

Summary
Drug efflux transporters are a major determinant of drug efficacy and toxicity. A canonical example is P-glycoprotein (P-gp), an efflux transporter that controls the intestinal absorption of diverse compounds. Despite a rich literature on the dietary and pharmaceutical compounds that impact P-gp activity, its sensitivity to gut microbial metabolites remains an open question. Surprisingly, we found that the cardiac drug-metabolizing gut Actinobacterium Eggerthella lenta increases drug absorption in mice. Experiments in cell culture revealed that E. lenta produces a soluble factor that post-translationally inhibits P-gp ATPase efflux activity. P-gp inhibition is conserved in the Eggerthellaceae family but absent in other Actinobacteria. Comparative genomics identified genes associated with P-gp inhibition. Finally, activity-guided biochemical fractionation coupled to metabolomics implicated a group of small polar metabolites with P-gp inhibitory activity. These results highlight the importance of considering the broader relevance of the gut microbiome for drug disposition beyond first-pass metabolism.
Subject areas: Pharmacology, Natural sciences, Biological sciences, Microbiology, Microbiome
Graphical abstract
Highlights
-
•
The gut Actinobacterium Eggerthella lenta increases drug absorption in mice
-
•
A secreted factor produced by E. lenta inhibits P-glycoprotein ATPase activity
-
•
P-glycoprotein inhibition is conserved within the Eggerthellaceae family
-
•
Activity-guided fractionation implicated a group of small polar metabolites
Pharmacology; Natural sciences; Biological sciences; Microbiology; Microbiome
Introduction
Drug metabolism and disposition depend upon both genetic and environmental factors that shape intestinal metabolism, transport across the intestinal epithelium, and first-pass metabolism in the liver.1,2 For the past decade, research at the interface of the microbiome and pharmacology has focused on the metabolism of drugs within the gastrointestinal tract.3,4,5 While this focus is understandable given the immense and poorly characterized enzymatic diversity within the microbiome, emerging data have begun to suggest that the gut microbiome has far broader impacts on drug disposition.6,7 Gut bacterial metabolism of common food and drug additives (i.e., excipients) increases drug bioavailability by lifting the inhibition of an intestinal drug influx transporter.8 However, it remains unclear if the diverse chemicals produced by the gut microbiome9 can also control drug influx and/or efflux transporters, and if so, whether or not these host-microbiome interactions have physiologically relevant consequences for drug bioavailability.7
Here, we begin to address this major gap in our knowledge. We opted to focus on P-glycoprotein (P-gp) (encoded by the ABCB1 gene in humans and Abcb1a in mice) due in part to its broad relevance for >300 known endogenous and exogenous substrates, including the cardiac drug digoxin and the anti-cancer agents doxorubicin and paclitaxel.10 Abcb1a can be differentially expressed between germ-free (GF) and conventionally raised (CONV-R) mice11,12; however, a recent study failed to detect a significant difference in Abcb1a transcript levels.13 Variation in the specific gut microbiota found between facilities could provide one potential reason for these discrepant results, consistent with experiments administering single pathogenic, probiotic, and commensal bacteria that can tune Abcb1a expression in either direction,11,14,15,16 experiments with broad-spectrum antibiotics,17 and a study implicating butyrate-producing bacteria in colonic P-gp protein levels.18 More importantly, the functional consequences of microbiota-driven changes in Abcb1a expression and the mechanisms responsible remain largely unexplored, with the exception of a recent paper implicating microbiota-dependent induction of Abcb1a expression in tacrolimus pharmacokinetics.17
Our prior work on the cardiac drug digoxin also motivated us to consider P-gp and its relationship to the gut microbiome. Digoxin is both a substrate for gut bacterial metabolism19 and a model substrate for P-gp efflux.20 We previously identified a two-gene operon (the cardiac glycoside reductase operon, cgr) which predicts strain-level variation in the metabolism of digoxin by the prevalent gut Actinobacterium Eggerthella lenta.21 The cgr2 gene was sufficient to catalyze digoxin reduction in a heterologous expression system, and biochemical characterization of the Cgr2 enzyme suggested that its substrate scope was restricted to cardenolides.22 Surprisingly, we also discovered that Cgr2 is necessary and sufficient for the induction of colonic T-helper 17 cells,23,24 suggesting that dietary and/or host substrates can be reduced by this enzyme. Yet despite these mechanistic insights into the metabolic activity of E. lenta, the relative impacts of bacterial metabolism versus other types of host-microbiome interactions on drug disposition remain poorly understood, even for a drug as well-characterized as digoxin.
Results
E. lenta increases drug absorption in mice
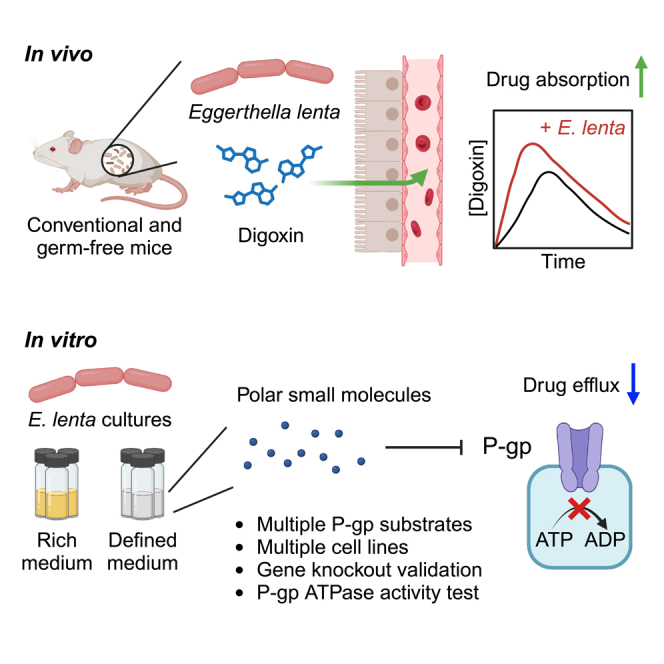
Given that E. lenta metabolizes the cardiac drug digoxin,21 we hypothesized that mono-colonization of GF mice with E. lenta DSM2243 would significantly decrease digoxin bioavailability by enhancing first-pass metabolism. 9-week-old mixed sex GF Swiss Webster mice were mono-colonized with E. lenta for 4 weeks followed by oral administration of 200 μg/kg digoxin. Colonization was confirmed by qPCR (Figure 1A). Surprisingly, E. lenta increased serum digoxin levels at the first time point (1-h post drug administration; Figure 1B), resulting in a higher maximum concentration (Cmax; Figure 1C) and lower time to reach the maximum concentration (Tmax; Figure 1D) relative to GF controls. Digoxin area under the curve (AUC) was unaffected (Figure 1E) due to significantly decreased digoxin concentrations at 4 and 5 h post-administration (Figure 1B). Taken together, these results suggest that E. lenta increases the rate of absorption of digoxin in gnotobiotic mice, balancing out the effects of bacterial drug metabolism.21
Figure 1.

E. lenta increases the rate of drug absorption
(A–E) 9-week-old GF mixed-sex Swiss Webster mice were mono-colonized with E. lenta DSM2243 for 4 weeks followed by a single oral administration of 200 μg/kg digoxin. (A) Quantification of E. lenta DSM2243 in grams of cecal content via qPCR. No amplification was detected (ND) in the entire GF group (n = 6–7 mice/group; Mann-Whitney test). (B) Serum digoxin was quantified at different time points in GF or E. lenta mono-colonized mice post-digoxin administration (n = 6–7 mice/group; mean± SEM; Wilcoxon test). From the pharmacokinetic (PK) curves, key PK parameters were quantified, such as (C) maximal serum concentration (Cmax), (D) the time at which concentrations are highest (Tmax), and (E) area under the curve (AUC).
(F–J) In a separate mouse model, 8- to 10-week-old female CONV-R Swiss Webster mice were gavaged 109 live E. lenta DSM2243 cells or sterile BHI+ media control for 7 consecutive days prior to oral administration of digoxin at 30 μg/kg. (F) E. lenta engraftment was quantified via qPCR using cecal content. The BHI-gavaged group had no detectable (ND) E. lenta (n = 6–7 mice/group; Mann-Whitney test).
(G) Serum digoxin was quantified by liquid chromatography-tandem mass spectrometry at different time points post-digoxin administration (n = 6–7 mice/group; mean± SEM; Wilcoxon test). From the PK curves, (H) Cmax, (I) Tmax, and (J) AUC were quantified (Wilcoxon test).
Next, we sought to test if the observed change in digoxin absorption in response to E. lenta was unique to GF mice, which have broad changes to gut physiology, including changes in the expression of numerous drug transporters relative to CONV-R-specific pathogen-free animals.12,13 To ensure consistent exposure to high levels of E. lenta, we orally gavaged 109 E. lenta colony-forming units or sterile media control for 7 consecutive days to 8- to 10-week-old female CONV-R Swiss Webster mice prior to the oral administration of digoxin. High levels of E. lenta were still detected in the cecum collected at the end of the experiment (Figure 1F). In this repeat experiment, we opted to use a lower dose of digoxin (30 μg/kg) to avoid saturating transporter effects for digoxin. The results were largely consistent with data from gnotobiotic mice (Figures 1A–1E). E. lenta significantly increased serum digoxin at the 1-h time point following drug administration (Figure 1G) and significantly increased Cmax (Figure 1H). There was a trend toward decreased Tmax (p value = 0.10, Wilcoxon test; Figure 1I). Overall AUC was unchanged, due to higher levels in the control mice at the later time points (Figure 1J). These results demonstrate that E. lenta increases the rate of digoxin absorption in the presence or absence of the mouse gut microbiota.
