

Abstract
The process of drug discovery is widely known to be lengthy and resource-intensive. Artificial Intelligence approaches bring hope for accelerating the identification of molecules with the necessary properties for drug development. Drug-likeness assessment is crucial for the virtual screening of candidate drugs. However, traditional methods like Quantitative Estimation of Drug-likeness (QED) struggle to distinguish between drug and non-drug molecules accurately. Additionally, some deep learning-based binary classification models heavily rely on selecting training negative sets. To address these challenges, we introduce a novel unsupervised learning framework called DrugMetric, an innovative framework for quantitatively assessing drug-likeness based on the chemical space distance. DrugMetric blends the powerful learning ability of variational autoencoders with the discriminative ability of the Gaussian Mixture Model. This synergy enables DrugMetric to identify significant differences in drug-likeness across different datasets effectively. Moreover, DrugMetric incorporates principles of ensemble learning to enhance its predictive capabilities. Upon testing over a variety of tasks and datasets, DrugMetric consistently showcases superior scoring and classification performance. It excels in quantifying drug-likeness and accurately distinguishing candidate drugs from non-drugs, surpassing traditional methods including QED. This work highlights DrugMetric as a practical tool for drug-likeness scoring, facilitating the acceleration of virtual drug screening, and has potential applications in other biochemical fields.
Keywords: drug-likeness, deep learning, unsupervised learning, chemical space
Introduction
Identifying compounds with drug-like properties is a key factor in the success of new drug development. Drug-likeness is an important criterion used to assess the potential of compounds for drug development. This indicator helps to screen out compounds that may fail in clinical trials at an early stage, which is significant for increasing the success rate of drug development and reducing costs [1, 2]. Since the factors determining drug-likeness are numerous and involve chemical properties closely related to the biological activity, metabolism and transport characteristics of the drug, such as molecular weight, lipophilicity and the number of hydrogen bond donors and acceptors, drug-likeness cannot be simply quantified through experimental means directly [3–8]. Over the years, researchers have developed various methods to characterize drug-likeness [9].
In the early stages of drug-likeness assessment, researchers established rules based on physicochemical properties, such as Lipinski’s ’Rule of Five’ (RO5), which define standard features of drug molecules, for example, a molecular weight of less than 500, a logP of less than 5, fewer than 5 hydrogen bond donors and fewer than 10 hydrogen bond acceptors [10]. Other rules, including the Ghose filter, Veber rules and Egan rules, also explored the correlation between drug-likeness and physicochemical properties and expanded the parameters considered by RO5 [11–13].
However, these rule-based methods have limitations. They provide a ’pass/fail’ assessment based on specific thresholds rather than quantifying drug-likeness, which means they cannot differentiate between compounds with varying degrees of drug-likeness [14, 15]. This means that although some compounds may only slightly exceed the set thresholds, they still have the potential to become effective drugs. This approach could lead to missing some promising drug candidates. Furthermore, drug development is a complex process that requires a balance between efficacy, safety and manufacturability, and the absence of a quantitative drug-likeness score can increase uncertainty in the decision-making process for drug development teams [16].
To address the limitations of drug-likeness rules based on physical and chemical properties, researchers have developed a scoring method called Quantitative Estimate of Drug-likeness (QED) [17]. QED combines eight physicochemical properties (molecular weight, LogP, H-bond donors, H-bond acceptors, charge, aromaticity, stereochemistry and solubility), generating a score between 0 and 1. The closer the score is to 1, the more drug-like the molecule. Due to its computational convenience, QED, as a molecular evaluation index, is widely used in the field of Artificial Intelligence (AI)-assisted drug discovery. In deep learning generative models (such as variational autoencoders (VAEs), generative adversarial networks (GANs), etc.), QED can serve as an optimization target or assessment metric, guiding the model to generate compounds with high drug-likeness. In reinforcement learning, QED can be used as a reward function, guiding the agent to design compounds with high drug-likeness. In the optimization problem considering multiple drug properties (such as activity, toxicity, pharmacokinetic properties, etc.), QED can serve as one of the optimization targets, helping to balance various properties and generate compounds with high drug-likeness [5, 18–23]. However, some studies have shown that QED cannot distinguish between drug and non-drug molecules [24–26], and the eight physicochemical properties selected by QED cannot fully cover all factors affecting whether a compound has drug-likeness. For example, some natural products with important biological activity, such as antibiotics or antitumor agents, may violate some traditional drug-likeness rules [27]. QED simplifies multiple properties into a single score, which may oversimplify the complexity of drug-likeness. Drug-likeness is determined by a variety of factors, including physicochemical properties, biological activity, pharmacokinetic properties, toxicity, etc. The interaction of these factors may be more important than any single factor [28]. This prompts researchers to explore alternative methods, such as deep learning techniques, to better predict drug-likeness.
Deep learning methods significantly differ from traditional methods in terms of feature extraction from raw data. The performance of deep learning models is largely influenced by the quantity and quality of the training data [29]. However, the challenge is that drug-likeness is not a directly measurable quantity, and, currently, there are no molecular annotation data related to it, making the development of related regression models difficult. Therefore, most of the deep learning methods currently used for drug-likeness prediction choose to use a binary classification approach, aiming to classify molecules as drugs or non-drugs. Hu et al. [30] used an autoencoder-based classifier with approximately 700 chemical descriptors that can be obtained from a given molecule to distinguish drugs and ZINC molecules. Their method focuses on using autoencoders to learn the low-dimensional representation of molecules and training a binary classifier with these representations. Beker et al. [24] adopted a method that uses both autoencoders, Mol2vec and Graph Convolutional Network (GCN) models to predict the drug-likeness of molecules. Their method first extracts the features of the molecules with autoencoders and Mol2vec, then processes these feature graphs with GCN, and finally predicts the drug-likeness of the molecules with a classifier. Sun et al. [31] used a Graph Convolutional Attention Network (D-GCAN) to predict the drug-likeness of molecular structures. They used GCAN to directly operate on molecular graphs to learn the features of the molecules and then used these features to predict the drug-likeness of the molecules.
Although these models achieve high classification accuracy, they also have their potential limitations. First, the binary classification method inevitably requires drug molecules as the positive set and non-drug molecules as the negative set. The positive set can be easily prepared using known drug molecules [25]. However, the selection of non-drug negative samples is challenging, and the preparation of a comprehensive negative set with chemical diversity is not easy, as true non-drug molecules can only be verified through clinical trials [25]. Second, binary classification models tend to learn features specifically used to distinguish between drug and non-drug molecules, rather than learning the features of molecules themselves. This means that the learned features will be significantly influenced by the negative set used for training, i.e. a binary classification model trained in a specific negative set may not be able to distinguish non-drug-like molecules that are fundamentally different from those in the negative set, thus limiting the model’s generalizability.
