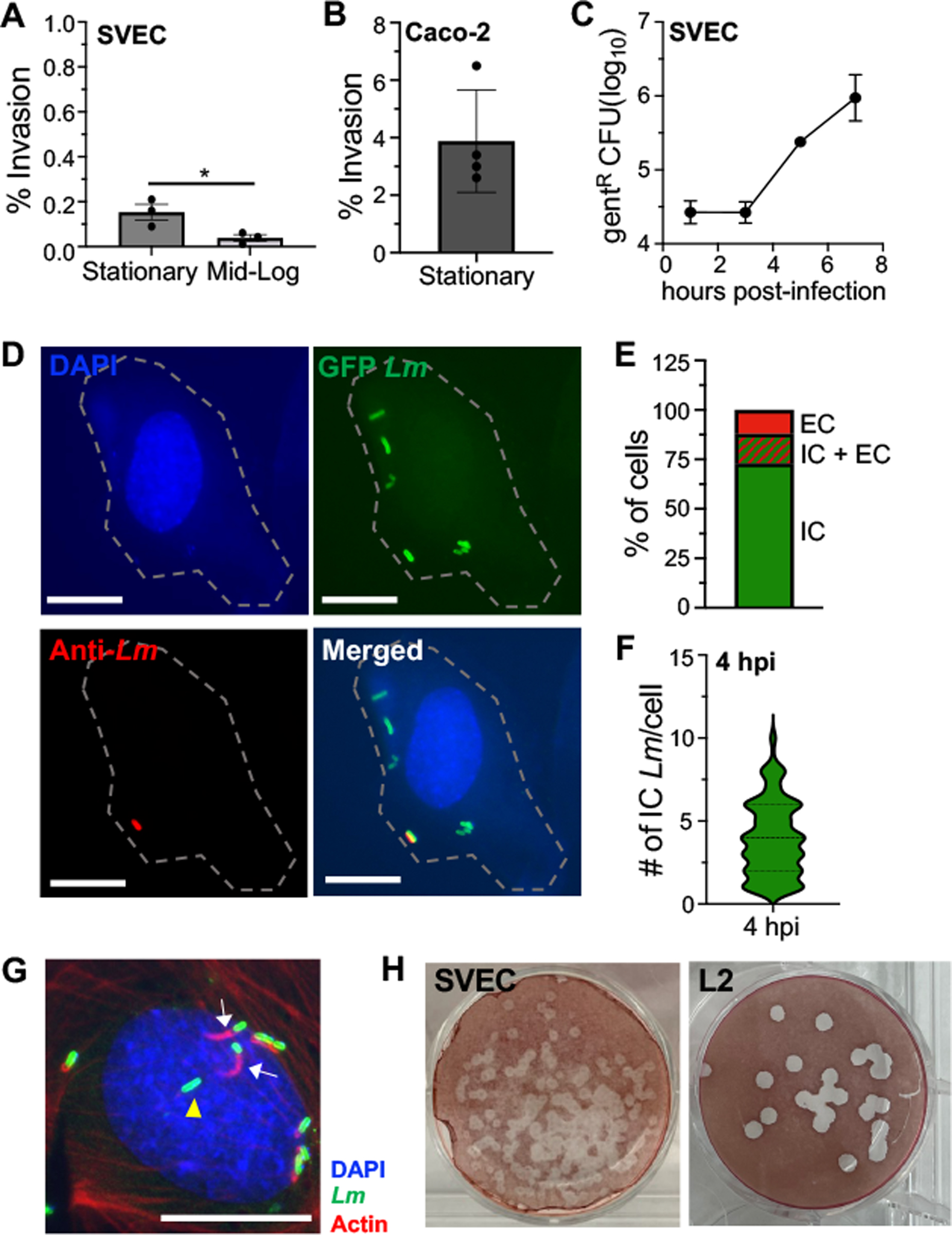
SVEC4–10 cells were infected with L. monocytogenes (Lm) at MOI=10. (A-B) Mean percentage (analyzed by Mann-Whitney) of Lm SD2000 inoculum that was gentamicin-resistant 1 hr post-infection (hpi) in (A) SVEC4–10 cells or (B) Caco-2 cells. (C) Intracellular growth assay using stationary phase Lm SD2000. For panels A and C, mean values (±SEM) for triplicate samples from one of two experiments is shown. For panel B, mean values (±SEM) for quadruplet samples from one experiments is shown. (D) Representative images for differential “in/out” staining of cells infected with stationary phase Lm SD2710 (constitutive GFP), fixed at 4 hpi, and stained with DAPI (nucleus/blue), and anti-Listeria antibody (Lm/red). Scale bar, 5 μm. Dashed line indicates the outline of infected cell from the brightfield image. (E) Stacked bar indicates the percentage of Lm-infected cells (n=300) with only intracellular bacteria (IC), both intracellular and extracellular (IC+ EC), or only extracellular bacteria (EC). (F) Violin plot indicates the number of intracellular Lm observed per cell (n=300). For panels C, D and E data from one of two independent experiments are shown. (G) Representative image for phalloidin staining of SVEC4–10 cells infected with Lm SD2710 at 4 hpi; white arrows indicate Lm associated with actin; yellow arrowhead indicates Lm not associated with actin. Scale bar, 10 μm. Two independent experiments (n=100 cells analyzed in each) were performed; in Exp. 1, 72% of Lm were associated with actin and in Exp. 2 80% were. (H) Representative images from one of three independent plaque assays using SVEC4–10 cells or L2 fibroblasts infected with Lm SD2000 and visualized 4 dpi.
To determine if the few bacteria taken up by SVEC4–10 could replicate inside the cells, we performed an intracellular growth assay using stationary phase L. monocytogenes. We found that at 3 hours post-infection, the number of gentamicin-resistant L. monocytogenes had not changed relative to the number of bacteria recovered at 1 hour post-infection (Fig. 1C). However, by 5 hours post-infection, gentamicin-resistant L. monocytogenes had increased exponentially and continued through the duration of the assay (Fig 1C). To confirm these results, we performed differential “in/out” staining and found that the majority (~85%) of infected SVEC4–10 cells contained intracellular bacteria 4 hours post-infection (Fig 1D and 1E). At this early time point, most cells contained on average only 4 bacteria per cell (Fig. 1F) further demonstrating that infection of the SVEC4–10 cells was inefficient and proceeded slowly.
Following vacuole escape, L. monocytogenes polymerize host actin in the cytoplasm, which creates a “cloud” of actin surrounding the bacteria. Once L. monocytogenes begin replicating, polarized expression of the bacterial surface protein ActA recruits the actin to one pole of the bacterium forming a “tail,” and resulting in directional movement of the bacteria [15, 29]. Therefore, to assess if the intracellular bacteria had escaped from the vacuole in SVEC4–10 cells and were replicating in the cytosol by 4 hours post-infection, we stained for F-actin (Fig. 1G). We found that approximately three-quarters of the intracellular bacteria visualized had either actin clouds or actin tails, suggesting that most of the L. monocytogenes were localized to the cytosol by 4 hours post-infection. To determine if L. monocytogenes could spread from the cytosol of one SVEC4–10 cell to the cytosol of an adjacent cell using actin-based motility we performed plaque assays. L2 fibroblasts were used as a positive control since it is well established that L. monocytogenes form large clear plaques in these cell monolayers [21, 30]. We found L. monocytogenes could create plaques within SVEC4–10 cells, although they were about half the size of those formed in L2 fibroblasts (Fig. 1H). Taken together, these results indicate that L. monocytogenes do invade SVEC4–10 cells, albeit inefficiently, and the few internalized bacteria can escape from the vacuole, replicate exponentially in the cytosol, and spread cell-to-cell.
3.2. Bulk LNSC isolated from either small or large intestine draining MLN support the exponential growth of intracellular L. monocytogenes.
Since LNSC constitute a small proportion of the MLN cellular composition, before attempting to purify the four stromal populations we first assessed if bulk CD45neg stromal cells isolated from the MLN could serve as an intracellular growth niche for L. monocytogenes. The small intestine (sMLN) and colon (cMLN) draining lymph nodes are distinct anatomic sites exposed to a varying composition and quantity of gut microbiota [22, 31] and we previously showed that Listeria infection persists longer in the colon than in the ileum following foodborne infection of mice [19, 32]. Although not yet extensively characterized, there is some evidence to suggest that the proportions of immune cell subsets can also differ between these two MLN sites [22]. Thus, we reasoned that the stromal populations within these two anatomical sites could also differ in their ability to support L. monocytogenes growth and opted to analyze the nodes separately.
Primary CD45neg cells were enriched by magnetic bead separation and then sort-purified from the sMLN and cMLN of uninfected BALBcBy/J mice (Fig. 2A) with a sort purity consistently above 99% in each experiment (Fig. 2B). Cells were infected with stationary phase L. monocytogenes directly ex vivo, and the amount of intracellular bacteria was determined at multiple time points. At 1 hour post-infection, we recovered similar amounts of gentamicin-resistant L. monocytogenes from the sMLN and the cMLN, with CD45neg cells at both sites internalizing approximately 1% of the L. monocytogenes inoculum (Fig. 2C). Gentamicin-resistant L. monocytogenes decreased slightly by 4 hours post-infection, but then increased exponentially in both MLN sites. This suggests that a substantial portion of the internalized bacteria were likely killed, and that the small subset of the bacteria that reached the cytosol required a few hours of replication to restore the bacterial burden. By 16 hours post-infection, a one log increase in bacterial burdens was observed (Fig. 2C). Notably, it took 16 hours to achieve these bacterial burdens, a growth rate slower than observed in SVEC4–10 cells where L. monocytogenes increased 100-fold over 8 hours (Fig. 1B). These results indicated that there was a CD45neg cell type in the MLN that did support exponential growth of L. monocytogenes when infected directly ex vivo. Since we did not observe any difference in the ability of cells isolated from either sMLN or cMLN to serve as an intracellular growth niche, for the remaining experiments in this study we pooled cells from all the gut draining MLN to increase cell yields from each mouse.
