

Abstract
Tumor tissues are chronically exposed to hypoxia owing to aberrant vascularity. Hypoxia induces metabolic alterations in cancer, thereby promoting aggressive malignancy and metastasis. While previous efforts largely focused on adaptive responses in glucose and glutamine metabolism, recent studies have begun to yield important insight into the hypoxic regulation of lipid metabolic reprogramming in cancer. Emerging evidence points to lipid droplet (LD) accumulation as a hallmark of hypoxic cancer cells. One critical underlying mechanism involves the inhibition of adipose triglyceride lipase (ATGL)-mediated intracellular lipolysis by a small protein encoded by hypoxia-inducible gene 2 (HIG2), also known as hypoxia inducible lipid droplet associated (HILPDA). In this review we summarize and discuss recent key findings on hypoxia-dependent regulation of metabolic adaptations especially lipolysis in cancer. We also pose several questions and hypotheses pertaining to the metabolic impact of lipolytic regulation in cancer under hypoxia and during hypoxia-reoxygenation transition.
Keywords: Lipolysis, hypoxia, HIF, hypoxia-inducible factor, oxygen, fatty acid, lipid droplet, ATGL, HIG2, hypoxia inducible gene 2, HILPDA
1. Introduction
Cancer cells survive within the hostile tumor microenvironment and maintain a rapid proliferation rate by acquiring alterations of metabolic pathways for carbohydrates, lipids, proteins and nucleic acids. “Aerobic glycolysis”, the so-called “Warburg effect”, is considered as a hallmark of nearly all highly expanding solid tumors [1, 2]. By generating ATP and lactate via the glycolytic pathway even under normoxia, this metabolic adaptation was hypothesized to benefit cancer cells with mitochondrial defects. However, later studies showed that most cancer cells are not impaired in mitochondrial function. The aerobic glycolysis not only supports rapid ATP production, but also provides carbons for biomass generation to sustain cell growth and produces reducing equivalents required for preserving redox balance [3, 4].
Maintenance of oxygen homeostasis is a key feature of adapting cancer cells. Several studies have shown that hypoxia-inducible factors (HIFs) mediate multiple protective mechanisms by reducing mitochondrial oxidative capacity and oxygen consumption in hypoxic cells [5–10]. Fatty acid (FA) oxidation consumes a large amount of oxygen and in a hypoxic environment, produces reactive oxygen species (ROS) that further exacerbate oxygen insufficiency. Elevated oxidative stress in turn causes damages to membrane lipids, deactivation of proteins and enzymes, and apoptotic cell death [11–13]. In response to limited oxygen supply, cancer cells often switch off FA oxidation through downregulation of intracellular lipolysis, while increasing accumulation of triglyceride lipid droplets (TG-LDs) [14–16]. LD accumulation and lipolytic inhibition not only provide fuel and sources of building blocks for the future growth, but also protect cancer cells against oxidative stress and lipotoxicity in hypoxia. Recent studies have yielded critical insight into a novel anti-lipolytic mechanism that is key for cancer cells to survive hypoxia. This mechanism represents a departure from the canonical metabolic pathways that have been targeted therapeutically to deprive cancer cells of necessary fuel/building blocks. In this review, we outline recent discoveries on reprogramming of FA metabolism in hypoxic cancer cells as well as the roles of lipolytic inhibition and LD formation as adaptive mechanisms to hypoxia.
2. Hypoxia and HIF signaling in cancer
Since the biochemical purification of the subunits of the heterodimeric HIF-1 (HIF-1α and HIF-1β) was reported by Semenza and colleagues in 1995 [17], oxygen sensing has been recognized to influence a host of physiologic processes including cell growth, energy metabolism, redox homeostasis, organ development and angiogenesis [18]. Changes in oxygen availability and adaptation to low oxygen environment at cellular, tissue and organismal levels also influence the development of several disorders such as cardiovascular disease, pulmonary hypertension, stroke, bacterial infections, inflammation and wound healing. A large body of existing evidence indicates that due to scarce oxygen diffusion through the abnormal and inefficient tumor vascular architecture, hypoxia occurs in 90% of solid tumors and plays a critical role in shaping the fate and aggressiveness of cancer cells [19, 20]. Tumor microenvironment is subject not only to chronic hypoxia (continuous low oxygen supply for several hours), but also to cycling (intermittent) hypoxia characterized by numerous spatial and temporal fluctuations of oxygen levels, alternating cycles of severe, moderate hypoxia followed by reoxygenation episodes when blood flow is restored [21]. Tumors exposed to cycling hypoxia have been reported to exhibit greater extent of pathological angiogenesis, intratumor inflammation, metastatic activity and drug resistance compared to chronic hypoxia [22–24]. Chronic hypoxia usually is the first type of hypoxia to occur during tumor development because of rapidly proliferating tumor cells whose distance from a functional blood vessel exceeds 100 μm [25, 26]. Under these oxygen-deprived conditions, HIFs play a crucial role in mediating cancer cell growth, migration, invasion and increased resistance to pharmacotherapies [27, 28].
Fluctuations in external oxygen gradients directly influence global nuclear gene expression programs in cancer cells, where the HIF-prolyl hydroxylase domain enzyme (PHD)-von Hippel-Lindau protein (pVHL) axis plays a crucial role as oxygen sensor at the molecular level [29–31]. In normoxia, the HIF-α subunits (HIF-1α; HIF-2α and HIF-3α) are hydroxylated on proline residues by PHD, permitting their ubiquitination by pVHL and consequent proteasomal degradation. Under hypoxic conditions where PHD hydroxylation is diminished, the stabilized HIF-α proteins translocate to the cell nucleus and dimerize with constitutively present HIF-1β. The HIF-α/β heterodimers bind specific DNA hypoxia response elements (HRE), activating the transcription of over 1,000 different genes and mediating a diverse array of signaling pathways in multiple disease processes [32]. The three HIF-α isoforms are encoded by different gene loci and differ in promoter usage and splicing patterns. Despite similar domain architecture and proteolytic regulation, HIF-α proteins exhibit different tissue distribution patterns. While HIF-1α is ubiquitously present in a variety of tissues, HIF-2α is selectively expressed in liver, duodenum, and to a lesser extent in kidney, heart and brain [33, 34]. HIF-3α, with a poorly characterized function, is expressed mainly in pulmonary alveolar epithelial cells [35]. In cancer cells, activities of HIFs are not only regulated by tumor oxygen levels, but also by mutations in oncogenes and tumor suppressor proteins. A notable example of HIF activation independently of tumor oxygen levels is loss-of-function mutations of VHL tumor suppressor gene associated with clear cell renal cell carcinoma (ccRCC), which accounts for 70–80% of all renal malignancies. A defining hallmark of ccRCC cells is the “clear” phenotype caused by the extensive cytoplasmic accumulation of lipids and glycogen [36].
3. Hypoxia and remodeling of glucose and glutamine metabolism in cancer
Metabolic adaptation to the hypoxic microenvironment has become a hallmark of cancer. In response to scarcity of oxygen and nutrients, cancer cells often fine-tune their metabolism to secure the amounts of energy and building blocks required for sustaining their growth and survival. The first and best characterized metabolic adaptation in cancer cells is the Warburg effect, which consists of switching ATP generation from oxidative phosphorylation to glycolysis even in the presence of sufficient oxygen supply [2]. In cancer cells with intact mitochondrial function, aerobic glycolysis is often favored over oxidative metabolism to gain three key metabolic benefits: rapid energy production, provision of metabolic intermediates and precursors for the biosynthesis of macromolecules, and generation of NADPH to help maintain the redox status [3, 4, 37]. These characteristic glycolytic phenotypes are largely controlled by HIF-1 [8], which activates expression of not only genes responsible for glucose uptake and glycolysis, but also enzymes involved in the inhibition of glucose oxidation such as pyruvate dehydrogenase kinase 1 (PDK1). PDK1 phosphorylates and inhibits pyruvate dehydrogenase (PDH) [38], thereby promoting lactate production through limiting the entry of glycolysis-derived pyruvate into the oxidative TCA cycle. In response to hypoxia, HIF-1 activation also acts to reduce oxygen consumption through downregulating expression of genes critical for mitochondrial oxidative metabolism [5, 6].
Cancer cells require large amounts of lipids for the synthesis of cellular membranes to sustain high proliferation rates. Over the last decade an increasingly number of reports have described how alterations in lipid metabolism contribute to tumor growth, survival, local invasion and metastatic development [39, 40]. FAs, either of exogenous origin or generated de novo, are paramount for anabolic and catabolic metabolism of cancer cells as either building blocks for more complex lipids or substrates for energy production [41]. For example, FAs were identified as a key regulator of pancreatic cancer cell proliferation in vitro, and n-6 polyunsaturated FAs were found to induce liver metastasis in a xenograft model [42]. A study conducted shortly after by Okumura et al. further showed that FAs derived from lipolysis in the peripancreatic adipose tissue promotes the invasiveness and chemoresistance of pancreatic ductal adenocarcinoma (PDAC) cells [43]. A more recent report by Iwamoto et al. demonstrated that hepatocellular carcinoma (HCC) cells implanted in fatty livers of mice fed a high fat diet accumulated LDs, and switched their metabolism from glycolysis to oxidative FA metabolism when treated with antiangiogenic drugs and under intermittent hypoxia. These orthotopic tumors were able to grow even in the presence of minimal number of microvessels [44].