To tackle the challenges above, we innovatively propose the DrugMetric model. We constructed a dataset based on potential drug candidates and selected three non-drug datasets with decreasing drug-likeness from ChEMBL, ZINC and GDB, informed by prior knowledge. Utilizing a Variational Autoencoder-Gaussian Mixture Model (VAE-GMM) architecture, we effectively delineated the chemical space distribution of these four datasets. By computing the distribution Distance, we not only clarified the relative distances between the chemical spaces of different datasets but also assigned drug-likeness labels to each molecule based on these distances. Finally, we employed ensemble learning techniques to enhance the predictive accuracy of our drug-likeness scoring model.
Our DrugMetric model has demonstrated exceptional performance across multiple tasks. Compared to the traditional QED method, DrugMetric exhibited higher accuracy in drug-likeness scoring tasks on Clinical Drug (CD), ChEMBL, ZINC and GDB dataset, anti-cancer dataset and the MoleculeNet molecular property prediction datasets. In distinguishing drugs from non-drugs, DrugMetric achieved AUC values of 0.83, 0.94 and 0.99 in three classification tasks, significantly outperforming existing methods. Furthermore, we conducted an in-depth analysis of DrugMetric’s potential and found a strong correlation with the hepatic microsomal stability data of candidate drugs. To enhance the accessibility of DrugMetric, we have made its code publicly available, allowing researchers to deploy the model locally and tailor it to their specific research needs.
Materials and methods
Datasets
The training datasets play a pivotal role in our study. The positive dataset, henceforth referred to as CD dataset, comprises known drug molecules from three reputable sources: (1) Clinical trial records from the PubChem database, a public repository for information on chemical substances and their biological activities [32]. (2) Molecules categorized as FDA-approved drugs, which have undergone extensive testing to confirm their safety and efficacy for human use [33]. (3) Drugs recorded in the World Drug Index (WDI), a directory of drugs that are marketed globally [34]. These sources were chosen based on their alignment with AI’s contributions to the identification and progression of potential drugs through the development pipeline.
For the negative dataset, we selected molecules from three distinct databases, each representing varying degrees of drug-likeness potential: - GDB17, a comprehensive virtual compound library generated computationally, contains molecules that are purely theoretical at this stage. - ZINC15, a collection of commercially available compounds that are often used in virtual screening during drug discovery processes. - ChEMBL, a curated database of bioactive molecules with experimentally measured bioactivity data. The negative set provides a gradient of drug-likeness, with GDB17 molecules being the least drug-like and ChEMBL molecules being the closest to drug-like properties.
To ensure the quality and relevance of data for our predictive models, we applied the following preprocessing steps to each dataset: 1. Removal of duplicate entries to maintain data integrity. 2. Exclusion of molecules exceeding a molecular weight of 1000 Dalton, as they typically face challenges related to bioavailability and membrane permeability. 3. Removal of molecules composed of fewer than six atoms to focus on compounds with sufficient complexity for potential biological activity. 4. Elimination of salts and preparations to avoid inaccuracies in computational models that predict solubility or stability.
To counteract the imbalance in dataset sizes, we employed random sampling to equilibrate the number of molecules across the three negative sets, ensuring that each has a representation equal to that of the CD dataset. This approach is based on the premise that balanced datasets can improve the performance and generalizability of machine learning models [25]. For a detailed breakdown of molecular counts in datasets used specifically for drug-likeness prediction tasks, refer to Table s4. We have updated the specific details of the datasets in to balance the positive dataset (CD), we sampled the three negative datasets (ChEMBL, ZINC and GDB) to match the number of molecules in the CD dataset, resulting in a 1:1:1:1 ratio across all four datasets.
Rationale and mechanism of DrugMetric
DrugMetric is a novel computational tool we have developed to assess the drug-likeness of molecules quantitatively. This tool aims to map the chemical properties of molecules to a score that reflects their potential as drug candidates. As illustrated in Fig. 1, DrugMetric employs a combination of a VAE and a GMM to analyze and differentiate the molecular drug-likeness within various datasets.
Figure 1.
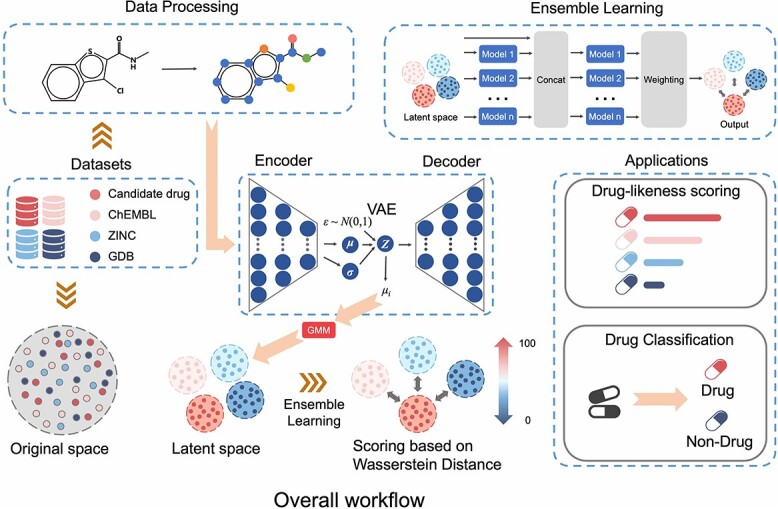
DrugMetric overall architecture. DrugMetric consists of four parts: data distribution acquisition through VAE; data distribution classification through Gaussian Mixture Model (GMM); setting of scoring labels based on inter-distribution distance; and selection of scoring model through ensemble learning.
The primary objective of DrugMetric is to categorize and score molecules by examining their distribution within a defined chemical space. It does this by leveraging the power of a VAE to capture the complex, high-dimensional distribution of molecular data and a GMM to classify these distributions into distinct clusters, each representing different grades of drug-likeness.
The VAE is predicated on the probabilistic framework of encoding and decoding. It transforms input data 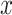 into a latent space representation
into a latent space representation 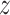 from which it seeks to reconstruct the original data. The mathematical construct that governs this process is known as the Evidence Lower Bound (ELBO), represented by Equation 1:
from which it seeks to reconstruct the original data. The mathematical construct that governs this process is known as the Evidence Lower Bound (ELBO), represented by Equation 1:
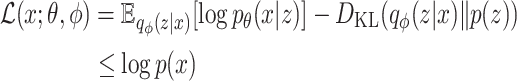 |
(1) |
The terms within the ELBO are delineated as follows:
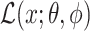 symbolizes the ELBO which the VAE seeks to maximize, indicating the model’s effectiveness in data encoding and reconstruction.
symbolizes the ELBO which the VAE seeks to maximize, indicating the model’s effectiveness in data encoding and reconstruction.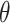 and
and 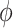 represent the parameters of the decoder and encoder, respectively.
represent the parameters of the decoder and encoder, respectively.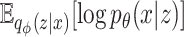 is the expected log-likelihood that quantifies the fidelity of the reconstruction from latent variables.
is the expected log-likelihood that quantifies the fidelity of the reconstruction from latent variables.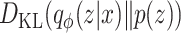 quantifies the Kullback–Leibler divergence, evaluating how the encoder’s approximate posterior distribution diverges from the prior distribution, thus acting as a regularization component.
quantifies the Kullback–Leibler divergence, evaluating how the encoder’s approximate posterior distribution diverges from the prior distribution, thus acting as a regularization component.
