

Abstract
Transcription initiation is an essential process to ensure proper function of any gene, yet we still lack a unified understanding of sequence patterns and rules that explains most transcription start sites in human genome. By predicting transcription initiation at base pair resolution from sequences with a deep learning-inspired explainable model called Puffin, we showed that a small set of simple rules can explain transcription initiation at most human promoters. We identified key sequence patterns that contribute to human promoter activity, each activating transcription with distinct position-specific effects. Furthermore, we explained the sequence basis of bidirectional transcription at promoters, identified the links between promoter sequence and gene expression variation across cell types, and explored the conservation of sequence determinants of transcription initiation across mammalian species.
One-Sentence Summary:
A small set of rules can explain how genome sequence drives transcription initiation in humans and across mammals.
Promoter sequences are responsible for transcription initiation and the central hub in integrating transcriptional regulatory information. Over several decades, a handful of core promoter elements including the TATA-box, the Initiator (Inr) motif, and several downstream motifs (MTE, DPE, DPR) have been identified in various species(1–3), but human promoters often possess none of these elements(3). While many sequence-specific transcription factor (TF) motifs appear near promoters(3–5), whether and how they contribute to promoter function have not been clearly defined. Additionally, for most human promoters, we do not have the knowledge of which base pairs contribute to their activity.
Our understanding of how sequence determines transcription start sites for the majority of human promoters thus remains incomplete(1–3, 6). This problem is especially challenging, because the transcription initiation process involves many factors, and a single base pair may have multiple functions (1–3, 7). Hence, a systematic approach that simultaneously dissects multiple types of sequence patterns, such as TF binding motifs, and their effects on transcription initiation is critical for solving this problem. Deep learning approaches have allowed simultaneously learning of complex dependencies between genomic sequence and its activities (8–14), but they do not directly allow dissection or validation of the underlying sequence patterns and effects that are learned.
Several fundamental questions remained open. What are the bases that contribute to any given promoter and determine transcriptional initiation signals at the base pair resolution? How do sequence patterns work together to determine transcription start sites? What is the impact of promoter sequence pattern composition on its function? What are the key factors that determine the strand-specificity of promoters? And finally, how conserved are sequence determinants of transcription initiation across species? Developing a strategy to answer these questions would allow us to analyze, predict, engineer, and control transcription initiation.
To address these questions and overcome the limitations of current methodologies, we developed Puffin, a deep learning-inspired explainable model, and showed that a few simple rules and sequence patterns are sufficient to explain base pair resolution transcription initiation signals at most promoters. With Puffin, we recapitulated prior findings and discovered new roles for known and unknown motifs, creating a unified view of transcription initiation at the sequence level. To facilitate interactive analysis of any promoter sequence, we have designed and built a user-friendly web server (https://tss.zhoulab.io) powered by Puffin to facilitate the understanding and manipulation of any promoter sequence of interest.
Results
Decoding sequence basis of transcription initiation at base pair resolution
Transcription initiation signals for most human promoters are not restricted to a single base pair but spread over a window(4). We hypothesized that the transcription initiation signals at base pair resolution reflect underlying sequence-based transcription initiation mechanisms. Therefore, we can deconvolve such mechanisms by dissecting how transcription initiation signals depend on sequence.
To this end, we first assembled the highest coverage transcription initiation maps at base pair resolution. We integrated transcription initiation signal datasets generated by five experimental techniques that precisely capture the 5’ end of transcripts and quantify the transcription initiation signal by counting the transcripts starting at each position: two variants of CAGE(15) and RAMPAGE(16) from the FANTOM and ENCODE projects that measure mostly mature transcripts and GRO/PRO-cap(17, 18) that measures only nascent transcripts. For each technique, we aggregated all samples to obtain the most robust estimate for the transcription initiation signal at single-base resolution on each strand (Data S1). FANTOM CAGE (6.7 billion reads) and PRO-cap (2.3 billion reads) have the highest coverage within each group of techniques and were prioritized for the presentation here (all analyses are consistent across techniques unless otherwise indicated).
To elucidate the sequence basis of transcription initiation, we developed base pair-resolution sequence models of transcription initiation signals (Fig. 1A–C), which include a pair of complementary models: the performance-focused Puffin-D, which is a deep learning model with an architecture that predicts strand-specific base pair resolution signals across the genome from a long sequence context of 100kb (Fig. S1, S2), and the interpretation-focused Puffin model, the focus of this manuscript, which extracted simple sequence rules that can explain transcription initiation at base pair resolution. The Puffin model was designed based on the analysis of sequence dependencies captured by the deep learning model Puffin-D (Supplementary Text).
Fig. 1. Dissect the sequence basis of transcription initiation with sequence models.
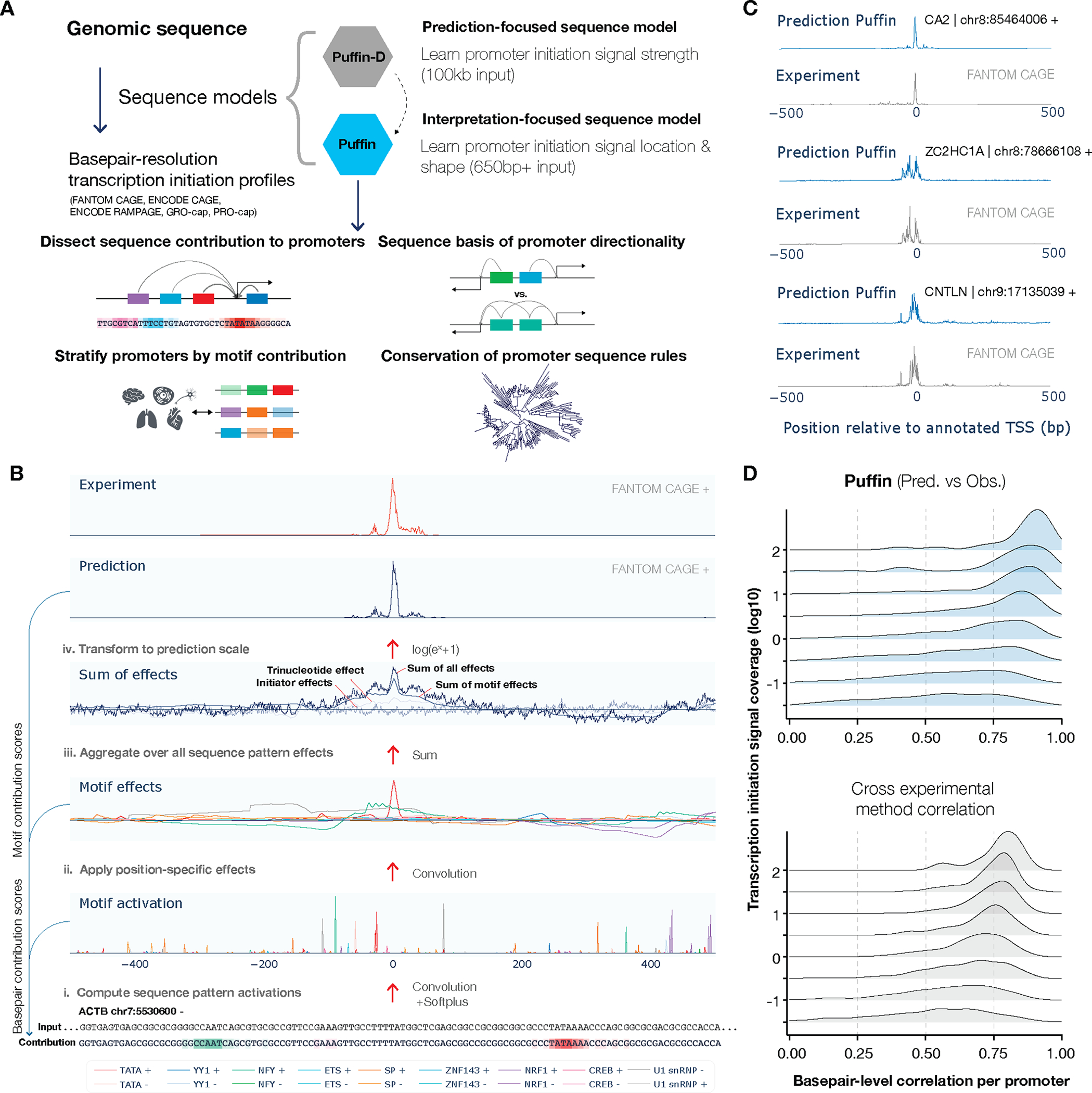
(A) Schematic overview of sequence-based models of transcription initiation. The prediction-focused Puffin-D and interpretation-focused Puffin models were trained to predict base pair-resolution transcription initiation signals from sequence. These sequence models enabled analyses of promoter motif composition, directionality, regulatory properties, and sequence rule conservation. (B) Step-by-step illustration of the Puffin promoter sequence model. The input sequence is first converted to activation scores of learned sequence patterns. Sequence pattern effects are computed next based on the activations. The motif effects are then summed together with initiator and trinucleotide sequence pattern effects, and transformed into the prediction. The computation of motif contribution scores and base pair contribution scores are illustrated on the left. The motif name legend is shown at the bottom. Motifs effects and the prediction shown were for FANTOM CAGE on the forward strand. (C) Example prediction of base pair resolution strand-specific transcription initiation signal from promoter sequences on chromosomes heldout from training. The x-axis indicates the position relative to the annotated transcription start site and the y-axis is shown in log10 scale. (D) Base pair-level correlation (x-axis) between Puffin (top panel) and experimental measurement (FANTOM CAGE) within 1kb window of each annotated TSS. Promoters were grouped by coverage level with the y-axis indicating the lower bound of each group (the upper bound of the group is the lower bound of the next group). The bottom panel shows the correlations between ENCODE CAGE and FANTOM CAGE, the most correlated pair of techniques, which also provides a reference for the expected decrease in correlation due to lower coverage.
Puffin shows that a simple set of rules can explain the majority of human promoter sequences (Fig. 1C–D). When evaluated on test chromosomes, Puffin achieves higher base pair-level correlation with experimental measurement than the most correlated pair of experimental techniques (Fig. 1C–D, Fig. S3). To focus on promoter sequence dependencies, Puffin uses only proximal sequence context for its prediction (+/− 325bp sliding window) and focuses on the shape but not the magnitude of the transcription initiation signal, because we expect the shape, or the relative intensities across base pairs, to be mostly dependent on local sequences while the magnitude can be affected by distal sequence which Puffin does not use.
Puffin predicts transcription initiation signal from sequence with two steps of computation. First, it computes base pair-resolution activation scores for all sequence patterns it learned via the first convolution layer. Activation score is analogous to motif match score with non-matches set to zero, and quantifies how well the sequence at a position matches with a sequence pattern (Methods). Then, the activating and repressive effects of each sequence pattern on transcription initiation were learned and applied via the second convolution layer. All sequence patterns’ effects are combined additively per base pair in log scale, which is equivalent to multiplicative combination in count scale (Methods, Fig. 1B). The output of this layer, which sums all sequence pattern effects per base pair, can be interpreted as log scale transcription initiation signal and is rescaled to final prediction (Methods).
The data-driven design of Puffin optimizes for interpretability and aims to capture most of the proximal sequence dependencies of transcription initiation with a small set of sequence patterns (Supplementary Text, Methods). The model learns three types of sequence patterns to capture different types of sequence dependencies (Fig. 2A–C, Fig. S4): 1. Motifs, the main sequence drivers of transcription initiation signals; 2. Initiators, which only tune the local base pair-level location preference for transcription initiation within these sequence patterns themselves; 3. Trinucleotides, which capture the residual sequence dependencies that were not captured by motifs and initiators. Thus, with a small number of sequence patterns (9 motifs + initiators + trinucleotides) and a simple additive/multiplicative rule (additive in log scale, multiplicative in count scale), we can predict transcription initiation signals at base pair resolution from sequence and the predictions are in strong agreement with experimental evidence (Fig. 2).
Fig. 2. Sequence patterns with position-specific effects on transcription initiation.