Human gut Actinobacterial metabolites inhibit P-gp efflux
Digoxin is a model substrate of the P-gp transporter,20 which is the only known mammalian mechanism that controls the absorption of digoxin. Drug-drug interactions resulting in altered P-gp function are known to disrupt digoxin pharmacokinetics, which can be dangerous due to its narrow therapeutic range.20 We hypothesized that E. lenta inhibits P-gp efflux, which would explain the elevated rate of digoxin absorption in mice (Figure 1). To avoid the many confounding factors in mice, we leveraged an established cell culture assay.15 Briefly, human enterocytes (T84 cells) were treated with rhodamine 123 (Rh123), a fluorescent P-gp substrate, to track efflux activity under different media conditions. As expected, the P-gp inhibitors cyclosporin A,15 vinblastine,15 and verapamil25 significantly increased intracellular Rh123 levels compared to the untreated or vehicle controls (Figure 2A). E. lenta cell-free supernatant (CFS) but not cell lysates resulted in significantly increased intracellular Rh123 accumulation (Figure 2A). The impact of E. lenta CFS was dose dependent (Figure 2B) and comparable in effect size to the purified pharmacological inhibitor controls (Figure 2A). These results suggest that one or more secreted metabolites produced by E. lenta inhibit P-gp.
Figure 2.

Eggerthella lenta inhibits P-gp efflux
(A) P-gp inhibition test of E. lenta DSM2243 cell pellet lysate and cell-free filter-sterilized supernatant (CFS) using rhodamine (Rh123) accumulation assay in T84 human enterocyte model (n = 3–4/condition, ANOVA with Tukey’s post hoc test). Vehicle (DMSO), cyclosporin A (CsA), vinblastine, and verapamil were included as controls.
(B) Dose-response test of E. lenta CFS on Rh123 accumulation (Pdose = 8.6e-13 ANOVA, n = 4 wells/condition).
(C) CFS Rh123 accumulation test on T84 ABCB1−/− knockout cells compared to parental T84 cells (see also Figure S1).
(D) CFS activity test in 3 other human enterocyte cell lines. Activity tests in T84 cells were also completed using accumulation of other P-gp substrates, such as (E) calcein-AM and (F) doxorubicin (DOX).
(G) Synergistic effects of E. lenta CFS and DOX were tested using MTT viability cell assay. DMSO was used as vehicle control. Unless otherwise noted, E. lenta was cultured in BHI+. n = 4/condition and two-way ANOVA with Sidak’s multiple testing correction. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
We used three independent approaches to validate the inhibition of P-gp by E. lenta CFS: (1) gene deletion, (2) alternative cell lines, and (3) alternative substrates. First, an ABCB1 knockout monoclonal cell line from parental T84 cells was generated via lentiviral delivery of cas9 and a guide RNA against ABCB1. Successful gene deletion was confirmed via sequencing of ABCB1 gene locus, western blot, and activity testing (Figure S1). Similar to the verapamil-positive control, incubation of E. lenta CFS no longer increased intracellular Rh123 levels in the ABCB1 knockout cell line, suggesting that the mechanism is P-gp dependent and consistent with the increased basal accumulation of Rh123 in ABCB1 knockout cells (Figure 2C). Second, incubation with E. lenta CFS led to high intracellular Rh123 concentrations in 3 other human enterocyte models (Caco-2, HCT-15, and HCT-116), indicating that inhibition is reproducible across cell lines (Figure 2D). Third, we validated the impact of E. lenta CFS on P-gp by measuring the accumulation of two additional P-gp substrates: calcein-AM26 (Figure 2E) and the anti-cancer drug doxorubicin26 (Figure 2F). Inhibition of P-gp efflux by E. lenta CFS led to a significant decrease in cancer cell viability in response to doxorubicin, consistent with the known P-gp inhibitor verapamil25 (Figure 2G). Taken together, these results indicate that one or more metabolites secreted by E. lenta robustly inhibit P-gp efflux across multiple cell lines.
E. lenta inhibits P-gp ATPase activity
Small molecules and bacterial toxins can inhibit P-gp function through multiple mechanisms (Figure 3A, and Table S1). Gene expression and protein levels did not explain the observed P-gp inhibition by E. lenta. ABCB1 transcript levels were significantly increased in response to E. lenta CFS in 3/4 cell lines tested (Figure 3B). A major mechanism by which cancer cells upregulate P-gp transcription is via activation of the pregnane X receptor (PXR).27 Using the human PXR luminescence reporter cell line,28 we found that the media or E. lenta CFS had no significant impact on PXR activation (Figure S2). However, in the presence of PXR agonist rifampicin, the media, and to a greater extent, CFS, both enhance PXR activation (Figure S2), suggesting that there must be endogenous PXR agonists produced by cell lines that induce P-gp transcription when incubated with E. lenta CFS. However, the increase in P-gp transcription did not translate to any changes in P-gp protein levels (Figure 3C), suggesting that the major mechanism of P-gp inhibition is post-translational in nature.
Figure 3.

E. lenta inhibits P-gp ATPase activity
(A) Possible mechanisms of P-gp inhibition reported in the literature (Table S1). Created with Biorender.com.
(B) Quantitative reverse-transcription PCR measurement of ABCB1 transcription in multiple human enterocytes with varying basal expression when treated with E. lenta CFS. GAPDH and beta-actin primers were used as loading controls. Amplification cycles (Ct) were normalized to untreated conditions for each cell line (n = 3 biological replicates/condition; two-way ANOVA with Sidak’s correction; see also Figure S2).
(C) Representative western blots of untreated, BHI sterile media, or E. lenta CFS-treated human enterocytes using C219 P-gp antibody and GAPDH antibody and densitometry quantifications normalized to untreated control for each cell line (n = 1–3 biological replicates/condition; Wilcoxon test).
(D) Cell-free P-gp vesicles were incubated with 3 kDa filtrate of E. lenta CFS prepared in EDM media in the presence of ATP. At the end of the incubation period, the amount of remaining ATP was quantified. Sodium orthovanadate (Na3VO4) and verapamil P-gp inhibitors were included as controls for an ATPase inhibitor and an activator, respectively. n = 4/condition; ANOVA with Tukey’s post hoc test.
To test the downstream impact of E. lenta on P-gp activity, we turned to a cell-free vesicle system wherein the ATPase activity of P-gp can be directly quantified29 (Figure 3D). The positive control29 sodium orthovanadate significantly blocked P-gp ATPase activity whereas verapamil significantly stimulated ATPase activity (Figure 3D). A fraction of small molecules <3 kDa from E. lenta CFS significantly increased free ATP in a dose-dependent manner (Figure 3D). These results suggest that E. lenta inhibits the ATPase activity of P-gp.
Comparative genomics reveals genes associated with P-gp inhibition
Next, we sought to determine the extent to which the P-gp inhibitory trait is conserved in gut bacteria. We leveraged our previously published strain collection,30 testing 24 E. lenta strains and 10 Coriobacteriia relatives for their ability to inhibit P-gp, using our T84 cell-based assay. All of the tested Eggerthellaceae strains had a comparable degree of inhibition, on par with our positive control verapamil (Figure 4A). However, no inhibitory activity was observed in Olsenella uli (a Coriobacteriia) and 15 other non-Coriobacteriia strains, highlighting that the activity is conserved in the Eggerthellaceae clade but not found in more distantly related members of the human gut microbiota (Figure 4A).
Figure 4.

Comparative genomics reveals genes associated with P-gp inhibitory activity
(A) Identification of genes shared among strains with P-gp inhibitory activity assessed via Rh123 accumulation assay. Strains cultured in BHI++ were considered P-gp inhibitory if Rh123 accumulation was significantly higher than the untreated control. Sterile BHI++ media and 10 μM verapamil were included as negative and positive controls, respectively (n = 3–8/strain, ANOVA with Tukey’s correction). ElenMatchR and NCBI BlastX were used to identify genes unique to P-gp inhibitory bacterial strains. A cladogram shows the phylogenetic relationships between all profiled strains (see also Tables S2 and S3).
(B) Functional annotation of 7 putative genes that were uniquely present in P-gp inhibitory strains (see also Figure S3).
(C) Locus diagram of modA and modB conserved genes in the E. lenta DSM2243 genome. Counts of transcripts from E. lenta DSM2243 mapped to the genome are shown.
Next, we sought to use comparative genomics to determine if gene presence/absence was associated with P-gp inhibition. Using ElenMatchR, a comparative genomic tool we previously developed to identify functional genes in E. lenta,30 we identified 36 genes shared among all 33 P-gp inhibitory Coriobacteriia but absent in Olsenella uli (Figure 4A). From these 36 hits, 7 genes were absent from the genomes of all 15 non-inhibitory non-Coriobacteriia organisms (Figure 4A and Table S2). All 7 of these hits were transcriptionally active in E. lenta DSM2243 (Table S3). Notably, two genes predictive of P-gp inhibition were found within the same genomic locus and annotated as impacting molybdate transport: modA (molybdate-binding protein) and modB (molybdenum transport system permease protein; Figure 4B and 4C).
Given that molybdate is a redox-active metal that complexes with diverse bacterial enzymes,31,32 we tested whether the modAB locus is necessary to produce P-gp inhibitors by generating ΔmodAB mutant in E. lenta DSM2243 (Figures S3A and S3B). Contrary to our hypothesis, ΔmodAB mutant still showed P-gp inhibitory activity similar to the wild-type E. lenta in our T84 cell-based assay, suggesting that modAB is not required to produce P-gp inhibitory molecules (Figure S3C). More work is needed to test our remaining 5 candidate genes, in addition to the molybdate-related genes moaA and moaC, which are downstream of modAB and expressed during in vitro growth (Figures 4B and 4C).
Polar metabolites from E. lenta CFS inhibit P-gp
Given that the P-gp inhibitory activity was detected in the CFS (Figure 2A), we used activity-guided fractionation to enrich for and better understand the chemical nature of the E. lenta P-gp inhibitor (Figure 5A). To narrow down the extracellular products, E. lenta CFS was filtered through a 3 kDa membrane. Both size fractions were tested in T84 and Caco-2 cells, which showed the activity to be in a fraction containing small molecules <3 kDa in size (Figure 5B). When the 3 kDa filtrate was separated into polar and nonpolar fractions via liquid-liquid extraction, the activity remained in the polar fraction (Figure 5C). The active polar fraction was further separated via size-exclusion chromatography and the activity was detected in fractions 3 and 4 (Figure 5D). These results indicate that E. lenta secretes one or more polar small molecules that inhibit P-gp.
Figure 5.