While normal cells preferentially use exogenous FAs for synthesis of structural lipids and as energy substrates, cancer cells upregulate the expression of fatty acid synthase (FASN) and acetyl-CoA carboxylase (ACC), thereby producing most of cellular FAs through de novo synthesis to support membrane phospholipid biosynthesis [45]. ACC catalyzes the carboxylation of acetyl-CoA - converted from glycolytic end-product pyruvate – to form malonyl-CoA.
Malonyl-CoA is further converted by FASN to long-chain FAs. Under normoxic conditions, citrate and acetyl-CoA used for de novo lipogenesis are primarily generated from glucose-derived pyruvate through the citrate shuttle and ATP citrate lyase in the cytosol. Hypoxic cancer cells, however, are able to increase de novo lipid synthesis using citrate produced from reductive carboxylation of glutamine. In particular, it was discovered that this reductive metabolism of glutamine is dependent on isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) [46, 47], which catalyzes the key step of isocitrate decarboxylation into α-ketoglutarate (α-KG). RCC cells with VHL deficiency and resultant pseudohypoxic response preferentially utilize reductive glutamine metabolism for lipid biosynthesis even in normoxic conditions [46, 48]. Furthermore, in a report by Bjornson et al., a global gene profiling of 361 hepatocellular carcinoma (HCC) tissues identified the mitochondrial acetyl-CoA synthetase 1 (ACSS1) as the top expressed gene in tumors exposed to hypoxia. ACSS1 is a key enzyme that converts acetate into acetyl-CoA. This finding suggests that in hypoxic conditions, cancer cells utilize mitochondrial acetate as carbon source to generate FAs and structural lipids required for membrane biosynthesis [49].
4. Hypoxia and lipid droplet accumulation in cancer
In most cell types, FAs can be oxidized to produce energy, esterified into neutral and phospholipids, or converted to various signaling molecules. LDs are a key organelle responsible for storing cellular surplus of FAs in esterified neural forms such as TGs and sterol esters. Studying the LD biology has revealed fundamental functions of LDs and has led to identification of numerous LD-associated structural proteins, enzymes and proteins regulating membrane trafficking [50–52]. Since the first reports describing LDs in mammary carcinoma and malignant lymphomas published in 1960s [53, 54], major breakthrough in cancer LD research has been achieved in providing evidence to demonstrate that LDs not only serve a neutral lipid storage depot, but also are implicated in all major steps of cancer development [15, 55]. The effects of LDs include advancement of cancer cell growth and proliferation, suppression of cancer cell death, evasion of tumor growth suppressing mechanisms, as well as promotion of angiogenesis, genome instability, inflammation, metabolic deregulation and evasion of host cancer immune suppression [56].
LD formation and TG synthesis are two tightly coupled processes under energy-rich conditions. The existing evidence is in support of a model that TG-LDs forms through de novo synthesis of TGs as a lens within the ER bilayer [57]. The initial step of TG synthesis is the conversion of exogenous or de novo synthetized FAs to fatty acyl-CoA by acyl-CoA synthetase (ACS). Next, fatty acyl-CoA and glycerol 3-phosphate are combined into TG in the ER via the classic glycerol phosphate pathway that consists of a series of key enzymes including glycerol-3-phosphate acyltransferase (GPAT), acylglycerolphosphate acyltransferase (AGPAT), phosphatidic acid phosphohydrolase (PAP or lipin), and diacylglycerol acyltransferase (DGAT). Subsequently newly formed LDs bud from the ER bilayer towards the cytosol. During nutrient shortage, LD-stored TGs can be hydrolyzed by LD-associated lipases to release FAs for energy production via mitochondrial β-oxidation and other metabolic pathways [58]. Regulation of intracellular lipolysis has been recently reported in various cancers and will be further discussed in this review. An alternative mechanism of LD breakdown is called lipophagy, and it consists of engulfment of portions of the LD by lipoautophagosomes and transport to the lysosomes for further degradation by lysosomal acid lipases (LAL) [59–61].
In 1977, Bush and colleagues first reported that cultured mouse fibroblasts accumulate TG-LDs in response to severe oxygen restriction [62]. Fast forward to now, increased accumulation of LDs is being recognized as a hallmark of hypoxic cancer cells of various origins [55]. A direct link between hypoxia and LDs has been extensively reported as HIFs regulate the transcription of various genes involved in FA metabolism [63]. Other studies found that in cancer cells, hypoxia increases FA uptake via upregulating FA binding proteins FABP3 and FABP7 [15], and to promote scavenging of FAs from serum lysophospholipids [64]. FABPs are a large family of cytoplasmic proteins essential for intracellular transport of FAs and other lipophilic molecules such as eicosanoids and retinoids. In addition to down-modulation of overall mitochondrial oxidative capacity, HIF-1 activation has been implicated in suppressing FA oxidation by decreasing the expression of carnitine palmitoyltransferase 1A (CPT1A) [65] as well as increasing TG synthesis by inducing Lipin 1 [66]. These events, which in combination lead to increased accumulation of TG-LDs, are strongly associated with greater tumor malignancy [67].
5. Regulation of cancer lipolysis by hypoxia
During instances of heightened metabolic demand, TA stores in LDs undergo a hydrolytic process termed lipolysis to release free FAs. In adipocytes, complete lipolysis is delicately regulated and is catalyzed sequentially by adipose triglyceride lipase (ATGL), hormone-sensitive lipase (HSL), and monoacylglycerol lipase (MGL) [58, 68–70]. A majority of the free FAs and glycerol derived from white adipocyte lipolysis are delivered to other peripheral tissues for energy production. Compared to the white adipocytes that store a single large TG-LD, cells from other tissues types (e.g. liver, skeletal muscle) and cancer only possess a limited capacity for TG synthesis and only contain many smaller LDs. In these nonadipocyte cells, FAs derived from intracellular lipolysis usually are oxidized internally to meet the local energy demand [58, 71].
5.1. ATGL-mediated lipolysis and its protein regulators
ATGL is the rate-limiting intracellular TG hydrolase that catalyzes the first step of lipolysis, converting TG to diacylglycerol (DG) and one free FA [58]. ATGL exhibits low or no lipase activity for other substrates, such as DG, MG or cholesteryl esters [68]. The human and mouse genes for ATGL/ Patatin Like Phospholipase Domain Containing 2 (PNPLA2) encode proteins that share 84% of amino acids. ATGL is expressed broadly in all tissues with the highest levels in white and brown adipose tissue. Studies using knockout mouse models have demonstrated the essential function of ATGL in adipose lipolysis as well as in the whole body lipid and energy metabolism [72–74]. In brown adipose tissue, liver and striated muscles, ATGL selectively channels hydrolyzed FAs to β-oxidation in mitochondria and for synthesis of lipid ligands for PPARα and PPARδ/β [75–77]. Consistently, ATGL deficiency causes accumulation of TG-LDs and impairs mitochondrial FA oxidation in these oxidative tissues [73, 77, 78].
Aside from the LD localization, the enzyme action of ATGL is decided by the presence of its coactivator CGI-58 (comparative gene identification-58; also known as ABHD5 (α/β hydrolase fold-containing protein 5))[79]. The patatin-like domain of ATGL interacts with CGI-58, while the C-terminal domain of this enzyme is responsible for its localization to LDs. In humans, mutations in ATGL and CGI-58 genes are causal for a group of neutral lipid storage disease characterized by excessive TG deposition in multiple nonadipose tissues [80, 81]. Earlier work by our laboratory identified a small protein encoded by G0/G1 Switch Gene 2 (G0S2) as a potent endogenous inhibitor of ATGL [82], providing another important piece to this complex lipolysis puzzle. G0S2 is a basic protein that contains two potential α helices separated by a hydrophobic domain (HD). Direct binding between the HD of G0S2 and the patatin-like catalytic domain of ATGL has been shown to convey inhibition of ATGL’s TG hydrolase activity in vitro and ATGL-mediated lipolysis in various cell types [83, 84]. An enzymatic study by Oberer and colleagues has further shown that a peptide derived from the human G0S2 HD inhibits ATGL noncompetitively in the nanomolar range [85]. Importantly, recent animal studies have yielded compelling evidence demonstrating the in vivo role of G0S2 as a major metabolic and energy regulator in adipose tissue, liver and cardiac muscle through its inhibitory action on ATGL [86–91].