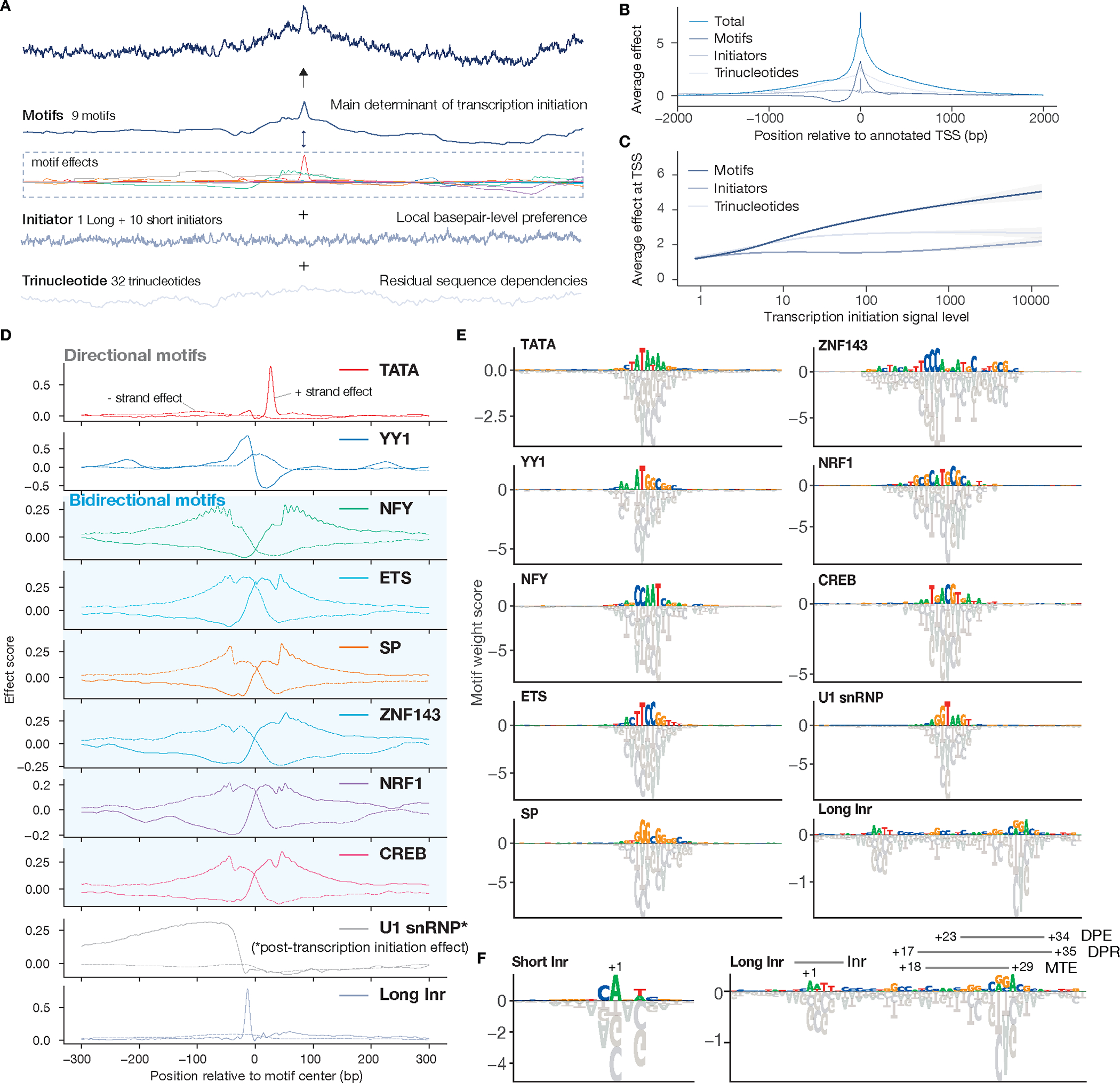
(A) Overview of the three sequence pattern types that the Puffin model learns. (B). The average position-specific effect of each sequence pattern type over the top 40,000 promoters by FANTOM CAGE signal (top panel). (C) The average sequence pattern effect at annotated TSS (y-axis) varies with promoter transcription initiation signal levels (x-axis). Generalized additive model fitted curves and 95% confidence intervals are shown. (D) Position-specific effects of all motifs. X-axis indicates position relative to motif center with positive values representing downstream of the motif and vice versa. The effect scores are obtained from the motif effect convolution layer weights. The bidirectional motifs are indicated with blue background and all other motifs are direction-specific. The dotted lines show the motif effect on reverse strand transcription initiation. U1 snRNP effects are post-transcription initiation effects. (E-F). All transcription initiation motifs learned by the Puffin model with assigned names. Known promoter motifs overlapping with the Long Inr motif are indicated in (F). The height of each base represents the motif score (convolution kernel weight) for that nucleotide.
Position- and strand-specific sequence effects on transcription initiation
The core of the transcription initiation sequence model is the position-specific effect curves of sequence patterns. The model has learned a distinct curve for each motif that represents the activation and repression effects on transcription initiation at different base pair positions relative to the motif (Fig. 2D). Training at base pair resolution allows Puffin to characterize and distinguish the fine details of motif-specific effect curves. Each position-specific effect curve can be considered as a transcriptional signature of the motif and likely reflects its mechanism of activating transcription.
All motifs included in the Puffin model are nonredundant motifs that are reproducibly discovered with >0.95 correlation across multiple training replicates (Methods, Data S2). Moreover, the same group of motifs was learned when training the model with cell type-specific data (Supplementary Text). While all motifs were learned from scratch using only sequence and transcription initiation signals, many of them match known transcription factor motifs(19). However, their position-specific effects on transcription initiation have not been characterized. Moreover, the position-specific motif effects characterized by Puffin are well supported by experimental perturbations and by evolutionary conservation, as described later.
We assigned each motif a name and an ID (Table S1). By symmetry of motif effects, the motifs can be divided into two groups: 1) a group of strand-specific or directional motifs that have strong effects on the forward strand and much weaker or no effect on the reverse strand, including TATA, YY1, U1 snRNP, and Long Initiator (Long Inr) (Fig. 2D–F); and 2) a group of non-strand-specific or bidirectional motifs with almost symmetrical effects on both strands, including SP, NFY, ETS, ZNF143, NRF1, and CREB (Fig. 2D–F, Fig. S5). We summarize below each group of motifs separately, as well as initiator sequence patterns and trinucleotide sequence patterns.
Direction-specific promoter motifs.
Puffin identified the well-established TATA motif as expected, and this motif also has the most position-specific and strand-specific effects. YY1 motif is also estimated to be highly strand-specific. YY1 motif has one of the most distinctive transcription initiation effect curves estimated by Puffin, activating transcription at the immediate upstream and ~220bp upstream of the motif (Fig. 2D). Of note, YY1 is known to bind promoter sequence and its role in promoter function has been suggested (5, 20), but its position-specific effect on transcription initiation has not been previously resolved. The U1 snRNP motif (or the 5’ splice site motif) was the only motif that was estimated to have a strong positive effect on the transcription initiation signals measured by CAGE/RAMPAGE that detect total mRNA, but not on the transcription initiation signals measured by PRO/GRO-cap that only detect nascent transcripts. This suggests that U1 snRNP motif exerts its impact on transcript abundance after transcription initiation, consistent with recent findings that it promotes transcription elongation(17, 21–23). The last direction-specific motif, the Long Initiator (Long Inr), resembles the initiator sequence patterns, thus we grouped the Long Inr with the initiators rather than motifs in this study.
Bidirectional promoter motifs.
The bidirectional motifs are expected to bind the trimeric NF-Y TF (NFY), KLF/SP family TFs (SP), ETS family TFs (ETS), the zinc finger TF ZNF143 (ZNF143), CREB/ATF family TFs (CREB), and the homodimeric TF NRF1 (NRF1), respectively. While each motif in the bidirectional group has a highly distinguishable position-specific effect pattern, they also share apparent similarities in position-specific effect profiles, suggesting similarities in their transcription activation mechanisms (Fig. 2D). The NFY motif showed the most distinctive position-specific effect curve with ~10.5bp periodicity. 10.5bp matches the helical periodicity of the DNA double-strand and may indicate a more rigid physical interaction between the NF-Y TF and the Pol II preinitiation complex. Similarly, albeit weaker, 10.5bp periodicity was also observed in the position-specific effect curves of other motifs. The CG-rich SP motif was the most common motif at promoters (Fig. S6, Table S2). The NRF1 motif was the only palindromic motif among all motifs with symmetric effects. As we will discuss later, bidirectional motifs are likely the basis of the bidirectional transcription initiation at most human promoters. Moreover, all bidirectional motifs activate transcription away from the motif on both strands, thus avoiding the formation of double-stranded RNA.
Initiators tune the local location preference for transcription initiation.
We named this type of sequence patterns after the initiator (Inr) element(24), one of the first core promoter elements identified. We refer to the initiator sequence pattern that matches the Inr as the Short Inr (Fig. 2F), because Puffin also identified a related Long Inr sequence pattern, which is an extended Short Inr that contains several downstream core promoter elements including MTE, DPE, and DPR (Fig. 2F). Similar to these elements, Long Inr may represent the nucleotide preferences of TFIID, which binds to the core promoter to position Pol II. Initiators are distinct from motifs as they only fine-tune transcription initiation signals at base pair resolution within the sequence pattern itself (Fig. 2A–B). Moreover, most base pairs affect initiator sequence pattern activation scores while only a small fraction affects motif activation scores. In addition to the canonical initiator (Inr) element-like sequence patterns including Long and Short Inr, other initiator sequence patterns explain about half of the variance of initiator sequence pattern effects. The sum of initiator effects is highly reproducible across training replicates (Fig. S7).
Trinucleotides capture residual local sequence dependencies.
Residual local sequence dependencies are captured by the trinucleotide sequence patterns which can represent all trinucleotide combinations of the four bases (A, C, G, and T) (Methods, Fig. S8–10, Supplementary Text). Both individual and total trinucleotide effects are highly reproducible across training replicates (Fig. S7,11–12). The trinucleotide patterns with the strongest contribution to transcription initiation are CpG-containing patterns (Fig. S8). The effects of CpG islands are likely explained by both CG-rich motifs like SP and trinucleotide effects in Puffin. We chose trinucleotide rather than shorter or longer sequence patterns to balance performance and interpretability of Puffin.
As shown by the average effect of each sequence pattern type (Fig. 2B), motifs, initiators, and trinucleotides sequence patterns define transcription start site (TSS) at different genomic distance scales. Motifs are the most important contributor to transcription initiation signals, and their effect ranges are the longest, up to 300bp from the motif. Moreover, promoters with higher transcriptional initiation signal levels are characterized by stronger contributions from motifs (Fig. 2C). Trinucleotides have mostly local effects within 50bp, but a broad region of a few kilobases surrounding the TSS is enriched in trinucleotide patterns preferable for transcription initiation (Fig 2B, Fig. S8–10). Initiator effects are the most local and they only fine-tune base-pair resolution transcription initiation signal. Overall, motifs, initiators, and trinucleotides capture different aspects of sequence dependencies, which together explain base pair resolution transcription initiation in most human promoters. Next, we focus on motifs and their roles in transcription initiation, gene regulation, and evolution.
Experimental perturbations validated position-specific motif effects on transcription initiation
To directly validate the motif effects using experimental data, we analyzed the effects of TF depletion on base pair-resolution transcription initiation signal and compared them with model predictions (Fig. 3A–C). Puffin has the capability of performing in silico knockout (KO) to predict the effects of depleting a specific TF by turning off the effects of the corresponding motif (Fig. 3A).
Fig. 3. Experimental TF and motif perturbation effects are recapitulated by Puffin prediction.