E. lenta secretes small polar metabolites that inhibit P-gp
(A) Activity-guided biochemical fractionation pipeline (created with BioRender.com); the fractions are considered active when it led to high Rh123 accumulation compared to the sterile media control. E. lenta CFS cultured in BHI+ was tested for activity in T84 cells after it was (B) filtered through a 3 kDa filter cartridge, (C) separated into polar and non-polar fractions via liquid-liquid extractions with MTBE:MeOH:H2O, and (D) separated into 5 fractions via size-exclusion chromatography. Verapamil and filter-sterilized CFS were included as positive controls. Similar fractionations and activity testing using E. lenta CFS cultured in EDM are shown in Figure S4n = 4/condition and two-way ANOVA with Sidak’s multiple testing correction. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
The arginine-supplemented brain heart infusion (BHI) media used to cultivate E. lenta is rich and undefined, potentially complicating further efforts to identify the active compounds via mass spectrometry.30,33 To address this issue, we switched to a recently developed defined minimal media that support robust growth of E. lenta (Eggerthella defined media, EDM; Table S4).33 Consistent with our results in BHI, we noted significant P-gp inhibitory activity in E. lenta CFS following growth in EDM (Figure S4). We then repeated our activity-guided fractionation steps (Figure 5A) and validated the activity in our T84 assay (Figure S4). The biochemical fractionations not only reveal the chemical nature of the P-gp inhibitor but also enrich the metabolites that could be detected via mass spectrometry.
Untargeted mass spectrometry of P-gp inhibitory fractions
Finally, we used untargeted metabolomics to identify putative P-gp inhibitors in our enriched CFS fraction. First, we compared culture supernatants from 3 E. lenta strains (strains DSM2243, 1356FAA, and W1BHI6) grown for 48 h in a defined medium (EDM) to sterile media controls. We selected E. lenta 1356FAA and E. lenta W1BHI6 given that they inhibit P-gp to a similar degree as E. lenta DSM2243 (Figure 4A), grow robustly in the EDM media (maximum OD600: 0.8–1.0), and are distant from DSM2243 in the phylogeny of E. lenta (Figure 4A). As expected,30,33 each strain had a distinct metabolomic profile (Figure 6A). Pairwise comparisons of the top 5,000 mass features from strain to sterile media identified 687 (DSM2243), 863 (1356FAA), and 503 (W1BHI6) features that were significantly enriched relative to media controls (≥3 fold-change, padj < 0.1). We then selected the subset of features that were consistently enriched in the active fractions of all 3 E. lenta strains (131 in total; Figure 6C and Table S5).
Figure 6.

Untargeted comparative metabolomics revealed metabolites associated with P-gp inhibition
(A and B) Principal component analysis of mass features detected following growth in (A) selective (EDM) and (B) rich (BHI++) media (n = 3–4 samples/condition/experiment). (A) Comparison of three E. lenta strains and sterile media controls following activity-guided fractionation (Figure 5A). (B) Comparison to organisms without P-gp inhibitory activity (Bifidobacterium longum, Clostridium innocuum; selected based on Figure 4A).
(C) Identification of candidate P-gp inhibitors. Enriched features were defined as ≥3 fold-change, padj < 0.1 using (A) unpaired Benjamini-Hochberg adjusted t tests relative to media controls and (B) one-way ANOVA with Dunnett’s test followed by Benjamini-Hochberg adjustment.
(D) Heatmap of the 35 consistently enriched mass features identified in C (see also Figure S5, Tables S5, and S6).
The EDM media was optimized for E. lenta,33 preventing us from including non-inhibitory control strains. Instead, we turned to rich media (BHI++), which enables a broader comparison of microbial metabolism with the downside of increasing the background of unidentified features due to the complexity of the media.30,33 We included the same 3 E. lenta strains previously tested in EDM in addition to 2 additional non-inhibitory control strains: Bifidobacterium longum DSM20219 and Clostridium innocuum DSM1286 (Figure 4A). Each strain was grown for 48 h in BHI++ media. An overall analysis of the metabolomic profiles of these strains revealed that all of the E. lenta strains grouped separately from the media controls and the two non-inhibitory strains (Figure 6B), consistent with their phylogenetic relationships (Figure 4A).
We restricted our search to the 131 previously identified features from the EDM metabolomics analyses, 35 of which were significantly enriched in supernatants from all 3 E. lenta strains relative to media controls (≥3 fold-change, padj < 0.1; Figure 6C and 6D). None of these 35 features were significant when comparing the two control strains to media controls. We were able to annotate 43% of these features (15/35) by comparing our high-resolution MS1 and MS2 spectra to libraries of known metabolites (see STAR methods). All of the annotated features corresponded to abundant E. lenta metabolites,33 including 13 multimeric salt adducts of citrulline, ornithine, and N-acetylglutamate (Figure 6D and Table S6). None of these metabolites inhibit P-gp, even at high concentrations (Figure S5). The remaining 20 features represent unknown metabolites, requiring further efforts to enable their identification and characterization.
Discussion
Our work uncovers how a prevalent gut Actinobacterium boosts absorption of digoxin, a cardiac glycoside with a narrow therapeutic window,20 by blocking a host drug efflux transporter. Mechanistic investigation revealed that E. lenta inhibits P-gp ATPase activity without affecting protein levels. The inhibitory activity was limited to the Coriobacteriia class and conserved across the Eggerthellaceae clade. Three distinct but complementary approaches, namely comparative genomics, activity-guided biochemical fractionations, and untargeted mass spectrometry, linked P-gp inhibition to a group of small polar metabolites that remain unidentified.
Our results emphasize the importance of considering the vast biosynthetic potential within the human microbiome9 for explaining inter-individual variations in drug absorption and metabolism. Seminal studies have shown that microbiome-derived bioactive molecules can directly interact with human cells or receptors (host-microbe interactions) or indirectly affect the host by impacting other members of the microbiome (microbe-microbe interactions).9 One example is the production of a metabolite structurally similar to the anti-cancer drug doxorubicin, a well-known P-gp substrate,9 that could compete for absorption of other P-gp substrates. As the microbiota is highly diverse and dynamic, inter-individual genetic differences in the microbiome could account for variations in drug disposition.6 By focusing on a prevalent member of the gut microbiota, this study demonstrates host-microbe interactions that resulted in altered drug absorption.
While bacterial metabolism of drugs is well documented, with examples ranging from bioactivation of the antibiotic prontosil to re-activation of anti-cancer drug irinotecan,3 little is known about whether and how gut microbes could alter drug absorption without biotransformation. We found that the same gut bacterial species, E. lenta, can have opposing effects on drug bioavailability, contributing to the intestinal metabolism of digoxin21,22 while also boosting drug absorption due to inhibition of P-gp efflux. E. lenta, which colonizes the entire intestinal tract,30 could be inhibiting P-gp in the proximal small intestine, which is the major site of oral digoxin absorption.20 The remaining digoxin in the gut lumen would then be subjected to bacterial metabolism in more distal regions; however, more work is needed to map out the specific site of conversion of digoxin to dihydrodigoxin.
E. lenta is unique to humans, has a distinctive metabolic niche, and performs several unusual chemical transformations,33 indicating that the mechanism described herein may represent a human-specific host-microbe interaction relevant to pharmacology. This host species specificity is also true for digoxin metabolism, as the bacterial genes that metabolize digoxin to dihydrodigoxin have only been detected in select E. lenta strains found in human gut microbiota.22
Our comparative genomics results shed light on the genes necessary for P-gp inhibition by E. lenta. Using the clustering cutoff of protein identity <50%, we identified 7 transcriptionally active genes that were uniquely present in the P-gp inhibitory strains. Among these 7 genes were modA and modB, two genes involved in molybdenum transport.34 Molybdenum, redox-active under physiological conditions, forms an active site for many bacterial enzymes involved in diverse redox chemistry.32,35,36,37 Thus, these genes involved in molybdenum were promising targets to test experimentally by generating knockout mutants in E. lenta. Contrary to our hypothesis, modA and modB were not necessary to produce P-gp inhibitors. Additional work is needed to test the remaining genes identified in the comparative genomics and perform more systematic functional genomic screens for the genetic determinants of P-gp inhibition.
Our untargeted metabolomics paired with biochemical fractionation revealed the nature of the putative P-gp inhibitors to be small molecules that are highly polar and remain active after treatment with heat, organic solvents, and proteinase. This suggests that the inhibitor must be structurally stable and non-proteinaceous in nature. Using untargeted metabolomics to further shed light on the identity of the putative P-gp inhibitors, we found that citrulline is a dominant metabolite present in the active fractions, which is a known byproduct of arginine metabolism in E. lenta. However, citrulline did not show P-gp inhibitory activity at a wide range of concentrations. Further efforts in purification and compound annotations are required to identify putative inhibitors for validation.
Limitations of the study
There are multiple limitations of our study. E. lenta-mediated P-gp inhibition was tested in GF and CONV-R mice using the P-gp substrate digoxin; synthetic communities38 or humanized mice39 could be used to test how E. lenta affects P-gp function in the context of other members of the human gut microbiota. Although digoxin is a sensitive and physiologically relevant P-gp probe, additional P-gp substrates could be used to assess generalizability. Our comparative genomics analysis identified 7 candidate genes, 5 of which have yet to be validated. It is also possible that bacteria in the Eggerthellaceae clade could encode different suites of genes to produce P-gp inhibitory molecules. By requiring that all species with P-gp inhibitory activity must encode the same gene(s) for P-gp inhibition, we may have missed many genes encoding putative P-gp inhibitors.
Through biochemical fractionations and metabolomics, we narrowed our candidate P-gp inhibitor to polar small molecules. Further purification efforts are required to isolate the metabolites and solve their structures by mass spectrometry and nuclear magnetic resonance analysis, which would allow for in-depth biochemical characterization and dose-response experiments in mice. Mechanistic insights into the role of E. lenta in drug absorption would be aided by follow-on work to quantify bacterial P-gp inhibitors in the mouse gut and other tissues, coupled to isogenic strains differing only in genes necessary for P-gp inhibition and/or digoxin metabolism.