5.2. Tumor suppressive role of intracellular lipolysis
A large body of evidence suggests that inhibition of intracellular lipolysis may promote cancer development. ATGL expression was found to be decreased in the tissues of human lung adenocarcinoma and lung squamous cell carcinoma compared to normal epithelium. Consistently, ablation of ATGL induces spontaneous pulmonary neoplasia in mice and a more aggressive phenotype in lung carcinoma cells [92]. Intestine-specific deletion of ATGL coactivator CGI-58 resulted in colorectal carcinogenesis [93]. Adipose-specific ATGL and HSL double-knock out induced development of liposarcoma in brown adipose tissue [94]. HSL was originally identified in white adipose tissue as a fasting-induced TG hydrolase [95]. However, later studies have shown that HSL mainly acts as a diacylglycerol lipase downstream of ATGL-mediate TG hydrolysis [69, 70]. Moreover, deficiency of HSL was recently reported to accelerate the development of pancreatic cancer in a conditional KrasG12D mouse model [96].
5.3. HIG2/HILPDA as a key regulator of ATGL-mediate lipolysis in hypoxic cancer
In the report by Bensaad et al., convincing data are provided to demonstrate that hypoxia induces HIF-1α-dependent accumulation of LDs in various tumor cell lines and that storage of neutral lipids in LDs constitutes an essential mechanism for survival during the hypoxia-reoxygenation transition [15]. This adaptive mechanism appears to require FA binding proteins FABP3/7 and LD structural protein Perilipin 2 as their singly depletion significantly decreased LD formation under hypoxia. Consequently, hypoxic cells became impaired in their ability to handle lipolytic products, leading to increased ceramide production and cell death [15]. Evidence derived from separate studies of glial and neural stem cells further implies that the capacity to accumulate LDs is positively linked to the ability of cells to survive oxidative stress in hypoxia [97, 98]. Given that FA oxidation and hypoxia both promote mitochondrial generation of reactive oxygen species (ROS) [11–13], a potential mechanism emerged in which TG-LDs accumulation in hypoxia is a result of lipolytic inhibition to limit the amount of free FAs available for oxidation. By using various ATGL CRISPR knockout CRC and RCC cell lines, we were able to obtain first piece of evidence that ATGL-mediated lipolysis is suppressed in hypoxic cancer cells [14]. A subsequent proteomic search for specific lipolytic regulators in hypoxic cancer cells led to our discovery of hypoxia-inducible gene 2 (HIG2) as a selective inhibitor of ATGL [14]. HIG2 is a 63-amino acid (~7-kDa) peptide encoded by a HIF-1 target gene named Hilpda (Hypoxia Inducible Lipid Droplet Associated).
Early reports described HIG2 as a PPARα target gene in liver and acts to promote hepatic TG deposition by reducing TG secretion [99]. In addition, HIG2-ablated hepatocytes were previously shown to exhibit increased TG turnover under normoxic conditions [100]. However, knockout mouse studies conducted later yielded results arguing against a direct involvement of HIG2 in lipolysis in the absence of oxygen shortage [101, 102]. Other studies suggested that HIG2 represent a LD associated protein upregulated during hypoxia [103]. Indeed, HIF-1α was shown to bind the promoter region of the Hilpda gene to activate transcription of HIG2 [103]. As a result, HIG2 expression pattern is consistent with that of HIF-1α. In macrophages, HIG2 was shown to be localized to the ER-LD interface, and HIG2 deletion abolished hypoxia-induced accumulation of TGs, cholesteryl esters and oxidized LDL. Conditional HIG2 knockout mice exhibited reduced plaque areas in aorta and macrophage content when crossed onto an apoE-deficient background [104]. These results suggest that HIG2 promotes lesion formation and progression of atherosclerosis. Furthermore, a most recent study by van Dierendonck et al. showed that HIG2 is essential for LD accumulation in adipose tissue macrophages, though the lipid storage mediated by HIG2 does not seem to influence adipose tissue inflammation or obesity phenotypes in mice fed with high fat diet [105].
Early studies have revealed that HIG2 contributes to the production of intact LDs in various cancer cells, including those from colorectal cancer (CRC), ovarian cancer, RCC and HCC [100, 103, 106]. The first hint on HIG2 protein function came from the alignment that revealed a homology between HIG2 and G0S2 HD sequences. Subsequent biochemical analysis showed that like G0S2, HIG2 inhibits ATGL through direct physical association [14, 107]. This inhibition can occur under both basal as well as CGI-58-stimulated conditions. Both interaction and co-localization with ATGL at the LD surface require a LY(V/L)LG motif in the HD of HIG2 [14]. In cells of various cancer types including RCC and CRC, hypoxia promotes TG-LD accumulation through HIG2-mediated ATGL inhibition [14, 16]. Lipolytic inhibition in turns triggers downregulation of PPARα target genes for mitochondrial activity and FA β-oxidation, leading to decreased ROS generation and apoptotic cell death (Figure 1). Consistently, when exposed to oleate and hypoxia, increased FA oxidation and excessive ROS generation caused most of HIG2 knockout cells to undergo capsase-3 dependent apoptosis. HIG2 knockout also led to a profound delay in tumor growth along with increased lipid peroxidation and apoptosis in xenograft tumor tissues. Though it was shown to be a weaker ATGL inhibitor than G0S2 in in vitro enzymatic assays [107], HIG2’s function appears to be ATGL-specific in vivo as co-deletion of ATGL was able to completely reverse the phenotypes caused by HIG2 deficiency [14]. In addition to hypoxia, nutrient deprivation was recently shown to upregulate HIG2 post-transcriptionally in CRC cells by a mechanism requiring autophagic flux and LD turnover [16]. This finding raises the possibility that HIG2 mediates the storage of autophagy-generated FAs as TGs in LDs. Additional insight into the function of HIG2 has been uncovered by lipidomic analysis showing that aside from promoting TG storage under hypoxia, HIG2 affects FA length, saturation and amounts of major phospholipids classes [16]. These results indicate a role of HIG2 in the regulation of fluidity, homeostasis and metabolism of membrane phospholipids.
Figure 1. Lipolytic inhibition by HIG2 is central for a survival strategy employed by hypoxic cancer cells.
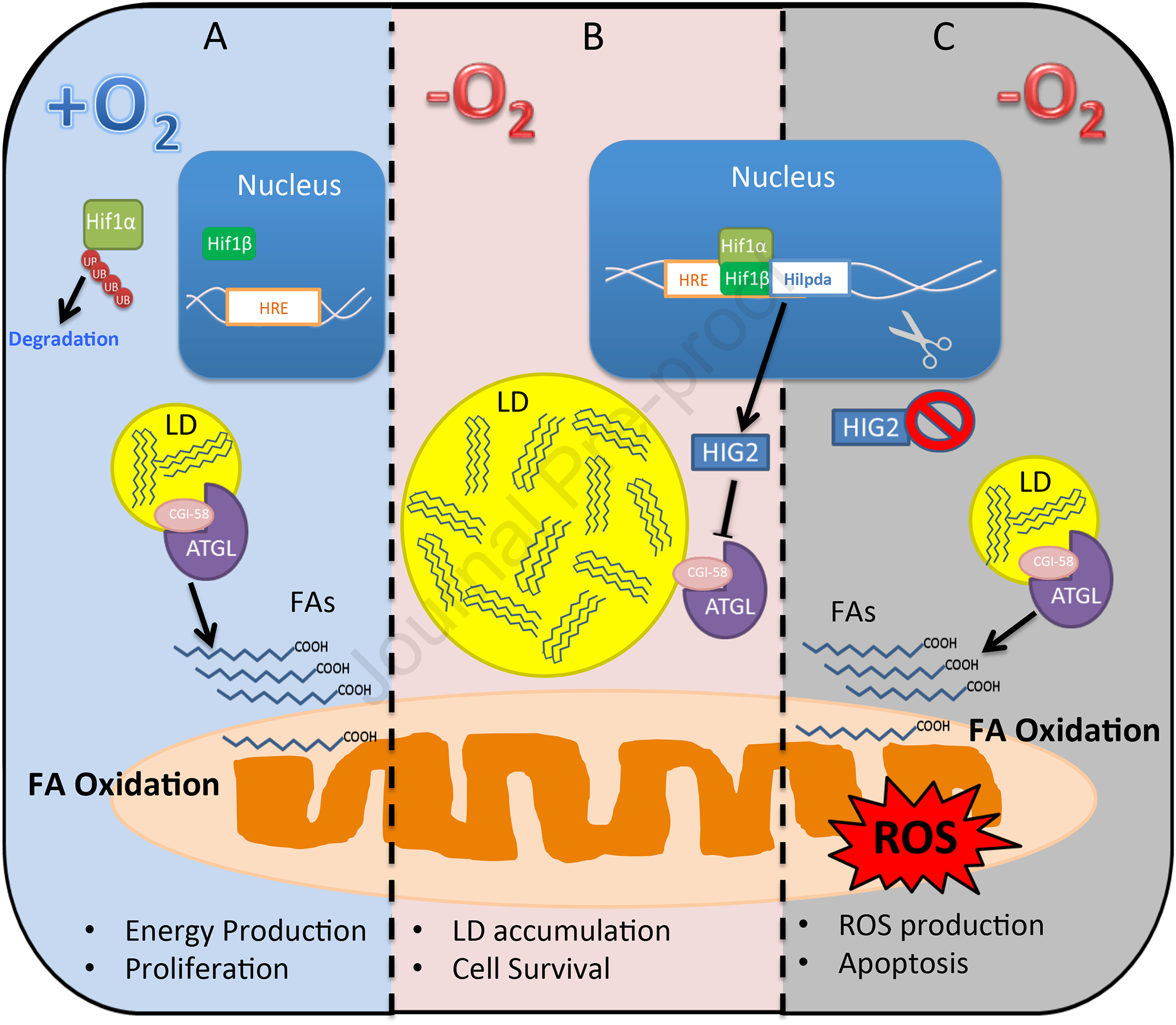
(A) In normoxic cells where HIF-1α protein stability is low due to increased ubiquitination and proteasomal degradation, ATGL-mediated lipolysis is active and downstream FA oxidation contributes to the overall energy production. (B) Hypoxic stabilization of HIF-1α protein leads to expression of the ATGL inhibitor HIG2, and subsequent lipolytic inhibition results in decreased FA oxidation along with improved oxygen homeostasis and cell viability. (C) HIG2 deficiency, induced either genetically or pharmacologically, forces increased lipolysis and FA oxidation that exacerbate ROS generation and apoptotic death in hypoxic cancer cells.
