

Abstract
The intravenous delivery of disease-relevant antigens (Ag) by polymeric nanoparticles (NP-Ags) has demonstrated Ag-specific immune tolerance in autoimmune and allergic disorders as well as allogeneic transplant rejection. NP-Ags are observed to distribute to the spleen, which has an established role in the induction of immune tolerance. However, studies have shown that the spleen is dispensable for NP-Ag-induced tolerance, suggesting significant contributions from other immunological sites. Here, we investigated the tolerogenic contributions of Kupffer cells (KCs) and liver sinusoidal endothelial cells (LSECs) to NP-Ag-induced tolerance in a mouse model of multiple sclerosis, experimental autoimmune encephalomyelitis (EAE). Intravenously delivered Ag-conjugated poly(lactide-co-glycolide) NPs (PLG-Ag) distributed largely to the liver, where they associated with both KCs and LSECs. This distribution was accompanied by CD4 T cell accumulation, clonal deletion, and PD-L1 expression by KCs and LSECs. Ex vivo co-cultures of PLG-Ag-treated KCs or LSECs with Ag-specific CD4 T cells resulted in PGE2 and IL-10 or PGE2 secretion, respectively. KC depletion and adoptive transfer experiments demonstrated that KCs were sufficient, but not necessary, to mediate PLG-Ag-induced tolerance in EAE. The durability of PLG-Ag-induced tolerance in the absence of KCs may be attributed to the distribution of PLG-Ags to LSECs, which demonstrated similar levels of PD-L1, PGE2, and T cell stimulatory ability. Collectively, these studies provide mechanistic support for the role of liver KCs and LSECs in Ag-specific tolerance for a biomaterial platform that is currently being evaluated in clinical trials.
Keywords: PLG, Nanoparticles, Immune tolerance, Immunomodulation, Kupffer cells, Liver sinusoidal endothelial cells
1. Introduction
The increasing frequency of autoimmune-, allergic-, and biologic-related hypersensitivities has created a demand for interventions that induce antigen (Ag)-specific immune tolerance [1,2]. No cures are available for these immune disorders, and symptoms are managed by avoidance of allergens or broadly immunosuppressive drugs that increase the risks for opportunistic infections and the development of cancer [3,4]. Specifically, therapies include steroids or non-steroidal anti-inflammatory agents (NSAIDS), antihistamines, and other methods of systemic immune suppression or immunodepletion that target T cell receptors, co-signaling molecules, cytokines, antibodies, or leukocyte trafficking [5]. A significant need exists for treatments that specifically reprogram pathological immune cells without compromising the ability of the immune system to perform its critical protective functions. Toward this goal, Ag delivery systems such as targeted peptide fusions, soluble Ag arrays, Ag-coupled splenocytes, and Ag-loaded nanoparticles (NP-Ags) have been investigated for their ability to induce Ag-specific immune responses [6–17].
As a class, NP-Ags have induced tolerance in models of Th1/17 and Th2-mediated diseases, allogeneic transplantation, and AAV- and biologic-mediated hypersensitivities due to their ability to deliver disease-relevant Ags to Ag presenting cells (APCs) without inducing immune activation [18–27]. NPs comprised of biodegradable polymers have been formulated with Ag incorporated by surface-conjugation, encapsulation, and conjugation to the polymer precursors of NPs [18, 28–30]. In several cases, the efficacy of NP-Ags was dependent on the incorporation of an immunomodulating agent (e.g. rapamycin or IL-10) [29,31]. Previous studies have identified highly negatively-charged Ag-containing poly(lactide-co-glycolide) (PLG) NPs (PLG-Ag) as an effective platform for tolerance induction in the absence of exogenous immunomodulators [5]. PLG-Ags have recently completed a phase 2 randomized, double-blind, placebo-controlled clinical trial for the treatment of celiac disease and are progressing to a phase 2b trial (ClinicalTrials.gov Identifier: NCT04530123) [32].
NP-Ag-induced tolerance strategies are frequently administered subcutaneously or intravenously (i.v.), where their biodistribution is largely dependent on NP size [33]. For i. v. delivery, NPs in the range of 300 nm–800 nm distribute to the liver and spleen and are processed by APCs in a fashion hypothesized to resemble the body’s natural clearance of apoptotic cells [5,34,35]. Highly negatively-charged PLG-Ag distribute to splenic macrophages resulting in internalization, Ag presentation, and anti-inflammatory signaling in the spleen [28]. While the spleen has historically been a site of immune tolerance, tolerance induction in splenectomized mice using PLG-Ags suggests other immunological sites play a contributing role [18]. In addition to the spleen, the liver has demonstrated a role in peripheral tolerance [36]. Based on the biodistribution of NPs to the liver and the known tolerogenic capability of liver cells, we hypothesize that i. v. administered PLG-Ag promote tolerance by influencing cellular responses in the liver.
The liver is considered a tolerogenic organ based on observations in organ transplantation, Ag-specific tolerance, and the persistence of liver-based infections [37]. The constant exposure of hepatic immune cells to gut-derived microbial endotoxins results in cells that have a higher threshold for immune activation and are naturally skewed toward a phenotype that maintains tolerance [36]. Namely, the macrophages of the liver, Kupffer cells (KCs), and liver sinusoidal endothelial cells (LSECs) have restrained immunogenic potential as APCs [36,38]. KCs are highly phagocytic, have subdued proinflammatory responses to TLR agonists, and evidence supports their role in peripheral tolerance [36]. Endocytic LSECs are capable of cross presentation, Foxp3+ regulatory T cell (Treg) induction, and have also been implicated in Ag-specific tolerance [39,40]. Previous studies of NP-induced tolerance demonstrated the size-dependent cellular distribution of NP-Ags to either LSECs or KCs, where 10 nm iron oxide NP-Ags selectively associated with LSECs and 500 nm latex NP-Ags selectively associated with KCs [40,41].
Herein, we investigated the role of the liver in PLG-Ag-mediated immune tolerance and the corresponding cellular contributions of KCs and LSECs. Initial studies analyzed the extent to which PLG-Ag accumulate within the liver and the hepatic populations associating with these particles. The phenotypes of KCs and LSEC were analyzed through expression of cell surface molecules, gene expression, and secretion of immunomodulatory factors. CD4 T cells were found to accumulate at and selectively die in the livers of OT-II mice receiving i. v. PLG-Ag loaded with cognate antigen, suggesting that PLG-Ag may induce tolerogenic effects through clonal deletion. In the mouse model of multiple sclerosis, relapsing-remitting experimental autoimmune encephalomyelitis (EAE), splenectomized mice with EAE were used to distinguish the role of the liver from the known contributions of the spleen in a disease model of PLA-Ag-induced tolerance. KC depletion using clodronate liposomes did not inhibit PLG-Ag-mediated tolerance, while KCs harvested from PLG-Ag-treated mice and transferred into naïve mice provided protection against the induction of EAE. These results indicate that KCs are sufficient, but not necessary, to mediate PLG-Ag-induced tolerance. Collectively, these studies highlight the contributions of the liver, and particularly KCs and LSECs, to the induction of PLG-Ag-mediated immune tolerance.
2. Results
2.1. Intravenously injected PLG NPs distribute to KCs and LSECs of the liver
The organ and cellular biodistribution was analyzed following intravenous administration of fluorescently labeled PLG particles (PLG-Cy5.5). These particles, and all particles used herein, were polymer-conjugate PLG nanoparticles, where the PLG is chemically conjugated to ligands (e.g., Cy5.5 or peptide Ags) prior to forming the nanoparticles using an emulsion process. These particles were in the size range of 380–420 nm and zeta potential of −42 to −36 mV (Table S1). The approach allows for the incorporation of precise Ag quantities and minimizes uncontrolled payload release [30]. The attachment of fluorophores was employed to track the biodistribution of PLG particles in vivo. The biodistribution of PLG-Cy5.5 was evaluated in the context of a dose response, as dose has been reported to influence PLG-Ag-induced tolerance [14,15]. The PLG-Cy5.5-derived organ fluorescence increased with particle dose and was greatest in the liver, with a lower presence in the lung and spleen (Fig. 1A). At therapeutically relevant doses of 1 and 2 mg [14], the PLG-Cy5.5 signal in the liver was 10-fold greater than in the lung and spleen. Within the liver, PLG-Cy5.5 association with KCs and LSECs also followed a dose response (Fig. 1B). LSECs had a greater frequency of PLG-Cy5.5 association than KCs indicating a broad distribution across LSECs. However, KCs had a greater median fluorescent intensity, which may indicate the internalization of larger particles or multiple particles per cell. At a dose of 2 mg, most KCs and LSECs were PLG-Cy5.5-positive. To confirm the connection between particle biodistribution and bioactive antigen delivery to KCs and LSECs, particles containing both Cy5.5. and model antigen OVA323–339 peptide (PLG-OVA,-Cy5.5) were injected i. v. at a dose of 2 mg. The livers were isolated and Cy5.5-labeled KCs and LSECs were sorted by FACS for co-culture with OVA-restricted T cells from OT-II mice. Both particle-associated KCs and LSECs induced the expression of CD25 on OT-II T cells confirming the in vivo bioactive delivery of Ag to KCs and LSECs by PLG particles (Figure S1). These data indicate that the liver is a significant site for PLG NP accumulation, after i. v. administration, and that these particles deliver bioactive antigen to both KCs and LSECs.
Fig. 1.
