

Abstract
The tumor microenvironment (TME) profoundly influences tumorigenesis, with gene expression in the breast TME capable of predicting clinical outcomes. The TME is complex and includes distinct cancer-associated fibroblast (CAF) subtypes whose contribution to tumorigenesis remains unclear. Here, we identify a subset of myofibroblast cancer associated fibroblasts (myCAF) that are senescent (senCAF) in mouse and human breast tumors. Utilizing the MMTV-PyMT;INK-ATTAC (INK) mouse model, we found that senCAF-secreted extracellular matrix specifically limits natural killer (NK) cell cytotoxicity to promote tumor growth. Genetic or pharmacologic senCAF elimination unleashes NK cell killing, restricting tumor growth. Finally, we show that senCAFs are present in Her2+, ER+, and triple negative breast cancer and in ductal carcinoma in situ (DCIS) where they predict tumor recurrence. Together, these findings demonstrate that senCAFs are potently tumor promoting and raise the possibility that targeting them by senolytic therapy could restrain breast cancer development.
Keywords: TME, tumor microenvironment, senescence, CAFs, breast cancer, stromal therapy
Introduction
The tumor microenvironment plays a pivotal role in tumorigenesis by providing critical reciprocal communication to tumor cells and the immune system. This critical interplay has attracted great interest as it represents a novel opportunity to target the support structure that enables tumor progression. The importance of the TME in breast cancer is underscored by studies showing that stromal gene expression in the TME can predict patient outcome (1). The TME is heterogenous, consisting of endothelial cells, adipocytes, immune cells, and fibroblasts, but also the acellular components of the extracellular matrix (ECM) such as collagen, hyaluronic acid, and fibronectin. Interestingly, in many cancers including breast tumors, cancer associated fibroblasts (CAF) are the most numerous cells in the TME (2,3). Recent single cell transcriptomic studies of CAFs from various tumor types revealed that CAFs are themselves heterogeneous and can have varied effects on tumorigenesis. These findings have increased the interest in further detailing how different CAF subtypes impact tumorigenesis.
Single cell RNA-sequencing analyses (scRNA-seq) of tumors from the spontaneous MMTV-PyMT mouse model revealed distinct CAF populations (4). Bartoschek et al., identified four CAF populations that were named based on their expression profiles, including cycling CAFs (cCAF), matrisome CAFs (mCAF), vascular CAFs (vCAF), and developmental CAFs (dCAF) (5). In pancreatic ductal adenocarcinoma models and orthotopic injection models of breast cancer, additional CAF subtypes have been described, including inflammatory CAFs (iCAF) that are characterized by expression of a large cadre of inflammatory cytokines, myCAFs that express αSMA, and antigen presenting CAFs (apCAF) that express MHC class II (6–9). Intriguingly, the pro- or anti-tumor effects of the different CAF subtypes is dictated by their tissue of origin, further underscoring the incredible heterogeneity of CAFs and the need to detail their impact on tumorigenesis. Moreover, CAFs also display a high degree of plasticity. For instance, TGF-β signaling drives differentiation of myCAFs (10) and TGF-β supplementation can lead to the blockade of JAK/STAT signaling and shift iCAFs to a myofibroblastic state (11), which further highlights the plasticity and complexity of CAFs and their crosstalk with the tumor microenvironment. Similar CAF subtypes have also been identified in human breast tumors (12,13), raising the possibility that they impact tumorigenesis.
Similar to CAFs, senescent fibroblasts promote tumorigenesis through numerous secreted factors that can directly drive tumor growth and modulate the immune response (14,15). Interestingly, activation of the p38MAPK-MK2 pathway, which controls expression of pro-tumorigenic factors collectively referred to as the senescence-associated secretory phenotype (SASP) in CAFs and senescent fibroblasts, governs the protumor capabilities of the stroma (16). Senescence can be induced by a wide array of stressors ranging from DNA damage to oxidative stress, to nutrient deprivation, to excessive cellular division (17). In response to these stressors, cells experience chronic p53 and Rb pathway activation, leading to the activation of senescence, which is typically characterized by expression of the cell cycle inhibitor CDKN2a (p16), permanent growth arrest, senescence-associated β-galactosidase (SA-βGal) expression, and SASP expression. Although the specific factors are dictated by the cell type undergoing senescence, the SASP is generally characterized by a plethora of inflammatory cytokines, ECM proteins and those that modify the ECM, and numerous growth factors (15). While senescence functions as a potent tumor suppressor when it arises within incipient tumor cells, the SASP can function in a cell extrinsic manner where it has been shown to both stimulate and limit tumorigenesis (18–23).
The varied effects of the SASP is likely due to the fact that the factors that constitute the SASP are both cell type and tissue type dependent (24). Indeed, when tumor cells senesce, they can secrete factors that recruit macrophages to limit tumor growth (18,20). More recently, senescing tumor cells were shown to upregulate MHCI and IFN signaling, resulting in CD8+ T cell killing of the senescent tumor cell (19). In a lung K-RAS driven model, senescing tumor cells were found to express TNFa and intracellular adhesion molecule-1, leading to increased NK cell surveillance and tumor cell killing (23). In contrast, senescing fibroblasts can stimulate tumorigenesis in part by secreting IL6, which leads to increased numbers of immature granulocytes that limit CD8+ T cell killing and increase tumor growth (14). However, whether senescent CAFs are present in human breast tumors and if so, what role they play, remains an open question. Intriguingly, Witkiewicz et. al., reported that high expression of p16 in the stromal compartment of DCIS of the breast specifically correlated with poor outcomes (25). Further analyses of the gene expression pattern in the stromal compartment of breast cancers revealed high expression of many p38MAPK-MK2-dependent factors that are associated with SASP (26). Together these finding raise the possibility that senescent stromal cells are present in breast cancer lesions and contribute to tumorigenesis.
Here using single cell transcriptomics analyses and multiplex immunohistochemistry (mIHC), we identify a senCAF population in human breast cancer subtypes including estrogen receptor positive (ER+), human epidermal growth factor receptor 2 positive (HER2+), and triple negative breast cancer (TNBC) and in the murine MMTV-PyMT model of breast cancer. Further, we find that initial biopsies from patients with recurrent DCIS harbor senCAFs, while senCAFs are absent in the DCIS lesions of patients that do not develop a local recurrence. In both the human and the murine samples, senCAFs express a wide array of ECM and ECM-modifying proteins. Using the MMTV-PyMT mouse model, we find that the ECM produced from senCAFs directly inhibits NK cell cytotoxicity and that the elimination of senCAFs reduces tumorigenesis in an NK cell dependent manner. Thus, our data demonstrate that senCAFs play an important role in promoting breast cancer tumorigenesis by suppressing NK cell antitumor responses.
Results
SenCAFs are present in breast cancer tissues
Human breast cancers contain heterogeneous populations of CAFs that exert varied effects on host immune responses. To further characterize CAFs, we queried a single cell scRNA-seq dataset of human breast cancer tissues including 26 samples spanning ER+, HER2+, and TNBC subtypes (13) (Fig. S1A–B). For our analysis, we focused on non-immune stromal cells and identified 3 PDGFRB-expressing CAF clusters and 5 endothelial cell clusters across all three subtypes of breast cancer samples (Fig. 1A and Fig. S1C–E). In agreement with previous literature on CAF heterogeneity in pancreas ductal carcinoma and breast cancer models (6–8), our analysis revealed the presence of 3 CAF populations—including a cluster of MCAM and ACTA2 double-positive vCAFs (5), a cluster of ACTA2 and PDGFRA double-positive myCAFs, and a cluster of PDGFRA and CXCL12 double-positive iCAFs (Fig. 1B).
Figure 1. Senescent CAFs are present in human breast tumors.
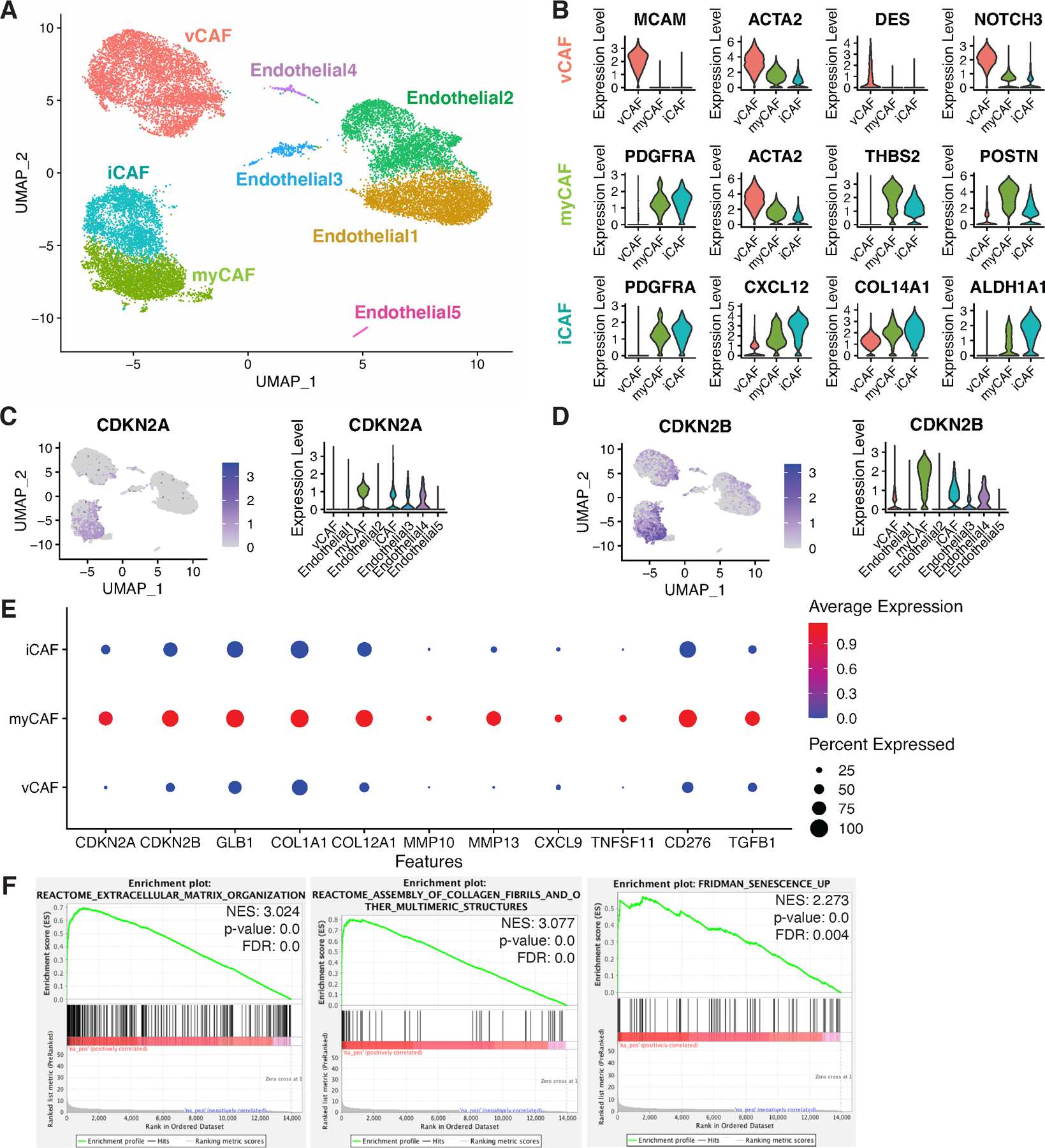
(A) UMAP of stromal cells from 26 human breast cancer samples of ER+ (n=11), HER2+ (n=5), and TNBC (n=10) cases. (B) Violin plots of CAF signature markers: vCAF—MCAM+, ACTA2+, DES+, NOTCH3+; myCAF—PDGFRA+, ACTA2+, THBS2+, POSTN+; iCAF—PDGFRA+, CXCLl12+, COL14A1+, ALDH1A1+. (C) Feature plot and violin plot for the expression of CDKN2A (p16) in selected stromal cell types. (D) Feature plot and violin plot for the expression of CDKN2B (p15) in selected stromal cell types. (E) Dot plot displaying gene expression parameters including percentage of cells (size of black dots) and expression levels (colored scale bar) of a designated gene within a specific population (y-axis). Features include senescence markers (CDKN2A (p16) and CDKN2B (p15) and SASP factors (GLB1, COL1A1, COL12A1, MMP10, MMP13, CXCL9, TNFSF11, CD276, and TGFB1). (F) Gene set enrichment analyses of the myCAF population compared to all other CAF populations show enrichment in pathways including extracellular matrix organization, collagen assembly, and Fridman senescence. Normalized Enrichment Score (NES), p-value, and False Discovery Rate (FDR) are shown in each plot.
Given we had found that breast stroma expresses SASP factors (27) and it was reported that CDKN2A (p16) is expressed in the stromal compartment of samples from recurrent DCIS patients (25), we next asked if senescent stromal cells were present in these breast cancer samples. To answer the question, we assessed the expression of p16, a key marker of senescence, across the clusters. Because p16 is poorly expressed, we utilized Adaptively-thresholded Low Rank Approximation (ARLA) imputation in our analyses, which is a validated approach to identify poorly expressed genes (28). Surprisingly, we found that the p16+ population expressed markers that were primarily restricted to myCAFs (Fig. 1C). Accordingly, these p16+ myCAFs also expressed elevated levels of other commonly reported senescent cell markers including p15 (coded by CDKN2B) (Fig. 1D) and various ECM-related SASP factors including collagen type I, collagen type XII, and MMP10 and 13 as well as SASP factors with immune-modulating capacity including CXCL9, RANKL (coded by TNFSF11), B7H3 (coded by CD276), and TGF-β (Fig. 1E) (17,29–35). In addition, compared to the other subsets of CAFs, myCAFs exhibited higher expression levels of another commonly reported senescent cell marker lysosomal β-d-galactosidase (coded by GLB1), whose activity is directly linked to the detection of senescence-associated beta-galactosidase (SA-βGal) in senescent and aging cells (Fig. 1E and S1F)(36,37). Finally, our differential gene expression (DGE) analysis and gene set enrichment analysis (GSEA) confirmed that, in agreement with their reported “myofibroblastic” nature, these p16+ myCAFs had enriched expression of genes related to ECM organization, collagen fiber assembly, and TGF-β signaling (6) (Fig. 1F and S1G). Significantly, senescence-related genes identified by Fridman et.al., were over-represented in myCAFs compared to the other CAF populations (Fig. 1F) (38).