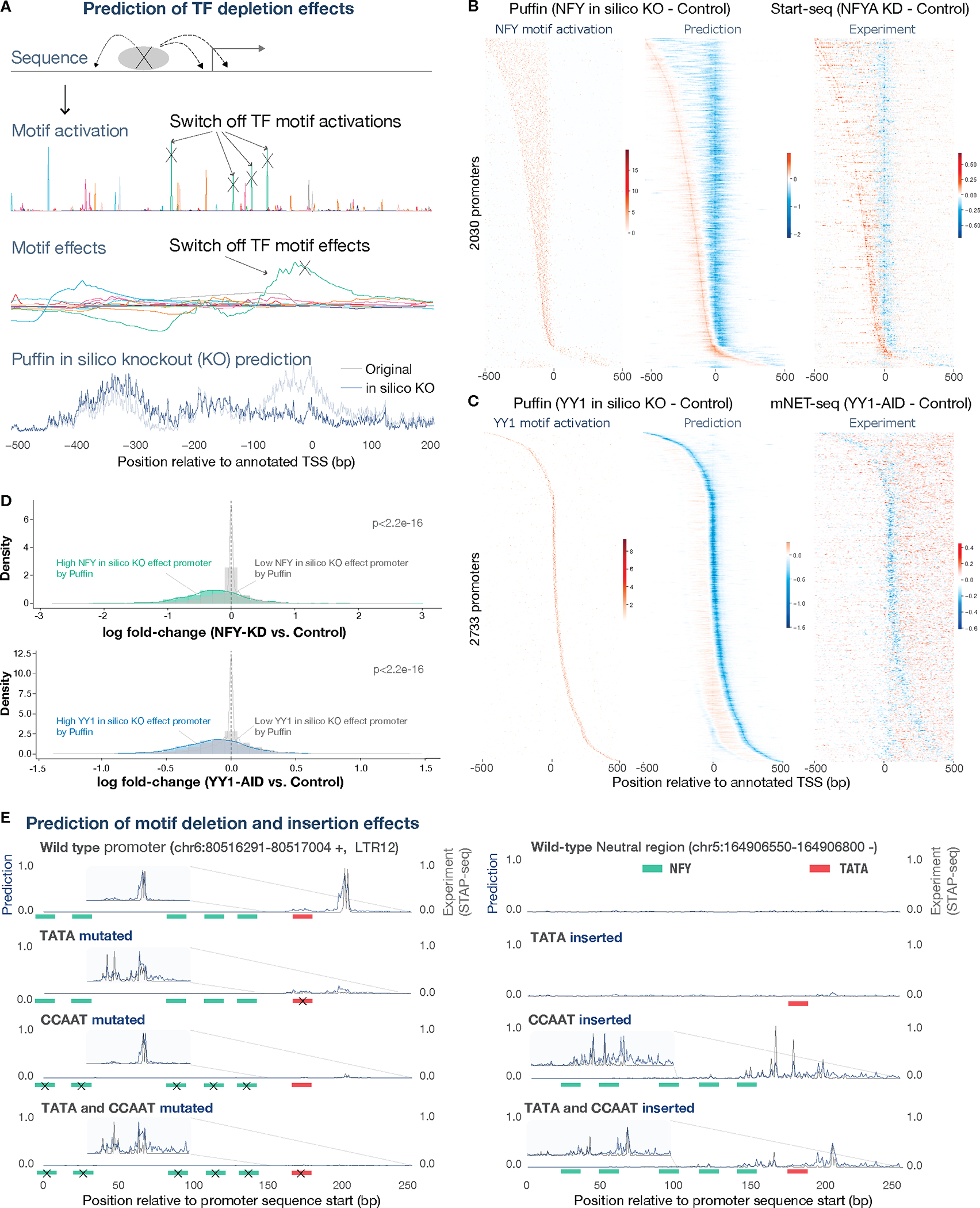
(A) Schematic illustration of predicting TF depletion effect by in silico knockout (KO) with Puffin. To predict the effects of TF depletion, we set the activation and consequently the effects of the corresponding motif to 0 in the Puffin model, and predict the transcription initiation signals. (B) In silico KO prediction for NFY (mid panel) compared with experimental measurement of NFYA knockdown with Start-seq (right panel). NFY motif activations are shown in the left panel. Promoters with strong NFY in silico KO effects were selected and sorted by the predicted shifted TSS positions. (C) In silico KO prediction for YY1 (mid panel) compared with the experimental measurement with mNET-seq after induced depletion of YY1 (right panel). NFY motif activations are shown in the left panel. Promoters with strong YY1 in silico KO effects were selected and sorted by the position with the strongest predicted decrease in transcription. The matrices shown in (B) and (C) heatmaps were smoothed with a small rectangular filter of size 3×3 for motif activation, 5×1 for prediction and 5×3 for experimental data. (D) Promoters with high NFY (top panel) or YY1 (bottom panel) contributions are significantly more influenced by corresponding TF depletion than those with low contributions (p-values derived from two-sided Wilcoxon rank sum test). (E) Example prediction and experimental measurements for TATA and NFY motif insertion and deletion experiments. All prediction and experimental values were shown in count scale and scaled to maximum 1. Only predictions and experimental data in the forward strand (expected transcription direction) are shown. Positions of inserted or deleted motifs were indicated by colored bars and deleted motifs were indicated by ‘x’ signs.
Puffin estimated that the depletion of NF-Y not only reduces transcription initiation at promoters with strong NFY contribution but also leads to an upstream shift of transcription start sites in some promoters (Fig. 3B). Consistent with these predictions, knockdown of NFYA(25) (encoding a subunit of NF-Y) showed an upstream shift of TSS at locations predicted by Puffin (Fig. 3B). Moreover, only promoters with strong predicted NFY in silico KO effects showed strong changes in transcription, whereas promoters without strong predicted effects were largely unaffected (Fig. 3D).
Puffin also predicted that depleting YY1 has a unique effect of reducing transcription initiation signal at the immediate upstream of the motif, and a weaker effect about 200bp upstream of the first peak (Fig. 2D). We validated this effect using experimental data of induced YY1 depletion by auxin-inducible degron (AID) (26). The effects of YY1 depletion were measured by mammalian native elongating transcript sequencing (mNET-seq) which generates nascent transcript profiles at base pair resolution (27). We observed the reduction of transcription initiation at the model-predicted positions, including at the weaker upstream peak (−220bp) of the YY1 motif effect (Fig. 3C). The decrease in transcription initiation was also specific to promoters with strong predicted YY1 in silico KO effects (Fig. 3D). Both NF-Y and YY1 depletion experiments were performed in mouse cells and the predictions were based on mouse genome sequence.
Next, we tested the model’s ability to predict the effects of editing genomic sequence on transcription initiation signals (Fig. 3E). First, we evaluated the predicted effects with transcription initiation signals measured by self-transcribing active core promoter sequencing (STAP-seq) (28). STAP-seq was applied to measure the effects of TATA and NFY motifs deletions from promoters carrying these motifs and insertions of these motifs into neutral sequences with no promoter activity (29). The Puffin prediction not only recapitulated effects on expression level caused by sequence alterations (promoter-level correlation 0.88, Fig. S13A), but also displayed close to 0.9 base pair-level correlation with experimental data for promoters with strong expression (Fig. S13B). For example, deletion of either TATA or NFY motifs strongly decreased transcription in an LTR12 promoter (chr6:80516291–80517004, Fig. 3E). Insertion of NFY motifs into a neutral sequence (chr5:164906550–164906800) alone was sufficient to drive transcription. Inserting both NFY and TATA motifs created a promoter with transcription initiation signals more concentrated at one genomic location, consistent with experimental data (Fig. 3E). Puffin prediction also captured the shape of the transcription initiation signal, as well as changes of the shape upon motif insertions or deletions (Fig. 3E). Moreover, we performed STAP-seq experiments to validate the effects of disrupting three additional motifs, including SP, NRF1 and ETS in the promoters that carry these motifs, and observed similar decreases of transcription initiation signals consistent with Puffin prediction for all cases (Fig. S14).
In addition, to test the effect of motif disruptions in the native genome, we developed a CRISPR-Cap assay to delete or shift the positions of motifs downstream of 16 promoters, followed by the analysis of the 5’ ends of capped transcripts (Fig. S15). Because CRISPR-Cap can only target motifs downstream of the TSS we focused on YY1 and Long Inr motifs. We obtained sufficient editing efficiency and coverage for 11 promoters and observed altered base pair-resolution transcription initiation signal shape consistent with model predictions, whereas the negative control motif edits predicted to have no effect did not impact the transcription initiation signals (Fig. S16). These results further validated the effects of YY1 and Long Inr motifs on transcription initiation in vivo. Furthermore, to test the ability of Puffin and Puffin-D in predicting transcriptional activities of promoter sequences inserted into the genome, we compared our predictions with experimental measurements of >15,000 promoters via fluorescence-based reporter assay and achieved >0.7 correlation (Fig. S17) for both models. Taken together, these experimental perturbations corroborated the Puffin-estimated position-specific motif effects on transcription initiation.
Human promoters display diverse motif compositions that affect expression patterns
Puffin allows quantifying motif contributions to each promoter based on effects from each motif type (Methods). By analyzing the statistics of motif contributions across 40,000 human promoters (Fig. 4A–B, Fig. S18, Table S3), we noted that promoters display very diverse motif combinations, and no motif is necessary for all promoters (Fig. 4A–B). We did not observe strongly preferred or underrepresented motif combinations relative to expectation based on single motif contribution (Fig. S18B), suggesting that motifs can be rather flexibly combined in human promoters. We estimated that the effective number of contributing motif types to each promoter is most commonly 2–3 (Fig. S18C).
Fig. 4. Motif compositions of promoters are linked to gene expression selectivity.
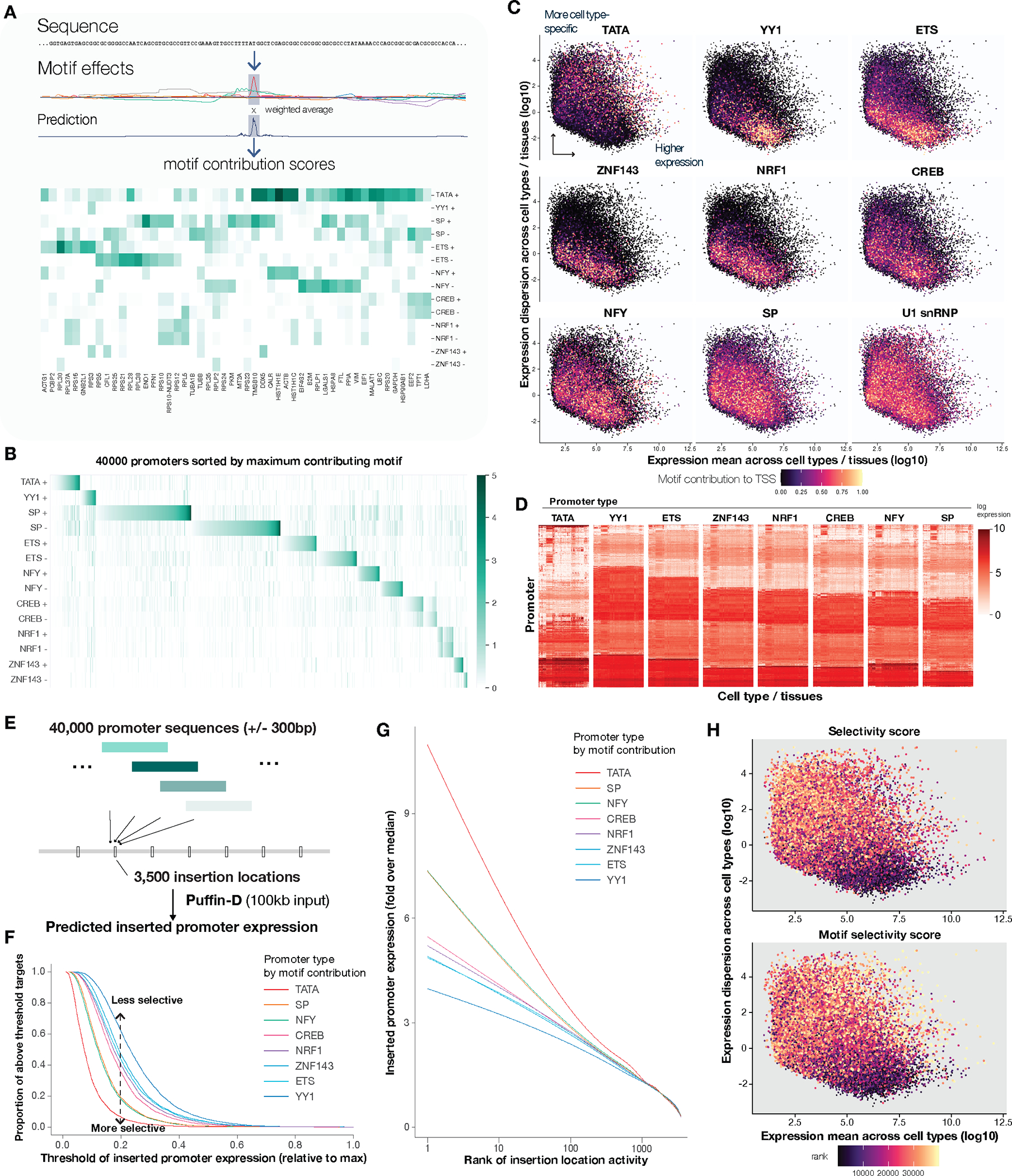
(A) Schematic illustration of motif contribution score. The motif contribution score is computed by the weighted average of motif effects within +/−20bp to the annotated TSS, the weighting function is the prediction. Example motif contribution scores of 100 promoters with the highest expression are shown. (B) Motif contribution scores across 40,000 promoters with the highest expression based on FANTOM CAGE, sorted first by the maximum contribution motif type and then by the contribution score of that motif. (C-D) Motif contribution has a strong impact on expression dispersion across cell types and tissues. Scatterplots (C) show log expression dispersion (y-axis) and log mean (x-axis) with each dot indicating a promoter. The dots are colored by the contribution score of a motif type in each subpanel. Expression matrices (cell type / tissue x promoter) for representative promoters per motif type (classified by maximum contributing motif type and filtered to promoters with top 2000 contribution scores from the classified motif type). (D) Promoter types defined by motif contribution displayed varied expression pattern distributions across cell type / tissues. Heatmap shows promoter by cell type/tissue expression matrix from FANTOM CAGE, represented by log size-factor normalized expression. (E) Schematic overview of the promoter insertion virtual screen with Puffin-D. 40,000 promoter sequences with 600bp length (+/−300bp to the annotated TSS) were inserted into 3500 target locations uniformly spaced over a 7Mb region (chr8:22964801–29964801). (F-G) Motif contribution affects selectivity to insertion targets in the virtual screen. (F) For each motif type (color), the top 1000 promoters by motif contribution scores were selected and the average predicted expression scores at each target location were computed, the proportions of target locations (y-axis) with predicted expression higher than the thresholds shown in the x-axis (scaled to proportion relative to the average of top-3 predicted expression levels across targets) were shown. (G) Generalized additive model-fitted curves of inserted promoter expression (scaled to fold over median expression across targets, y-axis) versus log rank of target activity (average target expression across 40,000 promoters) were shown. (H) The selectivity scores of promoters in the virtual screen (top panel) and motif selectivity scores (bottom panel) of promoters are linked to expression dispersion across cell types.