Nonetheless, our study emphasizes the broad impact of the microbiome on multiple aspects of drug disposition, including metabolism40 and absorption.7,8 Continued mechanistic dissection of these and other host-microbe interactions relevant to the treatment of disease will help to explain the often-unpredictable inter-individual variations in drug efficacy and toxicity and enable a new generation of microbiome-aware medicines.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-P-glycoprotein antibody [C219] | GeneTex | Clone C219; Cat#GTX23364; RRID: AB_367204 |
| Mouse anti-GAPDH monoclonal antibody | ThermoFisher | Clone GA1R; Cat#MA5-15738; RRID: AB_10977387 |
| Goat anti-mouse HRP-conjugated secondary antibody | Abcam | Cat#ab205719; RRID: AB_2755049 |
| Bacterial and virus strains | ||
| Collinsella aerofaciens ATCC25986 | Turnbaugh lab | ATCC 25986 |
| Adlercreutzia equolifaciens DSM19450 | Turnbaugh lab | DSMZ 19450 |
| Asaccharobacter celatus OB21GAM11 | Turnbaugh lab | N/A |
| Asaccharobacter celatus AP38TSA | Turnbaugh lab | N/A |
| Gordonibacter pamelaeae 3C | Turnbaugh lab | DSMZ 110924 |
| Gordonibacter pamelaeae DSM19378 | Turnbaugh lab | DSMZ 19378 |
| Gordonibacter urolithinfaciens DSM27213 | Turnbaugh lab | DSMZ 27213 |
| Gordonibacter species 28C | Turnbaugh lab | DSMZ 110925 |
| Eggerthella sinensis DSM16107 | Turnbaugh lab | DSMZ 16107 |
| Eggerthella lenta W1BHI6 | Turnbaugh lab | DSMZ 110923 |
| Eggerthella lenta AN51LG | Turnbaugh lab | DSMZ 110914 |
| Eggerthella sp. DSM 11863 | Turnbaugh lab | DSMZ 11863 |
| Eggerthella lenta 28B | Turnbaugh lab | DSMZ 110909 |
| Eggerthella lenta RC4/6F | Turnbaugh lab | DSMZ 110922 |
| Eggerthella lenta DSM 15644 | Turnbaugh lab | DSMZ 15644 |
| Eggerthella sp. DSM 11767 | Turnbaugh lab | DSMZ 11767 |
| Eggerthella lenta 32-6-I-6 NA | Turnbaugh lab | DSMZ 110910 |
| Eggerthella lenta FAA 1-1-60ABroad | Turnbaugh lab | DSMZ 110904 |
| Eggerthella lenta 14A | Turnbaugh lab | DSMZ 110907 |
| Eggerthella lenta 11C | Turnbaugh lab | DSMZ 110905 |
| Eggerthella lenta DSM 2243 | Turnbaugh lab | DSMZ 2243 |
| Eggerthella lenta ATCC 25559 | Turnbaugh lab | ATCC 25559 |
| Eggerthella lenta Valencia | Turnbaugh lab | N/A |
| Eggerthella lenta AB12n2 | Turnbaugh lab | DSMZ 110912 |
| Eggerthella lenta ElUC2 | Turnbaugh lab | DSMZ 110919 |
| Eggerthella lenta CC7/5 D5 2 | Turnbaugh lab | DSMZ 110915 |
| Eggerthella lenta CC8/6 D5 4 | Turnbaugh lab | DSMZ 110917 |
| Eggerthella lenta 22C | Turnbaugh lab | DSMZ 110908 |
| Eggerthella lenta MR1 #12 | Turnbaugh lab | DSMZ 110921 |
| Eggerthella lenta CC8/2 BHI2 | Turnbaugh lab | DSMZ 110916 |
| Eggerthella lenta AB8n2 | Turnbaugh lab | DSMZ 110913 |
| Eggerthella lenta ElUC5 | Turnbaugh lab | DSMZ 110920 |
| Eggerthella lenta 1-3-56FAA | Turnbaugh lab | DSMZ 110906 |
| Olsenella uli DSM7084 | Turnbaugh lab | DSMZ 7084 |
| Clostridium innocuum DSM1286 | Turnbaugh lab | DSMZ 1286 |
| Clostridium spiroforme DSM1552 | Turnbaugh lab | DSMZ 1552 |
| Eubacterium rectale DSM17629 | Turnbaugh lab | DSMZ 17629 |
| Clostridium scindens DSM5676 | Turnbaugh lab | DSMZ 5676 |
| Dorea longicatena DSM13814 | Turnbaugh lab | DSMZ 13814 |
| Ruminococcus obeum DSM25238 | Turnbaugh lab | DSMZ 25238 |
| Escherichia coli MG1655 | Turnbaugh lab | DSMZ 18039 |
| Prevotella copri DSM18205 | Turnbaugh lab | DSMZ 18205 |
| Bacteroides uniformis DSM6597 | Turnbaugh lab | DSMZ 6597 |
| Bacteroides thetaiotaomicron DSM2079 | Turnbaugh lab | DSMZ 2079 |
| Bacteroides vulgatus DSM1447 | Turnbaugh lab | DSMZ 1447 |
| Parabacteroides merdae DSM19495 | Turnbaugh lab | DSMZ 19495 |
| Akkermansia muciniphila DSM22959 | Turnbaugh lab | DSMZ 22959 |
| Bifidobacterium longum DSM20219 | Turnbaugh lab | DSMZ 20219 |
| Bifidobacterium adolescentis BD1 | Turnbaugh lab | N/A |
| Escherichia coli Stbl3 with sgRNA against P-gp | Genecopoeia | HCP267279-LvSG03-3-B |
| Escherichia coli with pCMV-dR8.91 packaging plasmid | Mukherjee lab | N/A |
| Escherichia coli with pMD2.G packaging plasmid | Mukherjee lab | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Rhodamine 123 | Sigma-Aldrich | Cat#83702 |
| Cyclosporin A | Sigma-Aldrich | Cat#30024 |
| Vinblastine sulfate | Sigma-Aldrich | Cat#V1377 |
| Verapamil hydrochloride | Sigma-Aldrich | Cat#V4629 |
| Bacto Brain Heart Infusion | Fisher Scientific | Cat#237500 |
| L-Arginine | Fisher Scientific | Cat#W381918 |
| DMEM/F-12 GlutaMAX | Thermo Fisher | Cat#10565042 |
| DMEM, high glucose, pyruvate | Thermo Fisher | Cat#11995065 |
| Fetal Bovine Serum, Heat Inactivated | Thermo Fisher | Cat#10-438-026 |
| Penicillin-Streptomycin (10,000 U/mL) | Life Technologies | Cat#15140122 |
| RIPA buffer, 10X | Sigma-Aldrich | Cat#20-188 |
| Mirus Bio | Thermo Fisher | Cat#MIR2304 |
| Doxorubicin hydrochloride | TCI Chemicals | Cat#D4193 |
| L-Citrulline | Sigma-Aldrich | Cat#C7629 |
| Sarcosine hydrochloride | Sigma-Aldrich | Cat#223107 |
| L-Ornithine monohydrochloride | Sigma-Aldrich | Cat#O2375 |
| N-Acetyl-L-glutamate | Thermo Fisher | Cat#B23621.14 |
| ATCC Vitamin Supplement | ATCC | Cat# MD-VS |
| ATCC Trace Mineral Supplement | ATCC | Cat# MD-TMS |
| Digoxin Injection, USP 500 mcg/2 mL | Westward | Cat#0641-1410-35 |
| Critical commercial assays | ||
| Invitrogen Vybrant Multidrug Resistance Assay kit (Calcein-AM) | Invitrogen | Cat#V13180 |
| CellTiter 96 Non-Radioactive Cell Proliferation Assay (MTT) | Promega | Cat#G4000 |
| Human MDR1 Vesicles | Thermo Fisher | Cat#GM0015 |
| Pgp-Glo Assay System | Promega | Cat#V3591 |
| PD MidiTrap G-10 | Sigma-Aldrich | Cat#GE28-9180-11 |
| QIAGEN Plasmid Midi Kit | Qiagen | Cat#12143 |
| ZymoBIOMICS 96 MagBead Kit (Lysis tubes) | Zymo Research | Cat#D4308 |
| GeneJET PCR Purification Kit | Thermo Scientific | Cat#K0701 |
| T4 polynucleotide kinase | Thermo Scientific | Cat#EK0031 |
| Deposited data | ||
| Metabolomics datasets | GitHub | GitHub: https://github.com/turnbaughlab/2024_Kyaw_iScience |
| RNA-Seq reads of Eggerthella lenta DSM2243 | Maini Rekdal et al.35 | NCBI: PRJNA507796 |
| Coriobacteriaceae genomes | Bisanz et al.30 | NCBI: PRJNA412637 |
| Other reference genomes | NCBI | NCBI: taxids: 562, 1680, 216816, 1522, 29347, 29348, 820, 821, 818, 239935, 46503, 39491, 40520, 88431, 165179 |
| Experimental models: Cell lines | ||
| T84 | ATCC | Cat#CCL-248 |
| Caco-2 | ATCC | Cat#HTB-37 |
| HCT-116 | ATCC | Cat#CCL-247 |
| HCT-15 | ATCC | Cat#CCL-225 |
| HEK293T | Shaeri Mukherjee lab | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: Swiss Webster | Taconic | Cat#SW |
| Mouse: Germ-free Swiss Webster | UCSF Gnotobiotic Core | N/A |
| Oligonucleotides | ||
| Primers | See Table S7 | N/A |
| Recombinant DNA | ||
| pCMV-dR8.91 packaging plasmid | Shaeri Mukherjee lab | N/A |
| pMD2.G packaging plasmid | Shaeri Mukherjee lab | N/A |
| HCP267279-LvSG03-3-B plasmid | Genecopoeia | HCP267279-LvSG03-3-B |
| Software and algorithms | ||
| R 4.1.1 | R | https://www.r-project.org/ |
| ggplot2 v3.3.6 | Wickham et al.41 | v3.3.6 |
| ape v5.6.2 | Paradis et al.42 | v5.6.2 |
| GraphPad PRISM 9.4.1 | GraphPad | https://www.graphpad.com/ |
| ElenMatchR | Bisanz et al.30 | https://github.com/jbisanz/ElenMatchR |
| BioRender | BioRender | https://biorender.com/ |
| Image Lab | Bio-Rad | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
| Other | ||
| Mouse diet: standard autoclaved diet for gnotobiotic | Lab diet | Cat# 5021 |
| Mouse diet: standard chow diet SPF | Lab diet | Cat#5058 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to the Lead Contact Peter Turnbaugh (Peter.Turnbaugh@ucsf.edu).
