

Abstract
Large-scale genomic initiatives, such as the Earth BioGenome Project, require efficient methods for eukaryotic genome annotation. Here we present an automatic gene finder, GeneMark-ETP, integrating genomic-, transcriptomic-, and protein-derived evidence that has been developed with a focus on large plant and animal genomes. GeneMark-ETP first identifies genomic loci where extrinsic data are sufficient for making gene predictions with “high confidence.” The genes situated in the genomic space between the high-confidence genes are predicted in the next stage. The set of high-confidence genes serves as an initial training set for the statistical model. Further on, the model parameters are iteratively updated in the rounds of gene prediction and parameter re-estimation. Upon reaching convergence, GeneMark-ETP makes the final predictions and delivers the whole complement of predicted genes. GeneMark-ETP outperforms gene finders using a single type of extrinsic evidence. Comparisons with gene finders MAKER2 and TSEBRA, those that use both transcript- and protein-derived extrinsic evidence, show that GeneMark-ETP delivers state-of-the-art gene-prediction accuracy, with the margin of outperforming existing approaches increasing in its application to larger and more complex eukaryotic genomes.
Massive sequencing of eukaryotic genomes, for example, the Earth BioGenome Project (Lewin et al. 2022), should be complemented with fast and accurate automatic tools of genome annotation. The development of such tools remains an active area of research. One of the long-standing challenges is to achieve optimal integration of the intrinsic evidence of protein-encoding by a nucleotide sequence with the extrinsic evidence of gene presence provided by endogenous RNA or known cross-species protein sequences. Gene-finding methods developed before the transition to massive genomic sequencing powered by high-throughput sequencing have been focused on using intrinsic evidence in the form of the Markov chain models reflecting the k-mer frequency patterns, probabilistic models of functional sites controlling translation and splicing, intron/exon length distributions, etc. All these probabilistic models served as elements of a generalized HMM (GHMM), for example, Genie (Kulp et al. 1996), GENSCAN (Burge and Karlin 1997), and GeneID (Parra et al. 2000). The bottleneck of the application of the early methods was a tedious preparation of the supervised species-specific training sets. The training automation was first made in GeneMark-ES, an ab initio gene finder implementing a hierarchical procedure of iterative unsupervised training (Lomsadze et al. 2005; Ter-Hovhannisyan et al. 2008). The accuracy of gene predictions made by GeneMark-ES in compact fungal and protist genomes has been high and is still difficult to improve even by adding currently available extrinsic evidence. Since the advent of high-throughput sequencing, the rapid accumulation of RNA and protein information has opened ample opportunities for the addition of extrinsic evidence (Guigo et al. 2006; Coghlan et al. 2008; Goodswen et al. 2012; Scalzitti et al. 2020). This addition proved to be critically important for increasing gene-prediction accuracy in large plant and animal genomes having low gene density. The accuracy improvement of fully automated methods is a twofold problem It concerns improvements of both training and prediction steps.
One kind of extrinsic evidence comes from known cross-species proteins. These proteins may guide predictions of the genes encoding proteins from the same family in a novel genome. Confident spliced alignment of a cross-species protein to a locus in the novel genome gives an excellent starting point for delineation of the full exon–intron structure of the true gene. Spliced alignments have been used in several gene finders, for example, in exonerate (Slater and Birney 2005), GenomeThreader (Gremme et al. 2005), and ProSplign (Kiryutin et al. 2007). Sequencing of expressed endogenous RNA provides another type of external evidence. Cufflinks (Trapnell et al. 2010), StringTie (Pertea et al. 2015; Kovaka et al. 2019), PsiCLASS (Song et al. 2019), etc. map short RNA-seq reads to the genome to identify exon/intron borders of the genes having detectable levels of expression. Notably, a method leveraging any type of extrinsic evidence is useful only for finding a subset of genes for which such evidence exists.
The goal of improving integration of intrinsic and extrinsic evidence has continued to motivate the creation of new gene-finding methods for more than two decades, for example, GAZE (Howe et al. 2002), Combiner (Allen et al. 2004), JIGSAW (Allen and Salzberg 2005), Evigan (Liu et al. 2008), EVidenceModeler (Haas et al. 2008), MAKER2 (Holt and Yandell 2011), IPred (Zickmann and Renard 2015), GeMoMa (Keilwagen et al. 2018), LoReAn (Cook et al. 2019), GAAP (Kong et al. 2019), FINDER (Banerjee et al. 2021), BRAKER1 (Hoff et al. 2016), BRAKER2 (Brůna et al. 2021), and TSEBRA (Gabriel et al. 2021).
GeneMark-ETP takes several steps of integration. First, GeneMarkS-T, the GHMM-based ab initio gene finder for transcripts (Tang et al. 2015), is used to predict protein-coding regions (CDSs) in transcripts assembled from RNA-seq reads by StringTie2 (Kovaka et al. 2019). Second, the predicted CDSs are validated or modified, if the protein level evidence suggests so, by the module that we call GeneMarkS-TP. Next, the CDSs predicted in the transcripts are mapped to the genome. We show that the resulting gene models have distinctly high Precision (Pr) and call them high-confidence genes (HC genes). The set of HC genes is used as an initial training set for the genomic GHMM used by GeneMark-ETP to predict genes situated in genomic regions between the HC genes. The extrinsic information (hints from spliced aligned proteins and mapped RNA-seq reads) is plugged in at this step into the Viterbi algorithm. The rounds of genome-wide gene prediction and model parameters estimation continue until convergence.
For benchmarking, we selected seven eukaryotic genomes, both GC-homogeneous and GC-inhomogeneous: Arabidopsis thaliana, Caenorhabditis elegans, Drosophila melanogaster, Solanum lycopersicum, Danio rerio, Gallus gallus, and Mus musculus. Along with GeneMark-ETP we tested GeneMark-ET; GeneMark-EP+; the pipelines BRAKER1, BRAKER2, and MAKER2; and TSEBRA. The tests showed the state-of-the-art performance of GeneMark-ETP, whose margin of improvement over other gene finders increased with the increase in the genome length.
Results
Introductory comments
Here, we introduce definitions of several terms used throughout the text. The goal of gene-finding algorithms is to predict protein-coding genes that may have several alternative transcripts. Upon RNA transcripts assembly (by StringTie2), transcripts representing one and the same gene (alternative RNA transcripts) are determined. The alternative mRNA transcripts may have identical CDS sequences but may differ in the UTR regions. We are concerned only with the protein (CDS) isoforms.
We measure gene-prediction accuracy at two levels, the gene level and the exon level. A protein-coding gene prediction is considered to be correct if at least one of the predicted CDS isoforms matches one of the annotated CDS isoforms. A protein-coding exon is predicted correctly if it matches exactly an annotated exon.
Most previous publications on gene-finding methods and their benchmarking have used the term “specificity” (Sn) for the measure defined as , where is the number of true positives and is the number of false positives. However, in the related and fast-growing field of machine learning, the term “specificity” has the meaning of true-negative rate defined as , where is the number of true negatives. To unify the designations, because of the request of the Genome Research editors, we have switched here to the term “precision” (Pr) for .
Thus, we characterize the prediction accuracy (at a gene or an exon level) by sensitivity (Sn), , and Pr, , where #TP and #FP are defined above, and #FN is the number of false-negative predictions.
As a single accuracy measure, we use the F1 score, the harmonic mean of Sn and Pr, F1 = 2 × Sn × Pr/(Sn + Pr) or . For convenience, the F1 score is multiplied by 100. Note that the latter formula for F1 is not equivalent to the former one if Sn and Pr are computed using different sets of positive examples (annotated CDSs). Therefore, to avoid ambiguities, we use the former formula in all the computations.
