

Abstract
The association of hormonal contraception with increased risk of inflammatory bowel disease (IBD) observed in females suggests involvement of ovarian hormones, such as estradiol, and the estrogen receptors in the progression of intestinal inflammation. Here, we investigated the effects of prophylactic SERM2 and estradiol supplementation in dextran sulfate sodium‐induced colitis using mice with intact ovaries and ovariectomized (OVX) female mice. We found that graded colitis score was threefold reduced in the OVX mice, compared to mice with intact ovaries. Estradiol supplementation, however, aggravated the colitis in OVX mice, increasing the colitis score to a similar level than what was observed in the intact mice. Further, we observed that immune infiltration and gene expression of inflammatory interleukins Il1b, Il6, and Il17a were up to 200‐fold increased in estradiol supplemented OVX colitis mice, while a mild but consistent decrease was observed by SERM2 treatment in intact animals. Additionally, cyclo‐oxygenase 2 induction was increased in the colon of colitis mice, in correlation with increased serum estradiol levels. Measured antagonist properties of SERM2, together with the other results presented here, indicates an exaggerating role of ERα signaling in colitis. Our results contribute to the knowledge of ovarian hormone effects in colitis and encourage further research on the potential use of ER antagonists in the colon, in order to alleviate inflammation.
Keywords: colitis, dextran sulfate, estradiol, estrogen receptor modulators, inflammatory bowel diseases, receptors, estrogen
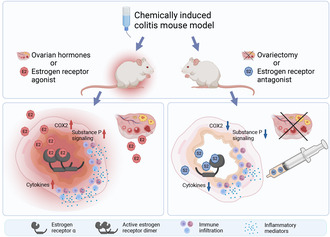
Hormonally intact females and E2 supplemented ovariectomized mice suffered from increased intestinal inflammation, while prophylactic treatment with ERα antagonist produced a mild but consistent anti‐inflammatory effect in hormonally intact mice and ovariectomy protected the mice from severe inflammation.
Abbreviations
- AUC
Area under the curve
- CCL2
C‐C motif chemokine ligand 2
- CD
Crohn's disease
- COX2
Cyclo‐oxygenase 2
- DAI
Disease activity index
- DSS
Dextran sulfate sodium
- E2
Estradiol
- ER
Estrogen receptor
- GR
Glucocorticoid receptor
- IBD
Inflammatory bowel disease
- IFN
Interferon
- IHC
Immunohistochemical staining
- IL
Interleukin
- MRC‐1
Mannose receptor 1
- OVX
Ovariectomized
- PGR
Progesterone receptor
- S2
SERM2
- TGF
Transforming growth factor
- TNF
Tumor necrosis factor
- UC
Ulcerative colitis
1. INTRODUCTION
The two major subtypes of chronic inflammatory bowel disease (IBD), Crohn's disease (CD) and ulcerative colitis (UC), are immunologically mediated diseases that have complex pathophysiology and etiology. 1 IBD incidence and prevalence had increased during the end of the 20th century in industrialized countries. 2 Ovarian hormones, including 17β‐estradiol (E2), have been associated with the occurrence of several chronic inflammatory diseases and an increased clearance of pathogens in females. 3 Further, global epidemiological data has revealed a higher prevalence of IBD and an earlier peak prevalence age in females. 4 CD occurs more often in males prepuberty and in females during the reproductive years, while UC prevalence is similar between sexes until after the age of 45, after which it is more common in males. 5 However, the use of oral contraceptives increases risk of both UC and CD. 6 Risk increase was reported especially with E2 containing contraceptives and was associated with the duration of use. 7
E2 and other estrogens signal through nuclear estrogen receptors (ER), which are expressed by genes ESR1 and ESR2 as genomically signaling subtypes ERα and ERβ. Besides reproductive tissues and bone, the ERs regulate metabolism, cardiovascular tone, and the immune system. ERα and ERβ are expressed in several cell types of both the innate and adaptive immune systems. 8 , 9 Decreased ERβ 10 , 11 , 12 and increased ERα 13 expressions have been reported in IBD. However, the effect of ER signaling in inflammation is not completely understood and both beneficial and detrimental effects have been observed in preclinical models. Alleviation of intestinal inflammation was reported in studies where mice were treated with E2. 14 , 15 On the contrary, ovariectomy (OVX) was observed to protect mice from colitis and associated cancer progression, while E2 supplementation exacerbated inflammation. 16 E2 treatment also induced ERα‐dependent proinflammatory activity in peritoneal macrophages. 17 When sex‐specific effects of ER signaling were evaluated, proinflammatory effects of ERα signaling were observed in female mice, but ERα‐independent exacerbation of inflammation in male colitis mice. 18 In another study, ERα inhibition improved recovery after DSS‐colitis in males. 19
Selective estrogen receptor modulators (SERMs), such as raloxifene and tamoxifen, have been utilized especially in treating and preventing breast cancer. These drugs are partial antagonist compounds; thus, they antagonize estrogen in mammary glands, while functioning as agonists in bone. When a ligand binds to the ER, ligand‐induced allosteric effects influence the transcription complex composition and this alters the response of hormone receptor modulators. Further, tissue‐specific responses can depend on receptor subtype ratio and by the coregulatory molecules expressed in the cell. 20 Previously, SERMs have been reported to modulate inflammation in animal models of IBD, 21 , 22 autoimmune encephalomyelitis, 23 , 24 and ankylosing spondylitis. 25 Additionally, clinical data suggests that tamoxifen may decrease C‐reactive protein levels in healthy females. 26
Here, we studied the role of ER ligands in acute DSS‐induced colitis in mice, which is a well‐established model of IBD. 27 Previously introduced hormone receptor modulator, SERM2, promoted an anti‐inflammatory macrophage phenotype and inhibited T cell proliferation. 28 In addition, we found that prophylactic administration of SERM2 ameliorated dextran sulfate sodium (DSS)‐induced colitis in male mice. 29 As the disease course of IBD is one of relapse and remission, it is crucial to treat relapse, but maintaining remission is also important. In this study, the aim was to investigate the role of long‐term supplementation of E2 and prophylactic SERM2 on acute colitis in female mice and to compare SERM2 effects to those of E2 in OVX animals. Another interest was to evaluate the feasibility of targeting the estrogen receptors in inflammation and to study the effects of estrogen receptor signaling on the severity of the developing colitis, since previously published reports have been conflicting. We found that ERα antagonism and hormone deprivation were associated with less severe colitis, while ER agonist E2 aggravated the disease.
2. MATERIALS AND METHODS
2.1. Mice and experimental design
Housing was carried out by the Central Animal Laboratory of the University of Turku. Eight‐week‐old female FVB/NRj mice (Janvier labs, France) were fed soy‐free diet and water ad libitum. All procedures were performed according to the animal study protocol approved by The State Provincial Office of South Finland (license numbers: ESAVI/3956/04.10.07/2016 and ESAVI/16359/2019).
2.1.1. Analysis of drug absorption and secretion
For serum analysis, SERM2 gavage at doses of 1, 10 and 30 mg/kg (n = 5) were repeated twice with a 72‐h interval. Samples were obtained 24 h after the second dose by collecting the blood at CO2 sacrifice through cardiac puncture. SERM2 was gavage‐fed to mice at doses of 1, 10 or 30 mg/kg (n = 3) in corn oil. Urine was collected in metabolic cages between intervals 0.5–6.5 h, 24–30 h and 48–54 h.
2.1.2. Colitis experiments
Mice were allocated to weight‐balanced treatment groups by a technician outside of the research group. All cages contained animals from within one treatment groups to avoid contamination. Treatment groups are described in Table 1. Researchers were not blinded during the experiment since they were administering the SERM2 and vehicle to the animals.
TABLE 1.
Treatment groups and number of mice used in the experiments.
| Intact | OVX | Vehicle | SERM2 | DSS | DSS + SERM2 | DSS + E2 | |
|---|---|---|---|---|---|---|---|
| Experiment 1 | X | n = 4 | n = 5 | n = 7 | n = 7 | ||
| Experiment 2 | X | n = 4 | n = 8 | n = 8 | n = 8 |
Two separate experiments were performed, the first in intact, and the second in OVX mice. SERM2 (Forendo Pharma, Finland, molecular weight 450 g/mol) was administered by oral gavage five (intact) and six times (OVX) in corn oil (Sigma Aldrich Ltd, St. Louis, MO) at a dose of 10 mg/kg with 72 h intervals. This dosing scheme was chosen based on SERM2 levels in urine and serum in healthy mice. Vehicle and DSS groups were gavaged with corn oil. Prior to ovariectomy, the mice were medicated s.c. with carprofen and buprenorphine (5 mg/kg and 50 μg/kg, respectively) and postoperative pain was managed for 3 days with s.c. carprofen. Isoflurane was used for anesthesia. Subcutaneous E2 implants (0.25 μg/day, Medrod, PreclinApps Ltd, Raisio, Finland) were placed at OVX, and animals were allowed recovery for 2 weeks before the induction of colitis.
The mice drank 2.5% DSS w/v (40 kDa, TdB Consultancy AB, Uppsala, Sweden) in autoclaved water for 7 days. Mice recovered from the inflammation‐inducing agent for 24 h (Figure 1A,F) before being sacrificed by CO2. Blood was collected by cardiac puncture. The colon length was measured, and uterus and ovaries were weighed.