Materials availability
This study does not contain newly generated materials
Data and code availability
-
•
Data: All data is available in the main text and the supplemental information and tables. Metabolomics datasets are available through our lab GitHub (https://github.com/turnbaughlab/2024_Kyaw_iScience). No new DNA, RNA, or protein sequence data were generated in this study.
-
•
Code: Comparative genomics analysis utilized the ElenMatchR tool30 and NCBI BlastX.
-
•
All other items: Previously published RNA-Seq reads of Eggerthella lenta DSM2243, Coriobacteriaceae genomes, and other reference genomes we used in this study are available in NCBI (NCBI: PRJNA507796; NCBI: PRJNA412637; NCBI taxids: 562, 1680, 216816, 1522, 29347, 29348, 820, 821, 818, 239935, 46503, 39491, 40520, 88431, 165179).
Experimental model and study participant details
Experimental design
The main objective of this study was to investigate whether and how Eggerthella lenta alters digoxin pharmacokinetics. We utilized a combination of gnotobiotic and conventional specific-pathogen-free mouse models, cell culture assays, comparative genomics, activity-guided biochemical fractionations, and untargeted mass spectrometry, which are all outlined below.
Mice
All mouse experiments were approved by and conformed to the standards of the University of California San Francisco Institutional Animal Care and Use Committee (Protocol number AN-184143). The mice were housed at temperatures ranging from 19°C to 24°C and humidity ranging from 30 to 70% with a 12 h/12 h light/dark cycle. No mice involved in previous procedures before experiments were used. Mice were randomly assigned to groups.
Gnotobiotic mouse studies
9-week-old mixed sex Swiss Webster germ-free mice were obtained from the Gnotobiotics Core Facility (gnotobiotics.ucsf.edu) at the University of California San Francisco. They were fed an autoclaved chow diet (Lab Diet 5021) and housed in gnotobiotic isolators (Class Biologically Clean) or in Iso-positive cages (Tecniplast). Mice were colonized by gavaging 200 μL of 109 CFU/mL Eggerthella lenta DSM2243 strains and colonization was confirmed via qPCR for an E. lenta specific marker gene (elenmrk1).30 They were colonized for 4 weeks prior to experiments. On the day of the pharmacokinetics experiment, pharmaceutical grade digoxin (250 μg/mL Westward) was freshly diluted in 0.3M glucose sterile solution and 200 μg/kg digoxin was orally administered to each mouse in 100–200 μL volume. 20–30 μL of blood was collected via tail vein bleeding method into a 96-well plate and allowed to clot on ice. After centrifugation at 2000 g for 10 min at 4°C, serum was collected and stored at −80°C until mass spectrometry analysis. These experiments were not adequately powered to analyze the influence of sex.
Specific-pathogen-free mouse studies
8-10-week-old female Swiss Webster conventional mice were obtained from Taconic (Model: SW-F). They were fed a chow diet (Lab Diet 5058). Mice were orally gavaged with 200 μL of 109 CFU/mL Eggerthella lenta DSM2243 or sterile media every day for 7 days. Daily gavage was required as E. lenta does not stably engraft in CONV-R mice.23 On day 8, the digoxin pharmacokinetics experiment was conducted using the protocol outlined above by gavaging freshly prepared 30 μg/kg digoxin in 100–200 μL volume to each mouse. Similarly, serum was stored at −80°C until mass spectrometry analysis.
Method details
Bacterial culturing
Bacterial isolate information can be found in the key resources table. All the bacterial strains were cultured at 37°C in an anaerobic chamber (Coy Laboratory Products) containing 2–5% H2, 20% CO2, and balance N2 gasses for 48 h. There were 3 different media used: (1) brain heart infusion (BHI; VWR 90003-040) media supplemented with 1% arginine (denoted BHI+), (2) BHI supplemented with 1% arginine, 0.05% L-cysteine-HCl, 5 μg/mL hemin, and 1 μg/mL vitamin K, and 0.0001% resazurin (denoted BHI++), and (3) Eggerthella defined media (denoted EDM) with media composition listed in Table S4.
Mammalian cell culture
Human colorectal cancer cell lines (T84, HCT-15, HCT-116, and Caco-2) were obtained from American Type Culture Collection and HEK-293T cell line was a gift from the Mukherjee lab. Except for HEK-293T, which was isolated from a female fetus, all remaining cell lines used were isolated from males. We did not perform further authentication of these cell lines. These cell lines except for Caco-2 were routinely cultured in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 GlutaMAX (DMEM/F12) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin antibiotic solution. Caco-2 cells were cultured in DMEM, high glucose, pyruvate media with 10% FBS and 1% penicillin-streptomycin. Cells were grown in humidified incubators at 37°C under 5% CO2. Media was replaced 2–3 times a week and cells were split (1:5 for T84 and Caco2 and 1:10 for the other cell lines) using trypsin when they reached 80–90% confluence.
Rhodamine (Rh123) accumulation assay
Human colorectal cancer cell lines were cultured in 24-well tissue culture-treated plates. Once the cells reached 100% confluence in monolayers, they were incubated with 20% (unless otherwise noted) of CFS or sterile bacterial media control in DMEM/F12 with FBS for 18–20 h. The media was then removed and the cells were incubated with fresh pre-warmed DMEM/F12 media containing fluorescent P-gp substrates (5μM Rhodamine 123) at 37°C under 5% CO2 for 30 min. 10μM verapamil hydrochloride, 20μM cyclosporin A, or 20μM vinblastine in 0.2% dimethyl sulfoxide (DMSO) were used as positive controls. After the media was removed, the cells were gently washed with 2mL of ice-cold phosphate buffer saline 3 times on ice. The final wash was carefully aspirated. The cells were then lysed with 300μL of RIPA buffer for 30 min and fluorescence was quantified using 100μL of supernatant lysate on the Biotek microplate reader (Tecan). To test P-gp inhibition with other substrates, the same procedures were performed with 50μM doxorubicin or 5μM Calcein-AM dye instead of 5μM Rhodamine 123. Fluorescence was converted to concentration using a standard curve and ANOVA or paired t test (see each figure legend) was used to test for statistical significance between the groups.
MTT proliferation assay
T84 cells were seeded at 20,000 cells/well in 50μL in 96-well tissue culture-treated plates in DMEM/F12 supplemented with 10% FBS. To test whether E. lenta CFS sensitizes the cells against doxorubicin, the cells were incubated for 72 h with 10% E. lenta DSM2243 CFS or sterile BHI+ media control and treated with either 2μM doxorubicin or vehicle DMSO control. 10μM verapamil was included as a positive control. After 3 days of incubation, the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) viability assay was performed according to manufacturer’s instructions (Promega). Briefly, 15μL MTT dye solution was added to the wells and incubated at 37°C under 5% CO2 for 4 h. 100μL of solubilization solution was added and incubated for 1 h at room temperature. Cell growth was assessed by measuring absorbance at 570 nm. The optical densities for each treatment group were compared using ANOVA or pairwise t-tests.
Differential expression via qPCR
Human colorectal cancer cell lines were cultured in 24-well tissue culture-treated plates. Once the cells reached confluence, 20% cell-free supernatant (CFS) or sterile bacterial media controls were added for 18–20 h. The media was removed and RNA was extracted using the PureLink RNA Mini extraction kit (Invitrogen), according to the manufacturer’s instructions. 5μL of 150–400 ng/μL extracted RNA was used to synthesize cDNA in 20μL reactions using iScript Reverse Transcription Supermix for RT-qPCR kit (Biorad). Diluted cDNA (1:20 in DNase-free water) was used to perform qPCR with SYBR Select Master Mix (Thermo Fisher Scientific) for 40 cycles. 30nM human ABCB1 exon-spanning primers were used to quantify P-gp expression along with human GAPDH and beta-actin primers (Table S7) as loading controls.
Western Blot analysis
Human colorectal cancer cell lines were cultured in 24-well tissue culture-treated plates. Once the cells reached 100% confluence in monolayers, they were incubated with 20% CFS or sterile bacterial media control for 18–20 h. The media was removed and the cells were washed twice with pre-warmed PBS. The cells were lysed on ice with 100μL of RIPA lysis buffer containing a cOmplete Mini Protease Inhibitor Cocktail tablet (Millipore Sigma). Proteins in the lysate were quantified using Pierce BCA Protein Assay kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. After normalizing protein concentrations in RIPA buffer, 30 μg of proteins in Laemmli sample buffer were loaded in a 10-well 4–15% Mini-PROTEAN gradient SDS-PAGE gel (Biorad) and ran for 15 min at 70V followed by 150V for 45 min in Tris-Glycine-SDS buffer at room temperature. Precision Plus Protein Dual Color standards were included in each gel. The proteins in the gel were then transferred to polyvinylidene difluoride membranes overnight at 23V at 4°C in the N-cyclohexyl-3-aminopropanesulfonic acid buffer. After blocking membranes in 5% milk, the membranes were incubated with C219 anti-ABCB1 primary antibody (Genetex) at 1:200 or anti-GAPDH primary antibody (Invitrogen) at 1:5000 in 5% milk in PBS-T blocking buffer overnight at 4°C. The membranes were washed 3 times and incubated with goat anti-mouse HRP-conjugated secondary antibody (Abcam) at 1:10,000 in a blocking buffer for 1 h at room temperature. After washing the membranes, they were treated with Clarity Max Western ECL substrates (Biorad) for 5 min and visualized in the ChemiDoc (Biorad). The densitometry analysis was performed using the Biorad ImageLab software.
P-gp vesicle assay
To test whether E. lenta modulates P-gp ATPase activity, vesicles containing P-gp (5 mg/mL) purchased from Thermo Fisher were tested for ATP consumption in P-gp Glo assay (Promega) according to the manufacturer’s instructions. Briefly, the vesicles were thawed rapidly at 37°C and immediately kept on ice. The vesicles were diluted to 1.25 mg/mL with Pgp-Glo assay buffer before each assay. For each assay 20μL of 1.25 mg/mL vesicles was mixed with 10-20μL of test agents (CFS, sterile media, 0.5mM verapamil, 0.25mM Na3VO4, or Pgp-Glo assay buffer control) in the presence of 10μL of 25mM MgATP in white flat-bottom 96-well plates. Pgp-Glo assay buffer was added if necessary to achieve the total volume of 50μL. The reaction was incubated for 1 h at 37°C under 5% CO2.
