

Summary
Desmoplastic small round cell tumor (DSRCT) is a rare, aggressive sarcoma driven by the EWSR1::WT1 chimeric transcription factor. Despite this unique oncogenic driver, DSRCT displays a polyphenotypic differentiation of unknown causality. Using single-cell multi-omics on 12 samples from five patients, we find that DSRCT tumor cells cluster into consistent subpopulations with partially overlapping lineage- and metabolism-related transcriptional programs. In vitro modeling shows that high EWSR1::WT1 DNA-binding activity associates with most lineage-related states, in contrast to glycolytic and profibrotic states. Single-cell chromatin accessibility analysis suggests that EWSR1::WT1 binding site variability may drive distinct lineage-related transcriptional programs, supporting some level of cell-intrinsic plasticity. Spatial transcriptomics reveals that glycolytic and profibrotic states specifically localize within hypoxic niches at the periphery of tumor cell islets, suggesting an additional role of tumor cell-extrinsic microenvironmental cues. We finally identify a single-cell transcriptomics-derived epithelial signature associated with improved patient survival, highlighting the clinical relevance of our findings.
Keywords: desmoplastic small round cell tumor, sarcoma, EWSR1::WT1, transcription factor, molecular and cellular heterogeneity, plasticity, single-cell RNA-sequencing, spatial transcriptomics, microenvironment, cancer-associated fibroblasts
Graphical abstract
Highlights
-
•
DSRCT cell heterogeneity is driven, at least in part, by transcriptional plasticity
-
•
Metabolic and lineage programs are two major components of DSRCT heterogeneity
-
•
Both cell-intrinsic and -extrinsic mechanisms drive transcriptional variability
-
•
ScRNA-seq-derived signatures predict outcome, highlighting intertumor heterogeneity
DSRCT is a rare sarcoma subtype of dismal prognosis driven by the aberrant transcription factor EWSR1::WT1. Using single-cell RNA-sequencing and an in vitro modeling of EWSR1::WT1 activity, Henon et al. show that DSRCT cells are characterized by partly overlapping cell-dependent and microenvironment-dependent transcriptional programs, whose variable expression has prognostic significance.
Introduction
Desmoplastic small round cell tumor (DSRCT) is a rare soft tissue sarcoma (STS) subtype related to the small round cell sarcomas (SRCSs) entity.1 DSRCT affects young adults, with a median age of 27 years and a 3 to 5:1 male-to-female sex ratio.2,3,4 The disease traditionally develops in the abdominopelvic cavity and spreads in the form of multiple peritoneal nodules and distant metastases.5,6 Despite aggressive multimodal therapeutic approaches, DSRCT is a devastating malignancy whose prognosis remains dismal, with a median overall survival of approximately two years.3,7,8,9
At the molecular level, DSRCT is characterized by the pathognomonic t(11; 22) (p13; q12) translocation, which fuses the N-terminal domain of EWSR1 with the C-terminal part of WT1. The resulting chimeric EWSR1::WT1 protein acts as an aberrant transcription factor (TF), activating various oncogenic pathways, such as cell proliferation, survival, and migration.10,11,12. EWSR1::WT1 represents the primary and unique driver of DSRCT, which otherwise displays a low mutational burden13,14,15,16,17,18 and very few recurrent secondary alterations (e.g., FGFR4, ARID1A, TERT, TP5314,15,16,18) or copy number variations.17 Histologically, DSRCT presents as nests of small round blue tumor cells surrounded by a desmoplastic stroma. Interestingly, despite tumor cell monotonous aspect, they exhibit a polyphenotypic differentiation, with positive immunohistochemical staining for epithelial, neural, mesenchymal, and myogenic markers. Intriguingly, recent findings in Ewing sarcoma, the prototypic EWSR1::FLI1-driven SRCS, revealed that variations in EWSR1::FLI1 activity influenced metabolic, proliferative, and migratory states of tumor cells.19,20 Such heterogeneity has yet to be explored in DSRCT.
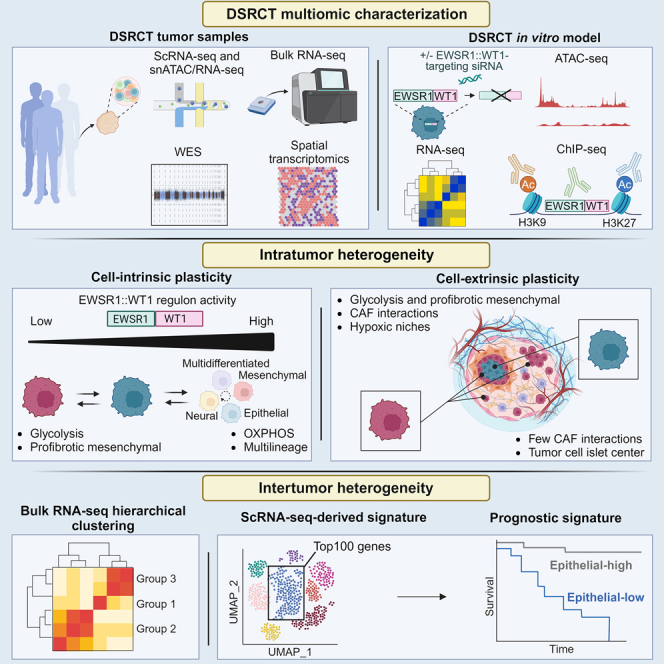
To gain insight into DSRCT heterogeneity, we characterized multiple patient samples by single-cell multi-omics and spatial and bulk transcriptomics, and integrated our findings with in vitro modeling characterizing DSRCT cells transcriptome and chromatin landscape upon EWSR1::WT1 modulation. By focusing on tumor cell-intrinsic plasticity, EWSR1::WT1-dependent transcriptome rewiring, and the interplay between DSRCT tumor cells and their microenvironment, we uncover mechanisms underlying DSRCT cell lineage and metabolic plasticity. We further identify the prognostic significance of a single-cell transcriptomics-derived epithelial signature in two independent cohorts of DSRCT patients.
Results
Single-cell RNA-sequencing deciphers DSRCT cellular composition
To comprehensively investigate DSRCT intra- and intertumor heterogeneity, we characterized: (1) 10 fresh human DSRCT samples and one juxtatumoral peritoneal sample using the droplet-based21 10x Genomics Chromium 3′ single-cell (sc)RNA-sequencing assay (3′ scRNA-seq); (2) one fresh tumor sample by single-nucleus (sn) Multiome assay (snMultiome), combining single-nucleus RNA-sequencing (snRNA-seq) and single-nucleus assay for transposase-accessible chromatin with sequencing (snATAC-seq); (3) 10 single-cell assays’ matched samples by bulk whole exome sequencing (WES); and (4) 29 archived frozen DSRCT samples by bulk RNA-seq (Figures 1A and 1B; Tables S1A and S1B).
Figure 1.

ScRNA-seq recapitulates DSRCT cellular composition
(A) DSRCT bulk and single-cell multiomic profiling.
(B) Patient and sample characteristics. L1: one prior treatment line; L2: two prior treatment lines.
(C) Uniform Manifold Approximation and Projection (UMAP) showing Int_sc dataset sample-of-origin.
(D) UMAP showing DSRCT signature score.
(E) UMAP showing DSRCT neotranscripts expression.
(F) UMAP (left panel) and barplot (right panel) highlighting DSRCT cell subpopulations.
(G) Heatmap showing expression Z-score of the top 50 DEGs of Int_sc cell types.
(H) H&E and IHC stainings for THY1, CD68/CD163, and CD3. The scale bar is displayed in the bottom left corner of each panel, representing 1 mm in the top panel and 200 µm in the four lower panels.
(I) DSRCT bulk RNA-seq cell subpopulation deconvolution.
To investigate intratumor heterogeneity, we integrated the 11 independent 3′ scRNA-seq datasets and generated a unique 3′ scRNA-seq dataset (“Int_sc” dataset). After quality control and filtering, we identified 51,671 high-quality cells, including 3,063 cells from the juxtatumoral peritoneal sample, with a median of 1,971 expressed genes per cell. Gene expression-based clustering allowed to identify different cell types (Figures 1C, 1D, and S1A; Tables S2A–S2C). Tumor cells (n = 44,781) (Table S2A), were confidently labeled based on the expression of two previously described and complementary DSRCT-specific genetic features: the top 10 DSRCT marker genes inferred from differential gene expression (DGE) analysis between DSRCT and a subset of sarcoma histotypes22 (Figure 1D), and DSRCT-specific neotranscripts23 (Figure 1E). Non-malignant cell types were labeled using expression of canonical markers. These included cancer-associated fibroblasts (CAFs) (n = 2,360; expressing COL1A1/SPARC/THY1), myeloid cells (n = 1,966; expressing C1QA/CD68/CD163), lymphoid (NK and T) cells (n = 1,126; expressing NKG7/IL7R/IL32/CD52/CD3E), mesothelial cells (n = 784; expressing CAV1/CALB2/MSLN), and endothelial cells (n = 654; expressing FABP4/VWF/PECAM1) (Figures 1F and 1G; Table S2A). After excluding the IC1 site#1 juxtatumoral sample, malignant cells represented 92% of detected cells, while the two major microenvironment cell types—CAFs and myeloid cells—represented less than 5% and 4% percent of total cells, respectively. Top marker genes of DSRCT malignant cells were highly concordant with the ones previously described (e.g., APOA1/CCL25/GJB2/GAL22) (Figure 1G). This single-cell derived cell type composition was in line with the known histological and immunohistochemical profile of DSRCT, harboring a paucicellular desmoplastic stroma mainly composed of CD68+/CD163+ macrophages, CD90+ (THY1) CAFs, and extremely rare lymphocytes (Figure 1H). To investigate cell type composition at a wider scale, we applied CIBERSORTx24 deconvolution to a cohort of 29 DSRCT samples profiled by bulk RNA-seq. The latter showed variable proportions of tumor cells, CAFs, and myeloid cells, while lymphoid cells, endothelial cells, and mesothelial cells consistently remained virtually undetectable (Figure 1I). We noted a discrepancy in the proportion of CAFs between the scRNA-seq dataset and the bulk RNA-seq deconvolution, which might either reflect sampling biases (frozen biopsy samples were mainly used for RNA-seq, whereas larger resection surgery specimens were used for scRNA-seq) and/or cell-type-dependent variable sensitivity to tissue dissociation used for single-cell suspension preparation.
DSRCT tumor cells harbor specific lineage- and metabolic-related states
We first focused on dissecting tumor cell heterogeneity, using gene expression-based clustering in individual and Int_sc datasets. Since we observed a high consistency of identified clusters between datasets—from both individual patients and synchronous distinct tumor sites—confirmed by the fact that nearly each of the Int_sc clusters contained cells from every sample, we further focused on the Int_sc dataset (Figures 1C, S1A, and 2A; Table S2B).
Figure 2.

DSRCT malignant cells show multilineage differentiation and metabolic states heterogeneity
(A) UMAP (left panel) and barplot (right panel) highlighting DSRCT malignant cell clusters in individual datasets.
(B) Heatmap showing expression Z score of the top 50 DEGs of each DSRCT malignant cell cluster.
(C) Top three GO pathways in DSRCT malignant cell clusters. Top 3 GO pathways based on gene ratio, after selection of the top 5 significantly enriched (p value <0.05) GO terms.
(D) Hierarchical clustering of HotSpot coexpressed gene modules. The top five genes driving module activity are shown.
(E) Single-cell level copy number variations (CNV) inference. Representative results of GR7 site#1.
(F) Bulk WES-derived CNV analysis on GR7 site#1.
(G) Representative IHC stainings for WT1, AE1/AE3, DES, and CD56 on a DSRCT sample. The scale bar representing 500 μm is displayed on the bottom left corner of each panel.
This allowed us to delineate 13 tumor cell clusters (Figures 2A and S1A): six lineage-, two metabolic-, and five pseudostate-related clusters. Lineage-related clusters included (1) one multilineage differentiation cluster (Multidiff_ASCL1 - 0), characterized by multilineage gene expression and the proneural ASCL1 marker gene; (2) two mesenchymal clusters (Mesenchymal_fibrosis - 8 and Mesenchymal_secretion - 11) respectively defined by overexpression of CHI3L1/IQCG/IGFBP5/ACTA2, and CCK/TNNT3/PTGDS; (3) one epithelial cluster (Epithelial_mesenchymal - 4), harboring concomitant expression of epithelial (e.g., CLDN3/CDH1/MUC16) and mesenchymal (e.g., MB/ACTA2) markers; (4) two neural/neuronal-related clusters: one neuronal/neuronal cluster (Neural_neuronal - 1) overexpressing IFI6/LY6H/NTRK3, and one neural stem cluster (Neural_stem - 19), expressing CCER2/NES/ELAVL3/4 (Figures 2A and 2B; Table S2B). Single-cell label transfer of a fetal development reference gene expression atlas25 (see STAR methods26) onto the Int_sc dataset suggested the coexistence of several developmental cell lineages within tumor cells (including neurons, epithelial, neuroendocrine, and smooth muscle cells), reminiscent of DSRCT polyphenotypic differentiation (Figure S1B).
The two metabolic clusters included (1) Metabolic glycolysis - 5 cluster, displaying high expression of genes involved in anaerobic glycolysis or Warburg effect (e.g., ENO1/ENO2/LDHA/NDUFA4L2); and (2) Metabolic_serine - 15 cluster, overexpressing genes involved in serine metabolism (e.g., PSAT1/ASNS/PHGDH) and aminoacyl-tRNA synthetases genes (e.g., YARS/CARS/GARS) (Figure 2B; Table S2B).
We identified five additional clusters, named “pseudostates,” related to either cell cycle or pathways of uncertain biological relevance. The latter included (1) one cycling cells’ cluster (Cycling cells - 3), expressing TOP2A/MKI67/TYMS; (2) two clusters expressing genes encoding ribosomal proteins (Ribosomal_catabolic – 2 and Ribosomal_IFN response – 6); and (3) two long non-coding RNA (lncRNA)-enriched clusters (Lnc_1 – 7 and Lnc_2 – 17), overexpressing lncRNAs frequently upregulated in cancer (e.g., MEG3/XIST/GRID2/MALAT1) (Figures 2A and 2B; Table S2B). Since the median number of genes per cell in Ribosomal and Lnc clusters was low (Table S2C), their biological relevance deserves cautious interpretation. Interestingly, few genes were strictly specific of one tumor cell cluster (Figures 2B; Table S2B), with a coexistence of shared and private marker genes possibly suggesting some tumor cell plasticity.
When focusing on GR4 and GR4_PC samples, arising from the same patient before and after chemotherapy, we observed an overall stability in the nature of the transcriptomic characteristics of cell populations, though the relative proportion of each cluster could vary (e.g., increase in cells belonging to the Cycling cells - 3 and Ribosomal catabolic - 2 clusters; Figures 1C and 2A).
To next explore functional pathways activation within each Int_sc cluster, we performed gene set enrichment analysis (GSEA) using Gene Ontology (GO) terms (Figure 2C). We found a significant enrichment in lineage- and metabolism-related pathways, corresponding to the previously identified clusters’ marker genes (Figure 2B) and to additional lineages such as myogenic cell states (data not shown). Intriguingly, LncRNA clusters were enriched in RNA splicing pathways, possibly linked to EWSR1 RNA- and SWI/SNF-binding ability.27
The study of coexpressed gene modules28 (see STAR methods) further supported the hypothesis of plastic transcriptional states (Figure 2D; Table S3). We identified three main module profiles. The first module category showed high expression in both neural (Neural_neuronal - 1, Neural_stem - 19) and Mesenchymal_fibrosis – 8 clusters (e.g., Modules 9, 8, 4, 5, 6, and 1, respectively enriched in lncRNAs, neural-, cytoskeleton-, interferon response-, transcription regulation-, and translation-related genes). By contrast, the second category comprised modules highly activated in only one or two clusters. For example, Module 7, enriched in epithelium development and cell adhesion (e.g., GJB2/CCL25/ITIH5), was exclusively activated in Epithelial_mesenchymal – 4 and Lnc_2 – 17 clusters; and Module 13, defined by glycolysis-related genes (e.g., LDHA/GAPDH/ENO1), was specific to Metabolic_glycolysis – 5. The third module category showed limited variability or specificity toward Int_sc clusters. Overall, the variable correlation between gene modules and DEG-derived clusters suggested a putative role for coregulators in fine-tuning the expression of shared or specific transcriptional programs.
DSRCT tumor cells show high gene expression entropy and do not follow consistent transcriptional trajectories
To explore whether DSRCT cells would follow a continuum of transcriptional states, we inferred transcriptional trajectories from gene expression in each scRNA-seq dataset. Using directed single-cell fate mapping combining trajectory inference and RNA velocity analysis,29 we could not find any consistent trajectory (data not shown). We next investigated whether DSRCT clusters would harbor various stemness levels using CytoTRACE30 and single-cell entropy.31 Most tumor cell clusters were predicted to be less differentiated than non-tumor cells, excepted pseudostate clusters (Ribosomal and LncRNA-enriched; Figures S1C and S1D). This suggested the coexistence of non-terminally differentiated cell states through which DSRCT cells might dynamically evolve, rather than unidirectional evolutionary trajectories.
Copy number variations are homogeneous across DSRCT tumor cell clusters and samples
The above results suggested a transcriptionally driven plasticity of DSRCT. To support this, we explored the contribution of genetic alterations to tumor cell heterogeneity. We inferred Copy Number Variations (CNVs)32 from scRNA-seq, and found high consistency with matched bulk WES (representative case in Figures 2E and 2F). Very few recurrent CNVs were identified across samples, in line with the known quiet genomic profile of DSRCT, at least in primary lesions.17 The most frequent alteration, in line with previous reports, was a chromosome 5 gain,17,22 but we did not identify chromosome 1q gain.17 Importantly, we found that the rare identified CNVs were highly consistent across single-cell clusters from each sample, thereby reinforcing the hypothesis that DSRCT cell heterogeneity results from transcriptional plasticity rather than somatic genetic (sub)clonal evolution.
The heterogeneity of DSRCT tumor cells is not associated with variable EWSR1::WT1 transcript expression level
Studies on Ewing sarcoma reported that variable EWSR1::FLI1 activity drives intratumor heterogeneity.19,20 We therefore hypothesized that similar mechanisms might operate in DSRCT.
We first investigated whether variable EWSR1::WT1 mRNA expression levels might drive distinct transcriptional programs. To this aim, we developed an in-house assay to specifically amplify EWSR1::WT1 transcripts from 10x-derived barcoded cDNAs (see STAR methods) (Figures S1E–S1H). EWSR1::WT1 expression variation was limited across cancer cells and did not correlate with DGE-derived clusters (Figure S1H), suggesting a limited role in DSRCT tumor cell heterogeneity. When exploring this at the protein level by immunohistochemistry (IHC) on a series of DSRCT samples, we found that, despite homogeneous staining of WT1 (used as a surrogate marker of EWSR1::WT1, since WT1 wild-type is reportedly not expressed in DSRCT12,33), AE1/AE3, Desmin (DES), and CD56 expression were heterogeneous across tumor cells (Figure 2G). This overall suggested that factors beyond EWSR1::WT1 expression levels—such as variable EWSR1::WT1 activity, DNA-binding sites, or fluctuating chromatin accessibility—might influence DSRCT’s polyphenotypic differentiation.
EWSR1::WT1 binds regulatory regions of genes involved in lineage-related programs and EGR1 consensus motifs
To investigate whether variations in EWSR1::WT1 functional activity may contribute to DSRCT heterogeneity, we first characterized in vitro the effects of EWSR1::WT1 silencing on the chromatin landscape (Figure S2).