Analysis of the Cancer Genome Atlas (TCGA) datasets showed that the expression of HIG2 mRNA is strongly associated with various solid tumors in humans including CRC and RCC [14]. HIG2 protein was found to be highly abundant in human RCC samples relative to the matched adjacent normal kidney tissues. The expression pattern of HIG2 also correlates with the tumor TG content [14]. Recently, Zou et al. reported a critical relationship between HIF2-dependent HIG2 expression and the vulnerability of clear cell RCC to ferroptosis [108]. One key piece of supporting evidence is that HIG2 expression promotes enrichment of polyunsaturated lipids, the rate-limiting substrates for lipid peroxidation. As G0S2 expression failed to elicit a similar effect, the involvement of HIG2 in ferroptosis of ccRCC is thought to be ATGL-independent. However, the fact that enrichment of polyunsaturated FAs increased in both TGs and phospholipids seems to argue against the possibility that HIG2 inhibits a separate TG lipase other than ATGL. Interestingly, both G0S2 and HIG2 were recently shown to possess an ATGL-independent role in promoting TG synthesis. While G0S2 harbors an intrinsic lysophosphatidic acid acyltransferase (LPAAT) activity [109], HIG2 appears to be capable of stimulating the function of DGAT1 [110]. However, it remains to be determined if HIG2 plays a similar lipogenic role in the settings of clear cell carcinoma.
6. Implications of lipolytic inhibition by HIG2/HILPDA
Hypoxia and pseudohypoxia associated with VHL deficiency potently induces expression of HIF-1α during the expanding growth of solid tumors. Increased HIF-1 activity is now known to trigger three major metabolic switches that support cell growth, proliferation and survival: (1) decreased glucose oxidation and increased glycolysis, (2) increased dependence on glutamine, and (3) inhibition of intracellular lipolysis. While glycolytic intermediates are channeled into pathways for synthesis of macromolecules (such as lipids, nucleotides, and proteins), glutamine is often favorably used through reductive carboxylation for de novo FA synthesis to support membrane biogenesis [46–48]. Lipolytic inhibition, on the other hand, leads to decreased FA oxidation and associated oxidative stress.
6.1. Impact on glucose and glutamine metabolism
Recently, increased storage of FAs as TGs is being recognized as another prominent feature of hypoxic and pseudohypoxic cancer cells [15, 55]. We and others discovered that the HIF-HIG2 antilipolytic pathway is critical for mediating this process [14, 16]. Specifically, HIG2 was found to directly binds to and inhibits ATGL at the LD surface, thereby blocking lipolytic breakdown of TGs. Although lipolytic liberation leads to increased FA oxidation and ROS production, HIG2 deletion incurs no effect on glucose consumption and glycolytic phenotypes in hypoxic cancer cells [14]. This departs from how glucose and FA metabolism is coordinately regulated in normoxic conditions. In the classical Randle cycle, increased FA oxidation occurs alongside inhibition of glucose uptake and utilization [111]. On the other hand, these results complement the existing model illustrating that HIF-1 represses glucose flux to the TCA cycle through mediating expression of PDK1, which inhibits pyruvate oxidation through phosphorylating and inactivating PDH [38]. In cancer cells located within the hypoxic regions of solid tumors, concurrent inhibition of ATGL by HIG2 and PDH by PDK1 should provide a survival advantage as inhibition of FA and glucose oxidation together should lead to substantially reduced oxygen consumption and mitochondrial ROS generation.
Whether and how lipolytic inhibition mediated by HIG2 impacts glutamine metabolism in hypoxic cancer cells remain to be determined. To this end, an arising question concerns why cancer cells would choose to synthesize more FAs from glutamine in hypoxia during the same time when FA uptake and storage in TG-LDs is upregulated. One explanation may be that FAs derived from different sources are metabolized in a highly partitioned manner. According to previous studies of normal oxidative cell types, exogenously acquired FAs are generally channeled through the LD-lipolysis-mitochondria route for oxidation [58]. In this context, LD storage and lipolytic action of ATGL constitute a buffering system to balance the uptake of exogenous FAs and their oxidative utilization. In proliferating cancer cells, however, membrane phospholipid (PL) biosynthesis was shown to mostly require FAs generated endogenously through de novo synthesis [112]. Moreover, a recent study found that lipogenesis through glutamine reduction is tightly regulated by intracellular citrate levels [48]. Based on these results, it is tempting to hypothesize that along with the prevention of ROS production to sustain cell survival, a simultaneous inhibition of FA oxidation by HIG2 and glucose/pyruvate oxidation by PDK1 leads to decreased generation of acetyl-CoA and citrate, thereby driving the reductive flux of glutamine by mass action to maintain lipogenesis and cell growth under hypoxia. Further investigation is needed to elucidate the interactions between the metabolic pathways of these three major energy substrates in hypoxic cancer cells.
6.2. Energy production upon reoxygenation following hypoxia
The abnormal vascularization greatly contributes to the fluctuation of oxygen levels inside solid tumors. The cyclic changes between hypoxia and reoxygenation due to the opening and closing of blood vessels have been associated with cancer metastasis [5, 56, 113]. While cancer cells exposed to hypoxia at the primary lesions mainly rely on glycolysis for energy production, detached cells are exposed to reoxygenation when they enter circulation and can potentially switch to reliance on mitochondrial substrate oxidation. In fact, FA oxidation was found recently to be essential to the development of anoikis resistance and metastatic phenotypes in various cancers [114–118]. Conceivably, tumor cells with a higher capacity to store TG-LDs in hypoxia and to mobilize them upon reoxygenation would be in a more advantageous position during detachment, invasion and metastasis. In accordance with this concept, the study by Bensaad et al. has shown that FAs stored in LDs during hypoxia are essential for ATP production in hypoxia-reoxygenation transition [15]. We speculate that the oxygen-sensing HIF-1-HIG2 pathway may be a crucial part of a mechanism conferring such a capacity. By inhibiting ATGL-mediated lipolysis, HIG2 acts to enhance TG stores downstream of HIF-1 activation in hypoxia. Upon reoxygenation, downregulation of the HIF-1-HIG2 pathway could lead to the liberation of ATGL activity, enabling TG hydrolysis to supply free FAs for β-oxidation in support of the energy need of invasion and metastasis. In human CRC, HIG2 expression was indeed found to be upregulated in liver and lung metastases as compared to the primary tumors [119].