Fig. 2. Bulk LNSC isolated from both small and large intestine draining MLN support exponential growth of intracellular L. monocytogenes.
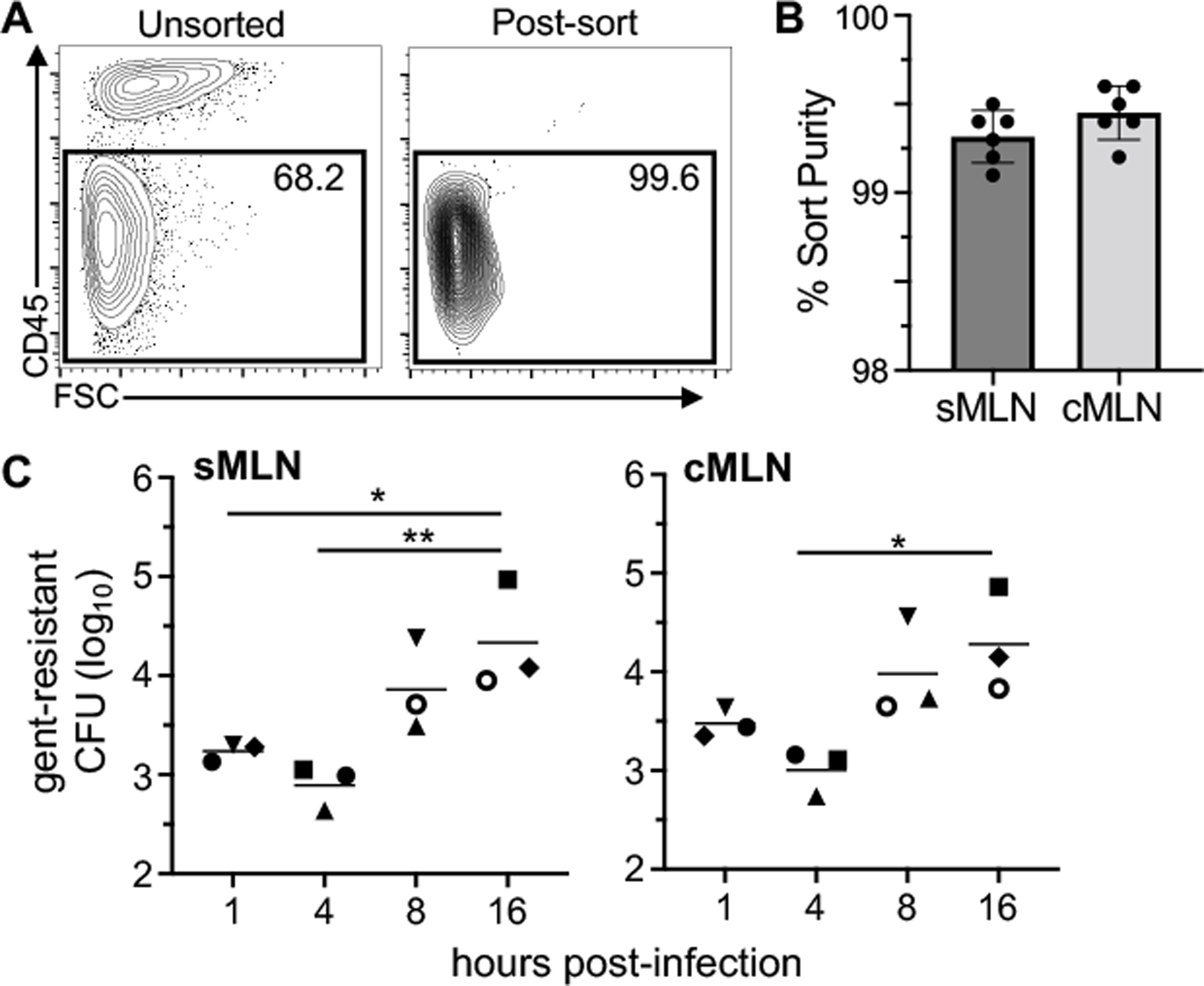
CD45neg cells were enriched from the MLN of uninfected BALBc/ByJ mice using magnetic bead separation and then sort-purified. (A) Representative dot plots indicate the purity of CD45neg cells after sorting. (B) Mean percent sort-purity (±SEM) of cells from the small intestine draining (sMLN) or colon draining MLN (cMLN). Each symbol represents the sort purity for pooled cells harvested from three animals that were used in a single experiment. (C) Sorted cells were infected with L. monocytogenes SD2000 (MOI=10) directly ex vivo and gentamicin-resistant L. monocytogenes were quantified. Pooled data from six separate experiments (each designated by a different symbol type) are shown; in each experiment, two time points were assessed. Horizontal lines indicate mean values for each time point. Statistical significance was determined by ANOVA with Tukeys multiple comparison test.
3.3. Primary fibroblastic reticular cells and blood endothelial cells support exponential growth of L. monocytogenes
To determine which primary LNSC subsets could support L. monocytogenes growth, we isolated four distinct stromal populations defined by cell surface expression of gp38 and CD31 (Fig. 3A and Fig. S1A). Although we could readily isolate sufficient FRC and DNC to perform in vitro infection experiments, we found that the two endothelial populations constituted less than 20% of MLN stromal cells (~7–8 × 103 cells/mouse), a cell yield insufficient for in vitro experiments. Pezoldt et al. found that the transcriptome of LEC and BEC isolated from naïve peripheral lymph nodes (PLN) such as the inguinal and axial nodes did not differ significantly from the transcriptional profile of LEC and BEC isolated from the MLN [33]. Therefore, to further increase the yield of these smaller populations, we also separately sorted cells harvested from PLN, and combined the isolated cells with the MLN populations only for the analysis of LEC and BEC.
Fig. 3. L. monocytogenes efficiently invade all stromal populations except primary blood endothelial cells when infected directly ex vivo.
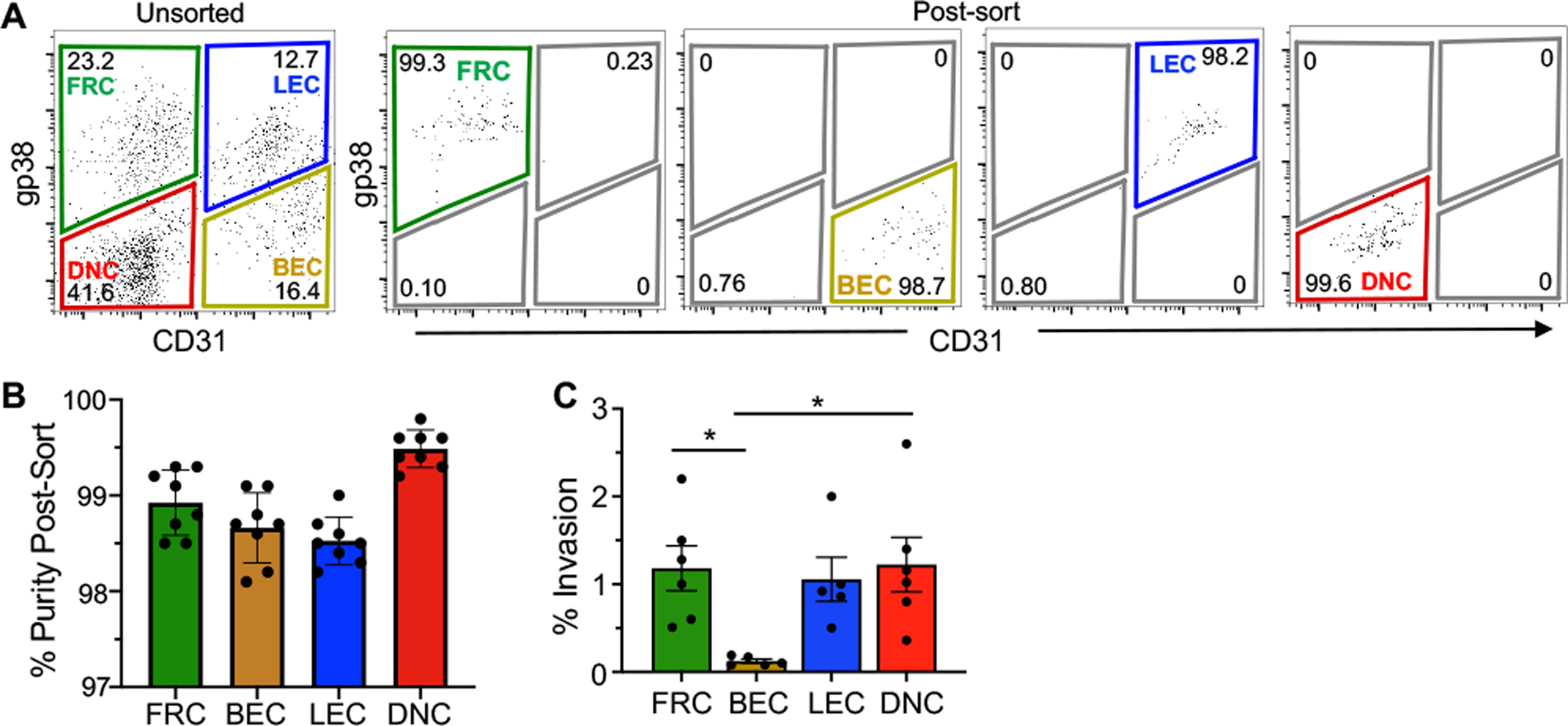
LNSC were sort-purified from the MLN and peripheral lymph nodes (PLN) of uninfected BALBc/ByJ mice. (A) Representative dot plots indicate the purity of stromal populations after sorting. (B) Mean percent sort-purity (±SEM) of each LNSC population; symbols represent individual experiments. (C) Sorted cells were infected ex vivo with with Lm SD2000 (MOI=10). Mean percent (±SEM) of the inoculum that was gentamicin resistant 1 hpi is shown; Pooled data from six separate experiments were analyzed by ANOVA with Tukeys multiple comparison test.