To identify genes and pathways regulated by EWSR1::WT1, we performed EWSR1::WT1 chromatin immunoprecipitation with sequencing (ChIP-seq) in the JN-DSRCT-1 cell line, using a WT1 C terminus antibody to specifically pull down EWSR1::WT1—WT1 wild-type not being expressed in JN-DSRCT-1 cells (Figure S2A, upper panel). We obtained high-quality coverage profiles (mean FRIP-score of 0.81%; peak-calling-independent mean relative strand correlation and normalized strand coefficient of 0.73 and 1.02, respectively34). This allowed us to identify 8,806 and 4,541 peaks for both WT1 ChIP replicates, corresponding to 8,782 and 4,507 consensus peaks regions, respectively. Of these, 1,587 were shared between WT1 C-terminal ChIP replicates #1 and #2; all consensus peak regions were used for further analysis. As previously reported,33 most EWSR1::WT1 target sequences were located within intergenic regions, followed by introns (Figure S2A, lower panel). Using control isotype ChIP to perform differential binding analysis, we identified previously described EWSR1::WT1 target genes (e.g., CCND1/FGFR4/EGR135) (Figure S2B; Table S4A) and further uncovered novel targets, including extracellular matrix-related genes (e.g., COL23A1/CHI3L1), neural-related genes (e.g., GAL/ADGRB1), and genes involved in fatty acid metabolisms (e.g., ACADVL/ECI2) or chromatin remodeling (e.g., DPF3/CTCFL) (Table S4A). GSEA on EWSR1::WT1 targets revealed an enrichment in critical developmental processes, including (1) multilineage tissue development (e.g., GOBP_embryonic heart tube development, GOBP_skin epidermis development, GOBP_spinal cord motor neuron differentiation); (2) stem cell differentiation and proliferation (e.g., GOBP_stem cell differentiation, GOBP_stem cell proliferation); and (3) regulation of fatty acid metabolic processes (e.g., GOBP_fatty acyl-CoA metabolic process, GOBP_sphingolipid biosynthetic process) (Figure S2C).
Since TF activity varies according to the recruitment of different transcriptional coregulators and to their cognate DNA sequence, we then explored EWSR1::WT1 binding motifs. As for wild-type WT1,36,37 the canonical EWSR1::WT1 binding motif varies according to the presence of the KTS amino acids between WT1 exons 9 and 10,35,38,39,40,41 and is related to EGR1 binding motifs.33 In line with previous reports describing EWSR1::WT1+KTS optimal binding site,41 we found that GGAGGA 6-mers were predominant within EWSR1::WT1 binding peaks (Figure S2D) and identified [GGA] repeats as a de novo motif. Indeed, eight de novo motifs were significantly enriched in EWSR1::WT1 binding regions, of which the top three (5′-GCGKGGGAGGVRGV-3′, 5′-CCACGCA-3′, and 5′-GGAGGAGRAGGAGGAA-3′) respectively best matched with WT1+/−KTS, EGR1/2/3, and ZNF263 TF motifs (Figure S2E). Despite not being specifically included within JASPAR2020 and HOCOMOCO-v11 databases, the top enriched motif was reminiscent of WT1 –KTS whereas the top 3, 4, and 5 motifs were highly concordant with the known WT1 +KTS motif, in line with the coexistence of both isoforms in the DSRCT-JN1 cell line identified by RNA-seq (data not shown). Among known TFs motifs, EGR1 was the most highly enriched EWSR1::WT1-binding motif, followed by other members of the EGR, ZNF, SP/KLF, FOX, E2F, ETS, and HOX TF families (Table S4B), suggesting either direct EWSR1::WT1 binding or collaboration with the latter TFs at these motif sites. This overall supports that EWSR1::WT1 has a de novo oncogenic activity and induces a significant epigenetic rewiring, primarily at [GGA] repeats, EGR1/2 binding sites, and domains involved in transcriptional activation (e.g., E2F) or cell fate (e.g., FOX, HOX).
EWSR1::WT1 modifies the chromatin landscape of DSRCT cells through direct and indirect mechanisms
To gain further insight into EWSR1::WT1 role on chromatin accessibility, we systematically characterized open chromatin regions using ATAC-seq (Assay for Transposase-Accessible Chromatin with high-throughput sequencing) in the JN-DSRCT-1 cell line upon modulation of EWSR1::WT1 expression (Figure S2F), and correlated it with ChIP-seq data. We identified 164,043 transposase-accessible DNA elements significantly modulated upon EWSR1::WT1 silencing, including 83,524 and 80,519 sites with decreased and increased accessibility, respectively, herein referred to as EWSR1::WT1 “on” and “off” ATAC-seq peaks (Figure S2G; Table S5A). No modification in WT1 accessibility was observed. Multiple EWSR1::WT1 “on” accessible genomic regions encoded top markers of scRNA-seq tumor cell clusters (e.g., GAL/CCL25/BAI1 alias ADGRB1), epigenetic modifying enzymes (e.g., TET2/SMARCA2/SMARCD1), and/or proteins involved in shaping the tumor microenvironment (e.g., ANGPT1) (Table S4A; Figure S2G). Interestingly, FOXA1/2 and androgen receptor (AR) showed significantly decreased accessibility upon EWSR1::WT1 silencing, questioning whether the previously reported potential cooperation between EWSR1::WT1 and AR42 may involve FOXA1/2.43,44,45
Motif enrichment analysis within EWSR1::WT1 “on” ATAC peaks showed a strong correlation with EWSR1::WT1 binding regions, including EGR1/2, FOXC2, FOXOA1/2, and SP/KLF TF motifs (Table S5B). Nearly two-thirds (953 of 1,318) of the EWSR1::WT1 ChIP-enriched peaks overlapped with EWSR1::WT1 “on” ATAC peaks (Figure S2H). Those corresponded to 857 unique genes, of which 262 (including CCND1, FGFR4, COL23A1, FOXA2, FOXD1) were significantly downregulated upon EWSR1::WT1 silencing (Tables S4A, S5A, and S6). These were not only related to multilineage cell development and lineage commitment, but also to Wnt signaling, intriguingly corresponding to pathways recently linked to the EWSR1::WT1-dependent DSRCT-specific AR retargeting at enhancers.42
Conversely, genomic regions with increased chromatin accessibility upon EWSR1::WT1 silencing were primarily enriched in FOS/JUN, BATF, BACH, and HOX TFs motifs (Table S5C). These regulated genes were involved in mesenchymal cell differentiation (e.g., ANKRD1/ANXA1/CDH2/FBN1) (Figure S2G; Table S5A), compatible with a putative pluripotent mesenchymal ancestor as DSRCT cell-of-origin. Intriguingly, the EWSR1::WT1 “off” ATAC peak set also included EGR1 regulatory regions (Figure S2G) although with less significant p value, in line with EGR1 expression upregulation identified upon EWSR1::WT1 silencing (Table S6). Strikingly, the latter results contrast with a previously published report35 that showed using a luciferase assay in transfected osteosarcoma Saos-2 cells that EWSR1::WT1–KTS activates EGR1 promoter through the EGR binding sequence, hence highlighting a more complex relationship between EWSR1::WT1+/−KTS and EGR1 expression in DSRCT models. Focusing on EGR1, we overall found that (1) EGR1 is a direct target of EWSR1::WT1 (Figure S2B), (2) EWSR1::WT1 targets EGR1 binding motifs (Figures S2D and S2E); and (3) EGR1 expression and regulatory regions accessibility are increased upon EWSR1::WT1 silencing (Figure S2G; Table S6). Taken together, this suggests that EWSR1::WT1 and EGR1 may compete at EGR1 binding sites, and that EWSR1::WT1 may dominate this competition in the absence of wild-type WT1, by reducing EGR1 promoter accessibility and expression.
To further investigate EWSR1::WT1 effects on the chromatin landscape, we analyzed by ChIP-seq in JN-DSRCT-1 cells the acetylation of histones H3K27 (H3K27ac) and H3K9 (H3K9ac), which are primarily associated with active enhancers and promoters, respectively. We found that 26% (2,992 of 8,667) and 46% (4,008 of 8,667) of EWSR1::WT1 target regions were co-occupied by H3K27ac and H3K9ac histone marks, respectively (Figure S2I; Tables S4C and S4D), suggesting an association with active transcriptional states.
Taken together, our findings suggest that EWSR1::WT1 drives DSRCT not only through direct aberrant regulation of transcriptional programs, but also possibly through a cooperation with oncogenic (e.g., AR) or pioneer (e.g., FOX family) TFs at their consensus sequences, and potential competition with alternate TFs (e.g., EGR1) at shared consensus sequences.
Chromatin landscape shapes DSRCT cell heterogeneity
Next, to investigate the link between EWSR1::WT1-dependent chromatin accessibility modulation and DSRCT transcriptional heterogeneity, we profiled one fresh tumor sample (GR11) using snMultiome assay (Figures 1A, 1B, and 3A). By combining gene expression and ATAC features for weighted nearest neighbors (WNN) clustering, we identified 13 clusters (Figure 3A). When compared to Int_sc dataset, neither DGE (Figure 3B; Table S7A) nor peak enrichment analyses (Table S7B) allowed us to confidently identify the previously described scRNA-seq clusters, except for WNN_cluster 9, WNN_cluster 4, and WNN_cluster 3/11/12, whose top marker genes/peaks were representative of non-malignant cells, a mitochondrial genes-enriched cluster, and cycling cells, respectively. Transferring cluster annotations from the 3′ scRNA-seq reference dataset on the snMultiome dataset using intronic reads only (“Int_sc_intron” dataset) enabled to better correlate snMultiome clusters with previously defined cell states (see STAR methods, Figure 3C). While several WNN clusters were tightly linked to a single Int_sc cluster (e.g., WNN_cluster 3/11/12 and Int_sc Cycling cells – 3, WNN_cluster 5 and Int_sc Metabolic_glycolysis – 5, WNN_cluster 10 and Int_sc Mesenchymal_fibrosis – 8), other pairwise correspondences were less straightforward (Figure 3C).
Figure 3.

EWSR1::WT1 activity and epigenetic reprogramming are one determinant of DSRCT heterogeneity
(A) UMAP of GR11 sample snMultiome WNN clustering.
(B) Heatmap showing expression Z score of the top 10 DEGs across WNN snMultiome clusters.
(C) Label transfer of Int_sc clusters on WNN snMultiome dataset.
(D) Barplot showing the top 10 enriched motifs in malignant versus non-malignant cells.
(E) UMAP showing EWSR1::WT1 chromatin accessibility signature score on snMultiome assay.
(F) UMAP showing EWSR1::WT1 targeted loci signature score on snMultiome assay.
(G) Graphical representation of single-cell EWSR1::WT1 regulon activity inference.
(H) UMAP showing EWSR1::WT1 regulon activity in Int_sc dataset.
(I) Boxplot representing EWSR1::WT1 regulon activity per Int_sc cluster.
(J) Heatmap-dotplot showing gene AUCs for the most specific regulons defined by regulon specific score (RSS), as well as EWSR1::WT1, AR, and EGR1 regulons.
Integration of preclinical modeling with single-cell multi-omics uncovers an EWSR1::WT1-dependent cell-intrinsic plasticity
We next analyzed differentially accessible snMultiome peaks between malignant and non-malignant cells (Figure 3D, upper panel). We found that motifs enriched in malignant cells corresponded to previously identified EWSR1::WT1 binding motifs (Table S4B), including EGR1/EGR3/SP2/ZNF263. Non-malignant cells were mainly enriched in FOS/JUN and BATF motifs (Figure 3D, lower panel), intriguingly correlating with those enriched in the EWSR1::WT1 “off” ATAC peaks dataset (Table S5C).
To explore in the snMultiome dataset whether EWSR1::WT1 effects on chromatin accessibility could affect DSRCT tumor cell heterogeneity, we first focused on the EWSR1::WT1 “on” ATAC peak set, from which we derived an EWSR1::WT1 chromatin accessibility signature score (see STAR methods). WNN_cluster 5, labeled as the anaerobic glycolysis pathway-enriched cluster, and WNN_cluster 10, labeled as the profibrotic mesenchymal cluster, displayed the lowest EWSR1::WT1 chromatin accessibility signature score among malignant cells (Figure 3E). This was consistent with data obtained using the EWSR1::WT1 ChIP-seq binding peaks dataset (Figure 3F), from which we similarly derived an EWSR1::WT1 targeted loci signature (see STAR methods).
Next, we integrated RNA-seq and ChIP-seq data of the JN-DSRCT-1 cell line upon EWSR1WT1 silencing to define a specific EWSR1::WT1 regulon used as a surrogate for EWSR1::WT1 TF activity. We identified 66 genomic regions mapping to 53 unique genes corresponding to direct targets of EWSR1::WT1 whose expression was significantly decreased upon silencing (see STAR methods, Figure 3G; Table S8A). As expected, the EWSR1::WT1 regulon activity score was almost undetectable in tumor microenvironment (TME) cells while significantly higher in tumor cells (Figures 3H and 3I). The highest scores were observed for lineage-related Epithelial_mesenchymal – 4, Mesenchymal_secretion – 11, Neural/neuronal – 1, Multidiff_ASCL1 – 0, and Neural_stem – 19 clusters (Figures 3H and 3I). By contrast, pseudostates-, metabolism-related (Metabolic_glycolysis - 5, Metabolic_serine – 15), and profibrotic mesenchymal (Mesenchymal_fibrosis – 8) clusters showed the lowest EWSR1::WT1 regulon activity, in line with previous results (Figures 3E and 3F).
Overall, these data support that EWSR1::WT1 activity contributes to shaping DSRCT cell states: while high EWSR1::WT1 activity (EWSR1::WT1high) relates to most multilineage differentiation states, low EWSR1::WT1 TF activity (EWSR1::WT1low) associates with cell states characterized by specific metabolic (e.g., anaerobic glycolysis) or profibrotic mesenchymal features.
DSRCT cell heterogeneity is driven by variable TF activity and cell state-specific regulons
To next explore whether variable TFs could cooperate with EWSR1::WT1 and foster DSRCT heterogeneity, we sought to identify binding motifs enriched in snMultiome clusters. When focusing on the activity of the 10 most differentially active known TFs motifs across WNN clusters (Figure S2J), we identified different families of development-related TFs, including SOX (WNN_cluster 2), FOX (WNN_cluster 12), and GATA (WNN_cluster 6), whose motifs were enriched in EWSR1::WT1 binding regions (Table S4B) and/or whose binding domains displayed variable accessibility upon EWSR1::WT1 modulation (Table S5B).
Interestingly, within EWSR1::WT1low cell states, WNN_cluster 10 (related to mesenchymal profibrotic tumor cells, Int_sc Mesenchymal_fibrosis – 8), displayed increased accessibility in interferon response factor TF motifs, potentially suggesting the contribution of extrinsic microenvironmental chemokine signaling to this phenotype. By contrast, WNN_cluster 5 (corresponding to the anaerobic glycolysis state), was enriched in AP-1 (e.g., FOS/JUN) motifs. In line with DSRCT biology, we also found that AR motif activity ranked within the top 10 enriched motifs in WNN_clusters 1, 6, and 8.
We next complemented our analyses by inferring enhancer-driven gene regulatory networks using SCENIC+46 on the snMultiome dataset. When exploring regulons within the entire snMultiome dataset or the malignant cells only, we identified 337 and 341 active TFs, among which 253 and 226 were activators (i.e., associated with increased chromatin accessibility), respectively. Top activator regulons in tumor cells (Figure 3J; Table S8B) were consistent with our motif-based analysis (Figure S2I). Lower EWSR1::WT1 regulon activity was detected in anaerobic glycolysis (WNN_cluster 5), mesenchymal profibrotic (WNN_cluster 10), and cycling cell (WNN_cluster 3/11/12) clusters, consistent with the EWSR1::WT1low cell states identified in the Int_sc dataset (Figure 3I). By contrast, EWSR1::WT1high clusters (WNN_cluster 0/1/4/6/8) displayed higher AR regulon activity, consistent with its role in DSRCT oncogenicity.42 Finally, we observed that EGR1 regulon activity was inversely correlated to the one of EWSR1::WT1 and AR regulons, supporting their potential antagonism and the heterogeneity of this balance among DSRCT cell states.
Altogether, our data show that DSRCT cell states are, at least in part, driven by varying EWSR1::WT1 activity—EWSR1::WT1high being linked to a higher and lower activity of AR and EGR1 regulons, respectively—and variable accessibility of cell fate-related TF domains.
DSRCT microenvironment displays immune-tolerant features
We next investigated whether tumor cell-extrinsic stimuli from the TME could also shape DSRCT heterogeneity. Overall, immune cell populations were scarce, representing only 6% of cells analyzed in the Int_sc dataset (Figures 1F, 1I, 4A, and S1A; Table S2A). Myeloid cells were the main subpopulation, representing 63% of all immune cells in the Int_sc dataset (Figures 1F and S1A; Table S2A). Further deconvolution of myeloid subpopulations using the MoMac-VERSE atlas47 (see STAR methods) identified a majority of macrophages and a minority of monocytes and dendritic cells (DCs) (Figure 4A, left panel). The dominant myeloid subpopulations were protumorigenic, harboring an M2 macrophage signature, including HES148,49 (27.7%), DC-like (11.9%), TREM250,51,52 (11.9%), and FTL53 (11.7%) macrophages or inflammatory monocyte (IL1B/VEGFA-positive, 8%). When focusing on lymphoid subpopulations, we identified seven clusters based on DGE analysis and known canonical markers54 (Figure 4A, right panel). The most abundant cell type was identified as CD4+ memory T cells (24.3%), overexpressing CD4/CD40LG/CD44/IL7R (CD4+ memory T cells – 0 cluster), followed by CD8+ cytotoxic effector T cells (23.1%, i.e., 0.6% of all Int_sc cells) overexpressing CD8A+/GZMB+/GZMK+/GZMA+/GZMH+ (CD8+ effector T cells – 1 cluster). Lymphoid cell expression profiles showed a low or null expression of immune checkpoints such as PDCD1 (encoding PD-1), HAVCR2 (encoding TIM-3), or LAG3.
Figure 4.

DSRCT microenvironment displays immunosuppressive features and distinct CAF subpopulations
(A) Int_sc infiltrating myeloid (left panel) and lymphoid (right panel) subpopulation proportions represented by pie charts.
(B) UMAP plots displaying CAFs according to patient ID (left panel), tissue of origin (middle panel), and subclustering (right panel).
(C) Heatmap highlighting expression Z score of the top 50 DEGs of CAFs subcluster.
(D) Violin plot showing expression profile of CAF subclusters for canonical markers.
(E) Violin plots displaying expression profile of CAF subclusters for immunosuppressive markers.
(F) DSRCT bulk RNA-seq CAF subpopulations deconvolution.
(G) Immunofluorescence triplex showing ACTA2 (red), MCAM (yellow), FAP (green), and DAPI staining on a DSRCT FFPE sample in distinct stromal areas. The #1 annotation designates desmoplastic stromal areas, #2 shows tumor cell islets periphery, and #3 indicates the tumor pseudocapsule. The ∗ sign shows tumor necrosis. The scale bar is displayed on the bottom left corner of each panel, representing 1 mm on the left panel and 100 μm on the three right panels.
Overall, DSRCT immune cell characterization suggested a predominantly immunotolerant microenvironment, with a majority of protumorigenic macrophages and low infiltration of effector cytotoxic CD8+ T cells.
DSRCT CAFs are composed of protumorigenic and immunosuppressive subpopulations
We next focused on CAFs. The initial Int_sc dataset clustering identified two CAF subpopulations, mainly arising from the tumor tissue samples and the IC1#1 juxtatumor peritoneal sample (Figures 1C, 1F, 4B left and middle panels, and S1A). Further CAFs subclustering defined seven distinct CAF subpopulations, which we manually annotated based on top marker genes and established CAF canonical markers55,56,57,58 (Figures 4B–4D). One additional cluster, potentially resulting from tumor cells’ misclustering, was labeled as Undetermined – 2.
CAF subclusters predominantly found within the juxtatumor sample (CD34/MYC fibroblasts – 0, Lipofibroblasts – 3, Adventitial fibroblasts – 7), and Inflammatory CAFs – 6 overexpressed genes from the complement pathway (e.g., C7) and the constitutive lipid droplet protein PLIN2. CD34/MYC fibroblasts – 0 also harbored pre-adipocyte/adipocyte stem cell markers (e.g., SCARA5/APOD/CXCL14),59 whereas Adventitial fibroblasts – 7 additionally expressed PI16/CD55/CD248, reminiscent of adventitial stromal cells found in vascular niches and producing extracellular matrix60,61 (Figures 4C and 4D).