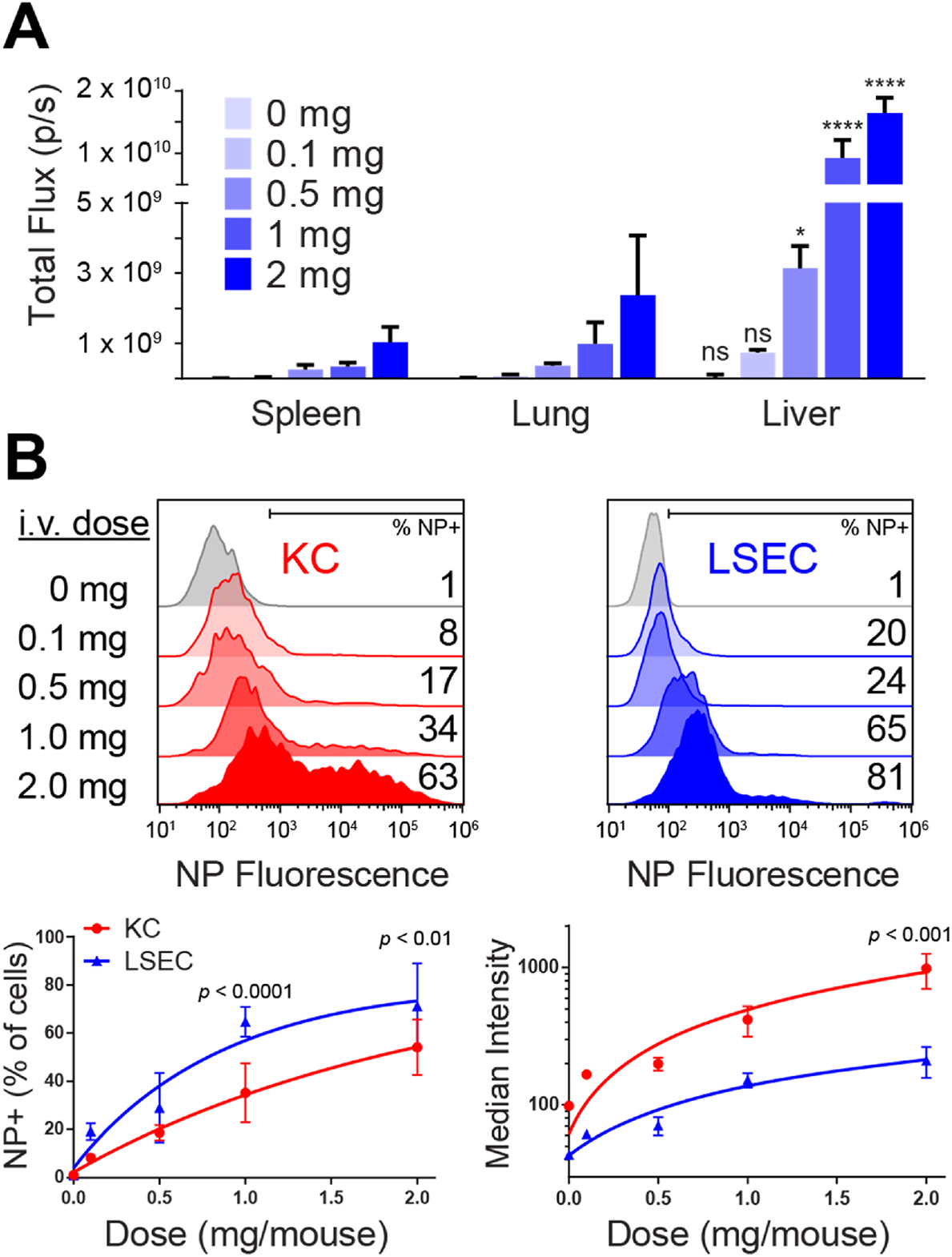
PLG nanoparticles distribute to KCs and LSECs in the liver. PLG-Cy5.5 were intravenously injected into C57BL/6 mice (n = 3) over a range of doses from 0 to 2 mg per mouse. (A) At 24 h, the mice were euthanized and PLG-Cy5.5 fluorescence was quantified in the spleens, lungs, and livers by IVIS imaging. (B) The frequency of PLG-Cy5.5 association with KCs and LSECs and median fluorescence intensity was measured by flow cytometry. At each dose in (A), statistical differences between the liver and the lung and spleen were determined using a two-way ANOVA with Tukey’s multiple comparisons test (*p < 0.05, ****p < 0.0001). Comparisons between KCs and LSECs (B) were determined by two-way ANOVA with Sidak’s multiple comparisons test. Error bars indicate SD and data are representative of 2 independent experiments.
2.2. PLG-Ag induce Ag-specific T cell accumulation and deletion in the liver
The number of CD4 T cells was quantified in the liver after intravenous PLG-Ag infusion. OT-II transgenic mice were utilized to identify the Ag-specific effect of PLG-OVA (containing 8 μg OVA323–339 peptide per mg) in comparison to PLG-PLP carrying irrelevant Ag (containing 8 μg PLP139–151 peptide per mg). Prior studies have demonstrated that PLG-Ag confer therapeutic tolerance in an antigen-specific manner; therefore, PLG-PLP serve as a treatment control in experiments involving OT-II mice with OVA-restricted TCRs [5,14,15,18,19,28]. Compared to treatment with PLG-PLP, mice injected with PLG-OVA experienced a 3-fold increase in CD4 T cells in the liver 24 h after injection (Fig. 2A). The total cell viability in the liver was unchanged between the two treatments, however, the increase in CD4 T cell number with PLG-OVA was accompanied by a 30% decrease in the T cell population viability (Fig. 2B). This result suggests that PLG-OVA caused an accumulation and deletion of T cells in the liver, and that this effect was Ag-specific.
Fig. 2.
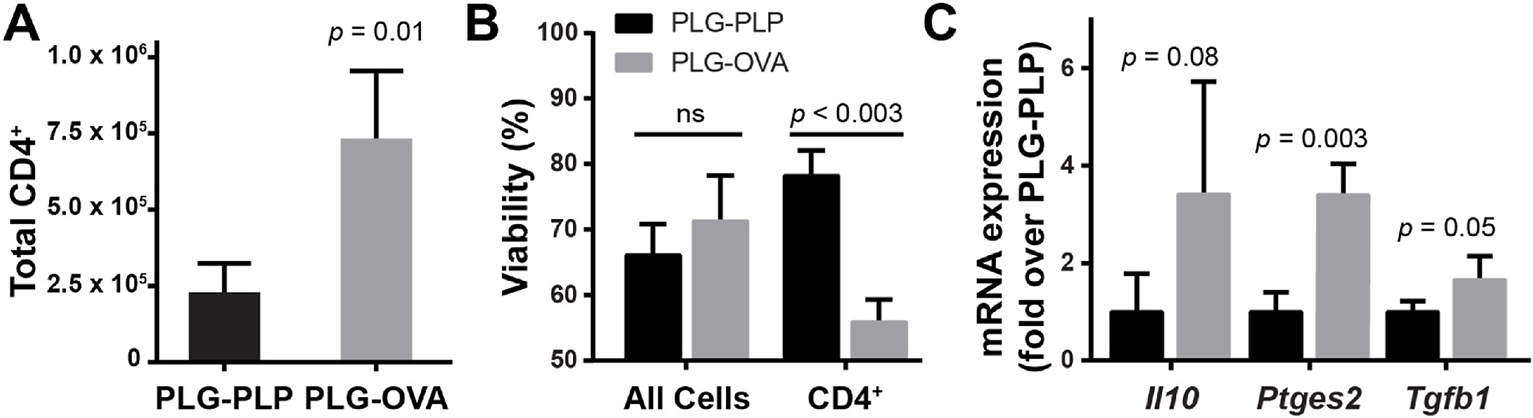
Intravenous injection of PLG-Ag results in Ag-restricted CD4 T cell accumulation and clonal deletion in the liver. OT-II mice (n = 3) were injected with 2 mg of PLG-OVA (8 μg OVA/mg), or irrelevant PLG-PLP (8 μg PLP/mg). 24 h post-injection, liver nonparenchymal cells were isolated. (A, B) CD4 T cell number per liver and viability were determined by flow cytometry. DAPI exclusion was used to evaluate viability. Statistical differences were determined by unpaired t-test (A) or two-way ANOVA with Tukey’s multiple comparisons test (B). (C) mRNA transcripts for Il10, Ptges2 and Tgfb1 were measured from liver non-parenchymal cells by RT-qPCR. The 2−ΔΔCT method was used to quantify relative mRNA expression between PLG-OVA- and PLG-PLP-treated mice with 18s-rRNA as an internal reference. Statistical differences were determined by unpaired t-tests. Error bars indicate SD and data are representative of 2 (A, B) and 3 (C) independent experiments.
2.3. Particles induce an Ag-specific tolerogenic phenotype in KCs and LSECs
KCs and LSECs were subsequently analyzed to understand their in vivo phenotype following PLG-Ag treatment. The mRNA transcripts of liver non-parenchymal cells (NPCs) were analyzed for the expression of genes involved in immunomodulatory signaling (Fig. 2C). Transcripts for TGF-β, IL-10, and prostaglandin E synthase (PTGES) protein production were analyzed as each have been implicated in liver-mediated tolerance. Again, OT-II mice were utilized to measure responses in the context of cognate Ag-presentation with PLG-PLP as a negative control. Ag-specific particle treatment (PLG-OVA) caused a significant increase in mRNA transcripts of Tgfb1 and Ptges2, the gene encoding for PTGES, which enzymatically produces PGE2. A non-significant (p = 0.08) increase in Il10 mRNA transcripts was observed between the Ag-relevant PLG-OVA and irrelevant PLG-PLP conditions. Together, these gene expression data may support a role for soluble factors in liver-mediated PLG-Ag-induced tolerance.
KC and LSEC costimulatory and coinhibitory molecule expression were then measured to determine if changes in receptor-mediated stimulatory potential were associated with PLG-Ag internalization and cognate Ag presentation. OT-II mice were injected with PLG-OVA and PLG-PLP and the liver NPC fraction was analyzed after 24 h. Both KCs (CD11b+F4/80+) and LSECs (CD146+) experienced no change in costimulatory CD80 molecules between the two treatment groups (Fig. 3). CD86 and CD40 were significantly upregulated on KCs and LSEC treated with PLG-OVA. Both KCs and LSECs showed a large and significant increase in the expression of coinhibitory molecule PD-L1 with PLG-OVA particle administration.
Fig. 3.