After incubation, the reaction was diluted 1:4 (12.5μL into 37.5μL of P-gp-Glo assay buffer) in a new white flat-bottom 96-well plate and 50μL of ATP detection reagents were added. Standard curves (3mM–0.375mM MgATP at 1:2 dilution) in different matrices (CFS, sterile media, Pgp-Glo assay buffer) were included. The mixture was incubated at room temperature for 20 min and luminescence was quantified on a plate reader (Tecan). Luminescence measurements were converted to ATP concentrations using matrix-specific standard curves.
Generation of P-gp knockout monoclonal cell line
Escherichia coli strains containing pCMV-dR8.91 and pMD2.G lentiviral packaging plasmids were a kind gift from the Mukherjee lab. An E. coli strain carrying the transfer plasmid (pCas9) with cas9 gene, eGFP, neomycin, and ampicillin marker genes and a separate E. coli strain carrying the transfer plasmid (pABCB1) with sgRNA against human ABCB1 gene, mCherry, puromycin, and ampicillin maker genes were purchased from Genecopoeia. To harvest the plasmids, these strains were cultured separately in LB media supplemented with 50μg/mL carbenicillin aerobically overnight with shaking at 37°C. The resulting turbid cultures were centrifuged to collect the cell pellets, which were then processed with Midi-Prep plasmid extraction kit (Qiagen) based on the manufacturer’s protocol. Plasmids were confirmed via enzymatic restriction digest.
4 μg of pCMV-dR8.91, 1 μg of pMD2.G, and 5 μg of either pCas9 or pABCB1 plasmids were added to a tube containing 30μL of LT1 transfection reagent (Mirus) and 1mL of Opti-MEM I Reduced Serum medium. The contents were vortexed and incubated at room temperature for 30 min. The mixture was added drop-by-drop into a 10 cm cell culture dish containing HEK293T cells at 90% confluency. The cells were incubated for 48 h and the supernatant containing recombinant lentiviral particles was filtered using 0.45 μm filters. The lentiviral filtrate was flash-frozen.
To transduce the cells, 0.5mL of lentiviral supernatant and 0.5mL of 20μg/mL polybrene transfection agent in cell culture media were added to T84 cells cultured in 12-well plates at 70% confluency. After 72 h of incubation, 10μg/mL of puromycin and 3 mg/mL of geneticin antibiotics were added to select for successfully transduced cells. After 10 days of antibiotics selection, GFP+/RFP+ double-positive cells from the resulting polyclonal pool were sorted individually into 96-well plates using a cell sorter at the Flow Cytometry Core. The sorted cells were expanded into monoclonal cell lines, which were tested for ABCB1 deletion via PCR, Western blot, and functional rhodamine accumulation assay.
Comparative genomics and associated transcriptomics
Comparative genomics among Coriobacteriia was performed using ElenMatchR.30 Genes were clustered at a minimum of 50% amino acid identity and 80% query coverage to allow for the identification of orthologs across genus boundaries. Hits from ElenMatchR were compared to the genomes of non-Coriobacteriia non-inhibitory strains using NCBI BlastX with identical cutoffs for amino acid identity and query coverage and a liberal E value threshold of 10 to ensure that all identified gene clusters were unique to P-gp inhibitory bacterial strains. Gene annotations were called using E. lenta DSM2243 genes corresponding to each orthologous gene cluster hit. Raw E. lenta DSM2243 RNA sequencing data in BHI was obtained from and filtered and aligned as described.35 Genes with RPKM >10 in all 3 replicates were called transcriptionally active.
E. lenta DSM 2243 ΔmodAB construction
To assemble the shuttle plasmid, 0.6-kb homologous sequences, both upstream and downstream of the modA and modB coding regions, were PCR-amplified from an E. lenta DSM2243 colony using the primer pairs modABUPRT_f/modABUPRT_r and modABDNRT_f/modABDNRT_r, which serve as repair templates (see Table S7 for primer sequences). The self-targeting crRNA plasmid pLRH3 backbone was amplified using the pLRHbb_f and pLRHbb_r primers. Following PCR amplification, amplicons were purified using the GeneJET PCR purification kit (Thermo Scientific) and quantified with a Nanodrop (Thermo Scientific). The plasmid backbone was subjected to a 12-h DpnI treatment (Thermo Scientific), followed by subsequent DNA purification and quantification. PCR products were mixed in equimolar amounts and phosphorylated using T4 polynucleotide kinase (Thermo Scientific). A ligation cycling reaction43 involving the bridging oligonucleotides BO_1 and BO_2 facilitated the ligation of the three PCR amplicons, which were subsequently electroporated into E. coli DH5α. Repair template insertion was identified using the primer pair pLRHmodABKOs1_f and plRHmodABKOs1_r. This intermediate plasmid was designated pLRHmodABKOs1. To clone the CRISPR spacer sequence, we assembled the CRISPR array from three gBlocks (IDT) that were joined through Golden Gate assembly. The first gBlock was created by nested PCR using primer pairs gblock1_f1 and gblock1_r1, and gblock1_f1-gblock1_r2. The second gBlock was PCR amplified using primer pairs gblock2_f and gblock2_r1, and gblock2_f and gblock2_r2, for constructing CRISPR arrays that specifically target either modA or modB, respectively. The third gBlock was amplified using gblock3_f and gblock3_r. All primer overhangs contained BsaI (GTGCTC) binding sites for Golden Gate Assembly. This ligated CRISPR array was then PCR amplified using primers crisprgblock_f and crisprgblock_r for integration into the pLRHmodABKOs1 backbone (amplified using s1_f and s1_r) through Gibson Assembly. Two μL of the Gibson product were transformed into E. coli DH5a cells (NEB) and individual colonies screened by colony PCR using primer pair pLRHmodABKO_f and pLRHmodABKO_r. Positive clones were subcultured and the final shuttle vectors, termed pLRHmodAKO and pLRHmodBKO, had their sequence integrity verified through whole plasmid sequencing at Primordium Labs.
We generated the E. lenta DSM2243 ΔmodAB knockout, as described.24 Briefly, 1 μg of pLRHmodAKO or pLRHmodBKO shuttle plasmid was electroporated into 100 μL of freshly prepared E. lenta DSM2243 competent cells utilizing a MicroPulse Electroporator (BioRad) set at parameters: 2.5kV voltage, 25 capacitance, 200-ohm resistance, and with 1 mm gap width cuvettes. Post-electroporation, cells were recovered in 1 mL of BHI+ medium supplemented with 10 mM sodium formate, for 3 h within a 37°C anaerobic chamber. Subsequently, cells were spread onto BHI+ agar, which contained 100 μg/mL kanamycin, and were incubated anaerobically at 37°C for three days. Individual colonies were isolated and propagated in liquid BHI+ medium containing 100 μg/mL kanamycin, and incubated for three days in a 37°C anaerobic chamber. These cultures were then plated on BHI+ agar containing 100 μg/mL kanamycin and 50 μM cumate. Clones emerging from crRNA-induced conditions underwent PCR screening using both internal (ΔmodABi_f, ΔmodABi_r) and flanking (ΔmodABf_f, ΔmodABf_r) primers. Sanger sequencing confirmed the deletion of modA and modB genes in E. lenta DSM2243.
Activity-guided biochemical fractionations
E. lenta DSM2243 cultures were grown anaerobically at 37°C in BHI++ or EDM media with sterile media control for 48 h. To test whether E. lenta-derived P-gp inhibitor was intracellular or extracellular, cell pellets and supernatants were separated via centrifugation to test for P-gp inhibitory activity using our Rh123 assay. The pellet was reconstituted in 15 mM HEPES buffer (pH 7.3), lysed using bead beating with Lysing Matrix E tubes (MP Bio) in a Mini Beadbeater 16 Disrupter 16 (BioSpec Products), and tested for activity. Supernatant was pH-adjusted to 7.6 with NaOH or HCl, filter-sterilized using 0.22 μm filters to create CFS, and tested for activity. To test whether the inhibitor was large cellular materials or small metabolites, CFS was further separated using 3 kDa filter cartridges. Large materials on the filter membrane were reconstituted in sterile media in the same volume. The flowthrough and reconstituted large materials were then tested for activity. To further separate the small metabolites based on polarity, 4mL of 3 kDa flowthrough was added to 12mL of M1 solvent (75% MTBE and 25% MeOH) and 8mL of M2 solvent (25% MeOH in water), vortexed for 1 min, and centrifuged at maximum speed for 5 min at 4°C. The polar aqueous layer was lyophilized down to 2mL while the non-polar organic layer was dried under a gentle stream of nitrogen gas in TurboVap. Their activity was tested after reconstituting in 0.5× volume of water to account for incomplete extractions and compound loss through prior steps. To fractionate based on size, 1mL of active polar fraction was loaded to G10 size exclusion midi-columns. Once the solution was absorbed into the column, water was added on top gently without disturbing the beads and allowed to flow through via gravity. 1mL fractions were collected and tested for activity.
Untargeted metabolomics
Untargeted liquid chromatography high-resolution mass spectrometry (LC-HRMS) analysis was performed on a Sciex Exion UPLC equipped with a BDS C8 hypersil column (Thermo, 150 × 4.6 mm, particle size 5 μm) and coupled to a Sciex TripleTOF 6600+ operated in negative mode electrospray ionization with IDA acquisition of MS2 (accumulation time 0.25 s, collision energy 35V (15V spread), acquisition window 100–2000 Da). A gradient of 0–100% acetonitrile over 14 min was used for compound separation.
The resulting untargeted metabolomics data was analyzed using Sciex MarkerView Software. We first extracted the top 5000 mass features from each group (sterile media control and 3 E lenta strains). Mass signals were aligned with a mass tolerance of 10.0 ppm and retention time tolerance of 0.2 min; only signals detected in at least 3 samples out of 16 were included. Enriched mass features in each E. lenta sample, compared to the medium control, were identified by fold-change calculations (≥3-fold-change in intensity) and unpaired t tests (Benjamini-Hochberg adjusted p-value < 0.1). Common features identified in all three E. lenta strains were further validated by manual inspection of peak integration and retention time alignment in Sciex Analyst software, yielding a total of 131 hits from the defined medium experiment.