Integration of extrinsic evidence in GeneMarkS-TP
At the first step of the GeneMark-ETP pipeline (Fig. 1; Supplemental Figs. S1–S6), short RNA-seq reads were assembled into transcripts by StringTie2 (Kovaka et al. 2019). Next, iterative unsupervised training of GHHM of GeneMarkS-T was performed on these assembled transcripts using the rounds of model parameter estimation and ab initio gene predictions (Tang et al. 2015).
Figure 1.

High-level diagram of the GeneMark-ETP processing of genomic, RNA-seq, and protein sequences. For more details, see Figure 2 and Supplemental Figures S1 through S6.
Running GeneMarkS-T is the first step in the GeneMarkS-TP module (Fig. 2). The CDSs predicted by GeneMarkS-T in assembled transcripts were translated, and the amino acid sequences were aligned with homologous cross-species proteins found by a database search. The alignments served as guides for corrections of the initially predicted CDSs (see Methods). The magnitudes of changes in Sn and Pr upon the corrections depended on the size of the protein database used by GeneMarkS-TP (Table 1). Corrections in initially predicted CDSs increased the Pr values, on average, by 25 percentage points, reaching close to or even higher than 90%. Thus, the refined CDSs were dubbed high-confidence CDSs (HC CDSs) (Table 1; Supplemental Table S1).
Figure 2.

The diagram illustrates the work of the GeneMarkS-TP module generating the CDSs and genes predicted with high confidence (see also Supplemental Figs. S1–S3).
Table 1.
Statistics characterizing the GeneMarkS-T initial gene predictions made in assembled transcripts as well as the HC genes predicted by the GeneMarkS-TP module
| Species | No. of genes initially predicted by GeneMarkS-T | Sn/Pr of GeneMarkS-T-predicted genes | Order excluded DB | Species excluded DB | ||
|---|---|---|---|---|---|---|
| No. of HC genes | Sn/Pr of HC genes | No. of HC genes | Sn/Pr of HC genes | |||
| C. elegans | 14,746 | 46.8/63.4 | 8062 | 35.7/88.4 | 11,399 | 51.7/90.6 |
| A. thaliana | 17,589 | 51.2/79.9 | 16,008 | 56.7/97.3 | 16,551 | 58.8/97.6 |
| D. melanogaster | 10,163 | 59.6/81.8 | 8109 | 55.0/94.7 | 9223 | 63.7/96.3 |
| S. lycopersicum | 19,526 | 68.0/78.4 | 17,231 | 75.1/95.3 | 17,489 | 75.8/95.2 |
| D. rerio | 22,992 | 59.6/59.9 | 16,918 | 67.0/88.5 | 16,573 | 66.9/90.4 |
| G. gallus | 17,381 | 49.6/47.0 | 12,473 | 74.4/89.1 | 12,564 | 74.0/88.4 |
| M. musculus | 15,819 | 49.6/63.2 | 13,057 | 63.5/93.2 | 12,965 | 63.9/94.5 |
Two versions of a reference protein database were used for each species: the “species excluded” and the “order excluded” (see section “Data sets”). Refinement made by GeneMarkS-TP reduced the number of initial predictions and produced a significant increase in Pr. In most of the genomes, there was also a positive change in Sn, especially in large inhomogeneous genomes of G. gallus and M. musculus. Additional data are provided in Supplemental Table S1.
We saw that both the numbers of GeneMarkS-T predictions (the HC CDS candidates) and the numbers of HC CDSs predicted by GeneMarkS-TP correlated with the numbers of the RefSeq annotated CDSs (Table 1; Supplemental Fig. S7). The HC CDSs could give accurate predictions of exon–intron structure coordinates for up to 60% of protein-coding genes present in the RefSeq genome annotations (Table 1).
The numbers of HC CDSs predicted with the larger “species excluded” databases did not differ significantly from the numbers of HC CDSs predicted with the use of the smaller “order excluded” databases (Table 1; Supplemental Fig. S7). Also, the Pr values of the HC CDSs did not change significantly upon transition from the “order excluded” to the “species excluded” database.
At the step preceding the generation of the final set of gene predictions, we have had a large set of gene candidates. The gene candidates could be divided into groups with different level of extrinsic support: (1) those with full extrinsic support, having all the exon borders supported by high-scoring hints; (2) those with partial extrinsic support, having high-scoring hints for some but not all exon borders; (3) those that were predicted ab initio and had an a posteriori detected match to a low-scoring hint of at least one exon border; and (4) those predicted ab initio and had no match to any extrinsic hints. Importantly, the HC CDSs belong to the first two categories. The meaning of an “a posteriori detected match to low-scoring hints” is that such hints were not used in the gene-prediction algorithm.
Now, to focus solely on results, we must skip further details related to the description of the pipeline (see Methods) (Figs. 1, 2; Supplemental Figs. S1–S6).
One could see that for all seven genomes, the Pr values of the candidate genes decreased significantly upon a decrease in the level of extrinsic support, thus indicating an increase in false-positive rates (Table 2).
Table 2.
Distribution of the gene-prediction candidates among the four groups that differ by the level of extrinsic support
| Species | Support for predicted gene candidates | Order excluded DB | Species excluded DB | ||
|---|---|---|---|---|---|
| No. of candidates | Precision % | No. of candidates | Precision % | ||
| C. elegans | Fully extrinsic | 7676 | 88.9 | 10,778 | 91.6 |
| Partially extrinsic | 4804 | 56.4 | 5417 | 54.4 | |
| Match to low-scoring hints | 4020 | 54.7 | 1548 | 45.2 | |
| No extrinsic match | 1298 | 24.9 | 778 | 18.0 | |
| A. thaliana | Fully extrinsic | 16,445 | 97.2 | 18,083 | 97.5 |
| Partially extrinsic | 4825 | 64.4 | 5807 | 55.7 | |
| Match to low-scoring hints | 1794 | 50.2 | 1360 | 30.1 | |
| No extrinsic match | 2964 | 27.9 | 1128 | 9.4 | |
| D. melanogaster | Fully extrinsic | 8059 | 95.1 | 9952 | 96.8 |
| Partially extrinsic | 2328 | 49.3 | 2751 | 44.9 | |
| Match to low-scoring hints | 1043 | 57.1 | 165 | 44.9 | |
| No extrinsic match | 1369 | 41.6 | 377 | 15.9 | |
| S. lycopersicum | Fully extrinsic | 17,639 | 95.2 | 18,420 | 95.0 |
| Partially extrinsic | 5174 | 47.3 | 5813 | 44.3 | |
| Match to low-scoring hints | 1577 | 38.4 | 1484 | 29.7 | |
| No extrinsic match | 4714 | 14.8 | 3703 | 9.2 | |
| D. rerio | Fully extrinsic | 15,691 | 89.8 | 15,501 | 92.6 |
| Partially extrinsic | 10,905 | 16.6 | 11,769 | 16.6 | |
| Match to low-scoring hints | 1973 | 11.4 | 1663 | 7.3 | |
| No extrinsic match | 12,534 | 0.8 | 11,879 | 0.3 | |
| G. gallus | Fully extrinsic | 11,856 | 89.3 | 11,547 | 89.9 |
| Partially extrinsic | 4857 | 19.6 | 5337 | 20.1 | |
| Match to low-scoring hints | 527 | 8.9 | 579 | 7.1 | |
| No extrinsic match | 11,332 | 0.4 | 11,352 | 0.3 | |
| M. musculus | Fully extrinsic | 13,556 | 94.6 | 13,769 | 96.2 |
| Partially extrinsic | 7376 | 20.6 | 7606 | 19.6 | |
| Match to low-scoring hints | 957 | 10.1 | 1155 | 7.3 | |
| No extrinsic match | 20,711 | 1.2 | 19,666 | 0.5 | |
In the “match to low-scoring hints” category, we placed the candidates that were predicted fully ab initio, whereas in a posteriori analysis, a match of at least one exon border to a low-scoring extrinsic hint was detected. The category “no extrinsic match” contains the ab initio predictions that have no match to any extrinsic hints (see text). Average gene-level Pr values are listed for each group. Protein databases are described in the section “Data sets.”