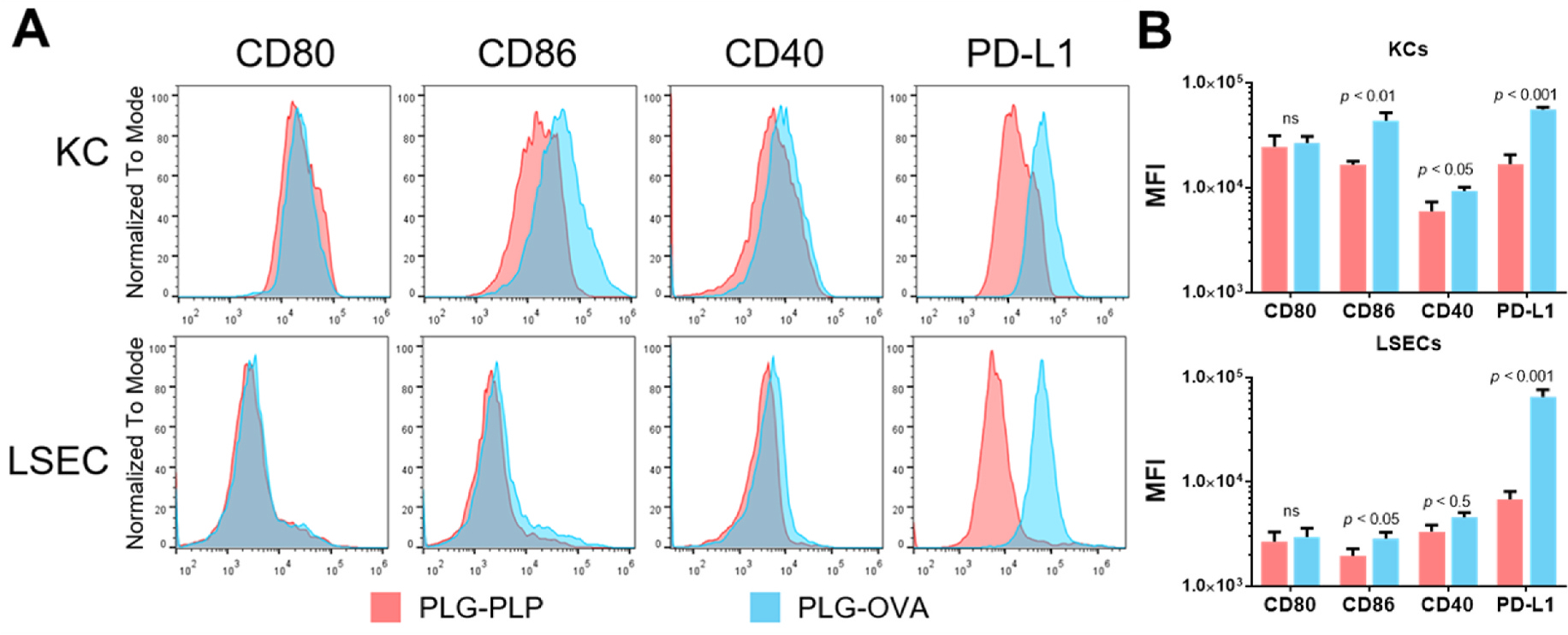
Delivery of PLG-Ag to KCs and LSECs results in Ag-specific upregulation of inhibitory molecule PD-L1. OT-II mice (n = 3) were injected with 2 mg of PLG-OVA or irrelevant PLG-PLP. After 24 h, liver non-parenchymal cells were isolated and analyzed by flow cytometry. (A) Histograms and (B) quantified median fluorescent intensity of costimulatory molecules CD80, CD86, and CD40, and coinhibitory molecule PD-L1 on KCs and LSECs. Statistical differences were determined by individual t-tests. Differences are indicated between PLG-OVA and PLG-PLP. Error bars represent SD and data are representative of 3 independent experiments.
2.4. KCs and LSECs present particle-derived Ag to T cells resulting in the secretion of regulatory factors
The CD4 T cell responses to Ag presentation by PLG-Ag-containing KCs and LSECs were compared ex vivo. Wild-type mice were injected with Cy5.5-labeled PLG-OVA. The KCs and LSECs from particle-treated mice (KC(PLG-OVA) and LSEC(PLG-OVA)) were sorted and co-cultured with naïve OVA-specific CD4 T cells from OT-II mice. Bone marrow-derived dendritic cells (BMDCs) and macrophages (BMMØs) were treated with soluble OVA and used as positive controls for Ag presentation. Where no OVA was present, the BMDCs and BMMØs were unable to stimulate T cells, which matches our previous observations that APCs receiving particles with irrelevant Ag (e.g. PLP) are unable to stimulate T cells [15]. Other reports showed that without cognate Ag, T cells are not stimulated by KCs or LSECs [36,42]. Blank particles were not tested because previous studies have shown that repeated delivery of blank particles can delay but not prevent disease onset, and do not prevent disease by mechanisms of tolerance [43,44]. In the presence of soluble OVA, BMDCs were efficient Ag presenters and induced the expression of the high-affinity IL-2 receptor, CD25, on 90% of T cells (Fig. 4A). BMMØs were less efficient Ag presenters, inducing an upregulation of CD25 on only 40% of T cells. Statistically less than BMDCs, and similar to BMMØs, KC(PLG-OVA) and LSEC(PLG-OVA) populations induced a moderate 30% and 20% expression of CD25 on co-cultured T cells, respectively. Under these in vitro conditions, no significant increase in the percentage of CD4+CD25+Foxp3+ T cells between the conditions was observed (data not shown).
Fig. 4.
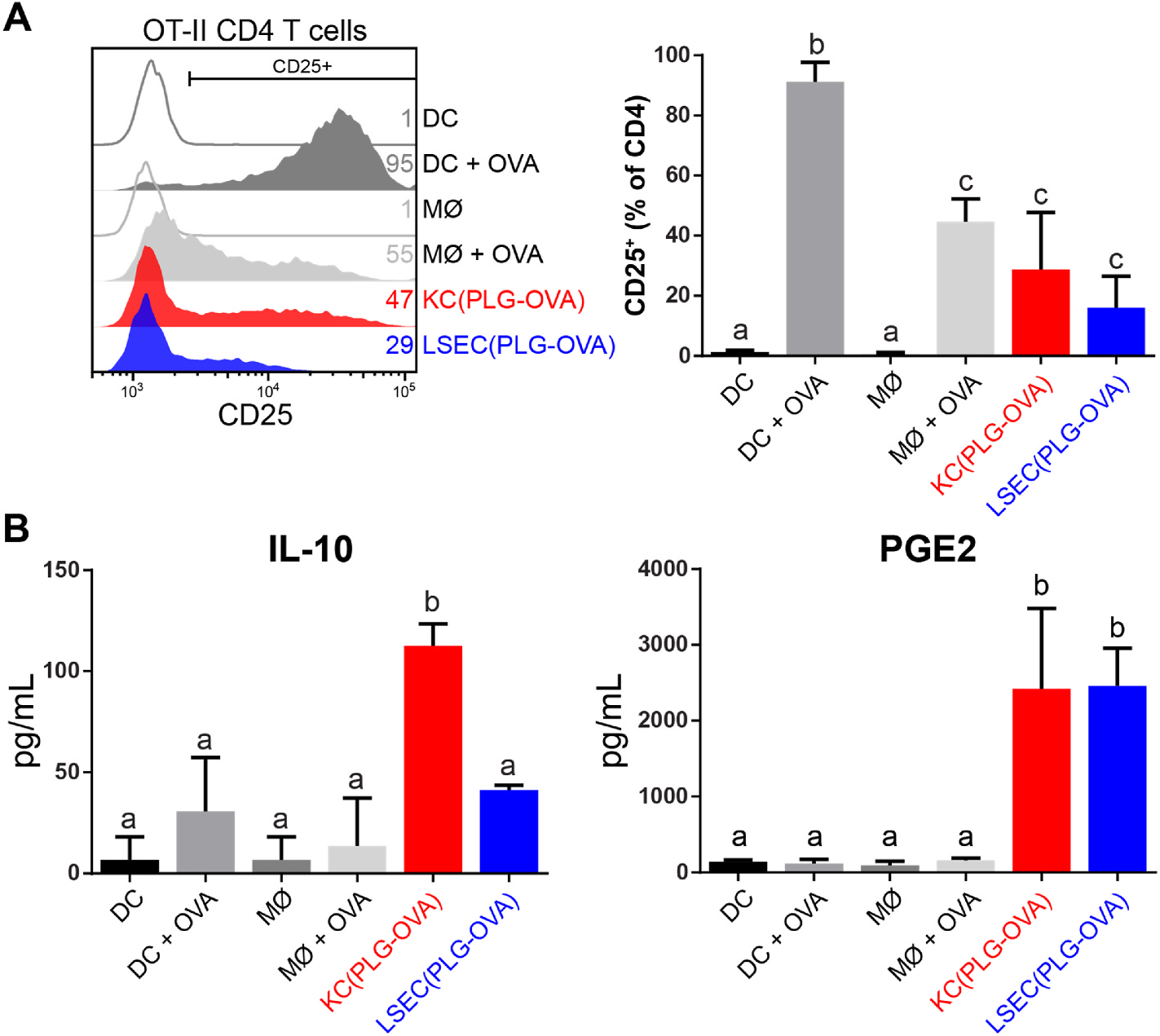
KCs and LSECs present particle-derived Ag resulting in moderate T cell expression of CD25 and secretion of immunomodulatory mediators. C57BL/6 mice (n = 3) were injected with 2 mg of PLG-OVA NPs. After 24 h, liver non-parenchymal cells were isolated and KCs and LSECs were sorted by MACS and co-cultured (105 cells) with naïve CD4 OT-II T cells (0.5 × 105 cells). Bone marrow-derived DCs and MØs were used as controls with and without the addition of soluble OVA peptide. (A) The efficiency of Ag presentation was measured by CD25 expression on live CD4 T cells using flow cytometry. (B) Soluble IL-10 and PGE2 were measured in the co-culture supernatants. Statistical differences were determined using one-way ANOVA with Tukey’s multiple comparisons tests. Nonsignificant differences are indicated by matching letters (p > 0.05). Error bars represent SD and data are representative of 5 (A) and 2 (B) independent experiments.
The supernatants of the APC:T cell co-culture assays were measured for the presence of soluble IL-10, PGE2, and TGF-β. IL-10 was produced in significant quantities in co-cultures with KC(PLG-OVA) as stimulators, but not in the BMDC and BMMØ controls or the LSEC(PLG-OVA) condition (Fig. 4B). PGE2 was secreted at significant levels in conditions containing KC(PLG-OVA) and LSEC(PLG-OVA), but not in any of the BMDC or BMMØs controls. TGF-β was not secreted at measurable levels in these conditions (data not shown). These data indicate that cognate Ag presentation by LSECs results in a secretion of PGE2, whereas cognate Ag presentation by KCs results in a secretion of both IL-10 and PGE2.
