

Summary
High salt (HS) consumption is a risk factor for multiple autoimmune disorders via disturbing immune homeostasis. Nevertheless, the exact mechanisms by which HS exacerbates rheumatoid arthritis (RA) pathogenesis remain poorly defined. Herein, we found that heightened phosphorylation of PDPK1 and SGK1 upon HS exposure attenuated FoxO1 expression to enhance the glycolytic capacity of CD4 T cells, resulting in strengthened Th17 but compromised Treg program. GSK2334470 (GSK), a dual PDPK1/SGK1 inhibitor, effectively mitigated the HS-induced enhancement in glycolytic capacity and the overproduction of IL-17A. Therefore, administration of GSK markedly alleviated HS-exacerbated RA progression in collagen-induced arthritis (CIA) model. Collectively, our data indicate that HS consumption subverts Th17/Treg homeostasis through the PDPK1-SGK1-FoxO1 signaling, while GSK could be a viable drug against RA progression in clinical settings.
Subject areas: Pharmacology, Natural sciences, Biological sciences, Physiology, Immunology
Graphical abstract
Highlights
-
•
High-salt (HS) intake positively correlates with RA severity
-
•
HS exacerbates Th17/Treg imbalance via PDPK1-SGK1-FoxO1 signaling
-
•
GSK rectifies HS-induced glycolytic reprogramming and Th17/Treg imbalance
-
•
GSK mitigates HS-exacerbated RA progression
Pharmacology; Natural sciences; Biological sciences; Physiology; Immunology
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disorder characterized by the symmetrical and progressive joint inflammation, ultimately leading to the degradation of articular cartilage, bone erosion and physical disability.1 The etiology of RA involves genetic predisposition as well as risk factors including cigarette smoking, obesity, microbial infections and so on.2 High salt (HS) diet has been acknowledged as a potential contributing factor for autoimmune diseases such as inflammatory bowel disease (IBD), experimental autoimmune encephalomyelitis (EAE), and systemic lupus erythematosus (SLE).3,4,5,6,7 Under the RA setting, elevated sodium intake in smokers has been linked to an increased likelihood of anti-citrullinated protein antibody (ACPA) positivity and the occurrence of RA. Additionally, RA patients presenting with bone erosion at the time of their initial diagnosis possess higher levels of sodium excretion compared to those patients absent of bone erosion.8,9,10,11 These preliminary clinical studies on RA patients collectively supported the notion of “salty truth” in autoimmune disorders.12 Nonetheless, compared to other disease conditions, the phenotypes and specific mechanisms linking salt consumption to RA severity are insufficiently addressed. Knowledge gaps exist regarding: (1) whether and how HS diet modifies RA progression in genetic predisposed individuals, (2) molecular consequences of HS intake among RA patients, and most critically (3) the pursuit of promising drug targets that can also be applied to RA patients who prefer salty taste. Considering the potency of dietary intervention in RA management13 and the prevalence of high-salt containing food in nowadays society, filling those knowledge gaps could lead to novel dietary recommendations and therapeutic strategies against RA, making this area of research scientifically vital and clinically relevant.
The immunopathology of RA is underscored by the imbalance between T helper 17 (Th17) cells and regulatory T (Treg) cells. Th17 cells instigate joint inflammation via releasing an array of pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin (IL)-17A and granulocyte-macrophage colony-stimulating factor (GM-CSF). IL-17A stimulates fibroblasts to produce receptor activator od nuclear factor-kappa B ligand (RANKL), which in turn promotes osteoclast maturation and the secretion of destructive enzymes like matrix metalloproteinase (MMP)-1 and MMP-3.14 Elevated levels of IL-17A in the serum and synovial fluid of RA patients positively correlate with autoantibody titer and disease activity.15,16,17,18 In sharp contrast, Treg cells are pivotal negative regulators in RA progression and a major source of anti-inflammatory IL-10. Unlike wild-type mice, Il-10 deficient mice develop more severe arthritis following type II collagen induction.19,20,21 Consistently, sodium butyrate inhibits collagen-induced arthritis (CIA) by enhancing IL-10 expression and Treg cell polarization, while anti-IL-10 neutralizing antibodies abolish the beneficial effects of sodium butyrate.22 A decline in the number or functionality of Treg cells leads to an exaggerated Th17 response, resulting in enhanced autoimmune reaction and joint inflammation.19,20,21,23 Worse still, Treg cells within RA patients tend to convert into pathogenic Th17 cells and accumulate in the inflammatory synovial membrane, which renders a Th17/Treg imbalance along with exacerbated disease progression.24 In animal models, the ablation of Th17 cells or their associated cytokines alleviates arthritis symptoms,25,26 while depletion of Treg cells or IL-10 exacerbates RA severity.19,20,27 Therefore, an imbalance between Th17 and Treg cells is crucial in RA initiation and progression.
Glucose metabolism has been identified as a master regulator of T cell function and differentiation.28 While Th17 and effector T cells mainly rely on aerobic glycolysis for energy demand, Treg cells and memory T cells depend on mitochondrial respiration and fatty acid oxidation.29 The serine/threonine kinase 3′-phosphoinositide dependent kinase 1 (PDPK1) is closely associated with glycolytic metabolism and acts as a key regulator of cell growth and proliferation.30,31 In CD4 T cells, PDPK1 integrates signals from the T cell receptor (TCR) and CD28, thereby participating in the activation of nuclear transcription factor-kappa B (NF-kB) signaling pathway.32 PDPK1 is also essential for the transition of thymocytes from the double-negative (DN) stage to the double-positive (DP) stage. However, its absence does not affect the thymic development of DP cells into single-positive (SP) cells.33 In rats with induced arthritis, PDPK1 overexpression accelerates disease progression by promoting fibroblast-like synoviocyte (FLS) proliferation.34 Conversely, inhibition of PDPK1 restrains the invasive capacity of human FLSs via reducing the expression of MMP-2 and MMP-9.35,36 These lines of evidence indicate that PDPK1 participates in RA progression through regulating the activity of FLS, yet its role in T cell immunometabolism in the context of RA remains unexplored.
PDPK1 activates several kinases of the protein kinase A-protein kinase G-protein kinase C (AGC) family, notably including serum- and glucocorticoid-induced protein kinase 1 (SGK1).30 SGK1 acts as a “sodium sensor” in T cells in response to extracellular salt fluctuation.37,38,39,40 This is biologically significant as SGK1 plays a critical part in IL-23R mediated inhibition of Treg cells and the promotion of Th17 program, both of which are pivotally engaged in the immunopathology of RA.41 The rationale of our study hinges on these observations and we determined to elucidate the mechanisms whereby increased salt intake exacerbates RA, with a focus on the PDPK1/SGK1 pathway. HS intake is operationally defined here as a daily consumption of sodium chloride exceeding 5 g, as the World Health Organization (WHO) recommends no more than 5 g daily salt intake, a threshold we adopted to categorize RA patients into low and HS diet groups based on their self-assessment.42 In accordance, we assess the potential of utilizing the dual PDPK1/SGK1 inhibitor, GSK2334470 (referred to as GSK hereafter), as a novel drug for RA treatment. Our study thus provides valuable insight into how HS diet modifies RA progression and offers a promising therapeutic strategy for RA treatment.
Results
The phosphorylated PDPK1 and SGK1 in CD4 T cells correlate with RA severity
To demonstrate the roles of PDPK1 and SGK1 in CD4 T cells of RA patients, peripheral CD4 T cells from RA patients and healthy controls (HCs) were isolated and subjected to western blot analysis. It was noted that the phosphorylated PDPK1 and SGK1 were higher in RA patients as compared to those of healthy individuals (Figures 1A–1C). Particularly, the phosphorylated PDPK1 (Figure 1D) and SGK1 (Figure 1E) levels exhibited a positive correlation with the DAS28-ESR score, an indicator of RA severity. We then decided to utilize PDPK1/SGK1 inhibitor GSK for the in vitro validation. At the concentration of 2.5 μmol/L, GSK displayed no observable impact on either CD4 T cell apoptosis (Figures S1A–S1C) or proliferation (Figure S1D). However, at the concentrations of 5 and 10 μmol/L, GSK notably augmented CD4 T cell apoptosis (Figures S1A–S1C) and hampered their proliferation (Figure S1D). Therefore, 2.5 μmol/L of GSK was applied in subsequent experiments. Specifically, exposure of RA derived CD4 T cells to HS further enhanced the phosphorylated levels of both PDPK1 and SGK1 (Figures 1F–1H), resulting in an increased Th17 proportion (Figures 1I and 1J) and a decline in Treg frequency (Figures 1K and 1L). In contrast, GSK treatment attenuated PDPK1 and SGK1 phosphorylation (Figures 1F–1H), by which it corrected the Th17 enhancement and Treg impediment triggered by HS treatment (Figures 1I–1L).