To validate protein expression of p16 in the CAFs of human breast cancer tissues, we performed mIHC for p16 and a series of CAF signature markers suggested by scRNA-seq. For these analyses, we utilized a human breast tumor tissue array consisting of 51 TNBC patient samples. In the pan-cytokeratin (panCK) negative stromal region of these samples, we identified iCAFs (COL14A1 and PDGFRα double-positive, orange arrows) and myCAFs (αSMA (encoded by ACTA2) and PDGFRα double-positive, hollow and solid white arrows), with the latter harboring a significant p16+ subpopulation (solid white arrows) (Fig. 2A). Quantification of p16-positivity in iCAFs and myCAFs across samples from 51 patients confirmed that p16 expression was largely restricted to the myCAF population in human breast cancer stroma (Fig. 2B).
Figure 2. p16 expression is present in myCAFs and the presence of p16+ myCAFs correlates with DCIS recurrence.
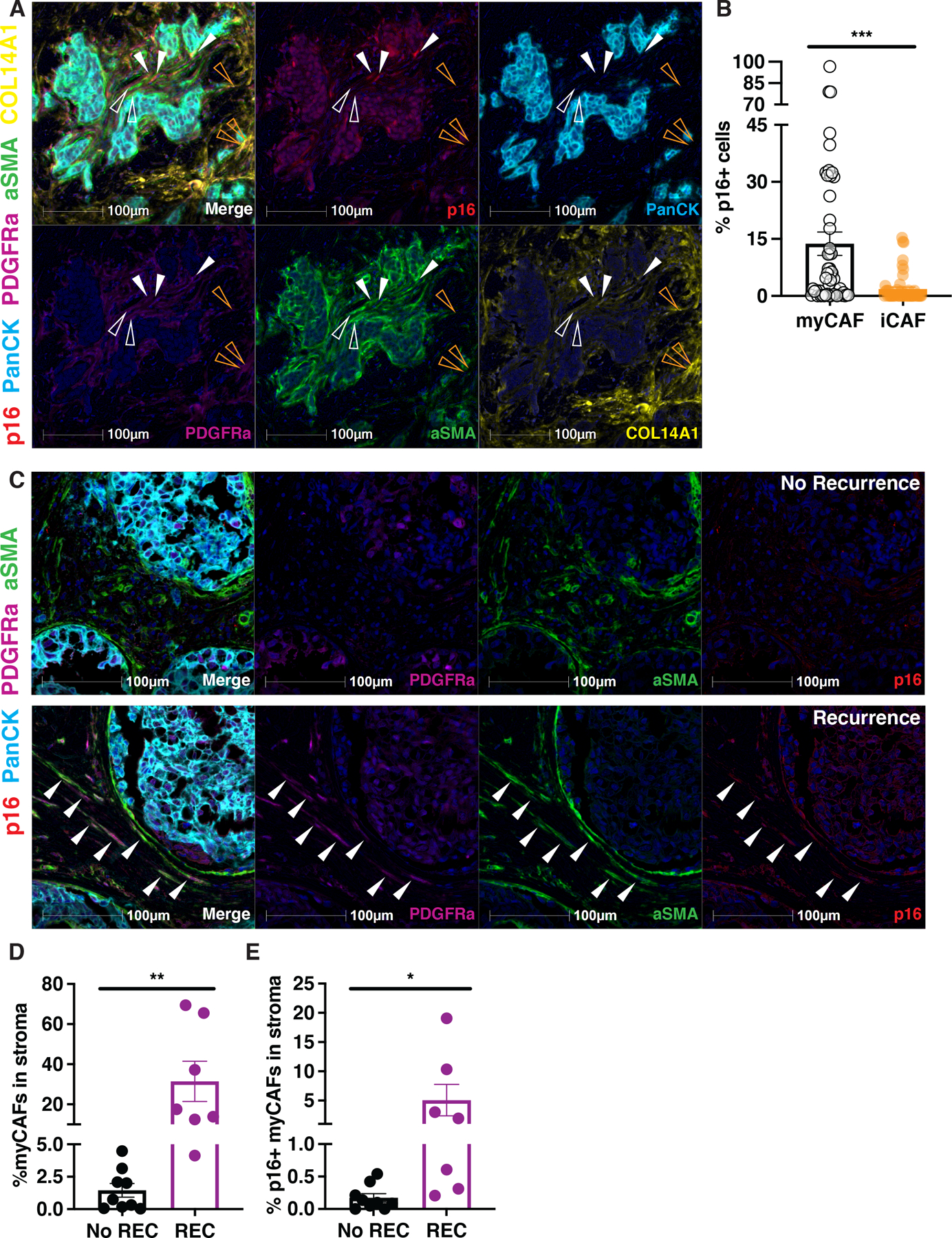
(A) mIHC were performed on 51 human breast cancer tumor tissue samples using p16 (red), pan-cytokeratin (PanCK; cyan), PDGFRa (magenta), alpha-smooth muscle actin (aSMA; green), and COL14A1 (yellow) specific antibodies and nuclei were counterstained with hematoxylin and shown as dark blue. Individual stains were pseudo colored as indicated and then merged (first panel). myCAFs are denoted with hollow (p16-) and solid (p16+) white arrowheads. iCAFs are denoted with hollow orange arrowheads. (B) Quantification of percentage of p16+ cells in myCAF and iCAF subpopulations using human breast cancer tumor tissue arrays (n=51). (C) mIHC were performed on tumor DCIS samples using p16 (magenta), aSMA (green), PDGFRa (red), and pan-cytokeratin (PanCK; cyan) specific antibodies and nuclei were counterstained with hematoxylin and shown as dark blue. Cells triple positive for p16, aSMA, and PDGFRa are denoted by white arrowheads. Individual stains were pseudo colored as indicated and then merged (first panel). (D) Quantification of myCAFs (PDGFRa+ aSMA+ cells) in stroma (PanCK− cells) from multiplex IHC of DCIS samples that recurred (REC, n=7) or did not recur (No REC, n=9). (E) Quantification of p16+ myCAFs (PDGFRa+ aSMA+ cells) in stroma (PanCK− cells) from multiplex IHC of DCIS samples that recurred (REC, n=7) or did not recur (No REC, n=9). Unpaired one-tailed student t-test performed for all statistical analyses shown in this figure. All numerical data are represented as mean ± SEM, *p<0.05, **p<0.01, ***p<0.001.
High p16 expression in the stromal compartment of human DCIS correlates with high recurrence rates, but the cell type(s) expressing p16 were not evaluated (25). We thus performed mIHC to ask if myCAFs were the source of the stromal p16 signal in these same DCIS samples. Indeed, we found that stromal p16 signal was largely restricted to myCAFs in the analyzed DCIS samples (Fig. 2C). More importantly, tissue samples from the recurrence group (REC) had significantly more myCAFs (Fig. 2D) and p16-expressing myCAFs (Fig. 2E) in their stroma. Together, our scRNA-seq and mIHC analysis of human samples demonstrate that p16+ myCAF is a bona fide senescent population within the stromal microenvironment of human breast tumors and thus we refer to them as senCAFs hereafter. Moreover, these data indicate that the presence of senCAFs correlates with tumor recurrence and poor outcomes.
Identification of senCAFs in MMTV-PyMT mammary tumors.
Senescent fibroblasts modulate tumorigenesis in a tissue and cell type dependent manner. Thus, we wanted to determine if senCAFs were present in a robust genetic engineered mouse model (GEMM) of breast cancer and determine what role they played in tumor progression. To this end, we turned to one of the most well-characterized GEMMs for human breast cancer studies, the murine mammary tumor virus polyomavirus middle T antigen mouse model (MMTV-PyMT, PyMT hereafter). Carcinoma stage mammary tumors were collected from 12-week-old nulliparous PyMT mice for tissue dissociation and downstream scRNA-seq analysis (39) (Fig. S2A–C). Non-tumor cell clusters were identified and re-clustered for refined analysis (Fig. S2D–H). Strikingly, CAFs in PyMT tumors could be categorized into the same 3 subpopulations as those found in the human samples. Specifically, PyMT tumors also harbored an Acta2 and Pdgfra double-positive myCAF population, a Pdgfra and Cxcl12 double-positive iCAF population, and an Mcam and Acta2 double-positive vCAF population, with all 3 populations positive for the pan-CAF marker Pdgfrb (Fig. 3A–B and Fig. S2D). We further examined the expression of p16 across all stromal cells identified in the dataset, including the 3 CAF clusters, a cluster of CD3+ immune cells, a cluster of antigen presenting cells (APCs), and 2 clusters of endothelial cells (Fig. S2E–F). Similar to our findings in the human dataset, high expression of p16 was limited to myCAFs (Fig. 3C and S2F). Moreover, p16+ myCAFs from PyMT tumors expressed SASP factors similar to those we found in the human tumor samples such as collagens, MMPs, CXCL9, RANKL, B7H3, and TGF-β (Fig. 3C). In accordance with our observations in the human dataset, other senescence-associated markers such as p15, BCL-W, and GLB1 were expressed at elevated levels by p16+ murine myCAFs (Fig. 3C and Fig. S2G–H) (36,37,40). Indeed, when we performed staining for SA-βGal on tumors from PyMT mice, we could identify positive signals in the stromal regions (Fig. S2I). To validate the identity of PyMT-derived p16+ myCAFs, we performed DGE and GSEA analyses. Both of these analyses confirmed that genes related to ECM modulation, collagen fiber assembly, and TGF-β signaling (Fig. 3D and S2J) were enriched specifically in the p16+ myCAF population compared to other CAFs. Remarkably, senescence-related genes reported by Fridman et. al., were also over-represented in p16+ myCAFs compared to the other two CAF populations (Fig. 3D) (38). Thus, our findings confirm that similar to what we found in human breast cancer samples, senCAFs are present in the PyMT transgenic mouse, indicating that the PyMT mouse model can be used to determine what role senCAFs play in breast cancer.
Figure 3. Senescent CAFs are present in MMTV-PyMT murine mammary tumors.
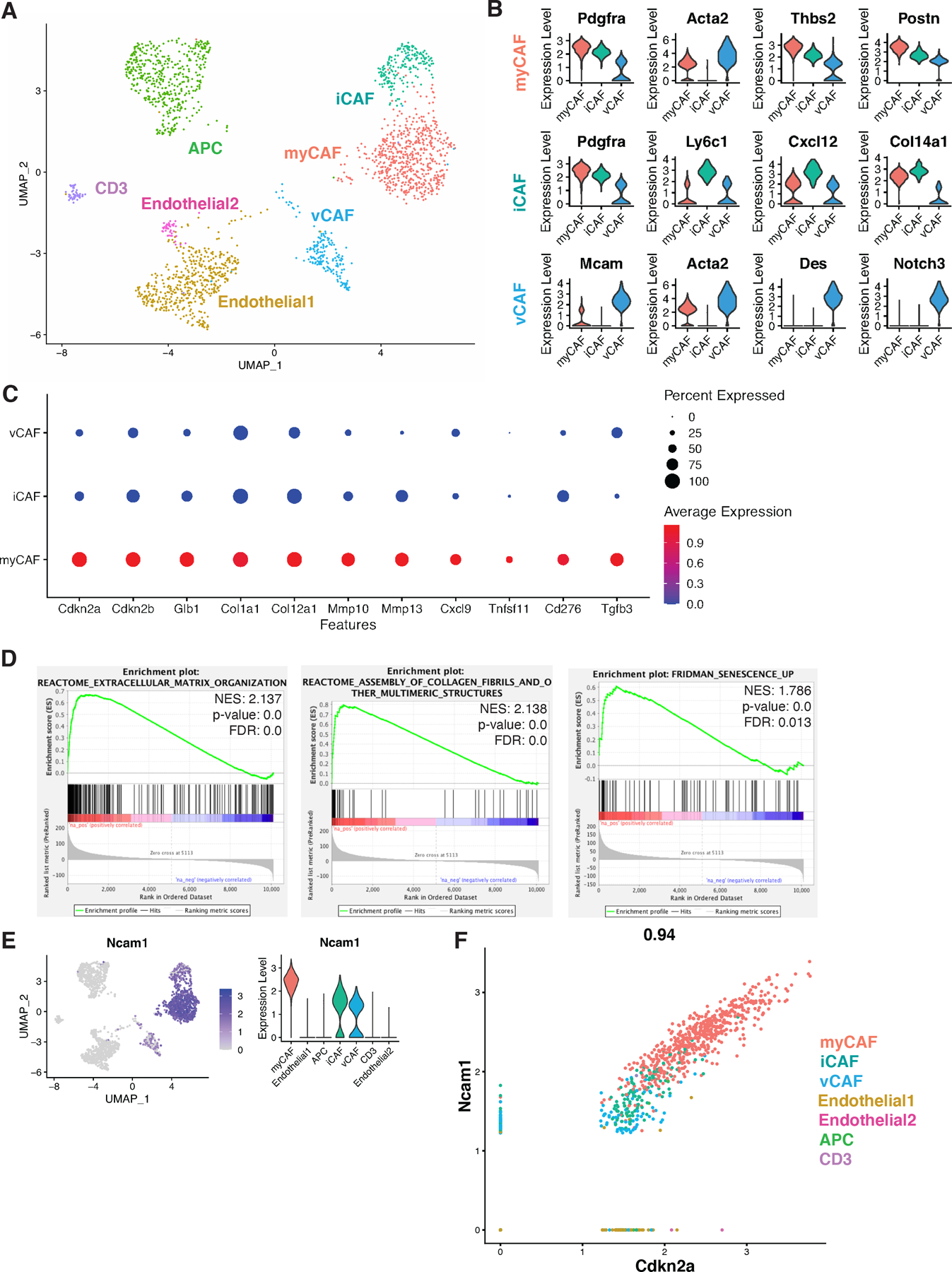
(A) UMAP of stromal cells from 12-week-old MMTV-PyMT tumors (n=2). (B) Violin plots of CAF signature markers: myCAF—Pdgfra+, Acta2+, Thbs2+, Postn+; iCAF—Pdgfra+, Ly6c1+, Cxcl12+, Col14a1+; vCAF—Mcam+, Acta2+, Des+, Notch3+. (C) Dot plot displaying gene expression parameters including percentage of cells (size of black dots) and expression levels (colored scale bar) of a designated gene within a specific population (y-axis). Features include senescence markers (Cdkn2a (p16) and Cdkn2b (p15)) and SASP factors (Glb1, Col1a1, Col12a1, Mmp10, Mmp13, Cxcl9, Tnfsf11, Cd276, and Tgfb3). (D) Gene set enrichment analyses of the myCAF population compared to vCAF and iCAF showed in pathways including extracellular matrix organization, collagen assembly, and Fridman senescence. NES, p-value, and FDR are shown in each plot. (E) Feature plot and violin plot for the expression of Ncam1 in selected stromal cell types. (F) Correlation curve of Ncam1 and Cdkn2a (p16) expression in mammary stromal cell populations.