FIGURE 1.
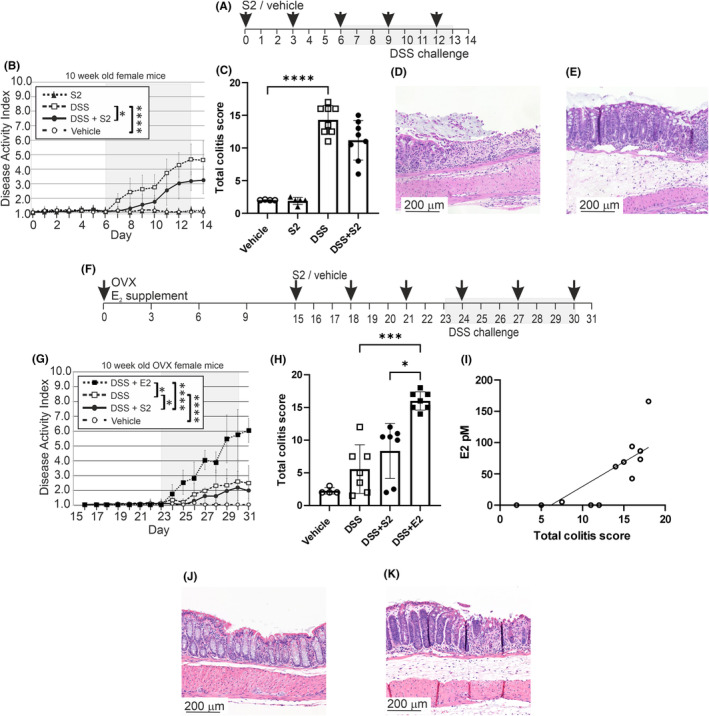
E2 and SERM2 effects on DSS colitis activity. Schematic diagram of intact mice colitis experiment, where shaded area represents 7‐day DSS challenge (A). In the intact mice, DAI AUC was increased by DSS (n = 8) compared to vehicle control (n = 4), and the DSS + SERM2 (“S2”, n = 8) was statistically significantly lower compared to DSS control (B). The total colitis score (endpoint DAI combined with the histological scoring) showed a similar pattern (C). Representative HE staining of intact DSS colon with severe erosion and immune infiltration (D) and DSS + SERM2 colon, where architecture is intact and immune infiltration is evident (E). Schematic diagram of OVX mice colitis experiment, where again, shaded area represents 7‐day DSS challenge (F). In the OVX animals DSS (n = 7) induced an increase of the DAI score AUC compared to vehicle control (n = 4). Compared to DSS‐group, DSS + SERM2 (n = 7) mice exhibited slightly decreased disease activity, but in the DSS + E2 (n = 7) mice, AUC was significantly increased (G). Total colitis score of OVX mice showed no difference between DSS control and DSS + SERM2, while in the DSS + E2 mice score was statistically significantly increased (H). E2 serum levels correlated with the total colitis score (I). Representative HE staining of OVX DSS colon with minor signs of inflammation (J). Representative HE staining of OVX DSS + E2 colon with edema, hyperproliferation, extensive immune infiltration and crypt loss (K). Data presented as the mean ± SD, statistical significance of AUC's and scoring data was calculated with ordinary one‐way ANOVA, Brown‐Forsythe and Welch ANOVA or Kruskal‐Wallis nonparametrical test according to distribution and variance. Statistical significance was defined as *p ≤ .05, **p ≤ .01, ***p ≤ .001, ****p ≤ .0001.
2.1.3. Disease activity index
Disease activity index (DAI) was used to quantify colitis severity each day throughout the experiment as described previously. 30 DAI was calculated as a sum of measured and graded body weight loss and graded intestinal symptoms and is described in Table 2.
TABLE 2.
Areas of interest and scoring of histological markers of inflammation.
| Disease and histological assessment | Score | Definition |
|---|---|---|
| Weight loss | 1 | 1 point per 5% of body weight loss |
| Stool consistency | 1 | Normal |
| 2 | Soft | |
| 3 | Slightly loose | |
| 4 | Liquid | |
| Blood in stool | 0 | None |
| 1 | Small amount of blood | |
| 2 | Blood throughout | |
| 3 | Clotted blood at anus | |
| 4 | Fresh blood on mice or on bedding | |
| Immune infiltration | 0 | No infiltration |
| 1 | Inflammatory cells in epithelia | |
| 2 | Moderate areas of infiltration | |
| 3 | Severe and acute inflammation | |
| Edema | 0 | No edema |
| 1 | Slight edema | |
| 2 | Moderate edema | |
| 3 | Extensive edema | |
| Erosion | 0 | No erosion |
| 1 | Some erosion of epithelium | |
| 2 | Moderate erosion of epithelium | |
| 3 | Extensive areas with eroded epithelia | |
| Hyperproliferation | 0 | No hyperproliferation |
| 1 | Slightly elongated crypts | |
| 2 | Moderately elongated crypts | |
| 3 | Extensive areas with hyperproliferation |
2.2. Histological and immunohistochemical analysis
Formaldehyde fixed distal colon was embedded in paraffin and cut in 4‐μm sections prior to hematoxylin and eosin staining. Samples were scanned using Pannoramic 250 and P1000 slide scanners (3DHistech) with 20X objective and then analyzed using Pannoramic viewer (Histech, version 1.15.4). Scoring was performed independently by two blinded investigators. A total colitis score was calculated by combining DAI at sacrifice and the histological score. The scoring method used is found in Table 2.
For immunohistochemistry, acidic heat‐induced antigen retrieval of the deparaffinized sections was performed, followed by protein blocking (5% normal goat serum) and incubating the primary antibodies (Table S1). After blocking of endogenous peroxidases, HRP‐labeled secondary antibody was used. DAB (Pierce™ DAB Substrate Kit) was used as chromogen, after which the tissue was counterstained by hematoxylin, mounted, and scanned by Pannoramic P1000 at 40x resolution. Antigen positive cells were counted from representative areas of middle or distal colon. All images were consistently color adjusted using FIJI. 31 A mean of five areas per sample was used to determine the average number of stained cells/0.3 mm2 in QuPath. 32
2.3. Gene expression
Distal colon samples were homogenized and total RNA and protein fractions were extracted using Nucleospin RNA and protein kit (Macherey‐Nagel, Germany) according to the manufacturer's protocol. Reverse transcription was performed using High‐Capacity cDNA Reverse Transcription Kit according to the manufacturer's protocol (Applied Biosystems, Foster City, CA, USA). RT‐qPCR was performed on a Taqman Universal Mastermix II, no. UNG (Applied Biosystems, Foster City, CA, USA) on a CFX96 thermal cycler (Bio‐rad Laboratories, Hercules, USA). Primers and TaqMan assays (Applied Biosystems, Foster City, CA, USA) are listed in Table S2. The ΔΔCt method was applied for quantification of gene expression using β‐actin or Rn18s gene as reference. The ΔEsr2/ΔEsr1 transcription‐level ratio was calculated using ΔCq fold change values.
2.4. Circulating cytokines
Blood samples were allowed to coagulate, and then centrifuged for 10 min at 1600 rpm to collect serum. Circulating cytokines were measured using Mouse Th1 Th2 10‐plex ELISA bead assay (Thermo Fisher Scientific, Vienna, Austria) according to manufacturer's instructions and using the Luminex 200 system (Luminex Corporation, Austin, TX, USA).
2.5. Mass‐spectrometric analyses
Mass‐spectrometric analyses were outsourced and methods were described in Appendix. Analysis of serum estradiol from intact, OVX, and OVX + E2 mice from the colitis and pharmacokinetics experiment (untreated control) can be found in Section 11.1.1. Drug absorption and secretion can be found in Section 11.1.2, and substance P levels in colon in Section 11.1.3.
2.6. Protein expression
Total extracted protein from distal colon (14 μg) in Laemmli sample buffer (Bio‐rad Laboratories, Hercules, USA) with 10% beta‐mercaptoethanol (Sigma) was separated by SDS‐PAGE and transferred onto PVDF membranes, which then was blocked using 2% BSA and incubated with primary and secondary antibodies (Table S1). SuperSignal™ West Pico PLUS Chemiluminescent Substrate (34577, Thermo Fisher) was used to induce the signal and images were acquired by Azure Sapphire biomolecular imager. Images were adjusted for contrast and brightness and analyzed using ImageJ 2.3.0/1.53q. The intensity was normalized to loading control intensities. In order to correct for intensities between membranes, a standard sample was used on all membranes. Representative‐pooled Western blots images are presented in Figure S1.
2.7. Receptor coactivator assays
Receptor activation was measured using LanthaScreen® TR‐FRET ER Coactivator Assays (Thermo Fisher) and a 12‐point, 3‐fold dilution series of SERM2, starting from 10 μM (ERs) or 200 μM (PR, GR). In order to determine the EC50 value, SERM2 dilution series was incubated together with a GST‐tagged recombinant ligand binding domain, a terbium‐labeled anti‐GST ab, and a fluorescein‐labeled coactivator peptide overnight. Time‐resolved fluorescence was measured using EnSight plate reader and Kaleido software (PerkinElmer) with a measurement height of 8 mm, 200 flashes, 100 μs delay time and 200 μs integration time. Three hundred and Forty nanometer excitation wavelength was used for measuring both 520 nm (fluorescein) and terbium (495 nm) emission. In order to determine the IC50 value, an agonist was added to all reactions. Kits, agonists, and concentrations were listed in Table S3.