In the genomes of D. rerio, G. gallus, and M. musculus, having the largest average length of introns and intergenic regions among the seven genomes, the gene candidates of category 4 had Pr values at the gene level <1.5%, thus indicating an exceedingly high false-positive rate. For these three large genomes, as well as for the genome of S. lycopersicum, an exclusion of gene candidates of category 4 led to a 21-percentage-point increase, on average, in the gene-level Pr, with a simultaneous 0.3-percentage-point decrease in Sn (Supplemental Table S2). On the other hand, such a pruning of the gene candidates generated in the analysis of the three compact genomes, A. thaliana, C. elegans, and D. melanogaster, would increase, on average, the gene-level Pr by 3.7 percentage points and decrease Sn by 1.7 percentage points. These observations suggest that when sufficiently long eukaryotic genomes (e.g., genomes >300 Mb) are considered, the gene candidates of category 4 should not be included into the lists of predicted genes (CDSs).
Yet another observation is that the numbers of predicted genes are close to the numbers of annotated genes, whereas the total numbers of predicted CDSs (including alternative CDSs) are consistently and significantly smaller than those in annotation (Table 3). Indeed, alternative CDSs are predicted in relatively small numbers on the whole-genome scale, because they are predicted only as CDS isoforms of the HC genes.
Table 3.
Numbers of the predicted and annotated genes as well as individual CDSs (including all alternative CDSs)
| Species | GeneMark-ETP | RefSeq annotation | ||
|---|---|---|---|---|
| No. of protein-coding genes | No. of predicted genomic CDSs | No. of protein-coding genes | No. of annotated genomic CDSs | |
| C. elegans | 18,820 | 19,806 | 19,969 | 28,544 |
| A. thaliana | 26,449 | 27,708 | 27,445 | 40,827 |
| D. melanogaster | 12,850 | 14,138 | 13,951 | 22,395 |
| S. lycopersicum | 24,420 | 26,341 | 25,158 | 31,911 |
| D. rerio | 28,608 | 31,961 | 25,610 | 42,929 |
| G. gallus | 17,275 | 21,433 | 17,279 | 38,534 |
| M. musculus | 23,956 | 27,686 | 22,405 | 58,318 |
Note that the genomic CDSs and the corresponding transcript CDSs are supposed to be identical in sequence. The “order excluded” reference databases were used by GeneMark-ETP (see section “Data sets”).
Assessment of the gene-prediction accuracy
The accuracy of previously developed gene-finding tools shows a consistent decline with the increase in the length of eukaryotic genome. This pattern is because of the increasing difficulty of accurately finding protein-coding sequences constituting a diminishing fraction of the whole genome, whereas noncoding regions are taking larger and larger genomic space. Indeed, for compact genomes, the values of gene-prediction Sn and Pr of the tools developed earlier and those developed later, including GeneMark-ETP, are relatively close and change incrementally (Fig. 3; Supplemental Fig. S8). However, for the large genomes, GeneMark-ETP makes significant improvements both for GC-homogenous S. lycopersicum and D. rerio and, especially, for GC-inhomogeneous G. gallus and M. musculus, for which we see the double-digit improvements of Sn and Pr (Fig. 4; Supplemental Fig. S8; Supplemental Tables S3, S4).
Figure 3.

Gene-level Sn and Pr of GeneMark-ETP and six other gene finders were computed for the three compact genomes: C. elegans, A. thaliana, and D. melanogaster. The dashed lines correspond to constant levels of (Sn + Pr)/2. The true positives were defined with respect to the complete set of annotated genes (either in RefSeq or Ensembl, which had identical annotations of these genomes). The “order excluded” protein databases were used.
Figure 4.

Gene-level Sn and Pr of gene predictions made by GeneMark-ETP and five other gene finders in the four large genomes: S. lycopersicum, D. rerio, G. gallus, and M. musculus. The Sn and Pr values for each genome were defined with respect to subsets of genes present in the RefSeq and Ensembl genome annotations (the intersections and the unions of annotations; see text). For these genomes, gene candidates lacking extrinsic support did not make it into the GeneMark-ETP list of predicted genes (see text). The “order excluded” protein databases were used.
The results of comparison with the gene finders using a single type of extrinsic evidence were as follows. Over GeneMark-ET (using RNA-seq reads mapping to augment the training process), the gene-level F1 was improved, on average, by 19.6, 47.8, and 66.3 points for the groups of compact, large homogeneous, and large inhomogeneous genomes, respectively. Over GeneMark-EP+ (using spliced aligned cross-species proteins), the increases in F1 were, respectively, 14.2, 33.9, and 55.7 points, noticeably lower numbers than the ones related to GeneMark-ET (Supplemental Table S3). GeneMark-ETP also showed improvements in F1 scores over BRAKER1 and BRAKER2, the pipelines containing GeneMark-ET and GeneMark-EP+, respectively. In comparison with BRAKER1, for the same three groups of genomes, the increases in F1 were, on average, by 9.7, 29.2, and 58.5 points. In comparison with BRAKER2, the F1 average improvements were 11.2, 21.9, and 47.6 points. The better performance of BRAKER2 in comparison with BRAKER1 is in line with the observed better performance of GeneMark-EP+ in comparison with GeneMark-ET, both of which are important parts of BRAKER2 and BRAKER1, respectively.
Next, we have considered gene predictions in which two sources of extrinsic evidence are accounted for. First, three combinations of the sets of gene predictions made separately by GeneMark-ET and GeneMark-EP+ could be constructed (see Methods). Still, GeneMark-ETP outperformed even the “best” combination of GeneMark-ET and GeneMark-EP+ gene predictions. For the D. melanogaster genome, taken as an example, GeneMark-ETP improved Sn by 7.5 percentage points and Pr by 13 percentage points over the “best” combination, in which Sn and Pr correspond to a “diamond” point (see Fig. 5). The gene-level F1 scores were improved by GeneMark-ETP over the F1 scores of the “best” combination of gene predictions by 10.1, 18.0, and 56.6 points for genomes of D. melanogaster, S. lycopersicum, and G. gallus, respectively.
Figure 5.
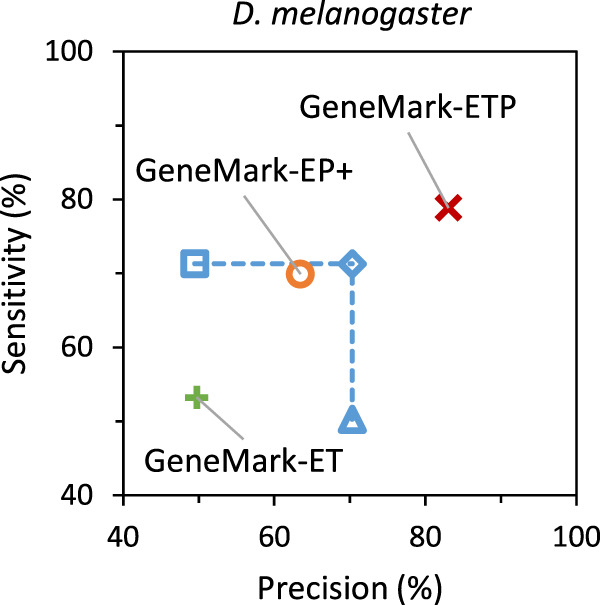
Gene-level Sn and Pr of the gene predictions in the D. melanogaster genome. The gene predictions were generated by GeneMark-ET (green), GeneMark-EP+ (orange), and GeneMark-ETP (red). We also show the Sn and Pr values for the Union (square) and the Intersection (triangle) of the sets of GeneMark-ET and GeneMark-EP+ gene predictions and the “best combination” of these two gene-prediction sets (diamond). The true positives were defined with respect to the annotation of the D. melanogaster genome. The “species excluded” database was used for the GeneMark-EP+ and GeneMark-ETP runs.