To evaluate how promoter composition is linked to gene expression properties, we analyzed promoter-level expression data across >200 cell types and tissues from the FANTOM project. We found that motifs have remarkable effects on expression variation across cell types (Fig. 4C–D). Promoters with higher TATA contributions were most likely to be cell type-specific or have high dispersion index across cell types and tissues. The dispersion index measures the variance of promoter expression divided by the mean and we used it to compare the cell type/tissue-specificities of promoters. The association between higher dispersion index and higher TATA contribution is consistent with previous studies (30). Moreover, the prevalence of TATA motif within cell type-specific promoters was not due to a preference for gene expression in any specific tissue (Fig. S19). Other motifs possess varying degrees of preference for ubiquitous expression patterns. For example, promoters with high YY1 contributions are most likely to be ubiquitously expressed or have low dispersion indices (Fig. 4C–D). High contributions from ETS, ZNF143, NRF1, and CREB motifs also favor more ubiquitous expression patterns. NFY and SP motifs are nearly neutral with a slight preference for more ubiquitous expression patterns. U1 snRNP, which we estimated to mostly affect transcript abundance after transcription initiation does not show a preference for ubiquitous or tissue-specific expression patterns. All promoter motifs are expected to bind at least one ubiquitously expressed TF. Because the link between promoter motif and cell type specificity cannot be explained by individual motif’s preference for specific tissues, we hypothesize that promoter motifs influence how promoters respond to cell type-specific transcriptional regulatory signals such as those mediated by distal enhancers.
To explore the potential mechanism of this link, we analyzed the promoter response to context sequences which we define as the flanking sequences to the promoters, and observed a similar relationship between motif contribution and promoter selectivity for context sequences. Specifically, we estimated promoter selectivity by inserting promoter sequences into different genomic locations and predicting the expression using the deep learning sequence model Puffin-D which takes 100kb sequence as input. In addition to accurately predicting the shape of transcription initiation signals, Puffin-D also excels at predicting the level of transcription initiation signals at the inserted promoters, with >0.9 promoter-level and gene-level correlation with experimental data (Fig. S1–2). Moreover, we showed that Puffin-D also predicts the effects of insertion locations when integrating promoters into the genome(31) (Table S4, Fig. S20). Thus, Puffin-D’s capability of utilizing long sequences makes it well-suited for studying the response of promoters to context sequences.
To estimate promoter selectivity, we inserted each of 40,000 human promoter sequences (600bp) into 3,500 locations that represent a diverse set of genomic contexts, and we predicted the levels of transcription initiation signals at the inserted promoters (Fig. 4E). More selective promoters are defined as being highly expressed in a smaller subset of genomic locations and thus are more selective to genomic context. Analysis of the 40,000 × 3,500 insertions revealed that motif contribution is strongly linked to promoter selectivity (Fig. 4E–H, Fig. S21). Notably, motifs displaying high and low selectivity are the same motifs that are linked to high and low expression dispersion indices across cell types/tissues (Fig. 4E–F). Moreover, promoter selectivity from the virtual insertion screen is predictive of expression dispersion index (Fig. 4G). By training a linear model to predict promoter selectivity from motif contribution, we obtained a sequence-based score of promoter selectivity, which is also predictive for high dispersion, tissue-specific expression patterns (Table S5).
Taken together, based on these results, we postulate that motif contributions determine the response curve of a promoter to external signals of transcriptional activation. For example, promoters with high TATA contribution are much more responsive to strong transcriptional activation signals than promoters with high YY1 contribution. This mechanism can also explain the similarities between promoter selectivity to context sequences and cell type-specific expression patterns. Hence, promoter sequence composition likely plays a key role in determining gene expression patterns in conjunction with distal regulatory sequences.
Sequence basis of bidirectional transcription at promoters
Bidirectional transcription initiation is observed at most human promoters when measuring nascent transcripts (17, 22, 32, 33); however, such bidirectional transcription shares a common sequence basis has remained an open question(34–36). With Puffin, we provide an explanation of the sequence basis of bidirectional transcription initiation (Fig. 2D and 5A). Specifically, bidirectional motifs with symmetric effects in both directions are the main contributors for most promoters (Table S2), leading to substantial transcription initiation on the reverse strand, specifically at the level of nascent transcripts. Directional motifs including TATA, YY1, and Long Inr contribute to preferential transcription initiation on a single strand, although they can have activating effects on the other strand, such as YY1, or be partly palindromic such as TATA. On the other hand, most promoters are strongly directional when measuring mature transcripts even if they are bidirectional when measuring nascent transcripts. As described above, U1 snRNP motif exhibited a unique mechanism by contributing to the production of mature transcripts unidirectionally likely via post-transcription initiation effects (Fig. 5A), consistent with previous studies(17, 21–23).
Fig. 5. Sequence-basis of bidirectional transcription initiation at human promoters.

(A) Schematic illustration of a sequence-based model of bidirectional transcription initiation. Directional motifs preferentially contribute to transcription initiation on the forward strand, and bidirectional motifs contribute to transcription initiation on both strands. Promoters with a high proportion of contribution from bidirectional motifs are bidirectional at the nascent transcript level. Most promoters are strongly directional at the mature transcript level, and the U1 snRNP motif contributes to a directional mature transcript outcome. (B). Base pair contribution analysis to an example promoter with bidirectional transcription initiation. The forward (+) and reverse strand (–) prediction and experimental measurements in log scale were shown in the top two panels. Reverse strand values were taken a negative sign. The bottom two panels showed per-motif base pair contribution scores to forward- and reverse- strand transcription respectively. All rows were scaled to maximum 1. (C) Base pair contribution scores for forward (top panel) and reverse (bottom panel) strand transcription of 8,216 promoters, sorted by reverse TSS position, scaled by the sum of positive base pair contribution scores per promoter and strand. The three columns are experimentally measured transcription initiation signals, predicted transcription initiation signals (PRO-cap), and base pair contribution scores (PRO-cap) respectively. (D) High correlations between forward and reverse strand base pair contribution scores (x-axis) for reverse TSS with distance to forward TSS (y-axis) within 300bp. The proportions of forward-reverse TSS pairs with less and greater than 0.75 correlation were indicated, for both pairs with less and greater than 300bp distance.
Beyond qualitative explanation, our model also allows quantifying the degree of shared sequence contribution for any forward-reverse TSS pair (Fig. 5B–D). To determine how much sequence contribution is shared between forward and reverse strand TSS pairs, we selected 8,216 promoters with high expression levels in both directions based on PRO-cap (Methods). Puffin prediction well recapitulated forward and reverse directional TSS positions (Fig. 5C), which allowed us to further analyze base pair level sequence contribution to transcription initiation on both strands. Comparing base pair contribution scores for both strands (Fig. 5D, Fig. S22) revealed high correlations for most promoters (87.7% with r > 0.75). Most of the reverse TSS that were within 300bp of the forward TSS have high correlations (92.6% with r> 0.75), while less shared sequence contribution was more common for reverse TSS that are further than 300bp away (60.7% with r > 0.75). Thus, most bidirectional TSS pairs in close proximity (<300bp) share a substantial proportion of contributing sequence.
Evolutionary conservation of promoter sequence determinant across mammal species
Finally, we examined whether the sequence dependencies captured by Puffin are conserved across mammalian species. Since high-coverage transcription initiation datasets are also available for mouse, we first assessed the cross-species generalization of transcription initiation models between human and mouse using the CAGE data from the FANTOM project(4) (Fig. 6A). The models trained on mouse data uncovered identical core motifs as the human model (Data S3). Predictions by the human model and mouse model on the same sequences are also highly similar (median correlation of 0.967, Fig. 6B). Moreover, applying the human Puffin model to mouse sequences achieved very close performance compared to the mouse model (Fig. 6B). Thus, the transcription initiation sequence dependencies learned by Puffin between human and mouse data are nearly interchangeable.
Fig. 6. Cross-species generalization and conservation of promoter sequence rules across mammals.