2.8. Experimental design and statistical analyses
All animal experiments, pharmacokinetics, the analyses of DAI, and total colitis score as well as the cytokine and immune‐related molecular analyses were conducted according to a preset plan. The criteria for excluding animals from the experiment were predetermined. If animals were to lose more than 20% weight or deteriorate in health, they would be euthanized and excluded. No animals were excluded from the animal experiment or from the DAI or histological analysis of inflammation. However, a few samples were excluded from further analysis because of sample quality issues or after statistical outlier analysis.
For statistical analysis, normal distribution and variance of the data were tested and according to this, the appropriate parametrical or nonparametrical test was chosen. For normally distributed data with equal variances, an ordinary one‐way ANOVA was performed. Normally distributed data with unequal variances was tested using Brown‐Forsythe and Welch ANOVA and data not normally distributed was tested using the Kruskal–Wallis test. Tukey's post hoc test was used for multiple comparisons of all groups. The area under curve was used to analyze differences in DAI. EC50 and IC50 analyses were performed using nonlinear regression curves where concentration was plotted against response with a four‐parameter variable slope. The association between serum E2 levels and Ptgs2 as well as COX2 was carried out using nonparametric Spearman's Rank Correlation Coefficient. Statistical outliers were identified using ROUT with Q = 1% for all the data. RT‐qPCR data was normalized to DSS group as a part of the ΔΔCt analysis and neuropeptide substance P peak areas were normalized to reference peptides. Statistical analyses were performed using GraphPad Prism v. 8 and 10. All data presented as mean ± SD, and each dot represents a biological replicate. Statistical significance in each analysis was defined as two‐tailed p‐value, *p ≤ .05, **p ≤ .01, ***p ≤ .001, ****p < .0001.
2.9. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY, 33 and are permanently archived in The Concise Guide to PHARMACOLOGY 2023/24: Nuclear hormone receptors. 34
3. RESULTS
3.1. Estradiol increased, and SERM2 reduced disease activity in DSS colitis
In order to investigate the effect of SERM2 in a female hormone environment, mice with intact ovaries were treated with SERM2 before and during the induction of colitis with DSS (Figure 1A). DSS elevated DAI area under curve (AUC) of intact mice compared to vehicle, and DSS + SERM2 reduced the AUC of DAI compared to DSS (vehicle 8.4 vs DSS 24.8, DSS vs DSS + SERM2 16.3, Figure 1B). Distal colon HE‐staining was analyzed to further assess the degree of inflammation. Total colitis score, combining endpoint DAI and histological score, was increased sevenfold by DSS compared to vehicle, but only a trend toward reduction could be observed in DSS + SERM2 compared to DSS (Figure 1C). Figure 1D,E depicts DSS as well as DSS + SERM2‐treated colons, where crypt loss, erosion, heavy immune infiltration, and edema were observed, but to a lesser extent in the SERM2‐treated group. In vehicle‐treated mice, the epithelium was intact, with scattered immune cells and no edema (Figure S2).
In order to investigate the effects of ER ligands in a hormone depleted environment, OVX mice were treated with SERM2 or E2 before and during the induction of colitis with DSS (Figure 1F). AUC of DAI was increased by DSS compared to vehicle (22.3 and 15.7, respectively), and AUC was decreased in DSS + SERM2 mice compared to DSS (19.3 and 22.3 respectively), while DSS + E2 increased AUC compared to DSS (36.0 and 22.3, respectively, Figure 1G). The total colitis score was moderately increased by DSS and in the DSS + E2 mice, total colitis score increased approximately threefold (Figure 1H). The effect of the OVX procedure and E2 supplementation was verified by measuring weights of the uteri, as well as by mass‐spectrometric analysis of serum E2 levels. Ovariectomy decreased serum E2 of the mice, and E2 supplementation statistically significantly increased the serum E2 levels compared to both intact and OVX mice (Figure S2). Serum estradiol levels were found to positively correlate with the total colitis score in OVX mice (r = .89, p = .0003, Figure 1I). Minor immune infiltration, but well‐preserved tissue architecture can be detected in the colon of DSS‐treated OVX mice (Figure 1J), while immune infiltration, edema, and hyperproliferation could be observed in the colon of DSS + E2 OVX mice (Figure 1K).
Weight loss was negligible, and none of the treatments, except OVX, affected the weight of the uteri and ovaries of the intact mice (Figure S2). Separate DAI and histological scoring in intact mice can be found in Figure S2. In the OVX mice, weight loss was evident in the DSS + E2 group (Figure S2). The uterus mass increased in SERM2‐treated mice, and E2 increased uterus weights further (Figure S2). Separate DAI and histological scoring in OVX mice can be found in Figure S2. Oral administration of 1, 10, and 30 mg/kg SERM2 resulted in absorption from the gut and measurable serum levels (1.60, 24.44, and 195.28 ng/mL, equaling to 3.6, 53.3, and 434.0 nM, respectively, Figure S3) and excretion to urine (Figure S3).
3.2. Estradiol, and to a lesser extent SERM2, induced colon shortening and immune infiltration in OVX mice
DSS‐induced inflammation caused shortening of the colon in mice with intact ovaries (Figure 2A). Additionally, immune infiltration was examined by IHC staining of monocyte and macrophage lineage cells (F4/80+) as well as T lymphocytes (CD3ε). Neither F4/80+ cells nor CD3ε + T cells in mucosa and submucosa were affected by DSS + SERM2 (Figure 2B,C). Infiltration of F4/80+ monocytes and macrophages (Figure 2D) and CD3ε + T cells (Figure 2E) can be observed in representative images of the inflamed area of the mucosa and in the submucosal edema.
FIGURE 2.
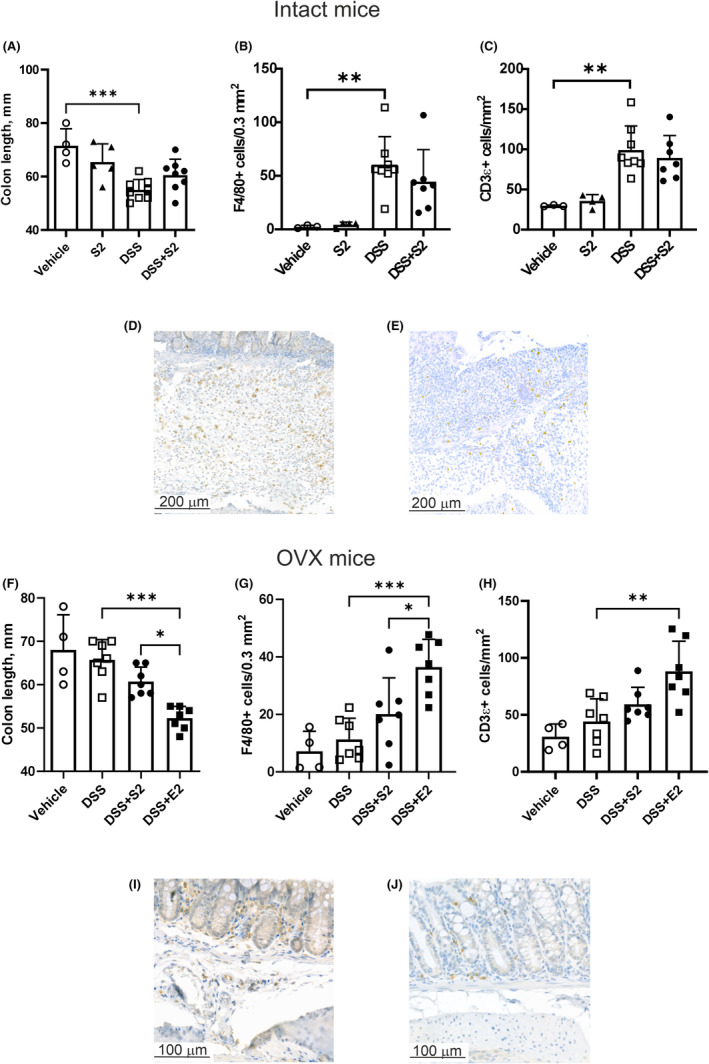
Colon shortening and immune infiltration analysis. Colon length was decreased by DSS (n = 8) in intact mice and SERM2 (n = 5, DSS + SERM2 n = 7) did not significantly alter the length (A). F4/80+ (B) and CD3ε + (C) cells increased with DSS in intact mice. Representative images of anti‐F4/80 (D) and anti‐CD3ε (E) immunostaining in intact DSS mice. In OVX mice, DSS (n = 7) did not affect colon length compared to vehicle (n = 4), but E2 (n = 7) reduced colon length both compared to DSS + SERM2 (n = 7) and DSS (F). F4/80+ (G) and CD3ε + (H) cell counts increased by DSS + E2 (n = 7) and trended toward an increase in DSS + SERM2. Representative images of F4/80 (I) and CD3ε (J) in the colons of OVX DSS mice. In figures B and C, one data point in DSS + SERM2 group is missing due to loss of sample. Data presented as mean ± SD, statistical significance was calculated with ordinary one‐way ANOVA, Brown–Forsythe and Welch ANOVA or Kruskal–Wallis nonparametrical test according to distribution and variance. Statistical significance was defined as *p ≤ .05, **p ≤ .01, ***p ≤ .001, ****p ≤ .0001.