2.5. KCs are dispensable for particle-induced tolerance
The respective roles of KCs and LSECs in PLG-Ag-induced immune tolerance were investigated in the context of autoimmunity using the relapsing-remitting EAE mouse model. The mice were splenectomized to distinguish the effects of the liver from the immunomodulatory contributions of the spleen. Mice were then treated with clodronate liposomes to deplete KCs and investigate the contribution of other hepatic APCs, namely LSECs, to PLG-Ag-induced tolerance (Figure S2). After 24 h, mice were injected with disease-relevant PLG-PLP or irrelevant PLG-OVA, then immunized with PLP/CFA 7 days later (Fig. 5A). The negative control group, receiving a sham splenectomy and PLG-OVA experienced acute and relapsing EAE symptoms (Fig. 5B). In contrast, the PLG-PLP reduced the severity of disease symptoms in both sham and splenectomized mouse groups. Injection of PLG-PLP in KC-depleted splenectomized mice resulted in a significant reduction of acute EAE symptoms and complete suppression of relapse symptoms suggesting a dispensable role for KCs in PLG-Ag-induced tolerance.
Fig. 5.
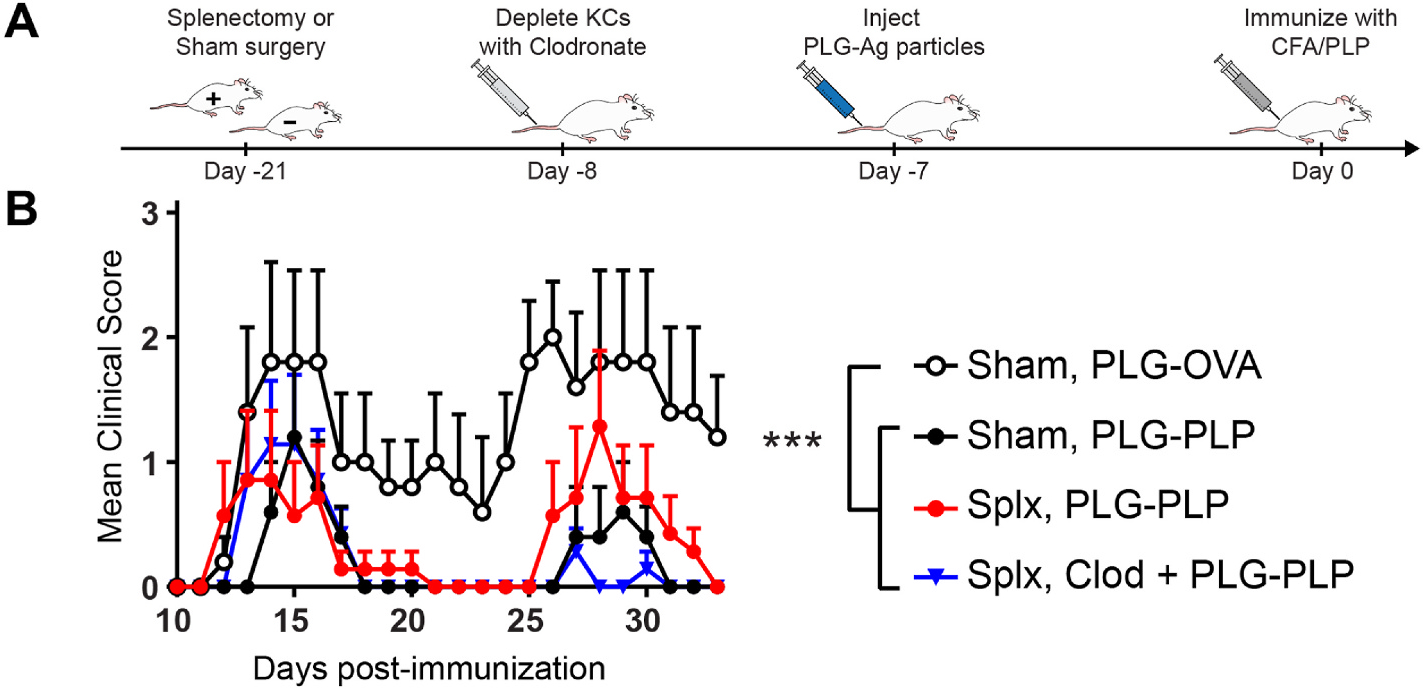
KCs are dispensable for PLG-Ag-induced tolerance in EAE. (A) Schematic representation of treatment scheme. On day −21, SJL mice were splenectomized (Splx; n = 7 per condition) or received a sham surgery (n = 5 per condition). After recovery, one splenectomized cohort was injected with 200 μL of 5 mg/mL clodronate liposomes (Clod) to deplete KCs. 24 h later, mice received 2 mg of PLG-PLP or irrelevant PLG-OVA. 7 days following NP treatment (day 0), mice were immunized with PLP/CFA to induce relapsing-remitting EAE. Mice were scored daily by blinded observers. (B) Mice treated with PLG-PLP displayed decreased clinical scores regardless of splenectomy status or KC depletion, demonstrating that KCs are dispensable to PLG-Ag-mediated tolerance in EAE. Statistical differences were determined using the Kruskal-Wallis test (one-way ANOVA nonparametric test) for the course of disease from day 11–33 (p < 0.05). Error bars indicate SEM and data are representative of 2 independent experiments. Splx, splenectomized; Clod, clodronate-treated.
2.6. KCs contribute to particle-induced tolerance
The protective effect of PLG-Ag in EAE despite clodronate depletion of KCs implicates other cells in this tolerogenic process, yet does not dismiss the role of KCs in PLG-Ag-mediated peripheral tolerance. An adoptive transfer model was used to test the sufficiency of KCs to mediate systemic tolerance in EAE. Previous studies have demonstrated that adoptive transfer of KCs and macrophages distribution to and functionally reconstitute the liver [45–49]. SJL mice were injected with Cy5.5-labeled PLG-PLP. The particle-containing KCs were isolated from these mice and were intravenously injected into naïve SJL recipients 7 days prior to immunization. Compared to mice that received no intervention, adoptive transfer of particle-containing KCs significantly mitigated EAE symptoms (Fig. 6). This result, combined with the clodronate depletion study, suggests that KCs promote PLG-Ag-induced immune tolerance, yet are not the sole contributor to this response.
Fig. 6.
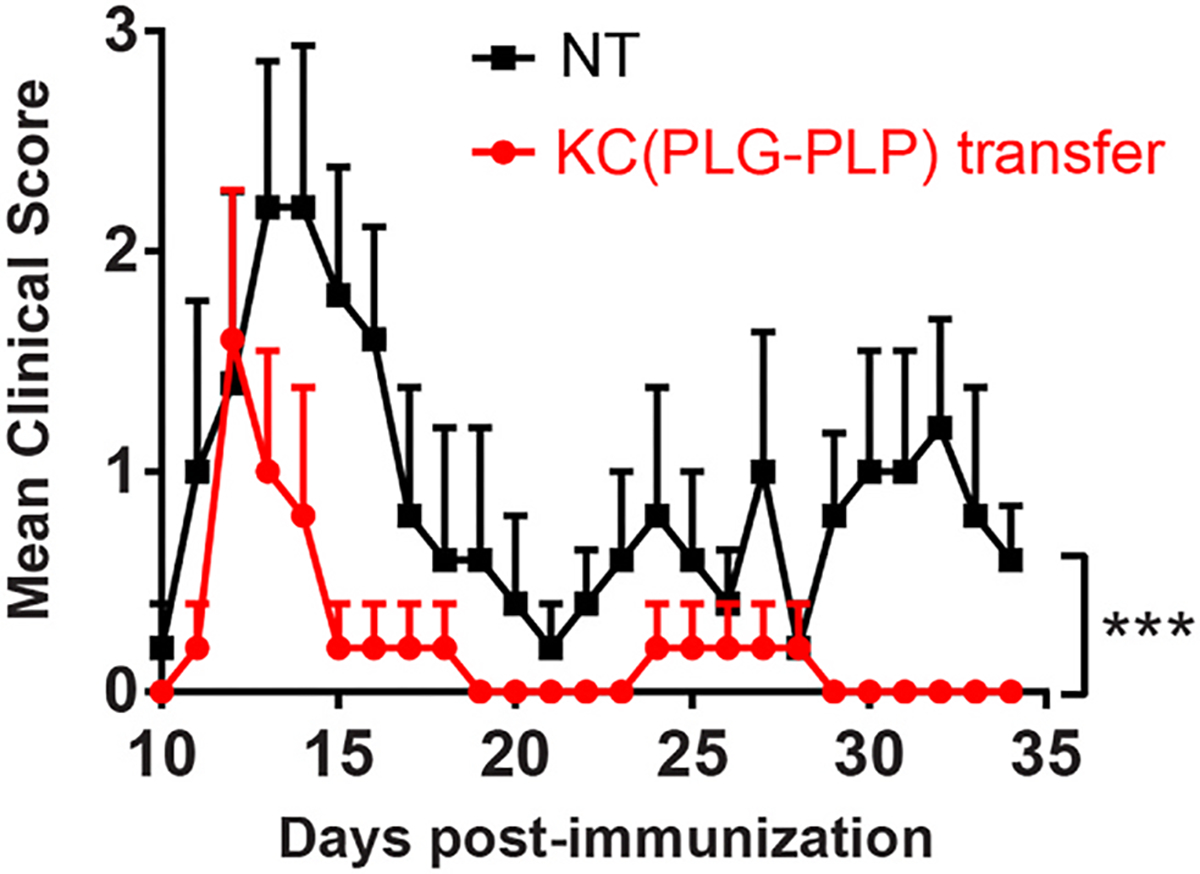
Adoptively transferred PLG-Ag-containing KCs are sufficient for inducing tolerance in the EAE model. SJL mice (n = 5) were injected with 2 mg of Cy5.5 labeled PLG-PLP. 24 h later, the liver non-parenchymal cells were isolated, and KCs were sorted based on particle-derived Cy5.5 fluorescence. Particle-containing KCs were adoptively transferred into naïve SJL mice (106 cells per mouse). The mice were immunized 7 days later with PLP/CFA and scored by blinded observers. Statistical differences were determined by the Mann-Whitney test (two-tailed nonparametric test) over the period from day 10–34 (***p < 0.001). Error bars indicate SEM and data are representative of 2 independent experiments.