In line with the known DSRCT-specific desmoplastic stroma, desmoplasia-related genes (e.g., MMP2/COL1A1/LOX/LOXL1/VEGFB)62 were highly expressed in all CAF subclusters (Figure 4E). CAF subpopulations mostly harbored immunosuppressive features, including T cell exclusion markers (e.g., MRC263) and M2 macrophage polarization signaling (e.g., CSF164,65/CXCL1266/IL667). Inflammatory CAFs – 6 further displayed high expression of genes involved in myeloid-derived suppressor cell recruitment,68 including CCL2/CXCL2/CXCL8 (Figure 4E). When deconvoluting CAFs subclusters on our DSRCT bulk RNA-seq dataset, we found that the most abundant subtypes were Desmoplastic CAFs - 1 and Myofibroblastic CAFs – 4 (Figure 4F).
To confirm our findings, we further characterized by IHC the three traditional CAF markers MCAM, FAP, and ACTA2 (alias αSMA) on eight DSRCT samples (Figure 4G). As illustrated in Figure 4G (representative example), MCAM+ CAFs were located at the vessel periphery, supporting their Pericyte-like CAFs – 5 features. Interestingly, ACTA2high/FAPlow/MCAM- CAFs, potentially corresponding to Myofibroblastic CAFs – 4, were mainly found in-between or at the periphery of the tumor cell islets. By contrast, FAPhigh CAFs (most likely Desmoplastic CAFs – 1) preferentially located within the prominent trabeculae of desmoplastic stroma and pseudocapsule (Figure 4G). The FAP/ACTA2 fluorescence ratio was also significantly higher in CAFs located in the pseudocapsule than in the desmoplastic area or at tumor cells’ islets periphery (paired t test p < 0.01 and p < 0.05, respectively).
Overall, our findings support that DSRCT TME predominantly has immunosuppressive features, and that CAF subpopulations may harbor a specific spatial organization.
DSRCT phenotypic and metabolic states harbor a specific spatial distribution
We further sought to characterize the heterogeneity of DSRCT cell subpopulations at the spatial level, and first assessed by immunofluorescence (IF) previously identified malignant cell and CAF markers of interest. We found that, in some DSRCT samples (e.g., GR2), DES/CHI3L1 expression was restricted to either the periphery of tumor cell islets, near THY1+ CAFs, at the invasive tumor front (Figures 2G and 5A), or in sparse tumor cells located within desmoplastic stromal areas. Interestingly, the corresponding scRNA-seq data (Figure 5B) revealed that the latter corresponded to mesenchymal DSRCT clusters (GR2 Mesenchymal_TNNT3 – 5, Mesenchymal_DES − 9, Neuronal_mesenchymal - 13, and Mesenchymal_stem – 14 clusters), expressing CHI3L1, DES, TNNT3, and MSLN (Figure 5B). In line with these findings, inference of cell-cell interactions between DSRCT tumor cells and CAFs from scRNA-seq data69 identified that the most ligand-receptor interactions occurred between CAFs and mesenchymal tumor cell clusters (Figure 5C).
Figure 5.

DSRCT heterogeneity is linked to tumor spatial organization
(A) Immunofluorescent staining highlighting mesenchymal DSRCT tumor cells (DES+ and/or CHI3L1+), and CAFs (THY1+) on GR2 sample.
(B) UMAP showing GR2 3′ scRNA-seq clustering (upper panel), and violin plot (bottom panel) showing the expression of (1) tumor cell mesenchymal markers CHI3L1, DES, TNNT3, and MSLN; and (2) the CAF-specific marker THY1.
(C) Ligand-receptor interactions (CellPhoneDB) between cell clusters in the GR2 sample, representative of all specimens.
(D) GR2 site#4 sample annotated H&E-stained slide used for Visium assay, and spatial representation of spots’ clusters.
(E) Heatmap highlighting expression Z score of the top 10 DEG across spots’ clusters from GR2 site#4 Visium assay.
(F) Spatial representation of CHI3L1, TNNT3, ENO1, DES, MSLN and THY1 expression levels in GR2 site#4 sample using the Visium assay.
(G) Spatial gene signatures scores for HALLMARK_HYPOXIA, GLYCOLYSIS, OXPHOS, and EWSR1::WT1 regulon in GR2 site#4 and GR7 site#2 samples.
(H) Median Spearman correlation coefficients between gene signatures and EWSR1::WT1 regulon across Visium assays (n = 6).
We next focused on previously identified protumorigenic and immune suppressive secreted growth factors (VEGFB), cytokines (IL6/CXCL2), chemokines (CXCL12), and extracellular matrix components (COL1A1) (Figure 4E). Using NicheNet,70 we inferred the most potent ligand-receptor interactions and downstream target genes (Figures S3A–S3C). We identified several potent interactions between CAF-derived ligands and tumor or microenvironment cell receptors, including targetable CXCL12-CXCR4, VEGFB-FAT1, or IL6-IL6R axes. Noteworthy, predicted downstream genes included EGR1, as well as genes involved in response to hypoxia (e.g., HIF1A/NFE2L2), migration (e.g., COL18A1/ERBB2), and proliferation (e.g., CCND1). This suggested that immunosuppressive signals that contribute to DSRCT desmoplasia and tumor growth may be activated through interactions between mesenchymal tumor cell clusters and stromal cells.
To further explore the relationship between tumor cell states and spatial organization, we performed spatial transcriptomics on six 3′ scRNA-seq matched samples using Visium (10x Genomics) (Figures 1A and 1B; Table S1A). By applying spot clustering and DGE analysis, we consistently found that previously defined stromal regions (i.e., desmoplastic areas, tumor pseudocapsule when present on the sample, and perivascular stroma) were characterized by distinct spot clusters. In GR2 site#4 sample, in addition to the perivascular stroma cluster (i.e., Strom_endothelial cells – 9) characterized by the expression of perivascular CAF markers (e.g., THY1, PDGFRB) and endothelial cells (e.g., ESM1) (Figures 5D and 5E), several clusters were identified within desmoplastic areas, suggesting spatial heterogeneity in extracellular matrix composition (overexpression of LUM, TIMP3, and MMP9 in Strom_SFRP4/LUM – 6 cluster), M2 infiltration (overexpression of CD68 in Strom_M2 – 1 cluster), or immunosuppressive signaling (overexpression of C3 and TGFBI in Strom_C3/TGFβ1 – 7 cluster) (Figures 5D and 5E). We also systematically identified tumor clusters overexpressing transcripts encoding histones, mainly within tumor cell islets. These tumor spots were spatially distinct from the ones enriched in mesenchymal features (e.g., DES/CHI3L1/TNNT3/MSLN) and/or glycolytic features (e.g., ENO1/ENO2/LDHA), which localized either at the periphery of tumor cell islets or within the invasive front (Figures 5D, 5F, and 5G), in line with our previous findings (Figure 5A). Tumor cells with mesenchymal and/or glycolytic features were localized within hypoxia-enriched areas.
Intriguingly, we observed a spatial anticorrelation between (1) the Hypoxia and Glycolysis HALLMARK signatures and (2) EWSR1::WT1 regulon activity and, to a weaker extent, the Oxidative Phosphorylation HALLMARK signature (Figures 5G and 5H). This suggested that DSRCT tumor cells located at the islet center display the highest EWSR1::WT1 activity and proliferative capacity, possibly relying on oxidative phosphorylation, as opposed to cells located within or next to the stroma, which present mesenchymal and/or anaerobic glycolysis features that may be, at least in part, driven by desmoplasia-induced hypoxia.71 Strikingly, when assessing the expression of the desmin (DES) mesenchymal marker by IF on DSRCT spheroids, we observed that desmin staining was restricted to the spheroid periphery, whereas WT1 staining was homogeneous (Figure S3D). This preclinical finding was in line with our previous observations on patients’ samples (Figures 2G and 5A) and suggested that both EWSR1::WT1-dependent (Figures 3H and 3I) and -independent signals may influence the expression of some lineage-related markers.
Since CAFs likely play an important role in DSRCT biology (Figures 4B–4G, 5A–5C, and S3A–S3C), we further sought to preclinically assess the protumorigenic potential of CAFs, using co-culture experiments. By using fresh patient material, we generated a DSRCT patient-derived xenograft (PDX) and isolated mouse CAFs from the TME. When co-culturing the latter with JN-DSRCT-1 cells, we observed a significant 50% increase in the number and size of colonies compared with CAF-free control (Figures S3E and S3F). Similarly, the volume of JN-DSRCT-1 spheroids, which are reported to better reflect in vivo tumors,12,72,73 co-cultured with PDX-isolated CAFs was 2.6-times higher at day 8 compared to CAF-free controls (p < 0.0001; Figure S3G).
Altogether, these results suggest that DSRCT heterogeneity results from a combination of cell-intrinsic mechanisms notably related to differentiation pathways, and cell-extrinsic microenvironmental stimuli notably linked to anaerobic glycolysis.
DSRCT heterogeneity correlates with prognosis
Having explored DSRCT intratumor heterogeneity, we sought to interrogate interpatient heterogeneity and the potential link between both. We performed a deconvolution of Int_sc tumor cell clusters in our DSRCT bulk RNA-seq cohort (N = 29). All single-cell clusters, except the Neural stem – 19, could be recurrently identified in all samples (Figure 6A).
Figure 6.

DSRCT shows interpatient heterogeneity and scRNA-seq-derived gene signatures define DSRCT patients’ prognostic groups
(A) DSRCT bulk RNA-seq Int_sc clusters deconvolution.
(B) DSRCT bulk RNA-seq samples hierarchical clustering highlighting three distinct subgroups.
(C) Kaplan-Meier overall survival plot according to DSRCT bulk RNA-seq hierarchical clustering subgroups. p values are calculated using the log rank test.
(D and E) Specificity and prognostic significance of the Cycling cells – 3 (D) and epithelial-mesenchymal) – 4 (E) signature scores. The signature specificity is assessed by comparing its value in a DSRCT bulk RNA-seq dataset versus other sarcoma subtypes. The Kaplan-Meier plot shows the overall survival according to High or Low 3′ scRNA-seq derived signatures’ scores. P values are calculated using the log rank test.
Hierarchical clustering of DSRCT bulk RNA-seq data cohort identified three subgroups (Figure 6B). DGE analysis between group 3—whose marker genes were GABRA1, ZIC3 and IGF2BP1—and group 1 or 2 showed an enrichment in genes encoding histones (e.g., H3C15/HIST2H3A, H3C8/HIST1H3G), chromatin remodeling factors (e.g., CENPA, ANO9), and cell cycle activators (e.g., AURKB) (Table S9A). Genes significantly upregulated in group 2—whose most DEGs were ARX and MIR202—were enriched in (1) epithelial cell differentiation (e.g., NOG, CDH3, CCL2), cell adhesion and extracellular matrix components (e.g., CLDN18, COL26A1, EMID1), antigen binding (e.g., IGHV3-30, SLC7A5, IL7R) and lymphocyte activation (e.g., CD19, CD22) when compared with group 1; and (2) epithelial cell differentiation (e.g., KRT80, CDH1, KRT17), cell adhesion and extracellular matrix components (e.g., CLDN18, EMILIN3, COL6A5, FBLN1), cytokine activity (e.g., CCL28, CCL14, CCL18, IL10), and response to hormone and xenobiotic stimulus (e.g., ADH1B, ADH1C, FOXA1, FOS, JUN) when compared with group 3. Finally, group 1 was enriched in microRNAs involved in translation repression (e.g., MIR148B, MIR326, MIR503) when compared with group 2, with no significant GO pathway enrichment compared with group 3, suggesting limited heterogeneity among these subgroups (Table S9A). Still, these subgroups had a significant prognostic value on overall survival (OS): patients from group 3 had the worse OS (15 months [95% CI: 12-NA]), compared with groups 1 and 2 (24 and 27 months, respectively [95% CI: 16-NA for both groups]; p = 0.0081) (Figure 6C).
We next interrogated the specificity toward DSRCT and the prognostic value of each gene signature characterizing an Int_sc 3′ scRNA-seq cluster. To do so, we first assessed the expression score of each signature in bulk RNA-seq data from DSRCT or alternate sarcoma subtypes (Table S9B). Overall, signatures related to pseudostates (e.g., Cycling cells – 3) or metabolism-related states (Metabolic_glycolysis – 5) were not histotype-specific (Figure 6D; Table S9C). By contrast, signatures associated with lineage (Multidiff_ASCL1 – 0, Neural_neuronal – 1, Epithelial_mesenchymal – 4, Mesenchymal_fibrosis – 8, and Mesenchymal_secretion – 11) showed some specificity toward DSRCT as compared with non-DSRCT sarcoma histotypes (Figure 6E; Table S9C).
We subsequently assessed the prognostic significance of each signature by Kaplan-Meier analysis in the DSRCT bulk RNA-seq cohort. As a positive control, a higher Cycling cells – 3 signature (comprising TOP2A, MKI67, TYMS, and CDK1) score was significantly associated with worse outcome (p = 0.029, Figure 6D). By contrast, several signatures were significantly associated with better OS, including lineage-related clusters (Multidiff_ASCL1 – 0 [p = 0.013], Neural_neuronal – 1 [p = 0.011], Epithelial_mesenchymal – 4 [p = 0.0042, Figure 6E], and Mesenchymal_secretion – 11 [p = 0.028]), and metabolism-related clusters (Metabolic_glycolysis – 5 [p = 0.0048], Metabolic_serine – 15 [p = 0.019]) (Table S9C). We next sought to revalidate the prognostic significance of Int_sc signatures on an independent bulk RNA-seq dataset of 21 DSRCT patient samples. This confirmed the prognostic value of the Epithelial_mesenchymal – 4 signature (comprising CDH1, MUC16, GJB2, and KRT7) (p = 7e−4; Table S9C), which appeared to be the most robust prognostic signature given its stability across cohorts and signature scoring methods (see STAR methods; Table S9C).
Overall, these data highlight the clinical relevance of scRNA-seq-derived signatures and uncover a DSRCT-specific epithelial signature, which associates with improved prognosis.
Discussion
While DSRCT is uniquely driven by the aberrant EWSR1::WT1 TF and presents as monotonous cells in histopathology, this tumor shows polyphenotypic differentiation and various patient outcomes, whose underlying mechanisms remains unknown.
Here, we find DSRCT cells display some degree of heterogeneity and plasticity,74,75 driven by both tumor cell-intrinsic (e.g., EWSR1::WT1 activity) and cell-extrinsic or microenvironmental factors. We identify three main components of this heterogeneity: (1) lineage plasticity, characterized by multiple coexisting and partially overlapping differentiation phenotypes related to epithelial, mesenchymal, and neural lineages; (2) metabolic switches between oxidative phosphorylation and anaerobic glycolysis or activation of serine metabolism; and (3) pseudostates including cell cycle-related states. By integrating data from human tumors with preclinical experiments, we propose a model where both variable EWSR1::WT1 target regions and transcriptional activity—rather than EWSR1::WT1 transcript expression level12—are important components of DSRCT heterogeneity.
Our findings suggest that DSRCT cancer cells evolve along a continuum of overlapping transcriptional states, which might be permitted by the presumed pluripotency of the cell-of-origin. Whether cell state transitions are reversible or definitive is unknown and may have therapeutic implications. Indeed, scRNA-seq-derived signatures assessed in two independent bulk RNA-seq cohorts allowed us to identify an epithelial signature significantly linked to patient survival. Beyond being useful in refining patient prognosis assessment in routine practice, this may have relevance for “state-gating” approaches.76 The latter aim at specifically targeting a given critical subpopulation, to destabilize the whole tumor ecosystem and/or rewire tumor transcriptional programs toward a more favorable state.
The role of chimeric TF activity in cell reprogramming, plasticity, and dedifferentiation has been studied in other sarcoma subtypes including clear cell sarcoma,77 synovial sarcoma,78 and the prototypic small round cell Ewing sarcoma,19,20 in which similar “OXPHOS versus glycolysis” metabolic variations have been reported. Which heterogeneity mechanisms are private to each TF-driven sarcoma and which ones are shared, remains to be defined. In DSRCT, our data suggest a specific role of cell-extrinsic signals arising from CAFs and/or a hypoxic tumor microenvironment, in favoring mesenchymal and glycolytic transcriptional programs, which predominated at the tumor islets periphery.
This is, to our knowledge, the first study characterizing DSRCT CAF subpopulations. Interestingly, herein defined DSRCT CAF subclusters correlated with the recently published pan-carcinoma single-cell CAF landscape.79 By identifying overlapping markers across DSRCT CAF subpopulations with lipogenic and/or stem cell features, our results question whether peritoneal adipose tissue may constitute a favorable niche for protumorigenic CAF differentiation and DSRCT development.80 This might be of clinical relevance, notably for therapeutic strategies targeting CAFs or CAF-tumor cell interactions.
Limitations of the study
Since DSRCT is an ultra-rare disease, we could profile only 12 samples from five patients by 3′scRNA-seq and one case by snMultiome. Similarly, we had access to only one DSRCT cell line for preclinical modeling. Revalidating key findings in independent datasets and performing further functional experiments in multiple preclinical models would therefore strengthen our message and should be envisioned. Also, since the 3′-end 10X Genomics technology does neither allow to distinguish between EWSR1::WT1 and wild-type WT1, nor assess the presence of variable isoforms (e.g., breakpoint variants or +/−KTS isoforms) across single cells, we could not assess whether isoform variability modulates EWSR1::WT1 activity and contributes to intratumor heterogeneity. This could best be addressed by single-cell long-read sequencing in future studies. Finally, our study generates hypotheses on EWSR1::WT1 gene regulatory network that should be confirmed notably using functional experiments further exploring the DSRCT chromatin landscape.