The encoder, parameterized by 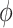 , maps the input data into distribution within the latent space, while the decoder, parameterized by
, maps the input data into distribution within the latent space, while the decoder, parameterized by 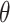 , reconstructs the data from this latent representation.
, reconstructs the data from this latent representation.
Following the training of the VAE on our datasets, we evaluate the generated molecules to ensure they align with the physicochemical properties of our training set. This alignment is critical as it confirms the VAE’s effectiveness in learning the essential features of drug-like molecules. Figure 2(A–F) compares ALERTS, FractionCSP3, MW, ALOGP, PSA and atom nums, demonstrating that our model successfully generalizes from the training data.
Figure 2.

(A–F) Comparison of physicochemical properties between training molecules and generated molecules. (A) ALERTS: a system for evaluating potential adverse drug reactions; (B) ALOGP: a parameter describing hydrophobicity FractionCSP3, the proportion of saturated carbon centers in a molecule; (C) synthetic accessibility: Synthetic accessibility refers to how easily a chemical compound can be synthesized based on the complexity of its synthesis steps and the availability of required materials.; (D) Hydrogen Bond Donor (HBD): HBD is an atom or group in a molecule that can donate a hydrogen atom to form a hydrogen bond with an electronegative acceptor; (E) PSA: polar surface area, a descriptor of molecular polarity; (F) atom nums: the number of atoms in a molecule. The generated molecules consistently mimic these properties of the training molecules. (G) GMM-based multi-class classification in the chemical space derived from the VAE-trained on four datasets. The AUC value of 0.67 reflects the discriminative ability of the GMM. (H) Selection and performance of the final drug-likeness prediction model using a three-layer Stacking and eight-fold Bagging ensemble learning strategy. The chosen model, WeightedEnsemble L3, uses an ensemble method based on weighted averages of multiple base models.
To encapsulate the molecular information efficiently, we utilize the VAE’s encoder to map each molecule to a Gaussian distribution, characterized by 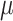 (mean) and
(mean) and 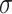 (standard deviation). However, for simplicity and stability in subsequent analysis, we extract only the mean vector
(standard deviation). However, for simplicity and stability in subsequent analysis, we extract only the mean vector 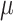 to represent the molecule’s position in chemical space.
to represent the molecule’s position in chemical space.
GMM to classify VAE-encoded molecule datasets
To enable the categorization of molecule datasets based on their drug-likeness levels, we employed a GMM on the latent space representations generated by a VAE. The latent space comprises molecular structures from four distinct datasets, each representing a unique level of drug-likeness. By configuring the GMM to recognize four Gaussian distributions within this space, we approach the task as a multi-class classification challenge, aiming to segregate the datasets according to their drug-likeness characteristics.
The model’s discriminative power was quantified using the Area Under the Curve (AUC) [35], which we calculated to be 0.67, as shown in Fig. 2(G). Our AUC value implies that the GMM can moderately distinguish between the different levels of drug-likeness in the combined chemical space.
Quantitative scoring of drug-likeness
After the GMM identifies the clusters, we assign a quantitative score to each molecule to represent its drug-likeness. This scoring is predicated on the molecule’s chemical proximity to a ’reference point’ determined by the Candidate drug dataset. For the calculation of this proximity, we utilize the Wasserstein distance [36], which is an effective metric for gauging dissimilarities between probability distributions that may not share common support. The Wasserstein distance, also known as Earth Mover’s Distance, is conceptually the cost necessary to transform one distribution into another. Its mathematical expression is given by
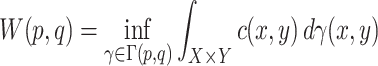 |
(2) |
where 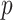 and
and 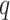 are the respective probability distributions,
are the respective probability distributions, 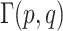 represents the set of all joint distributions with marginals
represents the set of all joint distributions with marginals 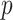 and
and 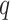 , and
, and 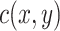 is a cost function that measures the ’distance’ between elements
is a cost function that measures the ’distance’ between elements 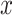 and
and 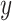 .
.
Given the mean vectors 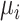 and covariance matrices
and covariance matrices 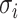 derived from the VAE-GMM process for the four datasets (CD, CHEMBL, ZINC, GDB), where
derived from the VAE-GMM process for the four datasets (CD, CHEMBL, ZINC, GDB), where 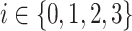 , the Wasserstein distance between two Gaussian distributions is notably simplified. For such distributions, the squared 2-Wasserstein distance can be computed as follows:
, the Wasserstein distance between two Gaussian distributions is notably simplified. For such distributions, the squared 2-Wasserstein distance can be computed as follows:
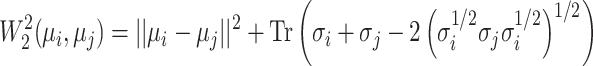 |
(3) |
The covariance matrices 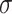 themselves are calculated using the formula:
themselves are calculated using the formula:
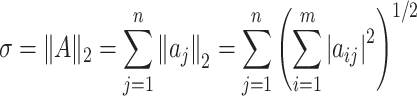 |
(4) |
where 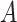 is a matrix with column vectors
is a matrix with column vectors 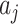 , and
, and 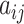 are the elements of
are the elements of 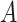 , with
, with 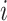 indexing rows and
indexing rows and 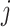 indexing columns.
indexing columns.
Scores are normalized to facilitate comparison across datasets, mapping them to a range from 0 to 100. A score of 100 corresponds to the reference Candidate drug dataset, while 0 signifies the greatest distance within the chemical space. The normalization formula is
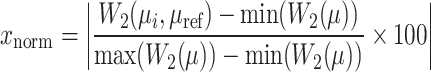 |
(5) |
where 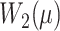 represents the Wasserstein distance for the molecule dataset, and
represents the Wasserstein distance for the molecule dataset, and 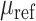 is the mean vector of the reference Candidate drug dataset.
is the mean vector of the reference Candidate drug dataset.
The normalized Wasserstein distances and their associated drug-likeness scores for molecules across the datasets are presented in Table 1. This scoring system offers a standardized approach for evaluating and contrasting the drug-likeness of compounds, thereby facilitating a more informed drug discovery process.
Table 1.
Normalized Wasserstein Distances and Drug-Likeness Scores
| Dataset | CD | ChEMBL | ZINC | GDB |
|---|---|---|---|---|
Normalized Score (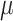 ) ) |
100 | 28.62 | 22.83 | 0 |
Normalized Score (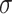 ) ) |
19.78 | 10.53 | 15.37 | 15.61 |
Employment of ensemble learning for optimal scoring model selection
The primary objective of this section is to develop a robust regression model that utilizes the drug-likeness scores derived from the previous stages as labels. This model aims to predict molecular drug-likeness with high accuracy by leveraging the strengths of ensemble learning techniques.