NCAM+/p16+ CAFs within PyMT tumors are senescent
To characterize the murine senCAFs and facilitate their isolation, we further examined the signature genes within the population. To our surprise, we found that Ncam1, which codes for a surface-expressed adhesion molecule NCAM, was highly upregulated in myCAFs in the PyMT dataset (Fig. 3E) and its expression level strongly correlated with that of Cdkn2a (Fig. 3F). To evaluate NCAM as a marker for senCAFs, we performed mIHC for p16, NCAM, and a series of markers that identify distinct CAF subpopulations in PyMT tumor sections. Consistent with our scRNA-seq results, our staining confirmed the existence of myCAF (αSMA and PDGFRα double-positive, hollow and solid white arrows), iCAF (PDGFRα and Ly6C1 double-positive, orange arrows), and vCAF (MCAM and αSMA double-positive, blue arrows) populations (Fig. 4A and S3A). More importantly, histology analysis demonstrated that p16-expression was limited to myCAFs and that all p16+ myCAFs (solid white arrows) were positive for NCAM (Fig. 4A). Finally, we also performed mIHC for p16 and Ki67 in PyMT tumor sections. Similar to what was reported in human breast cancer tissues (25), quantification of p16+ cells in tumor nests versus stromal areas (Fig. S3B) showed that the majority of p16+ tumor cells had high Ki67 positivity (hollow arrows) indicating that they were proliferative and thus not senescent. In contrast, the vast majority of p16+ stromal cells were negative for Ki67 (solid arrows) (Fig. 4B–C), indicating they were growth arrested. Together these analyses firmly establish that senCAFs are a unique population.
Figure 4. NCAM1 marks senescent CAFs in MMTV-PyMT tumors.
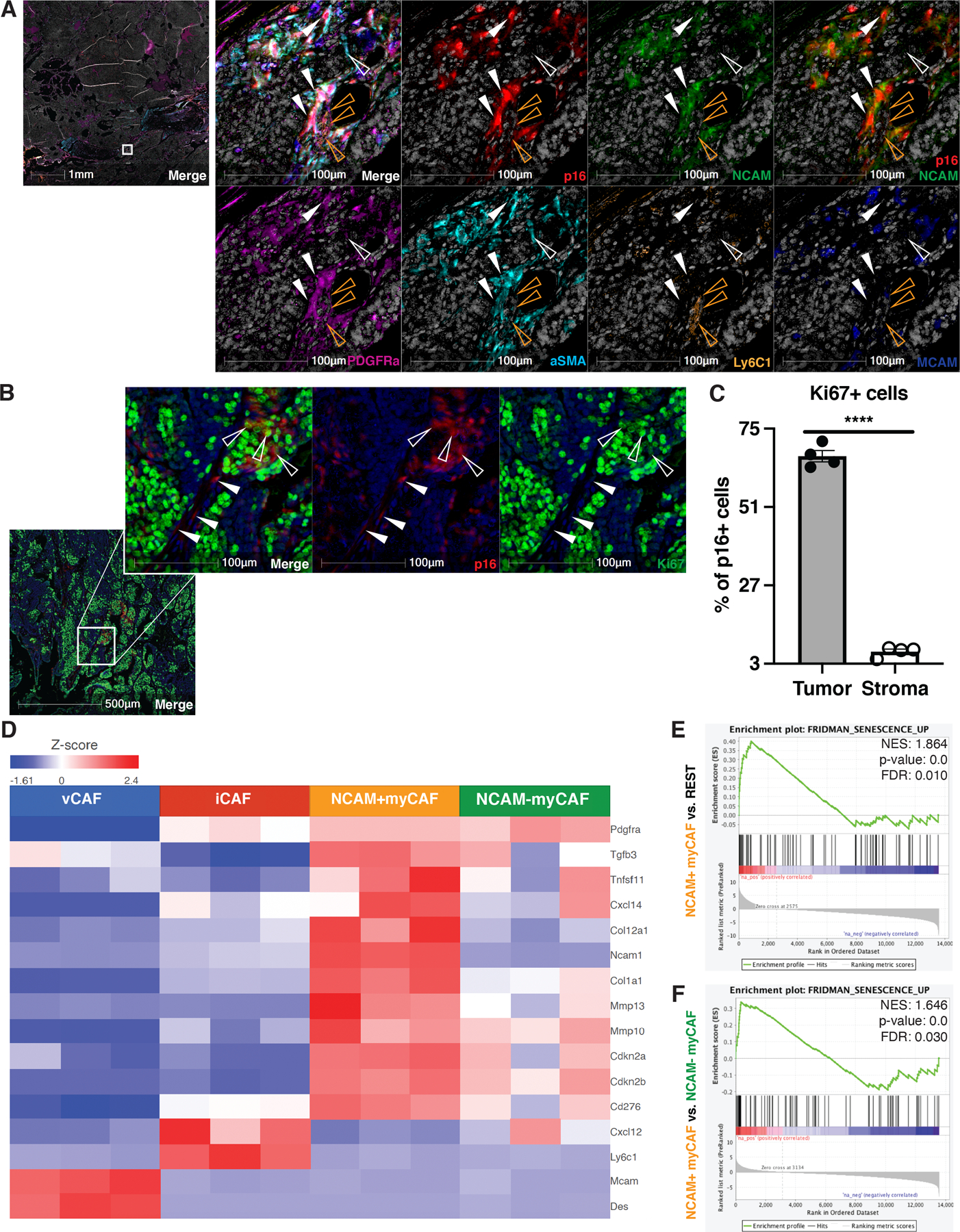
(A) mIHC analyses were performed on 10-week-old MMTV-PyMT tumors using p16 (red), NCAM (green), PDGFRα (magenta), alpha-smooth muscle actin (aSMA) (cyan), Ly6C1 (orange), and MCAM (blue) specific antibodies and nuclei were counterstain with hematoxylin and shown as white (left: 1x; right: 25x). Individual stains were pseudo colored as indicated and then merged (first panel). myCAFs are denoted with hollow (p16-) and solid (p16+) white arrowheads. iCAFs were mostly p16- and are denoted with hollow orange arrowheads. (B) mIHC analyses were performed on 10 weeks old MMTV-PyMT tumors using p16 (red) and Ki67 (green) and nuclei were counterstained with hematoxylin and shown as dark blue (left: 5x; right: 25x). Greater than 70% of p16+ tumor cells were also positive for Ki67 and are denoted with hollow arrowheads. p16+ stromal cells were mostly negative for Ki67 and are denoted with solid arrowheads. (C) Quantification of percentage of Ki67+ cells out of p16+ tumor or stromal compartments as shown in B (n=4). (D) Four CAF populations were sorted from 10-week-old MMTV-PyMT mammary tumors (n=3) and subjected to bulk RNA-Seq analyses. Shown is heatmap highlighting CAF markers, senescence markers, and SASP factors. (E) Gene set enrichment analyses of the NCAM+ myCAF population compared to the remaining 3 CAF populations showed enrichment of the Fridman senescence signature. (F) Gene set enrichment analyses of the NCAM+ myCAF compared to the NCAM− myCAF population also showed enrichment of the Fridman senescence signature. NES, p-value, and FDR are shown in each plot. Unpaired one-tail student t-test was performed for C. All numerical data are represented as mean ± SEM. **** p<0.0001.
Confident in our scRNA-seq-derived markers, we next sought to achieve a more extensive characterization of each CAF population through bulk RNA-sequencing (RNA-seq). To isolate the CAF populations, we used fluorescence-activated cell sorting (FACS) (antibody panel: CD45, CD31, EPCAM, PDGFRβ, Ly6C1, PDGFRα, MCAM, and NCAM), and found that among the different CAF populations, the expression of NCAM was largely restricted to myCAFs (Fig. S4A). Thus, we sorted iCAFs, vCAFs, NCAM-positive (NCAM+) and NCAM-negative (NCAM-) myCAF populations and carried out RNA-sequencing. Principle component analysis (PCA) revealed that the four CAF populations were distinct (Fig. S4B). Further, our DGE analysis revealed that NCAM+ myCAFs expressed numerous SASP genes (Fig. 4D) and confirmed their myofibroblastic identity (Fig. S4C). Further, we found that the Fridman gene set of cellular senescence was significantly enriched in NCAM+ myCAFs as compared to the remaining three CAF populations (Fig. 4E) and when we performed DGE analysis of NCAM+ versus NCAM- myCAFs (Fig. 4F). Collectively, our mIHC and bulk RNA-seq data reinforced our findings from scRNA-seq analyses and established NCAM as a genuine surrogate marker for senCAFs in mice; thus, we will refer to NCAM-negative myCAFs simply as myCAFs hereafter.
senCAFs contribute to breast cancer development
Depending on their cell of origin, SASP factors can have contradictory effects on tumor progression. While senescent tumor cells can facilitate immune-mediated tumor cell clearance (18–20), stromal SASP factors can directly promote tumor growth in vitro and in immunodeficient xenograft models (21,22) and recruit immature granulocytes capable of limiting CD8+ T cell killing and increasing tumor growth in the skin (14). The identification of senCAFs and their robust SASP signature led us to investigate whether these cells contributed to breast cancer tumorigenesis. To this end, we utilized the p16/INK4a Apoptosis Through Targeted Activation of Caspase (INK-ATTAC) mouse model (INK) developed by Baker et. al. (41,42). Briefly, the INK construct uses a minimal p16Ink4a promoter to drive the expression of an inducible suicide gene, which allows for systematic depletion of p16+ cells through the administration of AP20187 (AP) (41). For these studies, we mated the PyMT and the INK mouse lines and recruited both PyMT+/INK− (INK−) and PyMT+/INK+ (INK+) females to AP treatment as soon as the pups were weaned by 28–30 days of age (Fig. 5A). Weekly palpation of mammary fat pads (MFP) and subsequent caliper measurements of mammary tumors revealed that the AP treatment significantly delayed tumor onset in INK+ mice (Fig. 5B). To confirm the depletion of p16+ cells, we performed immunohistochemistry (IHC) of p16 on tissues collected from both 7- (Fig. 5C–D) and 10-week-old mice (Fig. S5A–B). Quantification of p16+ cells in tumor nests versus stromal areas (Fig. S3B) indicated that p16 expression was primarily detected in stromal cells but not tumor cells, which agreed with our observations in the PyMT scRNA-seq dataset (Fig. S2C). More remarkably, only p16+ stromal cells were reduced in INK+ mice while there was no difference in the number of p16+ tumor cells between the two genotypes (Fig. 5D and Fig. S5B), which suggests that senCAFs contribute to mammary tumorigenesis.
Figure 5. Senescent CAFs drive MMTV-PyMT tumorigenesis.
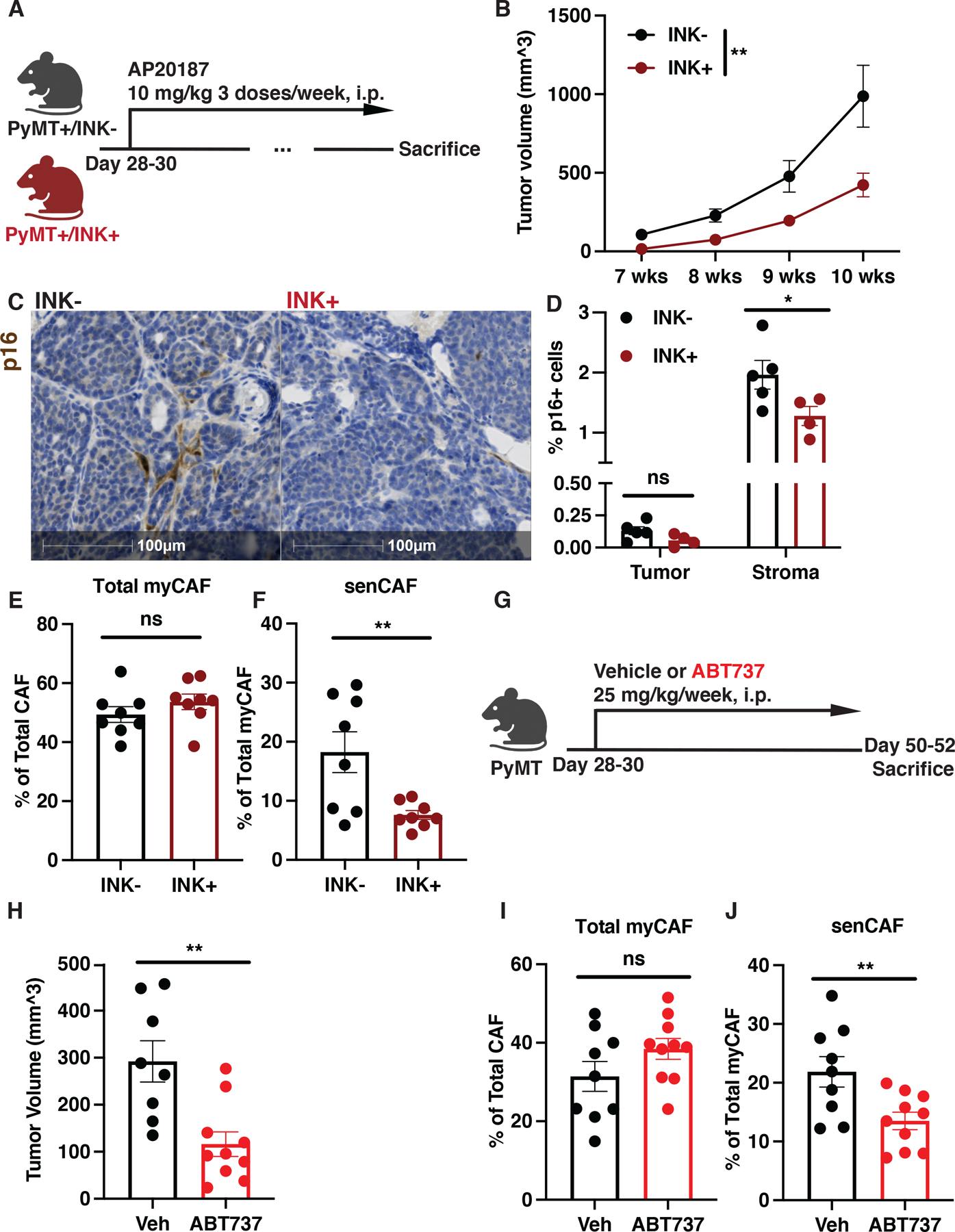
(A) AP treatment regimen of MMTV-PyMT (INK-) and MMTV-PyMTxINKATTAC+ (INK+) mice. (B) Tumor volume measured with calipers weekly for INK− (n=16) and INK+ (n=19) mice. 2-way ANOVA was performed to compare the tumor growth; **p<0.01. (C) Representative IHC images for p16 expression in 7-week-old INK- and INK+ mice show predominant staining in stromal cells. (D) Quantification of p16 staining in tumor versus stromal compartment shown in (C) indicates that only stromal p16+ cells are lost in INK+ mice (n=5) compared to INK− mice (n=4). (E) Quantification of total myCAF population in tumors from 7 weeks old INK− versus INK+ mice. n=8 for each group. (F) Quantification of senCAF populations in tumors from 7 weeks old INK− versus INK+ mice. n=8 for each group. (G) Senolytic (ABT737) treatment regimen of MMTV-PyMT mice. (H) Tumor volume measured with calipers at week 7 for vehicle (Veh, n=8) and ABT737 (n=10) treated mice. (I) Quantification of total myCAF population in MMTV-PyMT mice treated with vehicle (Veh, n=9) or ABT737 (n=10). (J) Quantification of senCAF populations in MMTV-PyMT mice treated with Veh (n=9) or ABT737 (n=10). Unpaired one-tail student t-test was performed for all panels except for B. All numerical data are represented as mean ± SEM. *p<0.05; **p<0.01; ns, not significant.