Conclusion
In conclusion, our study sheds light on DSRCT intra- and intertumor heterogeneity, which may have prognostic implications for patients and help customize therapies according to specific tumor cell states in this deadly disease.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-WT1 C-terminal (GTX15249) | GeneTex | Cat# 8089496 (discontinued) |
| Rabbit monoclonal anti-Acetyl-Histone H3 (Lys9) (C5B11) | Cell Signaling Technology | Cat# 9649; RRID: AB_823528 |
| Rabbit monoclonal anti-Acetyl-Histone H3 (Lys27) (D5E4) XP® | Cell Signaling Technology | Cat# 8173; RRID: AB_10949503 |
| Rabbit monoclonal anti-Thy1/CD90 (D3V8A) | Cell Signaling Technology | Cat# 13801; RRID: AB_2798316 |
| Rabbit polyclonal anti-CHI3L1 | Abcam | Cat# ab77528; RRID: AB_2040911 |
| Mouse monoclonal anti-Desmin | Dako (Agilent) | Cat# M0760; RRID: AB_2335684 |
| Sheep polyclonal anti-FAP | R and D Systems | Cat# AF3715; RRID: AB_2102369 |
| Rabbit polyclonal anti-Sheep Immunoglobulins/HRP | Agilent | Cat# P0163; RRID: AB_2892832 |
| Mouse monoclonal anti-MCAM, clone 5C4 | Origene | Cat# TA803548; RRID: AB_2626893 |
| Mouse monoclonal anti-ACTA2, clone 1A4 | Dako | Cat# M0851; RRID: AB_262054 |
| Rabbit monoclonal anti-c-MYC (Y69) | Roche | Cat# 790-4628 |
| Rabbit polyclonal anti-WT1 | Zytomed Systems | Cat# 523–3991; RRID: AB_2864626 |
| Mouse monoclonal antibody anti-cytokeratin AE1+AE3 | Diagnostic BioSystem Clinisciences | Cat# Mob190-05 |
| Polyclonal rabbit anti-human CD3 | Dako (Agilent) | Cat# A0452; RRID: AB_2335677 |
| Rabbit monoclonal anti-CD56 (MRQ-42) | Roche | Cat# 760-4596 |
| Mouse monoclonal anti-CD163, clone 10D6 | Diagnostic BioSystem Clinisciences | Cat# Mob460-05 |
| Mouse monoclonal anti-CD68 | Dako (Agilent) | Cat# M0876; RRID: AB_2074844 |
| Rabbit polyclonal anti-goat IgG H&L (Alexa Fluor 555) | Abcam | Cat# ab150146; RRID: AB_2895679 |
| Goat polyclonal anti-mouse IgG H&L (Alexa Fluor 488) | Abcam | Cat# ab150117; RRID: AB_2688012 |
| Biological samples | ||
| Human tumor fresh and frozen samples | Gustave Roussy and Institut Curie | N/A |
| Matching germline tissue from either peripheral blood mononuclear cells (PBMCs) or non-tumoral tissue | Gustave Roussy and Institut Curie | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM/F-12 | Gibco | Cat# 11320033 |
| Penicillin-Streptomycin | Gibco | Cat# 15140122 |
| Sodium Pyruvate | Gibco | Cat# 11360070 |
| Sodium Bicarbonate | Gibco | Cat# 25080094 |
| HEPES | Gibco | Cat# 15630080 |
| Lipofectamine™ RNAiMAX | Invitrogen | Cat# 13-778-075 |
| RPMI 1640 Medium, GlutaMAX™ Supplement | Gibco | Cat# 61870010 |
| MACS Tissue Storage Solution | Miltenyi Biotec | Cat# 130-100-008 |
| Deoxyribonuclease I from bovine pancreas | Sigma-Aldrich | Cat# DN25-100MG |
| Liberase™ TL (Thermolysin Low) Research Grade | Roche | Cat# 5401020001 |
| RBC Lysis Buffer (10X) | BioLegend | Cat# 420301 |
| Tris EDTA Buffer pH 9 | Genemed | Cat# 10-0046 |
| Perm Enzyme B | 10x Genomics | Cat# PN-3000602/3000553 |
| FFPE Post-Hyb Wash Buffer | 10x Genomics | Cat# PN-2000424 |
| Probe Ligation Enzyme | 10x Genomics | Cat# PN-2000426/2000425 |
| Post Ligation Wash Buffer | 10x Genomics | Cat# PN-2000420/2000419 |
| RNase Enzyme | 10x Genomics | Cat# PN-3000605/3000593 |
| Extension Enzyme | 10x Genomics | Cat# PN-2000427 |
| Dual Index Kit TS Set A | 10x Genomics | Cat# PN-3000511 |
| TWEEN 20 | Sigma-Aldrich | Cat# P1379 |
| SPRIselect Bead-Based Reagent | Beckman Coulter | Cat# 20389900 |
| Tagment DNA Enzyme and Buffer | Illumina | Cat# 20034197 |
| KAPA Pure Beads | Roche | Cat# 07983271001 |
| cOmplete Protease Inhibitor Cocktail | Roche | Cat# 11697498001 |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 04693132001 |
| Dynabeads Protein G for Immunoprecipitation | Thermo Fisher Scientific | Cat# 10009D |
| BOND Dewax Solution | Leica Biosystems | Cat# AR9222 |
| BOND Epitope Retrieval Solution 2 | Leica Biosystems | Cat# AR9640 |
| BOND Epitope Retrieval Solution 1 | Leica Biosystems | Cat# AR9961 |
| Opal anti-mouse + rabbit HRP | Akoya | Cat# ARH1001EA |
| SPECTRAL DAPI | Akoya | Cat# FP1490 |
| ProLong Diamond Antifade Mountant with DAPI | Thermo Fisher Scientific | Cat# P36962 |
| Cell Proliferation Staining Reagent - Deep Red Fluorescence | Abcam | Cat# ab176736 |
| MEM Non-Essential Amino Acids Solution | Gibco | Cat# 11140035 |
| Critical commercial assays | ||
| Chromium Next GEM Single Cell 3′ GEM, Library & Gel Bead Kit v3.1 | 10x Genomics | Cat# PN-1000121 |
| Chromium Next GEM Single Cell Multiome ATAC + Gene Expression Reagent Bundle | 10x Genomics | Cat# PN-1000283 |
| Qiagen AllPrep DNA/RNA kit | Qiagen | Cat# 80204 |
| Visium Spatial for FFPE Gene Expression Kit | 10x Genomics | Cat# 1000336 |
| DNeasy Blood & Tissue Kit | Qiagen | Cat# 69504 |
| QIAamp DNA FFPE Tissue Kit | Qiagen | Cat# 56404 |
| SureSelectXT Human All Exon V6 | Agilent | Cat# 5190-8863 |
| MinElute Reaction Cleanup Kit | Qiagen | Cat# 28204 |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | New England Biolabs | Cat# E7645S |
| High Sensitivity DNA Kit | Agilent | Cat# 5067-4626 |
| QIAquick PCR Purification Kit | Qiagen | Cat# 28104 |
| Tumor Dissociation Kit, human | Miltenyi Biotec | Cat# 130-095-929 |
| Tumor-Associated Fibroblast Isolation mouse kit (Miltenyi Biotech | Miltenyi Biotec | Cat# 130-116-474 |
| RNeasy FFPE Kit | Qiagen | Cat# 73504 |
| Deposited data | ||
| JASPAR 2020 human transcription factor motif database | N/A | https://jaspar2020.genereg.net/ |
| HOCOMOCO v11 | N/A | https://hocomoco11.autosome.org/ |
| DSRCT tumor- and cell-based genomic data | This paper | GSE263523 |
| Experimental models: Cell lines | ||
| Human: JN-DSRCT-1 | Pr Janet Shipley (Institute of Cancer Research, London) | RRID:CVCL_9W68 |
| Mouse Cancer-Associated Fibroblasts (CAFs) from PDX | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Mouse patient-derived xenograft (PDX) from a Nod SCID Gamma (NSG, Charles River) mouse | This paper | N/A |
| Oligonucleotides | ||
| siRNA EWSR1::WT1 (3′ GAT CTT GAT CTA GGT GAG A 5′) | This paper | N/A |
| siRNA CCND1 | Horizon Discovery « on » -TARGETplus Human CCND1 siRNA-smart pool | Cat# 003210-00-0005 |
| siRNA control non-targeting | Horizon Discovery « on » -TARGETplus Non-targeting siRNA#1 | Cat# D-001810-01-05 |
| Software and algorithms | ||
| R (v3.5.1, v4.3.0) | N/A | https://cran.r-project.org |
| Seurat (v3.1.4, v4.0.4, v4.1) | N/A | https://satijalab.org/seurat |
| Harmony (v1.0) | Korsunsky et al.80 | https://github.com/immunogenomics/harmony |
| gprofiler2 (v0.2.2) | N/A | https://cran.r-project.org/package=gprofiler2 |
| Hotspot (v1.0) | DeTomaso et al.29 | http://www.github.com/yoseflab/Hotspot |
| CellPhoneDB (v3.0.0) | Efremova et al.69 | https://github.com/Teichlab/cellphonedb |
| NicheNet (v1.1.1) | Browaeys et al.70 | https://github.com/saeyslab/nichenetr |
| inferCNV (v1.1.0) | Trinity CTAT Project | https://github.com/broadinstitute/infercnv |
| CytoTRACE (v0.3.3) | Gulati et al.31 | https://cytotrace.stanford.edu/ |
| RaceID (v0.2.6) | Grün et al.32 | https://github.com/dgrun/StemID |
| Velocyto (v0.17.16) | La Manno et al.81 | https://github.com/velocyto-team/velocyto.R |
| scVelo (v0.2.3) | Bergen et al.82 | https://github.com/theislab/scvelo |
| SCENIC+ (v0.1.dev447+gd4fd733) | González-Blas et al.83 | https://github.com/aertslab/scenicplus |
| Azimuth (v0.5.0) | Hao et al.26 | https://satijalab.github.io/azimuth/articles/run_azimuth_tutorial.html |
| MoMac-VERSE | Mulder et al.46 | https://macroverse.gustaveroussy.fr/2021_MoMac_VERSE/ |
| Cell Ranger ARC (v2.0.0) | 10x Genomics | https://support.10xgenomics.com/single-cell-multiome-atac-gex/software/overview/welcome |
| Signac (v1.5.0) | N/A | https://stuartlab.org/signac/ |
| ChromVAR (v1.23.0) | N/A | https://bioconductor.org/packages/release/bioc/html/chromVAR.html |
| STAR | N/A | https://github.com/alexdobin/STAR |
| Atropos | N/A | https://github.com/jdidion/atropos |
| Arriba | N/A | https://github.com/suhrig/arriba |
| nf-core/rnaseq (v3.3) | N/A | https://github.com/nf-core/rnaseq |
| DESeq2 | N/A | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| CIBERSORTx | Newman et al.24 | https://cibersortx.stanford.edu/ |
| Survminer (v0.4.9) | N/A | https://github.com/kassambara/survminer/ |
| AUCell (v 1.22.0) | Aibar et al.84 | https://bioconductor.org/packages/release/bioc/html/AUCell.html |
| Fiji ImageJ (v 2.1.0) | N/A | https://imagej.net/imagej-wiki-static/Fiji/Downloads |
| Space Ranger (v2.0.0) | 10x Genomics | https://www.10xgenomics.com/support/software/space-ranger/downloads/space-ranger-installation |
| Loupe Browser (v6.3.0) | 10x Genomics | https://www.10xgenomics.com/support/software/loupe-browser/latest |
| nf-core/sarek (v3.0.2) | N/A | https://github.com/nf-core/sarek |
| BWA | N/A | https://github.com/lh3/bwa |
| CNVkit | Talevich et al.85 | https://github.com/etal/cnvkit |
| nfcore/atacseq (v1.2.1) | Ewels et al.86 | https://github.com/nf-core/atacseq |
| FastQC | N/A | https://github.com/s-andrews/FastQC |
| Trim Galore | N/A | https://github.com/FelixKrueger/TrimGalore |
| Picard | N/A | https://github.com/broadinstitute/picard |
| BEDTools | N/A | https://github.com/arq5x/bedtools2 |
| Integrative Genomics Viewer (IGV) (v2.16.0) | N/A | https://software.broadinstitute.org/software/igv |
| deepTools | N/A | https://github.com/deeptools/deepTools |
| MACS2 | N/A | https://github.com/macs3-project/MACS |
| HOMER | N/A | http://homer.ucsd.edu/homer/ |
| MEME suite (v 1.8.0) | N/A | https://github.com/cinquin/MEME |
| nfcore/chipseq (v1.2.2) | Ewels et al.86 | https://github.com/nf-core/chipseq |
| ToppGene Suite | N/A | https://toppgene.cchmc.org/ |
| ChIP-Enrich (v 2.26.0) | Welch et al.87 | https://bioconductor.org/packages/release/bioc/html/chipenrich.html |
| OlyVIA (v2.9) | Olympus | https://www.olympus-lifescience.com/en/downloads/detail-iframe/?0[downloads][id] = 847249644 |
| QuPath (v0.3.0) | N/A | https://qupath.github.io/ |
| ImageJ (v1.53) | N/A | https://imagej.net/ij/download.html |
| Cell Ranger (v.3.0.2) | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/installation |
| Other | ||
| VS120 Virtual Slide Microscope | Olympus | Cat# VS120-S6-W |
| Qiagen TissueLyser II | Qiagen | N/A (discontinued) |
| NovaSeq 6000 sequencer | Illumina | N/A |
| S220 Focused-ultrasonicator | Covaris | Cat# 500217 |
| BOND RX Fully Automated Research Stainer | Leica Biosystems | Cat# 21.2821 |
| Invitrogen EVOS XL Core Imaging System | Thermo Fisher Scientific | Cat# 15339661 |
| ECLIPSE Ti2 | Nikon | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sophie Postel-Vinay (sophie.postel-vinay@gustaveroussy.fr).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Single-cell omics data (including scRNA-seq, snMultiome, and Visium assays) and DSRCT frozen tumor RNA-seq are publicly available on Gene Expression Omnibus (GEO) repository in count matrix format with the following accession number: GSE263523. Cell-line-based omics data (RNA-seq, ATAC-seq, ChIP-seq) are available processed and in fastq format using the same unique accession number, GSE263523.
-
•
Patient raw data reported in this study cannot be deposited in a public repository because of privacy concerns.
-
•
This paper does not report original code. All custom code used for the analyses was written with existing software as detailed in the STAR Methods section and is available upon request.
-
•
Any additional information required to reanalyze the data reported in this manuscript is available from the lead contact upon request.
Experimental model and study participant details
Human subjects and ethical considerations
This study was performed in accordance with European General Data Protection Regulation (GDPR) following Regulation (EU) 2016/679 of the European Parliament and of the Council of April 27, 2016.
Patients with DSRCT treated at Gustave Roussy and Institut Curie alive at the time of analysis gave their preoperative informed consent to allow the use of tumor residual samples for scientific purposes. The clinical characteristics of patients involved in this study are summarized in Table S1.
Cell line models
JN-DSRCT-1 cell line is a kind gift from Professor Janet Shipley (The Institute of Cancer Research, London).
JN-DSRCT-1 cell line was maintained in 2D adherent culture within DMEM/F-12 (Gibco) supplemented with 10% FBS, 1% Penicillin-Streptomycin (Gibco), 1% Sodium Pyruvate (Gibco), 1% Sodium Bicarbonate (Gibco), 1% Non-Essential Amino Acids (Gibco) and 1% HEPES (Gibco). Cell passaging was performed at 1/10 twice a week. Used cells were controlled for mycoplasma-free status.
Method details
Spheroid formation
JN-DSRCT-1 cells were grown as spheroids in 96-well ultra-low attachment cell culture plates in supplemented DMEM/F-12 medium. DSRCT cells were first seeded at a concentration of 1,000 cells per well before 15 min at 500 g centrifugation. The formation of the spheroid was assessed 24 h after the cells’ seeding.
Small interfering RNA knock-down
JN-DSRCT-1 cell line was transfected with a custom small interfering RNA (siRNA) targeting EWSR1::WT1 (3′ GAT CTT GAT CTA GGT GAG A 5′), CCND1 (Horizon Discovery « on » -TARGETplus Human CCND1 siRNA-smart pool, ref. L-003210–00-0005) or with a non-targeting siRNA (Horizon Discovery « on » -TARGETplus Non-targeting siRNA#1, ref. D-001810-01-05), according to manufacturer’s instructions. After cell seeding and obtention of 50% confluency, transfection was performed using Lipofectamine RNAiMAX (Invitrogen ref. 13778150), and the medium was replaced the day after. A 48-h silencing time point was used for each described experiment.
We performed western blotting with EWSR1 N-ter (C-9 clone, sc-48404), CCND1 (EPR224L clone, ab134175), and β-actin (BA3R clone, MA5-15739-HRP) antibodies to control EWSR1::WT1, CCND1 and ACTB expression upon EWSR1::WT1 siRNA-mediated silencing of the JN-DSRCT-1 cell line for the bulk RNA-seq and ATAC-seq experiments.
Human tumor samples 3′ single-cell RNA-sequencing
Tumor dissociation and scRNA-seq
Fresh tumor material from patients with DSRCT was profiled using the 3′-end single-cell RNA-sequencing (3′ scRNA-seq) 10x Genomics Chromium assay.
Briefly, fresh DSRCT tumor and peritumor material was collected into RPMI 1640 medium with GlutaMAX Supplement (GibcoTM, ref. 61870010) or MACS Tissue Storage Solution (Miltenyi Biotec, ref. 130-100-008) with a delay of less than 30 min after surgery. Tissue samples were further cut into small chunks and incubated at 37°C in RPMI medium containing DNAse I (Sigma, reference DN25-100MG) at 2.2 μg/mL (final concentration) and Liberase Thermolysin Low (TL) (Roche, ref. 5401020001) at 4.5 μg/mL (final concentration) during 20–30 min. After tissue dissociation, the mixture was washed in 1x PBS +0.04% BSA before centrifugation. To obtain a single-cell suspension, cells resuspended in 1x PBS +0.04% BSA were filtered twice with a 70 μm and 30 μm cell strainer. After another round of centrifugation, cells were resuspended in 1x PBS +0.04% BSA. When necessary, red blood cell lysis was performed with a 2 to 3 min incubation in 1X Red Blood Cell Lysis buffer (BioLegend, ref. 420301) protected from light before washing in 10 mL PBS and performing a last round of centrifugation.
Cell concentration and viability were controlled with an automated cell counter using trypan blue, and cells were loaded into the 10x Genomics cassette for a targeted cell recovery of 5,000 cells per sample following the recommendations from the manufacturer’s protocol.
Cell encapsulation, reverse transcription, and library generation were performed according to 10x Genomics standard protocols.81
Paired-end sequencing was performed on the Illumina NovaSeq 6000 sequencer for a targeted depth of 400 million reads per sample.
Data analysis
Single cell RNA-seq raw base call (BCL) files were demultiplexed and converted into FASTQ files by using the 10X Genomics Cell Ranger pipeline (v3.0.2) “mkfastq” command. FASTQ files were then processed with the Cell Ranger “count” command to perform quality control, barcode processing, and single-cell gene counting. Sequencing reads were aligned to the GRCh38 human reference genome (v3.0.0 Cell Ranger index). ScRNA-seq data were analyzed using the R package Seurat (R v3.5.1). Regularized negative binomial regression-based normalization was performed using sctransform.82 Cell subpopulations were clustered using a k-nearest neighbors graph method based on the Euclidean distance on PCA, followed by Louvain algorithm optimization. Visualization and exploration of the data were obtained with the Uniform Manifold Approximation and Projection (UMAP) non-linear dimensional reduction technique in Seurat.
Samples from synchronous distinct localizations were combined by merging the raw count matrices of individual Seurat objects. The integration of samples stemming from different patients and/or sampling timepoint was performed using the Harmony83 algorithm (https://github.com/immunogenomics/harmony).
Differential gene expression analysis was performed using the Mann-Whitney-Wilcoxon test. We identified the most differentially expressed genes of each cluster compared to the background, based on adjusted p value (<5%) and ranked them according to logarithmic fold change, and searched for enrichment of Gene Ontology (GO) terms to characterize each subpopulation. We performed GSEA using GO pathways (GO Biological Process, GOBP; GO Cellular Component, GOCC; and GO Molecular Function, GOMF) on the top 100 differentially expressed genes in each cluster (adjusted p-value <5%) using the R package gprofiler2.
Lastly, cluster-specific signatures were defined as the top 100 overexpressed genes from each predefined cluster and were further used for signatures’ scoring in bulk RNA-seq data (see below).
DSRCT neotranscripts expression
To quantify single-cell expression of DSRCT-specific neotranscripts, we ran CellRanger “count” using a custom index built by appending sequences of the neotranscripts to the reference transcriptome. Counts for neotranscripts were log-normalized, and the average log-normalized expression level was plotted with FeaturePlot.
Hotspot
Hotspot28 (http://www.github.com/yoseflab/Hotspot) was used to identify informative gene modules across clusters defined on the Harmony-integrated Seurat object (“Int_sc” clusters).
Hotspot is an algorithm that computes gene modules in three steps: finding informative genes with high local autocorrelation, evaluating the pairwise correlation between these genes, and clustering the results in a gene-gene affinity matrix. The Hotspot depth-adjusted negative binomial model was run using the count matrix and the 50 first principal components. A K-nearest neighbors (KNN) graph was then calculated using 30 neighbors, and the 500 genes with the highest significant autocorrelation (false discovery rate <0.05) were selected. Pairwise local correlation between these genes was computed, and gene modules were created by agglomerative clustering with a minimum number of genes per module set to 15 and a false discovery rate threshold of 0.05. Hotspot module scores for each cell were calculated by first centering the UMIs using the depth-adjusted negative binomial model. The centered values were then smoothed using the weighted average of their 30 nearest neighbors. These smoothed values were then modeled with PCA using the first principal component, and the cell loadings were reported as the module scores.
CellPhoneDB
To infer cell-cell communication between identified tumor and microenvironment cell clusters, we took advantage of CellPhoneDB69 (https://github.com/Teichlab/cellphonedb), an algorithm developed to investigate cellular crosstalk from a curated repository of interacting ligands and receptors.
NicheNet
We applied the NicheNet70 computational method (https://github.com/saeyslab/nichenetr) to further interrogate cell-cell interactions to DSRCT tumor cells, immune cells, and cancer-associated fibroblasts. NicheNet presents the advantage of computing the activity of ligand-receptor interactions by inferring their gene regulatory network at the single-cell level.