The 131 mass features were then searched in metabolomics data acquired from BHI++ cultures using Sciex Analyst software with mass tolerance of 10.0 ppm and retention time tolerance of 0.5 min. The retention time window was intentionally set wide to account for variations in retention time between two batches of mass spectrometry analysis. For example, the retention time of citrulline peaks shifted from 3.70 min (EDM experiment) to 4.0 min (BHI++ experiment). Detected signals were then aligned and their peak intensities (area under the peak) were extracted for comparison. A total of 35 mass hits were identified through enrichment (≥3-fold increase in intensity) in the positive groups (3 E lenta strains) compared to the control groups (medium control and 2 negative strains), with statistical significance relative to the medium control (one-way ANOVA with Dunnett’s test, p-value < 0.1, Benjamini-Hochberg adjusted). For compound annotation, the 35 hits were searched in MSDIAL database (>15,000 unique spectra in negative mode)44 and Human Metabolome Database (https://hmdb.ca/spectra/ms/search)45 with a mass tolerance of 10.0 ppm and matched MS/MS fragmentation patterns.
Quantification of serum digoxin
To quantify serum digoxin from the pharmacokinetics experiments, 5μL serum was combined with 100μL of 10 ng/mL oleandrin internal standard and 5μL of MeOH. For controls, 5μL of 0-200 ng/mL (1:2 dilution series) digoxin/dihydrodigoxin mix in MeOH was added to 5μL of blank serum along with 100μL of 10 ng/mL oleandrin. To all tubes, 1mL of 1:1 MTBE:ethyl acetate was added and vortexed for 2 min. After centrifuging for 10 min at 850 xg, the samples were dipped into the dry ice/isopropanol bath to freeze the aqueous layer. The liquid organic layer was transferred into new 2mL tubes and dried to completion under a gentle stream of nitrogen gas at 10psi at room temperature in a turbovap. The dried samples were reconstituted in 100μL of 65% MeOH in water and stored at −20°C. The extracted samples were analyzed in two methods.
In method A, electrospray ionization (ESI) triple-quadrupole liquid chromatography-mass spectrometry in negative-ion mode with multiple reaction monitoring was used to detect digoxin, dihydrodigoxin, and oleandrin. 10μL of the samples were injected onto a column (ODS Hypersil 4.6 × 100 mm, 5 μm particle size) using Shimadzu SIL-30AC injector set at 4°C. The column temperature was maintained at 50°C. Using 65% MeOH in water as a mobile phase in isocratic flow, each injection was run for 5 min. Digoxin, dihydrodigoxin, and oleandrin were expected to elute at 2.0 min, 2.1 min, and 3.6 min, respectively. Expected Q1 - Q3 m/z [M-H]- masses were as follows: 779.4 - 475.2 for digoxin, 781.4 - 521.1 for dihydrodigoxin, and 575.2 - 531.1 for oleandrin. Peak areas of the standard curves were converted to concentrations at ng/mL using weighted linear regression (1/x2).
In method B, the extracted samples were shipped on dry ice to the University of Washington Mass Spectrometry Center. Waters Xevo XS tandem mass spectrometry was used to detect digoxin, dihydrodigoxin, and oleandrin. 10μL of the samples were injected onto a column (Waters BEH C18, 2.1 × 50 mm, 1.9 μm particle size) and ran for 7 min. Gradient mobile phase was used (A: 0.1% formic acid in H2O and B: 0.1% formic acid in MeOH) at 50:50 for 0–4 min, 20:80 for 4–5 min, and 50:50 for 5.1–7 min at 0.3 mL/min. A post-column infusion of 5mM ammonium acetate was used at 10 μL/min. The expected retention times for digoxin, dihydrodigoxin, and oleandrin are 2.75 min, 2.6 min, and 3.41 min respectively. NH4-adducts [M + NH4]+ or digoxin (parent m/z: 798.6; daughter m/z: 651.6) and dihydrodigoxin (parent m/z: 800.6; daughter m/z: 408.3) were monitored. Oleandrin was monitored at 577.4 m/z for the parent compound and 373.2 m/z for the daughter compound. Peak areas of the standard curves were converted to concentrations at ng/mL using linear regression.
Quantification of E. lenta in mice
0.1–0.6 g of cecal content collected from mice at the end of the pharmacokinetics experiments was used to extract DNA using the ZymoBIOMICS DNA extraction kit according to the manufacturer’s instructions. 2μL of extracted DNA, 2μL of water, 5μL of iTaq Supermix reagents, and 1μL of 2 μM E lenta-specific primers with a FAM fluorophore probe (elnmrk1; see Table S7) were amplified in 4 technical replicates using the following parameters for 40 cycles: initial denaturation at 95°C for 5 min, denaturation at 95°C for 5 s, and anneal/extension at 60°C for 15 s. A 10-fold dilution series of DNA from E. lenta DSM2243 culture was included to construct a standard curve to convert amplification cycle (Cq) to genome equivalents using the formula that 1 ng of DSM2243 DNA = 2.7e5 genome equivalents.
Quantification and statistical analysis
Statistical analyses and data visualizations were performed using the R v4.1.1 or PRISM 9.4.1 software. As noted, statistical tests included Wilcoxon (analyses of E. lenta abundance and pharmacokinetic parameters), ANOVA (P-gp inhibition assays, qPCR, ATPase activity, metabolomics), and unpaired t tests (metabolomics). Sample size (n) is reported in the figure legends. Unless otherwise noted, values are mean ± SEM (standard error).
Acknowledgments
We are indebted to the other Turnbaugh lab members for their helpful suggestions during the preparation of this manuscript. We thank the Clardy lab at Harvard for their guidance in activity-guided biochemical fractionations, the Mukherjee lab at UCSF for providing E. coli strains with lentiviral packaging plasmids, the UCSF Gnotobiotics Core facility, the UCSF Parnassus Flow Cytometry core, the UCSF Quantitative Metabolite Analysis Center, and the University of Washington Mass Spectrometry Center. This project was supported by the National Institutes of Health (R01HL122593, R01AT011117, R01DK114034, R01AR074500, R21CA227232, PJT; F30CA257378, T.S.K.) and the Natural Sciences and Engineering Research Council of Canada (J.E.B.). P.J.T. is a Chan Zuckerberg Biohub-San Francisco Investigator, held an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund, and was a Nadia’s Gift Foundation Innovator who was supported in part by the Damon Runyon Cancer Research Foundation (DRR-42-16).
Author contributions
Conceptualization, J.E.B. and P.J.T.; investigation, T.S.K., C.Z., M.S., K.T., S.Z., L.A.R.H., L.R., J.J.N.G., K.Y., V.D., E.N.B., J.G.B., D.S.D., and J.E.B.; resources, P.J.T.; writing – original draft, T.S.K. and J.E.B.; writing – review and editing, P.J.T.; supervision, J.E.B. and P.J.T.
Declaration of interests
P.J.T. is on the scientific advisory boards for Pendulum, Seres, and SNIPR Biome; there is no direct overlap between the current study and these consulting duties.
Published: May 27, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.110122.
Contributor Information
Jordan E. Bisanz, Email: jordan.bisanz@psu.edu.
Peter J. Turnbaugh, Email: peter.turnbaugh@ucsf.edu.