To confirm our IHC results and ask if senCAFs were depleted upon AP treatment, we utilized our CAF antibody panel and analyzed PyMT tumors by flow cytometry. To assess the different CAF populations, PyMT tumors were isolated from 7-week-old INK+ and INK− mice and subjected to dissociation. Dissociated tumors were then stained according to the CAF panel described previously (Fig. S4A). We found that while the total number of myCAFs in the tumors was sustained (Fig. 5E), AP administration led to a significant reduction in the percentage of senCAFs in INK+ mice (Fig. 5F). In addition, we also noted that tumors from INK+ mice had fewer vCAFs (Fig. S5C); however, since vCAFs were not actively expressing p16 (Fig. 4D and S3A), the reduction was unlikely to be directly caused by INK suicide gene activation.
The survival of senescent cells largely relies on the activity of BCL-2 and related proteins, which are key modulators of apoptosis (40,43). Because our scRNA-seq analysis suggested that senCAFs expressed higher levels of BCL-W, which is a member of BCL-2 protein family, we next asked whether senCAFs could be pharmacologically depleted with the senolytic ABT737. ABT737 is a small molecule inhibitor of the BCL-2 family of proteins and has been reported to eliminate senescent cells effectively, including those in premalignant lesions (40,44). Therefore, we set out to determine if ABT737 could eliminate senCAFs and impact tumorigenesis (Fig. 5G). Similar to our findings with the INK transgenic mouse model, ABT737 treatment was sufficient to restrict tumor development in PyMT mice (Fig. 5H). IHC staining of p16 on the MFP tumors suggested that senCAFs were targeted by ABT737 (Fig. S5D–E). Moreover, we also investigated the CAF composition of the tumors via flow cytometry and confirmed that senCAFs were depleted upon ABT737 treatment (Fig. 5I–J). We noticed that tumors from ABT737 treated mice had fewer iCAFs (Fig. S5F); nevertheless, since iCAFs were negative for p16 (Fig. 4A and D), the reduction was likely not directly caused by the inhibition of BCL-2 family proteins. Together, our data illustrate that senCAFs support tumorigenesis and that they are susceptible to pharmacological therapy.
Elimination of senCAFs correlates with changes in tumor-infiltrating lymphocytes
SASP factors consist of soluble mitogens, cytokines, chemokines, and numerous ECM proteins, which can promote tumor growth by modulating the scaffolding of tissues (17,45). Among the list of SASP proteins expressed by senCAFs, inflammatory factors drew our attention. Extensive studies have shown that senescent cells modulate the recruitment and functions of various immune cells including T cells (14,46), which serve as important targets of current cancer immunotherapies (47). Indeed, DGE analyses indicated that senCAFs of both human and murine origin expressed elevated levels of inflammatory SASP factors, such as RANKL, whose overexpression can result in increases of immunosuppressive regulatory T cells (Tregs) through dendritic cell (DC) modulation (30), and TGF-β, a factor with extensive immunosuppressive effects on not only T cells, but also DCs, macrophages, neutrophils, and NK cells (33–35). Given the expression of these factors, we set out to evaluate whether the depletion of senCAFs, using either the INK GEMM or pharmacological tools, would induce any changes in the landscape of tumor-infiltrating lymphocytes, which are front line effectors in the killing of tumor cells.
We collected tumor samples from 7-week-old AP treated INK- and INK+ mice and from 7-week-old vehicle (Veh) or ABT737 treated mice, processed the samples for scRNA-seq, and conducted informatics analyses focusing on the CAF populations (Fig. S6A–B). Re-clustering of the non-immune stromal clusters of the datasets confirmed the presence of iCAFs, vCAFs, senCAFs, and myCAFs (Fig. S6C–D). We confirmed that senCAFs indeed expressed the highest level of p16 and NCAM among all CAFs (Fig. S6C–D). Next, we looked into the tumor-infiltrating T cell and NK cell populations (Fig. S6E–F). Interestingly, tumors from both AP treated INK+ mice and ABT737 treated mice harbored lower numbers of Tregs (Fig. S6G–H). Moreover, our examinations of functionally important genes revealed that Tregs from INK+ and ABT737 groups showed lower levels of CD25 (coded by Ir2ra) and FOXP3 expression (Fig. 6A–B), both of which are important for the differentiation and proliferation of Tregs and for maintaining their suppressor function (48,49). In addition, we found that tumor-infiltrating NK cells from INK+ and ABT737 treated mice expressed higher levels of CD11b (coded by Itgam) (Fig 6A–B), which marks more mature NKs (50,51), and FasL (Fig. 6A) or perforin (coded by Prf1) (Fig. 6B), which are key mediators of NK cells’ cytotoxicity (52). Together, our analyses indicate that the elimination of senCAFs led to a more robust anti-tumoral immune microenvironment.
Figure 6. Senescent CAFs alter NK cells activation status and infiltration to increase tumor formation.
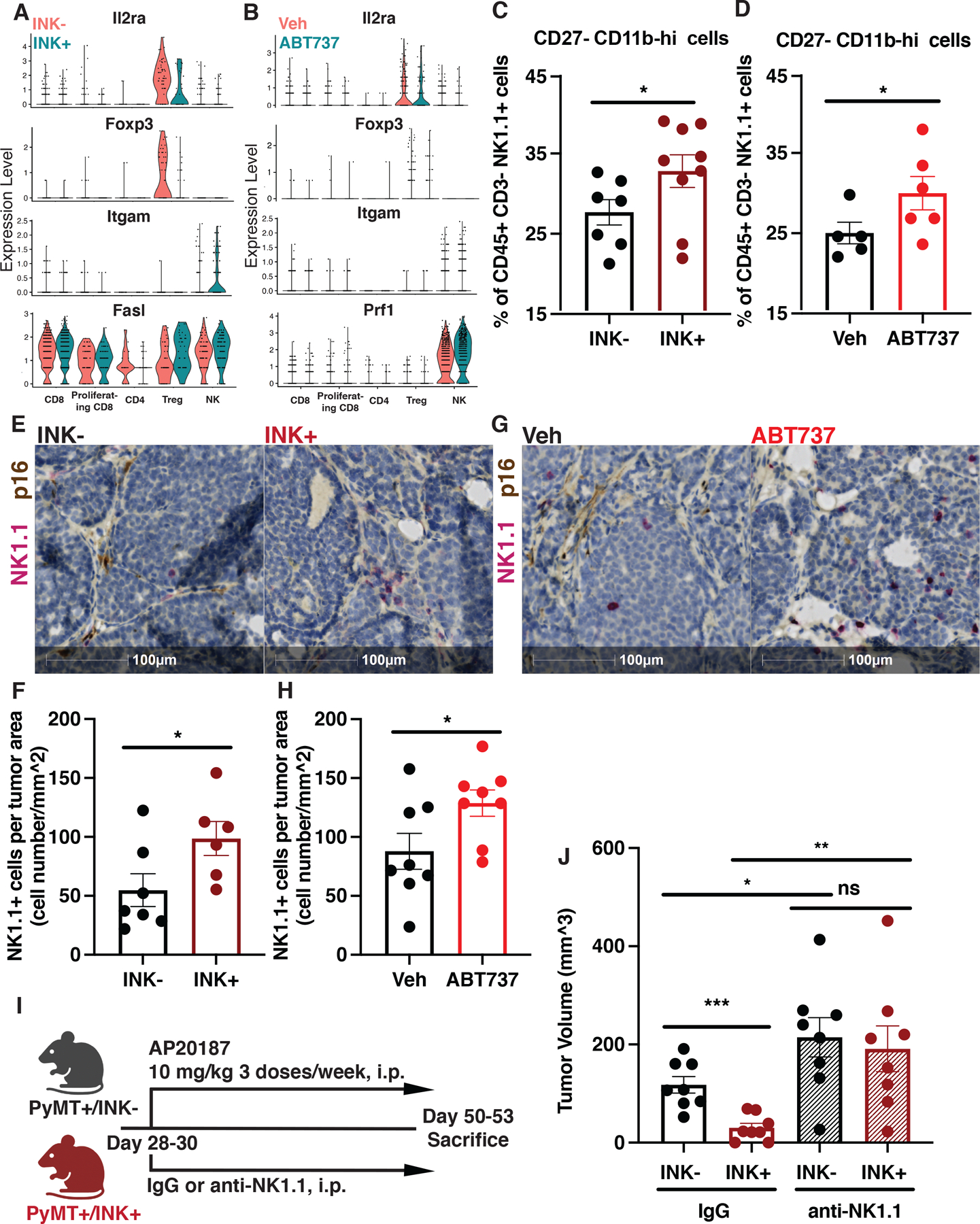
(A) Violin plot of indicated gene expression in different immune populations from 7-week-old AP treated MMTV-PyMT INK- and INK+ mice. (B) Violin plot of indicated gene expression in different immune populations from 7-week-old MMTV-PyMT mice treated with Veh or ABT737. (C) Flow cytometric quantification of intra-tumoral CD27-CD11b-hi NK cells in INK− (n=7) and INK+ (n=9) mice treated with AP. (D) Flow cytometric quantification of intra-tumoral CD27-CD11b-hi NK cells in MMTV-PyMT mice treated with Veh (n=5) or ABT737(n=6). (E) Representative IHC image for NK cells (NK1.1, red) and p16 (brown) in tumor sections from 7-week-old INK− (n=7) and INK+ (n=6) mice treated with AP. (F) Quantification of NK cells per tumor area from same mice shown in E. (G) Representative IHC image for NK cells (NK1.1, red) and p16 (brown) in tumor sections from 7-week-old MMTV-PyMT mice treated with Vehicle (Veh, n=8) or ABT737 (n=8). (H) Quantification of NK cells per tumor area from same mice shown in G. (I) NK cell depletion strategy in MMTV-PyMT (INK-) and MMTV-PyMTxINKATTAC+ (INK+) mice treated with AP. First dose of anti-NK1.1 or IgG isotype control antibody was administrated at 500 ug/mouse. The remaining doses were administrated every 4 days at 250 ug/mouse. (J) Tumor volume measured with calipers at week 7 for INK− versus INK+ mice depleted of NK cells (anti-NK1.1) or treated with control IgG, n=8. Unpaired one-tailed student t-test was performed for all statistical analyses shown in this figure. All numerical data are represented as mean ± SEM. *p<0.05; **p<0.01; ***p<0.001; ns, not significant.
senCAFs inhibit natural killer (NK) cells to promote tumor progression
To validate our scRNA-seq findings, we turned to flow cytometry to evaluate whether the depletion of senCAFs in PyMT mice would lead to any alterations in the status of tumor infiltrating T cells or innate immune cells with the capacity of modulating T cell activity, i.e. DCs, macrophages, granulocytes, and monocytes. Overall, MFP tumors from 7-week-old INK+ mice had already developed an antitumoral immune microenvironment compared to those from INK− mice. Specifically, compared to their INK− litter mates, MFP tumors collected from INK+ mice had fewer FOXP3+ CD4+ Tregs, fewer PD1+ Ki67− exhausted CD8 T cells, fewer CD11b+ Ly6C− Ly6G+ immature granulocytes, and more XCRI+ conventional type 1 DCs (cDC1) (Fig. S7A–B). In accordance with the genetic model, senCAF elimination with ABT737 also correlated with decreases in tumor-infiltrating immunosuppressive or immature immune cells, including FOXP3+ CD4+ Tregs, CD62L+ CD44− naïve CD8 T cells, and CD11b+ Ly6C-hi immature monocytes (Fig. S7C–D). To investigate whether T cells and the aforementioned T cell-related immune populations were directly modulated by senCAFs, we combined anti-CD4 and CD8 antibodies into the regimen of the PyMT/INK-AP system (Fig. S7E) to determine whether the presence of T cells was critical to senCAFs’ tumor promoting effect. Surprisingly, neither INK− nor INK+ mice exhibited significant changes in tumor burden upon the depletion of T cells (Fig. S7F), indicating that T cells were not responsible for the reduced tumor burdens that we observed in INK+ tumors. Gating strategies for the aforementioned flow cytometry panels can be found in Fig. S8A–C.
NK cells are emerging candidates for anticancer therapy. Interest in NK cells is due to their potent cytotoxicity against tumor cells, limited reactivity to healthy tissues, and the lack of a T cell receptor, which reduces graft-versus-host-disease (53). Because we failed to find a role for T cells in our model, we next analyzed the status of tumor-infiltrating NK cells via flow cytometry. Using transgenic GEMMs, we found that the elimination of senCAFs in INK+ mice led to no obvious increase in the quantity of NK cells within mammary tumors (Fig. S9A), but we observed a noticeable improvement in the maturation of the population. Specifically, we found that tumors from INK+ mice contained more CD27− CD11b-hi NK cells, which are the most mature and cytotoxic population of NK cells (Fig. 6C) (51,54). Additionally, we recapitulated this shift in NK cell maturation status using ABT737 (Fig. 6D and S9B). We observed no such changes in maturation status within the splenic NK cell population (Fig. S9 C–E), arguing that the reduced tumor burden that we observed upon senCAF elimination was a result of intra-tumoral interactions between senCAFs and NK cells rather than systematic effects.