Next, the comparisons were made with TSEBRA (Gabriel et al. 2021) and MAKER2 (Holt and Yandell 2011). TSEBRA combines predictions made independently by BRAKER1 and BRAKER2 and filters out less reliable predictions from the union of the sets of genes predicted by the two pipelines. The gene-level F1 values of GeneMark-ETP and TSEBRA were comparable in the group of compact genomes. However, the average F1 values of GeneMark-ETP were better by 8.2 points for the two large GC-homogeneous genomes and were better by 39.0 points for the two large GC-inhomogeneous genomes, respectively (Figs. 3, 4; Supplemental Table S4).
The MAKER2 pipeline runs AUGUSTUS, SNAP, and GeneMark-ES. As extrinsic evidence, MAKER2 uses transcript and protein data. The experiments performed with the genomes of D. melanogaster, D. rerio, and M. musculus representing compact, large GC-homogeneous, and GC-inhomogeneous genomes showed, respectively, 23.3-, 21.7-, and 27.8-point improvements in gene-level F1, made by GeneMark-ETP (Supplemental Table S5). All the gene finders used in MAKER2 were run with parameters that, arguably, corresponded to the best-case scenario of training (see Methods). We used specially designed reduced-size databases of the cross-species proteins as the runtime of MAKER2 with our minimal size “order excluded” databases turned out to be prohibitive (see Supplemental Methods, Sec. S5). Change to the smaller databases brought the F1 of GeneMark-ETP down in comparison with its run with “order excluded” databases by about 5.0 points for D. melanogaster and M. musculus but did not change F1 score for D. rerio.
Applicability of the BUSCO scores for assessment of the gene-prediction accuracy
To address possible questions about using the BUSCO scores (Manni et al. 2021) for assessment of gene-prediction accuracy, we computed the BUSCO completeness scores both for GeneMark-ETP predictions and for the RefSeq annotations (Supplemental Table S6). The BUSCO scores have reached >90% for all seven genomes, whereas the gene-level Sn, Pr, and the F1 scores have shown much broader distributions of scores and much larger deviations from the perfect 100 sealing (Figs. 3, 4; Supplemental Tables S3, S4). These results are not unexpected because the BUSCO technique does not expect “identity” between the predicted proteins and the BUSCO proteins that represent a subset of all the proteins in a species proteome. The rules we use in the assessment of the accuracy of gene prediction are more stringent; they require exact matches of predicted and annotated CDSs. Although BUSCO scores are overly optimistic if used for assessment of gene-prediction quality, they are certainly helpful for the coarse evaluation of completeness of genome assembly and annotation (Huang and Li 2023).
Discussion
The GeneMark-ETP features
The GeneMark-ETP algorithm is using several previously developed concepts and approaches, such as the construction of anchored exon borders (GeneMark-ET), generation of external hints from multiple protein spliced alignments (GeneMark-EP+), and unsupervised iterative training of GHMM (GeneMark-ES, GeneMark-ET, GeneMark-EP+). However, a distinct feature of GeneMark-ETP is the integration of transcript and protein information in the GeneMarkS-TP module that generates a set of high-confidence CDS predictions. The predicted HC CDSs are not changed in the subsequent steps of the algorithm when it makes iterative training and prediction of the CDSs in genomic regions between the HC genes.
We showed that the predicted HC genes had much higher Pr than the initial set of gene candidates (Table 1; Supplemental Fig. S7). The number of HC gene predictions made in each genome with the “order excluded” database did not differ significantly (except C. elegans) from the number of HC genes predicted with the “species excluded” database (Table 1; Supplemental Fig. S7). Also, the Pr values did not change significantly upon transition from the “order excluded” to the larger, “species excluded” databases. These observations indicated that more distant proteins carry significant information for refinements of the HC gene candidates. If for a novel genome, there is a limited number of sequenced and annotated genomes of closely related species, then the accuracy estimates made for gene predictions with the “order excluded” databases would be more realistic than the estimates obtained with the “species excluded” databases.
Gene-prediction Sn also changed in transition from the HC gene candidates to the HC genes. The pattern of change depended on the size of the protein database. For instance, if the “order excluded” databases were used, then among all the seven genomes only the HC genes predicted in the C. elegans or D. melanogaster genomes had a lower Sn in comparison with the ones of the HC gene candidates. However, the HC genes predicted with the larger “species excluded” database had a higher Sn than the HC gene candidates in all the seven genomes (Table 1).
Among groups of gene candidates segregated by the level of extrinsic support, we observed much higher Pr in the group having full extrinsic support than in the group having partial extrinsic support (Table 2). In the three compact genomes and in the tomato genome, the magnitude of the difference in Pr was 30–40 percentage points, whereas in the genomes of D. rerio, G. gallus, and M. musculus, the difference in Pr was more than 50 percentage points. The Pr values in the second and the third groups of gene candidates decreased as the size of the protein database increased (Table 2). This change is explained by the move of many gene candidates from these two groups to the group with full extrinsic support, when the “order excluded” database was changed to the “species excluded” one.
Another new feature was application of the two GHMMs: (1) the transcript level model used for gene prediction in assembled transcripts and (2) the genomic level one used for gene prediction in genomic DNA. The training of the transcript level GHMM did follow the path described for GeneMarkS-T (Tang et al. 2015). However, the training process of the genomic GHMM was organized differently in comparison with GeneMark-ES, -ET, -EP+. For those three tools, the initial values of parameters of genomic GHMM were defined by the functions approximating dependence of the k-mer frequencies on genomic GC content (Lomsadze et al. 2005, 2014; Brůna et al. 2020). In GeneMark-ETP, the training of genomic GHMM is preceded by identification of the HC genes by GeneMarkS-TP (see Methods). Thus, the sequences of the loci containing the HC genes are used as an initial training set for the genomic GHMM with subsequent iterations over the rounds of gene predictions in the non-HC segments and GHMM parameter re-estimation (see Methods) (Supplemental Fig. S5). In our experience, the genomic GHHM model derived from the set of HC genes would not change significantly in further iterations if more than 4000 HC genes were identified by GeneMarkS-TP.
The genomic GHMM training implemented in GeneMark-ES, -ET, and -EP+ used step-by-step unfreezing of the subsets of the GHMM parameters over the iterations. For instance, the transition probabilities between hidden states, that is, intron, exon, etc., as well as distributions of durations of hidden states, were fixed during the initial iterations, whereas the values of emission probabilities, derived from the k-mer frequencies, were free to change. In the last iterations, all the parameters were made free. Such a gradual unfreezing of the parameter set was shown to be unnecessary for GeneMark-ETP; all the GHMM parameters were estimated already in the first iteration.
Comparisons with other gene finders
As expected, in all the tests, GeneMark-ETP performed better than the tools using only a single type of extrinsic evidence: GeneMark-ET, GeneMark-EP+, BRAKER1, and BRAKER2 (see Results; Figs. 3, 4). One could notice that GeneMark-EP+, even running with the smaller “order excluded” database, outperformed GeneMark-ET. This observation was in part because GeneMark-ET uses RNA-seq-derived hints only in training but not in the prediction step. On the other hand, GeneMark-EP+ has a mechanism for enforcing high-scoring protein-derived external hints into gene predictions made by the Viterbi algorithm.
The magnitudes of improvement that GeneMark-ETP shows in the gene-level F1 scores in comparison with the best F1 among GeneMark-ET and GeneMark-EP+ follow already cited patterns of increase from shorter to longer genomes (Figs. 3, 4; Supplemental Table S3). F1 was improved by 11–19 points for compact genomes and by 26–60 points for large genomes, with the maximum for the G. gallus genome. The gaps in F1 values between GeneMark-ETP and GeneMark-EP+ are smaller when the larger “species excluded” database was used in comparison with the gaps observed upon the use of the “order excluded” database (Supplemental Table S3).