In the OVX mice, a reduction in colon length was observed in the DSS + E2 animals, compared to DSS and less pronounced compared also to DSS + SERM2 (Figure 2F). The DSS + SERM2‐induced increase in cell numbers was not statistically significant for neither F4/80+ (Figure 2G) nor CD3ε+, while the DSS + E2‐induced increase was (Figure 2H). F4/80+ and CD3ε + cells were approximately 20% and 50% decreased, respectively, in the OVX DSS‐treated animals (Figure 2I,J), compared to intact. Scattered F4/80+ and CD3ε + cells were visible in the lamina propria of vehicle‐treated animals (Figure S4). Secondary antibody controls for immunohistochemical staining in Figure S4 (F4/80) and Figure S4 (CD3ε and COX2).
3.3. SERM2 effects on immune mediators are opposite in mice with ovaries compared to OVX mice
Next, the gene expression of cell phenotype and immune activation markers was measured. SERM2 alone increased the transcription of mannose receptor 1 (Mrc1) mRNA, a marker of anti‐inflammatory macrophages (Figure 3A). Genes encoding for transforming growth factor (TGF) β and interleukin (IL)‐10 were elevated in DSS mice, while DSS + SERM2 reduced the expression of Tgfb compared to DSS (Figure 3B,C). The expression of the inflammatory cytokine tumor necrosis factor α gene (Tnf) was increased in colitis (Figure 3D), as were genes such as Il1b, Il6, and interferon γ (Ifng), but these had a consistent trend toward downregulation in DSS + SERM2 group (Figure 3E–G). Also, the regulatory T cell transcription factor Foxp3 increase was partially reversed by DSS + SERM2 (Figure 3H).
FIGURE 3.
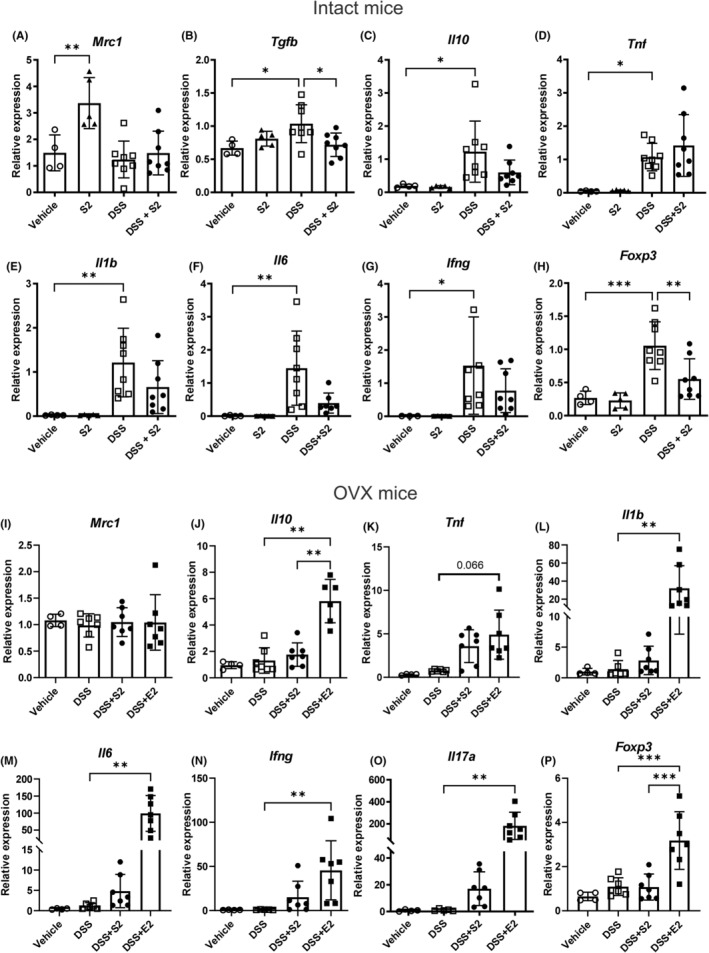
Immune activity‐associated gene expression in distal colon. Gene expression of Mrc1 was increased by SERM2 (“S2”, n = 5) in intact mice (A). Tgfb expression increased with DSS (n = 8) and decreased in the DSS + SERM2 (n = 8, B). Tnf (D) was not affected by DSS + SERM2, while Il10 (C), Il1b (E), Il6 (F), Ifng (G) expression increased by DSS and demonstrated consistent trends toward downregulation by DSS + SERM2, while only Foxp3 (H) the altered statistically significantly. In OVX mice, the treatment did not affect Mrc1 expression (I). Il10, Tnf, Il1b, Il6, Ifng, and Il17a (K–O) expressions were statistically significantly increased in the DSS + E2 group and trended toward an increase by DSS + SERM2, while FoxP3 increased only by DSS + E2 (P). Please note the two‐part y‐axis in figures (L, M, O). Statistical outliers were identified using ROUT with Q = 1% and one datapoint was removed in the DSS group of figure (F) and DSS + E2 group of figure (K). Data was normalized to DSS group as a part of the ΔΔCt analysis and presented as mean ± SD, statistical significance was calculated with ordinary one‐way ANOVA, Brown‐Forsythe and Welch ANOVA or Kruskal‐Wallis nonparametrical test according to distribution and variance. Statistical significance was defined as *p ≤ .05, **p ≤ .01, ***p ≤ .001, ****p ≤ .0001.
In OVX mice, Mrc1 was not altered by any treatment (Figure 3I) and anti‐inflammatory Il10 gene expression increased in the DSS + E2 mice compared to DSS and DSS + SERM2 (Figure 3J). OVX protected mice from DSS‐induced inflammatory signaling in the colon, and Tnf, Il1b, Il6, Ifng and Il17a increased by DSS + E2 and trended toward an increase by DSS + SERM2 (Figure 3K–O). Interestingly, inflammation‐limiting Foxp3 increased in the DSS + E2 mice compared to DSS but here, expression in DSS + SERM2 mice did not trend toward an increase (Figure 3P).
In the serum of intact mice, circulating IL‐5 was not elevated (Figure 4A), but IL‐6, IFNγ, TNFα and C‐C motif chemokine ligand (CCL) 2 were elevated in DSS mice (Figure 4B–E). SERM2 alone trended toward an increase of TNFα in serum, and this was further increased in DSS colitis (Figure 4D). Similar to gene expression in the colon, ovariectomy inhibited DSS‐induced changes in serum cytokine concentration. However, DSS + SERM2 and DSS + E2 after ovariectomy increased levels of IL‐5, IL‐6, IFNγ, and TNFα compared to DSS, although only IL‐6 and TNFα reached statistical significance (Figure 4F–I). Surprisingly, IL‐5 (Figure 4F) and TNFα (Figure 4I) serum levels were decreased after DSS treatment when compared to vehicle control. CCL2 was somewhat increased after DSS induction, but with major individual variation (Figure 4J).
FIGURE 4.
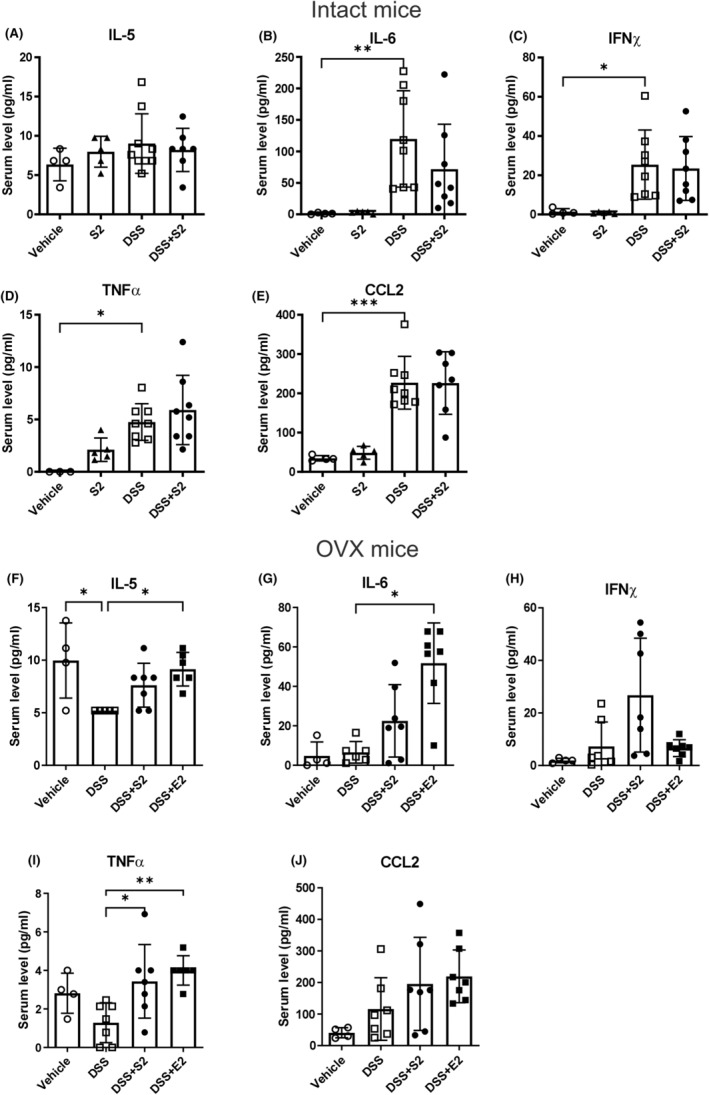
The serum cytokine response to DSS colitis. In intact animals, serum IL‐5 (A) was unaffected by treatment, while IL‐6, IFNγ, TNFα, and CCL2 increased by DSS (n = 8 vs vehicle n = 4, B–E). In OVX mice circulating IL‐5, IL‐6 and TNFα increased by both DSS + SERM2 (n = 7, “S2”) and DSS + E2 (n = 7), although SERM2 increase was statistically significant only in TNFα (F, G, I), The alterations in IFNγ and CCL2 (H, J) levels were not statistically significant. Statistical outliers were identified using ROUT with Q = 1% and one datapoint has been removed in each figure: A DSS + SERM2, D vehicle, F DSS and DSS + E2, G DSS. Data presented as mean ± SD, statistical significance was calculated with ordinary one‐way ANOVA, Brown–Forsythe and Welch ANOVA or Kruskal–Wallis nonparametrical test according to distribution and variance. Statistical significance was defined as *p ≤ .05, **p ≤ .01, ***p ≤ .001, ****p ≤ .0001.