Although we found no significant changes in the total number of NK cells in mammary tumors by flow cytometry (Fig. S9A), it remained possible that senCAFs impacted the localization of NK cells within the tissues. Therefore, we performed IHC staining of p16 and NK1.1 on tissues collected from treated PyMT mice. Quantification revealed that the depletion of senCAFs, using either INK+ mice or the senolytic ABT737, significantly increased NK cell infiltration into tumor epithelial cell nests (Fig. 6E–H and Fig. S3B). Co-staining with NK1.1 and EPCAM, to identify tumor cells revealed that NK cells were indeed located near tumor nests (Fig. S9F–I). To ask if senCAFs facilitated tumorigenesis via NK cell modulation, we administrated IgG control or anti-NK1.1 antibody to the INK- and INK+ mice treated with AP (Fig. 6I). Strikingly, INK- and INK+ mice depleted of NK cells showed no difference in tumor volume by 7 weeks of age (Fig. 6J), indicating that senCAFs exerted their tumor-supporting effect by hampering NK cell function. In summary, our data indicate that senCAFs limit tumor growth by compromising the maturation status of tumor-infiltrating NK cells and restraining NK cell penetration into tumors.
SenCAFs inhibit NK cell cytotoxicity through ECM deposition
To determine how senCAFs impact NK cells, we asked whether the two populations were in physical contact within tumors. Indeed, IHC staining indicated that a significant number of NK cells were in close proximity to p16+ stromal cells (Fig. 7A), suggesting that NK cells could be educated by senCAFs. Given the proximity of senCAFs and NK cells, we next asked which SASP factors might impact NK cells activity. SenCAF was the only depleted population using both INK model and ABT737 treatments while myCAF numbers were sustained (Fig. 5E–F and I–J); hence, we reasoned that factors differentially secretly between senCAFs and myCAFs led to modulated NK cell function. We thus revisited our RNA-seq analysis of senCAFs versus myCAFs. Top terms from our GSEA analysis suggested that, compared to myCAFs, senCAFs expressed increased collagen and enzymes that modify collagen fibers (Fig. 7B and S10A). To ask if the collagen was altered by the presence of senCAFs, we carried out second generation harmonic (SHG) imaging microscopy of PyMT tumor sections. Our SHG analyses revealed that areas surrounding senCAFs were surrounded by thicker collagen fibers compared to areas 50 um away from senCAFs (Fig. 7C). In addition, by performing CellChat analysis to profile the cell-cell communication between CAFs and tumor-infiltrating lymphocytes (55), we captured a prominent interaction between senCAFs and NK cells—specifically, the strongest collagen-related signaling originated from senCAFs and was primarily received by NK cells (Fig. 7D).
Figure 7. Senescent CAF-derived extracellular matrix limits NK tumor cell killing.
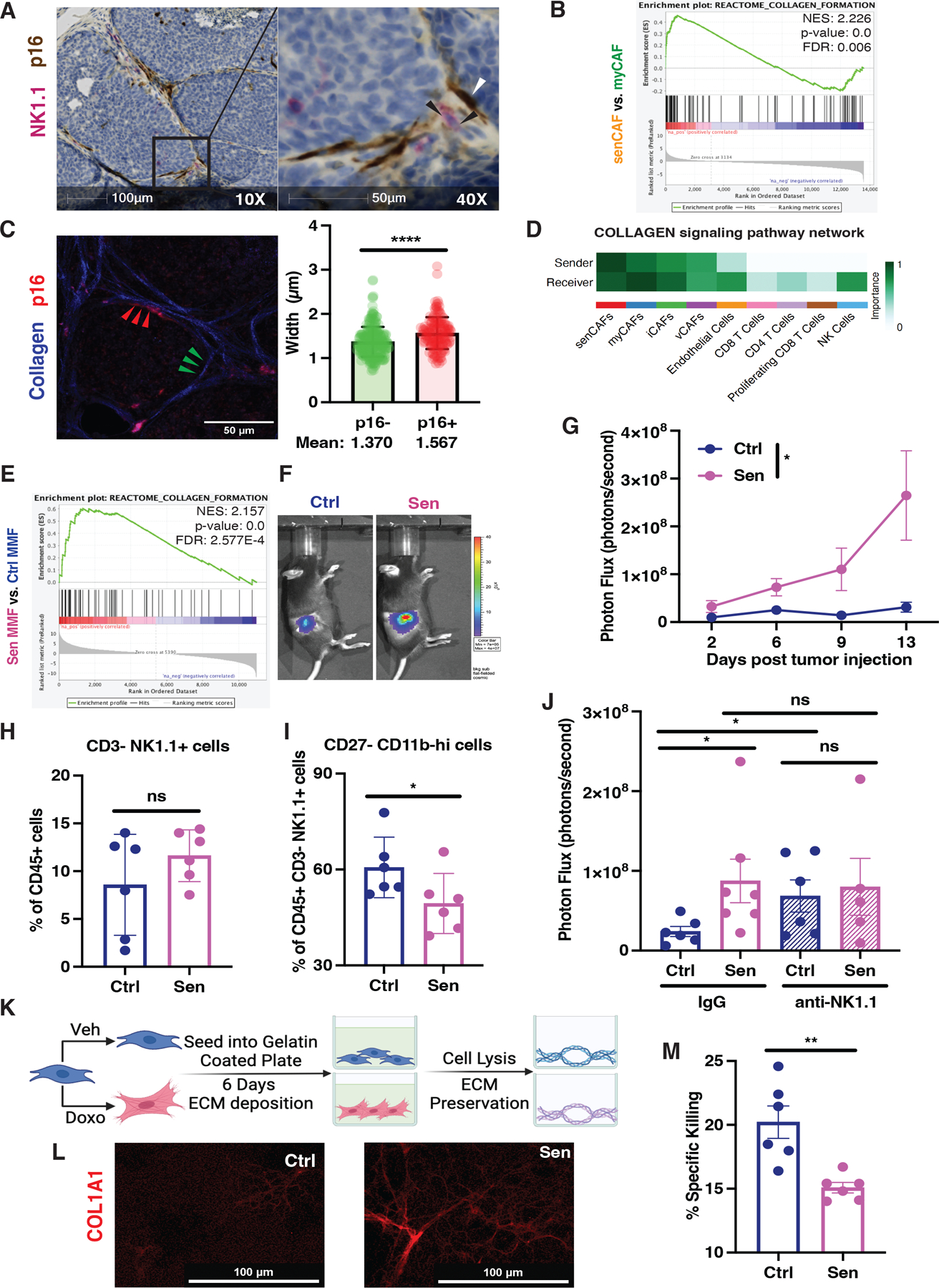
(A) Representative IHC image for NK cells (NK1.1, red) and p16 (brown) in tumor sections from 7 weeks old PyMT mice at 10X (left) and 40X (right) shows a close association between senCAFs and NK cells. (B) Gene set enrichment analyses of the senCAF population compared to the myCAF population showed enrichment of the collagen formation signature. NES, p-value, and FDR are shown in the plot. (C) Left: Representative image of Collagen (blue) and p16+ stromal cells (red). Collagen fibers imaged via second harmonic generation microscopy and analyzed via CT-FIRE/Curve align software. Fibers within 8μm of red p16+ stroma defined as p16+ (red arrowheads) and fibers at least 60μm away from p16+ stroma defined as p16- (green arrowheads). Right: Quantification of collagen fibers near p16- and p16+ stroma. Statistical difference determined via student t-test (n=3 mice, 3 FOV/mouse, unpaired student t-test, data are represented as mean ± SEM., ****p< 0.0001). (D) Cell chat analyses of collagen dialog between the different CAF and immune cell populations in tumors from 7-week-old PyMT mice. (E) Gene set enrichment analyses of the doxorubicin treated (i.e., senescent) compared to vehicle treated (i.e., control) fibroblasts also showed enrichment of collagen formation. NES, p-value, and FDR are shown. (F) 200,000 Py230 breast cancer cells labeled with luciferase were co-injected with 100,000 Ctrl or Sen fibroblasts and tumor growth was monitored by live animal bioluminescence imaging (BLI). Representative BLI of day 13 post tumor injection is shown. (G) Tumor growth was assessed by BLI on indicated time points (2-way ANOVA was performed to compare the tumor growth; data are represented as mean ± SEM. *p<0.05). (H) Quantification of CD3− NK1.1+ cells in Py230 tumors co-implanted with Ctrl (n=6) or Sen (n=6) fibroblasts on day 10 post tumor injection. (I) Quantification of CD27− CD11b-hi NK cells in Py230 tumors co-implanted with Ctrl (n=6) or Sen (n=6) fibroblasts on day 10 post tumor injection. (J) Tumor volume quantified with BLI for mice receiving Py230 tumor cells co-injected with Ctrl versus Sen fibroblasts under NK cell depletion treatment (day 13 post tumor inoculation). Py230-Ctrl fibroblasts group on IgG treatment, n=6; Py230-Sen fibroblasts group on IgG treatment, n=7; Py230-Ctrl fibroblasts group on anti-NK1.1 treatment, n=6; Py230-Sen fibroblasts group on anti-NK1.1 treatment, n=5. (K) Schematic of collagen deposition protocol. (L) Representative immunofluorescence of collagen 1A1 following the procedure outlined in K. (M) NK cell killing of tumor cells when plated on ECMs from Ctrl (n=6) or Sen (n=6) fibroblasts. Unpaired one-tailed student t-test was performed for all statistical analyses shown in this figure unless otherwise specified. All numerical data are represented as mean ± SEM. *p<0.05; **p<0.01; ns, not significant.
The ECM, an indispensable component of the tumor microenvironment, not only modulates the migration but can also regulate the function of immune cells. NK cells express various integrin- and non-integrin-based receptors for ECM proteins, some of which can directly inhibit the cytotoxicity of NK cells once they engage with their ligands (56–61). To directly assess senCAFs’ ECM deposition capacity, we turned to an in vitro system to model the interaction between senCAF produced ECM and NK cells. Previously, we established a mouse mammary fibroblast (MMF) cell line from the MFP of a heathy C57BL/6J mouse. The MMFs were treated with doxorubicin (Doxo) for 24 hours to induce senescence. RNA-seq analysis of the control (Ctrl) and senescent (Sen) MMFs showed that, similar to senCAFs, the Sen MMFs showed an enrichment in Fridman senescence-associated genes (Fig. S10B) and elevated expression of genes involved in collagen assembly (Fig7E and S10C). Indeed, in accordance with what we observed when comparing senCAFs to myCAFs (Fig S10D), Sen MMFs exhibited higher levels of Cdkn1a and reduced expression of cell cycle related genes including Hmgb1 and Mcms (Fig. S10E). In addition, a closer interrogation of our Sen MMFs’ gene expression profile revealed that they showed elevated expression of similar ECM proteins, including a plethora of collagens, as those found in senCAFs (Fig. S10D and E). When we co-implanted MMFs with Py230 or EO771 murine mammary tumor cells into the inguinal fat pads of mice, Sen MMFs displayed a robust tumor-promoting effect compared to Ctrl MMFs (Fig. 7F–G and S10F–G). Furthermore, our assessment of NK cells infiltrating into the implanted tumors revealed that, similar to what we observed in the spontaneous PyMT GEMM model, the presence of Sen MMFs led to a reduced percentage of CD27− CD27-hi NK cells (Fig. 7H–I). Most importantly, in agreement with our genetic model, Sen MMFs lost their tumor-promoting ability upon NK cell depletion (Fig. 7J). Together, our data indicate that senCAFs and Sen MMFs promote tumor development via modulating NK cells.
To determine the impact of MMF-secreted matrices on NK cell function, we seeded Ctrl or Sen MMFs into pre-coated 96-well culture plates and allowed them to deposit ECM for 6 days. Next, we removed the cells using mild detergent and performed thorough washing with PBS to preserve the assembled ECM (Fig. 7K). Immunofluorescent (IF) staining of the MMF-lysed wells showed that Sen MMFs deposited more COL1A1 than Ctrl MMFs (Fig. 7L). To examine the interaction between the MMF-derived ECM and NK cells, we executed ex vivo NK cell cytotoxicity assays using the ECM-containing wells. Briefly, splenic NK cells enriched from Poly I:C stimulated C57BL/6J mice were first allowed to interact with ECM derived from either Ctrl or Sen MMFs for 2 hours. RMA-S, a well-documented MHC-I-deficient target cell line, was then labeled with a violet cell trace dye (CTV) and incubated with NK cells in the presence of ECM for 16 hours. Strikingly, we found that Sen MMF-assembled ECM significantly impeded the killing capacity of NK cells (Fig. 7M), suggesting that ECM deposited by senCAFs directly suppresses NK cytotoxicity against tumor cells. Taken together, our data demonstrate that senCAFs promote tumorigenesis by producing and modulating ECM that negatively impacts NK cell killing capacity.
Discussion
The intriguingly strong correlation between senCAFs in DCIS and breast cancer recurrence implied that they play an important role in breast cancer tumorigenesis (25). Using both genetic and pharmacologic approaches, we show that the elimination of senCAFs significantly delays the onset of mammary tumors. Further, we show that the interaction between senCAFs and tumor-infiltrating NK cells not only hampers the infiltration of NK cells to the tumor epithelial cell nests, but also directly suppresses NK cells’ cytotoxicity against tumor cells. Our data suggest that ECM proteins secreted by senCAFs play critical roles in the tumor-promoting effect of the CAFs, which ultimately leads to compromised tumor immune surveillance.
To our knowledge, the impact of CAF-derived ECM on NK cell cytotoxicity has not been extensively investigated in breast cancer models. Interestingly, previous publications focusing on the plasticity of CAFs and NK cell maturation status have suggested the implicit interplay between the two populations within the tumor microenvironment. For example, TGF-β can impact the differentiation and maintenance of myCAF populations in pancreatic ductal adenocarcinoma models (10,11). Meanwhile, intra-tumoral administration of TGF-β neutralizing antibodies and interlukin-12 in PyMT mice results in enriched numbers of tumor-infiltrating CD27− CD11b-hi NK cells (54). More recent literature in the skin revealed that collagens and elastin in the stroma inhibit NK cell cytotoxicity; further, in agreement with our findings, stromal-derived ECM molecules can modulate NK cell signaling once they exit the circulation and contact the stroma (60).