Certainly, the new features implemented in GeneMark-ETP, as discussed in the previous section, were significant additional factors behind the better performance of GeneMark-ETP in comparison with GeneMark-ET, GeneMark-EP+, BRAKER1, and BRAKER2, especially for the four large genomes (Figs. 3, 4; Supplemental Table S4).
The TSEBRA pipeline selects a subset of all predictions made by either BRAKER1 or BRAKER2 by rules that attempt to increase Pr without compromising Sn (Gabriel et al. 2021). For compact genomes, A. thaliana, C. elegance, and D. melanogaster TSEBRA shows comparable performance with GeneMark-ETP, with TSEBRA having higher gene-level F1 for the first two species (by 1.5 and four points) and lower F1 (by 0.5 points) for the last one. However, for the larger genomes: S. lycopersicum, D. rerio, G. gallus, and M. musculus GeneMark-ETP improves F1 over TSEBRA by 6.4, 9.0, 45.6, and 29.4 points, respectively (Supplemental Table S4). The double digits of improvements in F1 are observed for the GC-inhomogeneous genomes G. gallus and M. musculus. All the above-mentioned novel features of GeneMark-ETP contributed to the improvement of the prediction accuracy, especially in the large genomes. Additionally, it should be stated that for large GC-inhomogeneous genomes, BRAKER1 and BRAKER2 use a single species-specific GHMM trained in a fashion that effectively makes GHMM for an average genome-specific GC content. GeneMark-ETP constructs three GC-specific models for each GC-inhomogeneous genome (see Methods). Also, GeneMark-ETP integrates the extrinsic RNA and protein information upon individual prediction of each gene, whereas in BRAKER1 and BRAKER2, these two channels of extrinsic information are disjointed.
The same novel features of GeneMark-ETP are behind the double-digit improvements in gene-level F1 compared with MAKER2 (Supplemental Table S5). Besides, a number of tools included into the MAKER2 pipeline have been improved since 2011 when MAKER2 was released. Thus, an update of the MAKER2 architecture by transition to more recently developed components would be quite useful.
Methods
Data sets
For computational experiments, we selected genomes of seven eukaryotic species. Among them were three model organisms, A. thaliana, C. elegans, and D. melanogaster, having well-studied GC-homogeneous and compact-in-size genomes. The larger genomes of S. lycopersicum and D. rerio were GC-homogenous, whereas the other large genomes of G. gallus and M. musculus were GC-inhomogeneous. In all cases, sequences of organelles, as well as contigs without chromosome assignment, were excluded (Table 4; Supplemental Table S7).
Table 4.
The table shows total numbers of protein-coding genes as well as individual CDSs (including alternative isoforms) annotated in the seven genomes (the annotation sources are described in Supplemental Table S7)
| Species | Genome length (Mb) | Reference annotation statistics | |||||
|---|---|---|---|---|---|---|---|
| No. of protein-coding genes | No. of CDSs | Introns per gene | |||||
| C. elegans | 100 | 19,969 | — | 28,544 | — | 4.8 | — |
| A. thaliana | 119 | 27,445 | — | 40,827 | — | 4.0 | — |
| D. melanogaster | 138 | 13,951 | — | 22,395 | — | 2.8 | — |
| S. lycopersicum | 807 | 25,158 | (15,138) | 31,911 | (15,150) | 4.4 | (4.3) |
| D. rerio | 1345 | 25,610 | (17,893) | 42,929 | (19,975) | 8.4 | (8.4) |
| G. gallus | 1050 | 17,279 | (10,736) | 38,534 | (12,733) | 9.0 | (9.2) |
| M. musculus | 2723 | 22,405 | (16,531) | 58,318 | (20,708) | 6.0 | (8.6) |
The numbers of genes and individual CDSs in the intersections of the NCBI RefSeq and the Ensembl annotations are given in parentheses (see Methods). Annotations of the C. elegans, A. thaliana, and D. melanogaster genomes are identical between RefSeq and Ensembl; therefore, the intersection sets have the same numbers of genes and CDSs as in the RefSeq annotation.
To generate the reference sets of proteins used as a source of extrinsic evidence, we used the OrthoDB v10.1 protein database (Kriventseva et al. 2019). For each of the seven species, we built an initial protein database (PD0) containing proteins from the kingdom, the phylum, or the class segment of OrthoDB, in which the given species belongs (Supplemental Table S8). For each species, we created two reference databases by removing from PD0 either (1) proteins of the species in question and its strains, that is, the database called “species excluded,” or (2) proteins of all the species from the same taxonomic order, that is, the database called “order excluded” (see also Brůna et al. 2020). These “order excluded” and “species excluded” databases were supposed to mimic practical scenarios when a species of interest would appear at either a larger or a smaller evolutionary distance from the species present in the protein database. The number of proteins in the databases created in our study ranged from 2.6 million to 8.3 million (Supplemental Table S8).
Data sets of RNA sequences, the sets of Illumina paired short reads, were selected from the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra). The read length varied between 75 and 151 nt. The total volume of the selected RNA-seq collections varied from 9 Gb for D. melanogaster to 83 Gb for M. musculus (Supplemental Table S9).
Algorithm overview
In the earlier-developed GeneMark-ES, -ET, and -EP+, the estimation of the parameters of GHHMs was performed by iterative unsupervised training (Lomsadze et al. 2005, 2014; Brůna et al. 2020). The final set of parameters was used in the final round of gene prediction. The model training and gene prediction are organized differently in GeneMark-ETP, which is also an automatic self-training tool (Fig. 1; Supplemental Methods).
GeneMarkS-TP: generation of HC gene predictions
The wealth of current RNA and protein sequence information precipitates development of a new algorithmic component of a whole-genome gene finder, the component conducting gene prediction in assembled transcripts with cross-species protein support. This component of GeneMark-ETP, named GeneMarkS-TP, is constructed as a pipeline built from several tools. HISAT2 (Kim et al. 2019) splice-aligns the short reads from the selected RNA-seq libraries to the species’ genome. StringTie2 (Kovaka et al. 2019) uses these data for transcript assembly. After filtering out the low-abundance transcripts, the assembled transcripts are merged by StringTie2 into a nonredundant set.
Initial gene predictions in transcripts and genomic DNA
A transcript may contain a protein-coding region (CDS) along with 5′ and 3′ UTRs. The predictions of the borders of a CDS sequence in a transcript are made by self-training GeneMarkS-T (Tang et al. 2015). Converting the predicting CDS sequence into a chain of protein-coding exons in the genomic DNA, known as genomic CDS, is efficiently performed by using data on RNA-seq reads anchored to the genome. This information is generated by StringTie2 upon the transcript's assembly. The genomic CDS is predicted by combining the coordinates of CDS identified in a transcript with the genomic coordinates of introns produced by StringTie2. The values of Sn and Pr for the set of predicted genomic CDSs, whose inference started with the identification of transcript CDSs by GeneMarkS-T, are shown in Table 1.
Refinement of gene predictions
The GeneMarkS-T predictions of complete CDSs in the RNA transcripts were shown to be quite accurate (Tang et al. 2015). On the other hand, predictions of 5′ incomplete CDSs (5′ partial genes) could be less precise. The 5′ partial gene predictions are refined in the GeneMarkS-TP module using protein data.
A 5′ partial CDS is supposed to start from the first nucleotide of a transcript. However, a true complete CDS may reside inside this 5′ partial CDS. To distinguish between the two possibilities, GeneMarkS-TP proceed as follows. Translations of the predicted 5′ partial CDS as well as the longest complete ORF having the same stop of translation as 5′ partial CDS serve as queries in the similarity search by DIAMOND (Buchfink et al. 2015). The protein target common for both searches (E-value < 10−3) is aligned to both queries. The strengths of sequence conservation along the pairwise alignments are analyzed (Supplemental Methods, condition S1). The 5′ partial CDS is confirmed if condition S1 is fulfilled. Otherwise, the predicted 5′ partial CDS is replaced by the shorter complete CDS (see Supplemental Methods, Sec. S1.1; Supplemental Table S10).