InferCNV
An inference of single cells’ copy number variations (CNV) was performed using InferCNV (https://github.com/broadinstitute/infercnv). Raw gene expression data were first extracted from each patient-integrated Seurat object. For each sample, normal reference cells were selected based on the expression of immune cells and/or cancer-associated fibroblast markers (Table S2). Tumor cells were grouped according to annotated Louvain clusters as defined above. The cutoff for the minimum average read count per gene was set to 0.1, as recommended for 10x data. The 'cluster_by_groups' setting was used to perform separate clustering for each cluster as defined in the cell annotations file. All other options were set to their default values. Each CNV was annotated as a gain or a loss to a p- or q-arm using the GRCh38 reference genome.
CytoTRACE
We used CytoTRACE30 to infer the degree of differentiation of DSRCT tumor cells using CytoTRACE R package version 0.1.0. CytoTRACE (Cellular (Cyto) Trajectory Reconstruction Analysis using gene Counts and Expression) aims at predicting the differentiation state of cells from scRNA-seq data by leveraging the number of detectably expressed genes per cell. Subsampling of 1,000 cells was used to run the CytoTRACE function as described in https://cytotrace.stanford.edu/. CytoTRACE results were then visualized on UMAPs colored according to the inferred degree of differentiation.
StemID
The cells’ degree of differentiation was predicted using StemID,31 which measures intracellular entropy based on cells’ median transcriptome entropy. The StemID tool was used as described in https://github.com/dgrun/StemID/blob/master/Reference_manual_RaceID2_StemID.pdf. Briefly, StemID relies on the concept that the multiplicity of states coexisting within a single cell is reflected by the uniformity of the transcriptome, approximated by Shannon’s entropy. StemID computes a StemID score, which reflects the level of multipotency.
CellRank
Spliced and unspliced read counts were generated using velocyto (v0.17.16) “run10x” function.88 Spliced and unspliced reads were then filtered and normalized using scVelo (v0.2.3)84 “filter_and_normalize” function [min_shared_counts = 20, n_top_genes = 2000]. A K-nearest-neighbor graph was built using 30 nearest neighbors on the 30 first principal components. RNA velocity scores were calculated using the scVelo “dynamical” model of transcriptional dynamics. Single-cell velocities were then projected onto UMAP embeddings. Cell-to-cell transition probabilities were calculated using the CellRank velocity kernel.29 Using scVelo dedicated functions, we could then compute predicted initial and terminal states, latent time, lineage probabilities, and driver genes, as well as a directed PAGA (partition-based graph abstraction) model.
SCENIC+
SCENIC+46 is a recent development of the SCENIC tool that takes advantage of multiomic data. It predicts genomic enhancers along with candidate upstream transcription factors (TF) and links these enhancers to candidate target genes. Specific TFs for each cell type or cell state are thus predicted based on the concordance of TF binding site accessibility, TF expression, and target gene expression as contained in multiomic data (scRNA-seq and scATAC-seq). We used SCENIC+ (v0.1.dev447+gd4fd733) for implementation.
Cell atlases used for label transfer
We used Azimuth26 to perform label transfer from Cao et al.25 fetal development cells atlas - which consists of 378,000 subsampled human fetal cells - on our generated scRNA-seq dataset. Results are shown for “l1” predicted annotations.
In addition, MoMac-VERSE atlas47 was used to deconvolute myeloid cell subpopulations. Briefly, MoMac-VERSE unifies dendritic cells (DCs), monocyte, and macrophage subpopulations across human tissues.
EWSR1::WT1 targeted scRNA-seq
Traditional 10x Genomics 3′ scRNA-seq data do not allow to explore this due to: (i) the potential confounding expression of WT1 wild-type – although WT1 wild type is supposedly repressed in DSRCT – which would be indistinguishable from EWSR1::WT1 since the 3′ reads are too short to cover the fusion breakpoint; and (ii) the insufficient sequencing depth for detection of lowly expressed transcripts (so-called “drop-out” phenomenon). To overcome this limitation, we designed an in-house assay to evaluate the expression level of EWSR1::WT1 transcripts at the single-cell level. The leftover barcoded cDNA libraries from the 10x Genomics protocol were used to selectively enrich for cDNA originating from the EWSR1::WT1 transcript. Briefly, a first polymerase chain reaction (PCR1) was done using either (i) an EWSR1::WT1 breakpoint specific biotinylated primer or (ii) a WT1 full-length specific biotinylated primer and a universal Read 1 specific primer (sequence added on the transcript during reverse transcription in the 10X Genomics protocol). The resulting PCR product was purified using Streptavidin beads. A second PCR (PCR2) using PCR1 as a matrix was done using a primer specific to the WT1 C-terminal end located at about 450 base pairs (bp) from the transcript end. The second primer was the same as the second one used in PCR1. The PCR2 product was purified and used as a matrix for a third PCR (PCR3) to add the P5 and P7 adapters using primer overhangs to construct an Illumina sequencing-compatible library.
The downstream bioinformatics analysis of EWSR1::WT1 targeted scRNA-seq relied on a negative selection method to consider potential unspecific amplification of WT1 full length with the EWSR1::WT1 targeted scRNA-seq assay.
Human tumor sample single-nuclei 3′ RNA and ATAC sequencing (snMultiome)
Tumor dissociation, nuclei isolation, and permeabilization
Fresh tumor material from one patient with DSRCT was profiled using the single-cell Multiome ATAC + Gene Expression 10x Genomics Chromium assay.
The fresh DSRCT tumor specimen was collected into 4°C MACS Tissue Storage Solution with a delay of less than 30 min after surgery. The tissue sample was then cut into small chunks before proceeding to nuclei isolation and nuclei permeabilization according to 10x Genomics Demonstrated Protocol for Nuclei Isolation from Complex Tissues for Single Cell Multiome ATAC + GEX Sequencing (CG000375 Rev B). Briefly, the tumor small chunks were incubated on ice in NP40 lysis buffer (Tris-HCl (pH 7.4) 10mM, NaCl 10mM, MgCl2 3mM, Nonidet P40 0.1%, DTT 1mM, RNase inhibitor 1 U/μL, in Nuclease-free Water) during 5 min. The suspension was then passed through a 70 μm strainer into a 2 mL Eppendorf tube before 500g centrifugation for 5 min at 4°C. The supernatant was removed, and the nuclei pellet was washed in 1 mL of PBS supplemented with 1% BSA, 1 U/μL RNAse inhibitor on ice for 5 min. After an additional 4°C centrifugation at 500g for 5 min, nuclei were resuspended in 1 mL PBS with 1% BSA and 1 U/mL RNAse inhibitor and counted using a Malassez counting chamber on Invitrogen EVOS XL imaging system after DAPI staining to confirm complete nuclei isolation from total cells and assess nuclei concentration. We further achieved nuclei permeabilization by performing an additional 5-min 500 g centrifugation at 4°C and incubating the nuclei pellet in 0.1X Lysis Buffer (1X Lysis Buffer: Tris-HCl (pH 7.4) 10 mM, NaCl 10 mM, MgCl2 3 mM, Tween 20 0.1%, Nonidet P40 0.1%, Digitonin 0.01%, BSA 1%, DTT 1 mM, RNase inhibitor 1 U/μL in Nuclease-free Water, diluted into Lysis Dilution Buffer: Tris-HCl (pH 7.4) 10 mM, NaCl 10 mM, MgCl2 3 mM, BSA 1%, DTT 1mM, RNase inhibitor 1 U/μL in Nuclease-free Water) on ice during 2 min. After washing the pellet in wash buffer (Tris-HCl (pH 7.4) 10mM, NaCl 10mM, MgCl2 3 mL, BSA 1%, Tween 20 0.1%, DTT 1mM, RNase inhibitor 1 U/μL, in Nuclease-free Water), the nuclei were resuspended in the appropriate volume of chilled Diluted Nuclei buffer (Nuclei Buffer (20X), DTT 1 mM, RNase inhibitor 1 U/mL in Nuclease-free Water) and counted using both a Malassez counting chamber and a Bio-Rad automated cell counter.
Chromatin transposition, ATAC, and RNA libraries construction
Next, the appropriate volume of cell nuclei suspension was extracted to target the effective encapsulation of 5,000 nuclei. Further steps were performed following the 10x Genomics Chromium Next GEM Single Cell Multiome ATAC + Gene Expression User Guide (CG000338).
Briefly, the transposition of the native chromatin was performed using 10x Genomics Chromium ATAC Enzyme B, which contains the transposase, and adapter sequences were simultaneously added to the ends of the DNA fragments. Gel beads-in-EMulsion (GEMs) generation and ATAC and Gene Expression libraries were constructed according to the 10x standard protocol.
Sequencing
The obtained ATAC and RNA libraries were sequenced on NovaSeq 150 base pairs with paired-end flow cells for a targeted depth of 400 million reads for the gene expression library and 500 million reads for the ATAC library.
Data analysis
Demultiplexing reads alignment, filtering, ATAC peak calling, and generation of feature-barcode matrices were done using Cell Ranger ARC.
We relied on Seurat v4.0.4 and Signac v1.5.0 packages for further downstream analyses. First, genome annotation was performed using the hg38 EnsDb—Hsapiens.v86 reference genome. Low-quality nuclei were then filtered out before peak calling on either the pseudo-bulk data or separately on each further defined cell clusters using MACS2. The number of counts per peak, features, and UMIs were then calculated before normalization using the sctransform method, which allows normalization and variance stabilization of molecular count data from sc and snRNA/ATAC-seq experiments. Nuclei clustering was then performed either based on RNA features, ATAC peaks features, or both features using the weighted nearest neighbors (WNN) method.26 Single-nuclei data and RNA/ATAC/WNN-derived clusters were visualized using Uniform Manifold Approximation and Projection (UMAP). Downstream analyses comprised differential gene expression and differential chromatin accessibility analyses, and motif enrichment analyses.
Motif enrichment analyses relied on the JASPAR 2020 human transcription factor motifs database using the ChromVAR pipeline, which calculates per cell motif activity Z-scores.
We performed 3′ scRNA-seq Int_sc dataset label transfer onto the snMultiome dataset. To increase the comparability of 3′ scRNA-seq Int_sc clusters and WNN snMultiome, we performed label transfer using exclusively Int_sc intronic reads, which derive from unprocessed nuclear transcripts and likely better reflect single-nuclei transcriptional profiles.
Human tumor samples bulk RNA sequencing
Samples collection and RNA sequencing
DSRCT frozen specimens collected and archived at Gustave Roussy Biological Resource Center from December 1991 to April 2021 constitute the DSRCT test cohort analyzed in this study.
According to applicable law, alive patients whose samples were to be used had to provide a non-opposition form to allow the use of their archived biological samples.
An external cohort from Institut Curie’s Unité de Génétique Somatique (UGS) comprising DSRCT RNA-seq data and additional selected soft tissue sarcomas RNA-seq data was used.
Ribbon sections of frozen samples were cut using a cryostat microtome after embedding within O.C.T. Next, frozen tissue was lysed using Qiagen TissueLizer II with 3 mm tungsten carbide beads according to the manufacturer's instructions.
RNA extraction was performed on tumor samples using the Qiagen AllPrep DNA/RNA kit standard protocol.
After RNA Integrity Number (RIN) quality control, RNA samples were used to construct total RNA sequencing libraries. Samples were sequenced on a NovaSeq sequencer with 150 base paired-end reads with a targeted depth of 30 million reads per sample.
Data analysis
The RNA sequencing data were analyzed according to nf-core/rnaseq pipeline. Raw reads quality control (QC) was performed using FastQC. Pseudoalignment was performed with Salmon on hg19 (GENCODE version 19). RNA counts were quantified using Salmon after adaptor trimming with Trim Galore!. Fusion transcripts prediction was performed with Arriba. Only the major EWSR1::WT1 fusion transcripts were reported.
After filtering out tumor samples outliers and/or low tumor cellularity samples (<5%) according to the corresponding pathology slide, hierarchical clustering was performed among Gustave Roussy (GR) DSRCT samples (test set) using Pearson’s correlation coefficient with Ward D2 linkage algorithm on the set of all sequenced genes.
Differential Gene Expression (DGE) analysis was performed using DESeq2 to compare transcriptomic profiles from DSRCT samples to those of a cohort of various STS subtypes.
Deconvolution of cell type composition was performed using CIBERSORTx24 (https://cibersortx.stanford.edu/) tool based on scRNA-seq clustering for cell types and malignant cell clusters. CIBERTSORTx uses scRNA-seq data as a reference to dissect cell-type-specific gene expression profiles in bulk datasets, thereby providing a more accurate inference of cell type abundances than traditional deconvolution methods using a fixed gene signature matrix.
To deconvolve cluster-specific cell subsets from DSRCT tumors bulk RNA-seq data, CIBERSORTx was used to derive a signature matrix from scRNA-seq data. Using the Seurat subset function, 500 or 1,000 cells were extracted from each cluster from the Harmony-integrated 3′ scRNA-seq dataset (Int_sc). Cluster-labeled cells served to obtain a single-cell reference matrix (scREF-matrix) used as an input on CIBERSORTx online server using the “Custom” option. Default values for replicates (n = 5), sampling (0.5), and Min. Expression (0.0) were used. Additional options for kappa (999), q-value (0.01), and number of barcode genes (300–500) were kept at default values. The imputation of cell subtypes fractions defined on the scREF-matrix was performed on transcripts per million (TPM) values from DSRCT tumors bulk RNA-seq data using the “Impute Cell Fractions” function with “Custom” option and ran in absolute mode. A total number of 500 permutations was performed to test for statistical significance.
Prognostic signatures generation and signature scores calculation
The top 100 upregulated genes within each Harmony-integrated sample cluster (“Int_sc” clusters) defined by Seurat graph-based clustering using k-NN (K-nearest neighbor) method were selected to define cluster-specific signatures. A score for each cluster-specific signature was then calculated from bulk RNA-seq data of the test cohort (n = 29) using two strategies: (i) the geometric mean of each gene expression expressed in transcripts per million (TPM), and (ii) the arithmetic mean of Z-scores of each gene expression expressed in variance-stabilizing transformed (VST) raw counts. First, to assess specificity of gene signatures for DSRCT, ordinary one-way ANOVA followed by Tukey’s multiple comparisons test was performed on signature values in DSRCT versus all 23 other subtypes of sarcomas. As every signature was significant using the ANOVA test (p < 0.05), Tukey’s multiple comparisons test was performed for all signatures. A number of significant p-values (p < 0.05) equal to or more than 18/23 (>75%) of comparisons was chosen as the threshold to call a signature « DSRCT-specific ».
To further evaluate the prognostic value of each signature, the DSRCT samples bulk RNA-seq dataset was segregated into “High” and “Low” signature scores using the optimal cutpoint method (“survminer” R package). We then performed a survival analysis for patients from whom the tumor samples were collected to compare “High” versus “Low” signature scores using the Kaplan-Meier model and tested for significance with the log rank test. Subsequently, we validated our findings on an external validation cohort comprising 21 RNA-seq samples from frozen DSRCT patient tumors.
EWSR1::WT1 regulon
We defined a de novo EWSR1::WT1 regulon, defined by a list of genes and corresponding genomic regions, which are specific to EWSR1::WT1 TF activity using bulk RNA-seq data on JN-DSRCT-1 with or without EWSR1::WT1 silencing, and EWSR1::WT1 ChIP-seq data.
To ensure its specificity, we intersected genes significantly downregulated upon EWSR1::WT1 silencing (please refer to JN-DSRCT-1 cell line bulk RNA sequencing section below) and genes corresponding to enriched peaks in EWSR1::WT1 ChIP-seq (please refer to JN-DSRCT-1 cell line Assay for Transposase-Accessible Chromatin using sequencing section below) with abs(logFC) > 2 and p < 0.01 for both datasets. EWSR1::WT1 regulon was used to calculate single cells’ scores using. AUCell89 in 3′ scRNA-seq and snMultiome.
Spatial transcriptomics with 10x Genomics Visium assay
Sample and library preparation
Assessment of RNA quality was first performed on selected DSRCT FFPE samples. According to the manufacturer’s instructions, ten sections of 10 μm of FFPE tissue were used for RNA isolation using the RNeasy FFPE kit (Qiagen). Samples with DV200 (percentage of RNA fragments longer than 200 nucleotides) > 30% were selected and used for further Visium spatial gene expression sequencing.
Using the human whole transcriptome probe set, spatial transcriptomics was performed with 10x Genomics Visium technology according to the Visium Spatial Gene expression for FFPE samples protocol.
A representative tissue area of 6.5 × 6.5 mm was previously selected on H&E (hematoxylin and eosin) slides.
The latter slide contained four capture areas, each composed of an array of ∼5,000 circular spots containing printed DNA oligos for mRNA capture, each composed of a PCR handle, a unique spatial barcode, a unique molecular identifier (UMI), and a poly-dT-VN tail. The resolution of 10x Visium spatial transcriptomics enabled to capture mRNA from 10 to 20 cells in each single 55 μm circular spot.
Five-μm sections of 3′ scRNA-seq matched DSRCT tumors FFPE samples were cut after dehydration and placed on 10x Genomics Visium Gene Expression slides as recommended. Visium slides were then incubated in a section dryer oven and kept in a desiccator at room temperature overnight. According to the 10x Visium general protocol, deparaffinization was performed within the next day by placing slides in a section dryer oven at 60°C for 2 h, followed by successive baths in xylene and ethanol gradient concentrations. Next, H&E staining and coverslipping were performed, and Visium slides were further imaged at 10× magnification using the Olympus VS120. Images were processed using Fiji ImageJ software.
After coverslip removal, slides were placed into the Visium cassette. Decrosslinking was performed by incubating Visium slides with Tris EDTA (TE) buffer (Ready-to-use, Genemed, Gentaur) in a thermocycler at 70°C for 60 min. A pre-hybridization step with Perm Enzyme B (10X Genomics, PN-3000602/3000553) and Tween 20 incubation for 15 min at room temperature was followed by the hybridization of the human whole transcriptome probe set panel on complementary target RNA transcripts on tissue sections. Briefly, this panel consists of a pair of specific probes for each targeted gene which contains Read2S and polyA sequences. After overnight incubation, several washes with FFPE Post-Hyb Wash Buffer (10x Genomics, PN-2000424) were performed, followed by the addition of a ligation enzyme (10x Genomics, PN-2000426/2000425) and incubation in a thermocycler at 37°C for 60 min to seal the junction of probes-RNA transcripts pairs. Probes’ ligation was followed by several washes with Post Ligation Wash Buffer (10x Genomics, PN-2000420/2000419). Finally, RNA digestion was performed to allow probes’ release. The latter step included samples’ incubation with RNase enzyme (10x Genomics, PN-3000605/3000593) at 37°C for 30 min and permeabilization with Perm Enzyme B for 40 min at 37°C to release the probes, which were then captured on the Visium slides surface. Probes’ extension was performed by incubating samples for 15 min at 45°C with the Extension enzyme (10X Genomics, PN-2000427) before elution using KOH 0.08M and 1M Tris-HCl to stabilize the reaction. Libraries were right after generated by adding to each sample 50 μL of Amp Mix (10X Genomics, PN-2000047) and 5 μL of dual index Kit/plate TS Set A well ID (10X Genomics, PN-3000511) and further amplified using the number of cycles determined by qPCR before libraries construction. Samples were then cleaned-up using SPRIselect reagent (Beckman Coulter) and stored in EB buffer at −20°C until sequencing. Quality and quantification of cDNA libraries were assessed using BioAnalyzer before sequencing.
Generated Visium libraries were pooled and loaded in a single SP Illumina flow cell. Sequencing was performed on NovaSeq PE 50 at a sequencing depth of 50k read pairs per spot covered with tissue using 10x Genomics recommended run parameters.
Data analysis
Space Ranger was used to perform the demultiplexing of Visium-prepared raw base call files generated by Illumina sequencers into FASTQ files using spaceranger mkfastq. Next, spaceranger count was used to perform tissue and fiducial alignment from the microscope slide image and barcode/UMI counting to generate feature-barcode matrices that were further used for downstream analyses. The number of spots covered with tissue was calculated using Loupe Browser. Overall, tissue sections from DSRCT samples encompassed a total of 4,236, 3,452, 3,583, 3,214, 3,537, and 3,686 spots containing included barcodes on the capture area for GR2, GR4, GR4_PC, IC1, GR7, and GR11 samples respectively.