Figure 1.

Phosphorylation levels of PDPK1 and SGK1 in CD4 T cells correlate with RA severity
(A) Expression levels of (p-)PDPK1 and (p-)SGK1 in CD4 T cells of RA (n = 19) and HC (n = 16) groups were detected by Western blot.
(B and C) Quantification of the phosphorylation levels of PDPK1 and SGK1 in CD4 T cells of RA and HC groups.
(D and E) Correlation analysis of disease activity index DAS28-ESR with the phosphorylation levels of PDPK1 and SGK1.
(F–H) CD4 T cells were isolated from RA patients and cultured for 3 days in the presence of HS (40 mmol/L) and/or GSK (2.5 μmol/L). Expression levels of (p-)PDPK1 and (p-)SGK1 in CD4 T cells of RA were detected by Western blot.
(I and J) CD4 T cells from RA patients were isolated and treated with HS and/or GSK for 3 days, and the proportion of Th17 cells was detected by flow cytometry.
(K and L) CD4 T cells from RA patients were isolated and treated with HS and/or GSK for 3 days, and the proportion of Treg cells was detected by flow cytometry. Statistical significance was calculated by unpaired Student’s t test and data are represented as mean ± SD. The correlation was determined by linear regression analysis for (D) and (E). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ns not significant.
Next, we divided RA patients into low salt (≤5g/day) and HS (>5g/day) diet group based on their daily salt consumption. Clinical variable analysis unveiled that patients in the HS diet group exhibited elevated phosphorylated levels for PDPK1 and SGK1, along with a higher number of affected joints and increased DAS28-ESR scores (p < 0.05). In contrast, no significant difference was identified between the two dietary groups in terms of clinical indices, such as the ESR level, disease duration, rheumatoid factor (RF) positivity, anti-cyclic citrullinated peptide (anti-CCP) antibody positivity, gender, and age (Table 1). Overall, these data suggest a possible connection between HS, PDPK1/SGK1, and disease activity of RA patients.
Table 1.
Comparison of clinical characteristics between RA patients on a low-salt diet versus RA patients on a high-salt diet
| Low-Salt Diet RA patients (n = 7) | High-Salt Diet RA patients (n = 12) | p-Value | |
|---|---|---|---|
| p-SGK1 Relative Expression (mean ± SD) | 0.54 ± 0.11 | 0.66 ± 0.10 | 0.020 |
| p-PDPK1 Relative Expression (mean ± SD) | 0.50 ± 0.06 | 0.57 ± 0.06 | 0.017 |
| Number of Tender Joints (mean ± SD) | 3.71 ± 1.11 | 6.17 ± 2.25 | 0.016 |
| Number of Swollen Joints (mean ± SD) | 1.71 ± 1.80 | 3.67 ± 2.50 | 0.089 |
| ESR (mean ± SD) | 31.3 ± 8.20 | 39.92 ± 10.31 | 0.076 |
| DAS28-ESR (mean ± SD) | 4.21 ± 0.59 | 5.04 ± 0.64 | 0.013 |
| Disease Duration (months, mean ± SD) | 6.00 ± 1.92 | 6.75 ± 2.01 | 0.435 |
| RF Positive (%) | 85.7 | 75.0 | 1.000 |
| anti-CCP Antibody Positive (%) | 100 | 75 | 0.263 |
| Female (%) | 71.4 | 83.3 | 0.603 |
| Age (years, mean ± SD) | 33.86 ± 5.70 | 33.17 ± 5.13 | 0.789 |
ESR: Erythrocyte Sedimentation Rate; DAS28-ESR: Disease Activity Score based on 28 Joints with ESR; RF: Rheumatoid Factor; anti-CCP Antibody: Anti-Cyclic Citrullinated Peptide Antibody. p < 0.05 indicates statistically significant difference.
GSK reverses the effects of HS on Th17 and Treg program
Based on the above findings, we next sought to address whether GSK affects the polarization of Th17 and Treg cells, particularly under a HS condition (Figure 2A). As expected, HS treatment markedly promoted Th17 differentiation, whereas GSK suppressed the Th17 program either used alone or in the context of HS treatment (Figures 2B and 2C). In contrast, HS substantially hindered the induction of Treg cells, which could be reversed by co-administration of GSK (Figures 2D and 2E). The Treg cells were next challenged under a Th17 polarizing condition. Strikingly, HS prominently bolstered the IL-17A levels in Foxp3+ Treg cells, while GSK prevented the ectopic expression of IL-17A with or without HS treatment (Figures 2F and 2G).
Figure 2.

GSK reverses the effects of HS on Th17 and Treg program in vitro
(A) Schematic representation of Th17 and Treg in vitro differentiation.
(B and C) The proportion of differentiated Th17 cells (n = 3).
(D and E) The proportion of differentiated Treg cells (n = 3).
(F and G) Naive CD4 T cells were cultured for 3 days under the Treg polarization condition. Then, Treg cells were harvested, washed and cultured for an additional 2 days under Th17 condition in the presence of HS and/or GSK. The proportion of IL-17A producing cells within the Foxp3+ Treg cells was shown (n = 3). Statistical significance was calculated by unpaired Student’s t test and data are represented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
In order to corroborate the in vitro observations, we adoptively transferred labeled Th17 cells into host mice pretreated with a control vehicle (Con), HS, GSK or HS + GSK, respectively (Figure 3A). Analysis of recipient-derived CD4 T cells revealed that mice in the HS group displayed an increased proportion of Th17 cells in both spleen (Figures 3B and 3C) and lymph nodes (Figure S2A) as compared to that of mice in the Con group, while mice in the GSK and HS + GSK groups were characterized by a lower Th17 frequency. Moreover, mice in the HS group exhibited a decreased proportion of Treg cells in the spleen (Figures 3D and 3E) and lymph nodes (Figure S2B), and the phenotypes could be partially rescued by GSK treatment. Consistently, in both spleen and lymph nodes, the Th17/Treg ratio within recipient-derived CD4 T cells increased in HS group, comparing to Con mice, and such increment could be abolished by GSK (Figures S2E and S2F). In line with these findings, the proportion of activated CD4 T cells was increased in the HS group but decreased in the GSK and HS + GSK groups in the spleen (Figures 3F and 3G) and lymph nodes (Figure S2C) as compared to that of mice in the control group. Similarly, in a separate analysis gated on donor cells, we found that the HS group had increased proportion of IL-17A producing cells in the spleen (Figures 3H and 3I) and lymph nodes (Figure S2D), while the GSK and HS + GSK groups showed a lower frequency.
Figure 3.

GSK reverses the effects of HS on Th17 and Treg program in vivo
(A) Schematic representation of the adoptive transfer model.
(B and C) The proportion of Th17 cells within splenic CD4 T cells of recipient mice (n = 5).
(D and E) The proportion of Treg cells within splenic CD4 T cells of recipient mice (n = 5).
(F and G) The proportion of activated CD4 T cells within splenic CD4 T cells of recipient mice (n = 5).
(H and I) The proportion of IL-17A producing donor cells in spleen (n = 3). Statistical significance was calculated by unpaired Student’s t test and data are represented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To further investigate the in vivo impact of HS and GSK on Treg cells, we generated the Treg lineage tracing mice (Foxp3Cre-eGFP; Rosa26flox-stop-flox-tdTomato, defined as Rosa-Red) as detailed in Figure 4A. The successful construction of Rosa-Red mice was confirmed by one-drop blood FACS staining (Figure 4B) and PCR genotyping (Figure 4C). Remarkably, the HS group displayed an elevated proportion of tdTomato+ GFP− unstable Treg cells within the total Treg population as compared to the control group, whereas GSK treatment reduced the frequency of unstable Tregs (Figures 4D and 4E). Similarly, in the lymph nodes, the proportion of unstable Tregs within the CD4 T cell population was decreased in the HS group, which was rescued by the GSK treatment (Figures 4D and 4F). Taken together, our findings confirmed that GSK rectifies HS-triggered Th17/Treg imbalance through inhibiting Th17 differentiation, promoting the Treg program, and enhancing Treg stability.