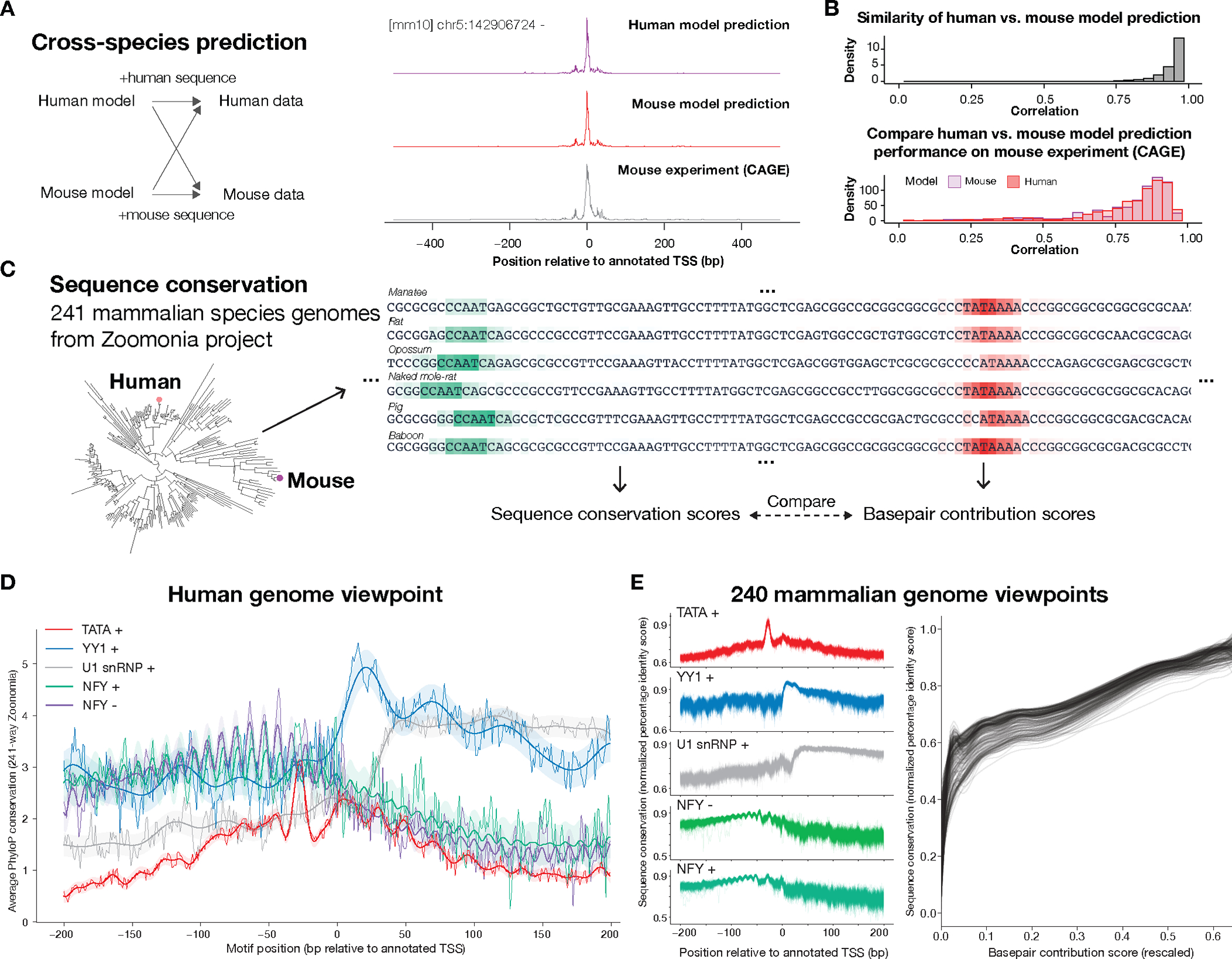
(A) Schematic illustration of cross-species prediction comparison of human and mouse Puffin transcription initiation models. Models trained on human and model data respectively are evaluated on predicting holdout promoters. Example predictions and experimental signals were shown. (B) Human and mouse models have highly similar predictions (top panel) and almost equal performances (bottom panel) on the mouse genome. The distribution of correlations between human and model predictions on a mouse is shown in the top panel and the base pair level correlation for the human model (red) and mouse model (purple) are shown in the bottom panel. All comparisons are made on mouse holdout sequences (Methods). (C) Schematic illustration of sequence rule conservation analysis across 241 mammalian species. For each species, motif-specific base pair contribution scores are computed and compared with sequence conservation scores. (D) Position-specific sequence conservation scores for motifs share a similar pattern as our estimated position-specific motif effects, from a human genome viewpoint. X-axis shows the position (bp) relative to annotated TSS (human genome viewpoint) and the y-axis shows average PhyloP scores for each motif, computed with the average weighting by motif activation scores. This TSS-centered pattern is mirrored versus the motif-centered view. The bold line and shades indicate posterior mean and 95% credible intervals of Gaussian process regression (Radial basis function kernel + White kernel + Periodic kernel for NFY, Radial basis function kernel + White kernel for other curves). (E) Position dependencies of sequence conservation scores are also observed from the viewpoint of each of the 240 non-human mammalian species (left panel), with normalized percentage identity score (y-axis, Methods) with other species as the conservation metric. Base pair contribution scores computed by Puffin (right panel, x-axis, scores were transformed to the power of 0.25) are strong predictors of sequence conservations measured by percentage identity scores (y-axis) in all 241 mammalian species.
We next analyzed the conservation across mammalian species using genomes of 240 species from the Zoonomia project(37) (Fig. 6C). We first tested the hypothesis that motifs located at positions where the motif effects at the TSS position are stronger are more evolutionarily conserved. Specifically, we computed the average evolutionary conservation PhyloP scores for each motif across all positions relative to TSS (Fig. 6D). The resulting position-specific evolutionary conservation curves indeed showed obvious similarities with position-specific motif effect curves. For example, TATA motifs are most conserved at ~30bp upstream of TSS, whereas YY1 motifs are most conserved immediately downstream of TSS. U1 snRNP motifs are most conserved starting from 50bp downstream of TSS. Moreover, the NFY motif’s ~10.5bp periodic effect patterns are reflected in evolutionary conservation scores (conservation curves for all motifs are shown in Fig. S23). These results are consistent with the motif’s estimated position-specific effects. Thus, evolutionary conservation provides another independent line of evidence for the position-specific motif effects.
We found similar conservation patterns from the viewpoint of any of the 241 mammalian species (Fig. 6E). To assess the conservation of transcription initiation sequence rules across species, we analyzed each of the other 240 mammalian genomes, using the genome sequence for each species as input and sequence identities with all other species per base as the conservation measure. We also found that the base pair-level contribution score by Puffin is a strong predictor of evolutionary conservation in all 241 species (Fig. 6E). Thus, we expect that sequence dependencies of transcription initiation captured by Puffin will be widely applicable across mammalian species.
Discussion
We have created a simple model that explains the transcription initiation activity of most human promoters at base pair resolution. This was achieved through the development of a deep learning-inspired explainable modeling approach, which allowed for learning a compact model that provides insights into the sequence basis of transcription initiation. Puffin both recapitulated known biology and provided new predictions that are well supported by experimental results and evolutionary conservation. We discovered that sequence determinants of transcription initiation can be effectively described using a small set of rules, ultimately making it tractable to systematically and quantitatively analyze transcription initiation.
Our model and analyses shed light on many questions related to promoter sequence and function that we set out to answer: for most human promoters, we can now identify individual base pairs and motifs that contribute to transcription initiation; a simple additive / multiplicative model of sequence pattern effects is sufficient to recapitulate most of the transcription initiation activity; we uncovered new connections between promoter sequence compositions, cell type-specific gene expression, and promoter selectivity; we provide an explanation for the sequence-basis of bidirectional transcription and strand preference at human promoters; lastly, we demonstrate that the sequence rules of promoters are conserved across mammalian species.
We anticipate that the sequence-level understanding of transcription initiation, especially the position-specific effect curves of motifs, and the understanding of molecular-level mechanisms will eventually converge, and future research will likely discover the molecular and structural underpinnings of each motif’s position-specific effects. The motif position-specific effect curves may also partially explain the variations of TF effects on different promoters. For example, YY1 and bidirectional motifs have both strong activating and repressive effects depending on the relative position between the motif and the TSS.
Together, our study provides systematic insights into the sequence determinants of transcription initiation in the human genome and beyond, as well as a powerful tool for understanding and engineering promoter sequences and gene regulation across species. Our findings also underscore the importance of explainable machine learning and computational modeling for unraveling sequence-based mechanisms governing transcriptional regulation, with the potential of discovering sequence rules for diverse genomic functions.
Materials and Methods
Processing of transcription initiation signal datasets
All transcription initiation datasets (FANTOM CAGE, ENCODE CAGE, ENCODE RAMPAGE, GRO-cap, PRO-cap) were downloaded from published datasets listed in Data S1. As individual signal profiles do not have enough coverage for providing accurate estimates at base pair resolution except for the most highly expressed promoters, we aggregated all signal profiles measured by the same technique. Specifically, the base pair-resolution count profiles were averaged after applying transformation where is the read count, with plus and minus strand profiles aggregated separately. The addition of the pseudocount can be interpreted as applying a uniform Dirichlet prior and obtaining the posterior mean. In this manuscript, we refer to this aggregated signal as the log scale signal and its inverse transformed value by as the count scale signal.
We next addressed the known bias for poly-T sequence in the FANTOM CAGE aggregated signal profile, which is specific to the HelioScope CAGE protocol used for FANTOM CAGE datasets(38), but does not affect ENCODE CAGE datasets. After analyzing FANTOM CAGE signal dependency on the number of consecutive ‘T’s across the genome (Fig. S24), we applied a filter with a threshold of >=8 consecutive ‘T’s, and masked the signals in [−6, +10) interval relative to the end of the poly-T sequence. The filtered FANTOM CAGE signal profile is replaced with ENCODE CAGE signal rescaled to the average signal level of FANTOM CAGE. The masked regions represent a minor fraction of the genome (0.5%).
We obtained human promoter annotation from the FANTOM-CAT catalog at the “robust” level(39) and removed promoters with inconsistent expression levels across datasets. We then rank all promoters by expression level in the aggregated FANTOM CAGE profile, quantified by the sum over +/− 20bp window surrounding annotated TSS in count scale (we refer to this ranking when we used top-N promoters in this manuscript). Specifically, for removing promoters with inconsistent expression levels, non-protein-coding gene promoters that are >40-fold lower in normalized expression value than FANTOM CAGE in both ENCODE CAGE and ENCODE RAMPAGE datasets, after scaling by total expression values across all promoters for each dataset, were removed. In addition, for performance evaluation, we selected high-confidence promoters that have consistent base pair-resolution transcription initiation profiles across experimental techniques. Specifically, for the 1000bp interval centered at the TSS position, only promoters with high correlations between the FANTOM CAGE profile and at least one other dataset (>0.5 for ENCODE CAGE, >0.4 for ENCODE RAMPAGE, GRO-cap, or PRO-cap datasets) were preserved. The filtered list of promoters with high-confidence promoter labels is provided in Table S6.
Interpretation-focused Puffin model for transcription initiation
The Puffin model consists of two learnable layers, a sequence pattern activation layer, and a transcription initiation effect layer (Fig. S4). Both layers were trained from scratch based on only sequence and transcription initiation signal data, without using any known motifs. Both layers’ computations can be represented by convolution layers. The convolution kernels of the first layer represent sequence patterns’ position-specific weights and the convolution kernels of the second layer represent sequence pattern activations’ position-specific effects on transcription initiation.
Since Puffin uses three sequence pattern types with different sizes (kernel size in the sequence pattern activation layer) and effect ranges (kernel size in the sequence pattern effect layer), each layer of the model contains several parallel convolution layers each corresponding to a sequence pattern type. Specifically, the sequence pattern sizes of motifs, initiators, and trinucleotides are 51bp, 15bp, and 3bp respectively, and the effect ranges are +/−300bp, +/−7bp, and +/−300bp respectively. The sequence pattern sizes and effect ranges specified in the model are the maximum that the model can learn and the effective sequence pattern size and effect ranges learned are usually lower. We note that the initial design of Puffin uses only the motif sequence patterns, and more parsimonious sequence patterns with smaller sizes or effect ranges including initiators and trinucleotides were introduced in a data-driven process to obtain a more compact and interpretable design while maintaining the performance (Supplementary Text).
The first layer, or sequence pattern activation layer, uses the softplus activation function to compute the sequence pattern activations. Moreover, the activations of each reverse-complement sequence pattern were also computed, which doubles the number of channels from the sequence pattern activation layer output. For the second layer, or sequence pattern effect layer, we used FFT-based convolution(40) for computing motif and trinucleotide effects because the large kernel size makes FFT-based convolution more efficient than regular convolution implementation. Finally, the sequence pattern effect layer computes sequence pattern effects for 10 targets, which corresponds to 5 different techniques on both forward and reverse strands, thus the sequence pattern effect layer learns a separate set of sequence pattern effects for each target.
The sum of sequence pattern effects per position were transformed by the softplus function to the final prediction. The softplus function is deliberately chosen here to make the input and output interpretable. In this formulation, the pre-activation sum of sequence pattern effects is comparable to where is the transcription initiation signal in count scale, because we train the model by fitting the scaled softplus function output to the log scale transcription initiation signal . Thus, the additive combination of sequence pattern effects in space can be considered as a multiplicative combination in space. Similarly, the final Puffin model prediction is comparable to natural log scale transcription initiation signal and can be converted to count scale using exponential minus 1 transform.
The main training loss function is the Kullback-Leibler(KL) divergence loss
where , , is the position index, and is set to 1e-10. The target is processed as introduced in the previous section. The KL divergence loss is sensitive to the shape but not the magnitude of the transcription initiation signal. This is intended as the overall abundance is dependent on not only the promoter. The prediction and target are both divided by the sum over the 4kb region before computing KL divergence loss. Since the KL-divergence loss does not constrain the scale of the prediction, we added an auxiliary loss that matches the exact transcription signal values. This auxiliary loss also increased the interpretability of the prediction by allowing it to be considered as a prediction of transcription initiation signals, even though the training emphasizes learning the shape rather than the magnitude of the transcription initiation signal. Specifically, the auxiliary loss is
where indicates the position and is 1e-10. We refer to this loss as the pseudo-Poisson loss as it has the same form as the Poisson loss function, which is derived from the Poisson negative log-likelihood after dropping constant terms, but the target values are not counts. The pseudo-Poisson loss is more robust to over-dispersion of the data than the Poisson loss. The factor is due to conversion from natural log scale to log10 scale, and is included for numerical stability. The auxiliary loss is weighted by a factor of 1e-3. The losses for all ten targets were averaged. Additional regularization terms were added to the loss function including L1 regularization to kernel weights in both layers, and L2 smoothness regularization between spatially adjacent kernel weights in the sequence pattern effect layer, for motifs and trinucleotides.