Graph Neural Networks (GNNs) have been acknowledged for their inherent capacity to process biochemical data effectively. However, recent investigations, including the work by Jiang et al. [37], indicate that the full potential of GNNs has yet to be realized in practice. Their study demonstrates that a synergy of GNNs (e.g. GCN) with well-established machine learning models (such as Support Vector Machines, Random Forests, and Extreme Gradient Boosting) can yield superior results in molecular property prediction tasks. This sentiment is echoed by Deng et al. [38] through their XGraphBoost method, which also validates the enhanced performance achieved by integrating traditional machine learning with GNNs.
These findings underpin our approach, suggesting that combining the innovative capabilities of GNNs with the robustness of traditional machine learning algorithms is a pragmatic strategy to fully harness their potential. The resulting hybrid models are anticipated to exhibit improved accuracy and reliability, particularly in the context of molecular property predictions.
To this end, we have adopted a three-layer Stacking coupled with an eight-fold Bagging ensemble learning strategy. Specifically, the training data are partitioned into three distinct layers, within which multiple foundational models are trained. Subsequently, the eight-fold Bagging technique is employed to further segregate the training data, ensuring that each foundational model is exposed to unique data subsets. This methodology is designed to bolster the model’s capacity for generalization while concurrently mitigating the likelihood of overfitting.
As depicted in Fig. 2H, the WeightedEnsemble L3 was selected as the ultimate model for predicting drug-likeness. This ensemble model operates on the principle of weighted averaging, where individual base models are assigned weights in proportion to their validation set performance. Such a weighting scheme empowers models demonstrating higher accuracy to exert greater influence on the ensemble’s final predictions, thereby elevating the predictive precision of the ensemble as a whole.
Experiments and results
Drug-Likeness scoring experiments
In this section, we detail our comparative analysis of DrugMetric and QED, two scoring systems designed to assess the potential drug-likeness of molecular compounds. Our goal is to elucidate the strengths and limitations of each system across various datasets and to provide insights into their relative effectiveness in aiding the drug discovery process.
Evaluation on diverse chemical datasets
To determine the efficacy of DrugMetric versus QED in predicting drug-likeness, we compared their scoring across four datasets: CD, ChEMBL, ZINC and GDB. These scores aim to quantify the potential for compounds within these datasets to qualify as drug candidates. In Fig. 3(A, B), DrugMetric’s scoring algorithm demonstrates a marked advantage over QED in evaluating the CD dataset, with scores of 67.15 compared to 51.27, respectively. This indicates a potential for DrugMetric to more accurately identify viable drug candidates within this set of compounds.
Figure 3.

Comparing DrugMetric Performance with QED across Datasets. (A,B) Shows the scores of DrugMetric and QED on the CD dataset, where DrugMetric (67.15) significantly outperforms QED (51.27). (C) Demonstrates the performance on a broad dataset of 195 anticancer drugs, with DrugMetric scoring an average of 70.01, significantly higher than QED’s 43.04. (D,E) Depicts their performance on MoleculeNet’s  molecular property prediction datasets, where DrugMetric exhibits superior accuracy and versatility, while QED demonstrates relatively weaker performance. These results underscore the potential and value of DrugMetric in practical applications.
molecular property prediction datasets, where DrugMetric exhibits superior accuracy and versatility, while QED demonstrates relatively weaker performance. These results underscore the potential and value of DrugMetric in practical applications.
Our analysis also shows that DrugMetric’s scores align with the expected drug-likeness hierarchy across the datasets: CD (67.15) > ChEMBL (41.45) > ZINC (30.41) > GDB (7.56). This gradation suggests that DrugMetric effectively differentiates between datasets with varying levels of drug-likeness and is consistent with the current understanding of these compound libraries.
Conversely, QED’s performance raises some concerns. Notably, it assigns the highest score to the ZINC dataset (72.99), which is counterintuitive given that ZINC, while rich in compounds with favorable physicochemical properties, may not necessarily comprise molecules with validated biological activity. This discrepancy highlights a potential overestimation by QED of the drug-likeness of ZINC’s compounds.
Additionally, QED’s lowest score for the CD dataset (51.27), which theoretically should contain highly drug-like molecules, suggests a limitation of the QED scoring system in recognizing complex or unique drug-like properties. This could lead to overlooking compounds with drug development potential.
Evaluation on anti-cancer drug dataset
Our study extends to an anti-cancer dataset comprising 195 drugs [39], chosen due to the critical nature of cancer therapeutics in global healthcare. Anti-cancer drugs often present unique challenges in drug-likeness evaluation due to their complex molecular structures designed to target specific biomarkers [40, 41]. Traditional scoring systems like QED may not fully account for these complexities, potentially underestimating the drug-likeness of compounds with unconventional structures or mechanisms.
In our analysis, as depicted in Fig. 3(C), DrugMetric consistently awarded higher scores to anti-cancer drugs, with an average of 70.01 compared to QED’s 43.04. This suggests that DrugMetric may possess a more refined algorithm capable of recognizing the specialized characteristics of anti-cancer drugs, thereby offering a more accurate assessment of their potential as therapeutic agents.
Evaluation on moleculenet datasets
To assess the generalizability of DrugMetric and QED, we evaluated their performance across nine MoleculeNet datasets [42], encompassing a range of molecular properties indicative of drug-likeness, such as biological activity, toxicity, solubility and pharmacokinetics.
DrugMetric demonstrated superior performance in datasets intrinsically linked to drug-like properties, including SIDER, BBBP, ToxCast, Tox21, ClinTox, FreeSolv and ESOL. The scoring trends observed (refer to Fig. 3DE) highlight DrugMetric’s proficiency in evaluating molecular properties that are directly relevant to drug discovery—particularly its precision and stability in scoring.
Conversely, in the qm7 and qm8 datasets, which focus primarily on quantum mechanical properties, the distinction between the scoring systems was less marked. This parity indicates that for molecular properties that extend beyond typical drug-like attributes, both DrugMetric and QED perform similarly. Nevertheless, it is noteworthy that DrugMetric’s core strength lies in its application to drug-likeness evaluation, where it demonstrated enhanced accuracy and practical utility.
Implications for drug discovery
The comparative analysis between DrugMetric and QED offers valuable insights for drug discovery, particularly when choosing a scoring system that can accurately reflect a compound’s potential as a drug candidate. DrugMetric’s superior performance across various datasets, including CD, ChEMBL, ZINC and GDB, reveals its robustness in identifying compounds with high drug-likeness, as supported by the scores presented in Fig. 3(A).
The ability of DrugMetric to adhere to the expected drug-likeness hierarchy—demonstrating the highest scores for compounds in the CD dataset and progressively lower scores for ChEMBL, ZINC and GDB—aligns with industry knowledge of these libraries. Such clear differentiation is critical in guiding researchers toward molecules with the highest potential for drug development, streamlining the selection process in the early stages of discovery.