Figure 4.

GSK rectifies high salt-induced Treg instability in vivo
(A) Schematic representation of constructing Rosa-Red mouse model.
(B) Peripheral one-drop blood flow cytometry was employed to validate Rosa-Red transgenic mice.
(C) PCR was employed for genotyping Rosa-Red mice.
(D–F) The proportion of unstable Treg cells (tdTomato+ GFP−) within the total Treg population in the spleen and lymph nodes of Rosa-Red mice. Statistical significance was calculated by unpaired Student’s t test and data are represented as mean ± SD. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
GSK ameliorates HS-exacerbated progression of arthritis
To assess the effects of HS and GSK on RA progression, we employed a CIA model in RA-susceptible DBA/1 mice. DBA/1 mice were fed with either a normal diet or a HS diet prior to the first immunization. Treatment with either GSK (25 mg/kg, every three days) or a control vehicle began immediately after the second immunization (Figure 5A). Our results showed that the HS diet significantly intensified arthritic symptoms, as evidenced by the elevated arthritis scores (Figure 5B), increased paw and ankle thickness (Figures 5C and 5D), and worsened joint pathology (Figures 5E–5G). Conversely, GSK treatment alleviated these arthritic symptoms and counteracted the adverse effects of HS diet on RA progression (Figures 5B–5G). In line with these observations, GSK-treated CIA mice displayed a reduced proportion of Th17 cells (Figures 5H, 5I, S3, andS4A) along with an increased proportion of Treg cells (Figures 5J, 5K; S4B) in the spleen and lymph nodes. In contrast, mice on HS diet exhibited an elevated frequency of Th17 cells and a reduced proportion of Treg cells that were efficiently reversed by GSK treatment in the spleen (Figures 5H–5K) and lymph nodes (Figures S4A and S4B). Consistently, in both spleen and lymph nodes, the Th17/Treg ratio increased in CIA mice fed with a high-salt diet when compared to Con mice, while GSK treatment could reverse such effect (Figures S4G and S4H). Similarly, the percentages of activated CD4 T cells both in the spleen (Figures 5L and 5M) and lymph nodes (Figure S4C) were significantly higher in the HS group, while GSK treatment efficiently dampened T cell activation.
Figure 5.

GSK ameliorates high salt-accelerated arthritis progression
(A) Schematic representation of CIA model.
(B) Arthritis score of CIA mice (n = 6).
(C) Paw thickness of CIA mice (n = 6).
(D) Ankle thickness of CIA mice (n = 6).
(E) Representative pictures of the hind paw joints of CIA mice at day 42.
(F) H&E staining of ankle joints of CIA mice. Scale bars, 100 μm.
(G) The histopathological score of knee joints of CIA mice.
(H and I) The proportion of Th17 cells within splenic CD4 T cells of CIA mice (n = 6).
(J and K) The proportion of Treg cells within splenic CD4 T cells of CIA mice (n = 6).
(L and M) The proportion of activated CD4 T cells within splenic CD4 T cells of CIA mice (n = 6). Statistical significance was calculated by unpaired Student’s t test and data are represented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Given that the Th17 and Treg frequencies were altered following treatment with GSK or HS, we then evaluated the serum levels of cytokines associated with these two T cell subsets. Strikingly, compared to control mice, pro-inflammatory cytokines TNF-α and IL-6, were reduced in the GSK group but elevated in the HS group. In contrast, the anti-inflammatory cytokine IL-10 was higher in the GSK group and lower in the HS group. Moreover, compared to the HS group, co-administration of GSK and HS resulted in the down-regulation of TNF-α and IL-6 but up-regulation of IL-10 (Figures S4D–S4F). Altogether, our results support that GSK serves as a promising candidate for RA treatment, and the therapeutic effect is achieved through mitigating the Th17/Treg imbalance, especially in the case of consumption of a HS diet.
GSK represses the glycolytic capacity of HS-insulted CD4 T cells
To elucidate the mechanisms by which HS and GSK influence the Th17/Treg balance, we carried out the following experiments using splenic CD4 T cells isolated from C57BL/6 mice (Figure 6A). Upon activation with plate-coated anti-CD3 and anti-CD28 antibodies (TCR stimulation), CD4 T cells were treated with HS, GSK or HS + GSK for 3 days. GSK significantly repressed the phosphorylation of PDPK1 and SGK1, while the opposite results were noted in HS-treated cells (Figures 6B–6D). As PDPK1 and SGK1 are pivotally engaged in the regulation of glycolytic process, we conducted glucose uptake assay and found GSK prominently counteracted the enhancing effect of HS (Figures S5A and S5B). Seahorse metabolic analysis further confirmed that GSK diminished both basal and maximal glycolytic rates that were boosted by HS (Figures 6E–6G). Next, CD4 T cells from RA patients were treated with HS, GSK, HS + GSK, and HS + anti-TNF-α for 3 days followed by Seahorse analysis. Of note, GSK similarly reversed the glycolysis-promoting function of HS while anti-TNFα only demonstrated a limited effect (Figures 6H–6J). Further, CD4 T cells from RA patients were cultured under conditions without high-salt stimulation and treated with either PBS, GSK or anti-TNF-α for 3 days, followed by Seahorse analysis. Remarkably, GSK reduced both the basal and maximal glycolytic rates of CD4 T cells, whereas anti-TNF-α displayed no perceptible impact on CD4 T cell glycolysis when compared to the PBS group (Figures 6K–6M). Collectively, these data indicate that HS challenge promotes the glycolytic capacity of CD4 T cells by activating the PDPK1-SGK1 signaling, which could be reversed by the addition of GSK.
Figure 6.

GSK down-tunes the glycolytic metabolism of high salt-insulted CD4 T cells
(A) Schematic representation of CD4 T cell culture.
(B) Expression levels of (p-)PDPK1 and (p-)SGK1 in CD4 T cells following treatment with GSK and/or HS (n = 3) were detected by western blot.
(C and D) Quantitative analysis of p-PDPK1 and p-SGK1 expression (n = 3).
(E) The glycolytic activity of CD4 T cells following GSK and/or HS treatment (n = 3).
(F) Basal glycolysis rates post-treatment (n = 3).
(G) Maximal glycolysis capacity of CD4 T cells following GSK and/or HS treatment (n = 3).
(H) The glycolytic activity of CD4 T cells following co-treatment with HS and GSK or anti-TNF-α (n = 3).
(I) Basal glycolysis rates of CD4 T cells following co-treatment with HS and GSK or anti-TNF-α (n = 3).
(J) Maximal glycolysis rates following co-treatment with HS and GSK or anti-TNF-α (n = 3).
(K) The glycolytic activity of CD4 T cells following treatment with PBS, GSK or anti-TNF-α (n = 3).
(L) Basal glycolysis rates of CD4 T cells following treatment with PBS, GSK or anti-TNF-α (n = 3).
(M) Maximal glycolysis rates following treatment with PBS, GSK or anti-TNF-α (n = 3). Statistical significance was calculated by unpaired Student’s t test and data are represented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, ns (not significant).
Blockade of FoxO1 impairs the immune regulatory effect of GSK
The above findings prompted us to check the expression of key molecules relevant to glycolysis and glucose uptake. Indeed, an HS insult induced the expression of HK2, LDHA and Glut1 both in mouse (Figures 7A and 7B) and human CD4 T cells (Figures 7C and 7D), which was reversed by the addition of GSK. It was interestingly noted that HS attenuated the expression of FoxO1, a crucial molecule downstream of SGK1 signaling, while its expression was restored upon the addition of GSK (Figures 7A–7D).
Figure 7.

Blockade of FoxO1 impairs the immune regulatory effects of GSK
(A and B) CD4 T cells were isolated from C57BL/6 mice and stimulated by anti-CD3/28 in the presence of GSK and/or HS or not for 3 days (A) Western blot analysis was employed to assess the expression levels of Glut1, HK2, LDHA, and FoxO1 in CD4 T cells from C57BL/6 mice (n = 3). (B) Quantitative analysis of Glut1, HK2, LDHA, and FoxO1 expression in CD4 T cells from C57BL/6 mice (n = 3). (C, D) CD4 T cells were isolated from peripheral blood of RA patients and stimulated by anti-CD3/28 in the presence of GSK and/or HS for 3 days.