The models were trained to predict from one-hot encoded sequence to transcription initiation signals on both strands for 5 experimental techniques. Specifically, the model was trained with the task of predicting transcription initiation signal in the 4kb region surrounding each annotated TSS with a random strand selected for each training sample. Top 40,000 high-confidence promoters ranked by expression level as described in the previous section were used. We divide the genome into the training set: all chromosomes except for chr8, 9, and 10, the validation set: chr10, and the test set: chr8 and 9. The training data was retrieved on-the-fly during training and the strand is selected randomly for each training sample.
We trained the Puffin model in three stages to make sure that all motifs included were reproducibly discovered across multiple training replicates. In the first stage, we trained 12 replicates with different random seeds. The first stage model differs from the final Puffin architecture in that it learns 40 motif sequence patterns and 10 initiator sequence patterns, and the SiLU activation function was used for the first layer because it facilitates motif discovery. A nonredundant set of 10 motifs that were reproducible (> 0.95 maximum cross-correlation across >7 replicates) were chosen as the consensus motifs. In the second stage, we use the final Puffin model architecture as described, and sequence patterns and effects were initialized by the consensus sequence patterns from the first stage. Since not all sequence patterns learned were located at the center of the 51bp motif kernel, in the third stage, we centered the motif sequence patterns and continued training.
The model can process variable-length input, and the raw model output size equals the input sequence length. But since predictions near the edge can be affected by padding, to completely remove the effect of padding, we recommend trimming predictions from each end by 325bp. Therefore, for example, to obtain N-bp prediction we use N + 650bp long sequence as an input.
For evaluation of the prediction performance, we computed Pearson correlation between experiment and prediction per promoter for 1kb windows centered at each annotated. High confidence promoters on test chromosomes among the top 100,000 promoters were used for evaluation. For downstream analysis, FANTOM CAGE prediction and contribution scores were used unless otherwise indicated.
Visualization of Puffin motifs and position-specific effect curves
For visualization of sequence patterns such as motifs, the position-specific weight matrices were directly obtained from the kernel weights of the first layer. We note that adding or subtracting a value to all four bases at the same position does not change the layer output (up to a constant which can be canceled out by bias term), thus for standardization we processed the motif position-specific weight matrices per position by first subtracting the mean and then subtracting 0.7x average absolute value per position (the later subtraction highlights the most positive base in visualization, otherwise typically more than one base were highlighted due to subtraction by mean). The processed position-specific weight matrices are visualized with the logomaker(41) package with each letter height indicating the absolute value of that nucleotide and positive and negative values shown above and below the axis respectively.
We visualize the position-specific effect curve of each motif in motif-centered coordinates, which were obtained by reversing the spatial dimension of the kernel weights of the sequence pattern effect layer.
Prediction-focused Puffin-D model for transcription initiation
Puffin-D is the prediction-focused architecture that captures sequence dependencies up to 100kb. Puffin-D is trained by randomly sampling 100kb intervals from the training chromosomes(42). Different from Puffin which is trained with KL divergence loss to predict the shape of the transcription initiation signal, Puffin-D is trained to predict the exact value of the transcription initiation signal. The training loss function is the pseudo-Poisson loss, which is the same as the auxiliary loss function for training Puffin without the factors. Thus Puffin-D prediction is interpreted as log10 scale transcription initiation signal where is the transcription initiation signal in count scale.
To utilize long-range sequence information efficiently, the Puffin-D architecture uses an architecture that iteratively propagates information across sequence locations via two upward-downward passes. The upward passes integrate long-range sequence information hierarchically and the downward passes distribute integrated information to local sequence representations (Fig. S25). Residual connections between the same levels of the upward and downward passes similar to U-Net(43) allow information at all spatial resolutions to be preserved. A new design different from U-Net is that we used two upward and downward passes to allow better mixing of global and local information. Individual convolution blocks were modified over previous sequence model architecture design(44) by adding size 1 convolution layers, replacing ReLU with SiLU activation function, and replacing max pooling with strided convolution.
To evaluate Puffin-D performance, we generated predictions for entire test chromosomes chr8 and chr9, with a sliding window step size of 50kb, and the center 50kb of each 100kb prediction was used. Regions within 1kb to unknown bases or 25kb to chromosome ends were excluded. In addition to base pair level correlation, we computed correlations at the transcript level and gene level. At the transcript level, we aggregated prediction and experimental signal at count scale within 400bp window to each annotated transcription start site; at gene level, we further summed all transcript-level prediction and experimental signal per gene.
Sequence contribution scores
An important advantage of the interpretation-focused Puffin architecture allows quantitatively analyzing sequence contribution to transcription initiation at motif and base pair levels. While base pair-level contribution scores can also be obtained via general-purpose deep learning model interpretation methods (45), the simple architecture allows a more tailored definition with simple interpretation. Here we describe the definition and interpretation for each motif and base pair-level contribution score:
Motif contribution score
Motif contribution score represents the amount of contribution from a motif type to any position or window in the sequence. The motif contribution to any position can be directly represented by motif effects in the Puffin model, because sequence pattern effects are additively combined, and the baseline for motif effects is zero due to regularization that drives the motif effect to zero at long distance. Thus, motif contribution is almost synonymous with motif effects in Puffin model, but motif contribution scores can also be computed by aggregating over a window in a weighted or unweighted fashion.
In this manuscript, the motif contribution scores for each promoter were computed by the weighted average motif effects within the 20bp window centered at the annotated TSS, and the weights are the predicted transcription initiation signals. We provide the full contribution scores in Table S2. For downstream analysis, we summed the motif contribution scores for forward and reverse directional motifs for bidirectional motifs, and for directional motifs, only the + direction motif contribution scores were retained. U1 snRNP (post-transcription initiation effect) and Long Inr (initiator sequence pattern) were usually excluded from motif contribution analysis, because U1 snRNP has mostly post-transcriptional initiation effects, and Long Inr effects are initiator-like and do not have zero baselines like other motifs.
Base pair contribution score to transcription initiation
We also refer to this score as the base pair contribution score, which represents the amount of contribution to transcription initiation from each base pair. Moreover, base pair contribution score can also be decomposed to per motif type scores, allowing dissecting base pair level sequence contribution by motifs. The base pair contribution score equals the sum of base pair contribution scores for all motifs.
Where indicates the base pair contribution score for motif and the base at position indicates the model prediction at the position and indicates the window of transcription initiation prediction that base pair contribution scores are computed for. For notational simplicity we use to denote the vector of motif activation across all positions for the motif indicates the gradient with respect to the true base at sequence position , while indicates the gradient with respect to any other base at sequence position . The base pair contribution score can be computed for any of the 10 targets that Puffin predicts by choosing the corresponding predictions.
The base pair contribution score can be computed with respect to transcription initiation signals in any position or window. In this manuscript we computed it for 1kb windows surrounding each annotated TSS, using 1650bp sequences. Moreover, in all analyses, we further scale the base pair contribution score as defined above by dividing the sum of positive base pair contribution scores within the 1kb window per promoter. This score was used to compare sequence contributions underlying bidirectional TSS pairs on both strands and to analyze the relationship between base pair contribution score and sequence conservation across species.
Base pair contribution score to motif activation
The base pair contribution score to motif activation represents the amount of contribution to motif activation from each base pair, which is computed using only the first layer of Puffin. This score was used in the analysis of position-specific evolutionary conservation patterns of each motif and notably it does not involve the sequence pattern effect layer or any learned motif effects to compute.
Puffin in silico knockout for TF perturbation effect prediction
As the Puffin model dissects sequence dependencies into effects from individual sequence patterns, which are mapped to TFs, we can simulate the effects of TF depletion by removing the effects from that motif using the Puffin model. We refer to this technique as “in silico KO”, which mimics the effect of acutely removing the TFs that bind to any motif. To perform in silico KO, we set the motif activation scores for the corresponding motif to 0 and continue the subsequent computations, which also set the effects of that motif effect to 0.
We evaluated in silico KO predictions by comparisons with experimental data from NF-Y and YY1-depletion datasets(25, 26). We first selected for promoters with a strong contribution from NFY and YY1 respectively, quantified by the sum of absolute predicted difference over the 1kb window centered at each TSS. A threshold of 20 and 55 were used for YY1 and NFY respectively. We demonstrated that the expression level of promoters above this threshold was significantly decreased compared to promoters without YY1 or NFY contribution with a threshold of 1, using a two-sided Wilcoxon rank sum test. The expression levels of promoters were quantified by −50 to +50bp for NFY Start-seq and −50 to +100bp for YY1 mNET-seq data (mNET-seq signals tend to be shifted downstream from the TSS). We then compared the predicted base pair resolution effect of TF depletion with experimental data for the selected promoters with heatmap visualization.
Puffin prediction of motif insertion and deletion effects
We compared Puffin prediction with the motif perturbation dataset published by(29), which measured the effects of TATA and NFY motif mutations in human promoters and insertions of these motifs into neutral sequences. The transcriptional activities of wildtype and mutants were measured using STAP-seq in 500 oligomers with 5 replicates for every sample. The oligomer sequence was obtained from the data and the surrounding sequence was retrieved from the human STAP-seq screening vector sequence (Addgene ID: 125150). Both base pair level and promoter-level prediction and experimental measurements were compared. Promoter-level quantification was computed by summing count scale predictions or signals over the 250bp oligonucleotide.
Puffin-D prediction of human TRIP promoter insertion
TRIP (Thousands of Reporters Intergrated in Parallel) insertion sequences were recreated according to Hong et al.(31) paper using the pCPL4 vector, promoter sequence coordinates, the locations of TRIP integrations and barcodes as provided by the authors. Predicted values were obtained by taking the log10 of the predicted ENCODE CAGE signal at count scale summed over a 2Kbp region surrounding the insertion site. The observed values are log-transformed ratios of RNA to DNA.
Puffin and Puffin-D predictions of K562 genome-integrated reporter assay
Sequences were reconstructed as described in Weingarten-Gabbay et al.(6) using a plasmid kindly provided by the authors. The prediction values for Puffin and Puffin-D are log10-transformed prediction for GRO-cap summed over the region surrounding the inserted oligomer at count scale. Puffin-D prediction of the inserted oligomers is normalized by the mCherry expression predicted by Puffin-D, similar to the experiment.
Promoter motif contribution and expression selectivity
Motif contribution scores for promoters were computed as described in the “Sequence contribution scores” section. Promoter-level expression values across >200 cell types and tissues were obtained from FANTOM CAGE profiles quantified by the sum over +/− 20bp at annotated TSS in count scale. CAGE profiles for samples for the same cell type or tissue were summed at count scales. The raw promoter-level counts for each cell type / tissue were then normalized by dividing the size factors estimated by the DESeq2 package(46). The mean and dispersion of promoter-level expression across cell types and tissues were then computed from normalized counts and compared with motif contribution scores.
For comparison of promoter expression patterns across promoter types by motif contribution, we selected the top 5% TSS by motif contribution score for each motif type among the top 40,000 TSS. The expression matrix of all 40,000 promoters were hierarchically clustered, and the row and column orders were preserved in the visualization of individual promoter types.
To estimate promoter selectivity to genome contexts, we performed an in silico insertion screen with Puffin-D model. For insertion sequences, we used 600bp centered at each of the top 40,000 promoters, and for target locations, we selected 3500 locations uniformly spaced in the genomic interval chr8:22964801–29963540 with the step size 2000bp. The target locations are on a chromosome that was held out from the model training. For each of the 40,000 × 3,500 in silico insertion experiments, we replaced 600bp of the target genome sequence with the 600bp insertion sequence. The prediction for the transcription initiation signal was made using the Puffin-D model. The predicted transcription initiation signal, or expression level, was measured by the mean FANTOM CAGE prediction in count scale within +/−20bp from the center position.
The selectivity score for each promoter was defined as the proportion of insertion targets with predicted expression levels below the threshold. The threshold for selectivity score computation for each TSS was defined as 1/3 of the mean of the top-3 predicted expression levels across target locations. Motif selectivity score is estimated by training an L2 regularized linear regression model to predict log-odds of selectivity score from motif contribution scores, and the regularization parameter was selected by leave-one-out cross-validation in closed form.