CD45neg cells from the lymph nodes of uninfected mice were enriched using magnetic bead separation and FACS-purified, with a sort purity consistently above 98% for each experiment (Fig. 3B). The four stromal populations were infected with L. monocytogenes directly ex vivo and the bacterial invasion efficiency was determined. As shown in Fig. 3C, primary FRC, LEC, and DNC populations internalized approximately 1% of the inoculum, a level similar to that seen for internalin-mediated invasion of L. monocytogenes in many transformed epithelial cell lines [27, 28]. However, BEC were significantly different from the rest of the cells, internalizing less than 0.2% of the L. monocytogenes, an invasion rate similar to SVEC4–10 cells (Fig. 1A and Fig. 3C).
To determine if the bacteria that were taken up by the stromal populations could replicate intracellularly, we assessed the amount of gentamicin-resistant bacteria at multiple time points. In FRC, there was a 10-fold increase in bacteria over time (Fig. 4A). In contrast, gentamicin-resistant L. monocytogenes increased almost 100-fold in BEC during the 16 hour time period. To verify the intracellular localization of L. monocytogenes within FRC and BEC, we performed differential “in/out” staining at 8 hours post-infection (Fig. 4B). FRC contained ten L. monocytogenes per cell on average (Fig. 4C). For BEC, we observed a bimodal distribution with half of the cells containing 25–35 intracellular bacteria per cell and the other half harboring less than 15 per cell (Fig. 4C). To confirm that L. monocytogenes were located within the cytoplasm of FRC and BEC, we also stained for F-actin at 8 hours post-infection (Fig. 4D). We found that 50–60% of L. monocytogenes were associated with either actin clouds or tails, suggesting that most of the internalized bacteria had localized to the cytosol by 8 hours post-infection in both FRC and BEC (Fig. 4E).
Fig. 4. Primary FRC and BEC infected ex vivo support exponential growth of intracellular L. monocytogenes.
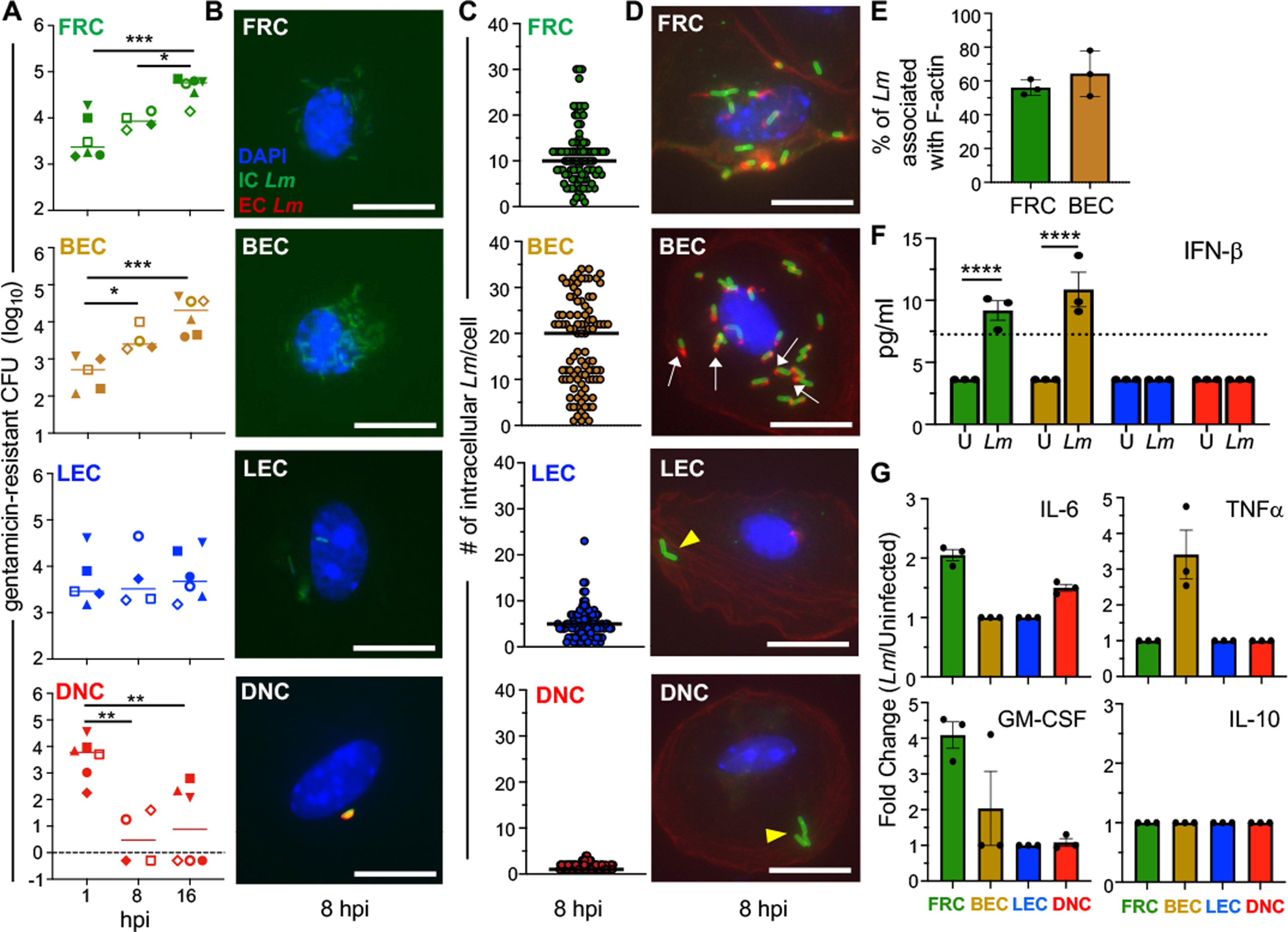
LNSC were sort-purified from the MLN and PLN of uninfected BALBc/ByJ mice and infected with Lm at an MOI of 10. (A) Gentamicin protection assay for cells infected with Lm SD2000; pooled data from eight separate experiments (each designated by a different symbol type) were analyzed by ANOVA with Tukeys multiple comparison test. In each experiment, total cells sorted from three mice were used at two time points. (B) Representative images for differential ”in/out” staining of sort-purified LNSC infected with Lm SD2710 (GFP+), fixed at 8 hpi and stained with DAPI (nuclei; blue), and anti-Lm antibody (red); scale bar, 5 μm. (C) Symbols indicate the number of intracellular Lm observed in a single infected cell; horizontal lines indicate median values for n=100 cells. For panels B and C, data from one of two independent experiments are shown. (D) Representative images for phalloidin staining of cells infected with Lm SD2710. White arrows indicate actin tails associated with Lm; yellow arrowheads indicate Lm not associated with actin. Scale bar, 5 μm. (E) Mean percent (±SEM) of intracellular Lm associated with actin. Each symbol represents results from a single experiment. (F) IFNβ present in supernatants of uninfected (U) and Lm SD2000-infected (Lm) cells (3 × 104 cells/well) at 16 hours was measured by multiplex immunoassay; symbols indicate mean values (+/− SEM) for duplicate samples from three separate experiments and were analyzed by one-way ANOVA. (G) Fold change of cytokine responses measured by multiplex immunoassay in Lm-infected cells relative to uninfected cells. Dashed lines indicate limits of detection.
Sort-purified primary LEC and DNC that were infected in vitro displayed a different pattern. The number of gentamicin-resistant L. monocytogenes recovered from LEC stayed relatively consistent over a 16-hour period (Fig. 4A), suggesting that the bacteria were surviving, but not replicating extensively in this cell type. LEC contained, on average, only five L. monocytogenes per cell at 8 hours post-infection (Fig. 4B and 4C). None of the bacteria observed in LEC were associated with F-actin (Fig. 4D), suggesting that L. monocytogenes did not escape the vacuole. Although DNC readily internalized L. monocytogenes, gentamicin-resistant bacteria decreased significantly over time (Fig. 4A). We found that DNC only harbored 1–2 bacteria per cell and did not contain any L. monocytogenes associated with F-actin at 8 hours post-infection (Fig. 4B, 4C and 4D). Thus, DNC did not support the growth or survival of intracellular L. monocytogenes.