(C) Western blot analysis was employed to assess the expression levels of Glut1, HK2, LDHA, and FoxO1 in CD4 T cells from RA patients (n = 3).
(D) Quantitative analysis of Glut1, HK2, LDHA, and FoxO1 expression in CD4 T cells from RA patients (n = 3).
(E–J) Naive CD4 T cells from C57BL/6 mice were polarized into Th17 cells or Treg cells in the presence of different combinations of HS, GSK, 2-DG and iFoxO1 for 3 days. For Treg stability assay, naive CD4 T cells were initially cultured for 3 days under Treg polarization condition. Then, Treg cells were harvested, washed and cultured for an additional 2 days under Th17 condition in the presence of HS, GSK, 2-DG or iFoxO1. (E, H) The proportion of Th17 cells within CD4 T cells following distinct stimulation (n = 3). (F, I) The proportion of Treg cells within CD4 T cells following distinct stimulation (n = 3). (G, J) The proportion of IL-17A producing cells within total Foxp3+ Treg cells following distinct stimulation (n = 3). Statistical significance was calculated by unpaired Student’s t test and data are represented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Given the role of FoxO1 played in GSK-mediated transcription of glycolytic effectors and IL-17A, we then embarked on the dependence of GSK on FoxO1-mediated glycolytic rewiring. To this end, CD4 T cells isolated from C57BL/6 mice were treated with FoxO1 inhibitor (iFoxO1) for 3 days. As expected, iFoxO1 significantly up-regulated the expression levels of Glut1, LDHA and HK2 (Figures S5C–S5F). We then polarized naive T cells to Th17 and Treg subsets under different combinations of HS, GSK, iFoxO1 and 2-DG (glycolysis inhibitor). Compared to the PBS control, the proportion of in vitro polarized Th17 cells increased upon HS treatment (Figures 7E and 7H). As both 2-DG and GSK inhibited Th17 program in the presence or absence of HS, iFoxO1 efficiently abolished the inhibitory effect of GSK (Figures 7E and 7H). Conversely, in respect to Treg induction, the proportion of in vitro polarized Treg cells decreased upon HS treatment (Figures 7F and 7I). However, 2-DG and GSK both restored the defective Treg program induced by HS, while iFoxO1 treatment diminished the rescuing effect of GSK (Figures 7F and 7I). We then checked Treg stability by probing the expression level of ectopic IL-17A. Compared to the PBS control, the expression levels of IL-17A in Treg cells elevated in the HS group (Figures 7G and 7J). While 2-DG and GSK restrained IL-17A ectopic expression in the presence or absence of HS, iFoxO1 treatment nullified the protective role of GSK (Figures 7G and 7J). Collectively, those data supported that GSK favorably rectifies the HS-triggered Th17/Treg imbalance, which depends on FoxO1-imposed restriction of glycolytic capacity and IL-17A production in CD4 T cells.
Discussion
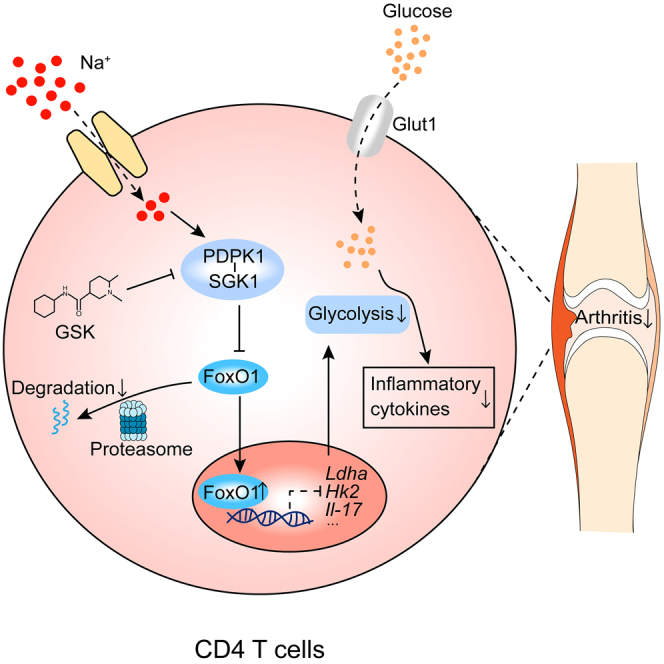
It was noted that mice fed a HS diet displayed worsened disease symptoms and elevated frequency of Th17 cells in the spleen of an EAE model.43 However, HS did not seemingly affect the quantity and function of dendritic cells (DCs), highlighting a direct impact of HS on the Th17 program. Herein in this report, we demonstrated evidence indicating that an HS insult significantly enhances the glycolytic capacity of CD4 T cells through the PDPK1-SGK1-FoxO1 axis, which subsequently disturbs the Th17/Treg program and promotes RA progression. Therefore, GSK, a PDPK1/SGK1 dual inhibitor, possesses the potency to effectively correct the Th17/Treg imbalance, thereby mitigating the detrimental outcomes resulting from an HS insult (Figure 8). We also provided preliminary clinical evidence that the elevated PDPK1/SGK1 phosphorylation level in CD4 T cells is highly correlated with daily salt intake and RA severity. It is noteworthy that our study only included a limited number of samples, follow-up studies with a larger RA cohort would be necessary.
Figure 8.

Graphical illustration
High salt stimulation turbocharges the glycolytic metabolism of CD4 T cells through the PDPK1-SGK1-FoxO1 signaling pathway, which subsequently disturbs the Th17/Treg balance and promotes the progression of RA. Utilization of PDPK1/SGK1 inhibitor, GSK, effectively corrects Th17/Treg imbalance to mitigate the detrimental outcomes of high salt insult.
Cellular metabolism is intricately connected to the functional adaptation of immune cells.44 Th17 cells predominantly rely on glycolysis for energy production, while Treg cells depend on mitochondrial metabolism and fatty acid oxidation for energy supply. The glycolytic capacity of Treg cells is significantly lower than that of Th17 cells.45 We now demonstrated that an HS insult promotes glycolytic capacity to impede Treg program. FoxO1, negatively regulated by SGK1, plays a dual role in Th17 and Treg subsets.46 On the one hand, FoxO1 promotes the expression of Foxp3 and other signature genes essential to Treg function.47,48 On the other hand, FoxO1 overexpression suppresses glycolytic capacity coupled with an attenuated Th17 program.49,50,51 Strikingly, extracellular HS environment elevates the intracellular Na+ level and disrupts Treg mitochondrial respiration through interfering with the electron transport chain (ETC), thereby impairing Foxp3 expression and enabling the ectopic expression of pro-inflammatory mediators.52 Furthermore, oxidative stress could be potently elicited by HS intake,53 and it is much likely that the metabolic rewiring and stress response are intertwined to mediate the pathologic consequences of HS stimulation.
Immune therapeutics have proven their value in RA treatment, especially for the currently developed biological agents.54 Although biologics such as IL-6 blockers and TNF-α neutralizing antibodies manifested notable efficacy in RA, a considerable population of patients, however, showed a mere suboptimal response.55 The insufficient responsiveness is ascribed to various factors,56 while the roles of an unhealthy lifestyle and bad dietary habits could not be excluded.57 Our study indicated that inhibition of PDPK1/SGK1 signaling by GSK significantly reduced the proportion of Th17 cells in HS-fed CIA mice, which was coupled with a marked decrease of circulating IL-6 and TNF-α levels, an increased proportion of Treg cells along with an enhanced IL-10 secretion. These findings support that targeting PDPK1/SGK1 signaling by GSK could be a viable strategy against RA in clinical settings.