3. Discussion
This study investigated the contributing roles of liver APCs to Ag-specific immune tolerance using a PLG NP Ag-delivery platform. The intravenous delivery of Ag by NPs has demonstrated system-wide tolerance induction in models of autoimmune, allergic, and allogeneic hypersensitivities [19,20,28]. Historically, the i. v. delivery of soluble Ag has demonstrated mixed tolerogenic effects; however, this approach requires orders of magnitude more antigen than presented here and the risk of anaphylaxis limits the safe implementation of this strategy [14,28,50]. Nanoparticles present a biomaterial platform for controlled delivery of Ag to immunomodulatory immune cells. The current investigation of PLG-Ag-induced tolerance in the liver was motivated by the observations that: 1) NPs distribute to the liver, 2) immune cells of the liver are semi-resistant to immune activation, and 3) PLG-Ag NPs have induced tolerance in splenectomized mice [18,36]. The liver is not traditionally regarded as a lymphoid organ; however, it contains its own complement of immune cells. Herein, within the context of biodegradable Ag-loaded PLG particles, the contributions of KCs and LSECs to immune tolerance were considered together. Specifically, KCs and LSECs demonstrated PLG-Ag association, Ag-specific phenotypic changes, arrest and deletion of T cells, and tolerogenic redundancy.
I.V. administration of PLG NPs resulted in an organ biodistribution primarily to the liver and cellular association with both KCs and LSECs. The 400–600 nm PLG NPs employed in this investigation associated with a majority of KCs and LSEC following i. v. injection of 2 mg. At the lower dose of 1 mg, less than half of KCs and LSECs were PLG NP-positive, which corresponds with previous reports of diminishing tolerogenic effect at lower doses [14]. A similar pattern of biodistribution within the liver has been detailed following direct injection of PLG NPs into the spleen for directed flow to the liver [51]. The study showed that similar PLG particles were captured by nearly all KCs and LSECs, half of hepatic stellate cells, and only 7% of hepatocytes. Others have investigated NP-induced tolerance using NPs that exclusively partitioned to specific liver APCs based on their size: 500 nm carboxylated latex particles preferentially target KCs as they are highly phagocytic, while 10 nm polymer-coated superparamagnetic iron oxide (SPIO) NPs preferentially targeted LSECs, where endocytosis through clathrin-mediated transport captures particles less than 200 nm [38,40,41]. The PLG NPs evaluated here, by virtue of their emulsion-based formulation, have a greater polydispersity than monodispersed latex and SPIO particles made by polymerization and precipitation, respectively. This broader size distribution of PLG NPs may account for the association with both KCs and LSECs enabling tolerance through multiple hepatic APCs. Other non-particle modalities, such as Ag-ligand fusions, have achieved a similar distribution of Ag to multiple hepatic cells. These conjugates of Ag fused to polymeric N-acetylgalactosamine (Ag-p(GalNAc)) or N-acetylglucosamine (Ag-p(GluNAc)) target the C-type lectin receptors on multiple liver APCs [17]. This targeting allowed Ag delivery to liver DCs, KCs, LSECs, and hepatocytes to induce Ag-specific tolerance by multiple mechanisms. Together, the use of PLG-Ag here, with intermediate size and polydispersity, results in the delivery of Ags across multiple liver APCs which may act to reinforce tolerance through multiple cell types.
The presentation of particle-derived Ag in the liver resulted in the Ag-specific arrest and tolerogenic reprogramming of T cells by clonal deletion. The delivery of cognate Ag by PLG-Ag caused a three-fold increase of Ag-specific CD4 T cells in the liver of OT-II mice, which was accompanied by a 30% reduction in T cell population viability. Importantly, the viability of other NPCs was not affected, indicating that this result represented Ag-specific clonal deletion. This result is supported by the observation that PLG-Ag treatment induced KC and LSEC expression of programmed death-ligand 1 (PD-L1), which has been shown to play a role in the regulation of autoreactive T cells (Fig. 3) [52]. The rapid arrest of T cells by Ag-treated KCs and LSECs has been previously observed in real-time, using intravital microscopy, and this arrest and deletion pattern of the liver has also been observed with both naïve and activated Th1 and Th2 T cells [41,53,54]. In addition to clonal deletion, other reports have demonstrated a capacity for regulatory T cell induction by KCs and LSECs. Ag-coupled latex particles have induced production of Foxp3+ Tregs and IL-10-producing regulatory Tr1 phenotypes by KCs [41]. Separately, Ag-coupled iron oxide particles induced tolerance that was attributed to Tregs and was abrogated by depleting Tregs [40]. Our previous work supports these additional mechanisms as the depletion of Tregs or IL-10 partially depleted PLG-Ag-induced tolerance in the EAE model [28]. In this work, we demonstrated the secretion of IL-10 in co-cultures of KCs and CD4 T cells which supports the Tr1 mechanism, but we did not observe significantly enhanced Treg induction in vitro without the addition of exogenous TGF-β (data not shown). Collectively, the delivery of PLG-Ag to liver APCs results in the arrest and clonal deletion of T cells which may act in parallel to other peripheral tolerance mechanisms of the liver involving Foxp3 Tregs and Tr1 T cells.
KCs and LSECs exhibited tolerogenic signal 1 and 2 phenotypes following PLG-Ag delivery and T cell interaction. KCs and LSECs from PLG-Ag-treated mice induced the expression of the high-affinity IL-2 receptor (CD25) on T cells indicating particle internalization, processing, and bioactive Ag presentation. The extent of CD25 expression induced on T cells by PLG-Ag-containing KCs and LSECs was suboptimal: similar to that of macrophages and lower than that of highly efficient DCs. Importantly, the presentation of T cell-specific Ag, but not irrelevant Ag, coincided with a marked upregulation of co-inhibitory molecule PD-L1 on KCs and LSECs, and significant increase in costimulatory molecules CD86 and CD40. The PD-1/PD-L1 pathway in the liver influences the balance between tolerance and immunity, where inhibition of PD-L1 on KCs can reactivate immunity against persistent viral infections and an increase in PD-L1 on LSECs suppresses Th1 and Th17 responses [53,55]. Previous work with PLG-Ag has corroborated a role for PD-1/PD-L1 signaling in particle-induced tolerance: Treatment with anti-PD-1 antibody limited tolerance in the EAE model and completely abolished tolerance in the BDC2.5 type 1 diabetes model [18,24]. PD-L1 expression by liver APCs is associated with tolerance induction, and here we showed Ag-specific enhancement of PD-L1 expression in PLG-Ag-treated KCs and LSECs [18].
The treatment of KCs and LSECs with PLG-Ag resulted in secretion of tolerogenic factors (signal 3) in co-cultures with Ag-specific T cells. Specifically, cultures with KC stimulators produced IL-10 and PGE2, and cultures stimulated with LSECs produced PGE2. Both of these soluble factors has been detected in the context of liver-based immune tolerance [42,56]. You et al. thoroughly demonstrated that KC-derived PGE2 is a suppressor of T cell activation and IL-2 production and, importantly, that PGE2 secretion diminished the ability of nearby APCs to activate T cells [36]. This secretion PGE2 may contribute to the observed low expression of CD25 in the co-cultured T cells. Our observation that IL-10 was exclusively secreted in co-cultures of KC and T cells matches other evidence that T cells become IL-10-producing after Ag delivery to KCs [41]. In the referenced study, IL-10 was present in 40% of T cells which suggesting an IL-10-producing regulatory Tr1 phenotype. Thus, the source of IL-10 production may have been the KCs, T cells, or both. Together, the secretion of anti-inflammatory mediators in the context of particle-treated KCs and LSECs contributes to a secretory microenvironment associated with T cell regulation.
KCs were sufficient, but not necessary, for mediating an Ag-specific tolerogenic response to PLG-Ag in the EAE model. Previous reports have indicated that NPs associate with KCs and LSECs upon intravenous injection [38,40,41], and we investigated the specific contribution of KCs to PLG-Ag-induced tolerance by depleting KCs and also by adoptively transferring PLA-Ag-positive KCs. Macrophages have previously been adoptively transferred in autoimmune, fibrosis, and transplantation models to assess their role in disease or therapeutic processes. Importantly, where LSECs are sessile, a majority of transferred macrophages localize to the liver within 24 h [45–48]. Remarkably, adoptive transfer of PLG-Ag-positive KCs reduced the severity of EAE disease symptoms. This result is in accordance with the tolerance-inducing ability of KCs, however, it is unclear whether the adoptively transferred PLG-Ag-positive KCs reconstituted their native effector behavior in the liver [41]. Importantly, clodronate liposome depletion of KCs in splenectomized mice did not abolish the tolerogenic effect of PLG-Ag. It is suspected that this effect results from the redundant tolerogenic capabilities of LSECs, which have demonstrated effective presentation of NP-derived Ag, inducible expression of PD-L1, low expression of costimulatory molecules, secretion of PGE2, and an induction of Tregs [39]. Together, these results suggest that KCs are independently capable of inducing tolerance, and that LSECs make contributions to this systemic tolerance.
In conclusion, hepatic KCs and LSECs make contributions to Ag-specific immune tolerance induced by antigen-loaded PLG nano-particles. The PLG NPs distributed largely to the liver where they associated with KCs and LSECs. PLG particles containing cognate Ag caused the arrest and clonal deletion of T cells in the liver within 24 h. In vivo, KC and LSEC costimulatory molecules were moderately affected by treatment, but inhibitory molecule PD-L1 was markedly upregulated in the context of relevant Ag delivery. KC interactions with T cells resulted in the secretion of PGE2 and IL-10, whereas interactions with LSECs produced PGE2 only. PLG-Ag-mediated tolerance in the EAE autoimmune model occurred despite the depletion of KCs, and tolerance could be induced by adoptively transferring PLG-Ag-containing KCs. These data suggest that KCs are sufficient, but not necessary, contributors to PLG-Ag-induced tolerance. Collectively, these studies contribute to identifying the mechanisms within the liver that induce tolerance for a biomaterial platform that is currently being evaluated in clinical trials.
4. Materials and methods
4.1. Materials
Acid-terminated 50:50 poly (D, l-lactide-co-glycolide) (PLG) (~0.17 dL/g inherent viscosity in hexafluoro-2-propanol) was purchased from Lactel Absorbable Polymers (Birmingham, AL). Poly(ethylene-alt-maleic anhydride) (PEMA) was purchased from Polysciences, Inc. (Warrington, PA). Amine-terminated ovalbumin peptide (NH2-OVA323–339), referred to as OVA, and proteolipid peptide (NH2-PLP139–151), referred to as PLP, were purchased from Genscript (Piscataway, NJ). Collagenase IV was purchased from Worthington Biochemical Corp. (Lakewood, NJ).