Base pair contribution scores for bidirectional promoters
TSS with high expression levels in both strands were selected based on the aggregated PRO-cap transcription initiation signal in count scale on both strands. Specifically, both the sum of the forward strand PRO-cap signal within −250 to +250bp window, and the sum of reverse strand signal within −500 to 0bp window are >50. Moreover, we selected promoters for which the maximum signal position on the reverse strand is upstream of the maximal signal position on the forward strand, the maximal signal position on the forward strand is within 50bp to the annotated TSS position, and the maximal signal position on the reverse strand is upstream of the annotated TSS position. Base pair contribution scores to transcription initiation for PRO-cap on both strands were computed and compared by correlation (within +/−500bp window to the annotated TSS).
Comparison of promoter sequence dependencies in human and mouse
To compare human and mouse sequence dependencies captured by Puffin, we trained Puffin models on mouse FANTOM CAGE data(4), aggregated with the same procedure as human FANTOM CAGE data. The consensus motifs from mouse training replicates were analyzed in the same process as for human and showed nearly identical motifs. We next compared predictions between human and mouse models applied to the mouse genome. To ensure that the comparison is appropriately done, we liftover the mouse TSS annotation from the FANTOM project from mm10 to hg38. The top 40,000 mouse promoters ranked in the same process as described for the human TSS were used for this analysis. The mouse models were trained on promoters for which the liftover coordinates were in human training chromosomes. Similarly, the evaluations were performed on mouse promoters for which the liftover coordinates in hg38 locate in the human holdout chromosomes. We compared human and mouse Puffin models after stage 1 training, and the average prediction across 12 training replicates was used for both human and mouse. Human and mouse model predictions for 1kb regions centered at annotated TSS that belong to the test set were also computed and compared with mouse FANTOM CAGE signals.
Evolutionary conservation across 241 mammalian genomes
We obtained 241-way mammalian genome alignment from the Zoonomia project and downloaded the PhyloP scores from the UCSC genome browser. To analyze position-specific evolutionary conservation for each motif from the human genome viewpoint, for every base pair position relative to the annotated TSS from −200bp to +200bp, we computed the weighted average PhyloP score across top 4000 promoters for each motif. The weight used is the base pair level motif activation score for that motif.
To analyze evolutionary conservation from the viewpoint of each of the other 240 genomes, human genome annotated TSS positions were liftover to each genome, and the 1650bp sequences centered at the TSS in each genome assembly were retrieved. Base pair contribution scores to motif activation and transcription initiation were computed separately for sequences from each genome. We used percentage identity with the rest of the 240 genomes as the raw sequence conservation score. Percentage identity scores per base pair were computed based on multi-sequence alignment generated by MAFFT(47). Since each species has a different distribution of evolutionary distance from other species, the raw percentage identity scores are not directly comparable in scale with each other. To make percentage identity scores comparable across species we applied quantile normalization by linearly scaling. The linear scaling matches each species’ 0.1 and 0.9 quantiles to the human quantiles. For each species, in addition to computing position-specific evolutionary conservation scores with normalized percentage identity scores, we also fitted species-specific curves representing the relationship between scaled base pair contribution scores (transformed to the power of 0.25) and normalized percentage identity scores by generalized additive model.
CRISPR-Cap assay for measuring effects of motif perturbation on transcription initiation sites
We designed single guide RNAs (sgRNAs) targeting TSS motifs containing the NGG PAM sequences (Table S7). Synthesized sgRNA oligos were mixed with purified CRISPR-Cas9 protein at 2:1 ratio to form RNP complexes in OptiMEM for 10 min. The RNP complexes were transfected into HEK293T cells by lipofectamine 3000. HEK293T was cultured in DMEM supplemented with 10% FBS at 37°C with 5% CO2. To maximize the efficiency of Cas9 editing, the second transfection was performed after 2 days. After two rounds of RNP transfection, total RNA was isolated by TRIzol. Then each 1ug of total RNA was treated with 2U DNase I for 15min at 37°C. DNase I was removed by TRIzol extraction and the RNA was diluted by nuclease-free water. Then 1ug of RNA was dephosphorylated by 0.5ul of calf intestinal alkaline phosphatase (CIP; NEB cat. no. M0525S). The reaction was cleaned up by TRIzol. Then the 5’ cap of RNA was removed using Cap-Clip Acid Pyrophosphatase (cat. no. C-CC15011H from CELLSCRIPT, CCAP), for each 1ug RNA with 0.05ul CCAP. The reaction was cleaned up by TRIzol. Then we ligated CCAP-treated RNA with 5’end RNA oligo (GUUCAGAGUUCUACAGUCCGACGAUCNNNNNNNN) with 1 ug RNA for 100uM oligo at 16°C for 16 h using 0.2 μl T4 RNA Ligase 1 (ssRNA Ligase, NEB, cat. no. M0204L), followed by TRIzol cleanup. 8nt of random nucleotides in the 3’ end of RNA oligo were used as unique molecular identifiers (UMI). First strand cDNA synthesis was performed using 1μl of Superscript IV (50°C for 60 min, 70°C for 15 min; Invitrogen, cat. no. 18080085) using a 20bp gene-specific RT primer (Table S7) for 2.5–5 μg of RNA in 20 μl. Then 1 μl of 10 mg/ml RNaseA was added to remove the RNA at 37°C for 15 min. Finally, cDNA was amplified for pair-end sequencing using Illumina NextSeq 2000 by the sequencing primer AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGA and gene-specific PCR primers (Table S7) with the Illumina adapter sequence (TTCAGACGTGTGCTCTTCCGATCTN20). All samples were pooled and amplified by Illumine sequencing adapters. The read 1 primer was replaced by Illumina small RNA TrueSeq primer for 5’ end RNA ligator (TCTACACGTTCAGAGTTCTACAGTCCGACGATC). The sequencing was performed using NextSeq 2000 by 80 (read1), 0 (index1), 8 (index2), and 150 (read2) with 200-cycle P3 kit. The reads were aligned to the hg38 reference genome by STAR(48). Only reads that covered the targeted sites of CRISPR sgRNA and allowed for genotyping were retained for downstream analysis. The distributions of 5’ end positions of transcripts with UMIs between WT reads and MUT promoter sequences were compared.
Experimental validation of the effects of motif disruption by STAP-seq
We designed three wildtype (WT) and three mutated (MUT) 240-bp promoter sequences to measure the effects of SP, ETS and NRF1 motifs deletions, respectively (Table S8). These sequences were cloned into pSTAP-seq_human-4xUAS vector (Addgene ID: 125150). Equal quantity of vectors were pooled for transfection into HEK293T cells using Lipofectamine 3000. After overnight incubation, we collected total RNA using TRIzol and constructed the library as previously described(28). Pair-end reads were aligned to the custom reference consisting of six plasmids containing the WT and MUT promoter sequences using Bowtie 2(49). The total read counts per position reflect the number of 5’ end positions of the aligned fragments.
Supplementary Material
Acknowledgments
The authors thank Ronald C. Conaway, Christopher Y. Park, and Kathleen M. Chen for feedback to this manuscript. This work is conducted on the high-performance computing facility BioHPC at the University of Texas Southwestern Medical Center.
Funding:
National Institutes of Health grant DP2GM146336 (JZ)
Cancer Prevention and Research Institute of Texas grant RR190071 (JZ)
National Institutes of Health grant R01DK111430 (JX)
National Institutes of Health grant R01CA230631 (JX)
National Institutes of Health grant R01CA259581 (JX)
Leukemia & Lymphoma Society scholar award (JX)
Footnotes
Competing interests: Authors declare that they have no competing interests.
Data and materials availability:
The code and model for Puffin and Puffin-D are available at https://github.com/jzhoulab/puffin. The code for reproducing the analyses in the manuscript is available at https://github.com/jzhoulab/puffin_manuscript. Both were deposited into Zenodo repository (50). A user-friendly website for using Puffin is available at https://tss.zhoulab.io.
The human GRCh38/hg38 reference genome was used for training the Puffin and Puffin-D models. The mouse models were trained with the mm10 reference genome. All coordinates in the manuscript refer to GRCh38/hg38 unless otherwise indicated. All transcription initiation signal datasets used for model training (FANTOM CAGE, ENCODE CAGE, ENCODE RAMPAGE, GRO-cap, PRO-cap) are listed in Data S1. The experimental validation datasets are obtained from NCBI GEO accessions GSE178982 (YY1-AID), GSE115110 (NFYA knockdown), and GSE156741 (TATA / NFY motif perturbation). The CRISPR-Cap and STAP-seq data have been deposited to GEO with the accession number GSE248771. The 241 Zoonomia genomes were obtained from the data portal https://zoonomiaproject.org/the-data/. All data used in this manuscript were also deposited into Zenodo repository (51).
References and Notes:
- 1.Smale ST, Kadonaga JT, The RNA polymerase II core promoter. Annu. Rev. Biochem. 72, 449–479 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Sandelin A, Carninci P, Lenhard B, Ponjavic J, Hayashizaki Y, Hume DA, Mammalian RNA polymerase II core promoters: insights from genome-wide studies. Nat. Rev. Genet. 8, 424–436 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Wang Y-L, Kassavetis GA, Kadonaga JT, Others, The punctilious RNA polymerase II core promoter. Genes Dev. 31, 1289–1301 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Forrest ARR, Kawaji H, Rehli M, Baillie JK, De Hoon MJL, Haberle V, Lassmann T, Kulakovskiy IV, Lizio M, Itoh M, Andersson R, Mungall CJ, Meehan TF, Schmeier S, Bertin N, Jørgensen M, Dimont E, Arner E, Schmidl C, Schaefer U, Medvedeva YA, Plessy C, Vitezic M, Severin J, Semple CA, Ishizu Y, Young RS, Francescatto M, Altschuler IA, Albanese D, Altschule GM, Arakawa T, Archer JAC, Arner P, Babina M, Rennie S, Balwierz PJ, Beckhouse AG, Pradhan-Bhatt S, Blake JA, Blumenthal A, Bodega B, Bonetti A, Briggs J, Brombacher F, Burroughs AM, Califano A, Cannistraci CV, Carbajo D, Chen Y, Chierici M, Ciani Y, Clevers HC, Dalla E, Davis CA, Detmar M, Diehl AD, Dohi T, Drabløs F, Edge ASB, Edinger M, Ekwall K, Endoh M, Enomoto H, Fagiolini M, Fairbairn L, Fang H, Farach-Carson MC, Faulkner GJ, Favorov AV, Fisher ME, Frith MC, Fujita R, Fukuda S, Furlanello C, Furuno M, Furusawa JI, Geijtenbeek TB, Gibson AP, Gingeras T, Goldowitz D, Gough J, Guhl S, Guler R, Gustincich S, Ha TJ, Hamaguchi M, Hara M, Harbers M, Harshbarger J, Hasegawa A, Hasegawa Y, Hashimoto T, Herlyn M, Hitchens KJ, Sui SJH, Hofmann OM, Hoof I, Hori F, Huminiecki L, Iida K, Ikawa T, Jankovic BR, Jia H, Joshi A, Jurman G, Kaczkowski B, Kai C, Kaida K, Kaiho A, Kajiyama K, Kanamori-Katayama M, Kasianov AS, Kasukawa T, Katayama S, Kato S, Kawaguchi S, Kawamoto H, Kawamura YI, Kawashima T, Kempfle JS, Kenna TJ, Kere J, Khachigian LM, Kitamura T, Klinken SP, Knox AJ, Kojima M, Kojima S, Kondo N, Koseki H, Koyasu S, Krampitz S, Kubosaki A, Kwon AT, Laros JFJ, Lee W, Lennartsson A, Li K, Lilje B, Lipovich L, Mackay-sim A, Manabe RI, Mar JC, Marchand B, Mathelier A, Mejhert N, Meynert A, Mizuno Y, De Morais DAL, Morikawa H, Morimoto M, Moro K, Motakis E, Motohashi H, Mummery CL, Murata M, Nagao-Sato S, Nakachi Y, Nakahara F, Nakamura T, Nakamura Y, Nakazato K, Van Nimwegen E, Ninomiya N, Nishiyori H, Noma S, Nozaki T, Ogishima S, Ohkura N, Ohmiya H, Ohno H, Ohshima M, Okada-Hatakeyama M, Okazaki Y, Orlando V, Ovchinnikov DA, Pain A, Passier R, Patrikakis M, Persson H, Piazza S, Prendergast JGD, Rackham OJL, Ramilowski JA, Rashid M, Ravasi T, Rizzu P, Roncador M, Roy S, Rye MB, Saijyo E, Sajantila A, Saka A, Sakaguchi S, Sakai M, Sato H, Satoh H, Savvi S, Saxena A, Schneider C, Schultes EA, Schulze-Tanzil GG, Schwegmann A, Sengstag T, Sheng G, Shimoji H, Shimoni Y, Shin JW, Simon C, Sugiyama D, Sugiyama T, Suzuki M, Suzuki N, Swoboda RK, ‘T Hoen PAC, Tagami M, Tagami NT, Takai J, Tanaka H, Tatsukawa H, Tatum Z, Thompson M, Toyoda H, Toyoda T, Valen E, Van De Wetering M, Van Den Berg LM, Verardo R, Vijayan D, Vorontsov IE, Wasserman WW, Watanabe S, Wells CA, Winteringham LN, Wolvetang E, Wood EJ, Yamaguchi Y, Yamamoto M, Yoneda M, Yonekura Y, Yoshida S, Zabierowski SE, Zhang PG, Zhao X, Zucchelli S, Summers KM, Suzuki H, Daub CO, Kawai J, Heutink P, Hide W, Freeman TC, Lenhard B, Bajic LVB, Taylor MS, Makeev VJ, Sandelin A, Hume DA, Carninci P, Hayashizaki Y, A promoter-level mammalian expression atlas. Nature (2014), doi: 10.1038/nature13182. [DOI] [Google Scholar]