Limitations of the study
Our study has several limitations. First, due to difficulties in obtaining specimens, we did not assess the expression levels and phosphorylation states of PDPK1 and SGK1 in synovial tissue of RA patients. Consequently, experiments with humanized animal model were not conducted. At last, though we preliminarily revealed the association between salt intake and RA severity, large-population based epidemiological survey and prospective cohort studies are demanded to verify the causal relationship by checking more immunological parameters.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human CD3 | Biolegend | Cat#300302; RRID:AB_314037 |
| Anti-human CD28 | Biolegend | Cat#302902; RRID:AB_314303 |
| Anti-mouse CD3 | Biolegend | Cat#100302; RRID:AB_312666 |
| Anti-mouse CD28 | Biolegend | Cat#102102; RRID:AB_312866 |
| FITC anti-mouse CD4 | Biolegend | Cat#100406; RRID:AB_312690 |
| Brilliant Violet 421™ anti-mouse IL-17A | Biolegend | Cat#506926; RRID:AB_10900442 |
| Alexa Fluor 647 anti-mouse Foxp3 | Biolegend | Cat#126408; RRID:AB_1089115 |
| PE anti-mouse CD44 | Biolegend | Cat#103008; RRID:AB_312958 |
| APC anti-mouse CD62L | Biolegend | Cat#104412; RRID:AB_313099 |
| PE/Cy7 anti-mouse CD25 | Biolegend | Cat#101916; RRID:AB_2616762 |
| Alexa Fluor 647 anti-human Foxp3 | Biolegend | Cat#320211; RRID:AB_430886 |
| PE anti-human IL-17A | Biolegend | Cat#512306; RRID:AB_961395 |
| Anti-PDPK1 (phospho S241) | Abcam | Cat#ab131098; RRID:AB_11159760 |
| Anti-HK2 | Abcam | Cat#ab209847; RRID:AB_2904621 |
| Anti-LDHA | Abcam | Cat#ab52488; RRID:AB_2134961 |
| Anti-PDPK1 | Santa Cruz Biotechnology | Cat#sc-17765; RRID:AB_626657 |
| Anti-β-Actin | Santa Cruz Biotechnology | Cat#sc-47778; RRID:AB_626632 |
| Anti-SGK1 | ABclonal | Cat#A1025; RRID:AB_2757776 |
| Anti-SGK1 (phospho S422) | Absin | Cat#abs147953; N.A |
| Anti-Glut1 | Proteintech | Cat#21829-1-AP; RRID:AB_10837075 |
| Anti-FoxO1 | proteintech | Cat#18592-1-AP; RRID:AB_10860103 |
| Biological samples | ||
| RA patients' peripheral blood samples | Tongji Hospital | N/A |
| Healthy Controls’ peripheral blood samples | Tongji Hospital | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| GSK2334470 (GSK) | MedChemExpress | HY-14981 |
| Protease inhibitor cocktail | Roche | 4693116001 |
| RIPA lysis buffer | Beyotime | P0013K |
| Recombinant murine IL-2 | Biolegend | #575402 |
| Recombinant murine IL-6 | Biolegend | #575702 |
| Recombinant murine TGF-β | Biolegend | #763102 |
| Recombinant human IL-2 | Biolegend | #791906 |
| Recombinant human IL-6 | Biolegend | #570806 |
| Recombinant human TGF-β | Biolegend | #583301 |
| Anti-TNF-α (Etanercept) | MedChemExpress | HY-108847 |
| Critical commercial assays | ||
| Mouse inflammation kit | BD Biosciences | 552364 |
| MojoSort™ Mouse CD4 Naive T cell Isolation Kit | Miltenyi Biotec | 130-104-453 |
| Annexin V-FITC/PI Apoptosis Detection Kit | Yeasen Biotechnology | 40302 |
| ECL kit | Biosharp Life Sciences | BL520A |
| CFSE | Thermo Fisher | C34554 |
| Experimental models: Organisms/strains | ||
| DBA/1 mice | Jackson Laboratory | N/A |
| C57BL/6 mice | Jackson Laboratory | N/A |
| Foxp3Cre-eGFP; Rosa26flox-stop-flox-tdTomato mice (Rosa-Red mice) | Jackson Laboratory | N/A |
| Software and algorithms | ||
| FlowJo software (v10.5.3) | BD Biosciences | https://www.bdbiosciences.com |
| GraphPad Prism 5.0 software | GraphPad Software Inc. | https://www.graphpad.com/; RRID:SCR_002798 |
| FCAP ArrayTM Software | BD Biosciences | https://www.bdbiosciences.com |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Cong-Yi Wang (wangcy@tjh.tjmu.edu.cn).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Human subjects
In the study, fresh peripheral blood samples were collected from treatment-naïve patients who were newly diagnosed with RA (n = 19) according to the 2010 American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) classification criteria for RA. Among the included RA patients, there were 15 females and 4 males. Healthy controls (n = 16, 13 females and 3 males) included in this study were matched for gender and age with RA patients. The average age of RA patients was 33 years, while the average age of HC individuals was 34 years. All participants are of Asian descent. Data collected from RA patients included age, gender, disease duration, daily salt intake, the number of tender joints in the 28 joints (TJC28), the number of swollen joints in the 28 joints (SJC28), the status of anti-CCP antibodies and RF, serum level of ESR and disease activity (Disease Activity Score-28 based ESR, DAS28-ESR). This study was approved by the Ethics Committee of Tongji Hospital, Tongji Medical College of Huazhong University of Science & Technology (project identification code: TJ -IRB20220973).
Experimental animal models
Collagen-induced arthritis (CIA) mouse model was established in male DBA/1 mice by injecting them with bovine type II collagen emulsified in complete Freund’s adjuvant, followed by a boost 21 days later with type II collagen emulsified in incomplete Freund’s adjuvant. The development of arthritis was monitored and arthritis scores were measured every 2 days. The Level of inflammation in each paw was graded on a scale from 0 to 4 as follows: 0, no inflammation; 1, detectable swelling in a single digit; 2, paw with swelling in more than one digit; 3, paw with swelling of all digits and instep; 4, severe swelling of the paw and ankle. The arthritis scores were the sum of the scores for all four paws. For the CIA model, mice on a high salt diet were fed sodium-rich chow containing 4% NaCl and provided with water containing 1% NaCl ad libitum from 7 days before the first immunization to 42 days thereafter. GSK2334470 (GSK) (25 mg/kg, every three days) or a vehicle control was administered to the mice via intraperitoneal injection from day 21 to day 42 following the second immunization. The vehicle for GSK consisted of 5% DMSO, 40% PEG300, 5% Tween-80 and 50% saline. On day 42 post-first immunization, blood samples, ankle joints, spleen and mesenteric lymph node (MLN) were collected from the mice. In the Adoptive transfer model C57BL/6 mice (6–8 weeks old) were fed with a standard chow diet and provided with water ad libitum, or they were fed a high salt diet (sodium-rich chow containing 4% NaCl and water containing 1% NaCl ad libitum) from day 0 to day 14. Concurrently, the mice were subjected to intraperitoneal injections of GSK (25 mg/kg, every three days) or vehicle control. Naive CD4 T cells from untreated C57BL/6 mice (8 weeks old) were polarized into Th17 cells in the presence of IL-6 (50 ng/mL) and TGF-β (2.5 ng/mL).58 The differentiated Th17 cells were then labeled with CFSE and transferred into the recipient mice that had been pretreated with a high salt diet and/or GSK via intraperitoneal injection upon the last dose of GSK administration (day 15). Spleens and MLNs were collected on day 18 for immunophenotyping.
We generated the Treg lineage tracing mice (Foxp3Cre-eGFP; Rosa26flox-stop-flox-tdTomato, defined as Rosa-Red) by crossing Foxp3Cre-eGFP mice with Rosa26flox-stop-flox-tdTomato mice. Treg lineage tracing Rosa-Red mice (6–8 weeks old) were either fed a standard chow diet and provided with water ad libitum, or placed on a high salt diet consisting of sodium-rich chow containing 4% NaCl and water with 1% NaCl ad libitum, and were given intraperitoneal injections of GSK (25 mg/kg, three times a week) or vehicle for 14 days. The spleen and MLN were collected on day 14 for flow cytometry analysis.
All animal experiments were conducted in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals and were approved by the Tongji Hospital Animal Care and Use Committee (project identification code: TJH-202206029).