Our quantification of tumor burden suggested that the effect of senCAFs was most prominent during the early tumorigenesis stage, as there was a significant delay in tumor onset. Moreover, our NK-depletion assay combined with the PyMT/INK model and MMF-tumor co-injection model highlighted that senCAFs’ tumor-promoting effect was mediated through NK cells. In parallel with our findings, Buqúe et.al. reported that NK cell depletion in a mammary tumor model driven by slow-release of medroxyprogesterone acetate combined with oral administration of the carcinogen 7,12-dimethylbenz[a]anthracene results in disadvantages in overall survival, tumor-free survival, but not time from disease detection to death (62). This finding suggests that the anti-tumor function of NK cells is primarily exerted during the establishment of mammary tumors, which concurs with our observations and supports the notion that senCAFs lead to compromised NK cell function in tumor immune surveillance during the tumor initiation stage. More importantly, such a notion provides an explanation for why the presence of senCAFs correlates with breast cancer recurrence (25).
Besides the impact of senCAFs on NK cell cytotoxicity, our data also showed that the elimination of senCAFs correlated with lower numbers of tumor infiltrating Tregs and more active CD8+ T cell status. Although we saw no significant changes in the tumor burden of PyMT mice upon the joint depletion of CD4+ and CD8+ T cells, which suggested that senCAFs did not contribute to tumorigenesis via direct modulation of T cells in the context of our PyMT/INK model, it remains possible that senCAFs drive increases in Tregs and impact T cell function. A recent study reported by Krishnamurty et.al. highlighted that, in mouse models of pancreatic cancer, the effector function of intra-tumoral CD8 T cells can be directly limited by a myofibroblastic CAF population that expresses high levels of LRRC15 (10). Interestingly, we found that senCAFs from PyMT tumors also showed elevated Lrrc15 expression (Fig S10D), suggesting that senCAFs may have an effect on T cell functions in other systems. Unfortunately, the PyMT model is poorly antigenic and thus the GEMM is a poor model for studying adaptive immune responses. Indeed, data reported from DeNardo et.al. showed that the loss of T cells had no impact on primary tumor development in PyMT mice (63). Thus, to investigate the impact of senCAFs on T cells, a mouse model that sustains tumor antigenicity, such as the doxycycline-inducible PyMT model or the MMTV-PyMT-ovalbumin model, should be introduced (64,65). On the other hand, as T cells also express receptors that lead to inhibitory signaling upon the binding with ECM proteins (58,59), it is possible that, should senCAFs play any role in suppressing the function of T cells, the effect could be mediated through ECM deposition as well. More importantly, Dominguez et.al. have shown that LRRC15+ myofibroblastic CAFs can be identified in numerous types of human cancers including breast cancer and that the signature of LRRC15+ CAFs predicts poor response to anti-PD-L1 therapy across several individual cancer types. This evidence raises the possibility that targeting senCAFs could potentiate the efficacy of T cell-based immunotherapy (66).
Senescence is thought to arise in cells in response to tissue and/or systemic level stress (15). If true, we would have expected to find evidence of senescence in all cell types including the various CAF subtypes. However, our extensive interrogation of human and murine breast tumors revealed the senCAFs are largely restricted to the myCAF subtype. This finding raises the intriguing possibility that rather than being a general response to stress, senescence is a specific program that is activated in response to stress. Identifying how this program is activated and how it differs across tissues will reveal why senescence is restricted to myCAFs and is an important area of future investigation.
Our work has identified a senescent CAF population that directly limits NK cell killing and promotes tumorigenesis. Given these cells are present in human DCIS that recur and human breast cancer samples of ER+, HER2+, and TNBC subtypes, our work suggests that targeting senescent CAFs with senolytics may benefit breast cancer patients. While we found that breast senCAFs promote tumor growth by limiting NK cell activity, in parallel with our findings, Belle et.al. find a senescent myofibroblastic CAF population in pancreatic cancer that contributes to tumor progression by modulating macrophages and dendritic cells, which impact T cell immunity. Although the mechanisms may vary across tissues, senescent CAFs display immunosuppressive and tumor-promoting effects across different types of cancer.
Methods and Materials
Animal studies
All mice were housed and all animal procedures were approved by Washington University’s Institutional Animal Care and Use Committee (IACUC).
Immunohistochemistry)/immunofluorescent staining and quantification
Specific information on vendor, catalog number, and dilution ratio of any antibody used for IHC or IF staining is listed in Supplementary Table 1.
Harvested tumors were fixed in 10% normal buffer formalin at room temperature for 24–48 hours, washed in phosphate buffered saline (PBS), and preserved in 70% ethanol at 4 Celsius until being processed into formalin-fixed paraffin-embedded (FFPE) blocks. FFPE sections of 5–7 μm thickness were stained on a Leica BOND Automated IHC/ISH Stainer. For p16 staining, mouse tumor sections were first stained with anti-CDKN2A (pH 6.0 antigen retrieval) in combination with rabbit anti-rat antibody using the BOND Polymer Refine Detection System. For p16-NK1.1 dual staining, mouse tumor sections were first stained with anti-CDKN2A as described above and then stained with anti-NK1.1, which was then developed using the BOND Polymer Refine Red Detection System. Slides were mounted and scanned using a Zeiss Axio Scan.Z1 slide scanner, and the resulting images were analyzed using Indica Lab’s HALO computational pathology software. For NK cell infiltration analyses, tumor and stromal areas were identified and measured by the tissue classifier tool using Indica Lab’s Halo software. Then, NK1.1+ cells were identified and counted using the multiplex IHC module using the same software.
For NK1.1-EPCAM dual staining, mouse tumor sections were first stained with anti-NK1.1(pH 6.0 antigen retrieval) and developed using the BOND Polymer Refine Detection System and then stained with anti-EPCAM, which was then developed using the BOND Polymer Refine Red Detection System. Slides were mounted and scanned using a Zeiss Axio Scan.Z1 slide scanner, and the resulting images were analyzed using Indica Lab’s HALO computational pathology software. For NK cell infiltration analyses, tumor and stromal areas were identified and measured by the tissue classifier tool using Indica Lab’s Halo software. Then, NK1.1+ cells were identified and counted using the multiplex IHC module using the same software.
mIHC staining on human or mouse tissue sections were performed on a Leica BOND Automated IHC/ISH Stainer using the BOND Polymer Refine Detection System in combination with goat anti-rabbit Fab fragment, rabbit anti-rat antibody, 3-Amino-9-ethylcarbazole (AEC) chromogenic substrate (Abcam ab64252) and hematoxylin (DAKO S3301). Human DCIS tissue sections were first stained with anti-CDKN2A antibody (pH 6.0 antigen retrieval). After the staining was finished, slides were mounted with aqueous mounting media (Vector H-5501) and scanned. The slides were then submerged in TBS-T buffer at 4° Celsius overnight to remove the coverslip. After 3 washes in diH2O, the slides were incubated in 50% ethanol for 5 minutes and then transferred to acidified (1% HCl) 70% ethanol and incubated for 10 minutes with agitation to remove AEC chromogen and hematoxylin. The slides were then incubated with 100% ethanol for 15 minutes with agitation and was rehydrated and stored in TBS-T at 4° Celsius until next staining cycle. Such staining-scanning-stripping cycle was repeated for each of the following markers in the listed order: αSMA (pH 6.0 antigen retrieval), PDGFRα (pH 8.0 antigen retrieval), and pan-Cytokeratin (pH 8.0 antigen retrieval). Human breast tumor tissue array sections were processed using the staining-scanning-stripping procedures as described above and stained for the following markers in the listed order: CDKN2A (pH 6.0 antigen retrieval), αSMA (pH 6.0 antigen retrieval), COL14A1 (pH 6.0 antigen retrieval), PDGFRα (pH 8.0 antigen retrieval), and pan-Cytokeratin (pH 8.0 antigen retrieval). Mouse tumor tissues were processed using the staining-scanning-stripping procedures as described above and stained for the following markers in the listed order to probe CAF subpopulations: Ly6C1 (pH 6.0 antigen retrieval), CDKN2A (pH 6.0 antigen retrieval), NCAM (pH 6.0 antigen retrieval), CDKN2A (pH 6.0 antigen retrieval), αSMA (pH 6.0 antigen retrieval), MCAM (pH 6.0 antigen retrieval), and PDGFRα (pH 8.0 antigen retrieval). Mouse tumor tissues were processed using the staining-scanning-stripping procedures as described above and stained for the following markers in the listed order to probe p16 and Ki67: CDKN2A (pH 6.0 antigen retrieval) and then Ki67 (pH 8.0 antigen retrieval). All scanned bright field images for mIHC staining were deconvoluted into pseudo-fluorescent images and merged using Indica Lab’s HALO computational pathology software.
IF staining on in vitro assembled ECMs was conducted as described below. Preserved ECM was first fixed using 4% paraformaldehyde at room temperature for 10 minutes and washed twice with PBS. ECM was then blocked with a blocking solution (5% normal goat serum and 2.5% bovine serum albumin in PBS) for 30 minutes and stained using anti-COL1A1 for 1 hour at room temperature. PBS was used to wash the ECM twice prior to the addition of an Alexa Fluor 594-conjugated secondary antibody. The ECM was incubated with the secondary antibody for 30 minutes at room temperature in the dark and then washed with PBS twice. The stained ECM was finally imaged using an inverted fluorescent microscope.
Second-harmonic imaging microscopy and data analysis
Tumor sections were first stained on a Leica BOND Automated IHC/ISH Stainer using the BOND Polymer Refine Red Detection System. Slides were unloaded from the BOND after the incubation of alkaline phosphatase-linked compact polymer and the Fast-Red signals were developed using a Vulcan Fast Red kit (Biocare Medical, Vulcan Fast Red Chromogen Kit 2) to obtain signals that can emit fluorescence in Texas Red channel.
SHG was performed on the prepared samples. SHG images were obtained on a Nikon A1RHD25 MP two-photon confocal microscope with a 40x objective. SHG images were obtained simultaneously with detectors equivalent to Texas Red to detect p16+ cells and images were overlapped. Collagen analysis was performed using CT-Fire analysis software (67). Collagen fibers across three FOV per tumor slice (x3 mice) were quantified for width (μm). To identify the effect of p16+ cells on local fibers, fibers within 8μm of red p16+ cells were quantified. Individual fibroblasts have been shown to communicate with each other by deforming collagen as far as 50μm (68–70). To ensure fibers are minimally affected by p16+ cells, fibers at least 60μm away from p16+ cells were quantified.
In vivo drug treatment, tumor measurement, and flow cytometry
The FVB PyMT mouse line was purchased from Jackson Laboratory. The C57BL/6J INK mouse line was a kind gift from Dr. Darren Baker and was backcrossed to FVB background.
PyMT+/INK- and PyMT+/INK+ mice were weaned by 4 weeks of age and recruited to studies. AP20187 (ApexBio, B1274) was delivered via intra peritoneal injections at a dosage of 10 mg/kg, 3 times per week. For ABT737 studies, PyMT+ mice were weaned and recruited by 4 weeks of age to be randomly assigned to treatment groups. ABT737 (ApexBio, A8193) or corresponding vehicle solution was delivered via intra peritoneal injections at a dosage of 25 mg/kg, once a week. Both AP20187 and ABT737/vehicle treated mice received the last drug injection within 24 hours prior to sacrifice. For any mice recruited for immune cell depletion assays, monoclonal antibodies (Supplementary Table 1) were first administrated at 500 ug/mouse via intra peritoneal injections. Following the first dose, antibodies were then administrated at 250 ug/mouse every four days. Mice recruited for all immune cell depletion assays were randomized before the assignment of antibody treatment groups.
All PyMT mice recruited to tumor studies were monitored for tumor burden. Briefly, mammary fat pad palpation and caliper measurement was performed every week under isoflurane anesthesia. Once a tumor reached measurable size (width and length both reached 3.5 mm), the measurements were recorded for volume calculation following the equation: Volume = Length ∙ Width2/2. For each mouse, tumor volume was reported as the sum of the volumes of all ten mammary glands. Because we are studying breast cancer we have restricted our analyses to females. The genotype of all mice were blinded during tumor assessment.
Mice were perfused with PBS-heparin solution upon sacrifice and tissue collection. Tumors were collected from the thoracic mammary glands of the mice and digested in DMEM F-12 media (Gibco 11320033) containing 2 mg/mL collagenase and 2 U/mL DNase for 25 minutes at 37° Celsius. Dissociated tissue suspension was then filtered through a 70 μm strainer and centrifuged at 1500 round per minute (rpm) for 5 minutes. Cell pellets were then washed using FACS buffer (1% bovine serum albumin, 1 mM EDTA, 0.05% sodium azide in PBS) and incubated with Fc receptor blocking antibody for 10 minutes on ice. Cells were stained according to the panels of interested populations (Fig. S7 and S8; Supplementary Table 1) for 30 minutes on ice. Stained samples were assayed on a BD LSRFortessa X20 flow cytometer (T cell panel, DC/myeloid panel, and CAF panel) or a Cytek Aurora 4L 16V-14B-10YG-BR full spectrum cytometer (NK panel). Collected flow cytometry data were analyzed using FlowJo Software.
Tumor sample preparation for sequencing and subsequent data processing
Tumor samples were processed and stained for FACS as described above (Supplementary Table 1). For CAF bulk RNA-seq, tumors were collected from 3 10-week-old PyMT mice. Tumor samples stained according to the CAF panel (Fig. S4A) and were sorted on a BD FACS AriaII 4 lasers cell sorter. About 5,000–10,000 cells were sorted for each CAF population and collected into Eppendorf tubes containing 2-Mecaptoethanol supplemented cell lysis buffer (Qiagen RNeasy Micro Kit 74004). RNA was then purified following the instructions of Qiagen RNeasy Micro Kit and submitted for sequencing at Genome Technology Access Center, where all samples were prepared according to library kit manufacturer’s protocol, indexed, pooled, and sequenced on an Illumina NovaSeq 6000. Count-per-million matrices generated post sequencing and were then normalized and filtered in Partek Bioinformatics software. DGE analyses were performed using Partek’s GSA function, and the resulted gene lists were pre-ranked and submitted for GSEA analyses based on p-values and log2 fold change values.