- 5.Xi H, Yu Y, Fu Y, Foley J, Halees A, Weng Z, Analysis of overrepresented motifs in human core promoters reveals dual regulatory roles of YY1. Genome Res. 17, 798–806 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weingarten-Gabbay S, Nir R, Lubliner S, Sharon E, Kalma Y, Weinberger A, Segal E, Systematic interrogation of human promoters. Genome Res. 29, 171–183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Boer CG, Vaishnav ED, Sadeh R, Abeyta EL, Friedman N, Regev A, Deciphering eukaryotic gene-regulatory logic with 100 million random promoters. Nat. Biotechnol. 38, 56–65 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou J, Troyanskaya OG, Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods. 12, 931–934 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou J, Theesfeld CL, Yao K, Chen KM, Wong AK, Troyanskaya OG, Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat. Genet. (2018), doi: 10.1038/s41588-018-0160-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen KM, Wong AK, Troyanskaya OG, Zhou J, A sequence-based global map of regulatory activity for deciphering human genetics. Nat. Genet. 54, 940–949 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bogard N, Linder J, Rosenberg AB, Seelig G, A deep neural network for predicting and engineering alternative polyadenylation. Cell. 178, 91–106.e23 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Avsec Ž, Weilert M, Shrikumar A, Krueger S, Alexandari A, Dalal K, Fropf R, McAnany C, Gagneur J, Kundaje A, Zeitlinger J, Base-resolution models of transcription-factor binding reveal soft motif syntax. Nat. Genet. 53, 354–366 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Avsec Ž, Agarwal V, Visentin D, Ledsam JR, Grabska-Barwinska A, Taylor KR, Assael Y, Jumper J, Kohli P, Kelley DR, Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods. 18, 1196–1203 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agarwal V, Shendure J, Predicting mRNA abundance directly from genomic sequence using deep convolutional neural networks. Cell Rep. 31, 107663 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Hayashizaki Y, Cap Analysis Gene Expression (CAGE). Cap-Analysis Gene Expression (Cage) (2019), pp. 1–6. [Google Scholar]
- 16.Moore JE, Zhang X-O, Elhajjajy SI, Fan K, Pratt HE, Reese F, Mortazavi A, Weng Z, Integration of high-resolution promoter profiling assays reveals novel, cell type–specific transcription start sites across 115 human cell and tissue types. Genome Res. 32, 389–402 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Core LJ, Martins AL, Danko CG, Waters CT, Siepel A, Lis JT, Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat. Genet. 46, 1311–1320 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwak H, Fuda NJ, Core LJ, Lis JT, Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science. 339, 950–953 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, Zheng H, Goity A, van Bakel H, Lozano JC, Galli M, Lewsey MG, Huang E, Mukherjee T, Chen X, Reece-Hoyes JS, Govindarajan S, Shaulsky G, Walhout AJM, Bouget FY, Ratsch G, Larrondo LF, Ecker JR, Hughes TR, Determination and inference of eukaryotic transcription factor sequence specificity. Cell (2014), doi: 10.1016/j.cell.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seto E, Shi Y, Shenk T, YY1 is an initiator sequence-binding protein that directs and activates transcription in vitro. Nature. 354, 241–245 (1991). [DOI] [PubMed] [Google Scholar]
- 21.Vlaming H, Mimoso CA, Field AR, Martin BJE, Adelman K, Screening thousands of transcribed coding and non-coding regions reveals sequence determinants of RNA polymerase II elongation potential. Nat. Struct. Mol. Biol. 29, 613–620 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Almada AE, Wu X, Kriz AJ, Burge CB, Sharp PA, Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature. 499, 360–363 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mimoso CA, Adelman KL, U1 snRNP increases RNA pol II elongation rate to enable synthesis of long genes. SSRN Electron. J. (2022), doi: 10.2139/ssrn.4296553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smale ST, Baltimore D, The “initiator” as a transcription control element. Cell. 57, 103–113 (1989). [DOI] [PubMed] [Google Scholar]
- 25.Oldfield AJ, Henriques T, Kumar D, Burkholder AB, Cinghu S, Paulet D, Bennett BD, Yang P, Scruggs BS, Lavender CA, Rivals E, Adelman K, Jothi R, NF-Y controls fidelity of transcription initiation at gene promoters through maintenance of the nucleosome-depleted region. Nat. Commun. 10, 3072 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsieh T-HS, Cattoglio C, Slobodyanyuk E, Hansen AS, Darzacq X, Tjian R, Enhancer–promoter interactions and transcription are largely maintained upon acute loss of CTCF, cohesin, WAPL or YY1. Nature Genetics. 54 (2022), pp. 1919–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nojima T, Gomes T, Grosso ARF, Kimura H, Dye MJ, Dhir S, Carmo-Fonseca M, Proudfoot NJ, Mammalian NET-seq reveals genome-wide nascent transcription coupled to RNA processing. Cell. 161, 526–540 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arnold CD, Zabidi MA, Pagani M, Rath M, Schernhuber K, Kazmar T, Stark A, Genome-wide assessment of sequence-intrinsic enhancer responsiveness at single-base-pair resolution. Nat. Biotechnol. 35, 136–144 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neumayr C, Haberle V, Serebreni L, Karner K, Hendy O, Boija A, Henninger JE, Li CH, Stejskal K, Lin G, Bergauer K, Pagani M, Rath M, Mechtler K, Arnold CD, Stark A, Differential cofactor dependencies define distinct types of human enhancers. Nature. 606, 406–413 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CAM, Taylor MS, Engström PG, Frith MC, Forrest ARR, Alkema WB, Tan SL, Plessy C, Kodzius R, Ravasi T, Kasukawa T, Fukuda S, Kanamori-Katayama M, Kitazume Y, Kawaji H, Kai C, Nakamura M, Konno H, Nakano K, Mottagui-Tabar S, Arner P, Chesi A, Gustincich S, Persichetti F, Suzuki H, Grimmond SM, Wells CA, Orlando V, Wahlestedt C, Liu ET, Harbers M, Kawai J, Bajic VB, Hume DA, Hayashizaki Y, Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 38, 626–635 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Hong CKY, Cohen BA, Genomic environments scale the activities of diverse core promoters. Genome Res. 32, 85–96 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seila AC, Calabrese JM, Levine SS, Yeo GW, Rahl PB, Flynn RA, Young RA, Sharp PA, Divergent transcription from active promoters. Science. 322, 1849–1851 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Core LJ, Waterfall JJ, Lis JT, Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 322, 1845–1848 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duttke SHC, Lacadie SA, Ibrahim MM, Glass CK, Corcoran DL, Benner C, Heinz S, Kadonaga JT, Ohler U, Human promoters are intrinsically directional. Mol. Cell. 57, 674–684 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andersson R, Chen Y, Core L, Lis JT, Sandelin A, Jensen TH, Human Gene Promoters Are Intrinsically Bidirectional. Mol. Cell. 60 (2015), pp. 346–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duttke SHC, Lacadie SA, Ibrahim MM, Glass CK, Corcoran DL, Benner C, Heinz S, Kadonaga JT, Ohler U, Perspectives on Unidirectional versus Divergent Transcription. Mol. Cell. 60 (2015), pp. 348–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zoonomia Consortium A comparative genomics multitool for scientific discovery and conservation. Nature. 587, 240–245 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawaji H, Lizio M, Itoh M, Kanamori-Katayama M, Kaiho A, Nishiyori-Sueki H, Shin JW, Kojima-Ishiyama M, Kawano M, Murata M, Ninomiya-Fukuda N, Ishikawa-Kato S, Nagao-Sato S, Noma S, Hayashizaki Y, Forrest ARR, Carninci P, FANTOM Consortium, Comparison of CAGE and RNA-seq transcriptome profiling using clonally amplified and single-molecule next-generation sequencing. Genome Res. 24, 708–717 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hon C-C, Ramilowski JA, Harshbarger J, Bertin N, Rackham OJL, Gough J, Denisenko E, Schmeier S, Poulsen TM, Severin J, Lizio M, Kawaji H, Kasukawa T, Itoh M, Burroughs AM, Noma S, Djebali S, Alam T, Medvedeva YA, Testa AC, Lipovich L, Yip C-W, Abugessaisa I, Mendez M, Hasegawa A, Tang D, Lassmann T, Heutink P, Babina M, Wells CA, Kojima S, Nakamura Y, Suzuki H, Daub CO, de Hoon MJL, Arner E, Hayashizaki Y, Carninci P, Forrest ARR, An atlas of human long non-coding RNAs with accurate 5’ ends. Nature. 543, 199–204 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oran Brigham E, Fast Fourier transform and its applications (Prentice-Hall, London, England, 1988). [Google Scholar]
- 41.Tareen A, Kinney JB, Logomaker: beautiful sequence logos in Python. Bioinformatics. 36, 2272–2274 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen KM, Cofer EM, Zhou J, Troyanskaya OG, Selene: a PyTorch-based deep learning library for sequence data. Nat. Methods (2019), doi: 10.1038/s41592-019-0360-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ronneberger O, Fischer P, Brox T, “U-Net: Convolutional Networks for Biomedical Image Segmentation” in Lecture Notes in Computer Science (Springer International Publishing, Cham, 2015), Lecture notes in computer science, pp. 234–241. [Google Scholar]
- 44.Zhou J, Sequence-based modeling of three-dimensional genome architecture from kilobase to chromosome scale. Nat. Genet. 54, 725–734 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Novakovsky G, Dexter N, Libbrecht MW, Wasserman WW, Mostafavi S, Obtaining genetics insights from deep learning via explainable artificial intelligence. Nat. Rev. Genet. 24, 125–137 (2023). [DOI] [PubMed] [Google Scholar]
- 46.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Katoh K, Standley DM, MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2. Nat. Methods. 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dudnyk K, Cai D, Shi C, Xu J, & Zhou J, Sequence basis of transcription initiation in human genome (Code and Model; ) (2024), , doi: 10.5281/zenodo.10655933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dudnyk K, Shi C, & Zhou J, Sequence basis of transcription Initiation in human genome manuscript (2023), , doi: 10.5281/zenodo.7954971. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code and model for Puffin and Puffin-D are available at https://github.com/jzhoulab/puffin. The code for reproducing the analyses in the manuscript is available at https://github.com/jzhoulab/puffin_manuscript. Both were deposited into Zenodo repository (50). A user-friendly website for using Puffin is available at https://tss.zhoulab.io.
The human GRCh38/hg38 reference genome was used for training the Puffin and Puffin-D models. The mouse models were trained with the mm10 reference genome. All coordinates in the manuscript refer to GRCh38/hg38 unless otherwise indicated. All transcription initiation signal datasets used for model training (FANTOM CAGE, ENCODE CAGE, ENCODE RAMPAGE, GRO-cap, PRO-cap) are listed in Data S1. The experimental validation datasets are obtained from NCBI GEO accessions GSE178982 (YY1-AID), GSE115110 (NFYA knockdown), and GSE156741 (TATA / NFY motif perturbation). The CRISPR-Cap and STAP-seq data have been deposited to GEO with the accession number GSE248771. The 241 Zoonomia genomes were obtained from the data portal https://zoonomiaproject.org/the-data/. All data used in this manuscript were also deposited into Zenodo repository (51).