In contrast, the inconsistencies observed in QED’s performance, particularly its overestimation of the ZINC dataset’s drug-likeness, underscore the need for DrugMetric that can discern beyond mere physicochemical properties. There is a significant demand in the pharmaceutical industry for tools that can capture the nuanced attributes of drug-like molecules, especially those with complex structures or mechanisms of action that are not adequately quantified by traditional scoring criteria.
Furthermore, the lower scores assigned by QED to the CD dataset, which is expected to contain highly drug-like molecules, raise concerns about its utility in current drug discovery paradigms. The risk of missing out on valuable drug candidates due to an underestimation of their drug-likeness underscores the importance of selecting a scoring system that is sensitive to the multifaceted nature of drug compounds.
Drug classification experiment
To comprehensively evaluate the classificatory ability of DrugMetric and other drug-likeness indices, we conducted a series of experiments.
To assess the efficacy of these scoring systems, we employed the Receiver Operating Characteristic (ROC) curve, a widely used tool in machine learning for diagnostic testing. The ROC curve effectively illustrates a scoring system’s ability to distinguish between drug-like and non-drug-like molecules by plotting the True Positive Rate (TPR) against the False Positive Rate (FPR). An ideal scoring system would yield an ROC curve that closely tracks the upper left corner of the graph, indicating high sensitivity (TPR) and specificity (1-FPR), with an Area Under the Curve (AUC) approaching the maximum value of 1. We define the TPR and FPR as follows: TPR, which reflects the proportion of true drug-like molecules correctly identified by the scoring system:
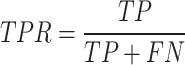 |
(6) |
FPR, which measures the proportion of non-drug-like molecules incorrectly classified as drug-like:
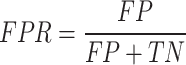 |
(7) |
Evaluation on diverse chemical datasets
In the initial, we utilized four datasets: CD, ChEMBL, ZINC and GDB. The purpose was to test DrugMetric alongside QED, as well as four renowned pharmaceutical evaluation criteria—GSK, Pfizer, Lipinski’s ’Rule of Five’ and the Golden Triangle—for comparative analysis. Specific details are provided in Supplementary Table S6.
The comparative ROC analysis revealed that the DrugMetric scoring system exhibited superior performance, achieving the highest AUC value of 0.99 in the comparison between the GDB and CD datasets (Fig. 4C). This result highlights DrugMetric’s exceptional ability to accurately categorize drug-like molecules. In contrast, the QED scoring system showed variable performance, with its lowest AUC at 0.21 when applied to the ZINC/CD dataset pairing, as shown in Fig. 4(B). The AUC values for the classical pharmaceutical criteria ranged from 0.29 to 0.56, indicating a moderate capability in the classification task.
Figure 4.

comparison of ROC curves of selected drug classification indicators. In addition to comparing the drug versus non-drug distinction performance of DrugMetric and QED, four rules used for drug screening were also tested: Pfizer, GSK, Lipinski, and Golden Triangle. (A) CD as the positive set, ChEMBL as the negative set. (B) CD as the positive set, ZINC as the negative set. (C) CD as the positive set, GDB as the negative set. In all three experiments, DrugMetric achieved the highest classification performance (0.83, 0.94, 0.99), whereas QED’s ability to distinguish between drugs and non-drugs was poorer than random guessing (0.44, 0.21, 0.29).
Comparation with ADMET-score
this experiment incorporated datasets provided by ADMET-score[43]: DrugBank, WITHDRAW[44] and ChEMBL. Due to the proprietary nature of ADMET-score, the analysis was based on pre-scored data. ADMET-score is a drug-likeness evaluation tool that aggregates data from 18 key ADMET parameters to create a comprehensive index. Although ADMET-score is not open-source, we utilized three datasets with available ADMET-score data for our analysis: DrugBank (n = 796), WITHDRAW (n = 240) and ChEMBL (n = 1954).
The WITHDRAW dataset includes 240 drugs that were withdrawn from the market primarily due to safety concerns. This dataset is particularly valuable for testing drug-likeness and safety profiles because it represents real-world scenarios where drugs that initially seemed promising were later found to have critical issues.
In the comparative analysis of ROC curves using DrugMetric, QED and ADMET-score, the performance of these scoring systems provides deep insights into the unique characteristics of the WITHDRAW dataset.
Firstly, in the comparison between DrugBank and WITHDRAW, all methods showed relatively low AUC scores (DrugMetric at 0.58, ADMET-score at 0.60 and QED at 0.52 Fig. 5C), which might initially seem disappointing. However, this result is actually very informative. The WITHDRAW dataset contains compounds that were once considered drug-like but were later removed from the market due to safety reasons. The moderate performance of these scoring systems suggests a potential gap in their ability to discern long-term safety issues from basic drug-like properties. This is particularly critical because a drug’s initial ’drug-like’ qualities (such as bioavailability, solubility, permeability) do not necessarily correlate with its safety profile, which often involves more complex, long-term biological interactions.
Figure 5.

Comparison of ROC curves for drug classification using DrugMetric, QED and ADMET-score across various dataset pairings. (A) DrugBank vs. ChEMBL, where DrugMetric achieved the highest AUC of 0.88, followed by QED with 0.79 and ADMET-score with 0.65. (B) ChEMBL versus WITHDRAW, with DrugMetric recording an AUC of 0.87, QED at 0.79 and ADMET-score at 0.70. (C) DrugBank vs. WITHDRAW, showing DrugMetric with an AUC of 0.58, QED at 0.52 and ADMET-score at 0.60. These results demonstrate the varying effectiveness of each scoring system in distinguishing drug-like molecules in different dataset contexts.
The results from the comparison between ChEMBL and WITHDRAW further clarify this point. Even though DrugMetric shows improved performance (AUC of 0.87 Fig. 5B) compared to its results against the DrugBank dataset, the scoring system’s ability to differentiate between generally safe, market-approved drugs (from ChEMBL) and those known for safety issues (from WITHDRAW) indicates a robustness in identifying compounds with higher initial drug-likeness without necessarily accounting for the subtler, often delayed toxicity profiles.
Evaluation on 17 FDA-approved but toxic molecules
We have broadened our analysis to include the ClinTox dataset, which distinguishes between FDA-approved drugs and those that failed clinical trials due to toxicity issues. Notably, within this dataset, 17 compounds are identified as both FDA-approved and toxic. We designed a binary classification task to differentiate FDA-approved compounds that exhibit clinical toxicity (positive samples) from those without reported toxicity issues (negative samples).
Figure 6 provides a visual comparison of the performance between two metrics, DrugMetric and QED, in identifying FDA-approved but clinically toxic molecules. The results clearly demonstrate that DrugMetric significantly outperforms QED. Specifically, DrugMetric achieves an ROC AUC of 0.83, indicating a robust capability to distinguish between the two categories of compounds. In contrast, QED registers a considerably lower AUC of 0.39, reflecting its limited effectiveness in this specific task. However, it is important to note that the small size of the test data could potentially limit the reliability of these results.