For scRNA-seq analysis of tumors from 7-week-old PyMT mice, 2 biological replicates were pooled for each treatment group. Dissociated tumors were then stained for CD45. Stained samples were sorted on a Sony Synergy 5 laser, 22 colors cell sorter to separate CD45+ and CD45− cell populations. The collected samples were processed for library generation following the instructions of 10x Genomics Chromium X instrument (10x Genomic, Pleasanton CA). The quality of the prepared libraries was assessed and submitted for sequencing at the Genome Technology Access Center. Output from Cell Ranger Software was imported into R Studio and analyzed using Seurat (71). For each Seurat object, genes expressed by less than 3 cells and cells expressing less than 1,000 or more than 8,000 genes were excluded. Cells with higher than 15% mitochondrial RNA content and cells with less than 100 or more than 250,000 counts were excluded as well. SCTransform was performed to normalize the expression matrix (72). PCA and UMAP dimensional reduction was performed using the first 25 PCA components. We then conducted FindNeighbors and FindClusters functions to cluster cells. The FindAllMarkers function was used to identify signature genes for each cluster. Lymphocytes populations including T cell clusters and NK cell clusters were re-clustered for in-depth analysis. CellChat analysis was performed to investigate the cell-cell communication between the lymphocytes and non-immune stromal cells including endothelial cells (Pecam1+) and CAF subpopulations (Krt8/Krt18/Krt14/Krt17/Epcam-, Pdgfrb+) (55).
For scRNA-seq analysis of tumors from 12-week-old PyMT mice, 2 biological replicates were included for the analysis. Dissociated tumors were sorted to obtain a live cell-enriched single cell suspension. The collected samples were processed for library generation following the instructions of 10x Genomics Chromium X instrument (10x Genomic, Pleasanton CA). Output from Cell Ranger Software was imported to R Studio and analyzed using Seurat (71). For each Seurat object, genes expressed by less than 3 cells and cells expressing less than 200 or more than 6000 genes were excluded. Cells with higher than 10% mitochondrial RNA content and cells with less than 100 or more than 20,000 counts were excluded as well. Log-based normalization (NormalizeData function) was performed to normalize the expression matrix, FindVariableFeatures and ScaleData arguments were then applied to the dataset. PCA and adaptively-threshold low-rank approximation (ALRA)1 was conducted as well (28). UMAP dimensional reduction was performed using the first 20 PCA components. We then conducted FindNeighbors and FindClusters functions to cluster cells at a resolution of 0.8. The FindAllMarkers function was used to identify signature genes for each cluster. Non-tumor cell populations (negative for Krt 8, 18, 14, 17, and Epcam) were re-clustered and filtered again to assure the lack of tumor cells in the new object. The resulted object was then subject to in-depth analysis of endothelial cells (positive for Pecam1) and CAF subpopulations (positive for Pdgfrb). DGE analyses between clusters were performed using MAST analysis and the obtained gene lists were pre-ranked for GSEA analysis based on p-values and log2 fold change values (73). All visualizations of gene expression were performed based on the ALRA assay of the dataset.
For scRNA-seq analysis of human breast cancer dataset (13), the matrix was downloaded from European Genome-Phenome Archive (EGA) EGAS00001005173. For the Seurat object, genes expressed by less than 3 cells and cells expressing less than 200 or more than 6000 genes were excluded. Log-based normalization (NormalizeData function) was performed to normalize the expression matrix, FindVariableFeatures and ScaleData arguments were then applied to the dataset. PCA and ALRA was conducted as well (28). UMAP dimensional reduction was performed using the first 20 PCA components. We then conducted FindNeighbors and FindClusters functions to cluster cells at a resolution of 0.1. The FindAllMarkers function was used to identify signature genes for each cluster. Non-tumor (negative for KRT8, 18, 14, 17, and EPCAM) non-immune cell populations were re-clustered for in-depth analysis of endothelial cells (positive for PECAM1) and CAF subpopulations (positive for PDGFRB). DGE analyses between clusters were performed using MAST analysis. Given that p-values of several genes were smaller than the lower limit of Excel’s computational power and were thus automatically rounder to 0, the MAST-derived gene lists were pre-ranked for GSEA analysis based on log2 fold change values (73). All visualizations of gene expression were performed based on the ALRA assay of the dataset.
Mouse mammary fibroblasts (MMFs) cell line establishment
Inguinal mammary fat pads were collected from a healthy C57BL/6J mouse and digested in sterile DMEM F-12 media (Gibco 11320033) containing 2 mg/mL collagenase and 2 U/mL DNase for 25 minutes at 37 Celsius. Tissue suspension was then filtered through a 70 μm strainer and centrifuged at 1500 round per minute (rpm) for 5 minutes. Cell pellets were then washed using sterile full DMEM F-12 media (10% fetal bovine serum and 1x penicillin-streptomycin (Gibco 15140122) in DMEM F-12). Cells were resuspended in full DMEM F-12 media and transferred to a tissue culture-treated petri dish and then kept in a 37 ° Celsius incubator at 5% CO2. After 30 minutes of cell attachment, supernatant was gently removed from the petri dish and replaced by fresh full-serum DMEM (DMEM (Sigma D6429) supplemented with 10% fetal bovine serum and 1x penicillin-streptomycin (Gibco 15140122)). The petri dish was hereafter kept at 37 Celsius 5% CO2 3% O2 and monitored regularly. Cells were lifted with 0.25% Trypsin-EDTA solution (Gibco, 25200072) and subcultivated once they reached confluence. Division rate of the cells gradually reached the slowest at the 4th to 5th passage with part of the cells survived the bottle neck and reattained sustainable robust growth rate beyond the 10th passage. These cells were then expanded and cryopreserved in large batches. Flow cytometry was conducted to assure the EPCAM/CD31/CD45-negativity of the cells (Fig. S11). Cells of the 12th passage were used in all experiments involving MMFs.
In vitro cell culture and MMF senescence induction
The EO771 and MMF cell lines were cultured in full-serum DMEM. The Py230 cell line was cultured in F-12K (ATCC 30–3004) supplemented with 5% fetal bovine serum, 0.1% MITO+ serum extender (Corning 355006), and 1x penicillin-streptomycin (Gibco 15140122). All of the cell lines were routinely tested as negative for mycoplasma. Tumor cells were cultured under 5% CO2 while MMFs were cultured under 5% CO2 and 3% O2. Upon senescence induction, MMFs were treated with 100 nM Doxo for 24 hours. After the drug treatment, Doxo-containing media was aspirated from the culture dish and the cell layer was carefully rinsed twice with PBS. Fresh media was added to the plate and was changed every 3 days. Senescence was fully established 7 days after the addition of Doxo.
Bulk RNA-seq analysis of MMF samples
Vehicle-treated control MMFs and Doxo-treated senescent MMFs were washed twice with PBS when cells were attached to the bottom of culture plates. 2-Mecaptoethanol supplemented cell lysis buffer (Qiagen RNeasy Mini Kit 74104) was directly added to the cell layers and the cell lysis suspension was collected. RNA was then purified following the instructions of Qiagen RNeasy Mini Kit and submitted for sequencing at the Genome Technology Access Center, where all samples were prepared according to library kit manufacturer’s protocol, indexed, pooled, and sequenced on an Illumina NovaSeq 6000. Count-per-million matrices generated post sequencing were then normalized and filtered in Partek Bioinformatics software. DGE analyses were performed using Partek’s GSA function, and the resulting gene lists were pre-ranked and submitted for GSEA analyses based on p-values and log2 fold change values.
In vitro ECM assembly
The ECM assembly procedure was developed based on previous literature (74). Gelatin solution (0.2% in PBS) was used to coat 96-well plates at 37° Celsius. The plates were then washed twice with PBS prior to the addition of glutaraldehyde (1% in PBS) for crosslinking reaction. After the plates were washed twice with PBS, 1M glycine solution was added into the plates to quench the crosslink reaction for 20 minutes at room temperature. The plates were then washed once with PBS. PBS supplemented with 1x penicillin-streptomycin was added to each well. The plates were finally sealed with parafilm and stored at 4° Celsius for a maximum of 3 days prior to cell seeding.
Once cellular senescence was fully established post Doxo treatment, MMFs were lifted using 0.25% Trypsin-EDTA solution (Gibco, 25200072) and prepared into cell suspension. Gelatin coated plates were retrieved from 4° Celsius and washed twice with PBS. Full-serum DMEM was added to the wells and the plates were kept at 37° Celsius for at least 1 hour to warm up and equilibrate the gelatin layer. MMFs were seeded into pre-warmed plates in full-serum DMEM (12,000 cells/well) and were allowed to attached to the bottom of the wells overnight. The next morning, media was aspirated and the wells were washed once with PBS prior to the addition of ECM assembly media (DMEM supplemented with 0.2% fetal bovine serum, 1x penicillin-streptomycin and 50 μg/mL freshly prepared sodium ascorbic acid, 200 μL per well) which was refreshed every other day. ECM assembly was performed under 5% CO2 and 3% O2 and was accomplished 6 days after the seeding of the MMFs. To preserve ECM, wells were first washed twice with PBS. Cell lysis solution (0.5% Triton and 20 mM NH4OH in PBS) was added to the wells and incubated for 5–8 minutes at 37° Celsius. PBS was then added to the wells to dilute the lysis solution. The plates were then sealed and store at 4° Celsius overnight. The next morning, the plates were washed three times with PBS. PBS supplemented with 1x penicillin-streptomycin was then added to each well. The plates were finally sealed with parafilm and stored at 4° Celsius for a maximum of 6 weeks.
NK cytotoxicity assay
The experimental setup for NK cytotoxicity assays was developed based on previous literature (52). RMA-S cell line (mycoplasma positive) was a kind gift from Dr. Roberta Faccio. A 96-well plate with preserved ECM was first retrieved from 4° Celsius and washed once with PBS. NK media (interleukin-15 and 2-Mecaptoethanol supplemented full RPMI media (RPMI (Gibco 11875–093) supplemented with 10% fetal bovine serum, 1x penicillin-streptomycin, 1mM sodium pyruvate (Corning 25–000-CI), 1x MEM nonessential amino acids (Corning 25–025-CI), 10 mM HEPES buffer (Corning 25–060-CI), and 1X GlutaMAX (Gibco 3050–061)) was added to each well (50 μL per well). The plates were transferred to a 37° Celsius incubator for later use. Spleens were harvested from mice stimulated with Poly I:C (300 ug/mouse) 20–24 hours prior to the sacrifice. NK cells were enriched from the spleens using a negative selection enrichment kit (Stemcell Technologies Cat# 19855). NK cells were suspended in the NK media at 2×106 cells/mL and 100 μL cell suspension was added to each well (100 μL NK media was used instead for target cell-only wells). The plate was briefly centrifuged to 200 rcf and then incubated at 37° Celsius for 2 hours. Meanwhile, RMA-S cells were labeled with CellTrace Violet and resuspended at 0.8×106 cells/mL. By the end of NK-ECM incubation, 50 μL RMA-S suspension was added to each well. The plate was then briefly centrifuged to 200 rcf and kept in a 37° Celsius incubator for 16 hours. After the incubation, each well was stained with 8 μL of 7-AAD and analyzed on a BD LSRFortessa flow cytometer. The % Specific Killing value was calculated as % [7-AAD+ of CTV+ cells] (Effector+Target) well – [%7-AAD+ of CTV+ cells] Target only well.
Orthotopic co-implantation of MMF and tumor cells and subsequent bioluminescence imaging (BLI)
MMFs (8×106 cells/mL) and firefly luciferase-expressing Py230 or EO771 tumor cells (16×106 cells/mL) were suspended in a 50–50 mixture of PBS and Matrigel (Corning 356231). Each wildtype C57Bl/6 mouse was surgically implanted with 50 μL of the cell suspension to its inguinal MFP. For any mice recruited for immune cell depletion assays, monoclonal antibodies (Supplementary Table 1) were first administrated at 500 ug/mouse via intra peritoneal injections 2 days prior to the MMF-tumor co-injection. Following the first dose, antibodies were then administrated at 250 ug/mouse every four days. BLI was performed routinely after the orthotopic injection and the imaging results were analyzed using Live Imaging software (PerkinElmer). All mice utilized in the orthotopic co-injection experiments were randomly assigned to receive either control or senescent MMFs along with tumor cells. All mice utilized in the immune cell depletion assays were first randomly assigned to receive either IgG or anti-NK1.1 antibody and then randomized to receive either control or senescent MMFs along with tumor cells.
Statistical Analyses
All data were analyzed and plotted using Prism. Please refer to figure legends for details on statistical tests used for any specific analysis.
Supplementary Material
Statement of Significance.
senCAFs limit NK cell-mediated killing thereby contributing to breast cancer progression. Thus, targeting senCAFs could be a clinically viable approach to limit tumor progression.
Acknowledgements:
We would like to thank Dr. Todd Fehniger and Pamela Wong for their support and guidance in NK cytotoxicity assays. We thank Dr. Edna Cukierman for advice on in vitro ECM generation. We also thank Siteman Flow Cytometry Core for help with sorting; Julie Prior and Katie Duncan in the Molecular Imaging Center for their help with bioluminescence imaging; Dr. Roberta Faccio and Seunghyun Lee for providing RMA-S cell lines and suggestions on in vitro NK cytotoxicity assays; the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help with sequencing, alignment, and genomic data analysis; Washington University Center for Cellular imaging (WUCCI), which is supported by Washington University School of Medicine, The Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (CDI-CORE-2015–505 and CDI-CORE-2019–813), and the Foundation for Barnes-Jewish Hospital (3770 and 4642) for support on performing second harmonic generation imaging. In addition, we would like to acknowledge Dr. Lorenzo Galluzzi for his suggestions on NK cell flow cytometry panel.