To confirm that our results for LEC and BEC were not altered pooling cells isolated from different lymph nodes, we performed a single experiment comparing MLN cells to PLN cells. As expected, BEC isolated from both type of nodes supported exponential growth of Listeria over 16 hours while LEC showed no change in bacterial burdens (Fig. S1B). For the more plentiful FRC and DNC we were able to demonstrate that cells isolated from PLN were nearly identical to those isolated from MLN. FRC and DNC internalized nearly 1% of the L. monocytogenes inoculum (Fig. S1C), and gentamicin-resistant bacteria increased over time in FRC, and decreased in DNC (Fig. S1D). Thus, the observed growth phenotypes of L. monocytogenes in LNSC appeared to be consistent irrespective of whether sMLN, cMLN, or PLN cells were used, indicating a broader applicability of these findings across different lymph nodes.
3.4. Pro-inflammatory cytokine responses of LNSC correlate with susceptibility to infection.
An important function of stromal cells is the secretion of chemokines and cytokines to maintain homeostasis and cell migration within lymph nodes, but it is not currently known if these cells produce pro-inflammatory mediators in response to L. monocytogenes that could promote clearance of the infection. To assess this, the four distinct stromal populations were FACS-sorted, infected with L. monocytogenes, and supernatants were collected from both uninfected control cells and infected cells 16 hours later. Given the small number of cells seeded in each sample well, we expected to find relatively low cytokine output, but detectable using a multiplex immunoassay. We first assessed the production of type I interferons, cytokines that are induced when Listeria localized to the cell cytosol either release DNA detected by RIG-I or secrete c-di-AMP to trigger STING-dependent signaling [34, 35]. Both FRC and BEC produced detectable IFNβ in response to L. monocytogenes (Fig. 4F), further confirming that these cell types contained cytosolic bacteria. Neither LEC or DNC contained cytosolic Listeria at 8 hours post-infection (Fig. 4D), so the lack of type I IFN secretion in these cell types was expected (Fig. 4F). Even bacteria that cannot escape from endocytic vacuoles can trigger a TLR-dependent cytokine responses, so we also assessed the production of IL-6, TNFα, and GM-CSF. We found that infected FRC and BEC displayed a proinflammatory phenotype, while the infected LEC and DNC remained relatively unchanged compared to cells at steady state (Fig. 4G and Fig. S3 A–D). None of the stromal populations produced IL-10, suggesting that the infection did not illicit an anti-inflammatory response.
3.5. FRC and BEC have robust chemokine responses in response to L. monocytogenes.
Stromal cell-derived homeostatic chemokines play a crucial role in compartmentalizing the lymph node by guiding the precise localization of immune cells to distinct anatomical regions. In their absence, the lymph node architecture is disrupted, resulting in a dysfunctional immune response [36]. Using the same multiplex immunoassay as described above, we found that production of the gut-homing chemokines CCL19 and CCL22 did not change significantly in any of the four stromal populations, suggesting that L. monocytogenes does not alter the homeostatic function of these cells in the MLN (Fig. S2).
We next assessed the ability of these stromal cells to produce recruitment chemokines in response to L. monocytogenes infection (Fig. 5). We found that FRC produced the most robust chemokine response during in vitro infection of the FACS-sorted cells. Chemokines involved in neutrophil recruitment (CXCL1 and CXCL5), dendritic cell and monocyte migration (CCL2 and CCL7), and both T and B cell recruitment (CXCL13 and CXCL11), were all significantly increased in L. monocytogenes-infected FRC (Fig. 5A). Infected BEC also significantly increased production of a smaller subset of chemokines that included CXCL5, CCL2 and CXCL11 (Fig. 5B). On the contrary, uninfected LEC secreted low, but detectable amounts of CCL11 and CXCL13, but there was no induction following exposure to L. monocytogenes (Fig. 5C). Surprisingly the DNC did produce slightly more chemokines following infection, despite there being no evidence of replicating L. monocytogenes within these cells (Fig. 5D and S2). This may reflect the fact that microbial ligands that can trigger TLR mediated chemokine production are more likely to be present when the bacteria are degraded. Together, these results suggest that both FRC and BEC can contribute significantly in attracting immune cells to the MLN following foodborne L. monocytogenes infection.
Fig. 5. FRC, BEC and DNC produce chemokines in response to L. monocytogenes exposure.
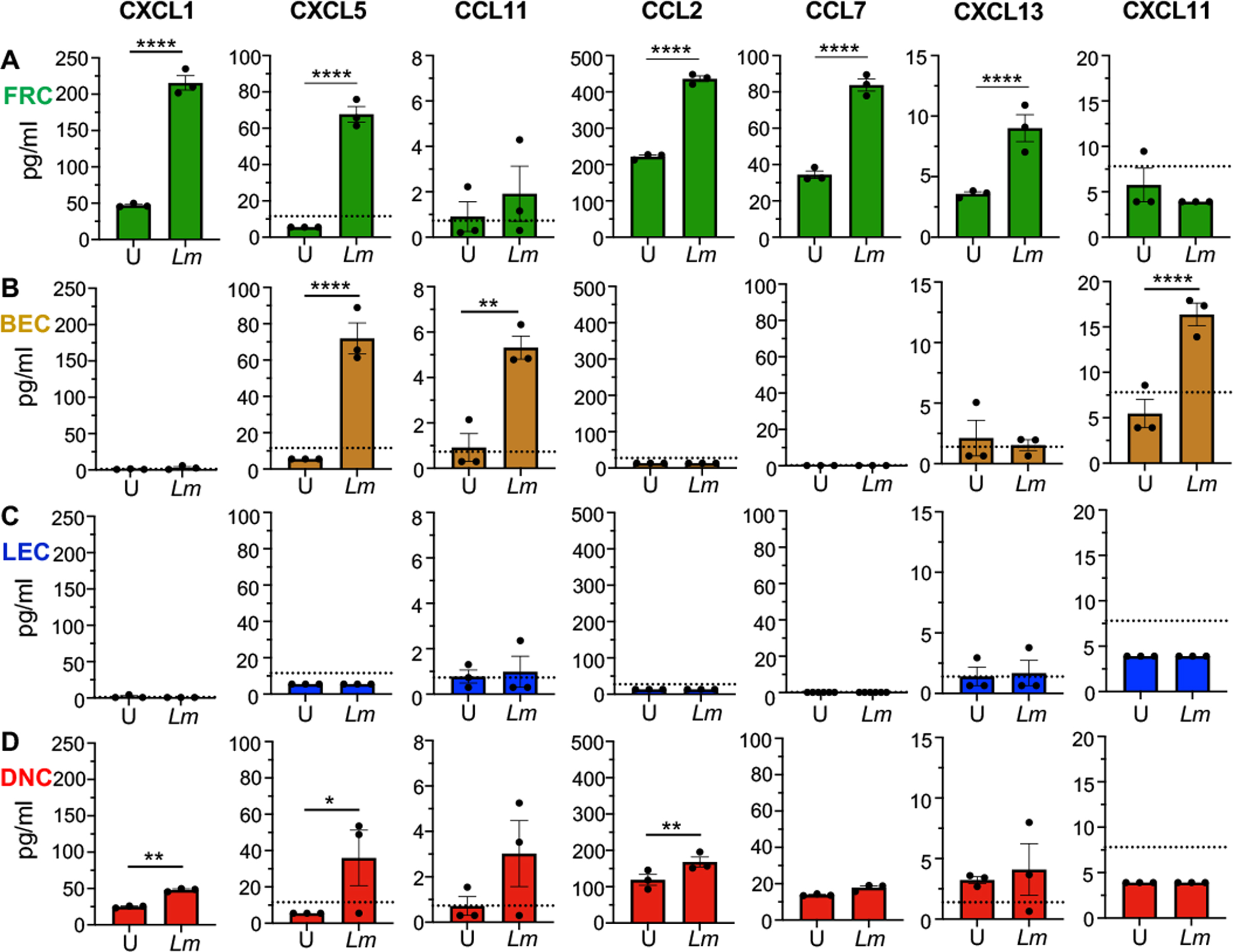
Supernatants were collected from uninfected (U) and Lm SD2000-infected (Lm) stromal cells (3 × 104 cells/well) at 16 hours for subsequent multiplex immunoassay. Selected chemokine responses of (A) FRC (B) BEC (C) LEC and (D) DNC are shown; symbols indicate mean values (+/− SEM) for duplicate samples from three separate experiments; dotted lines indicate limits of detection. Bars indicate mean values for the three biological replicates. Statistical significance was determined by one-way ANOVA. Not shown are 7 chemokines (CCL27, CCL3, CCL4, CXCL16, CXCL10, CCL12, and CCL17) and 9 cytokines (IFNα, IFNγ IL-1β, IL-2, IL-4, IL-7, RANK-L, M-CSF, and VEGF-A) which were either undetected or unchanged after Lm infection.
3.6. L. monocytogenes associate with stromal cells during foodborne infection of mice.
The results described above indicated that FRC and BEC could serve as an intracellular growth niche for L. monocytogenes when the bacteria were added directly to purified cells in a controlled in vitro environment. We next questioned whether infection of either of these stromal subsets occurred in vivo following foodborne transmission of L. monocytogenes. To begin to address this, we first confirmed that gp38 and CD31 could effectively be used to distinguish the four LNSC subsets in the context of a bacterial infection since the surface levels of some proteins do change in inflammatory environments. MLN were harvested at 48- and 72- hours post-infection, timepoints when L. monocytogenes are consistently recovered from the lymph nodes of all orally infected mice [19, 32]. As shown in Fig. 6A, expression profiles of gp38 and CD31 did not change in the MLN of infected mice, but the relative abundance of each stromal cell population was altered following foodborne infection (Fig. 6B). Notably, the two cell types found to support intracellular growth of L. monocytogenes ex vivo, FRC and BEC, increased in both percentage (Fig. 6C) and absolute number (Fig. 6B) of cells in the MLN at 48- and 72- hours post-infection. In contrast, the DNC population decreased significantly following foodborne infection, while the LEC population remained unchanged.