Selecting HC CDS candidates: CDSs with uniform protein similarity support
Complete and partial CDSs with uniform protein similarity support are selected from the set of CDSs predicted in assembled transcripts. A complete CDS is said to have uniform protein support if a pairwise alignment of the predicted protein to a known protein satisfies condition S2 (see Supplemental Methods). A complete CDS with uniform protein similarity support is a candidate for complete HC CDS.
One more candidate may be present in the same transcript if it is possible to extend the predicted CDS to the “longest” ORF. If the protein product of this longest ORF has a uniform protein similarity support (Supplemental Fig. S9, condition S2), then the extended CDS is yet another candidate for complete HC CDS, likely to be an alternative isoform of the same gene.
The uniform protein similarity support could also be found for a predicted 5′ partial CDSs. If a condition like condition S2 is fulfilled for a pairwise alignment of the C-terminal of the protein translation of the 5′ partial CDS with a database protein (Supplemental Figs. S10, S11), then the 5′ partial CDS becomes a candidate for a partial HC CDS.
Selecting HC CDS candidates: CDSs without uniform protein similarity support
Some genes that do not have uniform protein similarity support may still be qualified as HC CDS candidates. If a predicted complete CDS satisfies all the following conditions—(1) the length of the CDS is >299 nt, (2) the 5′ UTR contains an in-frame stop codon triplet, and (3) the exons mapped to genomic DNA do not create a conflict with the ProtHint hints (see Supplemental Methods, see Sec. S2)—then the CDS is designated as a candidate HC CDS. Notably, if a CDS lacks a stop codon, it cannot be an HC CDS candidate.
Selection of HC CDSs and HC genes from the candidates; generating GeneMarkS-TP output
Alternative transcripts of individual genes are identified by StringTie2, which segregates the whole set of assembled transcripts, complete and incomplete, into groups associated with individual protein-coding genes. We note that a protein-coding gene is defined here as a set of alternative CDSs coming from the same locus. Alternative mRNA transcripts of a gene may encode identical proteins and have differences only in UTRs. Such alternative transcripts are not considered.
If a gene has only one mRNA transcript (no alternatives), then the HC CDS candidate becomes an HC CDS, and the gene is called an HC gene. If more than one alternative transcript with an HC CDS candidate exist, then we follow the procedure described in Supplemental Methods, Sec. S3 to select HC CDSs from the candidates, and the gene in question becomes the HC gene with the selected set of HC CDS isoforms. Finally, the set of HC genes that may consist of several alternative HC CDSs, either complete or partial, represents the output of GeneMarkS-TP. This set of HC genes was shown to have a significantly higher Pr than the set of initial gene predictions made by GeneMarkS-T (Supplemental Table S10).
The genomic GHMM training
Single-step GHMM training
An initial training set for the genomic GHMM is constructed from the set of predicted HC genes. This set, in a GC-inhomogeneous genome, may have significant variation in GC content. To characterize a genome as GC-homogenous, we check if there exists a 9% wide interval of the GC content distribution that would contain at least 70% of the HC genes selected for training. If there is no such interval, which is a sign that the genomic GC content distribution is wide, the genome is characterized as GC-inhomogeneous (Supplemental Fig. S12).
In a GC-homogeneous genome, the sequences of the predicted HC genes are extended by 1000 nt in both the 5′ and 3′ directions (making a set of the HC loci). If an HC gene has alternative isoforms, the one with the longest CDS is selected for the training set. The extended sequences, including introns and segments of intergenic regions, are used for the initial estimation of the genomic GHMM parameters. In a GC-inhomogeneous genome, the sequences of the HC loci are split into three bins: low GC, mid GC, and high GC. The mid GC bin (with the default width of 9%) (see Supplemental Fig. S12) is defined by the 9% GC interval that contains the largest number of the HC loci sequences selected for training. Setting up the mid GC interval immediately determines the borders of the low and high GC bins. The sets of the HC loci assigned to the three bins are used to train the GC-specific GHMMs. Note that GeneMarkS-T was developed for gene prediction in GC-inhomogeneous transcriptomes. This tool is using a set of GC-specific models (Tang et al. 2015).
Extended GHMM training for a genome with homogeneous GC
The logic of extended training of the genomic GHMM is similar but not identical to iterative training used in GeneMark-ET and GeneMark-EP+ (Lomsadze et al. 2014; Brůna et al. 2020).
In a GC-homogeneous genome, the initial GHMM parameters derived from the sequences of the HC loci are used for the first round of gene predictions. The predictions made in the genomic sequences situated between the HC genes, the non-HC segments, are of special interest (Supplemental Figs. S5, S6). These predictions could be refined in subsequent iterations, whereas the predicted HC genes would not change.
The gene predictions made in the non-HC segments are used in ProtHint to generate protein-based hints, as in GeneMark-EP+ (Brůna et al. 2020). An additional set of hints comes from RNA-seq reads mapped to genome by HISAT2 (Kim et al. 2019). All over, there are the following categories of hints: (1) RNA-seq- and ProtHint-derived hints to intron borders that agree with each other, (2) high-score ProtHint hints to intron borders, (3) RNA-seq-based hints to intron borders that may or may not coincide with the intron borders predicted ab initio, and (4) hints representing partial HC genes that could be extended into the non-HC segments. Note that partial HC genes can appear only at the borders of non-HC segments, and technically, we consider partial HC genes to belong to non-HC segments.
The hints of the first three categories point to elements of a multiexon gene without indication that these elements may or may not belong to the same gene. Hints of the fourth category represent “chains” of introns that should belong to the same gene. The requirement of corroboration of RNA-seq hints of category 3 by ab initio predictions allows to filter out the “false-positive” intron hints mapped from expressed noncoding RNA. The whole set of selected hints is now ready for enforcement in a run of GeneMark.hmm. The hints are enforced in the gene predictions made in the non-HC segments. The predicted genes are included in the updated training set. The iterations stop when the identity of the training sets in two consecutive iterations reaches 99%. The number of iterations observed in our experiments was rarely more than three. Frequently, the convergence had been reached in the second iteration. If the number of HC genes is large (more than 4000), then, based on our experience, a single iteration is sufficient for convergence. The set of genes predicted in the non-HC segments along with the set of the HC genes made the final set of genes predicted by GeneMark-ETP.
Extended GHMM training for a genome with inhomogeneous GC
The extended GHMM training for GC-inhomogeneous genomes works as follows. The three initial GC-specific GHMMs are trained on the sequences of the HC loci from the corresponding bins. These GHMMs are used to predict genes in the non-HC segments associated with the same GC bin as the GC-specific model. From this point on, the extended training of the three GC-specific GHHMs is performed on the non-HC segments segregated into the three bins. These trainings are conducted separately following the logic described for the GC-homogeneous case. The GC-specific GHMMs are used in the rounds of iterative gene prediction and parameters estimation that occur until convergence (Supplemental Fig. S6). If the number of the HC genes in a bin is large (more than 4000), then a single iteration is sufficient for parameter estimation of the GHMM associated with this bin.
In general, GeneMark-ETP could be run in the “GC-inhomogeneous” mode on any genome. However, for a “true” GC-homogeneous genome, this choice would increase runtime and sometimes even decrease gene-prediction accuracy owing to splitting the overall training set into smaller subsets. Therefore, the degree of GC-heterogeneity is assessed before the GHMM training.