Method details
Cell culture and polarization
For Th17 or Treg polarization, naive CD4 T cells were isolated from the spleens of C57BL/6 mice using a mouse naive CD4 T cell isolation kit (Miltenyi Biotec, 130-104-453). Naive CD4 T cells were cultured for 3 days with anti-CD3 and anti-CD28 stimulation either in Th17 polarization condition (IL-6 50 ng/mL, TGF-β 2.5 ng/mL) or Treg polarization condition (IL-2 10 ng/mL, TGF-β 5 ng/mL). For Treg stability assay, naive CD4 T cells were initially cultured for 3 days with the stimulation of anti-CD3 and anti-CD28 under Treg polarization condition, and the differentiated Treg cells were harvested, washed and cultured for an additional 2 days under Th17 condition. In some wells, the indicated stimulant(s) and chemical compound(s) were added in the final concentrations of 40 mmol/L HS,6 2.5 μmol/L GSK, 1 mmol/L 2-DG, and 25 nmol/L iFoxO1.
T cell proliferation and apoptosis assay
Naive CD4 T cells from spleens of C57BL/6 mice were labeled with CFSE (Life Technologies, Carlsbad, CA) and cultured for 3–5 days under the stimulation of plate-coated anti-CD3 and anti-CD28. The mean fluorescence intensity (MFI) of CFSE was analyzed by flow cytometry. Similarly, naive CD4 T cells from spleens of C57BL/6 mice were isolated and cultured for 24 h for the T cell apoptosis assay. The apoptotic ratio was measured by the Annexin V-FITC/PI staining, employing the Annexin V-FITC/PI Apoptosis Detection Kit (Yeasen, 40302) according to the manufacturer’s instructions.
Western blot analysis
Western blot was conducted as previously reported.59 Cells were isolated and lysed with cold RIPA lysis buffer from Beyotime (Shanghai, China), which was enriched with a protease inhibitor cocktail from Roche (IN, USA). Proteins were separated on SDS-PAGE gels and subsequently transferred to PVDF membranes. These membranes were then probed with primary antibodies. β-actin was served as the loading control to ensure consistent protein loading across the samples. Following the primary antibody incubation, the membranes were exposed to the corresponding HRP-conjugated secondary antibodies. For visualization, the membranes were treated using an ECL kit (Biosharp Life Sciences, BL520A). The protein bands were viewed and analyzed for relative intensities using the GelView 6000Plus Smart Chemiluminescence Imaging System.
Flow cytometry analysis
A single-cell suspension from spleen or lymph nodes was prepared. For cell surface staining, cells were incubated with specified antibodies and incubated for 30 min on ice. Subsequent intracellular staining was performed using the Transcription Factor Buffer Set alongside the selected antibody cocktails. To identify intracellular cytokines, cells were stimulated with the Cell Activation Cocktail for a duration of 4–6 h. The antibodies used included FITC anti-mouse CD4 (#100406), APC anti-mouse CD62L (#104412), PE/Cy7 anti-mouse CD25 (#101916), Alexa Fluor 647 anti-mouse Foxp3 (#126408), Brilliant Violet 421 anti-mouse IL-17A (#506926), and PE anti-mouse CD44 (#103008). All antibodies were derived from Biolegend (San Diego, CA, USA). A consistent antibody dilution of 1/200 was used to ensure optimal results. After the staining procedure, the data were acquired with the MACSQuant Analyzer 10 from Miltenyi Biotec and processed using the FlowJo software (v10.5.3). In this study, CD4+ IL-17A+ cells are denoted as Th17 60 and CD4+ Foxp3+ cells are denoted as Treg.61
Measurement of serum cytokines
Serum cytokines were measured using the mouse inflammation kits (BD Biosciences, 552364) according to the manufacturer’s instructions. Briefly, the capture beads, samples and PE-conjugated detection antibodies were incubated together at room temperature for 2 h. Data were collected with flow cytometry, and the results were generated using FCAP Array Software.
Quantification and statistical analysis
The correlation was determined by linear regression analysis. All other data are expressed as mean ± SD. Student’s t test was applied for comparisons between two groups. Once more than two groups were compared, one-way or two-way analysis of variance was employed. In all cases, p < 0.05 was considered as statistically significant. Statistical analyses were conducted using the GraphPad Prism 5.0 software (GraphPad Software Inc., San Diego, CA, USA).
Study approval
All animal care and experimental procedures were approved by the Animal Care and Use Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology (TJH-202206029), and conducted in accordance with NIH guidelines. The studies in human samples were approved by the Ethics Committee of Tongji Hospital, Tongji Medical College of Huazhong University of Science and Technology (TJ-IRB20220973).
Acknowledgments
We thank Wen-Ye Mo for animal breeding. We also thank the engaged patients and healthy volunteers in this study.
This study was supported by the National Key R&D Program of China (2022YFA0806101) from the Ministry of Science and Technology of the People's Republic of China, the National Natural Science Foundation of China (81920108009, 82130023, 82100892, 82070808, 81873656, 82100823, 82100931, 91749207, 81770823, 82200923, 82270885 and 81800068), the Postdoctoral Science Foundation of China (2023M731200), the Fundamental Research Funds for the Central Universities (YCJJ20230235), the Key Laboratory of Endocrine and Metabolic Diseases of Shanxi Province (202104010910009), the Integrated Innovative Team for Major Human Diseases Program of Tongji Medical College, Huazhong University of Science and Technology, and the Innovative Funding for Translational Research from Tongji Hospital.
Author contributions
L.L.D., F.S., and C.Y.W. designed the study; Q.M., M.B., S.J.R., F.S., C.L.Y., J.H.L., W.Y.L., Q.J.C., J.W.Z., F.X.W., T.W., Y.L., and X.L. performed the experiments; Q.M., M.B., S.J.R., S.Z., F.X., Q.L.Y., Z.Y.Z., and F.S. provided acquisition, analysis, and interpretation of data; Q.M., M.B., S.J.R., F.S., L.L.D., and C.-.Y.W. evaluated the general conclusions and wrote the manuscript. All authors read and approved the publication of this manuscript.
Declaration of interests
The authors declare no competing interests.
Published: April 26, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109798.
Contributor Information
Fei Sun, Email: phil_sunfei@163.com.
Ling-Li Dong, Email: tjhdongll@163.com.
Cong-Yi Wang, Email: wangcy@tjh.tjmu.edu.cn.