Figure 6.
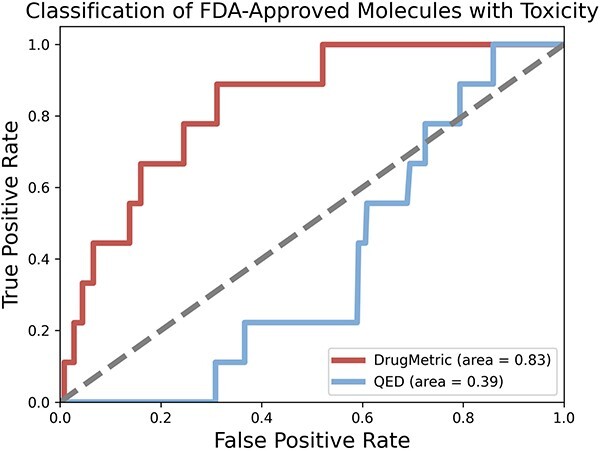
ROC curve comparison of DrugMetric and QED in identifying FDA-approved but clinically toxic molecules from the ClinTox dataset. The analysis targeted a subset of 17 FDA-approved toxic compounds. DrugMetric outperformed QED with an ROC AUC of 0.83 compared to 0.39 for QED.
In summary, DrugMetric emerges as an exceptionally effective scoring system for classifying drug candidate molecules, demonstrating superior performance over QED and other traditional metrics within the context of this study. The insights derived from our comprehensive analysis can significantly aid researchers in selecting the most appropriate scoring systems and refining their predictive models, thereby potentially enhancing the efficiency and success of the drug discovery processes. Yet, it also indicates limitations in detecting subtler, often delayed toxicity profiles.
Correlation analysis: liver microsomal stability and drugmetric of CDK2/4/6 inhibitors
Hepatic microsomes are organelles within liver cells that contain a variety of enzymes, including the cytochrome P450 enzymes, which are major pathways for drug metabolism [45]. The rate of drug metabolism determines the drug’s half-life, impacting its dosage and administration frequency [46]. Assessing drug stability in hepatic microsomes can therefore predict the metabolic rate in vivo citeobach1999prediction. This study evaluated the hepatic microsomal stability of 52 CDK2/4/6 kinase inhibitors in the cellular assay phase. The half-life (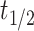 ) and clearance rate (
) and clearance rate ( ) of the drugs were determined using first-order kinetic equations. Liquid chromatography-tandem mass spectrometry methods quantified the concentration of compounds in the hepatic microsomes. The half-life is the time required for the drug concentration to reduce by half, calculated as follows:
) of the drugs were determined using first-order kinetic equations. Liquid chromatography-tandem mass spectrometry methods quantified the concentration of compounds in the hepatic microsomes. The half-life is the time required for the drug concentration to reduce by half, calculated as follows:
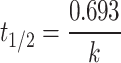 |
(8) |
Where the elimination rate constant ( ) is derived from the slope of the linear regression of the natural logarithm of concentration versus time. The volume of distribution (
) is derived from the slope of the linear regression of the natural logarithm of concentration versus time. The volume of distribution (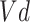 ) is inversely related to the drug’s microsomal concentration and is a critical determinant of the clearance rate. C represents the microsomal compound concentration:
) is inversely related to the drug’s microsomal concentration and is a critical determinant of the clearance rate. C represents the microsomal compound concentration:
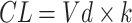 |
(9) |
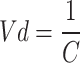 |
(10) |
Data regarding the half-lives of these inhibitors in both human and mouse microsomes were collected. To determine the association between DrugMetric scores and hepatic microsomal stability, molecules were ranked based on their DrugMetric scores, and both the top 10 (TOP-10) and bottom 10 (BOTTOM-10) molecules were selected for further analysis.
As shown in Table 2, the experimental results indicated that the TOP-10 molecules have an average half-life of 44.40 min in human-derived liver microsomes and 56.53 min in mouse-derived liver microsomes. Conversely, the BOTTOM-10 molecules have an average half-life of 30.32 min in human microsomes and 24.18 min in mouse microsomes. This suggests that molecules with higher DrugMetric scores may have slower metabolic rates, potentially leading to longer therapeutic effects. It is also observed that the same molecule may have different half-lives in human and mouse microsomes, possibly reflecting interspecies metabolic differences—an important factor to consider in drug development and pharmacokinetic studies.
Table 2.
Comparison of hepatic stability for high versus low ChemAIrank Molecules
| Mouse Half-life (mins) | Human Half-life (mins) | |
|---|---|---|
| TOP-10 | 56.53 | 44.40 |
| BOTTOM-10 | 24.18 | 30.32 |
Web server development
In order to achieve a more intuitive and user-friendly presentation of results, we developed the DrugMetric web server (Fig.7) using Streamlit [47]. Additionally, users can quickly deploy the service locally via the publicly available code repository.
Figure 7.

DrugMetric web server interface. DrugMetric web server allows users to type in or upload their mol file with multiple SMILES sequences and the backend model will check the validity of the molecule and predict the durg-likeness score.
In the DrugMetric web server, users can upload their molecular data for computations like drug-likeness scoring. The server will display the screened candidate compounds in an interactive visualized form, allowing users to conduct comparative analysis.
Conclusion
In conclusion, our study has demonstrated that DrugMetric, a computational tool leveraging a VAE coupled with a GMM, offers a significant advancement in assessing the drug-likeness of molecular entities. By meticulously curating datasets from diverse sources such as PubChem, FDA records and the WDI, and applying rigorous preprocessing steps including deduplication, molecular weight filtering and balancing via random sampling, we have established a robust foundation for model training and validation. DrugMetric not only surpasses the QED system in scoring drug-like properties but also exhibits superior performance compared to traditional pharmaceutical evaluation criteria.
The innovative use of ensemble learning, integrating GNNs with established machine learning techniques, has resulted in a regression model—WeightedEnsemble L3—that predicts molecular drug-likeness with high precision. This model’s superiority is evidenced by its judicious combination of multiple foundational models through stacking and bagging, thereby enhancing generalizability and mitigating overfitting.
DrugMetric is capable of accurately ranking compounds in various datasets, including CD, ChEMBL, ZINC and GDB, in alignment with industry expectations. This marks a crucial advancement in molecular drug-likeness prediction. By effectively distinguishing between molecules of varying drug-likeness and providing a quantitative measure that reflects the complex characteristics of drug-like molecules, DrugMetric is poised to become a key tool for researchers.
Our findings further reveal that DrugMetric scores correlate well with the hepatic microsomal stability of CDK2/4/6 kinase inhibitors, suggesting that higher scores are indicative of slower metabolic rates and potentially extended therapeutic effects. This correlation is particularly valuable in informing the pharmacokinetic profiling of drug candidates and underscores the necessity of accounting for interspecies variations in drug metabolism during the development process.