Fig. 6. L. monocytogenes associate with stromal cells following foodborne infection.
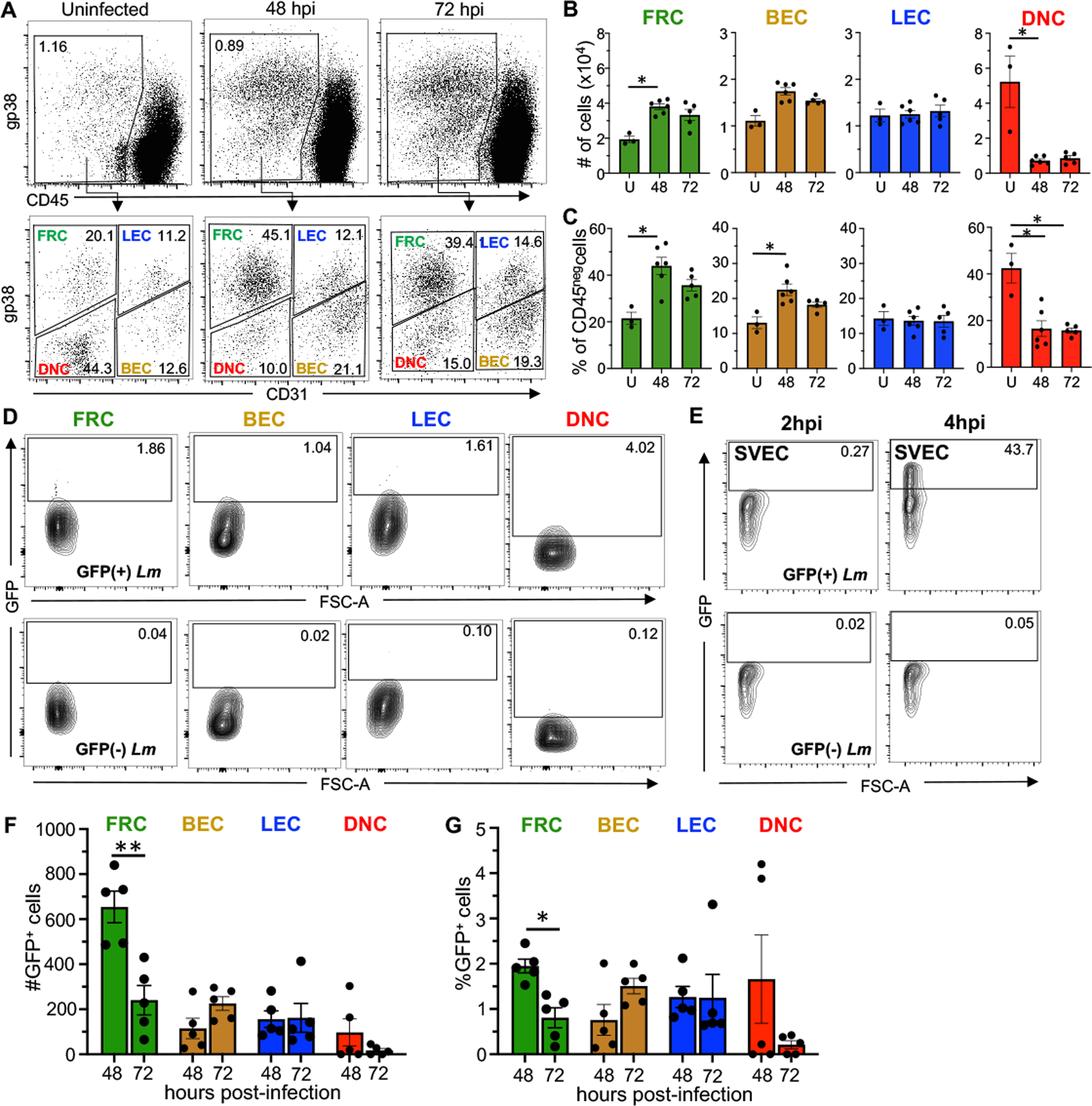
BALBc/ByJ mice were infected with 5 × 108 CFU of Lm SD2710 (GFP+) and MLN were harvested either 48 or 72 hpi. (A) Representative dot plots comparing stromal populations in the MLN of uninfected versus infected mice. (B) Total number and (C) Percentage of stromal cells (mean ± SEM) in the MLN of uninfected (U), 48 hpi, or 72 hpi mice analyzed by Mann-Whitney test. (D) Representative dot plots showing gating strategy to identify GFP positive (Lm-associated) cells. All plots were pre-gated as shown in panel A. (E) Representative dot plots showing gating strategy to identify GFP positive (Lm-associated) SVEC4–10 cells at 2 and 4 hpi (n=2) (F) Total number and (G) Percentage (±SEM) of GFP+ cells in each stromal population. Each symbol represents results from a single experiment (one infected and one uninfected mouse per experiment). Statistical significance was determined by Mann-Whitney test.
To determine if stromal cells were associated with L. monocytogenes during foodborne infection, we infected mice with L. monocytogenes that constitutively expressed GFP and harvested the MLN at 48 and 72 hours post-infection. The percentage of GFP-positive LNSC was determined by comparison to LNSC isolated from mice fed L. monocytogenes that lacked GFP. As shown in Fig. 6D, all of the GFP-positive LNSC had only low levels of fluorescence. As a positive control, we infected SVEC4–10 cells in vitro with the same Listeria strains and assessed their fluorescence by flow cytometry at 2 and 4 hours post-infection. At 2 hours post-infection, we expected to find a very small number of Listeria associated with each cell (see Fig. 1C) and in fact, the GFP signal was very weak (Fig. 6E). In contrast, by 4 hours post-infection when the number of intracellular bacteria was expected to have increased exponentially (Fig. 1C), the GFP signal was much stronger. Thus, we interpret a weakly GFP-positive cell as identified by flow cytometry to be a cell that is infected with only one or perhaps a few Listeria.
Overall, we found that less than 5% of the stromal cells were infected with L. monocytogenes following foodborne infection. FRC were associated with L. monocytogenes at both time points during infection, however there were significantly fewer bacteria associated with FRC at 72 hours post-infection, suggesting that FRC infection peaks early and then begins to decline (Fig. 6F). The inverse was observed for BEC, with more GFP+ cells detected at 72 hours, although the difference was not statistically significant (Fig. 6F and 6G). Approximately 1% of the LEC were associated with Listeria at both time points (Fig. 6G). DNC gave variable results at 48 hours post-infection, with a relatively high level of GFP+ cells in two animals, but no GFP+ cells in the other three (Fig. 6F and 6G). These results suggest that the bacteria may associate with DNC for only a short period of time early during infection, which would be consistent with the ex vivo infections showing a continual decrease in the number of CFU detected.
We next questioned whether the L. monocytogenes associated with these cells were intracellular or extracellular bacteria. To test this, we infected animals with GFP-expressing L. monocytogenes and harvested MLN at 48, 60, and 72 hours post-infection. We performed differential ‘in/out’ staining and analyzed the sorted populations by immunofluorescence microscopy. As expected, FRC, LEC, and BEC all contained intracellular bacteria (Fig. 7A). When we assessed how many L. monocytogenes were found within each cell, FRC and BEC had significantly more intracellular bacteria (8–12) per cell at 72 hours post-infection than at earlier timepoints (Fig. 7B). Additionally, we found that that the percentage of GFP positive FRC significantly decreased by 72 hours post-infection, while the opposite phenotype was observed in BEC (Fig. 7C). Thus, these results confirmed the infection patterns we observed using the flow cytometric approach shown Fig. 6E. In contrast, the amount of intracellular L. monocytogenes in LEC and the percentage of GFP+ cells stayed relatively consistent throughout infection. Unsurprisingly, we did not observe any extracellular or intracellular L. monocytogenes associated with DNC, indicating that these cells were not well infected in vivo (Fig. 7A).
Fig. 7. Live intracellular L. monocytogenes can be found within stromal populations following foodborne infection.