Supplemental information
References
- 1.Sparks J.A. Rheumatoid Arthritis. Ann. Intern. Med. 2019;170:ITC1–ITC16. doi: 10.7326/AITC201901010. [DOI] [PubMed] [Google Scholar]
- 2.Deane K.D., Demoruelle M.K., Kelmenson L.B., Kuhn K.A., Norris J.M., Holers V.M. Genetic and environmental risk factors for rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 2017;31:3–18. doi: 10.1016/j.berh.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carranza-León D.A., Oeser A., Marton A., Wang P., Gore J.C., Titze J., Stein C.M., Chung C.P., Ormseth M.J. Tissue sodium content in patients with systemic lupus erythematosus: association with disease activity and markers of inflammation. Lupus. 2020;29:455–462. doi: 10.1177/0961203320908934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dent E.L., Broome H.J., Sasser J.M., Ryan M.J. Blood pressure and albuminuria in a female mouse model of systemic lupus erythematosus: impact of long-term high salt consumption. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020;319:R448–R454. doi: 10.1152/ajpregu.00070.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kleinewietfeld M., Manzel A., Titze J., Kvakan H., Yosef N., Linker R.A., Muller D.N., Hafler D.A. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. 2013;496:518–522. doi: 10.1038/nature11868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu C., Yosef N., Thalhamer T., Zhu C., Xiao S., Kishi Y., Regev A., Kuchroo V.K. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496:513–517. doi: 10.1038/nature11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu H., Huang X., Qiu H., Zhao M., Liao W., Yuan S., Xie Y., Dai Y., Chang C., Yoshimura A., Lu Q. High salt promotes autoimmunity by TET2-induced DNA demethylation and driving the differentiation of Tfh cells. Sci. Rep. 2016;6 doi: 10.1038/srep28065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang X., Sundström B., Alfredsson L., Klareskog L., Rantapää-Dahlqvist S., Bengtsson C. High sodium chloride consumption enhances the effects of smoking but does not interact with SGK1 polymorphisms in the development of ACPA-positive status in patients with RA. Ann. Rheum. Dis. 2016;75:943–946. doi: 10.1136/annrheumdis-2015-209009. [DOI] [PubMed] [Google Scholar]
- 9.Marouen S., du Cailar G., Audo R., Lukas C., Vial G., Tournadre A., Barrat E., Ribstein J., Combe B., Morel J., Daien C.I. Sodium excretion is higher in patients with rheumatoid arthritis than in matched controls. PLoS One. 2017;12 doi: 10.1371/journal.pone.0186157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salgado E., Bes-Rastrollo M., de Irala J., Carmona L., Gómez-Reino J.J. High Sodium Intake Is Associated With Self-Reported Rheumatoid Arthritis: A Cross Sectional and Case Control Analysis Within the SUN Cohort. Medicine. 2015;94 doi: 10.1097/MD.0000000000000924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sundström B., Johansson I., Rantapää-Dahlqvist S. Interaction between dietary sodium and smoking increases the risk for rheumatoid arthritis: results from a nested case-control study. Rheumatology. 2015;54:487–493. doi: 10.1093/rheumatology/keu330. [DOI] [PubMed] [Google Scholar]
- 12.Sharif K., Amital H., Shoenfeld Y. The role of dietary sodium in autoimmune diseases: The salty truth. Autoimmun. Rev. 2018;17:1069–1073. doi: 10.1016/j.autrev.2018.05.007. [DOI] [PubMed] [Google Scholar]
- 13.Shekhar K.V., Pathak M.M., Pisulkar G. Diet and Lifestyle Impact on Rheumatoid Arthritis: A Comprehensive Review. Cureus. 2023;15 doi: 10.7759/cureus.48625. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Chabaud M., Garnero P., Dayer J.M., Guerne P.A., Fossiez F., Miossec P. Contribution of interleukin 17 to synovium matrix destruction in rheumatoid arthritis. Cytokine. 2000;12:1092–1099. doi: 10.1006/cyto.2000.0681. [DOI] [PubMed] [Google Scholar]
- 15.Paulissen S.M.J., van Hamburg J.P., Davelaar N., Vroman H., Hazes J.M.W., de Jong P.H.P., Lubberts E. CCR6(+) Th cell populations distinguish ACPA positive from ACPA negative rheumatoid arthritis. Arthritis Res. Ther. 2015;17:344. doi: 10.1186/s13075-015-0800-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Penatti A., Facciotti F., De Matteis R., Larghi P., Paroni M., Murgo A., De Lucia O., Pagani M., Pierannunzii L., Truzzi M., et al. Differences in serum and synovial CD4+ T cells and cytokine profiles to stratify patients with inflammatory osteoarthritis and rheumatoid arthritis. Arthritis Res. Ther. 2017;19:103. doi: 10.1186/s13075-017-1305-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang T., Li S., Yang Y., Zhang K., Dong S., Wang X., Liu X., Ren Y., Zhang M., Yan X., et al. T helper 17 and T helper 1 cells are increased but regulatory T cells are decreased in subchondral bone marrow microenvironment of patients with rheumatoid arthritis. Am. J. Transl. Res. 2016;8:2956–2968. [PMC free article] [PubMed] [Google Scholar]
- 18.Zizzo G., De Santis M., Bosello S.L., Fedele A.L., Peluso G., Gremese E., Tolusso B., Ferraccioli G. Synovial fluid-derived T helper 17 cells correlate with inflammatory activity in arthritis, irrespectively of diagnosis. Clin. Immunol. 2011;138:107–116. doi: 10.1016/j.clim.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 19.Finnegan A., Kaplan C.D., Cao Y., Eibel H., Glant T.T., Zhang J. Collagen-induced arthritis is exacerbated in IL-10-deficient mice. Arthritis Res. Ther. 2003;5:R18–R24. doi: 10.1186/ar601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johansson A.C.M., Hansson A.S., Nandakumar K.S., Bäcklund J., Holmdahl R. IL-10-deficient B10.Q mice develop more severe collagen-induced arthritis, but are protected from arthritis induced with anti-type II collagen antibodies. Journal of Immunology (Baltimore, Md. 2001;167:3505–3512. doi: 10.4049/jimmunol.167.6.3505. [DOI] [PubMed] [Google Scholar]
- 21.Park M.-J., Lee S.-H., Kim E.-K., Lee E.-J., Baek J.-A., Park S.-H., Kwok S.-K., Cho M.-L. Interleukin-10 produced by myeloid-derived suppressor cells is critical for the induction of Tregs and attenuation of rheumatoid inflammation in mice. Sci. Rep. 2018;8:3753. doi: 10.1038/s41598-018-21856-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hui W., Yu D., Cao Z., Zhao X. Butyrate inhibit collagen-induced arthritis via Treg/IL-10/Th17 axis. Int. Immunopharm. 2019;68:226–233. doi: 10.1016/j.intimp.2019.01.018. [DOI] [PubMed] [Google Scholar]
- 23.Avdeeva A., Rubtsov Y., Dyikanov D., Popkova T., Nasonov E. Regulatory T cells in patients with early untreated rheumatoid arthritis: Phenotypic changes in the course of methotrexate treatment. Biochimie. 2020;174:9–17. doi: 10.1016/j.biochi.2020.03.014. [DOI] [PubMed] [Google Scholar]
- 24.Komatsu N., Okamoto K., Sawa S., Nakashima T., Oh-hora M., Kodama T., Tanaka S., Bluestone J.A., Takayanagi H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014;20:62–68. doi: 10.1038/nm.3432. [DOI] [PubMed] [Google Scholar]
- 25.Schinocca C., Rizzo C., Fasano S., Grasso G., La Barbera L., Ciccia F., Guggino G. Role of the IL-23/IL-17 Pathway in Rheumatic Diseases: An Overview. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.637829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yasuda K., Takeuchi Y., Hirota K. The pathogenicity of Th17 cells in autoimmune diseases. Semin. Immunopathol. 2019;41:283–297. doi: 10.1007/s00281-019-00733-8. [DOI] [PubMed] [Google Scholar]
- 27.Morgan M.E., Sutmuller R.P.M., Witteveen H.J., van Duivenvoorde L.M., Zanelli E., Melief C.J.M., Snijders A., Offringa R., de Vries R.R.P., Toes R.E.M. CD25+ cell depletion hastens the onset of severe disease in collagen-induced arthritis. Arthritis Rheum. 2003;48:1452–1460. doi: 10.1002/art.11063. [DOI] [PubMed] [Google Scholar]
- 28.Gerriets V.A., Rathmell J.C. Metabolic pathways in T cell fate and function. Trends Immunol. 2012;33:168–173. doi: 10.1016/j.it.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palmer C.S., Ostrowski M., Balderson B., Christian N., Crowe S.M. Glucose metabolism regulates T cell activation, differentiation, and functions. Front. Immunol. 2015;6:1. doi: 10.3389/fimmu.2015.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bayascas J.R. Dissecting the role of the 3-phosphoinositide-dependent protein kinase-1 (PDK1) signalling pathways. Cell Cycle. 2008;7:2978–2982. doi: 10.4161/cc.7.19.6810. [DOI] [PubMed] [Google Scholar]