Processing of repetitive elements
Transposable elements (TEs), particularly families of retrotransposons with thousands of copies of similar TE sequences, occupy substantial portions of eukaryotic genomes. Errors in gene prediction may be caused by the presence of repetitive elements with a composition similar to that of protein-coding regions (Yandell and Ence 2012; Tørresen et al. 2019). Identification of the repetitive sequences is performed independently from gene finding. We generate species-specific repeat libraries de novo using RepeatModeler2 (Flynn et al. 2020). Repetitive sequences are then identified in genomic sequence by RepeatMasker (https://www.repeatmasker.org). Some of the predicted repeats may overlap with protein-coding genes (Bayer et al. 2018). To conduct gene finding in genomic sequence with “soft masked” repeats, a bonus function with a species-independent parameter was introduced (Stanke et al. 2008). For a sequence designated as repetitive by a repeat finder, this function increased the likelihood of emission from noncoding state. Here, we have introduced a species-specific penalty function to decrease the likelihood of a sequence emission from a protein-coding state inside a region identified as a repetitive one (Equation 1). The parameter of this function, q, is defined (trained) for each genome. Technically, we introduce a state for an overlap of a repeat and CDS and compute the probability of a sequence (of length n) to be emitted from such a state:
| (1) |
The genome-specific parameter q is estimated after obtaining the set of HC genes in the first round of the GHMM training (see Supplemental Methods; Supplemental Fig. S13; Supplemental Table S11).
Computation of accuracy measures
Assessment of the gene-prediction accuracy in each genome was performed as follows. The test set for computing the Sn values was the set of genes having identical annotations in NCBI RefSeq and in Ensembl, the intersection set (Table 4). To compute the Pr values, we used the union of genes annotated by NCBI RefSeq and Ensembl. In all the tests, regions of annotated pseudogenes were excluded from consideration. Because annotations of the A. thaliana, C. elegans, and D. melanogaster genomes were the same at NCBI and Ensembl, then, for each of them, the union and the intersection sets coincided with each other and with the full complement of annotated genes.
GeneMark-ET, GeneMark-EP+, and combinations of their sets of gene predictions
The gene finders GeneMark-ET and GeneMark-EP+ were designed to use a single source of extrinsic support, either RNA-seq reads or protein database. They were run to get reference points on what accuracy could be achieved with a single source of evidence. We also considered combinations of the sets of genes predicted separately by GeneMark-ET and GeneMark-EP+.
The union of the two sets of gene predictions would have increased Sn and decreased Pr. The intersection of the two sets would have increased Pr and decreased Sn (the case of D. melanogaster) (Fig. 5). The “best” combination of the members of the two sets could be made by removing false positives from the union set or adding true positives made by either method to the intersection set. When the union set is changed by taking away the incorrect predictions, the point for the Union in Figure 5 moves horizontally to the right. If one adds correct predictions made by one of the tools but not the other to the intersection set, the Intersection point in Figure 5 moves up vertically. The crossing of the two lines corresponds to the “best” combination of the members of the two sets of predicted genes in terms of (Sn + Pr)/2 (Fig. 5). The described changes cannot be made when a gene finder is running on a novel genome because information on the true and false positives is not immediately available. Nevertheless, such modifications could be made for gene predictions in genomes with known annotations.
Running BRAKER1, BRAKER2, TSEBRA, and MAKER2
To make comparisons with the transcript-supported BRAKER1 (Hoff et al. 2016) and protein-supported BRAKER2 (Brůna et al. 2021), we ran BRAKER1 and BRAKER2 with the same RNA-seq libraries and the same protein databases, respectively, as the ones used in experiments with GeneMark-ETP. Also, we ran TSEBRA (Gabriel et al. 2021), which selects a subset of all the gene predictions made separately by BRAKER1 and BRAKER2 and filters out less reliably predicted genes. TSEBRA was shown to achieve higher accuracy than either BRAKER1 or BRAKER2 running alone.
The execution of MAKER2 has some degree of freedom as the rules of training AUGUSTUS, SNAP, and GeneMark.hmm are not precisely specified in the MAKER2 publication (Holt and Yandell 2011). Therefore, we wanted to present the accuracy figures that would, arguably, correspond to the upper limit achieved by the optimal training of MAKER2. The selected models for AUGUSTUS and SNAP were either the models provided by the code developers (available with the respective software distribution) or the models generated by supervised training on the genes annotated by NCBI (RefSeq). As an exception, SNAP training was performed on the Ensembl-annotated D. rerio genome. Both MAKER2 and GeneMark-ETP use the GeneMark.hmm gene-finding algorithm. MAKER2 uses models for GeneMark.hmm self-trained by GeneMark-ES using no extrinsic evidence. We assumed that more precise models would be obtained if extrinsic evidence was also used in the training. Therefore, we trained the GeneMark.hmm models on a set of the HC genes (determined by GeneMark-ETP). To get the accuracy figures, the gene predictions made for the genomes of D. melanogaster, D. rerio, and M. musculus by MAKER2 were processed in the same way as the gene predictions made by GeneMark-ETP or other gene finders. The repeat coordinates and RNA-seq data sets were the same for MAKER2 and GeneMark-ETP (see Supplemental Materials). We compiled reduced-size protein databases for running the experiments with MAKER2 because the runtime of MAKER2 sharply increases with the increase in the volume of protein data. The minimal size of the “order excluded” database used in the experiments described for the seven genomes was 2.5 million proteins. The maximum size database that was used in the experiments for comparison of GeneMark-ETP with MAKER2 was about 300,000 proteins (see Supplemental Methods, Sec. S5). The same reduced-in-size protein sets were used for both GeneMark-ETP and MAKER2. It should be noted that for a GC-inhomogeneous genome of M. musculus GeneMark-ETP used the models with the GC-specific parameters. In MAKER2, by design, the GC-specific parameters were used in AUGUSTUS but not in SNAP or GeneMark.hmm.
Runtime of GeneMark-ETP
The runtime of GeneMark-ETP depends linearly on the genome size and is comparable to that of GeneMark-EP+. The runtime dependence on the size of protein database is also linear. The size of RNA-seq libraries is critical for the HISAT2 runtime but not for the rest of the GeneMark-ETP operations with RNA sequences. We do not include the runtimes of HISAT2 and StringTie2 into the runtime of GeneMark-ETP. To give examples of absolute runtimes, on a machine with 64 CPU cores, GeneMark-ETP finished operations on the genomes of D. melanogaster, D. rerio, and M. musculus in 3, 12, and 18 hr, respectively. In these experiments, we used the “order excluded” protein databases and RNA reads sets described in Supplemental Tables S8 and S9.
Software availability
GeneMark-ETP is available at https://exon.gatech.edu/GeneMark/license_download.cgi and as Supplemental Software. All scripts and data used to generate figures and tables in this paper are available at GitHub (https://github.com/gatech-genemark/GeneMark-ETP-exp) and as Supplemental Code. GeneMark-ETP was included in the recently developed pipeline BRAKER3 (Gabriel et al. 2024).
Supplementary Material
Acknowledgments
The National Institutes of Health (GM128145 to M.B.) supported this work. Georgia Tech provided funding for the open access charge.
Author contributions: T.B., A.L., and M.B. designed the project; T.B. and A.L. designed and wrote the code, prepared all the initial data, and conducted computational experiments; and T.B., A.L., and M.B. wrote and edited the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278373.123.
Freely available online through the Genome Research Open Access option.
Competing interest statement
GeneMark-ETP and its partner GeneMark.hmm are distributed under the Creative Commons license. Licensing GeneMark.hmm by commercial companies may create a conflict of interest for A.L. and M.B. T.B. declares having no competing interests.