Ultimately, the efficacy of DrugMetric in enhancing the drug discovery pipeline is clear. Its methodological rigor, high predictive accuracy, and consistent performance across diverse datasets suggest that it can serve as a reliable guide in the prioritization and selection of promising drug candidates, potentially expediting the journey from concept to clinic.
Key Points
We introduce DrugMetric, a novel unsupervised learning framework combining a VAE and a GMM that enhances the precision and reliability of drug-likeness evaluation while addressing the limitations of existing methods.
DrugMetric employs a strategy that scores drug-likeness based on chemical space distance, leveraging unlabeled data to overcome the challenges of traditional methods.
DrugMetric consistently surpasses traditional drug-likeness scoring methods, proving its robustness and higher accuracy through comprehensive evaluations on various datasets, with implications for improving the drug discovery pipeline.
Supplementary Material
Acknowledgments
The authors thank the anonymous reviewers for their valuable suggestions.
Author Biographies
Bowen Li obtained his Master in Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences. He works as an algorithm engineer in Hangzhou Institute of Medicine, Chinese Academy of Sciences. His main research interest is computational biology.
Zhen Wang is currently a postdoc fellow at the Hangzhou Institute of Medicine, Chinese Academy of Sciences. He received his PhD. from the Department of Electronic Science and Technology, School of Electrical and Information Engineering, Hunan University. His research interests focus on artificial intelligence, drug discovery, and computational biology.
Ziqi Liu is a master's student at the Hangzhou Institute of Advanced Study, University of Science and Technology of China, with research interests in AIassisted drug design and molecular structure prediction.
Yanxin Tao is a PhD. candidate in Hangzhou Institute of Medicine, Chinese Academy of Sciences. His research interests focus on organic chemistry, medicinal chemistry and drug discovery.
Chulin Sha is an associate professor at the Hangzhou Institute of Medical Sciences, Chinese Academy of Sciences. Her research interest is bioinformatics.
Min He is a professor at Hangzhou Institute of Medical Sciences, Chinese Academy of Sciences. Her research interest is deep learning.
Xiaolin Li is a professor at Hangzhou Institute of Medical Sciences, Chinese Academy of Sciences. His research interest is artificial intelligence and drug discovery.
Contributor Information
Bowen Li, Hangzhou Institute of Medicine, Chinese Academy of Sciences, Hangzhou, 310018 Zhejiang, China.
Zhen Wang, Hangzhou Institute of Medicine, Chinese Academy of Sciences, Hangzhou, 310018 Zhejiang, China; College of Electrical and Information Engineering, Hunan University, Changsha, 410082 Hunan, China.
Ziqi Liu, Hangzhou Institute of Medicine, Chinese Academy of Sciences, Hangzhou, 310018 Zhejiang, China; Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou, 310024 Zhejiang, China.
Yanxin Tao, Hangzhou Institute of Medicine, Chinese Academy of Sciences, Hangzhou, 310018 Zhejiang, China.
Chulin Sha, Hangzhou Institute of Medicine, Chinese Academy of Sciences, Hangzhou, 310018 Zhejiang, China.
Min He, Hangzhou Institute of Medicine, Chinese Academy of Sciences, Hangzhou, 310018 Zhejiang, China; College of Electrical and Information Engineering, Hunan University, Changsha, 410082 Hunan, China.
Xiaolin Li, Hangzhou Institute of Medicine, Chinese Academy of Sciences, Hangzhou, 310018 Zhejiang, China; ElasticMind Inc, Hangzhou, 310018 Zhejiang, China.
Funding
This work is supported in part by funds from the National Key Research and Development Program of China (2022YFC3600902).
Author contributions
B.L. and X.L. conceived the research project. C.S., M.H. and X.L. supervised and advised the research project. B.L. and Z.W. designed and implemented the DrugMetric framework. B.L., Z.L. and Z.W. conducted the computational analyses. Y.T. provided the experimental data. B.L., Z.W., Z.L. and X.L. wrote the manuscript. All the authors discussed the experimental results and commented on the manuscript.
References
- 1. Ursu O, Rayan A, Goldblum A. et al.. Understanding drug-likeness. Wiley Interdiscip Rev Comput Mol Sci 2011;1:760–81. 10.1002/wcms.52 [DOI] [Google Scholar]
- 2. Leeson PD, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discov 2007;6:881–90. 10.1038/nrd2445 [DOI] [PubMed] [Google Scholar]
- 3. Kirchmair J, Göller AH, Lang D. et al.. Predicting drug metabolism: experiment and/or computation? Nat Rev Drug Discov 2015;14:387–404. 10.1038/nrd4581 [DOI] [PubMed] [Google Scholar]
- 4. Chen H, Engkvist O, Wang Y. et al.. The rise of deep learning in drug discovery. Drug Discov Today 2018;23:1241–50. 10.1016/j.drudis.2018.01.039 [DOI] [PubMed] [Google Scholar]
- 5. Zhavoronkov A, Ivanenkov YA, Aliper A. et al.. Deep learning enables rapid identification of potent ddr1 kinase inhibitors. Nat Biotechnol 2019;37:1038–40. 10.1038/s41587-019-0224-x [DOI] [PubMed] [Google Scholar]
- 6. Vamathevan J, Clark D, Czodrowski P. et al.. Applications of machine learning in drug discovery and development. Nat Rev Drug Discov 2019;18:463–77. 10.1038/s41573-019-0024-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stokes JM, Yang K, Swanson K. et al.. A deep learning approach to antibiotic discovery. Cell 2020;180:688–702.e13. 10.1016/j.cell.2020.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Paul D, Sanap G, Shenoy S. et al.. Artificial intelligence in drug discovery and development. Drug Discov Today 2021;26:80–93. 10.1016/j.drudis.2020.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clark DE, Pickett SD. Computational methods for the prediction of ‘drug-likeness’. Drug Discov Today 2000;5:49–58. 10.1016/S1359-6446(99)01451-8 [DOI] [PubMed] [Google Scholar]
- 10. Lipinski CA, Lombardo F, Dominy BW. et al.. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 1997;23:3–25. 10.1016/S0169-409X(96)00423-1 [DOI] [PubMed] [Google Scholar]
- 11. Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Comb Chem 1999;1:55–68. 10.1021/cc9800071 [DOI] [PubMed] [Google Scholar]
- 12.Plinski EF, Plinska S. Veber’s rules in terahertz light. Res. Sq. 2020, preprint. 10.21203/rs.2.22281/v1. [DOI]
- 13. Egan WJ, Merz KM, Baldwin JJ. Prediction of drug absorption using multivariate statistics. J Med Chem 2000;43:3867–77. 10.1021/jm000292e [DOI] [PubMed] [Google Scholar]
- 14. Patrick Walters W, Murcko MA. Prediction of ‘drug-likeness’. Adv Drug Deliv Rev 2002;54:255–71. 10.1016/S0169-409X(02)00003-0 [DOI] [PubMed] [Google Scholar]
- 15. Kelder J, Grootenhuis PDJ, Bayada DM. et al.. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm Res 1999;16:1514–9. 10.1023/A:1015040217741 [DOI] [PubMed] [Google Scholar]
- 16. Hughes JD, Blagg J, Price DA. et al.. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg Med Chem Lett 2008;18:4872–5. 10.1016/j.bmcl.2008.07.071 [DOI] [PubMed] [Google Scholar]
- 17. Bickerton GR, Paolini GV, Besnard J. et al.. Quantifying the chemical beauty of drugs. Nat Chem 20112012;4:90–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Olivecrona M, Blaschke T, Engkvist O. et al.. Molecular de-novo design through deep reinforcement learning. J Chem 2017;9:1–14. 10.1186/s13321-017-0235-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Popova M, Isayev O, Tropsha A. Deep reinforcement learning for de novo drug design. Sci Adv 2018;4. 10.1126/sciadv.aap7885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou Z, Kearnes S, Li L. et al.. Optimization of molecules via deep reinforcement learning. Sci Rep 2019;9. 10.1038/s41598-019-47148-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma C, Zhang X. GF-VAE: A Flow-based Variational Autoencoder for Molecule Generation. In Proceedings of the 30th ACM International Conference on Information & Knowledge Management, CIKM ‘21, pages 1181–1190, Virtual Event, Queensland, Australia, 2021. Association for Computing Machinery.