6.3. Possible functional interaction between HIG2/HILPDA and G0S2
ATGL-mediated intracellular lipolysis is tightly coupled to FA oxidation, a catabolic process that depends on the supply of both FAs and oxygen. G0S2 is mainly expressed in normal metabolic tissues such as liver, adipose tissue and striated muscles [83]. In comparison, HIG2 is abundantly expressed in various solid tumors as well as macrophages [104, 105]. While G0S2 expression is controlled by PPARγ and LXRα and is responsive to increased FA levels [120, 121], endogenous level of HIG2 is low under normoxia and its expression is activated by HIF-1 in response to oxygen depletion [14, 103]. In macrophages but not cancer cells, mRNA expression of HIG2 can also be upregulated upon exposure to ectopic lipids [16, 104, 105]. Given the short half life of these small inhibitory proteins [122], the available data are in support of a proof-of-concept that ATGL together with G0S2 and HIG2 constitute a gauge and valve system capable of reacting to the rapid changes in the ratio of intracellular free FAs to O2. With the ability to adjust the rates of ATGL-mediated lipolysis and downstream FA oxidation, such a system renders cells capable of storing TGs during exposure to excessive amounts of FAs as well as evading oxidative stress when oxygen becomes scarce. Currently, no data is available with regard to the functional relationship between G0S2 and HIG2, or a context in which they are co-expressed and contribute to common phenotypes. Although G0S2 was originally thought to be a cell cycle regulator [123, 124], evidence is still lacking to establish its function in cancer settings. An earlier study by Kioka et al. showed that G0S2 promotes hypoxia tolerance by positively regulating oxidative phosphorylation and ATP synthesis in cardiomyocytes [125]. However, G0S2 expression does not appear to increase in hypoxic cancer cells [14]. Whether HIG2 plays a similar role in cancer also remains to be determined.
7. Conclusions
A large volume of evidence has been reported on the crucial role of metabolic reprogramming during cancer development. Cancer cells often are exposed to nutrient-deprived and poorly oxygenated tumor microenvironment. As a consequence, cancer cells rely on metabolic adaptations for maintaining energy production, acquiring building blocks to support cell growth, and synthesizing signaling molecules for various tumor-promoting activities. Rewiring of FA metabolism under hypoxia is particularly important in lipid-rich solid tumors such as RCC, CRC and fatty liver-associated HCC as FA oxidation consumes a large amount of oxygen and is a major contributor to ROS generation. The recently obtained evidence supports a novel concept that inhibition of TG breakdown or lipolysis by HIG2 is one of the crucial metabolic adaptations that hypoxic cancer cells deploy to maintain redox homeostasis through limiting FA. We propose that HIG2 is a metabolic oncogenic factor, which exerts tumor-promoting activity by inhibiting the complex ATGL/CGI-58 under hypoxia. Despite the progress, however, several major questions remain to be answered: whether and how is TG accumulation linked to glucose and glutamine metabolism in cancer cells? Why would hypoxic cancer cells need to simultaneously upregulate LD storage and de novo lipogenesis? Is TG-LD accumulation controlled by HIF-1-dependent mechanism relevant in tumor development in vivo? Do TG storage in hypoxia and hydrolysis in reoxygenation impact the energy supply required for cancer invasion and metastasis? Do TG-LDs, because of their anti-oxidant role, contribute to the resistance of cancer cells to pro-oxidant and anti-angiogenic therapeutics? Future research is needed to uncover the full spectrum of the interplay between lipolytic and antilipolitic mechanisms in specific tumor settings, and how it influences the overall metabolic behavior of cancer in hypoxia. Novel insight if produced will likely open up additional diagnostic and therapeutic avenues that improve outcomes for patients with solid tumors.
Disclosure Statement:
We wish to confirm that there are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Warburg O, On respiratory impairment in cancer cells, Science, 124 (1956) 269–270. [PubMed] [Google Scholar]
- [2].Warburg O, On the origin of cancer cells, Science, 123 (1956) 309–314. [DOI] [PubMed] [Google Scholar]
- [3].Vander Heiden MG, Cantley LC, Thompson CB, Understanding the Warburg effect: the metabolic requirements of cell proliferation, Science, 324 (2009) 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Semenza GL, Artemov D, Bedi A, Bhujwalla Z, Chiles K, Feldser D, Laughner E, Ravi R, Simons J, Taghavi P, Zhong H, ‘The metabolism of tumours’: 70 years later, Novartis Found Symp, 240 (2001) 251–260; discussion 260–254. [PubMed] [Google Scholar]
- [5].Masson N, Ratcliffe PJ, Hypoxia signaling pathways in cancer metabolism: the importance of co-selecting interconnected physiological pathways, Cancer & metabolism, 2 (2014) 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, Dang CV, Semenza GL, HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity, Cancer cell, 11 (2007) 407–420. [DOI] [PubMed] [Google Scholar]
- [7].Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC, HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption, Cell metabolism, 3 (2006) 187–197. [DOI] [PubMed] [Google Scholar]
- [8].Semenza GL, Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics, Oncogene, 29 (2010) 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rankin EB, Giaccia AJ, The role of hypoxia-inducible factors in tumorigenesis, Cell death and differentiation, 15 (2008) 678–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gordan JD, Simon MC, Hypoxia-inducible factors: central regulators of the tumor phenotype, Current opinion in genetics & development, 17 (2007) 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bleier L, Drose S, Superoxide generation by complex III: from mechanistic rationales to functional consequences, Biochimica et biophysica acta, 1827 (2013) 1320–1331. [DOI] [PubMed] [Google Scholar]
- [12].Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT, Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing, Cell metabolism, 1 (2005) 401–408. [DOI] [PubMed] [Google Scholar]
- [13].Schonfeld P, Wojtczak L, Fatty acids as modulators of the cellular production of reactive oxygen species, Free radical biology & medicine, 45 (2008) 231–241. [DOI] [PubMed] [Google Scholar]
- [14].Zhang X, Saarinen AM, Hitosugi T, Wang Z, Wang L, Ho TH, Liu J, Inhibition of intracellular lipolysis promotes human cancer cell adaptation to hypoxia, Elife, 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, Pinnick KE, Wigfield S, Buffa FM, Li JL, Zhang Q, Wakelam MJ, Karpe F, Schulze A, Harris AL, Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation, Cell reports, 9 (2014) 349–365. [DOI] [PubMed] [Google Scholar]
- [16].VandeKopple MJ, Wu J, Auer EN, Giaccia AJ, Denko NC, Papandreou I, HILPDA regulates lipid metabolism, lipid droplet abundance and response to microenvironmental stress in solid tumors, Mol Cancer Res, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang GL, Jiang BH, Rue EA, Semenza GL, Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension, Proc Natl Acad Sci U S A, 92 (1995) 5510–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lee JW, Ko J, Ju C, Eltzschig HK, Hypoxia signaling in human diseases and therapeutic targets, Exp Mol Med, 51 (2019) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vaupel P, Mayer A, Hockel M, Tumor hypoxia and malignant progression, Methods Enzymol, 381 (2004) 335–354. [DOI] [PubMed] [Google Scholar]
- [20].Cosse JP, Michiels C, Tumour hypoxia affects the responsiveness of cancer cells to chemotherapy and promotes cancer progression, Anticancer Agents Med Chem, 8 (2008) 790–797. [DOI] [PubMed] [Google Scholar]
- [21].Michiels C, Tellier C, Feron O, Cycling hypoxia: A key feature of the tumor microenvironment, Biochim Biophys Acta, 1866 (2016) 76–86. [DOI] [PubMed] [Google Scholar]
- [22].Semenza GL, Ruvolo PP, Introduction to tumor microenvironment regulation of cancer cell survival, metastasis, inflammation, and immune surveillance, Biochim Biophys Acta, 1863 (2016) 379–381. [DOI] [PubMed] [Google Scholar]
- [23].Hockel M, Vaupel P, Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects, J Natl Cancer Inst, 93 (2001) 266–276. [DOI] [PubMed] [Google Scholar]
- [24].Span PN, Bussink J, Biology of hypoxia, Semin Nucl Med, 45 (2015) 101–109. [DOI] [PubMed] [Google Scholar]