4.2. PLG-ligand bioconjugation and characterization
Acid-terminated PLG (37.8 mg, 0.009 mmol, 4200 g/mol) was dissolved in 2 mL of N,N-dimethylformamide (DMF) (Sigma, St. Louis, MO). N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) (9.0 mg, 0.047 mmol, 5X to PLG) (Sigma, St. Louis, MO) was dissolved in 0.5 mL of DMF and added dropwise to the PLG solution. N-hydroxysuccinimide (NHS) (5.5 mg, 0.047 mmol, 5X to PLG) (ThermoFisher, Waltham, MA) was dissolved in 0.5 mL DMF and added dropwise. The reaction was stirred for 15 min at room temperature. Antigenic peptide (1.2X to PLG) was dissolved in 1 mL of DMSO and 0.5 mL of DMF and stirred. Triethylamine (5X to peptide) was added to the peptide solution, and the resulting mixture was added dropwise to the stirring PLG solution. The reaction proceeded overnight at room temperature. The resulting polymer was isolated using 3500 molecular weight cut-off membrane dialysis in 4 L of distilled water replaced 6 times over 2 days. The dialyzed polymer was washed with MilliQ water three times by centrifugation at 7000×g. Finally, the polymer was lyophilization for 2 days. Coupling efficiency of peptide or fluorophore to PLG was determined by 1H NMR analysis in DMSO-d6.
4.3. Nanoparticle preparation and characterization
PLG-Ag NPs were prepared using Ag-PLG bioconjugates using an oil-in-water (o/w) emulsion solvent evaporation (SE) technique as previously described in publications [30]. Briefly, 400 mg of the acid-terminated PLG and Ag-PLG polymer conjugate (based on the 4200 g/mol MW of PLG and the coupling efficiency of Ag-PLG for a total Ag concentration 8 μg/mg) was dissolved in 2 mL of dichloromethane (DCM). This organic phase was added to 10 mL of 1% PEMA and sonicated at 100% amplitude for 30 s using a Cole-Parmer Ultrasonic processor (Model XPS130). The emulsion was added to 200 mL of magnetically stirred 0.5% PEMA overnight to allow for DCM evaporation. The nanoparticles were collected by centrifugation at 7000×g for 15 min and washed twice with 0.1 M sodium bicarbonate buffer and a final wash using MilliQ water. Sucrose (4% w/v) and mannitol (3% w/v) were added as cryoprotectants and the particles were then lyophilized for 48 h before use. The size and zeta potential of the nanoparticles were determined in MilliQ water by dynamic light scattering (DLS) using a Malvern Zetasizer ZSP (Worcestershire, UK). Cy5.5-amine (Lumiprobe, Hunt Valley, MD) was conjugated to acid-terminated PLG and incorporated into PLG NPs at 1% w/w to create fluorescent PLG-Cy5.5 NPs, as previously described [30]. Ag-containing particles were formulated with a loading of 8 μg Ag/mg. NP size and zeta potentials are reported in Table S1.
4.4. Mice
Female C57BL/6J and OT-II mice (B6. Cg-Tg(TcraTcrb)425Cbn/J) (6–8 weeks old) were purchased from The Jackson Laboratories (Bar Harbor, ME). Swiss Jim Lambert (SJL/J) mice (6–8 weeks old) were purchased from Envigo Laboratories (Indianapolis, IN). All mice were housed under specific pathogen-free conditions in the University of Michigan Unit for Laboratory Animal Medicine and all mice procedures and experiments were compliant with the protocols of the University of Michigan Animal Care and Use Committee.
4.5. Antibodies and flow cytometry
All antibodies were purchased from BioLegend (San Diego, CA). Flow cytometric data were collected using a Beckman Coulter CytoFLEX S Research Flow Cytometer. Cells were blocked per the manufacturers’ instructions with anti-CD16/32 antibody prior to staining with the following extracellular antibodies: anti-F4/80 (clone BM8), anti-CD11b (clone M1/70), anti-CD146 (clone ME-9F1), anti-CD80 (clone 16–10A1), anti-CD86 (clone GL-1), anti-CD40 (cone 3/23), anti-PD-L1 (clone 10F.9G2). Viability was assessed with 4′,6-Diamidino-2-Phenylindole, Dilactate (DAPI) (Biolegend). CD45+F4/80+CD11b + KCs and CD45− CD146+ LSECs were separated by fluorescence activated cell sorting (FACS) using a Beckman Coulter MoFlo Astrios EQs. Analysis was performed using FlowJo software (FlowJo, San Jose, CA).
4.6. Liver cell isolation
Liver non-parenchymal cells (NPCs) were isolated as described previously by Bourgognon et al. [57]. Briefly, mice were anaesthetized with isoflurane. The liver portal vein was cannulated with a butterfly needle and perfused with 15 mL of a 0.5 mM ethylene glycol tetraacetic acid solution followed by 20 mL of 100 U/mL collagenase IV solution. Liver cells were strained through a 70 μm nylon strainer and parenchymal cells were removed by centrifugation at 50g for 2 min (four times). Non-parenchymal cells in the supernatant were further enriched using a 25/50% Percoll gradient (GE Healthcare, Uppsala, Sweden). KCs and LSECs were separated using FACS (KC: CD45+CD11b+F4/80+ and LSEC: CD45− CD146+) or MACS (KC: F4/80 positive selection and LSEC: CD146 positive selection).
4.7. Bone marrow-derived antigen presenting cells
Bone marrow was harvested from the tibia and femurs of C57BL/6J mice to differentiate BMDCs and BMMØs. Cell media consisted of RPMI 1640 supplemented with GlutaMAX (Life Technologies, Carlsbad, CA), penicillin (100 units/mL), streptomycin (100 mg/mL), 10% heat-inactivated fetal bovine serum (FBS) (Invitrogen Corporation, Carlsbad, CA). For BMMØs, the media was further supplemented with 20% L929 (ATCC, Manassas, VA) conditioned media on days 0, 3, and 6, and for BMDCs, 50 mM β-mercaptoethanol and 20 ng/mL of granulocyte-macrophage colony-stimulating factor (GM-CSF) (Peprotech, Rocky Hill, NJ) was added according to the Lutz protocol [58]. The BMMØs were lifted using Versene (ThermoFisher, Waltham, MA) and the BMDCs were loosely adherent and washed with gentle pipetting.
4.8. Nanoparticle biodistribution
C57BL/6J mice were injected with 0, 0.1, 0.5, 1, or 2 mg of Cy5.5-labeled PLG NPs in PBS. After 24 h, mice were euthanized and lungs, livers, and spleens were isolated. NP-derived fluorescence was measured using an in vivo imaging system (PerkinElmer IVIS® Spectrum, Waltham, MA). Liver cells were subsequently isolated by ex vivo incubation with collagenase IV and enriched for NPCs as described above. Cells were blocked, stained, and analyzed by flow cytometry to measure PLG-Cy5.5 signal in viable cells. Viability was assessed using DAPI exclusion dye.
4.9. In vivo T cell arrest and liver cell phenotyping
OT-II mice were injected with 2 mg of PLG-OVA323–339 or PLG-PLP139–151 (8 μg Ag/mg NP). 24 h later, liver NPCs were isolated. CD4 T cell viability was assessed by flow cytometry with DAPI exclusion dye, and the number of T cells was quantified with CountBright Absolute Counting Beads (Invitrogen Corporation, Carlsbad, CA). KC and LSEC costimulatory (CD80, CD86, CD40) and coinhibitory molecules (PD-L1) were evaluated by flow cytometry. mRNA transcripts for Tgfb1, Il10, and Ptges2 were quantified using reverse-transcription followed by quantitative polymerase chain reaction (RT-qPCR). mRNA was isolated using a High Pure RNA Isolation Kit (Roche, Indianapolis, IN). cDNA was synthesized using iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) according to manufacturer’s protocol using 1 μg RNA per sample. qPCR was conducted with iQ SYBR Green Supermix (Bio-Rad). Fluorescence incorporation and threshold cycle quantification was determined using a CFX Connect Real-time PCR Detection System (Bio-Rad). The primers (ThermoFisher, Waltham, MA) used were: 18s-rRNA (internal control) forward 5′-GCAATTATTCCCCATGAACG-3′ and reverse 5′-GGCCTCACTAAACCATCCAA-3′Tgfb1: forward 5′-CAGAAATACAGCAACAATTCC-3′ and reverse 5′-CTGAAGCAATAGTTGGTGTC-3′. Il10: forward 5- ATGCAGGACTTTAAGGGTTACTTGGGTT-3′ and reverse 5′- ATTTCGGAGAGAGGTACAAACGAGGTTT-3′. Ptges2: forward 5′- AATGTCCACAGCTCAGCCTC-3′ and reverse 5′-CTCAGGACTCTGGAGGGACA-3′. The 2−ΔΔCT method was used to quantify relative mRNA expression between PLG-OVA- and PLG-PLP-treated mice with 18s-rRNA as an internal control [59].
4.10. Detection of cytokine production by ELISA
Enzyme-linked immunosorbent assays (ELISA) for were used to measure murine TGF-β and IL-10 in the cell culture supernatants. A competitive binding assay was used to measure PGE2 (Cayman Chemical Company, Ann Arbor, MI). Assays were performed by the University of Michigan Cancer Center Immunology Core.
4.11. KC and LSEC Co-culture with T cells
C57BL/6J mice were injected with 2 mg of PLG-OVA323–339 (8 μg OVA323–339/mg NP). 24 h later, liver NPCs were isolated, stained, and sorted by MACS into KC and LSEC populations. MACS was conducted with anti-F4/80 and anti-CD146 microbeads. Liver APCs were seeded into round-bottom 96-well plates at 105 cells/well. BMDCs and BMMØs were used as controls and were seeded at 5 × 104 cells/well. Naïve CD4 T cells were isolated from OT-II mice using a naïve CD4 T cell isolation kit (Miltenyi, San Diego, CA) and co-incubated with the APCs at 5 × 104 cells/well. OVA323–339 was added to positive controls at 100 ng/mL. After 4 days, CD4 T cells were analyzed for CD25 expression and the supernatants were analyzed for TGF-β, IL-10, and PGE2 using ELISA or competitive binding assay.