We also wanted to expand the observation of inflammatory signaling to other cell types, such as neurons and epithelial cells, in order to further investigate the mechanism of the hormonally aggravated colitis. In intact mice, an increase in Ptgs2 (gene for COX2) by DSS and a decrease by DSS + SERM2 compared to DSS were observed, but as in cytokine gene expression, these failed to reach statistical significance (Figure 5A). However, a similar pattern but with statistically significant alterations could be detected in COX2 protein expression (Figure 5B). The distribution of COX2 expression was observed to be similar in intact and OVX mice, and localized to neurons, endothelia, epithelia, and immune cells (not shown), but also in gut‐associated lymphoid tissue as well as ulcers (Figure 5C). Gene expression of Tacr1, which is the receptor for neuroimmune peptide substance P, was increased statistically significantly by DSS, and the increase was partially prevented by DSS + SERM2 (Figure 5D). In OVX mice, the transcription of Ptgs2 was highly elevated by DSS + E2 (Figure 5E), while a statistically significant increase in COX2 protein was produced by both DSS + SERM2 and DSS + E2 compared to DSS, (Figure 5F). The strongest statistically significant association of serum E2 and the measured inflammation markers, was with COX2 gene and protein expression in OVX mice (r = .78, p = .0068 Ptgs2 and r = .69, p = .0326 COX2; Figure 5G,H, respectively). Tacr1 expression was not affected by DSS in OVX mice, contrary to what was seen in intact females. DSS + SERM2 elevated, and DSS + E2 further increased Tacr1 mRNA compared to DSS (Figure 5I). However, neuropeptide substance P did not respond significantly to treatment (Figure 5J). Substance P localized to neurons, epithelia, and immune cells both in intact and OVX mice (Figure 5K).
FIGURE 5.
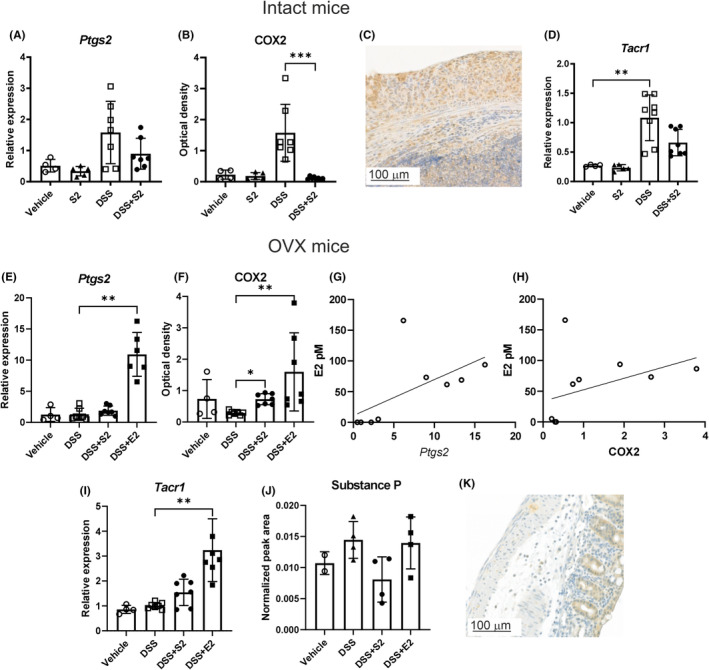
COX2 and substance P expression in distal colon. DSS + SERM2 (“S2”, n = 7) trended toward a decrease in Ptsg2 expression compared to DSS (n = 7, A) while COX2 protein levels measured by Western blots showed a similar pattern, though statistically significant (B). Representative image of COX2 immunostaining of the colon in intact and OVX mice (C). The expression of Tacr1 was statistically significantly increased by DSS compared to vehicle (n = 4) and trended toward decrease by DSS + SERM2 (D). In OVX mice the transcription of Ptgs2 was highly elevated in the DSS + E2 group (n = 8, E) and COX2 protein levels were elevated by both DSS + SERM2 and DSS + E2 (F). COX2 gene (G) and protein (H) expression correlated with serum E2 levels. Tacr1 levels were increased by DSS + E2 (I), while neuropeptide substance P was not affected by treatments (J). Representative image of substance P immunostaining in the colon of intact and OVX mice (K). Gene expression data was normalized to DSS group as a part of the ΔΔCt analysis and substance P peak area was normalized to reference peptide. Statistical outliers were identified using ROUT with Q = 1% and one datapoint was removed in DSS and DSS + SERM2 group of figures (A) and DSS in figure (B). Data presented as mean ± SD, statistical significance was calculated with ordinary one‐way ANOVA, Brown‐Forsythe and Welch ANOVA or Kruskal‐Wallis nonparametrical test according to distribution and variance. Statistical significance was defined as *p ≤ .05, **p ≤ .01, ***p ≤ .001, ****p ≤ .0001. The correlation of E2 with Ptgs2 and COX2 was calculated using Spearman's Rank Correlation Coefficient analysis.
3.4. SERM2 exerts antagonistic effects at ovarian hormone receptors and gene expression was influenced by both inflammation and receptor ligands
Receptor agonist and antagonist profile for SERM2 at ERα, ERβ, the progesterone receptor (PGR), and the glucocorticoid receptor (GR) were determined in order to identify the mechanism of the anti‐inflammatory effects. In biochemical nuclear receptor coactivator assays, SERM2 exerted antagonist effects at ERα and ERβ (IC50 88 nM and IC50 7.3 μM, respectively, Figure 6A) and PGR (IC50 7.3 μM, Figure 6B). Agonist effects were not detected for neither ERs at up to a 10 μM concentration (Figure S5) nor PGR and GR at up to a 200 μM concentration (Figure S5). GR antagonism was not detected at similar SERM2 levels (Figure S5).
FIGURE 6.
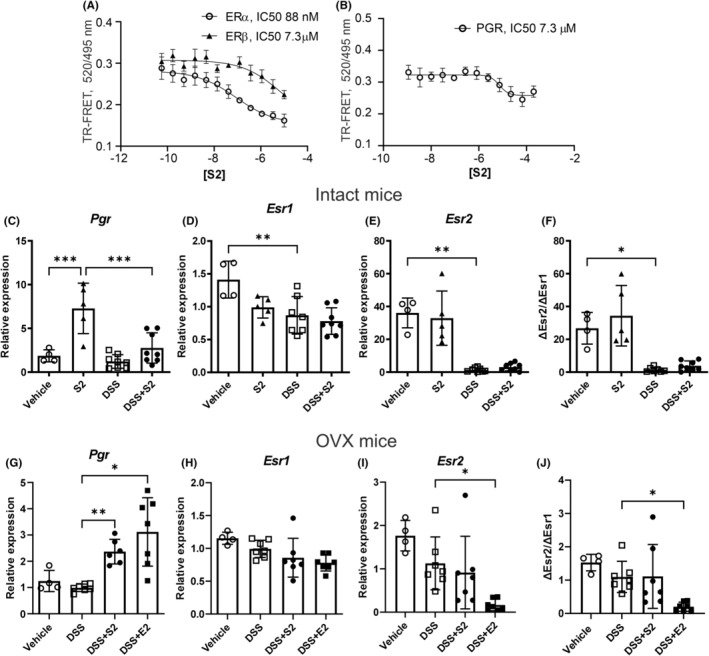
SERM2 IC50 values at ER and PGR and hormone receptor expression in the distal colon. In coactivator assays, SERM2 was observed to antagonize ERα, ERβ (A) and PGR (B). In both intact and OVX mice, Pgr was upregulated by both SERM2 (“S2”) and E2 compared to DSS (C, G). Esr1 was decreased by DSS compared to vehicle (D). Esr2 was downregulated in the DSS treated intact mice (E), and so was ΔEsr2/ΔEsr1 ratio (F). In OVX mice Esr1 was not affected by treatment (H). In the OVX mice DSS + E2 decreased both Esr2 expression, as well as the ΔEsr2/ΔEsr1 expression ratio (I, J). Treatment groups intact: Vehicle n = 4, SERM2 n = 5, DSS and DSS + SERM2 n = 8. Gene expression data was normalized as a part of the ΔΔCt analysis. Statistical outliers were identified using ROUT with Q = 1% and one datapoint has been removed in DSS‐SERM2 in figure (G). Data presented as mean ± SD, statistical significance of (C–J) was calculated with ordinary one‐way ANOVA, Brown‐Forsythe and Welch ANOVA or Kruskal–Wallis nonparametrical test according to distribution and variance. IC50 values were determined using a four‐parameter non‐linear regression analysis. Statistical significance was defined as *p ≤ .05, **p ≤ .01, ***p ≤ .001, ****p ≤ .0001.