- 31.Sun F., Wang F.-X., Zhu H., Yue T.-T., Yang C.-L., Luo J.-H., Luo X., Zhou H.-F., Rong S.-J., Lu W.-Y., et al. SUMOylation of PDPK1 Is required to maintain glycolysis-dependent CD4 T-cell homeostasis. Cell Death Dis. 2022;13:181. doi: 10.1038/s41419-022-04622-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park S.-G., Schulze-Luehrman J., Hayden M.S., Hashimoto N., Ogawa W., Kasuga M., Ghosh S. The kinase PDK1 integrates T cell antigen receptor and CD28 coreceptor signaling to induce NF-kappaB and activate T cells. Nat. Immunol. 2009;10:158–166. doi: 10.1038/ni.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hinton H.J., Alessi D.R., Cantrell D.A. The serine kinase phosphoinositide-dependent kinase 1 (PDK1) regulates T cell development. Nat. Immunol. 2004;5:539–545. doi: 10.1038/ni1062. [DOI] [PubMed] [Google Scholar]
- 34.Fan C., Cui X., Chen S., Huang S., Jiang H. LncRNA LOC100912373 modulates PDK1 expression by sponging miR-17-5p to promote the proliferation of fibroblast-like synoviocytes in rheumatoid arthritis. Am. J. Transl. Res. 2020;12:7709–7723. [PMC free article] [PubMed] [Google Scholar]
- 35.Ma J.-D., Jing J., Wang J.-W., Yan T., Li Q.-H., Mo Y.-Q., Zheng D.-H., Gao J.-L., Nguyen K.-A., Dai L. A novel function of artesunate on inhibiting migration and invasion of fibroblast-like synoviocytes from rheumatoid arthritis patients. Arthritis Res. Ther. 2019;21:153. doi: 10.1186/s13075-019-1935-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun C., Sun Y., Jiang D., Bao G., Zhu X., Xu D., Wang Y., Cui Z. PDK1 promotes the inflammatory progress of fibroblast-like synoviocytes by phosphorylating RSK2. Cell. Immunol. 2017;315:27–33. doi: 10.1016/j.cellimm.2016.10.007. [DOI] [PubMed] [Google Scholar]
- 37.Amato R., Menniti M., Agosti V., Boito R., Costa N., Bond H.M., Barbieri V., Tagliaferri P., Venuta S., Perrotti N. IL-2 signals through Sgk1 and inhibits proliferation and apoptosis in kidney cancer cells. J. Mol. Med. 2007;85:707–721. doi: 10.1007/s00109-007-0205-2. [DOI] [PubMed] [Google Scholar]
- 38.Basnet R., Gong G.Q., Li C., Wang M.-W. Serum and glucocorticoid inducible protein kinases (SGKs): a potential target for cancer intervention. Acta Pharm. Sin. B. 2018;8:767–771. doi: 10.1016/j.apsb.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen S., Grigsby C.L., Law C.S., Ni X., Nekrep N., Olsen K., Humphreys M.H., Gardner D.G. Tonicity-dependent induction of Sgk1 expression has a potential role in dehydration-induced natriuresis in rodents. J. Clin. Invest. 2009;119:1647–1658. doi: 10.1172/JCI35314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perrotti N., He R.A., Phillips S.A., Haft C.R., Taylor S.I. Activation of serum- and glucocorticoid-induced protein kinase (Sgk) by cyclic AMP and insulin. J. Biol. Chem. 2001;276:9406–9412. doi: 10.1074/jbc.M007052200. [DOI] [PubMed] [Google Scholar]
- 41.Wu C., Chen Z., Xiao S., Thalhamer T., Madi A., Han T., Kuchroo V. SGK1 Governs the Reciprocal Development of Th17 and Regulatory T Cells. Cell Rep. 2018;22:653–665. doi: 10.1016/j.celrep.2017.12.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Powles J., Fahimi S., Micha R., Khatibzadeh S., Shi P., Ezzati M., Engell R.E., Lim S.S., Danaei G., Mozaffarian D., Global Burden of Diseases Nutrition and Chronic Diseases Expert Group NutriCoDE Global, regional and national sodium intakes in 1990 and 2010: a systematic analysis of 24 h urinary sodium excretion and dietary surveys worldwide. BMJ Open. 2013;3 doi: 10.1136/bmjopen-2013-003733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jörg S., Kissel J., Manzel A., Kleinewietfeld M., Haghikia A., Gold R., Müller D.N., Linker R.A. High salt drives Th17 responses in experimental autoimmune encephalomyelitis without impacting myeloid dendritic cells. Exp. Neurol. 2016;279:212–222. doi: 10.1016/j.expneurol.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 44.Saravia J., Raynor J.L., Chapman N.M., Lim S.A., Chi H. Signaling networks in immunometabolism. Cell Res. 2020;30:328–342. doi: 10.1038/s41422-020-0301-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cluxton D., Petrasca A., Moran B., Fletcher J.M. Differential Regulation of Human Treg and Th17 Cells by Fatty Acid Synthesis and Glycolysis. Front. Immunol. 2019;10:115. doi: 10.3389/fimmu.2019.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hedrick S.M., Hess Michelini R., Doedens A.L., Goldrath A.W., Stone E.L. FOXO transcription factors throughout T cell biology. Nat. Rev. Immunol. 2012;12:649–661. doi: 10.1038/nri3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang H., Tindall D.J. Dynamic FoxO transcription factors. J. Cell Sci. 2007;120:2479–2487. doi: 10.1242/jcs.001222. [DOI] [PubMed] [Google Scholar]
- 48.Ouyang W., Beckett O., Ma Q., Paik J.-h., DePinho R.A., Li M.O. Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat. Immunol. 2010;11:618–627. doi: 10.1038/ni.1884. [DOI] [PubMed] [Google Scholar]
- 49.Gibson S.A., Yang W., Yan Z., Qin H., Benveniste E.N. CK2 Controls Th17 and Regulatory T Cell Differentiation Through Inhibition of FoxO1. J. Immunol. 2018;201:383–392. doi: 10.4049/jimmunol.1701592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lainé A., Martin B., Luka M., Mir L., Auffray C., Lucas B., Bismuth G., Charvet C. Foxo1 Is a T Cell-Intrinsic Inhibitor of the RORγt-Th17 Program. J. Immunol. 2015;195:1791–1803. doi: 10.4049/jimmunol.1500849. [DOI] [PubMed] [Google Scholar]
- 51.Wilhelm K., Happel K., Eelen G., Schoors S., Oellerich M.F., Lim R., Zimmermann B., Aspalter I.M., Franco C.A., Boettger T., et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature. 2016;529:216–220. doi: 10.1038/nature16498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Côrte-Real B.F., Hamad I., Arroyo Hornero R., Geisberger S., Roels J., Van Zeebroeck L., Dyczko A., van Gisbergen M.W., Kurniawan H., Wagner A., et al. Sodium perturbs mitochondrial respiration and induces dysfunctional Tregs. Cell Metabol. 2023;35 doi: 10.1016/j.cmet.2023.01.009. [DOI] [PubMed] [Google Scholar]
- 53.Krajina I., Stupin A., Šola M., Mihalj M. Oxidative Stress Induced by High Salt Diet-Possible Implications for Development and Clinical Manifestation of Cutaneous Inflammation and Endothelial Dysfunction in Psoriasis vulgaris. Antioxidants. 2022;11 doi: 10.3390/antiox11071269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McInnes I.B., Schett G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet (London, England) 2017;389:2328–2337. doi: 10.1016/S0140-6736(17)31472-1. [DOI] [PubMed] [Google Scholar]
- 55.Smolen J.S., Landewé R., Bijlsma J., Burmester G., Chatzidionysiou K., Dougados M., Nam J., Ramiro S., Voshaar M., van Vollenhoven R., et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann. Rheum. Dis. 2017;76:960–977. doi: 10.1136/annrheumdis-2016-210715. [DOI] [PubMed] [Google Scholar]
- 56.Reynolds G., Cooles F.A.H., Isaacs J.D., Hilkens C.M.U. Emerging immunotherapies for rheumatoid arthritis. Hum. Vaccines Immunother. 2014;10:822–837. doi: 10.4161/hv.27910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schäfer C., Keyßer G. Lifestyle Factors and Their Influence on Rheumatoid Arthritis: A Narrative Review. J. Clin. Med. 2022;11 doi: 10.3390/jcm11237179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou H.-F., Wang F.-X., Sun F., Liu X., Rong S.-J., Luo J.-H., Yue T.-T., Xiao J., Yang C.-L., Lu W.-Y., et al. Aloperine Ameliorates IMQ-Induced Psoriasis by Attenuating Th17 Differentiation and Facilitating Their Conversion to Treg. Front. Pharmacol. 2022;13 doi: 10.3389/fphar.2022.778755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yue T., Sun F., Wang F., Yang C., Luo J., Rong S., Zhou H., Xiao J., Wang X., Zhou Q., et al. MBD2 acts as a repressor to maintain the homeostasis of the Th1 program in type 1 diabetes by regulating the STAT1-IFN-γ axis. Cell Death Differ. 2022;29:218–229. doi: 10.1038/s41418-021-00852-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang W.-L., Qiu W., Zhang T., Xu K., Gu Z.-J., Zhou Y., Xu H.-J., Yang Z.-Z., Shen B., Zhao Y.-L., et al. Nsun2 coupling with RoRγt shapes the fate of Th17 cells and promotes colitis. Nat. Commun. 2023;14:863. doi: 10.1038/s41467-023-36595-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tang X., Shang Y., Yang H., Song Y., Li S., Qin Y., Song J., Chen K., Liu Y., Zhang D., Chen L. Targeted delivery of Fc-fused PD-L1 for effective management of acute and chronic colitis. Nat. Commun. 2024;15:1673. doi: 10.1038/s41467-024-46025-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.