4.12. EAE, splenectomy, and KC depletion/adoptive transfer
Relapsing-remitting experimental autoimmune encephalomyelitis (EAE), a mouse model of autoimmune multiple sclerosis, was induced by immunization with encephalitogenic PLP139–151 peptide as previously described [60]. 6–8 week female SJL/J mice were immunized by subcutaneous administration of 100 μL of 1 mg/mL PLP139–151/complete Freund’s adjuvant (CFA) emulsion distributed over 3 spots on the nape and hind flanks. The PLP/CFA (1 mg PLP/mL) emulsion was prepared by sonicating (Cole-Parmer Ultrasonic processor Model XPS130) one volume of 4 mg/mL M. tuberculosis H37Ra (Difco, Detroit, MI) in incomplete Freund’s adjuvant (Difco, Detroit, MI) with 1 volume of 2 mg/mL PLP139–151 in PBS. Disease severity in individual mice was assessed by blinded observers using a 0 to 5 point scale: 0 = no disease, 1 = limp tail or hind limb weakness, 2 = limp tail and hind limb weakness, 3 = partial hind limb paralysis, 4 = complete hind limb paralysis, 5 = moribund. In each EAE study, PLG-Ags were injected 7 days prior to immunization. In KC-depletion studies, mice were splenectomized as described previously [61]. After 13 days of recovery, KCs were depleted by injecting 200 μL (5 mg/mL) of clodronate liposomes (Encapsula, Brentwood, TN) 24 h before injection with PLG-PLP. Depletion was confirmed by flow cytometry analysis of liver NPCs (Figure S2). In adoptive transfer experiments, SJL/J mice were injected with 2 mg of PLG-PLP,-Cy5.5 (8 μg PLP/mg NP). 24 h later, liver NPCs were isolated, stained, and particle-positive KC(PLG-PLP) were sorted and injected into recipient mice at 106 kC(PLG-PLP) cells/mouse.
4.13. Statistical analyses
Results are reported as mean ± standard deviation (SD) or standard error of mean (SEM) as indicated in figure captions. Students’ t-test was used to determine the significance of parametric data between two groups. Significant differences between cytokine expressions were determined by one-way ANOVA along with Tukey’s multiple comparison test. Student’s t-test was used to compare fold-change differences in RT-qPCR analysis and differences in costimulatory molecule expression. Unless noted, p < 0.05 was considered to be statistically significant. In EAE experiments, differences between disease courses of more than two treatment groups were analyzed for statistical significance using the Kruskal-Wallis test (one-way ANOVA nonparametric test) (p < 0.05). Statistical differences between two EAE treatment groups were determined by the Mann-Whitney (two-tailed nonparametric) test (p < 0.05).
Supplementary Material
Acknowledgements
We thank the University of Michigan Rogel Cancer Center Immunology Core, the University of Michigan Flow Cytometry Core, and the University of Michigan Biointerfaces Institute for technical support. This work was supported in part by NIH grant EB-013198 (to L.D.S.). LMC is supported by the University of Michigan Rackham Predoctoral Fellowship. M.N.S. is supported by NIH grant T32GM007863. Research reported in this publication was supported by National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers R01AI155678 and R01AI148076.
Footnotes
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
Lonnie Shea reports a relationship with Cour Pharmaceuticals Development Company that includes: consulting or advisory and funding grants. Ryan Pearson reports a relationship with Cour Pharmaceuticals Development Company that includes: consulting or advisory. Stephen Miller reports a relationship with Cour Pharmaceuticals Development Company that includes: consulting or advisory and funding grants.
Credit author statement
Liam M. Casey: Conceptualization, Investigation, Data curation, Formal analysis, Writing – original draft, Writing – review & editing, Kevin R. Hughes: Investigation, Michael N. Saunders: Writing – review & editing, Stephen D. Miller: Conceptualization, Supervision, Funding acquisition, Ryan M. Pearson: Supervision, Lonnie D. Shea: Conceptualization, Supervision, Funding acquisition, Writing – review & editing.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.biomaterials.2022.121457.
References
- [1].Bach J-F, The effect of infections on susceptibility to autoimmune and allergic diseases, N. Engl. J. Med. 347 (2002) 911–920. [DOI] [PubMed] [Google Scholar]
- [2].Galvão VR, Castells MC, Hypersensitivity to biological agents—updated diagnosis, management, and treatment, J. Allergy Clin. Immunol. Pract. 3 (2015) 175–185. [DOI] [PubMed] [Google Scholar]
- [3].Calabrese L, Molloy E, Progressive multifocal leucoencephalopathy in the rheumatic diseases: assessing the risks of biological immunosuppressive therapies, Ann. Rheum. Dis. 67 (2008) iii64–iii65. [DOI] [PubMed] [Google Scholar]
- [4].Adami J, Gäbel H, Lindelöf B, Ekström K, Rydh B, Glimelius B, Ekbom A, Adami H-O, Granath F, Cancer risk following organ transplantation: a nationwide cohort study in Sweden, Br. J. Cancer 89 (2003) 1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pearson RM, Casey LM, Hughes KR, Miller SD, Shea LD, In vivo reprogramming of immune cells: technologies for induction of antigen-specific tolerance, Adv. Drug Deliv. Rev. 114 (2017) 240–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lorentz KM, Kontos S, Diaceri G, Henry H, Hubbell JA, Engineered binding to erythrocytes induces immunological tolerance to E. coli asparaginase, Sci. Adv. 1 (2015), e1500112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kontos S, Kourtis IC, Dane KY, Hubbell JA, Engineering antigens for in situ erythrocyte binding induces T-cell deletion, Proc. Natl. Acad. Sci. Unit. States Am. 110 (2013) E60–E68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hartwell BL, Pickens CJ, Leon M, Northrup L, Christopher MA, Griffin JD, Martinez-Becerra F, Berkland C, Soluble antigen arrays disarm antigen-specific B cells to promote lasting immune tolerance in experimental autoimmune encephalomyelitis, J. Autoimmun. 93 (2018) 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sestak JO, Sullivan BP, Thati S, Northrup L, Hartwell B, Antunez L, Forrest ML, Vines CM, Siahaan TJ, Berkland C, Codelivery of antigen and an immune cell adhesion inhibitor is necessary for efficacy of soluble antigen arrays in experimental autoimmune encephalomyelitis, Mol. Ther.-Meth. Clin. Develop. 1 (2014) 14008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Northrup L, Sestak JO, Sullivan BP, Thati S, Hartwell BL, Siahaan TJ, Vines CM, Berkland C, Co-delivery of autoantigen and b7 pathway modulators suppresses experimental autoimmune encephalomyelitis, AAPS J. 16 (2014) 1204–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jenkins MK, Schwartz RH, Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo, J. Exp. Med. 165 (1987) 302–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kheradmand T, Wang S, Bryant J, Tasch JJ, Lerret N, Pothoven KL, Houlihan JL, Miller SD, Zhang ZJ, Luo X, Ethylenecarbodiimide-fixed donor splenocyte infusions differentially target direct and indirect pathways of allorecognition for induction of transplant tolerance, J. Immunol. 189 (2012) 04–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Luo X, Pothoven KL, McCarthy D, DeGutes M, Martin A, Getts DR, Xia G, He J, Zhang X, Kaufman DB, ECDI-fixed allogeneic splenocytes induce donor-specific tolerance for long-term survival of islet transplants via two distinct mechanisms, Proc. Natl. Acad. Sci. Unit. States Am. 105 (2008) 14527–14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hunter Z, McCarthy DP, Yap WT, Harp CT, Getts DR, Shea LD, Miller SD, A biodegradable nanoparticle platform for the induction of antigen-specific immune tolerance for treatment of autoimmune disease, ACS Nano 8 (2014) 2148–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Casey LM, Pearson RM, Hughes KR, Liu JM, Rose JA, North MG, Wang LZ, Lei M, Miller SD, Shea LD, Conjugation of transforming growth factor beta to antigen-loaded poly (lactide-co-glycolide) nanoparticles enhances efficiency of antigen-specific tolerance, Bioconjugate Chem. 29 (2017) 813–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].LaMothe RA, Kolte PN, Vo T, Ferrari JD, Gelsinger TC, Wong J, Chan VT, Ahmed S, Srinivasan A, Deitemeyer P, Tolerogenic Nanoparticles induce antigen-specific regulatory T cells and provide therapeutic efficacy and transferrable tolerance against experimental autoimmune encephalomyelitis, Front. Immunol. 9 (2018) 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wilson DS, Damo M, Hirosue S, Raczy MM, Brünggel K, Diaceri G, Quaglia-Thermes X, Hubbell JA, Synthetically glycosylated antigens induce antigen-specific tolerance and prevent the onset of diabetes, Nat. Biomed. Eng. 3 (2019) 817–829. [DOI] [PubMed] [Google Scholar]