In addition, the expression of hormone receptors was measured. In intact mice, SERM2 upregulated Pgr expression in both DSS‐induced colitis and healthy control groups (Figure 6C). In vehicle‐treated mice, DSS statistically significantly decreased both Esr1 and Esr2, and SERM2 induced a trend toward decrease of Esr1 expression (Figure 6D,E). As the colitis‐related decrease in Esr2 was more prominent, the ΔEsr2/ΔEsr1 expression ratio decreased with DSS (Figure 6F). In the OVX experiment, both SERM2 and E2 increased Pgr transcription (Figure 6G). Transcription of Esr1 was not significantly altered by the treatments (Figure 6H). In the OVX mice, DSS + E2 decreased Esr2 expression (Figure 6I). A trend toward reduction of ΔEsr2/ΔEsr1 transcription‐level ratio by DSS could be observed in the OVX mice, and this was significantly exaggerated in the DSS + E2 mice (Figure 6J). However, inflammation‐related alteration in estrogen receptor expression was less pronounced in OVX, compared to intact mice (Figure 6F,J).
4. DISCUSSION
In this study, we demonstrate how developing acute colitis is enhanced by estrogen receptor activation in female mice and how ERα inhibition could be a possibility to limit inflammation. Altogether, the results indicate that ovarian hormones, such as estradiol, can substantially aggravate inflammation, as the induced colitis in OVX mice was notably milder compared to intact mice. Further, supraphysiological E2 supplementation led to a response similar to that of the mice with endogenous ovarian hormones. E2 has similarly been reported to exaggerate inflammation in models with longer duration E2 supplementation 16 , 17 but contrary to experiments where E2 was administered simultaneously with induced inflammation. 14 , 15 Here, SERM2 in intact mice and E2 in OVX mice oppositely affected the measured symptoms and histological signs of inflammation, as well as the expression of molecules conveying inflammation. The SERM2‐induced decrease was consistently observed in most of the immune‐related molecules measured from the colons of intact mice. Nonetheless, large individual variation resulted in the SERM2 effect failing to reach statistical significance.
The mild but consistent prophylactic effect of SERM2 in intact mice was likely due to its antagonist properties at ERα in the colon, partially inhibiting the effects of the endogenous ligand. In the literature, ERα signaling has been reported to induce inflammation both when pharmacologically activated, and in genetically modified animals. 18 , 19 Also, the coactivator assay analysis indicated an antagonist effect of SERM2 on ERα. The 53‐nM serum concentration of SERM2 was higher compared to what was measured in cell‐based assays 29 and also in the active range for antagonism at ERα based on the biochemical assay presented here. However, the bioavailability of lipophilic SERM2 requires further research.
Micromolar ERβ and PGR antagonism was observed, but the measured serum levels indicated that SERM2 did not reach the required inhibitory concentration. It is likely that antagonizing ERβ would have been detrimental in colitis, as ERβ activation or a high ERβ to ERα ratio has been suggested to inhibit inflammatory processes in the colon, 11 , 35 , 36 and ERβ has been reported to negatively regulate ERα‐activated transcription at least in bone, liver, and uterus. 37 , 38 , 39 Based on this, one could ask whether ERα inhibition through pharmacological antagonism might induce similar anti‐inflammatory effects as ERβ activation. However, the colon Pgr upregulation indicates that typically to SERMs, in some alternative cell population, SERM2 might have acted as an agonist at ERα 40 also in the intact animals. Albeit, this did not affect inflammatory status.
Nonetheless, in contrast to the effects in mice with the intact ovaries, in OVX mice, without the presence of the endogenous agonists, SERM2 itself exerted a weak agonist effect on ERα in both the colon and uterus, a feature that has been reported for hormone receptor modulators 41 and corresponding to what was previously observed in vitro. 28 This weak ERα activation could be observed as colon shortening by DSS + SERM2, and also in the expression of inflammatory cytokine genes as well as serum cytokine levels in OVX mice. Although there was a slight improvement of the AUC of DAI scores by DSS + SERM2 in the OVX mice, this was not in line with the other results. It is noteworthy that as OVX conferred protection from colitis, the inflammation was very low also in the DSS‐treated control mice.
Increased expression of cytokines like IL‐6, IFNγ, and IL‐17 reflected the hormonally aggravated inflammation both in intact and OVX + E2 mice. Surprisingly, the expression pattern of TNFα did not follow that of the other inflammatory cytokines, neither in intact nor OVX animals. This might be time point‐related, as TNFα is secreted as an early myeloid response to inflammatory stimuli, or to the pleiotropic effects at later timepoints. 42 , 43 Expression of genes encoding anti‐inflammatory IL‐10, TGFβ, and regulatory T cell transcription factor Foxp3 was simultaneously increased to counteract the acute colitis. Intestinal inflammation is restrained by anti‐inflammatory signaling, such as IL‐10 from regulatory T cells and scavenging macrophages, and this is well recognized as important in maintaining mucosal immune homeostasis. 44 In healthy mice, anti‐inflammatory macrophage marker Mrc1 was upregulated, as SERM2 treatment was previously associated with MRC‐1 expression increase. 28 , 29 The expansion of homeostasis‐maintaining macrophages could possibly be one factor explaining the prophylactic effect of SERM2 in mice with endogenous ovarian hormones, even though the effect was diminished after 7 days of colitis.
Hormonally modulated inflammation was reflected in COX2 and substance P signaling of epithelial and neuronal cells, in addition to the immune cells. COX2 expression has been reported to be hormone regulated in uterine tissue and in endothelial cells, 45 , 46 and besides in immune cells, COX2 is expressed in neurons, 47 epithelial, and stromal cells. 48 It is noteworthy, that COX2 was the only inflammation marker statistically significantly correlating with serum E2 levels in the OVX and supplemented animals. This might indicate that COX2 is an important hormonally regulated mediator of inflammation in this model. Even though it was observed in neurons, endothelia, and epithelia, the increase in colitis appeared to be associated with activity at ulcer sites and in gut‐associated lymphoid tissue. The correlation of substance P or Tacr1 to serum E2 was not significant, even though substance P and its receptor have also been indicated to be hormonally regulated and involved in reproduction. 49 , 50 , 51 Substance P from gastrointestinal neurons, which signals to the immune system, has been associated with IBD. 52 , 53 , 54 It is possible that even though substance P did not alter significantly after treatment, nevertheless the increase in its receptor augmented the substance P response in the colon.
Together, the alterations in inflammatory molecules suggest that ER signaling augments inflammation, which has also been reported elsewhere in mice 18 , 19 and in humans. 6 , 7 , 55 Albeit, we cannot exclude disease model‐specific effects behind the results presented here. Nor can we fully exclude SERM2 activity at ERβ, PGR, or the G protein‐coupled estrogen receptor (GPER), which was previously reported to be expressed in human male PBMC‐derived macrophages, 28 and has been reported to alleviate colitis in male mice. 56 Similarly to the ligands of nuclear ER's, GPER agonist G1 has been suggested to modulate immune cells and inflammation in both experimental models and human studies. Further, GPER signaling has been indicated to produce sex‐specific responses. However, there is some discrepancy also in the reports of the inflammatory and anti‐inflammatory effects of GPER. 57
Comparing these results to our previous observations of colitis in male mice, 29 the DSS‐induced colitis was less severe both in intact and OVX female mice. Male mice suffering from pronounced inflammation after DSS treatment has also been reported elsewhere. 14 , 18 The results presented here offer insight on the effects of ovarian hormones in intestinal acute inflammation of female mice and support further research focusing on tissue‐specific ERα antagonists as treatment to alleviate intestinal inflammation. In order to investigate the long‐term effects of estradiol or prophylactic SERMs, experiments mimicking the relapse‐remission pattern of inflammatory bowel disease could be conducted.
5. CONCLUSIONS
In this study, ovariectomy protected mice from developing inflammation in the colon, while hormonally intact females and E2 supplemented OVX mice were more prone to colitis, possibly via COX2, which correlated with serum estradiol levels. Prophylactic treatment with ERα antagonist SERM2 produced a mild but consistent anti‐inflammatory effect. In this model, acute inflammation in the colon seems to be exacerbated by long‐term ERα signaling, and alleviated when ER signaling is inhibited, either by OVX or by antagonist treatment.