Supplemental information
References
- 1.Altman R.B. PharmGKB: a logical home for knowledge relating genotype to drug response phenotype. Nat. Genet. 2007;39:426. doi: 10.1038/ng0407-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brunton L., Chabner B.A., Knollmann B.C. Twelfth. McGraw-Hill Publishing Company; 2011. (Goodman and Gilman’s the Pharmacological Basis of Therapeutics). [Google Scholar]
- 3.Zimmermann M., Zimmermann-Kogadeeva M., Wegmann R., Goodman A.L. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature. 2019;570:462–467. doi: 10.1038/s41586-019-1291-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lam K.N., Alexander M., Turnbaugh P.J. Precision Medicine Goes Microscopic: Engineering the Microbiome to Improve Drug Outcomes. Cell Host Microbe. 2019;26:22–34. doi: 10.1016/j.chom.2019.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Javdan B., Lopez J.G., Chankhamjon P., Lee Y.-C.J., Hull R., Wu Q., Wang X., Chatterjee S., Donia M.S. Personalized Mapping of Drug Metabolism by the Human Gut Microbiome. Cell. 2020;181:1661–1679.e22. doi: 10.1016/j.cell.2020.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bisanz J.E., Spanogiannopoulos P., Pieper L.M., Bustion A.E., Turnbaugh P.J. How to Determine the Role of the Microbiome in Drug Disposition. Drug Metab. Dispos. 2018;46:1588–1595. doi: 10.1124/dmd.118.083402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kyaw T.S., Turnbaugh P.J. Tiny Gatekeepers: Microbial Control of Host Drug Transporters. Clin. Pharmacol. Ther. 2022;112:443–445. doi: 10.1002/cpt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zou L., Spanogiannopoulos P., Pieper L.M., Chien H.-C., Cai W., Khuri N., Pottel J., Vora B., Ni Z., Tsakalozou E., et al. Bacterial metabolism rescues the inhibition of intestinal drug absorption by food and drug additives. Proc. Natl. Acad. Sci. USA. 2020;117:16009–16018. doi: 10.1073/pnas.1920483117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sugimoto Y., Camacho F.R., Wang S., Chankhamjon P., Odabas A., Biswas A., Jeffrey P.D., Donia M.S. A metagenomic strategy for harnessing the chemical repertoire of the human microbiome. Science. 2019;366 doi: 10.1126/science.aax9176. [DOI] [PubMed] [Google Scholar]
- 10.Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z., et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hooper L.V., Wong M.H., Thelin A., Hansson L., Falk P.G., Gordon J.I. Molecular analysis of commensal host-microbial relationships in the intestine. Science. 2001;291:881–884. doi: 10.1126/science.291.5505.881. [DOI] [PubMed] [Google Scholar]
- 12.Larsson E., Tremaroli V., Lee Y.S., Koren O., Nookaew I., Fricker A., Nielsen J., Ley R.E., Bäckhed F. Analysis of gut microbial regulation of host gene expression along the length of the gut and regulation of gut microbial ecology through MyD88. Gut. 2012;61:1124–1131. doi: 10.1136/gutjnl-2011-301104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu Z.D., Selwyn F.P., Cui J.Y., Klaassen C.D. RNA-Seq Profiling of Intestinal Expression of Xenobiotic Processing Genes in Germ-Free Mice. Drug Metab. Dispos. 2017;45:1225–1238. doi: 10.1124/dmd.117.077313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saksena S., Goyal S., Raheja G., Singh V., Akhtar M., Nazir T.M., Alrefai W.A., Gill R.K., Dudeja P.K. Upregulation of P-glycoprotein by probiotics in intestinal epithelial cells and in the dextran sulfate sodium model of colitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2011;300:G1115–G1123. doi: 10.1152/ajpgi.00027.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siccardi D., Mumy K.L., Wall D.M., Bien J.D., McCormick B.A. Salmonella enterica serovar Typhimurium modulates P-glycoprotein in the intestinal epithelium. Am. J. Physiol. Gastrointest. Liver Physiol. 2008;294:G1392–G1400. doi: 10.1152/ajpgi.00599.2007. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y., Liu Y., Sidhu A., Ma Z., McClain C., Feng W. Lactobacillus rhamnosus GG culture supernatant ameliorates acute alcohol-induced intestinal permeability and liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2012;303:G32–G41. doi: 10.1152/ajpgi.00024.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Degraeve A.L., Haufroid V., Loriot A., Gatto L., Andries V., Vereecke L., Elens L., Bindels L.B. Gut microbiome modulates tacrolimus pharmacokinetics through the transcriptional regulation of ABCB1. Microbiome. 2023;11:138. doi: 10.1186/s40168-023-01578-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Foley S.E., Tuohy C., Dunford M., Grey M.J., De Luca H., Cawley C., Szabady R.L., Maldonado-Contreras A., Houghton J.M., Ward D.V., et al. Gut microbiota regulation of P-glycoprotein in the intestinal epithelium in maintenance of homeostasis. Microbiome. 2021;9:183. doi: 10.1186/s40168-021-01137-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saha J.R., Butler V.P., Jr., Neu H.C., Lindenbaum J., Lindenbaum J. Digoxin-inactivating bacteria: identification in human gut flora. Science. 1983;220:325–327. doi: 10.1126/science.6836275. [DOI] [PubMed] [Google Scholar]
- 20.Elmeliegy M., Vourvahis M., Guo C., Wang D.D. Effect of P-glycoprotein (P-gp) Inducers on Exposure of P-gp Substrates: Review of Clinical Drug-Drug Interaction Studies. Clin. Pharmacokinet. 2020;59:699–714. doi: 10.1007/s40262-020-00867-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haiser H.J., Gootenberg D.B., Chatman K., Sirasani G., Balskus E.P., Turnbaugh P.J. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science. 2013;341:295–298. doi: 10.1126/science.1235872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koppel N., Bisanz J.E., Pandelia M.-E., Turnbaugh P.J., Balskus E.P. Discovery and characterization of a prevalent human gut bacterial enzyme sufficient for the inactivation of a family of plant toxins. Elife. 2018;7 doi: 10.7554/eLife.33953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alexander M., Ang Q.Y., Nayak R.R., Bustion A.E., Sandy M., Zhang B., Upadhyay V., Pollard K.S., Lynch S.V., Turnbaugh P.J. Human gut bacterial metabolism drives Th17 activation and colitis. Cell Host Microbe. 2022;30:17–30.e9. doi: 10.1016/j.chom.2021.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong X., Guthrie B.G.H., Alexander M., Noecker C., Ramirez L., Glasser N.R., Turnbaugh P.J., Balskus E.P. Genetic manipulation of the human gut bacterium Eggerthella lenta reveals a widespread family of transcriptional regulators. Nat. Commun. 2022;13:7624. doi: 10.1038/s41467-022-33576-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ledwitch K.V., Gibbs M.E., Barnes R.W., Roberts A.G. Cooperativity between verapamil and ATP bound to the efflux transporter P-glycoprotein. Biochem. Pharmacol. 2016;118:96–108. doi: 10.1016/j.bcp.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang X., Yang P., Shen J., Osaka E., Choy E., Cote G., Harmon D., Zhang Z., Mankin H., Hornicek F.J., Duan Z. Prevention of multidrug resistance (MDR) in osteosarcoma by NSC23925. Br. J. Cancer. 2014;110:2896–2904. doi: 10.1038/bjc.2014.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harmsen S., Meijerman I., Febus C.L., Maas-Bakker R.F., Beijnen J.H., Schellens J.H.M. PXR-mediated induction of P-glycoprotein by anticancer drugs in a human colon adenocarcinoma-derived cell line. Cancer Chemother. Pharmacol. 2010;66:765–771. doi: 10.1007/s00280-009-1221-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koji T., Ewa M., Vanden Huevel J.P., Bruce S. 2017. Profiling Drug Activity of Human and Ortholog Xenobiotic-Sensing Receptors: PXR, CAR, AhR, and FXR. Indigobiosciences.https://indigobiosciences.com/profiling-drug-activity-human-ortholog-xenobiotic-sensing-receptors-pxr-car-ahr-fxr/ [Google Scholar]
- 29.Duan Z., Choy E., Hornicek F.J. NSC23925, identified in a high-throughput cell-based screen, reverses multidrug resistance. PLoS One. 2009;4 doi: 10.1371/journal.pone.0007415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bisanz J.E., Soto-Perez P., Noecker C., Aksenov A.A., Lam K.N., Kenney G.E., Bess E.N., Haiser H.J., Kyaw T.S., Yu F.B., et al. A Genomic Toolkit for the Mechanistic Dissection of Intractable Human Gut Bacteria. Cell Host Microbe. 2020;27:1001–1013.e9. doi: 10.1016/j.chom.2020.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhong Q., Kobe B., Kappler U. Molybdenum Enzymes and How They Support Virulence in Pathogenic Bacteria. Front. Microbiol. 2020;11 doi: 10.3389/fmicb.2020.615860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maini Rekdal V., Nol Bernadino P., Luescher M.U., Kiamehr S., Le C., Bisanz J.E., Turnbaugh P.J., Bess E.N., Balskus E.P. A widely distributed metalloenzyme class enables gut microbial metabolism of host- and diet-derived catechols. Elife. 2020;9 doi: 10.7554/eLife.50845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Noecker C., Sanchez J., Bisanz J.E., Escalante V., Alexander M., Trepka K., Heinken A., Liu Y., Dodd D., Thiele I., et al. Systems biology elucidates the distinctive metabolic niche filled by the human gut microbe Eggerthella lenta. PLoS Biol. 2023;21 doi: 10.1371/journal.pbio.3002125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rech S., Wolin C., Gunsalus R.P. Properties of the periplasmic ModA molybdate-binding protein of Escherichia coli. J. Biol. Chem. 1996;271:2557–2562. doi: 10.1074/jbc.271.5.2557. [DOI] [PubMed] [Google Scholar]
- 35.Maini Rekdal V., Bess E.N., Bisanz J.E., Turnbaugh P.J., Balskus E.P. Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science. 2019;364 doi: 10.1126/science.aau6323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bess E.N., Bisanz J.E., Yarza F., Bustion A., Rich B.E., Li X., Kitamura S., Waligurski E., Ang Q.Y., Alba D.L., et al. Genetic basis for the cooperative bioactivation of plant lignans by Eggerthella lenta and other human gut bacteria. Nat. Microbiol. 2020;5:56–66. doi: 10.1038/s41564-019-0596-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hille R. The Mononuclear Molybdenum Enzymes. Chem. Rev. 1996;96:2757–2816. doi: 10.1021/cr950061t. [DOI] [PubMed] [Google Scholar]
- 38.Cheng A.G., Ho P.-Y., Aranda-Díaz A., Jain S., Yu F.B., Meng X., Wang M., Iakiviak M., Nagashima K., Zhao A., et al. Design, construction, and in vivo augmentation of a complex gut microbiome. Cell. 2022;185:3617–3636.e19. doi: 10.1016/j.cell.2022.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Turnbaugh P.J., Ridaura V.K., Faith J.J., Rey F.E., Knight R., Gordon J.I. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009;1 doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spanogiannopoulos P., Bess E.N., Carmody R.N., Turnbaugh P.J. The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism. Nat. Rev. Microbiol. 2016;14:273–287. doi: 10.1038/nrmicro.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wickham H. Springer-Verlag; New York: 2016. ggplot2: Elegant Graphics for Data Analysis. [Google Scholar]
- 42.Paradis E., Schliep K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics. 2019;35:526–528. doi: 10.1093/bioinformatics/bty633. [DOI] [PubMed] [Google Scholar]
- 43.Chandran S. Rapid Assembly of DNA via Ligase Cycling Reaction (LCR) Methods Mol. Biol. 2017;1472:105–110. doi: 10.1007/978-1-4939-6343-0_8. [DOI] [PubMed] [Google Scholar]
- 44.Tsugawa H., Cajka T., Kind T., Ma Y., Higgins B., Ikeda K., Kanazawa M., VanderGheynst J., Fiehn O., Arita M. MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods. 2015;12:523–526. doi: 10.1038/nmeth.3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wishart D.S., Guo A., Oler E., Wang F., Anjum A., Peters H., Dizon R., Sayeeda Z., Tian S., Lee B.L., et al. HMDB 5.0: the Human Metabolome Database for 2022. Nucleic Acids Res. 2022;50:D622–D631. doi: 10.1093/nar/gkab1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data: All data is available in the main text and the supplemental information and tables. Metabolomics datasets are available through our lab GitHub (https://github.com/turnbaughlab/2024_Kyaw_iScience). No new DNA, RNA, or protein sequence data were generated in this study.
-
•
Code: Comparative genomics analysis utilized the ElenMatchR tool30 and NCBI BlastX.
-
•
All other items: Previously published RNA-Seq reads of Eggerthella lenta DSM2243, Coriobacteriaceae genomes, and other reference genomes we used in this study are available in NCBI (NCBI: PRJNA507796; NCBI: PRJNA412637; NCBI taxids: 562, 1680, 216816, 1522, 29347, 29348, 820, 821, 818, 239935, 46503, 39491, 40520, 88431, 165179).