

Abstract
The liver, the largest internal organ and a metabolic hub, undergoes significant declines due to aging, affecting mitochondrial function and increasing the risk of systemic liver diseases. How the mitochondrial three-dimensional (3D) structure changes in the liver across aging, and the biological mechanisms regulating such changes confers remain unclear. In this study, we employed Serial Block Face-Scanning Electron Microscopy (SBF-SEM) to achieve high-resolution 3D reconstructions of murine liver mitochondria to observe diverse phenotypes and structural alterations that occur with age, marked by a reduction in size and complexity. We also show concomitant metabolomic and lipidomic changes in aged samples. Aged human samples reflected altered disease risk. To find potential regulators of this change, we examined the Mitochondrial Contact Site and Cristae Organizing System (MICOS) complex, which plays a crucial role in maintaining mitochondrial architecture. We observe that the MICOS complex is lost during aging, but not Sam50. Sam50 is a component of the sorting and assembly machinery (SAM) complex that acts in tandem with the MICOS complex to modulate cristae morphology. In murine models subjected to a high-fat diet, there is a marked depletion of the mitochondrial protein SAM50. This reduction in Sam50 expression may heighten the susceptibility to liver disease, as our human biobank studies corroborate that Sam50 plays a genetically regulated role in the predisposition to multiple liver diseases. We further show that changes in mitochondrial calcium dysregulation and oxidative stress accompany the disruption of the MICOS complex. Together, we establish that a decrease in mitochondrial complexity and dysregulated metabolism occur with murine liver aging. While these changes are partially be regulated by age-related loss of the MICOS complex, the confluence of a murine high-fat diet can also cause loss of Sam50, which contributes to liver diseases. In summary, our study reveals potential regulators that affect age-related changes in mitochondrial structure and metabolism, which can be targeted in future therapeutic techniques.
Keywords: Aging, 3D Structure, Mitochondria, Metabolism, MICOS Complex, Liver Disease
Graphical Abstract:
Liver aging causes metabolic, lipidomic, and mitochondrial structural alterations, reflecting age-dependent losses in the MICOS complex. Diet-dependent losses of the SAM complex underlie genetic disease associations and mitochondrial structure.
INTRODUCTION:
Mitochondria are required for various purposes including oxidative phosphorylation, calcium homeostasis, and biochemical pathways (1). Mitochondria structure is crucial to many mitochondrial functions (2, 3). Unlike many other organelles, mitochondria have independent fusion and fission dynamics, which allow them to dynamically respond to their environment and adapt a variety of phenotypes beyond the spherical or tubular shapes that are typically associated with mitochondria (2, 4, 5). It is well understood that across aging, changes in morphology and size can impair mitochondrial functions and structures (6–8). Typically, these declines are measured through transmission electron microscopy (TEM), which offers high x- and y-resolutions (9, 10) providing insights into mitochondrial ultrastructure such as cristae, the folds of the inner mitochondrial membrane (11). However, a nascent research avenue is studying 3D phenotypes and exploring novel mitochondria structures (2, 12), such as donut-shaped mitochondria (13, 14), nanotunnels (15, 16), and megamitochondria (17). These structures require imaging techniques, such as serial block face-scanning electron microscopy (SBF-SEM), which allows 3D reconstruction of mitochondria (18–22). For many organs and experimental models, 3D reconstruction has not yet been performed, thus creating a gap in knowledge of 3D mitochondrial structural changes and molecular regulators that are present across the aging process in various tissues. Previously, we have demonstrated changes in mitochondria in other tissues, such as cardiac and skeletal muscle tissue (23, 24). Here, we aim to use 3D reconstruction to understand mitochondrial remodeling in the aging murine liver—which serves as the metabolic hub for the body.