AUTHOR CONTRIBUTIONS
Participated in research design and conducted original experiments: Määttä. J. Polari, L. Anttila, S. Performed laboratory or histology analyses: Hjelt, A. Wiklund, Al‐Ramahi, D. Contributed new reagents or analytic tools: Määttä, J. Toivola, D.M. Performed data analysis: Hjelt, A., Polari, L. Al‐Ramahi, D. (mass‐spectrometry of SERM2) Rokka, A. (mass‐spectrometry of substance P). Wrote or contributed to the writing of the manuscript: Hjelt, A. Polari, L. Määttä, J.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ETHICS STATEMENT
All animal research was conducted according to the national guidelines provided by the European Union (Directive 2010/63/EU) and was approved by The State Provincial Office of South Finland.
Supporting information
Figure S1.
{kind=link}
Figure S2.
{kind=link}
Figure S3.
{kind=link}
Figure S4.
{kind=link}
Figure S5.
{kind=link}
Data S1.
ACKNOWLEDGMENTS
This work has been supported by Business Finland (Former TEKES), decisions # 473/31/2015, 253/31/2019 and the Novo Nordisk Foundation, Pioneer Innovator Grant # NNF21OC0068904, Research Council of Finland 332582/315139, the InFLAMES Flagship Programme of the Research Council of Finland (337531 and 357911), Sigrid Juselius Foundation, Svenska Kulturfonden (187699), Victoriastiftelsen grant number 20240472, and Åbo Akademi University Centers of Excellence in Cellular Mechanostasis. We kindly thank Forendo Pharma Ltd (now Organon R&D Finland) for providing the experimental molecule SERM2. We acknowledge Heidi Liljenbäck at the Turku Center for Disease Modeling for the surgical procedures and post‐operative care of the animals. We thank Merja Lakkisto and Joel Nyström for excellent technical assistance. Mass spectrometry analyses were performed at the Turku Center for Chemical and Molecular Analytics, Bioanalytical Laboratory and Turku Proteomics Facility, and University of Turku and Åbo Akademi University. The facilities are supported by Biocenter Finland.
Hjelt A, Anttila S, Wiklund A, et al. Estrogen deprivation and estrogen receptor α antagonism decrease DSS colitis in female mice. Pharmacol Res Perspect. 2024;12:e1234. doi: 10.1002/prp2.1234
Anja Hjelt and Santeri Anttila have equally contributed to this paper.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Chang JT. Pathophysiology of inflammatory bowel diseases. N Engl J Med. 2020;383(27):2652‐2664. doi: 10.1056/NEJMra2002697 [DOI] [PubMed] [Google Scholar]
- 2. Kaplan GG, Windsor JW. The four epidemiological stages in the global evolution of inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2021;18(1):56‐66. doi: 10.1038/s41575-020-00360-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Straub RH. The complex role of estrogens in inflammation. Endocr Rev. 2007;28(5):521‐574. doi: 10.1210/er.2007-0001 [DOI] [PubMed] [Google Scholar]
- 4. Wang R, Li Z, Liu S, Zhang D. Global, regional and national burden of inflammatory bowel disease in 204 countries and territories from 1990 to 2019: a systematic analysis based on the global burden of disease study 2019. BMJ Open. 2023;13(3):e065186. doi: 10.1136/bmjopen-2022-065186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shah SC, Khalili H, Gower‐Rousseau C, et al. Sex‐based differences in incidence of inflammatory bowel diseases—pooled analysis of population‐based studies from Western countries. Gastroenterology. 2018;155(4):1079‐1089. doi: 10.1053/J.GASTRO.2018.06.043 [DOI] [PubMed] [Google Scholar]
- 6. Ortizo R, Lee SY, Nguyen ET, Jamal MM, Bechtold MM, Nguyen DL. Exposure to oral contraceptives increases the risk for development of inflammatory bowel disease: a meta‐analysis of case‐controlled and cohort studies. Eur J Gastroenterol Hepatol. 2017;29(9):1064‐1070. doi: 10.1097/MEG.0000000000000915 [DOI] [PubMed] [Google Scholar]
- 7. Pasvol TJ, Bloom S, Segal AW, Rait G, Horsfall L. Use of contraceptives and risk of inflammatory bowel disease: a nested case‐control study summary background: how contraceptive formulation, dose, duration of therapy and mode. Aliment Pharmacol Ther. 2022;55:318‐326. doi: 10.1111/apt.16647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kovats S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell Immunol. 2015;294(2):63‐69. doi: 10.1016/j.cellimm.2015.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hoffmann JP, Liu JA, Seddu K, Klein SL. Sex hormone signaling and regulation of immune function. Immunity. 2023;56(11):2472‐2491. doi: 10.1016/J.IMMUNI.2023.10.008 [DOI] [PubMed] [Google Scholar]
- 10. Pierdominici M, Maselli A, Varano B, et al. Linking estrogen receptor β expression with inflammatory bowel disease activity. Oncotarget. 2015;6(38):40443‐40451. doi: 10.18632/ONCOTARGET.6217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Linares PM, Algaba A, Urzainqui A, et al. Ratio of circulating estrogen receptors Beta and Alpha (ERβ/ERα) indicates endoscopic activity in patients with Crohn's disease. Dig Dis Sci. 2017;62(10):2744‐2754. doi: 10.1007/S10620-017-4717-5/FIGURES/4 [DOI] [PubMed] [Google Scholar]
- 12. Li H, Chen H, Chen L, Shen D, Xu X. Expression of oestrogen receptor beta was negatively correlated with disease activity in patients with Crohn's disease involving the terminal ileum. Steroids. 2019;141:36‐40. doi: 10.1016/J.STEROIDS.2018.09.013 [DOI] [PubMed] [Google Scholar]
- 13. Jacenik D, Cygankiewicz AI, Mokrowiecka A, Małecka‐Panas E, Fichna J, Krajewska WM. Sex‐ and age‐related estrogen signaling alteration in inflammatory bowel diseases: modulatory role of estrogen receptors. Int J Mol Sci. 2019;20(13):1‐18. doi: 10.3390/IJMS20133175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bábíčková J, Tóthová Ľ, Lengyelová E, et al. Sex differences in experimentally induced colitis in mice: a role for estrogens. Inflammation. 2015;38(5):1996‐2006. doi: 10.1007/s10753-015-0180-7 [DOI] [PubMed] [Google Scholar]
- 15. Song CH, Kim N, Sohn SH, et al. Effects of 17β‐estradiol on colonic permeability and inflammation in an azoxymethane/dextran sulfate sodium‐induced colitis mouse model. Gut Liver. 2018;12(6):682‐693. doi: 10.5009/GNL18221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Heijmans J, Wielenga MCB, Rosekrans SL, et al. Oestrogens promote tumorigenesis in a mouse model for colitis‐associated cancer. Gut. 2014;63(2):310‐316. doi: 10.1136/GUTJNL-2012-304216 [DOI] [PubMed] [Google Scholar]
- 17. Calippe B, Douin‐Echinard V, Laffargue M, et al. Chronic estradiol administration in vivo promotes the proinflammatory response of macrophages to TLR4 activation: involvement of the phosphatidylinositol 3‐kinase pathway. J Immunol. 2008;180(12):7980‐7988. [DOI] [PubMed] [Google Scholar]
- 18. Goodman WA, Havran HL, Quereshy HA, Kuang S, De Salvo C, Pizarro TT. Estrogen receptor α loss‐of‐function protects female mice from DSS‐induced experimental colitis. Cell Mol Gastroenterol Hepatol. 2018;5(4):630‐633. doi: 10.1016/J.JCMGH.2017.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gao X, Fan W, Tan L, et al. Soy isoflavones ameliorate experimental colitis by targeting ERα/NLRP3 inflammasome pathways. J Nutr Biochem. 2020;83:108438. doi: 10.1016/J.JNUTBIO.2020.108438 [DOI] [PubMed] [Google Scholar]
- 20. Riggs BL, Hartmann LC. Drug therapy selective estrogen‐receptor modulators‐mechanisms of action and application to clinical practice. N Engl J Med. 2003;348:618‐629. [DOI] [PubMed] [Google Scholar]
- 21. Chadwick CC, Chippari S, Matelan E, et al. Identification of pathway‐selective estrogen receptor ligands that inhibit NF‐κB transcriptional activity. Proc Natl Acad Sci USA. 2005;102(7):2543‐2548. doi: 10.1073/PNAS.0405841102/SUPPL_FILE/05841TABLE3.HTML [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Greish K, Taha S, Jasim A, et al. Styrene maleic acid encapsulated raloxifene micelles for management of inflammatory bowel disease. Clin Transl Med. 2017;6(1):e28. doi: 10.1186/S40169-017-0157-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bebo BF, Dehghani B, Foster S, Kurniawan A, Lopez FJ, Sherman LS. Treatment with selective estrogen receptor modulators regulates myelin specific T‐cells and suppresses experimental autoimmune encephalomyelitis. Glia. 2009;57(7):777‐790. doi: 10.1002/GLIA.20805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Elloso MM, Phiel K, Henderson RA, Harris HA, Adelman SJ. Suppression of experimental autoimmune encephalomyelitis using estrogen receptor‐selective ligands. J Endocrinol. 2005;185(2):243‐252. doi: 10.1677/JOE.1.06063 [DOI] [PubMed] [Google Scholar]