- [18].McCarthy DP, Yap JW-T, Harp CT, Song WK, Chen J, Pearson RM, Miller SD, Shea LD, An antigen-encapsulating nanoparticle platform for TH1/17 immune tolerance therapy, Nanomed. Nanotechnol. Biol. Med. 13 (2017) 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Smarr CB, Yap WT, Neef TP, Pearson RM, Hunter ZN, Ifergan I, Getts DR, Bryce PJ, Shea LD, Miller SD, Biodegradable antigen-associated PLG nanoparticles tolerize Th2-mediated allergic airway inflammation pre-and postsensitization, Proc. Natl. Acad. Sci. Unit. States Am. 113 (2016) 5059–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bryant J, Hlavaty KA, Zhang X, Yap W-T, Zhang L, Shea LD, Luo X, Nanoparticle delivery of donor antigens for transplant tolerance in allogeneic islet transplantation, Biomaterials 35 (2014) 8887–8894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lim H-H, Yi H, Kishimoto TK, Gao F, Sun B, Kishnani PS, A pilot study on using rapamycin-carrying synthetic vaccine particles (SVP) in conjunction with enzyme replacement therapy to induce immune tolerance in Pompe disease, Mol. Genet. Metab. Rep. 13 (2017) 18–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hlavaty KA, McCarthy DP, Saito E, Yap WT, Miller SD, Shea LD, Tolerance induction using nanoparticles bearing HY peptides in bone marrow transplantation, Biomaterials 76 (2016) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Meliani A, Boisgerault F, Hardet R, Marmier S, Collaud F, Ronzitti G, Leborgne C, Verdera HC, Sola MS, Charles S, Vignaud A, van Wittenberghe L, Manni G, Christophe O, Fallarino F, Roy C, Michaud A, Ilyinskii P, Kishimoto TK, Mingozzi F, Antigen-selective modulation of AAV immunogenicity with tolerogenic rapamycin nanoparticles enables successful vector re-administration, Nat. Commun. 9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Prasad S, Neef T, Xu D, Podojil JR, Getts DR, Shea LD, Miller SD, Tolerogenic Ag-PLG nanoparticles induce tregs to suppress activated diabetogenic CD4 and CD8 T cells, J. Autoimmun. 89 (2018) 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Freitag TL, Podojil JR, Pearson RM, Fokta FJ, Sahl C, Messing M, Andersson LC, Leskinen K, Saavalainen P, Hoover LI, Gliadin nanoparticles induce immune tolerance to gliadin in mouse models of celiac disease, Gastroenterology 158 (6) (2020) 1667–1681, 10.1053/j.gastro.2020.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tostanoski LH, Chiu Y-C, Gammon JM, Simon T, Andorko JI, Bromberg JS, Jewell CM, Reprogramming the local lymph node microenvironment promotes tolerance that is systemic and antigen specific, Cell Rep. 16 (2016) 2940–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hess KL, Oh E, Tostanoski LH, Andorko JI, Susumu K, Deschamps JR, Medintz IL, Jewell CM, Engineering immunological tolerance using quantum dots to tune the density of self-antigen display, Adv. Funct. Mater. 27 (2017) 1700290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Getts DR, Martin AJ, McCarthy DP, Terry RL, Hunter ZN, Yap WT, Getts MT, Pleiss M, Luo X, King NJ, Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis, Nat. Biotechnol. 30 (2012) 1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Maldonado RA, LaMothe RA, Ferrari JD, Zhang A-H, Rossi RJ, Kolte PN, Griset AP, O’Neil C, Altreuter DH, Browning E, Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance, Proc. Natl. Acad. Sci. Unit. States Am. 112 (2015) E156–E165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Pearson RM, Casey LM, Hughes KR, Wang LZ, North MG, Getts DR, Miller SD, Shea LD, Controlled delivery of single or multiple antigens in tolerogenic nanoparticles using peptide-polymer bioconjugates, Mol. Ther. 25 (2017) 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cappellano G, Woldetsadik AD, Orilieri E, Shivakumar Y, Rizzi M, Carniato F, Gigliotti CL, Boggio E, Clemente N, Comi C, Subcutaneous inverse vaccination with PLGA particles loaded with a MOG peptide and IL-10 decreases the severity of experimental autoimmune encephalomyelitis, Vaccine 32 (2014) 5681–5689. [DOI] [PubMed] [Google Scholar]
- [32].Kelly CP, Murray JA, Leffler DA, Getts DR, Bledsoe AC, Smithson G, First MR, Morris A, Boyne M, Elhofy A, TAK-101 nanoparticles induce gluten-specific tolerance in celiac disease: a randomized, double-blind, placebo-controlled study, Gastroenterology 161 (1) (2021) 66–80, 10.1053/j.gastro.2021.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Liao W-Y, Li H-J, Chang M-Y, Tang AC, Hoffman AS, Hsieh PC, Comprehensive characterizations of nanoparticle biodistribution following systemic injection in mice, Nanoscale 5 (2013) 11079–11086. [DOI] [PubMed] [Google Scholar]
- [34].Decuzzi P, Godin B, Tanaka T, Lee S-Y, Chiappini C, Liu X, Ferrari M, Size and shape effects in the biodistribution of intravascularly injected particles, J. Contr. Release 141 (2010) 320–327. [DOI] [PubMed] [Google Scholar]
- [35].Pearson RM, Podojil JR, Shea LD, King NJ, Miller SD, Getts DR, Overcoming challenges in treating autoimmuntity: development of tolerogenic immune-modifying nanoparticles, Nanomed. Nanotechnol. Biol. Med. 18 (2019) 282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].You Q, Cheng L, Kedl RM, Ju C, Mechanism of T cell tolerance induction by murine hepatic Kupffer cells, Hepatology 48 (2008) 978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Heymann F, Tacke F, Immunology in the liver—from homeostasis to disease, Nat. Rev. Gastroenterol. Hepatol. 13 (2016) 88. [DOI] [PubMed] [Google Scholar]
- [38].Sorensen KK, McCourt P, Berg T, Crossley C, Le Couteur DG, Wake K, Smedsrod B, The scavenger endothelial cell-a new player in homeostasis and immunity, Am. J. Physiol. Heart Circ. Physiol. 303 (12) (2012) R1217–R1230, 10.1152/ajpregu.00686.2011. [DOI] [PubMed] [Google Scholar]
- [39].Carambia A, Freund B, Schwinge D, Heine M, Laschtowitz A, Huber S, Wraith DC, Korn T, Schramm C, Lohse AW, TGF-β-dependent induction of CD4+ CD25+ Foxp3+ Tregs by liver sinusoidal endothelial cells, J. Hepatol. 61 (2014) 594–599. [DOI] [PubMed] [Google Scholar]
- [40].Carambia A, Freund B, Schwinge D, Bruns OT, Salmen SC, Ittrich H, Reimer R, Heine M, Huber S, Waurisch C, Nanoparticle-based autoantigen delivery to Treg-inducing liver sinusoidal endothelial cells enables control of autoimmunity in mice, J. Hepatol. 62 (2015) 1349–1356. [DOI] [PubMed] [Google Scholar]
- [41].Heymann F, Peusquens J, Ludwig-Portugall I, Kohlhepp M, Ergen C, Niemietz P, Martin C, van Rooijen N, Ochando JC, Randolph GJ, Liver inflammation abrogates immunological tolerance induced by Kupffer cells, Hepatology 62 (2015) 279–291. [DOI] [PubMed] [Google Scholar]
- [42].Knolle P, Uhrig A, Hegenbarth S, Löser E, Schmitt E, Gerken G, Lohse A, IL-10 down-regulates T cell activation by antigen-presenting liver sinusoidal endothelial cells through decreased antigen uptake via the mannose receptor and lowered surface expression of accessory molecules, Clin. Exp. Immunol. 114 (1998) 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Saito E, Kuo R, Pearson RM, Gohel N, Cheung B, King NJ, Miller SD, Shea LD, Designing drug-free biodegradable nanoparticles to modulate inflammatory monocytes and neutrophils for ameliorating inflammation, J. Contr. Release 300 (2019) 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Getts DR, Terry RL, Getts MT, Deffrasnes C, Müller M, van Vreden C, Ashhurst TM, Chami B, McCarthy D, Wu H, Therapeutic inflammatory monocyte modulation using immune-modifying microparticles, Sci. Transl. Med. 6 (2014), 219ra217–219ra217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].MacPhee P, Schmidt E, Groom A, Evidence for Kupffer cell migration along liver sinusoids, from high-resolution in vivo microscopy, Am. J. Physiol. Gastrointest. Liver Physiol. 263 (1992) G17–G23. [DOI] [PubMed] [Google Scholar]
- [46].Parsa R, Andresen P, Gillett A, Mia S, Zhang X-M, Mayans S, Holmberg D, Harris RA, Adoptive transfer of immunomodulatory M2 macrophages prevents type 1 diabetes in NOD mice, Diabetes 61 (2012) 2881–2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bai L, Liu X, Zheng Q, Kong M, Zhang X, Hu R, Lou J, Ren F, Chen Y, Zheng S, Liu S, Han Y-P, Duan Z, Pandol SJ, M2-like macrophages in the fibrotic liver protect mice against lethal insults through conferring apoptosis resistance to hepatocytes, Sci. Rep. 7 (2017) 10518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Pan G, Zhao Z, Tang C, Ding L, Li Z, Zheng D, Zong L, Wu Z, Soluble fibrinogen-like protein 2 ameliorates acute rejection of liver transplantation in rat via inducing Kupffer cells M2 polarization, Canc. Med. 7 (2018) 3168–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wu J, Li J, Salcedo R, Mivechi NF, Trinchieri G, Horuzsko A, The proinflammatory myeloid cell receptor TREM-1 controls kupffer cell activation and development of hepatocellular carcinoma, Cancer Res. 72 (2012) 3977–3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Smith CE, Eagar TN, Strominger JL, Miller SD, Differential induction of IgE-mediated anaphylaxis after soluble vs. cell-bound tolerogenic peptide therapy of autoimmune encephalomyelitis, Proc. Natl. Acad. Sci. U. S. A. 102 (2005) 9595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Park J-K, Utsumi T, Seo Y-E, Deng Y, Satoh A, Saltzman WM, Iwakiri Y, Cellular distribution of injected PLGA-nanoparticles in the liver, Nanomed. Nanotechnol. Biol. Med. 12 (2016) 1365–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Francisco LM, Sage PT, Sharpe AH, The PD-1 pathway in tolerance and autoimmunity, Immunol. Rev. 236 (2010) 219–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Carambia A, Frenzel C, Bruns OT, Schwinge D, Reimer R, Hohenberg H, Huber S, Tiegs G, Schramm C, Lohse AW, Inhibition of inflammatory CD4 T cell activity by murine liver sinusoidal endothelial cells, J. Hepatol. 58 (2013) 112–118. [DOI] [PubMed] [Google Scholar]
- [54].Klugewitz K, Blumenthal-Barby F, Schrage A, Knolle PA, Hamann A, Crispe IN, Immunomodulatory effects of the liver: deletion of activated CD4+ effector cells and suppression of IFN-γ-Producing cells after intravenous protein immunization, J. Immunol. 169 (2002) 2407–2413. [DOI] [PubMed] [Google Scholar]
- [55].Dolina JS, Sung S-SJ, Novobrantseva TI, Nguyen TM, Hahn YS, Lipidoid nanoparticles containing PD-L1 siRNA delivered in vivo enter Kupffer cells and enhance NK and CD8+ T cell-mediated hepatic antiviral immunity, Mol. Ther. Nucleic Acids 2 (2013) e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Knolle PA, Gerken G, Local control of the immune response in the liver, Immunol. Rev. 174 (2000) 21–34. [DOI] [PubMed] [Google Scholar]
- [57].Bourgognon M, Klippstein R, Al-Jamal KT, Kupffer cell isolation for nanoparticle toxicity testing, JoVE (102) (2015) e52989, 10.3791/52989. JoVE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lutz MB, Kukutsch N, Ogilvie AL, Rößner S, Koch F, Romani N, Schuler G, An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow, J. Immunol. Methods 223 (1999) 77–92. [DOI] [PubMed] [Google Scholar]
- [59].Livak KJ, Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method, Methods 25 (2001) 402–408. [DOI] [PubMed] [Google Scholar]
- [60].Miller SD, Karpus WJ, Davidson TS, Experimental autoimmune encephalomyelitis in the mouse, Curr. Protoc. Im. 88 (2010), 15.11. 11–15.11. 20. [DOI] [PubMed] [Google Scholar]
- [61].Reeves J, Reeves P, Chin LT, Survival surgery: removal of the spleen or thymus, Curr. Protoc. Im. 2 (1992), 1.10. 11–11.10. 11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.