Downstream analyses relied on the Seurat package (v4.1) and included spots’ clustering (k-nearest neighbors graph method with Louvain algorithm optimization), differential gene expression analysis, and spatially variable features’ exploration. We used an anchor-based label transfer workflow85 using 3′ scRNA-seq matching samples identified clusters as a reference. To account for spatially resolved pathway activations, we calculated the average expression levels of selected hallmark gene signatures substracted by the aggregated expression of control gene sets using the AddModuleScore function from Seurat.
Whole exome sequencing
Sample preparation
DSRCT tumor frozen samples were collected for whole exome sequencing (WES), corresponding to those used for 3′ scRNA-seq, along with matching germline tissue from either peripheral blood mononuclear cells (PBMCs) or non-tumoral tissue. DNA extraction was performed using Qiagen DNeasy Blood and Tissue Kit or QIAamp DNA FFPE Tissue Kit for non-tumoral FFPE samples when patient-derived PBMCs could not be obtained.
Sequencing
After BioAnalyzer quality control, genomic DNA was sheared, and exons were captured by the Agilent SureSelect Human All Exon V6 kit. Sequencing libraries were prepared and sequenced with paired-end sequencing (150bp) on an Illumina NovaSeq 6000 sequencer with 40 million reads per sample.
Data processing
WES data were processed using the nf-core/sarek pipeline (https://github.com/nf-core/sarek). Following alignment to GRCh38 with BWA, allowing up to 4% of mismatches, BAM files were cleaned according to the Genome Analysis Toolkit (GATK) recommendations, namely duplicate marking and base quality score recalibration.
Downstream analyses
Copy number variation between normal and matched tumor tissue was computed using CNVkit90 (https://github.com/etal/cnvkit).
JN-DSRCT-1 cell line bulk RNA sequencing
After outlier sample removal, differential gene expression analysis between H48 EWSR1::WT1-silenced and non-silenced conditions was performed using DESeq2.
Sample preparation
JN-DSRCT-1 cells were silenced for EWSR1::WT1 as described above (please refer to the Small interfering RNA (siRNA) Knock-Down (KD) section). Cells were collected at 4°C in PBS after 48-hour silencing. Cell pellets were lyzed and RNA extracted using Qiagen RNEasy kit according to manufacturer's instructions. The experiment was performed in triplicate.
Sequencing
RNA quality control was performed before library construction based on RNA Integrity Number (RIN) evaluated on Agilent 2100 Bioanalyzer. Sequencing libraries were prepared after rRNA removal. Samples were sequenced on a NovaSeq sequencer with 150 base paired-end reads with a targeted depth of 30 million reads per sample.
Data processing
The RNA sequencing data were analyzed according to nf-core/rnaseq pipeline. Raw reads quality control (QC) was performed using FastQC. Pseudoalignment was performed with Salmon on hg19 (GENCODE version 19). RNA counts were quantified using Salmon after adaptor trimming with Trim Galore!.
Downstream analyses
After outline sample removal, differential gene expression analysis between H48 EWSR1::WT1-silenced and non-silenced conditions was performed using DESeq2.
JN-DSRCT-1 cell line assay for transposase-accessible chromatin using sequencing
Assay for transposase-accessible chromatin
JN-DSRCT-1 cells were grown to target ∼80% confluence in 6-well plates 48 h after EWSR1::WT1 or CCND1 siRNA-mediated knockdown, or transfection with a non-targeting siRNA. Independent biological duplicates were performed for this experiment.
Although working on cell lines, assay for transposase-accessible chromatin (ATAC) was performed according to Corces et al.91 Omni-ATAC protocol optimized from the standard Buenrostro et al.86 protocol. Briefly, cells were harvested to target 100,000 cells per sample. Cell lysis was performed to extract nuclei by incubating cells for 3 min on ice in cold lysis buffer (Resuspension buffer (10mM Tris-HCl, pH 7.5; 10mM NaCl; 3mM MgCl2); 0.1% NP-40; 0.1% Tween 20; 1% Digitonin). The suspension was then washed in Wash buffer (Resuspension buffer with 10% Tween 20) before centrifugation at 500g for 10 min at 4°C to isolate pellets (nuclei). DNA transposition was performed by incubating nuclei for 45 min a 37°C in a thermomixer at 1,000 revolutions per minute (rpm) in a transposition reaction mix containing 2X Tagment DNA (TD) buffer, 1X PBS, with 0.1% Tween 20, 0.01% Digitonin, 10% Tn5 Transposase (Tagment DNA Enzyme 1). DNA purification was then performed using the Qiagen MinElute Reaction Cleanup kit. Library amplification was performed using the NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs) while determining the total number of PCR cycles by qPCR. Next, libraries were purified with KAPA Pure Beads (Roche) before assessing the library quality on Agilent BioAnalyzer with the High Sensitivity DNA kit. Libraries were finally sequenced at a depth of 200 million reads per sample on NovaSeq with paired-end 150 bp reads.
Data analysis
ATAC-seq data were analyzed according to nf-core/atacseq pipeline92. Raw reads quality control (QC) was performed using FastQC, followed by adapter trimming with Trim Galore! After read mapping to the reference genome (GRCh38) using BWA, duplicate reads were discarded using picard. BigWig files were generated using BEDTools for IGV visualization. Genome-wide immunoprecipitation enrichment relative to input was performed using deepTools. Broad and narrow peaks were called using MACS2 and were annotated relative to gene features using HOMER. Finally, differential binding analysis was performed using DESeq2. Motif analyses were subsequently performed on significantly enriched peaks using the MEME suite. Enrichment in known transcription factors was evaluated among the JASPAR2020 database using AME. EWSR1::WT1 de novo motifs were explored using DREME.
JN-DSRCT-1 cell line WT1 C-terminal chromatin immunoprecipitation with sequencing
Chromatin immunoprecipitation, library generation, and sequencing
JN-DSRCT-1 cells were grown to ∼80% confluence in 15 cm dishes. To perform chromatin immunoprecipitation (ChIP), chromatin was first crosslinked for 15 min at room temperature (RT) by adding methanol-free formaldehyde (1% final) in the culture media. Formaldehyde was quenched by adding a glycine solution at a final concentration of 125 mM for 5 min RT incubation. After two washes with ice-cold PBS supplemented with Complete Protease Inhibitor Cocktail (Roche), cells were scraped and centrifuged for 5 min at 300g before freezing at 80°C for subsequent utilization. After thawing on ice, cells were resuspended in Farnham lab (FL) buffer (5 mM PIPES pH 8; 85 mM KCl; 0.5% Igepal CA-630) supplemented with Complete Protease Inhibitor Cocktail, EDTA-free (Roche) at a final concentration of 1 million cells/mL. Furthermore, nuclei extraction was performed according to the NEXSON (Nuclei EXtraction by Sonication) protocol87. Briefly, cell suspensions were sonicated in 12x12 1mL milliTUBES with AFA Fiber using the Covaris S220 Focused-ultrasonicator at peak power 75W, duty factor 2%, and 200 cycles/burst at 4°C for 2 min. Isolated nuclei were then resuspended in 1 mL of shearing buffer (10 mM Tris-HCl pH 8; 0.1% SDS; 1 mM EDTA) supplemented with Complete Protease Inhibitor Cocktail EDTA-free (Roche) and chromatin was sheared using the Covaris S220 Focused-ultrasonicator at peak power 140W, duty factor 5%, 200 cycles/burst during 20 min at 4°C. When needed, sheared chromatin was kept at −80°C for subsequent use the day after. An aliquot of sheared chromatin was incubated overnight with proteinase K at 65°C to reverse the crosslink for the quality control of adequate shearing. DNA was then purified using the Qiagen PCR purification kit and analyzed on a Bioanalyzer (DNA High Sensitivity kit) for size distribution.
Immunoprecipitation was performed on sheared chromatin diluted with 10X dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2mM EDTA, 16.7 mM Tris pH8, 167mM NaCl) by incubating WT1 C-terminal (Genetex GTX15249) targeting antibody at 0.5 mg/mL final concentration, H3K9ac-targeting antibody (Cell Signalling Cat #9649) at 1:50 dilution, H3K27ac-targeting antibody (Cell Signalling Cat #8173) at 1:100 dilution, or rabbit isotype at 1 mg/mL final concentration overnight at 4°C. The next day, Dynabeads Protein G beads (Thermo Fisher Scientific) were blocked with PBS-BSA 0.5% at RT for 30 min. Antibody-incubated chromatin was added upon blocked Dynabeads to capture immune complexes. Immunoprecipitates were then washed twice using successively low saft buffer (0.1% SDS, 1% Triton X-100, 2mM EDTA, 20mM Tris pH8, 150mM NaCl), high salt buffer (0.1% SDS, 1% Triton X-100, 2mM EDTA, 20mM Tris pH8, 500mM NaCl), LiCl wash buffer (250mM LiCl, 1% NP-40, 1% Na-Deoxycholate, 10mM Tris pH8, 1mM EDTA) and TE 1X buffer. Precipitated chromatin was eluted from beads by heating at 65°C in Elution buffer (25mM Tris pH 7.5, 5mM EDTA, 0.5% SDS). Crosslink was reverted from eluted samples by overnight incubation at 65°C with Proteinase K. DNA was purified on Qiagen PCR purification columns. Library generation was finally performed according to the NEBNext Ultra II DNA Library Prep Kit for Illumina protocol. Quality control of amplified library profiles was performed on Agilent Bioanalyzer, and samples were sequenced at 200 million reads depth on NovaSeq with 150 base paired-end reads.
Independent biological duplicates were performed for WT1 C-terminal ChIP while a single assay was performed for H3K9ac and H3K27ac ChIP.
ChIP-seq data analyses
Peak calling, gene annotation, and differential binding analysis
ChIP-seq data analyses were performed according to nf-core/chipseq pipeline92. Briefly, a raw read QC (FastQC) was performed before adapter trimming with Trim Galore! Reads were further mapped to the reference genome (GRCh38) using BWA, and duplicate reads were discarded using picard. BigWig files were generated using BEDTools to allow IGV visualization of fragments. The distribution of peaks was annotated using HOMER. Gene-wide immunoprecipitation enrichment relative to input was performed using deepTools. Broad and narrow peaks were called using MACS2 and were annotated relative to gene features using HOMER. Finally, differential binding analysis was performed using DESeq2.
ChIP gene set enrichment analysis
Gene set enrichment (GSE) analysis was performed on WT1 ChIP-seq data using two methods. First, peak-associated genes with a peak fold enrichment >2 were selected to generate a gene list which was used as an input for ToppFun analysis (ToppGene Suite) focusing on Gene Ontology gene sets. Secondly, we performed GSE testing on a list of ChIP-seq-derived genomic regions, defined as the differentially enriched broad peaks’ genomics ranges derived from DESeq2 analysis on WT1 ChIP compared to isotype. For the latter study, we took advantage of the ChIP-Enrich package93, which relies on four steps to account for biases induced by the properties of ChIP-seq data, including the increasing of Type I error secondary to multiple testing (various numbers of peaks for a single gene), and the gene-length bias. Briefly, ChIP-Enrich analysis is divided into four steps: first, we defined loci of interest as the regions spanning the midpoints between the transcription start sites (TSSs) of adjacent genes so that each peak genomic range is assigned to the gene with the nearest TSS. Secondly, the proportion of each gene locus covered by ChIP-seq peaks is calculated. Thirdly, a logistic regression is performed for each GO gene set, including a correction for locus length. Finally, p-values for enrichment or depletion are adjusted for each GO gene set for multiple testing.
EWSR1::WT1 ChIP-seq signature
An EWSR1::WT1 ChIP-seq signature (n = 176 genes) reflecting EWSR1::WT1 transcriptional activity was inferred by selecting peaks corresponding to genes displaying a fold enrichment >5 compared to isotype control ChIP (DESeq2). The calculation of a ChIP-seq signature score at the single-cell level on 3′scRNAseq Harmony integrated data was performed using the AddModuleScore function (Seurat), which computes the average expression level of a program of interest, subtracted by the aggregated expression of randomly selected control features.
K-mer overrepresentation analysis
We searched for k-mer enrichment within the 100 bp-wide sequences around EWSR1::WT1 specific ChIP-seq peak summits. EWSR1::WT1 specific ChIP-seq peaks were inferred from significantly (p-value<0.05) enriched peaks in EWSR1::WT1 ChIP compared to rabbit isotype ChIP using DESeq2. We compared the frequency of all 6-mer oligonucleotides (n = 46 = 4,096) found within EWSR1::WT1 ChIP-seq peak summits to their frequency within the whole genome.
Motif enrichment analysis
Motif analyses were subsequently performed on significantly enriched peaks using the MEME suite. Enrichment in known transcription factors was evaluated among the JASPAR202094 database using AME. EWSR1::WT1 de novo motifs were explored using STREME and were matched to known TF motifs from JASPAR2020 and HOCOMOCO-v11 using the TomTom motif comparison tool. HOCOMOCO v11 database contains two human WT1 wild type motifs WT1_HUMAN.H11MO.1.B and WT1_HUMAN.H11MO.1.C, corresponding to the short and long version of pooled WT1 +/− KTS motifs from GSE81009 dataset, respectively.
ATAC-seq and ChIP-seq integration
We analyzed overlapping peaks between EWSR1::WT1 specific binding sites inferred from isotype versus EWSR1::WT1 differential binding analysis (“EWSR1::WT1 ChIP module”) and EWSR1::WT1 induced differentially accessible peaks generated from EWSR1::WT1-silenced versus non-silenced DSRCT cell lines derived samples ATAC-seq data (“EWSR1::WT1 ATAC module”).
Immunohistochemistry
DSRCT cases, including those corresponding to the samples processed for scRNA-seq, were selected by an expert sarcoma pathologist. Serial sections of FFPE tissue were cut in 3-μm thickness sections for further fluorescent or DAB immunohistochemitry (IHC) as described below.
Fluorescent stainings
Fluorescent multiplex stainings were performed on BOND RX automated stainer (Leica Biosystems). Dewaxing was performed with BOND Dewax Solution. Antigen retrieval was performed using BOND Epitope Retrieval Solution 2 (pH = 9) for 20 min at 100°C. Protein blocking was performed with PKI blocking (Akoya) for 5 min.
Antigen detection was performed using the Opal system (Akoya) with Opal anti-mouse and anti-rabbit HRP polymer.
Between each sequence, antigen stripping was performed using BOND Epitope Retrieval Solution 1 (pH = 6) for 20 min at 100°C.
A counter-coloration was done with DAPI (Akoya) for both multiplex assays. After staining, tissue sections were submitted to serial gradients of xylene and mounted manually with a coverslip using ProLong Diamond.
The following antibodies and respective conditions were used for THY1/CHI3L1/Desmin triplex: anti-THY1/CD90 (1:1000, rabbit IgG D3V81 clone, Cell Signaling #13801), detected with OPAL480 fluorophore at 1:75; anti-CHI3L1 (1:1600, polyclonal rabbit, Abcam ab77528), detected with OPAL570 fluorophore at 1:100; anti-Desmin (1:200, monoclonal mouse D33 clone, DAKO M0760), detected with OPAL690 at 1:150.
The following antibodies and respective conditions were used for FAP/MCAM/ACTA2 triplex: anti-FAP (1:200, polyclonal sheep, R&S AF3715), detected by rabbit anti-sheep (P0163, DAKO) and OPAL520 fluorophore at 1:50; anti-MCAM (1:600, mouse OTI5C4 clone, Origene TA803548), detected with OPAL570 at 1:600; anti-ACTA2 (1:1000, mouse 1A4 clone, DAKO M0851), detected with OPAL690 at 1:150.
Mono DAB stainings
Mono DAB stainings were performed using the automated BenchMark ULTRA stainer.
Dilution and antibodies for mono DAB IHC stainings were as follows: anti-c-Myc (fixed concentration pre-filled syringe, monoclonal anti-rabbit, Y69 clone, Roche 790–4628), anti-WT1 (1:100, polyclonal rabbit, Zytomed AB_2864626), anti-Desmin (1:40, monoclonal mouse D33 clone, DAKO M0760), anti-AE1/AE3 (1:75, monoclonal mouse, Diagnostic BioSystem Clinisciences Mob190-05), anti-CD3 (1:100, polyclonal rabbit, DAKO A0452), anti-THY1 (alias CD90, 1:750, rabbit IgG D3V81 clone, Cell Signaling #13801), and CD56 (alias NCAM, fixed concentration pre-filled syringe, monoclonal rabbit, MRQ-42 clone, Roche 760–4596).
Revelations were performed using the UV DAB kit, and counter coloration was done with hematoxylin and bluing reagent.
Dual chromogenic stainings
Dual chromogenic CD68/CD163 staining was performed on an automated Discovery Ultra stainer using the following antibody references and dilutions: anti-CD163 (1:100, Diagnostic BioSystem Clinisciences Mob460-05) followed by anti-mouse HRP and DAB incubation, and anti-CD68 (1:200, PG-MI clone, DAKO M0876), followed by anti-mouse HRP and DS Discovery purple incubation.
Counter coloration was performed with hematoxylin and bluing reagent.
Image analysis
Slides were scanned on the Olympus VS120 automated slide scanner, followed by visualization with OlyVIA v2.9. Images were analyzed using QuPath v0.3.0 and ImageJ v1.53.
CAFs co-culture
Colony formation assay
JN-DSRCT-1 cells were co-cultured with mouse Cancer-Associated Fibroblasts (CAFs) isolated from a mouse patient-derived xenograft (PDX). Briefly, PDX was collected from a Nod SCID Gamma (NSG, Charles River) mouse and dissociated with the Tumor Dissociation kit (Miltenyi Biotech). Mouse fibroblasts were isolated with the Tumor-Associated Fibroblast Isolation mouse kit (Miltenyi Biotech). Isolated cells were seeded into three wells of a 6-well plate in DMEM/F12 complemented medium, supplemented with 1X Insulin-Tranferrin-Selenium (ITS). The day after, a Transwell device of 0.4 μm was inserted into each well (with or without CAFs), and JN-DSRCT-1 cells were seeded (500 cells per well) for the colony-forming assay.
Both 6-well plates seeded with DSRCT tumor cells and Transwell seeded CAFs were washed with PBS stained with crystal violet 0.5% in methanol.
Plates and Transwell inserts were scanned, and the total colony area was calculated using ImageJ. The staining was then solubilized in 100% Methanol for 20 min, and triplicate aliquots were collected to measure absorbance at 570 nm.
3D spheroids co-culture growth test
JN-DSRCT-1 cells were plated on ultra-low adherence 96-well plates at a concentration of 1,000 cells per well in DMEM/F12 before 15 min of centrifugation at 500 x G. Plates were incubated at 37°C + 5% CO2 and the formation of the spheroid was assessed 72 h after seeding. PDX-derived CAFs were first isolated as previously described using the Tumor-Associated Fibroblast Isolation mouse kit (Miltenyi Biotech) and stained using Cell Proliferation Staining Reagent according to the manufacturer’s protocol (Abcam - Deep Red Fluorescence).
Stained CAFs (n = 500) were added to wells containing JN-DSRCT-1 spheroids. Pictures were acquired every two days to assess the spheroids’ growth on Invitrogen EVOS XL using the product of their two largest diameters. On day 15, live cells spheroids were finally pictured on a spinning disk microscope (Nikon Eclipse Ti2 equipped with Leica/Gataca CSU-W1 confocal scanner unit, Live-SR module, and incubation chamber) using a 20× oil objective. Spheroids were then measured using ImageJ software v1.53, and tumor cells were distinguished from CAFs based on deep red fluorescence, negative for JN-DSRCT-1 cells and positive for CAFs. The experiment was performed in triplicates.