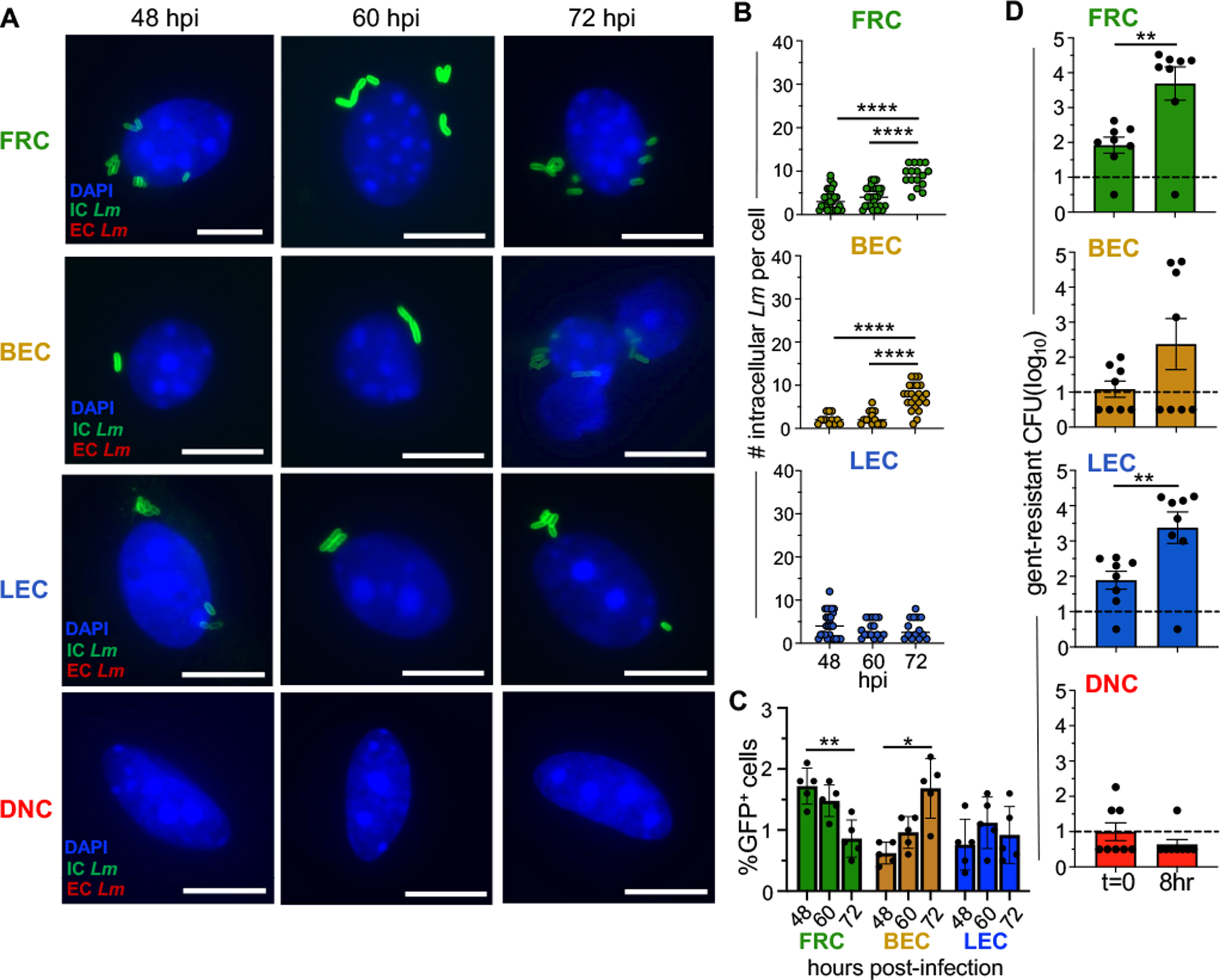
BALBc/ByJ mice were infected with 5 × 108 CFU of Lm SD2710 and stromal populations were sort-purified from pooled MLN at 48, 60 and 72 hpi. (A) Representative images for differential ”in/out” staining of stromal populations infected with Lm SD2710 at 48– 60- and 72-hours post-infection. Scale bar, 5 μm (B) Symbols represent individual cells in five experiments. Horizontal lines indicate mean values. Statistical significance was determined by ANOVA with Tukeys multiple comparison. At least 1500 cells of each population were visualized. (C) Average percentage (±SEM) of GFP+ cells in each stromal population. Each symbol represents results from a single experiment. Statistical significance was determined by Two-way ANOVA with Tukey’s multiple comparisons. (D) Symbols indicate the number of gentamicin-resistant L. monocytogenes found within stromal populations at 48 hpi immediately post-sort (t=0) or after an 8 h incubation. Bars indicate mean value (±SEM); Dashed line indicate the limit of detection. Statistical significance was determined by Mann-Whitney analysis.
To determine if these intracellular L. monocytogenes were viable and capable of replicating, we sort-purified infected cells from mice 48 hours after foodborne challenge and then incubated them at 37°C in the presence of gentamicin to allow for the growth of any intracellular bacteria. One-half of each population was incubated in gentamicin for 20 minutes and then lysed to determine how many intracellular L. monocytogenes were within the cells immediately post-sort. The remaining cells were incubated in gentamicin for an additional 8 hours to assess intracellular replication ex vivo. Notably, gentamicin-resistant L. monocytogenes significantly increased in the FRC population within 8 hours compared to the bacteria recovered immediately post-sort (Fig. 7D), a result similar to FRC exposed to L. monocytogenes ex vivo (Fig. 4A). We found that only half of the mice harbored BEC that contained gentamicin-resistant bacteria, however, the bacteria within this subset increased 100-fold within 8 hours. LEC recovered from infected mice also harbored intracellular L. monocytogenes, and surprisingly, the number of gentamicin-resistant bacteria in LEC increased significantly during ex vivo incubation. This was in stark contrast to our findings using sort purified cells that were infected in vitro, suggesting that there may be a signal in vivo that alters these cells into a growth niche for L. monocytogenes. Consistent with our previous findings, few animals had DNC that contained gentamicin-resistant L. monocytogenes (Fig. 7D). Together, these results suggest that viable, replicating intracellular L. monocytogenes can be found within MLN stromal cells following foodborne infection.
4. Discussion
In this study, we demonstrated that two LNSC subsets, FRC and BEC, could support exponential growth of L. monocytogenes and that replicating intracellular bacteria were found within these cells following foodborne infection of mice. Prior to this report, it was long thought that macrophages were the primary cell type in the MLN that served as a growth niche for L. monocytogenes. Macrophages are very likely to be a major site of intracellular growth in lymphoid tissues until they become activated by IFNγ [16, 38, 39], but our data indicate that other cells within the MLN also need to be considered. Intracellular replication and subsequent cell-to-cell spread of L. monocytogenes is essential for rapid egress from the MLN [15], but the critical cell type(s) in the MLN that are required for bacterial access to the bloodstream are still unresolved. Although L. monocytogenes can replicate in a wide variety of transformed cell lines, we previously showed that neither Ly6Chi monocytes [16] nor conventional dendritic cells [17] isolated from the MLN were permissive for the intracellular replication. Identifying all the possible intracellular growth niches for L. monocytogenes in the MLN is essential to gain insights into the in vivo progression and systemic spread of these facultative intracellular pathogens.
Studies investigating the involvement of LNSC in the clearance of bacterial infections are limited. LNSC are difficult to work with and are often overlooked since they comprise only about 1% of the cells found in lymph nodes [1]. However, LNSC are perfectly positioned to interact with infiltrating pathogens as they traffic through the lymph node, and there is some evidence to suggest that these cells can be important for bacterial virulence. For example, St John and Abraham showed that interactions between Salmonella-derived LPS and BEC in the mesenteric lymph node (MLN) resulted in the upregulation of suppressor cytokine signaling-3 expression, which in turn, disrupted secretion of homeostatic chemokines CCL21 and CXCL13 produced by BEC [40]. The absence of these chemokines disrupted immune cell trafficking, which allowed the bacteria to persist. In another study, RNA-Seq analysis revealed that LNSC can upregulate MHC-II machinery in response to LPS [1]. Although this typically results in the generation of T regulatory cells or T cell apoptosis during steady state conditions [1], it is not yet clear how the presentation of antigen via MHC-II by LNSC would influence the inflammatory environment during L. monocytogenes infection. Both these studies suggest that the role of LNSC can shift significantly during infection.
The primary virulence strategy of L. monocytogenes is to spread from cell-to-cell to avoid extracellular immune defenses. However, the lymph node contains an abundance of circulating immune cells that lack permanent cellular junctions and thus, do not readily allow for this mode of spread. FRC, which are present in nearly every lymph node compartment, may provide a three-dimensional network for spread of L. monocytogenes [6, 41]. Given that FRC are in close proximity to one another and other cells within the lymph node, one could speculate that L. monocytogenes may preferentially target FRC early during the infection, allowing the bacteria to spread to different regions of the node while avoiding extracellular immune recognition. In fact, FRC have been shown to be a known key target within lymphoid tissues for several different viral infections [42–46]. This idea is also consistent with our previous observation that cell-to-cell spread is required for rapid egress of L. monocytogenes from the MLN [15], indicating that the critical cellular niche must be closely situated to other cells.
We found that L. monocytogenes replicated with a rapid growth rate within BEC, but the bacteria did not invade this subset very efficiently. Surprisingly, there was a notable bimodal distribution in the amount of intracellular L. monocytogenes found within BEC at 8 hpi. One possibility for this result could be that specific subsets of BEC exhibit varying susceptibilities to L. monocytogenes infections. Alternatively, there might be a slight delay in bacterial vacuole escape, resulting in delayed exponential replication in some cells. BEC are positioned directly underneath FRC, and together, they create the high endothelial venules located in the T cell zone of the lymph node. Thus, the escape of bacteria from an infected BEC could provide direct access to the bloodstream, and we show here that the BEC are more heavily infected later during the course of the infection when the bacteria begin to spread systemically. We speculate that in vivo L. monocytogenes may initially invade the perivascular FRC and then use actin-based motility to spread into the underlying BEC rather than directly invade BEC. Supporting this idea, a study by Davis et al. showed a similar phenomenon in the MLN of Ebola virus-infected African green monkeys [42]. Electron microscopy images revealed that virally infected FRC surrounding high endothelial venules contained inclusions and budding virions that were later found in the underlying endothelium and bloodstream. Spreading from infected FRC into BEC could serve as an effective strategy for L. monocytogenes to also spread systemically.