- 22.Lee S, Jo J, Hwang SJ. Exploring Chemical Space with Score-based Out-of-distribution Generation. In Proceedings of the 40th International Conference on Machine Learning, ICML'23, pages 18872–18892, Honolulu, Hawaii, USA, 2023. PMLR.
- 23.Jin W, Barzilay R, Jaakkola T. Junction Tree Variational Autoencoder for Molecular Graph Generation. In Proceedings of the 35th International Conference on Machine Learning, ICML'18, pages 2323–2332, Stockholm, Sweden, 2018. PMLR.
- 24. Beker W, Wołos A, Szymkuć S. et al.. Minimal-uncertainty prediction of general drug-likeness based on Bayesian neural networks. Nat Mach Intell 2020;2:457–65. 10.1038/s42256-020-0209-y [DOI] [Google Scholar]
- 25. Lee K, Jang J, Seo S. et al.. Drug-likeness scoring based on unsupervised learning. Chem Sci 2022;13:554–65. 10.1039/D1SC05248A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cai C, Lin H, Wang H. et al.. Midruglikeness: subdivisional drug-likeness prediction models using active ensemble learning strategies. Biomolecules 2022;13:29. 10.3390/biom13010029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Feher M, Schmidt JM. Property distributions: differences between drugs, natural products, and molecules from combinatorial chemistry. J Chem Inf Comput Sci 2003;43:218–27. 10.1021/ci0200467 [DOI] [PubMed] [Google Scholar]
- 28. Lipinski CA, Lombardo F, Dominy BW. et al.. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 2012;64:4–17. 10.1016/j.addr.2012.09.019 [DOI] [PubMed] [Google Scholar]
- 29. LeCun Y, Bengio Y, Hinton G. Deep learning. Deep learning nature 2015;521:436–44. 10.1038/nature14539 [DOI] [PubMed] [Google Scholar]
- 30. Qiwan H, Feng M, Lai L. et al.. Prediction of drug-likeness using deep autoencoder neural networks. Front Genet 2018;9:585. 10.3389/fgene.2018.00585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sun J, Wen M, Wang H. et al.. Prediction of drug-likeness using graph convolutional attention network. Bioinformatics 2022;38:5262–9. 10.1093/bioinformatics/btac676 [DOI] [PubMed] [Google Scholar]
- 32. Kim S, Thiessen PA, Bolton EE. et al.. Pubchem substance and compound databases. Nucleic Acids Res 2016;44:D1202–13. 10.1093/nar/gkv951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bobo D, Robinson KJ, Islam J. et al.. Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date. Pharm Res 2016;33:2373–87. 10.1007/s11095-016-1958-5 [DOI] [PubMed] [Google Scholar]
- 34. Langer T, Eder M, Hoffmann RD. et al.. Lead identification for modulators of multidrug resistance based on in silico screening with a pharmacophoric feature model. Arch Pharm 2004;337:317–27. 10.1002/ardp.200300817 [DOI] [PubMed] [Google Scholar]
- 35. Huang J, Ling CX. Using auc and accuracy in evaluating learning algorithms. IEEE Trans Knowl Data Eng 2005;17:299–310. 10.1109/TKDE.2005.50 [DOI] [Google Scholar]
- 36. Fournier N, Guillin A. On the rate of convergence in wasserstein distance of the empirical measure. Probab Theory Relat Fields 2015;162:707–38. 10.1007/s00440-014-0583-7 [DOI] [Google Scholar]
- 37. Jiang D, Zhenxing W, Hsieh CY. et al.. Could graph neural networks learn better molecular representation for drug discovery? A comparison study of descriptor-based and graph-based models. J Chem 2021;13:1–23. 10.1186/s13321-020-00479-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Deng D, Chen X, Zhang R. et al.. XGraphBoost: extracting graph neural network-based features for a better prediction of molecular properties. J Chem Inf Model 2021;61:2697–705. 10.1021/acs.jcim.0c01489 [DOI] [PubMed] [Google Scholar]
- 39. Pantziarka P, Rica Capistrano I, De Potter. et al.. An open access database of licensed cancer drugs. Front Pharmacol 2021;12:236. 10.3389/fphar.2021.627574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 41. Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer 2012;12:278–87. 10.1038/nrc3236 [DOI] [PubMed] [Google Scholar]
- 42. Zhenqin W, Ramsundar B, Feinberg EN. et al.. MoleculeNet: a benchmark for molecular machine learning. Chem Sci 2018;9:513–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guan L, Yang H, Cai Y. et al.. Admet-score–a comprehensive scoring function for evaluation of chemical drug-likeness. Medchemcomm 2019;10:148–57. 10.1039/C8MD00472B [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Siramshetty VB, Nickel J, Omieczynski C. et al.. WITHDRAWN-a resource for withdrawn and discontinued drugs. Nucleic Acids Res 2016;44:D1080–6. 10.1093/nar/gkv1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Peter F, Guengerich.. Cytochrome p450 and chemical toxicology. Chem Res Toxicol 2008;21:70–83. 10.1021/tx700079z [DOI] [PubMed] [Google Scholar]
- 46.Rowland M. Tozer TN. Clinical Pharmacokinetics and Pharmacodynamics, Wolters Kluwer Lippicott, Philadelphia, 1995.
- 47.Shukla S, Maheshwari A, Johri P. Comparative Analysis of ML Algorithms & Stream Lit Web Application. In Proceedings of the 2021 3rd International Conference on Advances in Computing, Communication Control and Networking (ICAC3N), pages 175–180, Greater Noida, India, 2021. IEEE.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.