- [25].Thomlinson RH, Gray LH, The histological structure of some human lung cancers and the possible implications for radiotherapy, Br J Cancer, 9 (1955) 539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Helmlinger G, Yuan F, Dellian M, Jain RK, Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation, Nat Med, 3 (1997) 177–182. [DOI] [PubMed] [Google Scholar]
- [27].Xuan Y, Wang YN, Hypoxia/IL-1alpha axis promotes gastric cancer progression and drug resistance, J Dig Dis, 18 (2017) 511–520. [DOI] [PubMed] [Google Scholar]
- [28].Luo W, Wang Y, Hypoxia Mediates Tumor Malignancy and Therapy Resistance, Adv Exp Med Biol, 1136 (2019) 1–18. [DOI] [PubMed] [Google Scholar]
- [29].Hu CJ, Sataur A, Wang L, Chen H, Simon MC, The N-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1alpha and HIF-2alpha, Molecular biology of the cell, 18 (2007) 4528–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tian H, McKnight SL, Russell DW, Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells, Genes & development, 11 (1997) 72–82. [DOI] [PubMed] [Google Scholar]
- [31].Semenza GL, Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy, Trends in pharmacological sciences, 33 (2012) 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Simon MC, The Hypoxia Response Pathways - Hats Off!, N Engl J Med, 375 (2016) 1687–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Morello E, Sutti S, Foglia B, Novo E, Cannito S, Bocca C, Rajsky M, Bruzzi S, Abate ML, Rosso C, Bozzola C, David E, Bugianesi E, Albano E, Parola M, Hypoxia-inducible factor 2alpha drives nonalcoholic fatty liver progression by triggering hepatocyte release of histidine-rich glycoprotein, Hepatology, 67 (2018) 2196–2214. [DOI] [PubMed] [Google Scholar]
- [34].Wiesener MS, Jurgensen JS, Rosenberger C, Scholze CK, Horstrup JH, Warnecke C, Mandriota S, Bechmann I, Frei UA, Pugh CW, Ratcliffe PJ, Bachmann S, Maxwell PH, Eckardt KU, Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs, FASEB J, 17 (2003) 271–273. [DOI] [PubMed] [Google Scholar]
- [35].Li QF, Wang XR, Yang YW, Lin H, Hypoxia upregulates hypoxia inducible factor (HIF)-3alpha expression in lung epithelial cells: characterization and comparison with HIF-1alpha, Cell Res, 16 (2006) 548–558. [DOI] [PubMed] [Google Scholar]
- [36].Rini BI, Campbell SC, Escudier B, Renal cell carcinoma, Lancet, 373 (2009) 1119–1132. [DOI] [PubMed] [Google Scholar]
- [37].Koppenol WH, Bounds PL, Dang CV, Otto Warburg’s contributions to current concepts of cancer metabolism, Nature reviews. Cancer, 11 (2011) 325–337. [DOI] [PubMed] [Google Scholar]
- [38].Kim JW, Tchernyshyov I, Semenza GL, Dang CV, HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia, Cell metabolism, 3 (2006) 177–185. [DOI] [PubMed] [Google Scholar]
- [39].Beloribi-Djefaflia S, Vasseur S, Guillaumond F, Lipid metabolic reprogramming in cancer cells, Oncogenesis, 5 (2016) e189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Liu Q, Luo Q, Halim A, Song G, Targeting lipid metabolism of cancer cells: A promising therapeutic strategy for cancer, Cancer Lett, 401 (2017) 39–45. [DOI] [PubMed] [Google Scholar]
- [41].Rohrig F, Schulze A, The multifaceted roles of fatty acid synthesis in cancer, Nat Rev Cancer, 16 (2016) 732–749. [DOI] [PubMed] [Google Scholar]
- [42].Yu M, Liu H, Duan Y, Zhang D, Li S, Wang F, Four types of fatty acids exert differential impact on pancreatic cancer growth, Cancer Lett, 360 (2015) 187–194. [DOI] [PubMed] [Google Scholar]
- [43].Okumura T, Ohuchida K, Sada M, Abe T, Endo S, Koikawa K, Iwamoto C, Miura D, Mizuuchi Y, Moriyama T, Nakata K, Miyasaka Y, Manabe T, Ohtsuka T, Nagai E, Mizumoto K, Oda Y, Hashizume M, Nakamura M, Extra-pancreatic invasion induces lipolytic and fibrotic changes in the adipose microenvironment, with released fatty acids enhancing the invasiveness of pancreatic cancer cells, Oncotarget, 8 (2017) 18280–18295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Iwamoto H, Abe M, Yang Y, Cui D, Seki T, Nakamura M, Hosaka K, Lim S, Wu J, He X, Sun X, Lu Y, Zhou Q, Shi W, Torimura T, Nie G, Li Q, Cao Y, Cancer Lipid Metabolism Confers Antiangiogenic Drug Resistance, Cell Metab, 28 (2018) 104–117 e105. [DOI] [PubMed] [Google Scholar]
- [45].Koundouros N, Poulogiannis G, Reprogramming of fatty acid metabolism in cancer, Br J Cancer, 122 (2020) 4–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, Kelleher JK, Vander Heiden MG, Iliopoulos O, Stephanopoulos G, Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia, Nature, 481 (2011) 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ, Reductive carboxylation supports growth in tumour cells with defective mitochondria, Nature, 481 (2011) 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gameiro PA, Yang J, Metelo AM, Perez-Carro R, Baker R, Wang Z, Arreola A, Rathmell WK, Olumi A, Lopez-Larrubia P, Stephanopoulos G, Iliopoulos O, In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation, Cell metabolism, 17 (2013) 372–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bjornson E, Mukhopadhyay B, Asplund A, Pristovsek N, Cinar R, Romeo S, Uhlen M, Kunos G, Nielsen J, Mardinoglu A, Stratification of Hepatocellular Carcinoma Patients Based on Acetate Utilization, Cell Rep, 13 (2015) 2014–2026. [DOI] [PubMed] [Google Scholar]
- [50].Greenberg AS, Coleman RA, Kraemer FB, McManaman JL, Obin MS, Puri V, Yan QW, Miyoshi H, Mashek DG, The role of lipid droplets in metabolic disease in rodents and humans, The Journal of clinical investigation, 121 (2011) 2102–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Krahmer N, Farese RV Jr., Walther TC, Balancing the fat: lipid droplets and human disease, EMBO molecular medicine, 5 (2013) 905–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kory N, Farese RV Jr., Walther TC, Targeting Fat: Mechanisms of Protein Localization to Lipid Droplets, Trends in cell biology, 26 (2016) 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Aboumrad MH, Horn RC Jr., Fine G, Lipid-secreting mammary carcinoma. Report of a case associated with Paget’s disease of the nipple, Cancer, 16 (1963) 521–525. [DOI] [PubMed] [Google Scholar]
- [54].Wright DH, Lipid content of malignant lymphomas, J Clin Pathol, 21 (1968) 643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Koizume S, Miyagi Y, Lipid Droplets: A Key Cellular Organelle Associated with Cancer Cell Survival under Normoxia and Hypoxia, International journal of molecular sciences, 17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hanahan D, Weinberg RA, Hallmarks of cancer: the next generation, Cell, 144 (2011) 646–674. [DOI] [PubMed] [Google Scholar]
- [57].Wilfling F, Haas JT, Walther TC, Farese RV Jr., Lipid droplet biogenesis, Current opinion in cell biology, 29 (2014) 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zechner R, Zimmermann R, Eichmann TO, Kohlwein SD, Haemmerle G, Lass A, Madeo F, FAT SIGNALS--lipases and lipolysis in lipid metabolism and signaling, Cell metabolism, 15 (2012) 279–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Schott MB, Weller SG, Schulze RJ, Krueger EW, Drizyte-Miller K, Casey CA, McNiven MA, Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes, J Cell Biol, 218 (2019) 3320–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Schulze RJ, Drizyte K, Casey CA, McNiven MA, Hepatic Lipophagy: New Insights into Autophagic Catabolism of Lipid Droplets in the Liver, Hepatol Commun, 1 (2017) 359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Schulze RJ, Sathyanarayan A, Mashek DG, Breaking fat: The regulation and mechanisms of lipophagy, Biochim Biophys Acta Mol Cell Biol Lipids, 1862 (2017) 1178–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Gordon GB, Barcza MA, Bush ME, Lipid accumulation of hypoxic tissue culture cells, Am J Pathol, 88 (1977) 663–678. [PMC free article] [PubMed] [Google Scholar]
- [63].Huang, Li T, Li X, Zhang L, Sun L, He X, Zhong X, Jia D, Song L, Semenza GL, Gao P, Zhang H, HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression, Cell Rep, 8 (2014) 1930–1942. [DOI] [PubMed] [Google Scholar]