References
- Allen JE, Salzberg SL. 2005. JIGSAW: integration of multiple sources of evidence for gene prediction. Bioinformatics 21: 3596–3603. 10.1093/bioinformatics/bti609 [DOI] [PubMed] [Google Scholar]
- Allen JE, Pertea M, Salzberg SL. 2004. Computational gene prediction using multiple sources of evidence. Genome Res 14: 142–148. 10.1101/gr.1562804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Bhandary P, Woodhouse M, Sen TZ, Wise RP, Andorf CM. 2021. FINDER: an automated software package to annotate eukaryotic genes from RNA-seq data and associated protein sequences. BMC Bioinformatics 22: 205. 10.1186/s12859-021-04120-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer PE, Edwards D, Batley J. 2018. Bias in resistance gene prediction due to repeat masking. Nat Plants 4: 762–765. 10.1038/s41477-018-0264-0 [DOI] [PubMed] [Google Scholar]
- Brůna T, Lomsadze A, Borodovsky M. 2020. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genom Bioinform 2: lqaa026. 10.1093/nargab/lqaa026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brůna T, Hoff KJ, Lomsadze A, Stanke M, Borodovsky M. 2021. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom Bioinform 3: lqaa108. 10.1093/nargab/lqaa108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B, Xie C, Huson DH. 2015. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12: 59–60. 10.1038/nmeth.3176 [DOI] [PubMed] [Google Scholar]
- Burge C, Karlin S. 1997. Prediction of complete gene structures in human genomic DNA. J Mol Biol 268: 78–94. 10.1006/jmbi.1997.0951 [DOI] [PubMed] [Google Scholar]
- Coghlan A, Fiedler TJ, McKay SJ, Flicek P, Harris TW, Blasiar D, the nGASP Consortium, Stein LD. 2008. nGASP: the nematode genome annotation assessment project. BMC Bioinformatics 9: 549. 10.1186/1471-2105-9-549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DE, Valle-Inclan JE, Pajoro A, Rovenich H, Thomma B, Faino L. 2019. Long-read annotation: automated eukaryotic genome annotation based on long-read cDNA sequencing. Plant Physiol 179: 38–54. 10.1104/pp.18.00848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG, Feschotte C, Smit AF. 2020. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117: 9451–9457. 10.1073/pnas.1921046117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel L, Hoff KJ, Brůna T, Borodovsky M, Stanke M. 2021. TSEBRA: transcript selector for BRAKER. BMC Bioinformatics 22: 566. 10.1186/s12859-021-04482-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel L, Brůna T, Hoff KJ, Ebel M, Lomsadze A, Borodovsky M, Stanke M. 2024. BRAKER3: fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS, and TSEBRA. Genome Res (this issue) 34: 769–777. 10.1101/gr.278090.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodswen SJ, Kennedy PJ, Ellis JT. 2012. Evaluating high-throughput ab initio gene finders to discover proteins encoded in eukaryotic pathogen genomes missed by laboratory techniques. PLoS One 7: e50609. 10.1371/journal.pone.0050609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremme G, Brendel V, Sparks ME, Kurtz S. 2005. Engineering a software tool for gene structure prediction in higher organisms. Inform Software Tech 47: 965–978. 10.1016/j.infsof.2005.09.005 [DOI] [Google Scholar]
- Guigo R, Flicek P, Abril JF, Reymond A, Lagarde J, Denoeud F, Antonarakis S, Ashburner M, Bajic VB, Birney E, et al. 2006. EGASP: the human ENCODE genome annotation assessment project. Genome Biol 7 Suppl 1: S2.1–S2.31. 10.1186/gb-2006-7-s1-s2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, White O, Buell CR, Wortman JR. 2008. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol 9: R7. 10.1186/gb-2008-9-1-r7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff KJ, Lange S, Lomsadze A, Borodovsky M, Stanke M. 2016. BRAKER1: unsupervised RNA-seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 32: 767–769. 10.1093/bioinformatics/btv661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt C, Yandell M. 2011. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12: 491. 10.1186/1471-2105-12-491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe KL, Chothia T, Durbin R. 2002. GAZE: a generic framework for the integration of gene-prediction data by dynamic programming. Genome Res 12: 1418–1427. 10.1101/gr.149502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N, Li H. 2023. compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics 39: btad595. 10.1093/bioinformatics/btad595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keilwagen J, Hartung F, Paulini M, Twardziok SO, Grau J. 2018. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics 19: 189. 10.1186/s12859-018-2203-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. 2019. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37: 907–915. 10.1038/s41587-019-0201-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryutin B, Souvorov A, Tatusova T. 2007. ProSplign: protein to genomic alignment tool. In Research in Computational Molecular Biology, 11th Annual International Conference, RECOMB 2007, Oakland, CA, USA, April 21–25, 2007, Proceedings (ed. Speed T, Huang H). (Lecture Notes in Computer Science, 4453.) Springer. [Google Scholar]
- Kong J, Huh S, Won JI, Yoon J, Kim B, Kim K. 2019. GAAP: a genome assembly + annotation pipeline. Biomed Res Int 2019: 4767354. 10.1155/2019/4767354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovaka S, Zimin AV, Pertea GM, Razaghi R, Salzberg SL, Pertea M. 2019. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol 20: 278. 10.1186/s13059-019-1910-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriventseva EV, Kuznetsov D, Tegenfeldt F, Manni M, Dias R, Simão FA, Zdobnov EM. 2019. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res 47: D807–D811. 10.1093/nar/gky1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulp D, Haussler D, Reese MG, Eeckman FH. 1996. A generalized hidden Markov model for the recognition of human genes in DNA. Proc Int Conf Intell Syst Mol Biol 4: 134–142. [PubMed] [Google Scholar]
- Lewin HA, Richards S, Lieberman Aiden E, Allende ML, Archibald JM, Balint M, Barker KB, Baumgartner B, Belov K, Bertorelle G, et al. 2022. The Earth BioGenome Project 2020: starting the clock. Proc Natl Acad Sci USA 119: e2115635118. 10.1073/pnas.2115635118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Mackey AJ, Roos DS, Pereira FC. 2008. Evigan: a hidden variable model for integrating gene evidence for eukaryotic gene prediction. Bioinformatics 24: 597–605. 10.1093/bioinformatics/btn004 [DOI] [PubMed] [Google Scholar]
- Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, Borodovsky M. 2005. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res 33: 6494–6506. 10.1093/nar/gki937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomsadze A, Burns PD, Borodovsky M. 2014. Integration of mapped RNA-seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res 42: e119. 10.1093/nar/gku557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manni M, Berkeley MR, Seppey M, Simão FA, Zdobnov EM. 2021. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol Biol Evol 38: 4647–4654. 10.1093/molbev/msab199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra G, Blanco E, Guigó R. 2000. GeneID in Drosophila. Genome Res 10: 511–515. 10.1101/gr.10.4.511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. 2015. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33: 290–295. 10.1038/nbt.3122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalzitti N, Jeannin-Girardon A, Collet P, Poch O, Thompson JD. 2020. A benchmark study of ab initio gene prediction methods in diverse eukaryotic organisms. BMC Genomics 21: 293. 10.1186/s12864-020-6707-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater GS, Birney E. 2005. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6: 31. 10.1186/1471-2105-6-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Sabunciyan S, Yang G, Florea L. 2019. A multi-sample approach increases the accuracy of transcript assembly. Nat Commun 10: 5000. 10.1038/s41467-019-12990-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Diekhans M, Baertsch R, Haussler D. 2008. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24: 637–644. 10.1093/bioinformatics/btn013 [DOI] [PubMed] [Google Scholar]
- Tang S, Lomsadze A, Borodovsky M. 2015. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res 43: e78. 10.1093/nar/gkv227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. 2008. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res 18: 1979–1990. 10.1101/gr.081612.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tørresen OK, Star B, Mier P, Andrade-Navarro MA, Bateman A, Jarnot P, Gruca A, Grynberg M, Kajava AV, Promponas VJ, et al. 2019. Tandem repeats lead to sequence assembly errors and impose multi-level challenges for genome and protein databases. Nucleic Acids Res 47: 10994–11006. 10.1093/nar/gkz841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515. 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandell M, Ence D. 2012. A beginner's guide to eukaryotic genome annotation. Nat Rev Genet 13: 329–342. 10.1038/nrg3174 [DOI] [PubMed] [Google Scholar]
- Zickmann F, Renard BY. 2015. IPred: integrating ab initio and evidence based gene predictions to improve prediction accuracy. BMC Genomics 16: 134. 10.1186/s12864-015-1315-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.