Our data suggest that FRC and BEC may play a significant role in enhancing both the innate and adaptive immune response against L. monocytogenes. In addition to producing a variety of chemokines for the recruitment of immune cells into the node, we found that FRC produced a significant amount of IL-6 in response to L. monocytogenes. Brown et al. showed that FRC-derived IL-6 enhanced IL-2 and TNF-α production, as well as chromatin remodeling in activated CD8+ T cells [47]. L. monocytogenes is known to elicit a robust CD8 T cell response, and these cells play a pivotal role in resolving the infection. Outside the lymph node, cytokine production by stromal cells was shown to block reinfection of intestinal epithelial cells. Disson et al. found that L. monocytogenes that invaded Peyer’s patches in gut triggered the production of IL-11 by gp38+ stromal cells in the underlying lamina propria [48]. This activated STAT3-dependent secretion of IFNγ, resulting in increased epithelial proliferation, decreased goblet cell maturation, and importantly, decreased accessibility to E-cadherin which blocked further InlA-mediated invasion of the intestinal villi. IL-11 was not included in our multiplex immunoassay, however, the idea that critical inflammatory mediators may be produced by infected stromal cells and that this can influence the course of L. monocytogenes survival and dissemination is likely to apply to other sites in the body such as the lymph node.
We observed a noteworthy difference in the ability of LEC to support intracellular growth of L. monocytogenes when comparing cells infected during foodborne challenge of mice and cells purified from naïve mice and then infected ex vivo. L. monocytogenes are auxotrophic for several nutrients that can be provided by the host. For example, we previously demonstrated that exogenous lipoate was severely limited in the extracellular environment of the MLN and Listeria could only obtain this nutrient within the cytosol of a mammalian cell [15]. It is possible that LEC have limited quantities of an essential nutrient at steady state that L. monocytogenes require for growth. The metabolic profile of LEC changed significantly when the cells were co-cultured with a breast cancer cell line [49], so it is possible that soluble factors produced during the inflammatory response to infection alter the cells in a way that promotes cytosolic replication of L. monocytogenes. Alternatively, the absence of L. monocytogenes growth noted in vitro might stem from a stress response triggered during the processing of the tissue and manipulation of the cells to achieve highly purified populations. However, it is worth noting that this was not observed in the case of FRC and BEC.
During L. monocytogenes foodborne infection, there was a significant shift in the proportions of FRC and BEC in the mesenteric lymph nodes. Since these cells have a critical structural role it makes sense that they would need to expand to accommodate the increased number of phagocytes and lymphocytes in the node. Likewise, other studies have shown that FRC and BEC both increase in response to viral [50, 51] and parasitic infections (Dubey et al., 2016). However, Pezoldt et al. found that during a Y. pseudotuberculosis bacterial infection, FRC decreased in number by 3 days post-infection [33]. This might be attributed to the fact that Y. pseudotuberculosis has been shown to cause caseous necrosis of the MLN [52]. Interestingly, LEC proportions were unchanged following L. monocytogenes infection. Lucas et al. showed that both type I and type II interferons can inhibit LEC division following various inflammatory stimuli [53]. Thus, the lack of change in the LEC population in our study could be due to interferon release in response to L. monocytogenes.
DNC are certainly the most understudied of all the stromal populations, but this study suggests that they may play a role during L. monocytogenes infection. Although DNC did not support the intracellular growth of L. monocytogenes ex vivo, they still produced cytokines and chemokines in response to the bacteria. These cells upregulated expression of IL-6, CXCL5 and CCL11, indicating that the DNC also contribute to the pro-inflammatory environment during L. monocytogenes infection. The DNC population decreased significantly during foodborne infection of mice. We speculate that DNC may consist of immature FRC or hematopoietic cells that have downregulated CD45 expression, and that the DNC may further differentiate in the setting of an inflammatory response such as occurs during L. monocytogenes infection. Identifying distinguishable surface markers on DNC in the future would significantly improve investigations into this particular cell subset.
The migration of L. monocytogenes through the spleen has been well-studied using the intravenous model of inoculation. It was previously shown that L. monocytogenes is quickly filtered from the bloodstream into the marginal sinus of the spleen, a compartment that resembles the subcapsular sinus in the MLN, in that it is organized by stromal cells and resident macrophages that quickly phagocytize L. monocytogenes [54–60]. Within 24 hours, the bacteria are localized within the periarteriolar lymphoid sheaths surrounded by T cells [61, 62]. Although dendritic cells are implicated in the process, the exact mechanism is not well understood [63]. Only recently have research efforts shifted towards exploring the mechanism L. monocytogenes uses to spread systemically following foodborne transmission and the critical cellular niche in the MLN required for this escape [15–17, 64]. It is now clear from this study that both FRC and BEC can support the growth of L. monocytogenes, and any model of how L. monocytogenes overcomes the MLN barrier should take these potential growth niches into account. We predict that L. monocytogenes readily invade perivascular FRC, replicate in the cytosol, and use actin-based motility to spread into adjacent BEC. The rapid exponential intracellular growth we observed in BEC could lead to cellular lysis or membrane damage that would allow for bacteria to escape into the bloodstream. However, many unanswered questions remain that are essential for refining this model. Future studies should focus on determining how L. monocytogenes migrate through the lymph node from the afferent lymphatics to the T cell zone and what role, if any, FRC play in this process.
Supplementary Material
Acknowledgments
We thank Jennifer Strange and Siva K. Gandhapudi for technical assistance in the University of Kentucky Flow Cytometry and Immune Monitoring Core Facility. This work was supported by National Institute of Health grant R21AI151482 to S.E.F.D.
Abbreviations:
- BEC
blood endothelial cells
- BHI
brain heart infusion
- CFU
colony forming unit
- cMLN
colon-draining mesenteric lymph nodes
- DNC
double negative cells
- FRC
fibroblastic reticular cells
- LEC
lymphoid endothelial cells
- LNSC
lymph node stromal cells
- sMLN
small intestine draining mesenteric lymph nodes
Footnotes
Disclosures
The authors declare no conflict of interest.
References
- 1.Malhotra D, et al. , Transcriptional profiling of stroma from inflamed and resting lymph nodes defines immunological hallmarks. Nat Immunol, 2012. 13(5): p. 499–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rodda LB, et al. , Single-Cell RNA Sequencing of Lymph Node Stromal Cells Reveals Niche-Associated Heterogeneity. Immunity, 2018. 48(5): p. 1014–1028 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kapoor VN, et al. , Gremlin 1(+) fibroblastic niche maintains dendritic cell homeostasis in lymphoid tissues. Nat Immunol, 2021. 22(5): p. 571–585. [DOI] [PubMed] [Google Scholar]
- 4.Jarjour M, et al. , Fate mapping reveals origin and dynamics of lymph node follicular dendritic cells. J Exp Med, 2014. 211(6): p. 1109–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dubey LK, et al. , Lymphotoxin-Dependent B Cell-FRC Crosstalk Promotes De Novo Follicle Formation and Antibody Production following Intestinal Helminth Infection. Cell Rep, 2016. 15(7): p. 1527–1541. [DOI] [PubMed] [Google Scholar]
- 6.Link A, et al. , Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat Immunol, 2007. 8(11): p. 1255–65. [DOI] [PubMed] [Google Scholar]
- 7.Jalkanen S and Salmi M, Lymphatic endothelial cells of the lymph node. Nat Rev Immunol, 2020. 20(9): p. 566–578. [DOI] [PubMed] [Google Scholar]
- 8.Xiang M, et al. , A Single-Cell Transcriptional Roadmap of the Mouse and Human Lymph Node Lymphatic Vasculature. Front Cardiovasc Med, 2020. 7: p. 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gowans JL and Knight EJ, The Route of Re-Circulation of Lymphocytes in the Rat. Proc R Soc Lond B Biol Sci, 1964. 159: p. 257–82. [DOI] [PubMed] [Google Scholar]
- 10.Marchesi VT and Gowans JL, The Migration of Lymphocytes through the Endothelium of Venules in Lymph Nodes: An Electron Microscope Study. Proc R Soc Lond B Biol Sci, 1964. 159: p. 283–90. [DOI] [PubMed] [Google Scholar]
- 11.Low S, et al. , Role of MAdCAM-1-Expressing High Endothelial Venule-Like Vessels in Colitis Induced in Mice Lacking Sulfotransferases Catalyzing L-Selectin Ligand Biosynthesis. J Histochem Cytochem, 2018. 66(6): p. 415–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gedde MM, et al. , Role of listeriolysin O in cell-to-cell spread of Listeria monocytogenes. Infect Immun, 2000. 68(2): p. 999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marquis H, Doshi V, and Portnoy DA, The broad-range phospholipase C and a metalloprotease mediate listeriolysin O-independent escape of Listeria monocytogenes from a primary vacuole in human epithelial cells. Infect Immun, 1995. 63(11): p. 4531–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dabiri GA, et al. , Listeria monocytogenes moves rapidly through the host-cell cytoplasm by inducing directional actin assembly. Proc Natl Acad Sci U S A, 1990. 87(16): p. 6068–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tucker JS, et al. , Egress of Listeria monocytogenes from Mesenteric Lymph Nodes Depends on Intracellular Replication and Cell-to-Cell Spread. Infect Immun, 2023: p. e0006423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones GS and D’Orazio SE, Monocytes Are the Predominant Cell Type Associated with Listeria monocytogenes in the Gut, but They Do Not Serve as an Intracellular Growth Niche. J Immunol, 2017. 198(7): p. 2796–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones GS, Smith VC, and D’Orazio SEF, Listeria monocytogenes Replicate in Bone Marrow-Derived CD11c(+) Cells but Not in Dendritic Cells Isolated from the Murine Gastrointestinal Tract. J Immunol, 2017. 199(11): p. 3789–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wollert T, et al. , Extending the host range of Listeria monocytogenes by rational protein design. Cell, 2007. 129(5): p. 891–902. [DOI] [PubMed] [Google Scholar]