- [64].Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, Thompson CB, Rabinowitz JD, Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids, Proc Natl Acad Sci U S A, 110 (2013) 8882–8887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Du W, Zhang L, Brett-Morris A, Aguila B, Kerner J, Hoppel CL, Puchowicz M, Serra D, Herrero L, Rini BI, Campbell S, Welford SM, HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism, Nat Commun, 8 (2017) 1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Mylonis I, Sembongi H, Befani C, Liakos P, Siniossoglou S, Simos G, Hypoxia causes triglyceride accumulation by HIF-1-mediated stimulation of lipin 1 expression, J Cell Sci, 125 (2012) 3485–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Laurenti G, Benedetti E, D’Angelo B, Cristiano L, Cinque B, Raysi S, Alecci M, Ceru MP, Cifone MG, Galzio R, Giordano A, Cimini A, Hypoxia induces peroxisome proliferator-activated receptor alpha (PPARalpha) and lipid metabolism peroxisomal enzymes in human glioblastoma cells, J Cell Biochem, 112 (2011) 3891–3901. [DOI] [PubMed] [Google Scholar]
- [68].Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, Neuberger G, Eisenhaber F, Hermetter A, Zechner R, Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase, Science, 306 (2004) 1383–1386. [DOI] [PubMed] [Google Scholar]
- [69].Bezaire V, Mairal A, Ribet C, Lefort C, Girousse A, Jocken J, Laurencikiene J, Anesia R, Rodriguez AM, Ryden M, Stenson BM, Dani C, Ailhaud G, Arner P, Langin D, Contribution of adipose triglyceride lipase and hormone-sensitive lipase to lipolysis in hMADS adipocytes, J Biol Chem, 284 (2009) 18282–18291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Haemmerle G, Zimmermann R, Hayn M, Theussl C, Waeg G, Wagner E, Sattler W, Magin TM, Wagner EF, Zechner R, Hormone-sensitive lipase deficiency in mice causes diglyceride accumulation in adipose tissue, muscle, and testis, J Biol Chem, 277 (2002) 4806–4815. [DOI] [PubMed] [Google Scholar]
- [71].Lass A, Zimmermann R, Oberer M, Zechner R, Lipolysis - a highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores, Progress in lipid research, 50 (2011) 14–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, Heldmaier G, Maier R, Theussl C, Eder S, Kratky D, Wagner EF, Klingenspor M, Hoefler G, Zechner R, Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase, Science, 312 (2006) 734–737. [DOI] [PubMed] [Google Scholar]
- [73].Ahmadian M, Abbott MJ, Tang T, Hudak CS, Kim Y, Bruss M, Hellerstein MK, Lee HY, Samuel VT, Shulman GI, Wang Y, Duncan RE, Kang C, Sul HS, Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype, Cell metabolism, 13 (2011) 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kienesberger PC, Lee D, Pulinilkunnil T, Brenner DS, Cai L, Magnes C, Koefeler HC, Streith IE, Rechberger GN, Haemmerle G, Flier JS, Zechner R, Kim YB, Kershaw EE, Adipose triglyceride lipase deficiency causes tissue-specific changes in insulin signaling, J Biol Chem, 284 (2009) 30218–30229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Wu JW, Wang SP, Alvarez F, Casavant S, Gauthier N, Abed L, Soni KG, Yang G, Mitchell GA, Deficiency of liver adipose triglyceride lipase in mice causes progressive hepatic steatosis, Hepatology, 54 (2011) 122–132. [DOI] [PubMed] [Google Scholar]
- [76].Ong KT, Mashek MT, Bu SY, Mashek DG, Hepatic ATGL knockdown uncouples glucose intolerance from liver TAG accumulation, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 27 (2013) 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Haemmerle G, Moustafa T, Woelkart G, Buttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D, Kienesberger PC, Zierler K, Schreiber R, Eichmann T, Kolb D, Kotzbeck P, Schweiger M, Kumari M, Eder S, Schoiswohl G, Wongsiriroj N, Pollak NM, Radner FP, Preiss-Landl K, Kolbe T, Rulicke T, Pieske B, Trauner M, Lass A, Zimmermann R, Hoefler G, Cinti S, Kershaw EE, Schrauwen P, Madeo F, Mayer B, Zechner R, ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1, Nature medicine, 17 (2011) 1076–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Ong KT, Mashek MT, Bu SY, Greenberg AS, Mashek DG, Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning, Hepatology, 53 (2011) 116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Lass A, Zimmermann R, Haemmerle G, Riederer M, Schoiswohl G, Schweiger M, Kienesberger P, Strauss JG, Gorkiewicz G, Zechner R, Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome, Cell Metab, 3 (2006) 309–319. [DOI] [PubMed] [Google Scholar]
- [80].Pena-Penabad C, Almagro M, Martinez W, Garcia-Silva J, Del Pozo J, Yebra MT, Sanchez-Manzano C, Fonseca E, Dorfman--Chanarin syndrome (neutral lipid storage disease): new clinical features, The British journal of dermatology, 144 (2001) 430–432. [DOI] [PubMed] [Google Scholar]
- [81].Schweiger M, Lass A, Zimmermann R, Eichmann TO, Zechner R, Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5, Am J Physiol Endocrinol Metab, 297 (2009) E289–296. [DOI] [PubMed] [Google Scholar]
- [82].Yang X, Lu X, Lombes M, Rha GB, Chi YI, Guerin TM, Smart EJ, Liu J, The G(0)/G(1) switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase, Cell Metab, 11 (2010) 194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Zhang X, Heckmann BL, Campbell LE, Liu J, G0S2: A small giant controller of lipolysis and adipose-liver fatty acid flux, Biochim Biophys Acta, 1862 (2017) 1146–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Heckmann BL, Zhang X, Xie X, Liu J, The G0/G1 switch gene 2 (G0S2): regulating metabolism and beyond, Biochimica et biophysica acta, 1831 (2013) 276–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Cerk IK, Salzburger B, Boeszoermenyi A, Heier C, Pillip C, Romauch M, Schweiger M, Cornaciu I, Lass A, Zimmermann R, Zechner R, Oberer M, A Peptide Derived from G0/G1 Switch Gene 2 Acts as Non-competitive Inhibitor of Adipose Triglyceride Lipase, The Journal of biological chemistry, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Zhang X, Xie X, Heckmann BL, Saarinen AM, Czyzyk TA, Liu J, Targeted disruption of g0/g1 switch gene 2 enhances adipose lipolysis, alters hepatic energy balance, and alleviates high-fat diet-induced liver steatosis, Diabetes, 63 (2014) 934–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Heckmann BL, Zhang X, Xie X, Saarinen A, Lu X, Yang X, Liu J, Defective adipose lipolysis and altered global energy metabolism in mice with adipose overexpression of the lipolytic inhibitor G0/G1 switch gene 2 (G0S2), The Journal of biological chemistry, 289 (2014) 1905–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].El-Assaad W, El-Kouhen K, Mohammad AH, Yang J, Morita M, Gamache I, Mamer O, Avizonis D, Hermance N, Kersten S, Tremblay ML, Kelliher MA, Teodoro JG, Deletion of the gene encoding G0/G 1 switch protein 2 (G0s2) alleviates high-fat-diet-induced weight gain and insulin resistance, and promotes browning of white adipose tissue in mice, Diabetologia, 58 (2015) 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Ma T, Lopez-Aguiar AG, Li A, Lu Y, Sekula D, Nattie EE, Freemantle S, Dmitrovsky E, Mice lacking G0S2 are lean and cold-tolerant, Cancer biology & therapy, 15 (2014) 643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Chen PR, Shin S, Choi YM, Kim E, Han JY, Lee K, Overexpression of G0/G1 Switch Gene 2 in Adipose Tissue of Transgenic Quail Inhibits Lipolysis Associated with Egg Laying, International journal of molecular sciences, 17 (2016) 384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Heier C, Radner FP, Moustafa T, Schreiber R, Grond S, Eichmann TO, Schweiger M, Schmidt A, Cerk IK, Oberer M, Theussl HC, Wojciechowski J, Penninger JM, Zimmermann R, Zechner R, G0/G1 Switch Gene 2 Regulates Cardiac Lipolysis, The Journal of biological chemistry, 290 (2015) 26141–26150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Al-Zoughbi W, Pichler M, Gorkiewicz G, Guertl-Lackner B, Haybaeck J, Jahn SW, Lackner C, Liegl-Atzwanger B, Popper H, Schauer S, Nusshold E, Kindt AS, Trajanoski Z, Speicher MR, Haemmerle G, Zimmermann R, Zechner R, Vesely PW, Hoefler G, Loss of adipose triglyceride lipase is associated with human cancer and induces mouse pulmonary neoplasia, Oncotarget, 7 (2016) 33832–33840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Ou J, Miao H, Ma Y, Guo F, Deng J, Wei X, Zhou J, Xie G, Shi H, Xue B, Liang H, Yu L, Loss of abhd5 promotes colorectal tumor development and progression by inducing aerobic glycolysis and epithelial-mesenchymal transition, Cell reports, 9 (2014) 1798–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Wu JW, Preuss C, Wang SP, Yang H, Ji B, Carter GW, Gladdy R, Andelfinger G, Mitchell GA, Epistatic interaction between the lipase-encoding genes Pnpla2 and Lipe causes liposarcoma in mice, PLoS genetics, 13 (2017) e1006716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Vaughan M, Effect of Pitressin on Lipolysis and on Phosphorylase Activity in Rat Adipose Tissue, Am J Physiol, 207 (1964) 1166–1168. [DOI] [PubMed] [Google Scholar]