3D immunofluorescence
JN-DSRCT-1 cells were seeded and grown in 3D as previously described. An immunofluorescence assay was performed to evaluate the staining pattern of elected markers within the spheroids. After 15 days of growth in ultra-low attachment (ULA) plates, spheroids were fixed for 30 min in PBS-PFA 4%, permeabilized with Triton X-100 0.5% for 10 min and blocked with PBS-BSA 0.1%, Triton X-100 0.2% and Tween 20 0.5%. Spheroids were then incubated within 1:500 or 1:100 diluted primary antibodies (anti-WT1 C-terminal (rabbit polyclonal Genetex GTX15249), or anti-Desmin (mouse monoclonal D33 clone, Dako M0760)) overnight at RT. After three rounds of PBS washing, spheroids were further incubated overnight with secondary antibody solutions (Rabbit anti-goat Alexa Fluor 555, 1:2000 and Goat anti-mouse Alexa Fluor 488, 1:2000). The day after, a DAPI staining was finally performed by incubating spheroids 30 min in a 100 μg/mL DAPI solution. Before image acquisition, spheroids were submerged within a sucrose and urea solution to allow tissue clearing with minimal tissue shrinkage. Spheroids were then pictured on a spinning disk microscope (Nikon Eclipse Ti2 equipped with Leica/Gataca CSU-W1 confocal scanner unit, Live-SR module, and incubation chamber) using a 20× oil objective and appropriate wavelength.
Quantification and statistical analysis
The tests used for statistical analyses are described in the dedicated paragraphs within the main text and the STAR Methods section, and have been performed using R.
Acknowledgments
We dedicate this research to patients and their families. We thank patients and families for their donations to Gustave Roussy Sarcoma Tumor Board and Sarcoma Research program which contributed to this work, and especially L. family and “Un élan pour Lucas.” C.H. was supported by the “Philanthropia Foundation” PhD grant. J.V. was supported by Institut Curie and the “La Ligue contre le Cancer” PhD grant.
This work was also partly funded by program grants to S.P.V. (Fondation ARC PGA1- RF20190208576, European Research Council ERC TargetSWitch – 101077864, Inserm ATIP-Avenir/La Ligue Contre le Cancer 2018, SFCE/Imagine for Margo, Cancéropôle Île-de-France - Recherche et Innovation en Cancérologie-RIC-01-IGR, Agence Nationale pour la Recherche ANR tremplin-ERC(9)2020), and by program grants to Gustave Roussy (INCa-DGOS-Inserm_12551 SIRIC2, INCa-DGOS-Inserm-ITMO Cancer_18002 SIRIC EpiCURE). We also thank FIGHT KIDS CANCER (FKC) Representatives KickCancer, Imagine for Margo, Fondatioun Kriibskrank Kanner, and CRIS Cancer Foundation for awarding the 2024 FIGHT KIDS CANCER & St. Baldrick’s Foundation Arceci Innovation Award to S.P.V. The independent selection process of this Innovation Award, funded by FKC, was administered by the St. Baldrick's Foundation. We also thank patients and families for their donations to our research project.
Author contributions
Conceptualization, C. Henon, J.V., T.E., S.W., and S.P.-V.; methodology, C. Henon, J.V., T.E., N.G., C.N., M.G., N.D., M.E.M.D.C., V.M., N.S., A.M., A.K.-K., M.L., C.A., R.C., S.W., and S.P.-V.; software, C. Henon, J.V., and L.C.-D., validation, C. Henon, J.V., M.G., and T.E.; formal analysis, C. Henon, J.V., L.C.-D., and N.S.; investigation, C. Henon, J.V., T.E., N.G., M.G., N.D., N.H., M.E.M.D.C., V.M., and N.S.; resources, C. Henon, B.V., R.B., A.L.C., M.F., and C. Honoré; data curation, C. Henon and J.V.; writing – original draft: C. Henon; writing – review & editing, C. Henon, J.V., C.N., M.G., J.J.W., S.W., and S.P.-V.; visualization, C. Henon and J.V.; supervision, O.D., J.J.W., F.M.-G., S.W., and S.P.-V.; project administration, C. Henon and S.P.-V.; funding acquisition, C. Henon, J.V., M.F., C. Honoré, S.W., and S.P.-V.
Declaration of interests
The authors declare no competing interests.
Published: May 22, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101582.
Supplemental information
(A) Single-cell assays. AI: Adriamycin-Ifosfamide; IE: Ifosfamide-Etoposide; VAC: Vincristine-Actinomycin D-Cyclophosphamide; WES: Whole Exome Sequencing.
(B) DSRCT Bulk RNA-seq test cohort (N = 29). AI: Doxorubicin-Ifosfamide; API: Doxorubicin-CDDP-Ifosfamide; HIPEC: Hyperthermic Intra Peritoneal Chemotherapy; IVA: Ifosfamide-Vincristine-Doxorubicin; IVADo: Ifosfamide-Vincristine-Actinomycin-Doxorubicin; L1: First line; L2: Second line; TEMIRI: Temozolomide-Irinotecan; VAC: Vincristine-Actinomycin D-Cyclophosphamide; VDC: Vincristine-Doxorubicin-Cyclophosphamide; VIDE: Vincristine-Ifosfamide-Doxorubicin-Etoposide; VIP: Vincristine-Ifosfamide-CDDP; WES: Whole Exome Sequencing.
(A) Top 50 DEGs across main cell populations in Int_sc dataset.
(B) Top 50 DEGs across nearest neighbors' defined Int_sc clusters.
(C) Int_sc dataset clusters quality control metrics.
(A) Differentially bound genomics regions between EWSR1::WT1 and isotype ChIP-seq. The 262 genes significantly upregulated upon EWSR1::WT1 silencing intersected with genes corresponding to EWSR1::WT1 ″off" ATAC peaks and EWSR1::WT1 ChIP-enriched peaks are shown in orange.
(B) Motif enrichment analysis in significantly enriched peaks in EWSR1::WT1 ChIP assay.
(C) H3K27ac and EWSR1::WT1 co-occupied regions.
(D) H3K9ac and EWSR1::WT1 co-occupied regions.
(A) Differentially accessible genomics regions between EWSR1::WT1-silenced versus EWSR1::WT1-non-silenced JN-DSRCT-1 cell line. The 262 genes significantly upregulated upon EWSR1::WT1 silencing intersected with genes corresponding to EWSR1::WT1 ″off" ATAC peaks and EWSR1::WT1 ChIP-enriched peaks are shown in orange.
(B) Motif enrichment analysis in significantly enriched peaks in EWSR1::WT1-non-silenced JN-DSRCT-1 cells (EWSR1-W1 ″on").
(C) Motif enrichment analysis in significantly enriched peaks in EWSR1::WT1- -silenced JN-DSRCT-1 cells (EWSR1-W1 ″off").
The 262 genes significantly upregulated upon EWSR1::WT1 silencing intersected with genes corresponding to EWSR1::WT1 ″off" ATAC peaks and EWSR1::WT1 ChIP-enriched peaks are shown in orange.
(A) Top 50 DEGs across snMultiome WNN clusters.
(B) Top 50 differentially accessible peaks across snMultiome WNN clusters.
(A) EWSR1::WT1 regulon.
(B) Top 10 SCENIC+ regulons per snMultiome WNN cluster. RSS relates to Regulon Specificity Score.
(A) Differential gene expression (DGE) analysis between DSRCT bulk RNA-seq groups defined by hierarchical clustering. Significantly enriched genes for each group paired comparison is displayed.
(B) Sarcoma bulk RNA-seq external cohort. The number of samples per histotype is shown in the second column.
(C) Specificity and prognostic value of scRNA-seq signatures. The second column displays the proportion of significant p values (p < 0.05) over all 23 paired Tukey’s comparison tests performed on gene signature values (transcripts per million [TPM] geometric mean, see STAR methods) for DSRCT versus all other sarcoma subtypes. Each signature was significant (p < 0.05) for one-way ANOVA test. A number equal to or more than 18 of 23 (>75%) of comparisons was chosen as the threshold to call a signature « DSRCT-specific », i.e., signatures of clusters 0, 1, 4, 7, 8, and 11. The third column displays prognostic value and log rank p values for the test (N = 29) and validation (N = 21) DSRCT cohorts using the first scoring strategy (i.e., TPM geometric mean, see STAR methods) while the fourth column displays the same values using the second scoring strategy (VST-normalized raw count arithmetic mean, see STAR methods).
References
- 1.Cidre-Aranaz F., Watson S., Amatruda J.F., Nakamura T., Delattre O., de Alava E., Dirksen U., Grünewald T.G.P. Small round cell sarcomas. Nat. Rev. Dis. Prim. 2022;8:66. doi: 10.1038/s41572-022-00393-3. [DOI] [PubMed] [Google Scholar]
- 2.Gerald W.L., Ladanyi M., de Alava E., Cuatrecasas M., Kushner B.H., LaQuaglia M.P., Rosai J. Clinical, pathologic, and molecular spectrum of tumors associated with t(11;22)(p13;q12): desmoplastic small round-cell tumor and its variants. J. Clin. Oncol. 1998;16:3028–3036. doi: 10.1200/JCO.1998.16.9.3028. [DOI] [PubMed] [Google Scholar]
- 3.Wong H.H., Hatcher H.M., Benson C., Al-Muderis O., Horan G., Fisher C., Earl H.M., Judson I. Desmoplastic small round cell tumour: characteristics and prognostic factors of 41 patients and review of the literature. Clin. Sarcoma Res. 2013;3:14. doi: 10.1186/2045-3329-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lettieri C.K., Garcia-Filion P., Hingorani P. Incidence and Outcomes of Desmoplastic Small Round Cell Tumor: Results from the Surveillance, Epidemiology, and End Results Database. J. Cancer Epidemiol. 2014;2014 doi: 10.1155/2014/680126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas R., Rajeswaran G., Thway K., Benson C., Shahabuddin K., Moskovic E. Desmoplastic small round cell tumour: the radiological, pathological and clinical features. Insights Imaging. 2013;4:111–118. doi: 10.1007/s13244-012-0212-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morani A.C., Bathala T.K., Surabhi V.R., Yedururi S., Jensen C.T., Huh W.W., Prasad S., Hayes-Jordan A. Desmoplastic Small Round Cell Tumor: Imaging Pattern of Disease at Presentation. Am. J. Roentgenol. 2019;212:W45–W54. doi: 10.2214/AJR.18.20179. [DOI] [PubMed] [Google Scholar]
- 7.Giani C., Radaelli S., Miceli R., Gandola L., Sangalli C., Frezza A.M., Provenzano S., Pasquali S., Bertulli R., Fiore M., et al. Long-term survivors with desmoplastic small round cell tumor (DSRCT): Results from a retrospective single-institution case series analysis. Cancer Med. 2023;12:10694–10703. doi: 10.1002/cam4.5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shoushtari A.N., Qin L.-X., Kuk D., Magnan H.D., Modak S., Wolden S.L., Cho E.J., Pallos V., Grubman O., Viny A.D., et al. Predictors of overall survival in patients diagnosed with desmoplastic small round cell tumor (DSRCT) J. Clin. Oncol. 2014;32:10582. doi: 10.1200/jco.2014.32.15_suppl.10582. [DOI] [Google Scholar]
- 9.Waqar S.H.B., Ali H. Changing incidence and survival of desmoplastic small round cell tumor in the USA. SAVE Proc. 2022;35:415–419. doi: 10.1080/08998280.2022.2049581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bandopadhayay P., Jabbour A.M., Riffkin C., Salmanidis M., Gordon L., Popovski D., Rigby L., Ashley D.M., Watkins D.N., Thomas D.M., et al. The oncogenic properties of EWS/WT1 of desmoplastic small round cell tumors are unmasked by loss of p53 in murine embryonic fibroblasts. BMC Cancer. 2013;13:585. doi: 10.1186/1471-2407-13-585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gedminas J.M., Chasse M.H., McBrairty M., Beddows I., Kitchen-Goosen S.M., Grohar P.J. Desmoplastic small round cell tumor is dependent on the EWS-WT1 transcription factor. Oncogenesis. 2020;9:41. doi: 10.1038/s41389-020-0224-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bleijs M., Pleijte C., Engels S., Ringnalda F., Meyer-Wentrup F., van de Wetering M., Clevers H. EWSR1-WT1 Target Genes and Therapeutic Options Identified in a Novel DSRCT In Vitro Model. Cancers. 2021;13:6072. doi: 10.3390/cancers13236072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bulbul A., Fahy B.N., Xiu J., Rashad S., Mustafa A., Husain H., Hayes-Jordan A. Desmoplastic Small Round Blue Cell Tumor: A Review of Treatment and Potential Therapeutic Genomic Alterations. Sarcoma. 2017;2017 doi: 10.1155/2017/1278268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chow W.A., Yee J.-K., Tsark W., Wu X., Qin H., Guan M., Ross J.S., Ali S.M., Millis S.Z. Recurrent secondary genomic alterations in desmoplastic small round cell tumors. BMC Med. Genet. 2020;21:101. doi: 10.1186/s12881-020-01034-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu C.-C., Beird H.C., Lamhamedi-Cherradi S.-E., Soeung M., Ingram D., Truong D.D., Porter R.W., Krishnan S., Little L., Gumbs C., et al. Multi-site desmoplastic small round cell tumors are genetically related and immune-cold. npj Precis. Oncol. 2022;6:21. doi: 10.1038/s41698-022-00257-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Devecchi A., De Cecco L., Dugo M., Penso D., Dagrada G., Brich S., Stacchiotti S., Sensi M., Canevari S., Pilotti S. The genomics of desmoplastic small round cell tumor reveals the deregulation of genes related to DNA damage response, epithelial–mesenchymal transition, and immune response. Cancer Commun. 2018;38 doi: 10.1186/s40880-018-0339-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nacev B.A., Sanchez-Vega F., Smith S.A., Antonescu C.R., Rosenbaum E., Shi H., Tang C., Socci N.D., Rana S., Gularte-Mérida R., et al. Clinical sequencing of soft tissue and bone sarcomas delineates diverse genomic landscapes and potential therapeutic targets. Nat. Commun. 2022;13:3405. doi: 10.1038/s41467-022-30453-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Slotkin E.K., Bowman A.S., Levine M.F., Dela Cruz F., Coutinho D.F., Sanchez G.I., Rosales N., Modak S., Tap W.D., Gounder M.M., et al. Comprehensive Molecular Profiling of Desmoplastic Small Round Cell Tumor. Mol. Cancer Res. 2021;19:1146–1155. doi: 10.1158/1541-7786.MCR-20-0722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franzetti G.-A., Laud-Duval K., van der Ent W., Brisac A., Irondelle M., Aubert S., Dirksen U., Bouvier C., de Pinieux G., Snaar-Jagalska E., et al. Cell-to-cell heterogeneity of EWSR1-FLI1 activity determines proliferation/migration choices in Ewing sarcoma cells. Oncogene. 2017;36:3505–3514. doi: 10.1038/onc.2016.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aynaud M.-M., Mirabeau O., Gruel N., Grossetête S., Boeva V., Durand S., Surdez D., Saulnier O., Zaïdi S., Gribkova S., et al. Transcriptional Programs Define Intratumoral Heterogeneity of Ewing Sarcoma at Single-Cell Resolution. Cell Rep. 2020;30:1767–1779.e6. doi: 10.1016/j.celrep.2020.01.049. [DOI] [PubMed] [Google Scholar]
- 21.Zheng G.X.Y., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J., et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8 doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sydow S., Versleijen-Jonkers Y.M.H., Hansson M., van Erp A.E.M., Hillebrandt-Roeffen M.H.S., van der Graaf W.T.A., Piccinelli P., Rissler P., Flucke U.E., Mertens F. Genomic and transcriptomic characterization of desmoplastic small round cell tumors. Genes Chromosomes Cancer. 2021;60:595–603. doi: 10.1002/gcc.22955. [DOI] [PubMed] [Google Scholar]
- 23.Vibert J., Saulnier O., Collin C., Petit F., Borgman K.J.E., Vigneau J., Gautier M., Zaidi S., Pierron G., Watson S., et al. Oncogenic chimeric transcription factors drive tumor-specific transcription, processing, and translation of silent genomic regions. Mol. Cell. 2022;82:2458–2471.e9. doi: 10.1016/j.molcel.2022.04.019. [DOI] [PubMed] [Google Scholar]
- 24.Newman A.M., Steen C.B., Liu C.L., Gentles A.J., Chaudhuri A.A., Scherer F., Khodadoust M.S., Esfahani M.S., Luca B.A., Steiner D., et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019;37:773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao J., O’Day D.R., Pliner H.A., Kingsley P.D., Deng M., Daza R.M., Zager M.A., Aldinger K.A., Blecher-Gonen R., Zhang F., et al. A human cell atlas of fetal gene expression. Science. 2020;370 doi: 10.1126/science.aba7721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boulay G., Sandoval G.J., Riggi N., Iyer S., Buisson R., Naigles B., Awad M.E., Rengarajan S., Volorio A., McBride M.J., et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell. 2017;171:163–178.e19. doi: 10.1016/j.cell.2017.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeTomaso D., Yosef N. Hotspot identifies informative gene modules across modalities of single-cell genomics. Cell Syst. 2021;12:446–456.e9. doi: 10.1016/j.cels.2021.04.005. [DOI] [PubMed] [Google Scholar]
- 29.Lange M., Bergen V., Klein M., Setty M., Reuter B., Bakhti M., Lickert H., Ansari M., Schniering J., Schiller H.B., et al. CellRank for directed single-cell fate mapping. Nat. Methods. 2022;19:159–170. doi: 10.1038/s41592-021-01346-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gulati G.S., Sikandar S.S., Wesche D.J., Manjunath A., Bharadwaj A., Berger M.J., Ilagan F., Kuo A.H., Hsieh R.W., Cai S., et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science. 2020;367:405–411. doi: 10.1126/science.aax0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grün D., Muraro M.J., Boisset J.-C., Wiebrands K., Lyubimova A., Dharmadhikari G., van den Born M., van Es J., Jansen E., Clevers H., et al. De Novo Prediction of Stem Cell Identity using Single-Cell Transcriptome Data. Cell Stem Cell. 2016;19:266–277. doi: 10.1016/j.stem.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tickle T.I., Georgescu C., Brown M., Haas B. 2019. inferCNV of the Trinity CTAT Project.https://github.com/broadinstitute/inferCNV [Google Scholar]
- 33.Hingorani P., Dinu V., Zhang X., Lei H., Shern J.F., Park J., Steel J., Rauf F., Parham D., Gastier-Foster J., et al. Transcriptome analysis of desmoplastic small round cell tumors identifies actionable therapeutic targets: a report from the Children’s Oncology Group. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-69015-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Landt S.G., Marinov G.K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S., Bernstein B.E., Bickel P., Brown J.B., Cayting P., et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J., Nau M.M., Yeh J.C., Allegra C.J., Chu E., Wright J.J. Molecular Heterogeneity and Function of EWS-WT1 Fusion Transcripts in Desmoplastic Small Round Cell Tumors. Clin. Cancer Res. 2000;6:3522–3529. [PubMed] [Google Scholar]
- 36.Rauscher F.J., Morris J.F., Tournay O.E., Cook D.M., Curran T. Binding of the Wilms’ tumor locus zinc finger protein to the EGR-1 consensus sequence. Science. 1990;250:1259–1262. doi: 10.1126/science.2244209. [DOI] [PubMed] [Google Scholar]
- 37.Ullmark T., Järvstråt L., Sandén C., Montano G., Jernmark-Nilsson H., Lilljebjörn H., Lennartsson A., Fioretos T., Drott K., Vidovic K., et al. Distinct global binding patterns of the Wilms tumor gene 1 (WT1) −KTS and +KTS isoforms in leukemic cells. Haematologica. 2017;102:336–345. doi: 10.3324/haematol.2016.149815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang H.-J., Park J.H., Chen W., Kang S.I., Moroz K., Ladanyi M., Lee S.B. EWS–WT1 Oncoprotein Activates Neuronal Reprogramming Factor ASCL1 and Promotes Neural Differentiation. Cancer Res. 2014;74:4526–4535. doi: 10.1158/0008-5472.CAN-13-3663. [DOI] [PubMed] [Google Scholar]
- 39.Palmer R.E., Lee S.B., Wong J.C., Reynolds P.A., Zhang H., Truong V., Oliner J.D., Gerald W.L., Haber D.A. Induction of BAIAP3 by the EWS-WT1 chimeric fusion implicates regulated exocytosis in tumorigenesis. Cancer Cell. 2002;2:497–505. doi: 10.1016/s1535-6108(02)00205-2. [DOI] [PubMed] [Google Scholar]