Financial Support:
This work was supported by NIH grants R01 AG059244, CA217208 (S.A. Stewart), The U.S. Army Medical Research Acquisition Activity, 820 Chandler Street, Fort Detrick, MD 21702-5014, is the awarding and administrating acquisition office, and this was supported in part by the Office of the Assistant Secretary of Defense for Health Affairs, through the Breast Cancer Research Program, under award No. BC181712. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense. This work was also supported by the Siteman Cancer Center Investment Program (NCI Cancer Center Support Grant P30CA091842, Fashion Footwear Association of New York, and the Alvin J. Siteman Cancer Center Siteman Investment Program (supported by The Foundation for Barnes-Jewish Hospital, Cancer Frontier Fund) to S.A. Stewart. J.Y. was supported by Cancer Biology Pathway of Siteman Cancer Center, which was funded by an NCI T32 grant CA113275, and NIH F31 grant CA271721-01. J.M.B. was supported by F31 DK122633. V.A.M was supported by F32 CA275212-01. S.P. and A.P.D. were supported by R01 CA21720605. G.D.L was supported by R01 CA223758 and CA254060. A. K. W. and E. S. K. were supported by R01CA247362 and received sponsored research funding from Bristol Meyer Squibb and Blueprint Medicine. E. C. was supported by NIH MSTP T32 GM7200-49 under the Medical Scientist Training Program of Washington University School of Medicine.
Footnotes
Conflict of Interest Disclosure Statement: No authors have any conflicts to disclose.
Data Availability
The human breast cancer dataset is reported by Wu et.al. (13) and is available in the European Genome-Phenome Archive under the accession code EGAS00001005173. The scRNA-seq dataset for 12-weeks-old PyMT mice is available in GEO database under the accession code GSE199515. The scRNA-seq dataset for 7-weeks-old PyMT mice and the RNA-seq dataset for CAFs and MMFs are available in GEO database under the accession number GSE229356.
References
- 1.Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14:518–27. [DOI] [PubMed] [Google Scholar]
- 2.Xing F Cancer associated fibroblasts (CAFs) in tumor microenvironment. Frontiers in Bioscience. 2010;15:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kubo N, Araki K, Kuwano H, Shirabe K. Cancer-associated fibroblasts in hepatocellular carcinoma. World J Gastroenterol. 2016;22:6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lavie D, Ben-Shmuel A, Erez N, Scherz-Shouval R. Cancer-associated fibroblasts in the single-cell era. Nat Cancer. 2022;3:793–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartoschek M, Oskolkov N, Bocci M, Lövrot J, Larsson C, Sommarin M, et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat Commun. 2018;9:5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. Journal of Experimental Medicine. 2017;214:579–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019;9:1102–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sebastian A, Hum NR, Martin KA, Gilmore SF, Peran I, Byers SW, et al. Single-Cell Transcriptomic Analysis of Tumor-Derived Fibroblasts and Normal Tissue-Resident Fibroblasts Reveals Fibroblast Heterogeneity in Breast Cancer. Cancers (Basel). 2020;12:1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hosein AN, Huang H, Wang Z, Parmar K, Du W, Huang J, et al. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight. 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krishnamurty AT, Shyer JA, Thai M, Gandham V, Buechler MB, Yang YA, et al. LRRC15+ myofibroblasts dictate the stromal setpoint to suppress tumour immunity. Nature. 2022;611:148–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019;9:282–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu SZ, Roden DL, Wang C, Holliday H, Harvey K, Cazet AS, et al. Stromal cell diversity associated with immune evasion in human triple‐negative breast cancer. EMBO J. 2020;39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu SZ, Al-Eryani G, Roden DL, Junankar S, Harvey K, Andersson A, et al. A single-cell and spatially resolved atlas of human breast cancers. Nat Genet. 2021;53:1334–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruhland MK, Loza AJ, Capietto A-H, Luo X, Knolhoff BL, Flanagan KC, et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun. 2016;7:11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faget DV, Ren Q, Stewart SA. Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer. 2019;19:439–53. [DOI] [PubMed] [Google Scholar]
- 16.Alspach E, Flanagan KC, Luo X, Ruhland MK, Huang H, Pazolli E, et al. p38MAPK Plays a Crucial Role in Stromal-Mediated Tumorigenesis. Cancer Discov. 2014;4:716–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coppé J-P, Desprez P-Y, Krtolica A, Campisi J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annual Review of Pathology: Mechanisms of Disease. 2010;5:99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marin I, Boix O, Garcia-Garijo A, Sirois I, Caballe A, Zarzuela E, et al. Cellular Senescence Is Immunogenic and Promotes Antitumor Immunity. Cancer Discov. 2023;13:410–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen H-A, Ho Y-J, Mezzadra R, Adrover JM, Smolkin R, Zhu C, et al. Senescence Rewires Microenvironment Sensing to Facilitate Antitumor Immunity. Cancer Discov. 2023;13:432–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bavik C, Coleman I, Dean JP, Knudsen B, Plymate S, Nelson PS. The Gene Expression Program of Prostate Fibroblast Senescence Modulates Neoplastic Epithelial Cell Proliferation through Paracrine Mechanisms. Cancer Res. 2006;66:794–802. [DOI] [PubMed] [Google Scholar]
- 22.Liu D, Hornsby PJ. Senescent Human Fibroblasts Increase the Early Growth of Xenograft Tumors via Matrix Metalloproteinase Secretion. Cancer Res. 2007;67:3117–26. [DOI] [PubMed] [Google Scholar]
- 23.Ruscetti M, Leibold J, Bott MJ, Fennell M, Kulick A, Salgado NR, et al. NK cell–mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science (1979). 2018;362:1416–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018;28:436–53. [DOI] [PubMed] [Google Scholar]
- 25.Witkiewicz AK, Rivadeneira DB, Ertel A, Kline J, Hyslop T, Schwartz GF, et al. Association of RB/p16-Pathway Perturbations with DCIS Recurrence. Am J Pathol. 2011;179:1171–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faget DV., Luo X, Inkman MJ, Ren Q, Su X, Ding K, et al. p38MAPKa stromal reprogramming sensitizes metastatic breast cancer to immunotherapy. Cancer Discov. 2023;13:1454–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murali B, Ren Q, Luo X, Faget DV., Wang C, Johnson RM, et al. Inhibition of the Stromal p38MAPK/MK2 Pathway Limits Breast Cancer Metastases and Chemotherapy-Induced Bone Loss. Cancer Res. 2018;78:5618–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Linderman GC, Zhao J, Roulis M, Bielecki P, Flavell RA, Nadler B, et al. Zero-preserving imputation of single-cell RNA-seq data. Nat Commun. 2022;13:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101. [DOI] [PubMed] [Google Scholar]
- 30.Loser K, Mehling A, Loeser S, Apelt J, Kuhn A, Grabbe S, et al. Epidermal RANKL controls regulatory T-cell numbers via activation of dendritic cells. Nat Med. 2006;12:1372–9. [DOI] [PubMed] [Google Scholar]
- 31.Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell. 2018;33:463–479.e10. [DOI] [PubMed] [Google Scholar]
- 32.Tominaga K, Suzuki HI. TGF-β Signaling in Cellular Senescence and Aging-Related Pathology. Int J Mol Sci. 2019;20:5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tran DQ. TGF-β: the sword, the wand, and the shield of FOXP3+ regulatory T cells. J Mol Cell Biol. 2012;4:29–37. [DOI] [PubMed] [Google Scholar]
- 34.Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The polarization of immune cells in the tumour environment by TGFβ. Nat Rev Immunol. 2010;10:554–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilson EB, El-Jawhari JJ, Neilson AL, Hall GD, Melcher AA, Meade JL, et al. Human Tumour Immune Evasion via TGF-β Blocks NK Cell Activation but Not Survival Allowing Therapeutic Restoration of Anti-Tumour Activity. PLoS One. 2011;6:e22842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martínez-Zamudio RI, Robinson L, Roux P-F, Bischof O. SnapShot: Cellular Senescence Pathways. Cell. 2017;170:816–816.e1. [DOI] [PubMed] [Google Scholar]
- 37.Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, et al. Senescence-associated β-galactosidase is lysosomal β-galactosidase. Aging Cell. 2006;5:187–95. [DOI] [PubMed] [Google Scholar]
- 38.Fridman AL, Tainsky MA. Critical pathways in cellular senescence and immortalization revealed by gene expression profiling. Oncogene. 2008;27:5975–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Attalla S, Taifour T, Bui T, Muller W. Insights from transgenic mouse models of PyMT-induced breast cancer: recapitulating human breast cancer progression in vivo. Oncogene. 2021;40:475–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun. 2016;7:11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature. 2016;530:184–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ovadya Y, Krizhanovsky V. Strategies targeting cellular senescence. Journal of Clinical Investigation. 2018;128:1247–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kolodkin-Gal D, Roitman L, Ovadya Y, Azazmeh N, Assouline B, Schlesinger Y, et al. Senolytic elimination of Cox2-expressing senescent cells inhibits the growth of premalignant pancreatic lesions. Gut. 2022;71:345–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krtolica A, Parrinello S, Lockett S, Desprez P-Y, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proceedings of the National Academy of Sciences. 2001;98:12072–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kale A, Sharma A, Stolzing A, Desprez P-Y, Campisi J. Role of immune cells in the removal of deleterious senescent cells. Immunity & Ageing. 2020;17:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Durgeau A, Virk Y, Corgnac S, Mami-Chouaib F. Recent Advances in Targeting CD8 T-Cell Immunity for More Effective Cancer Immunotherapy. Front Immunol. 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Plitas G, Rudensky AY. Regulatory T Cells: Differentiation and Function. Cancer Immunol Res. 2016;4:721–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williams LM, Rudensky AY. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol. 2007;8:277–84. [DOI] [PubMed] [Google Scholar]
- 50.Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, Walzer T. Maturation of mouse NK cells is a 4-stage developmental program. Blood. 2009;113:5488–96. [DOI] [PubMed] [Google Scholar]
- 51.Fu B, Wang F, Sun R, Ling B, Tian Z, Wei H. CD11b and CD27 reflect distinct population and functional specialization in human natural killer cells. Immunology. 2011;133:350–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wong P, Wagner JA, Berrien-Elliott MM, Schappe T, Fehniger TA. Flow cytometry-based ex vivo murine NK cell cytotoxicity assay. STAR Protoc. 2021;2:100262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shimasaki N, Jain A, Campana D. NK cells for cancer immunotherapy. Nat Rev Drug Discov. 2020;19:200–18. [DOI] [PubMed] [Google Scholar]
- 54.Krneta T, Gillgrass A, Chew M, Ashkar AA. The breast tumor microenvironment alters the phenotype and function of natural killer cells. Cell Mol Immunol. 2016;13:628–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan C-H, et al. Inference and analysis of cell-cell communication using CellChat. Nat Commun. 2021;12:1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vyas M, Peigney D, Demehri S. Extracellular matrix-natural killer cell interactome: an uncharted territory in health and disease. Curr Opin Immunol. 2022;78:102246. [DOI] [PubMed] [Google Scholar]
- 57.Lebbink RJ, de Ruiter T, Kaptijn GJA, Bihan DG, Jansen CA, Lenting PJ, et al. Mouse leukocyte-associated Ig-like receptor-1 (mLAIR-1) functions as an inhibitory collagen-binding receptor on immune cells. Int Immunol. 2007;19:1011–9. [DOI] [PubMed] [Google Scholar]
- 58.Lebbink RJ, de Ruiter T, Adelmeijer J, Brenkman AB, van Helvoort JM, Koch M, et al. Collagens are functional, high affinity ligands for the inhibitory immune receptor LAIR-1. Journal of Experimental Medicine. 2006;203:1419–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jansen CA, Cruijsen CWA, de Ruiter T, Nanlohy N, Willems N, Janssens-Korpela P-L, et al. Regulated expression of the inhibitory receptor LAIR-1 on human peripheral T cells during T cell activation and differentiation. Eur J Immunol. 2007;37:914–24. [DOI] [PubMed] [Google Scholar]
- 60.Bunting MD, Vyas M, Requesens M, Langenbucher A, Schiferle EB, Manguso RT, et al. Extracellular matrix proteins regulate NK cell function in peripheral tissues. Sci Adv. 2022;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Van Laethem F, Donaty L, Tchernonog E, Lacheretz-Szablewski V, Russello J, Buthiau D, et al. LAIR1, an ITIM-Containing Receptor Involved in Immune Disorders and in Hematological Neoplasms. Int J Mol Sci. 2022;23:16136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buqué A, Bloy N, Perez-Lanzón M, Iribarren K, Humeau J, Pol JG, et al. Immunoprophylactic and immunotherapeutic control of hormone receptor-positive breast cancer. Nat Commun. 2020;11:3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, et al. CD4+ T Cells Regulate Pulmonary Metastasis of Mammary Carcinomas by Enhancing Protumor Properties of Macrophages. Cancer Cell. 2009;16:91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bos PD, Plitas G, Rudra D, Lee SY, Rudensky AY. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. Journal of Experimental Medicine. 2013;210:2435–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Engelhardt JJ, Boldajipour B, Beemiller P, Pandurangi P, Sorensen C, Werb Z, et al. Marginating Dendritic Cells of the Tumor Microenvironment Cross-Present Tumor Antigens and Stably Engage Tumor-Specific T Cells. Cancer Cell. 2012;21:402–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dominguez CX, Müller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15+ Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2020;10:232–53. [DOI] [PubMed] [Google Scholar]
- 67.Bredfeldt JS, Liu Y, Pehlke CA, Conklin MW, Szulczewski JM, Inman DR, et al. Computational segmentation of collagen fibers from second-harmonic generation images of breast cancer. J Biomed Opt. 2014;19:16007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Abhilash AS, Baker BM, Trappmann B, Chen CS, Shenoy VB. Remodeling of fibrous extracellular matrices by contractile cells: predictions from discrete fiber network simulations. Biophys J. 2014;107:1829–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patil G, Vuong TD, Kale S, Valliyodan B, Deshmukh R, Zhu C, et al. Dissecting genomic hotspots underlying seed protein, oil, and sucrose content in an interspecific mapping population of soybean using high-density linkage mapping. Plant Biotechnol J. 2018;16:1939–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen X, Chen D, Ban E, Toussaint KC, Janmey PA, Wells RG, et al. Glycosaminoglycans modulate long-range mechanical communication between cells in collagen networks. Proc Natl Acad Sci U S A. 2022;119:e2116718119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Franco‐Barraza J, Beacham DA, Amatangelo MD, Cukierman E. Preparation of Extracellular Matrices Produced by Cultured and Primary Fibroblasts. Curr Protoc Cell Biol. 2016;71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The human breast cancer dataset is reported by Wu et.al. (13) and is available in the European Genome-Phenome Archive under the accession code EGAS00001005173. The scRNA-seq dataset for 12-weeks-old PyMT mice is available in GEO database under the accession code GSE199515. The scRNA-seq dataset for 7-weeks-old PyMT mice and the RNA-seq dataset for CAFs and MMFs are available in GEO database under the accession number GSE229356.