- [96].Xu M, Chang HH, Jung X, Moro A, Chou CEN, King J, Hines OJ, Sinnett-Smith J, Rozengurt E, Eibl G, Deficiency in hormone-sensitive lipase accelerates the development of pancreatic cancer in conditional KrasG12D mice, BMC Cancer, 18 (2018) 797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Bailey AP, Koster G, Guillermier C, Hirst EM, MacRae JI, Lechene CP, Postle AD, Gould AP, Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila, Cell, 163 (2015) 340–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, Li Z, Hui J, Graham BH, Quintana A, Bellen HJ, Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration, Cell, 160 (2015) 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Mattijssen F, Georgiadi A, Andasarie T, Szalowska E, Zota A, Krones-Herzig A, Heier C, Ratman D, De Bosscher K, Qi L, Zechner R, Herzig S, Kersten S, Hypoxia-inducible lipid droplet-associated (HILPDA) is a novel peroxisome proliferator-activated receptor (PPAR) target involved in hepatic triglyceride secretion, The Journal of biological chemistry, 289 (2014) 19279–19293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].DiStefano MT, Danai LV, Roth Flach RJ, Chawla A, Pedersen DJ, Guilherme A, Czech MP, The Lipid Droplet Protein Hypoxia-inducible Gene 2 Promotes Hepatic Triglyceride Deposition by Inhibiting Lipolysis, The Journal of biological chemistry, 290 (2015) 15175–15184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].DiStefano MT, Roth Flach RJ, Senol-Cosar O, Danai LV, Virbasius JV, Nicoloro SM, Straubhaar J, Dagdeviren S, Wabitsch M, Gupta OT, Kim JK, Czech MP, Adipocyte-specific Hypoxia-inducible gene 2 promotes fat deposition and diet-induced insulin resistance, Molecular metabolism, 5 (2016) 1149–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Dijk W, Mattijssen F, de la Rosa Rodriguez M, Loza Valdes A, Loft, Mandrup S, Kalkhoven E, Qi L, Borst JW, Kersten S, Hypoxia-inducible lipid droplet-associated (HILPDA) is not a direct physiological regulator of lipolysis in adipose tissue, Endocrinology, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Gimm T, Wiese M, Teschemacher B, Deggerich A, Schodel J, Knaup KX, Hackenbeck T, Hellerbrand C, Amann K, Wiesener MS, Honing S, Eckardt KU, Warnecke C, Hypoxia-inducible protein 2 is a novel lipid droplet protein and a specific target gene of hypoxia-inducible factor-1, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 24 (2010) 4443–4458. [DOI] [PubMed] [Google Scholar]
- [104].Maier A, Wu H, Cordasic N, Oefner P, Dietel B, Thiele C, Weidemann A, Eckardt KU, Warnecke C, Hypoxia-inducible protein 2 Hig2/Hilpda mediates neutral lipid accumulation in macrophages and contributes to atherosclerosis in apolipoprotein E-deficient mice, FASEB J, 31 (2017) 4971–4984. [DOI] [PubMed] [Google Scholar]
- [105].van Dierendonck X, de la Rosa Rodriguez MA, Georgiadi A, Mattijssen F, Dijk W, van Weeghel M, Singh R, Borst JW, Stienstra R, Kersten S, HILPDA Uncouples Lipid Droplet Accumulation in Adipose Tissue Macrophages from Inflammation and Metabolic Dysregulation, Cell Rep, 30 (2020) 1811–1822 e1816. [DOI] [PubMed] [Google Scholar]
- [106].Cui C, Fu K, Yang L, Wu S, Cen Z, Meng X, Huang Q, Xie Z, Hypoxia-inducible gene 2 promotes the immune escape of hepatocellular carcinoma from nature killer cells through the interleukin-10-STAT3 signaling pathway, J Exp Clin Cancer Res, 38 (2019) 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Padmanabha Das KM, Wechselberger L, Liziczai M, De M la Rosa Rodriguez, G.F. Grabner, C. Heier, R. Viertlmayr, C. Radler, J. Lichtenegger, R. Zimmermann, J.W. Borst, R. Zechner, S. Kersten, M. Oberer, Hypoxia-inducible lipid droplet-associated protein inhibits adipose triglyceride lipase, J Lipid Res, 59 (2018) 531–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, Tseng YY, Deasy R, Kost-Alimova M, Dancik V, Leshchiner ES, Viswanathan VS, Signoretti S, Choueiri TK, Boehm JS, Wagner BK, Doench JG, Clish CB, Clemons PA, Schreiber SL, A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis, Nat Commun, 10 (2019) 1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Zhang X, Xie X, Heckmann BL, Saarinen AM, Gu H, Zechner R, Liu J, Identification of an intrinsic lysophosphatidic acid acyltransferase activity in the lipolytic inhibitor G0/G1 switch gene 2 (G0S2), FASEB J, 33 (2019) 6655–6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].de la Rosa Rodriguez MA, Kersten S, Regulation of lipid droplet homeostasis by hypoxia inducible lipid droplet associated HILPDA, Biochim Biophys Acta Mol Cell Biol Lipids, (2020) 158738. [DOI] [PubMed] [Google Scholar]
- [111].Hue L, Taegtmeyer H, The Randle cycle revisited: a new head for an old hat, Am J Physiol Endocrinol Metab, 297 (2009) E578–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Scaglia N, Tyekucheva S, Zadra G, Photopoulos C, Loda M, De novo fatty acid synthesis at the mitotic exit is required to complete cellular division, Cell Cycle, 13 (2014) 859–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Rankin EB, Giaccia AJ, Hypoxic control of metastasis, Science, 352 (2016) 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, Berenguer A, Prats N, Toll A, Hueto JA, Bescos C, Di Croce L, Benitah SA, Targeting metastasis-initiating cells through the fatty acid receptor CD36, Nature, 541 (2017) 41–45. [DOI] [PubMed] [Google Scholar]
- [115].Wright HJ, Hou J, Xu B, Cortez M, Potma EO, Tromberg BJ, Razorenova OV, CDCP1 drives triple-negative breast cancer metastasis through reduction of lipid-droplet abundance and stimulation of fatty acid oxidation, Proc Natl Acad Sci U S A, 114 (2017) E6556–E6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Lee CK, Jeong SH, Jang C, Bae H, Kim YH, Park I, Kim SK, Koh GY, Tumor metastasis to lymph nodes requires YAP-dependent metabolic adaptation, Science, 363 (2019) 644–649. [DOI] [PubMed] [Google Scholar]
- [117].Wang YN, Zeng ZL, Lu J, Wang Y, Liu ZX, He MM, Zhao Q, Wang ZX, Li T, Lu YX, Wu QN, Yu K, Wang F, Pu HY, Li B, Jia WH, Shi M, Xie D, Kang TB, Huang P, Ju HQ, Xu RH, CPT1A-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis, Oncogene, 37 (2018) 6025–6040. [DOI] [PubMed] [Google Scholar]
- [118].van Weverwijk A, Koundouros N, Iravani M, Ashenden M, Gao Q, Poulogiannis G, Jungwirth U, Isacke CM, Metabolic adaptability in metastatic breast cancer by AKR1B10-dependent balancing of glycolysis and fatty acid oxidation, Nat Commun, 10 (2019) 2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Kim SH, Wang D, Park YY, Katoh H, Margalit O, Sheffer M, Wu H, Holla VR, Lee JS, DuBois RN, HIG2 promotes colorectal cancer progression via hypoxia-dependent and independent pathways, Cancer Lett, 341 (2013) 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Zandbergen F, Mandard S, Escher P, Tan NS, Patsouris D, Jatkoe T, Rojas-Caro S, Madore S, Wahli W, Tafuri S, Muller M, Kersten S, The G0/G1 switch gene 2 is a novel PPAR target gene, Biochem J, 392 (2005) 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Heckmann BL, Zhang X, Saarinen AM, Schoiswohl G, Kershaw EE, Zechner R, Liu J, Liver X receptor alpha mediates hepatic triglyceride accumulation through upregulation of G0/G1 Switch Gene 2 expression, JCI insight, 2 (2017) e88735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Heckmann BL, Zhang X, Saarinen AM, Liu J, Regulation of G0/G1 Switch Gene 2 (G0S2) Protein Ubiquitination and Stability by Triglyceride Accumulation and ATGL Interaction, PloS one, 11 (2016) e0156742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Siderovski DP, Blum S, Forsdyke RE, Forsdyke DR, A set of human putative lymphocyte G0/G1 switch genes includes genes homologous to rodent cytokine and zinc finger protein-encoding genes, DNA Cell Biol, 9 (1990) 579–587. [DOI] [PubMed] [Google Scholar]
- [124].Russell L, Forsdyke DR, A human putative lymphocyte G0/G1 switch gene containing a CpG-rich island encodes a small basic protein with the potential to be phosphorylated, DNA Cell Biol, 10 (1991) 581–591. [DOI] [PubMed] [Google Scholar]
- [125].Kioka H, Kato H, Fujikawa M, Tsukamoto O, Suzuki T, Imamura H, Nakano A, Higo S, Yamazaki S, Matsuzaki T, Takafuji K, Asanuma H, Asakura M, Minamino T, Shintani Y, Yoshida M, Noji H, Kitakaze M, Komuro I, Asano Y, Takashima S, Evaluation of intramitochondrial ATP levels identifies G0/G1 switch gene 2 as a positive regulator of oxidative phosphorylation, Proc Natl Acad Sci U S A, 111 (2014) 273–278. [DOI] [PMC free article] [PubMed] [Google Scholar]