- 25. Jeong H, Kim IY, Bae EK, Jeon CH, Ahn KS, Cha HS. Selective estrogen receptor modulator lasofoxifene suppresses spondyloarthritis manifestation and affects characteristics of gut microbiota in zymosan‐induced SKG mice. Sci Rep. 2021;11(1):1‐12. doi: 10.1038/s41598-021-91320-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cushman M, Costantino JP, Tracy RP, et al. Tamoxifen and cardiac risk factors in healthy women. Arterioscler Thromb Vasc Biol. 2001;21(2):255‐261. doi: 10.1161/01.ATV.21.2.255 [DOI] [PubMed] [Google Scholar]
- 27. Katsandegwaza B, Horsnell W, Smith K. Inflammatory bowel disease: a review of pre‐clinical murine models of human disease. Int J Mol Sci. 2022;23(16):1‐19. doi: 10.3390/IJMS23169344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Polari L, Wiklund A, Sousa S, et al. SERMs promote anti‐inflammatory signaling and phenotype of CD14+ cells. Inflammation. 2018;41(4):1157‐1171. doi: 10.1007/s10753-018-0763-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Polari L, Anttila S, Helenius T, et al. Novel selective estrogen receptor modulator ameliorates murine colitis. Int J Mol Sci. 2019;20(12):1‐14. doi: 10.3390/ijms20123007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. ten Hove T, van den Blink B, Pronk I, Drillenburg M, Peppelenbosch MP, van Deventer SJH. Dichotomal role of inhibition of p38 MAPK with SB 203580 in experimental colitis. Gut. 2002;50:507‐512. doi: 10.1136/gut.50.4.507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schindelin J, Arganda‐Carreras I, Frise E, et al. Fiji: an open‐source platform for biological‐image analysis. Nat Methods. 2012;9(7):676‐682. doi: 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bankhead P, Loughrey MB, Fernández JA, et al. QuPath: open source software for digital pathology image analysis. Sci Rep. 2017;7(1):16878. doi: 10.1038/s41598-017-17204-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res. 2018;46(D1):D1091‐D1106. doi: 10.1093/NAR/GKX1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Alexander SPH, Cidlowski JA, Kelly E, et al. The concise guide to PHARMACOLOGY 2023/24: nuclear hormone receptors. Br J Pharmacol. 2023;180(S2):S223‐S240. doi: 10.1111/BPH.16179 [DOI] [PubMed] [Google Scholar]
- 35. Hases L, Indukuri R, Birgersson M, et al. Intestinal estrogen receptor beta suppresses colon inflammation and tumorigenesis in both sexes. Cancer Lett. 2020;492:54‐62. doi: 10.1016/J.CANLET.2020.06.021 [DOI] [PubMed] [Google Scholar]
- 36. Fan W, Ding C, Liu S, et al. Estrogen receptor β activation inhibits colitis by promoting NLRP6‐mediated autophagy. Cell Rep. 2022;41(2):111454. doi: 10.1016/J.CELREP.2022.111454 [DOI] [PubMed] [Google Scholar]
- 37. Pettersson K, Delaunay F, Gustafsson JA. Estrogen receptor β acts as a dominant regulator of estrogen signaling. Oncogene. 2000;19(43):4970‐4978. doi: 10.1038/SJ.ONC.1203828 [DOI] [PubMed] [Google Scholar]
- 38. Lindberg MK, Movérare S, Skrtic S, et al. Estrogen receptor (ER)‐β reduces ERα‐regulated gene transcription, supporting a “Ying Yang” relationship between ERα and ERβ in mice. Mol Endocrinol. 2003;17(2):203‐208. doi: 10.1210/ME.2002-0206 [DOI] [PubMed] [Google Scholar]
- 39. Weihua Z, Saji S, Mäkinen S, et al. Estrogen receptor (ER) β, a modulator of ERα in the uterus. Proc Natl Acad Sci U S A. 2000;97(11):5936‐5941. doi: 10.1073/PNAS.97.11.5936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Petz LN, Ziegler YS, Schultz JR, Kim H, Kemper JK, Nardulli AM. Differential regulation of the human progesterone receptor gene through an estrogen response element half site and Sp1 sites. J Steroid Biochem Mol Biol. 2004;88:113‐122. doi: 10.1016/j.jsbmb.2003.11.008 [DOI] [PubMed] [Google Scholar]
- 41. Spitz IM, Bardin CW. Mifepristone (RU 486)—a modulator of progestin and glucocorticoid action. N Engl J Med. 1993;329(6):404‐412. [DOI] [PubMed] [Google Scholar]
- 42. Zakharova M, Ziegler HK. Paradoxical anti‐inflammatory actions of TNF‐α: inhibition of IL‐12 and IL‐23 via TNF receptor 1 in macrophages and dendritic cells. J Immunol. 2005;175(8):5024‐5033. doi: 10.4049/JIMMUNOL.175.8.5024 [DOI] [PubMed] [Google Scholar]
- 43. Steeland S, Libert C, Vandenbroucke RE. A new venue of TNF targeting. Int J Mol Sci. 2018;19(5):1442. doi: 10.3390/IJMS19051442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wei HX, Wang B, Li B. IL‐10 and IL‐22 in mucosal immunity: driving protection and pathology. Front Immunol. 2020;11:548633. doi: 10.3389/FIMMU.2020.01315/BIBTEX [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brazil M. Oestrogen activates COX2 pathway. Nat Rev Drug Discov. 2005;4(1):16. doi: 10.1038/nrd1615 [DOI] [Google Scholar]
- 46. Tamura M, Deb S, Sebastian S, Okamura K, Bulun SE. Estrogen up‐regulates cyclooxygenase‐2 via estrogen receptor in human uterine microvascular endothelial cells. Fertil Steril. 2004;81(5):1351‐1356. doi: 10.1016/j.fertnstert.2003.09.076 [DOI] [PubMed] [Google Scholar]
- 47. Zhang J, Chen C. Endocannabinoid 2‐arachidonoylglycerol protects neurons by limiting COX‐2 elevation. J Biol Chem. 2008;283(33):22601‐22611. doi: 10.1074/JBC.M800524200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ishikawa TO, Oshima M, Herschman HR. Cox‐2 deletion in myeloid and endothelial cells, but not in epithelial cells, exacerbates murine colitis. Carcinogenesis. 2011;32(3):417‐426. doi: 10.1093/CARCIN/BGQ268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Villablanca AC, Hanley MR. 17β‐estradiol stimulates substance P receptor gene expression. Mol Cell Endocrinol. 1997;135(2):109‐117. doi: 10.1016/S0303-7207(97)00193-7 [DOI] [PubMed] [Google Scholar]
- 50. Okamura H, Yokosuka M, Hayashi S. Induction of substance P‐immunoreactivity by estrogen in neurons containing estrogen receptors in the Anterovental periventricular nucleus of female but not male rats. J Neuroendocrinol. 1994;6(6):609‐615. doi: 10.1111/J.1365-2826.1994.TB00626.X [DOI] [PubMed] [Google Scholar]
- 51. García‐Ortega J, Pinto FM, Prados N, et al. Expression of tachykinins and tachykinin receptors and interaction with kisspeptin in human granulosa and cumulus cells. Biol Reprod. 2016;94(6):124‐125. doi: 10.1095/BIOLREPROD.116.139881/2863883 [DOI] [PubMed] [Google Scholar]
- 52. Tavano F, di Mola FF, Latiano A, et al. Neuroimmune interactions in patients with inflammatory bowel diseases: disease activity and clinical behavior based on substance P serum levels. J Crohns Colitis. 2012;6(5):563‐570. doi: 10.1016/J.CROHNS.2011.11.004 [DOI] [PubMed] [Google Scholar]
- 53. Patel M, Subas SV, Ghani MR, et al. Role of substance P in the pathophysiology of inflammatory bowel disease and its correlation with the degree of inflammation. Cureus. 2020;12(10):e11027. doi: 10.7759/CUREUS.11027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Renzi D, Pellegrini B, Tonelli F, Surrenti C, Calabro A. Substance P (neurokinin‐1) and neurokinin a (neurokinin‐2) receptor gene and protein expression in the healthy and inflamed human intestine. Am J Pathol. 2000;157(5):1511‐1522. doi: 10.1016/S0002-9440(10)64789-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Khalili H, Higuchi LM, Ananthakrishnan AN, et al. Hormone therapy increases risk of ulcerative colitis but not Crohn's disease. Gastroenterology. 2012;143(5):1199‐1206. doi: 10.1053/J.GASTRO.2012.07.096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang Q, Li Z, Liu K, et al. Activation of the G protein‐coupled estrogen receptor prevented the development of acute colitis by protecting the crypt cell. J Pharmacol Exp Ther. 2021;376(2):281‐293. doi: 10.1124/jpet.120.000216 [DOI] [PubMed] [Google Scholar]
- 57. Prossnitz ER, Barton M. The G protein‐coupled oestrogen receptor GPER in health and disease: an update. Nat Rev Endocrinol. 2023;19(7):407‐424. doi: 10.1038/s41574-023-00822-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Figure S2.
Figure S3.
Figure S4.
Figure S5.
Data S1.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.