- 19.Jones GS, et al. , Intracellular Listeria monocytogenes comprises a minimal but vital fraction of the intestinal burden following foodborne infection. Infect Immun, 2015. 83(8): p. 3146–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones GS and D’Orazio SE, Listeria monocytogenes: cultivation and laboratory maintenance. Curr Protoc Microbiol, 2013. 31: p. 9B 2 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marquis H, Tissue culture cell assays used to analyze Listeria monocytogenes. Curr Protoc Microbiol, 2006. Chapter 9: p. 9B 4 1–9B 4 25. [DOI] [PubMed] [Google Scholar]
- 22.Houston SA, et al. , The lymph nodes draining the small intestine and colon are anatomically separate and immunologically distinct. Mucosal Immunol, 2016. 9(2): p. 468–78. [DOI] [PubMed] [Google Scholar]
- 23.Fletcher AL, et al. , Reproducible isolation of lymph node stromal cells reveals site-dependent differences in fibroblastic reticular cells. Front Immunol, 2011. 2: p. 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bou Ghanem EN, et al. , Oral transmission of Listeria monocytogenes in mice via ingestion of contaminated food. J Vis Exp, 2013(75): p. e50381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bou Ghanem EN, Myers-Morales T, and D’Orazio SEF, A mouse model of foodborne Listeria monocytogenes infection. Curr Protoc Microbiol, 2013. 31: p. 9B 3 1–9B 3 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O’Connell KA and Edidin M, A mouse lymphoid endothelial cell line immortalized by simian virus 40 binds lymphocytes and retains functional characteristics of normal endothelial cells. J Immunol, 1990. 144(2): p. 521–5. [PubMed] [Google Scholar]
- 27.Gaillard JL, et al. , In vitro model of penetration and intracellular growth of Listeria monocytogenes in the human enterocyte-like cell line Caco-2. Infect Immun, 1987. 55(11): p. 2822–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monk IR, et al. , Directed evolution and targeted mutagenesis to murinize Listeria monocytogenes internalin A for enhanced infectivity in the murine oral infection model. BMC Microbiol, 2010. 10: p. 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kocks C, et al. , Polarized distribution of Listeria monocytogenes surface protein ActA at the site of directional actin assembly. J Cell Sci, 1993. 105 (Pt 3): p. 699–710. [DOI] [PubMed] [Google Scholar]
- 30.Sun AN, Camilli A, and Portnoy DA, Isolation of Listeria monocytogenes small-plaque mutants defective for intracellular growth and cell-to-cell spread. Infect Immun, 1990. 58(11): p. 3770–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kho ZY and Lal SK, The Human Gut Microbiome - A Potential Controller of Wellness and Disease. Front Microbiol, 2018. 9: p. 1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bou Ghanem EN, et al. , InlA promotes dissemination of Listeria monocytogenes to the mesenteric lymph nodes during food borne infection of mice. PLoS Pathog, 2012. 8(11): p. e1003015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pezoldt J, et al. , Neonatally imprinted stromal cell subsets induce tolerogenic dendritic cells in mesenteric lymph nodes. Nat Commun, 2018. 9(1): p. 3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woodward JJ, Iavarone AT, and Portnoy DA, c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science, 2010. 328(5986): p. 1703–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stetson DB and Medzhitov R, Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity, 2006. 24(1): p. 93–103. [DOI] [PubMed] [Google Scholar]
- 36.Muller G, Hopken UE, and Lipp M, The impact of CCR7 and CXCR5 on lymphoid organ development and systemic immunity. Immunol Rev, 2003. 195: p. 117–35. [DOI] [PubMed] [Google Scholar]
- 37.Melton-Witt JA, et al. , Oral infection with signature-tagged Listeria monocytogenes reveals organ-specific growth and dissemination routes in guinea pigs. Infect Immun, 2012. 80(2): p. 720–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Biroum N, Listericidal activity of non-stimulated and stimulated human macrophages in vitro. Clin Exp Immunol, 1977. 28(1): p. 138–45. [PMC free article] [PubMed] [Google Scholar]
- 39.Shaughnessy LM and Swanson JA, The role of the activated macrophage in clearing Listeria monocytogenes infection. Front Biosci, 2007. 12: p. 2683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.St John AL and Abraham SN, Salmonella disrupts lymph node architecture by TLR4-mediated suppression of homeostatic chemokines. Nat Med, 2009. 15(11): p. 1259–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Katakai T, et al. , Lymph node fibroblastic reticular cells construct the stromal reticulum via contact with lymphocytes. J Exp Med, 2004. 200(6): p. 783–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davis KJ, et al. , Pathology of experimental Ebola virus infection in African green monkeys. Involvement of fibroblastic reticular cells. Arch Pathol Lab Med, 1997. 121(8): p. 805–19. [PubMed] [Google Scholar]
- 43.Twenhafel NA, et al. , Pathology of experimental aerosol Zaire ebolavirus infection in rhesus macaques. Vet Pathol, 2013. 50(3): p. 514–29. [DOI] [PubMed] [Google Scholar]
- 44.Steele KE, Anderson AO, and Mohamadzadeh M, Fibroblastic reticular cell infection by hemorrhagic fever viruses. Immunotherapy, 2009. 1(2): p. 187–97. [DOI] [PubMed] [Google Scholar]
- 45.Mueller SN, et al. , Viral targeting of fibroblastic reticular cells contributes to immunosuppression and persistence during chronic infection. Proc Natl Acad Sci U S A, 2007. 104(39): p. 15430–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ng CT, et al. , Immortalized clones of fibroblastic reticular cells activate virus-specific T cells during virus infection. Proc Natl Acad Sci U S A, 2012. 109(20): p. 7823–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brown FD, et al. , Fibroblastic reticular cells enhance T cell metabolism and survival via epigenetic remodeling. Nat Immunol, 2019. 20(12): p. 1668–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Disson O, et al. , Peyer’s patch myeloid cells infection by Listeria signals through gp38(+) stromal cells and locks intestinal villus invasion. J Exp Med, 2018. 215(11): p. 2936–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Acevedo-Acevedo S, et al. , Metabolomics revealed the influence of breast cancer on lymphatic endothelial cell metabolism, metabolic crosstalk, and lymphangiogenic signaling in co-culture. Sci Rep, 2020. 10(1): p. 21244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abe J, et al. , Lymph node stromal cells negatively regulate antigen-specific CD4+ T cell responses. J Immunol, 2014. 193(4): p. 1636–44. [DOI] [PubMed] [Google Scholar]
- 51.Gregory JL, et al. , Infection Programs Sustained Lymphoid Stromal Cell Responses and Shapes Lymph Node Remodeling upon Secondary Challenge. Cell Rep, 2017. 18(2): p. 406–418. [DOI] [PubMed] [Google Scholar]
- 52.El-Maraghi NR and Mair NS, The histopathology of enteric infection with Yersinia pseudotuberculosis. Am J Clin Pathol, 1979. 71(6): p. 631–9. [DOI] [PubMed] [Google Scholar]
- 53.Lucas ED, et al. , Type 1 IFN and PD-L1 Coordinate Lymphatic Endothelial Cell Expansion and Contraction during an Inflammatory Immune Response. J Immunol, 2018. 201(6): p. 1735–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hardy J, Chu P, and Contag CH, Foci of Listeria monocytogenes persist in the bone marrow. Dis Model Mech, 2009. 2(1–2): p. 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mitchell LM, et al. , Distinct responses of splenic dendritic cell subsets to infection with Listeria monocytogenes: maturation phenotype, level of infection, and T cell priming capacity ex vivo. Cell Immunol, 2011. 268(2): p. 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Edelson BT, et al. , CD8alpha(+) dendritic cells are an obligate cellular entry point for productive infection by Listeria monocytogenes. Immunity, 2011. 35(2): p. 236–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Neuenhahn M, et al. , CD8alpha+ dendritic cells are required for efficient entry of Listeria monocytogenes into the spleen. Immunity, 2006. 25(4): p. 619–30. [DOI] [PubMed] [Google Scholar]
- 58.Conlan JW, Early pathogenesis of Listeria monocytogenes infection in the mouse spleen. Journal of Medical Microbiology, 1996. 44(4): p. 295–302. [DOI] [PubMed] [Google Scholar]
- 59.Aichele P, et al. , Macrophages of the splenic marginal zone are essential for trapping of blood-borne particulate antigen but dispensable for induction of specific T cell responses. Journal of Immunology, 2003. 171(3): p. 1148–1155. [DOI] [PubMed] [Google Scholar]
- 60.den Haan JM, Mebius RE, and Kraal G, Stromal cells of the mouse spleen. Front Immunol, 2012. 3: p. 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aoshi T, et al. , Bacterial entry to the splenic white pulp initiates antigen presentation to CD8+ T cells. Immunity, 2008. 29(3): p. 476–86. [DOI] [PubMed] [Google Scholar]
- 62.Berg RE, et al. , Relative contributions of NK and CD8 T cells to IFN-gamma mediated innate immune protection against Listeria monocytogenes. Journal of Immunology, 2005. 175(3): p. 1751–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perez OA, et al. , CD169(+) macrophages orchestrate innate immune responses by regulating bacterial localization in the spleen. Sci Immunol, 2017. 2(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Imperato JN, et al. , Mucosal CD8 T Cell Responses Are Shaped by Batf3-DC After Foodborne Listeria monocytogenes Infection. Front Immunol, 2020. 11: p. 575967. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.