- 40.Wong J.C., Lee S.B., Bell M.D., Reynolds P.A., Fiore E., Stamenkovic I., Truong V., Oliner J.D., Gerald W.L., Haber D.A. Induction of the interleukin-2/15 receptor β-chain by the EWS–WT1 translocation product. Oncogene. 2002;21:2009–2019. doi: 10.1038/sj.onc.1205262. [DOI] [PubMed] [Google Scholar]
- 41.Reynolds P.A., Smolen G.A., Palmer R.E., Sgroi D., Yajnik V., Gerald W.L., Haber D.A. Identification of a DNA-binding site and transcriptional target for the EWS-WT1(+KTS) oncoprotein. Genes Dev. 2003;17:2094–2107. doi: 10.1101/gad.1110703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lamhamedi-Cherradi S.-E., Maitituoheti M., Menegaz B.A., Krishnan S., Vetter A.M., Camacho P., Wu C.-C., Beird H.C., Porter R.W., Ingram D.R., et al. The androgen receptor is a therapeutic target in desmoplastic small round cell sarcoma. Nat. Commun. 2022;13:3057. doi: 10.1038/s41467-022-30710-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao N., Zhang J., Rao M.A., Case T.C., Mirosevich J., Wang Y., Jin R., Gupta A., Rennie P.S., Matusik R.J. The role of hepatocyte nuclear factor-3 alpha (Forkhead Box A1) and androgen receptor in transcriptional regulation of prostatic genes. Mol. Endocrinol. 2003;17:1484–1507. doi: 10.1210/me.2003-0020. [DOI] [PubMed] [Google Scholar]
- 44.Yu X., Gupta A., Wang Y., Suzuki K., Mirosevich J., Orgebin-Crist M.-C., Matusik R.J. Foxa1 and Foxa2 Interact with the Androgen Receptor to Regulate Prostate and Epididymal Genes Differentially. Ann. N. Y. Acad. Sci. 2005;1061:77–93. doi: 10.1196/annals.1336.009. [DOI] [PubMed] [Google Scholar]
- 45.Jin H.-J., Zhao J.C., Wu L., Kim J., Yu J. Cooperativity and equilibrium with FOXA1 define the androgen receptor transcriptional program. Nat. Commun. 2014;5:3972. doi: 10.1038/ncomms4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bravo González-Blas C., De Winter S., Hulselmans G., Hecker N., Matetovici I., Christiaens V., Poovathingal S., Wouters J., Aibar S., Aerts S. SCENIC+: single-cell multiomic inference of enhancers and gene regulatory networks. Nat. Methods. 2023;20:1355–1367. doi: 10.1038/s41592-023-01938-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mulder K., Patel A.A., Kong W.T., Piot C., Halitzki E., Dunsmore G., Khalilnezhad S., Irac S.E., Dubuisson A., Chevrier M., et al. Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity. 2021;54:1883–1900.e5. doi: 10.1016/j.immuni.2021.07.007. [DOI] [PubMed] [Google Scholar]
- 48.Inoue T., Abe C., Kohro T., Tanaka S., Huang L., Yao J., Zheng S., Ye H., Inagi R., Stornetta R.L., et al. Non-canonical cholinergic anti-inflammatory pathway-mediated activation of peritoneal macrophages induces Hes1 and blocks ischemia/reperfusion injury in the kidney. Kidney Int. 2019;95:563–576. doi: 10.1016/j.kint.2018.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shang Y., Coppo M., He T., Ning F., Yu L., Kang L., Zhang B., Ju C., Qiao Y., Zhao B., et al. The transcriptional repressor Hes1 attenuates inflammation via regulating transcriptional elongation. Nat. Immunol. 2016;17:930–937. doi: 10.1038/ni.3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Binnewies M., Pollack J.L., Rudolph J., Dash S., Abushawish M., Lee T., Jahchan N.S., Canaday P., Lu E., Norng M., et al. Targeting TREM2 on tumor-associated macrophages enhances immunotherapy. Cell Rep. 2021;37 doi: 10.1016/j.celrep.2021.109844. [DOI] [PubMed] [Google Scholar]
- 51.Nakamura K., Smyth M.J. TREM2 marks tumor-associated macrophages. Signal Transduct. Targeted Ther. 2020;5:233. doi: 10.1038/s41392-020-00356-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Molgora M., Esaulova E., Vermi W., Hou J., Chen Y., Luo J., Brioschi S., Bugatti M., Omodei A.S., Ricci B., et al. TREM2 Modulation Remodels the Tumor Myeloid Landscape Enhancing Anti-PD-1 Immunotherapy. Cell. 2020;182:886–900.e17. doi: 10.1016/j.cell.2020.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fan Y., Zhang J., Cai L., Wang S., Liu C., Zhang Y., You L., Fu Y., Shi Z., Yin Z., et al. The effect of anti-inflammatory properties of ferritin light chain on lipopolysaccharide-induced inflammatory response in murine macrophages. Biochim. Biophys. Acta. 2014;1843:2775–2783. doi: 10.1016/j.bbamcr.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 54.van der Leun A.M., Thommen D.S., Schumacher T.N. CD8+ T cell states in human cancer: insights from single-cell analysis. Nat. Rev. Cancer. 2020;20:218–232. doi: 10.1038/s41568-019-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pelon F., Bourachot B., Kieffer Y., Magagna I., Mermet-Meillon F., Bonnet I., Costa A., Givel A.-M., Attieh Y., Barbazan J., et al. Cancer-associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat. Commun. 2020;11:404. doi: 10.1038/s41467-019-14134-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Costa A., Kieffer Y., Scholer-Dahirel A., Pelon F., Bourachot B., Cardon M., Sirven P., Magagna I., Fuhrmann L., Bernard C., et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell. 2018;33:463–479.e10. doi: 10.1016/j.ccell.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 57.Zhang T., Ren Y., Yang P., Wang J., Zhou H. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. Cell Death Dis. 2022;13:897. doi: 10.1038/s41419-022-05351-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Geng X., Chen H., Zhao L., Hu J., Yang W., Li G., Cheng C., Zhao Z., Zhang T., Li L., Sun B. Cancer-Associated Fibroblast (CAF) Heterogeneity and Targeting Therapy of CAFs in Pancreatic Cancer. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.655152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vijay J., Gauthier M.-F., Biswell R.L., Louiselle D.A., Johnston J.J., Cheung W.A., Belden B., Pramatarova A., Biertho L., Gibson M., et al. Single-cell analysis of human adipose tissue identifies depot and disease specific cell types. Nat. Metab. 2020;2:97–109. doi: 10.1038/s42255-019-0152-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Buechler M.B., Pradhan R.N., Krishnamurty A.T., Cox C., Calviello A.K., Wang A.W., Yang Y.A., Tam L., Caothien R., Roose-Girma M., et al. Cross-tissue organization of the fibroblast lineage. Nature. 2021;593:575–579. doi: 10.1038/s41586-021-03549-5. [DOI] [PubMed] [Google Scholar]
- 61.Merrick D., Sakers A., Irgebay Z., Okada C., Calvert C., Morley M.P., Percec I., Seale P. Identification of a mesenchymal progenitor cell hierarchy in adipose tissue. Science. 2019;364 doi: 10.1126/science.aav2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Monteran L., Erez N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019;10:1835. doi: 10.3389/fimmu.2019.01835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jenkins L., Jungwirth U., Avgustinova A., Iravani M., Mills A., Haider S., Harper J., Isacke C.M. Cancer-Associated Fibroblasts Suppress CD8+ T-cell Infiltration and Confer Resistance to Immune-Checkpoint Blockade. Cancer Res. 2022;82:2904–2917. doi: 10.1158/0008-5472.CAN-21-4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boulakirba S., Pfeifer A., Mhaidly R., Obba S., Goulard M., Schmitt T., Chaintreuil P., Calleja A., Furstoss N., Orange F., et al. IL-34 and CSF-1 display an equivalent macrophage differentiation ability but a different polarization potential. Sci. Rep. 2018;8:256. doi: 10.1038/s41598-017-18433-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jones C.V., Ricardo S.D. Macrophages and CSF-1. Organogenesis. 2013;9:249–260. doi: 10.4161/org.25676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sánchez-Martín L., Estecha A., Samaniego R., Sánchez-Ramón S., Vega M.Á., Sánchez-Mateos P. The chemokine CXCL12 regulates monocyte-macrophage differentiation and RUNX3 expression. Blood. 2011;117:88–97. doi: 10.1182/blood-2009-12-258186. [DOI] [PubMed] [Google Scholar]
- 67.Casella G., Garzetti L., Gatta A.T., Finardi A., Maiorino C., Ruffini F., Martino G., Muzio L., Furlan R. IL4 induces IL6-producing M2 macrophages associated to inhibition of neuroinflammation in vitro and in vivo. J. Neuroinflammation. 2016;13:139. doi: 10.1186/s12974-016-0596-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li B.-H., Garstka M.A., Li Z.-F. Chemokines and their receptors promoting the recruitment of myeloid-derived suppressor cells into the tumor. Mol. Immunol. 2020;117:201–215. doi: 10.1016/j.molimm.2019.11.014. [DOI] [PubMed] [Google Scholar]
- 69.Efremova M., Vento-Tormo M., Teichmann S.A., Vento-Tormo R. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc. 2020;15:1484–1506. doi: 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- 70.Browaeys R., Saelens W., Saeys Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods. 2020;17:159–162. doi: 10.1038/s41592-019-0667-5. [DOI] [PubMed] [Google Scholar]
- 71.Jacobetz M.A., Chan D.S., Neesse A., Bapiro T.E., Cook N., Frese K.K., Feig C., Nakagawa T., Caldwell M.E., Zecchini H.I., et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62:112–120. doi: 10.1136/gutjnl-2012-302529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zanoni M., Piccinini F., Arienti C., Zamagni A., Santi S., Polico R., Bevilacqua A., Tesei A. 3D tumor spheroid models for in vitro therapeutic screening: a systematic approach to enhance the biological relevance of data obtained. Sci. Rep. 2016;6 doi: 10.1038/srep19103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Magrath J.W., Kang H.-J., Hartono A., Espinosa-Cotton M., Somwar R., Ladanyi M., Cheung N.-K.V., Lee S.B. Desmoplastic small round cell tumor cancer stem cell-like cells resist chemotherapy but remain dependent on the EWSR1-WT1 oncoprotein. Front. Cell Dev. Biol. 2022;10 doi: 10.3389/fcell.2022.1048709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pérez-González A., Bévant K., Blanpain C. Cancer cell plasticity during tumor progression, metastasis and response to therapy. Nat. Cancer. 2023;4:1063–1082. doi: 10.1038/s43018-023-00595-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Davies A., Zoubeidi A., Beltran H., Selth L.A. The Transcriptional and Epigenetic Landscape of Cancer Cell Lineage Plasticity. Cancer Discov. 2023;13:1771–1788. doi: 10.1158/2159-8290.CD-23-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Burkhardt D.B., San Juan B.P., Lock J.G., Krishnaswamy S., Chaffer C.L. Mapping Phenotypic Plasticity upon the Cancer Cell State Landscape Using Manifold Learning. Cancer Discov. 2022;12:1847–1859. doi: 10.1158/2159-8290.CD-21-0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Möller E., Praz V., Rajendran S., Dong R., Cauderay A., Xing Y.-H., Lee L., Fusco C., Broye L.C., Cironi L., et al. EWSR1-ATF1 dependent 3D connectivity regulates oncogenic and differentiation programs in Clear Cell Sarcoma. Nat. Commun. 2022;13:2267. doi: 10.1038/s41467-022-29910-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jerby-Arnon L., Neftel C., Shore M.E., Weisman H.R., Mathewson N.D., McBride M.J., Haas B., Izar B., Volorio A., Boulay G., et al. Opposing immune and genetic mechanisms shape oncogenic programs in synovial sarcoma. Nat. Med. 2021;27:289–300. doi: 10.1038/s41591-020-01212-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Luo H., Xia X., Huang L.-B., An H., Cao M., Kim G.D., Chen H.-N., Zhang W.-H., Shu Y., Kong X., et al. Pan-cancer single-cell analysis reveals the heterogeneity and plasticity of cancer-associated fibroblasts in the tumor microenvironment. Nat. Commun. 2022;13:6619. doi: 10.1038/s41467-022-34395-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhu K., Cai L., Cui C., de Los Toyos J.R., Anastassiou D. Single-cell analysis reveals the pan-cancer invasiveness-associated transition of adipose-derived stromal cells into COL11A1-expressing cancer-associated fibroblasts. PLoS Comput. Biol. 2021;17 doi: 10.1371/journal.pcbi.1009228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chromium Single Cell 3’ Reagent Kits User Guide (v3.1 Chemistry) -User Guide -Library Prep -Single Cell Gene Expression -Official 10x Genomics Support https://support.10xgenomics.com/single-cell-gene-expression/library-prep/doc/user-guide-chromium-single-cell-3-reagent-kits-user-guide-v31-chemistry.
- 82.Hafemeister C., Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bergen V., Lange M., Peidli S., Wolf F.A., Theis F.J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 2020;38:1408–1414. doi: 10.1038/s41587-020-0591-3. [DOI] [PubMed] [Google Scholar]
- 85.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single cell data. bioRxiv. 2018 doi: 10.1101/460147. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Buenrostro J.D., Wu B., Chang H.Y., Greenleaf W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015;109:21.29.1–21.29.9. doi: 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Arrigoni L., Richter A.S., Betancourt E., Bruder K., Diehl S., Manke T., Bönisch U. Standardizing chromatin research: a simple and universal method for ChIP-seq. Nucleic Acids Res. 2016;44:e67. doi: 10.1093/nar/gkv1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.La Manno G., Soldatov R., Zeisel A., Braun E., Hochgerner H., Petukhov V., Lidschreiber K., Kastriti M.E., Lönnerberg P., Furlan A., et al. RNA velocity of single cells. Nature. 2018;560:494–498. doi: 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.-C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Talevich E., Shain A.H., Botton T., Bastian B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016;12 doi: 10.1371/journal.pcbi.1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Corces M.R., Trevino A.E., Hamilton E.G., Greenside P.G., Sinnott-Armstrong N.A., Vesuna S., Satpathy A.T., Rubin A.J., Montine K.S., Wu B., et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods. 2017;14:959–962. doi: 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ewels P.A., Peltzer A., Fillinger S., Patel H., Alneberg J., Wilm A., Garcia M.U., Di Tommaso P., Nahnsen S. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020;38:276–278. doi: 10.1038/s41587-020-0439-x. [DOI] [PubMed] [Google Scholar]
- 93.Welch R.P., Lee C., Imbriano P.M., Patil S., Weymouth T.E., Smith R.A., Scott L.J., Sartor M.A. ChIP-Enrich: gene set enrichment testing for ChIP-seq data. Nucleic Acids Res. 2014;42:e105. doi: 10.1093/nar/gku463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fornes O., Castro-Mondragon J.A., Khan A., van der Lee R., Zhang X., Richmond P.A., Modi B.P., Correard S., Gheorghe M., Baranašić D., et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020;48:D87–D92. doi: 10.1093/nar/gkz1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Single-cell assays. AI: Adriamycin-Ifosfamide; IE: Ifosfamide-Etoposide; VAC: Vincristine-Actinomycin D-Cyclophosphamide; WES: Whole Exome Sequencing.
(B) DSRCT Bulk RNA-seq test cohort (N = 29). AI: Doxorubicin-Ifosfamide; API: Doxorubicin-CDDP-Ifosfamide; HIPEC: Hyperthermic Intra Peritoneal Chemotherapy; IVA: Ifosfamide-Vincristine-Doxorubicin; IVADo: Ifosfamide-Vincristine-Actinomycin-Doxorubicin; L1: First line; L2: Second line; TEMIRI: Temozolomide-Irinotecan; VAC: Vincristine-Actinomycin D-Cyclophosphamide; VDC: Vincristine-Doxorubicin-Cyclophosphamide; VIDE: Vincristine-Ifosfamide-Doxorubicin-Etoposide; VIP: Vincristine-Ifosfamide-CDDP; WES: Whole Exome Sequencing.
(A) Top 50 DEGs across main cell populations in Int_sc dataset.
(B) Top 50 DEGs across nearest neighbors' defined Int_sc clusters.
(C) Int_sc dataset clusters quality control metrics.
(A) Differentially bound genomics regions between EWSR1::WT1 and isotype ChIP-seq. The 262 genes significantly upregulated upon EWSR1::WT1 silencing intersected with genes corresponding to EWSR1::WT1 ″off" ATAC peaks and EWSR1::WT1 ChIP-enriched peaks are shown in orange.
(B) Motif enrichment analysis in significantly enriched peaks in EWSR1::WT1 ChIP assay.
(C) H3K27ac and EWSR1::WT1 co-occupied regions.
(D) H3K9ac and EWSR1::WT1 co-occupied regions.
(A) Differentially accessible genomics regions between EWSR1::WT1-silenced versus EWSR1::WT1-non-silenced JN-DSRCT-1 cell line. The 262 genes significantly upregulated upon EWSR1::WT1 silencing intersected with genes corresponding to EWSR1::WT1 ″off" ATAC peaks and EWSR1::WT1 ChIP-enriched peaks are shown in orange.
(B) Motif enrichment analysis in significantly enriched peaks in EWSR1::WT1-non-silenced JN-DSRCT-1 cells (EWSR1-W1 ″on").
(C) Motif enrichment analysis in significantly enriched peaks in EWSR1::WT1- -silenced JN-DSRCT-1 cells (EWSR1-W1 ″off").
The 262 genes significantly upregulated upon EWSR1::WT1 silencing intersected with genes corresponding to EWSR1::WT1 ″off" ATAC peaks and EWSR1::WT1 ChIP-enriched peaks are shown in orange.
(A) Top 50 DEGs across snMultiome WNN clusters.
(B) Top 50 differentially accessible peaks across snMultiome WNN clusters.
(A) EWSR1::WT1 regulon.
(B) Top 10 SCENIC+ regulons per snMultiome WNN cluster. RSS relates to Regulon Specificity Score.
(A) Differential gene expression (DGE) analysis between DSRCT bulk RNA-seq groups defined by hierarchical clustering. Significantly enriched genes for each group paired comparison is displayed.
(B) Sarcoma bulk RNA-seq external cohort. The number of samples per histotype is shown in the second column.
(C) Specificity and prognostic value of scRNA-seq signatures. The second column displays the proportion of significant p values (p < 0.05) over all 23 paired Tukey’s comparison tests performed on gene signature values (transcripts per million [TPM] geometric mean, see STAR methods) for DSRCT versus all other sarcoma subtypes. Each signature was significant (p < 0.05) for one-way ANOVA test. A number equal to or more than 18 of 23 (>75%) of comparisons was chosen as the threshold to call a signature « DSRCT-specific », i.e., signatures of clusters 0, 1, 4, 7, 8, and 11. The third column displays prognostic value and log rank p values for the test (N = 29) and validation (N = 21) DSRCT cohorts using the first scoring strategy (i.e., TPM geometric mean, see STAR methods) while the fourth column displays the same values using the second scoring strategy (VST-normalized raw count arithmetic mean, see STAR methods).
Data Availability Statement
-
•
Single-cell omics data (including scRNA-seq, snMultiome, and Visium assays) and DSRCT frozen tumor RNA-seq are publicly available on Gene Expression Omnibus (GEO) repository in count matrix format with the following accession number: GSE263523. Cell-line-based omics data (RNA-seq, ATAC-seq, ChIP-seq) are available processed and in fastq format using the same unique accession number, GSE263523.
-
•
Patient raw data reported in this study cannot be deposited in a public repository because of privacy concerns.
-
•
This paper does not report original code. All custom code used for the analyses was written with existing software as detailed in the STAR Methods section and is available upon request.
-
•
Any additional information required to reanalyze the data reported in this manuscript is available from the lead contact upon request.