The liver is the largest internal organ in vertebrates, performing essential biological functions such as detoxification, metabolism of drugs and nutrients, storage of minerals, and synthesis of plasma (25). The liver facilitates digestion and metabolism, which has an impact on the gastrointestinal and endocrine systems (26). Additionally, the liver generates carrier proteins crucial for growth and reproduction and participates in the metabolism of sex hormones (27). Therefore, studying liver metabolism will provide insights into many crucial biological processes including lipid and glucose homeostasis and detoxification pathways. One of the principal factors that deteriorate the liver is aging (26). After the age of 45, the percentage of mortality linked to liver disease rises significantly in humans (28). With aging comes a progressive expansion of the liver’s neural fat and cholesterol volumes and a rise in blood cholesterol, high-density lipoprotein cholesterol, and neutral fat levels (29). Volume alterations, polyploidy nuclei, the accumulation of dense bodies containing lipofuscin, a reduction in smooth endoplasmic reticulum area, and most importantly, a decrease in the quantity and functionality of mitochondria are all associated with aging in liver cells (29). During aging, lipids are also readily accumulated by the liver, raising the risk of cirrhosis, steatohepatitis, and non-alcoholic fatty liver disease (NAFLD), among other hepatic conditions (30, 31). Lipids are necessary for several essential mitochondrial functions, including metabolism and biogenesis, mitochondrial dynamics, oxidative phosphorylation, architecture, and structure of mitochondrial membranes (32). Additionally, the production of energy, regulation of cell death, and bioenergetics are all significantly impacted by the lipid content of the inner mitochondrial membrane (IMM) (32). Thus, as liver cells undergo mitochondrial dysfunction during aging, mitochondrial alterations may contribute to the age-related risk of systematic diseases (25, 33). Thus, within this study, we combine our murine approach of looking at mitochondrial structure with the examination of gross liver structure, lipogenesis, and genetically regulated gene expression in human cohorts.
Age-related changes in mitochondrial function are characterized by dynamic structural alterations within the mitochondria. Mitochondria are important for generating ATP through the electron transport chain in their cristae, folds of the IMM (11). Age-related liver disease may decrease ATP synthesis (34), resulting decreased energy allocation for essential functions. Beyond this, mitophagy, mitochondria DNA (mtDNA) mutations, disruptions in calcium homeostasis, and reactive oxygen species (ROS) have been implicated in many liver pathologies, such as alcohol-related liver disease, non-alcoholic fatty liver disease, chronic hepatitis B and hepatocellular carcinoma (35, 36). Through mitochondrial quality control mechanisms, oxidative stress, mtDNA, mitochondrial structure are highly interdependent (37, 38). Notably, changes in mitochondrial structure are often driven by changes in mitochondrial dynamic proteins (4, 39), which coordinate fusion and fission processes, allowing mitochondria to increase volume or repair mtDNA defects through fusion. In contrast, fission can form new mitochondria and remove damaged mitochondria (40). The organization of proteins belonging to the dynamin-related GTPase family and their adaptors controls this mitochondrial remodeling (41). The key regulators of mitochondrial dynamics are mitofusins 1 and 2 (MFN1 and MFN2) and optic atrophy 1 (OPA1). MFN 1 and 2 regulate outer mitochondrial membrane (OMM) fusion, whereas OPA1 regulates IMM fusion (42, 43). In contrast, fission is typically associated with dynamin-related protein 1 (DRP-1). DRP1 redistribution from the cytosol to the OMM is necessary for mitochondrial fission (42, 44–46). Dysregulation of these finely tuned dynamics can support pathophysiology; for example, in liver cancer, upregulation of Mfn1 and Opa1 play a role in cancer tumor cell growth mechanisms (5). Conversely, increased Drp1 and impaired Mfn1 expression were hallmarks of chronic alcohol exposure in mice; while other models show Drp1 downregulation upon chronic alcohol exposure (35). Deletion of mitochondrial fission factor, a recruitment factor of DRP1, in hepatocytes, has similarly led to altered mitochondrial morphology concomitant with enhanced susceptibility to NAFLD (47), but conversely, blocking fission can ameliorate hepatic steatosis and oxidative stress in NAFLD (48). This underscores that while traditional fusion and fission dynamic proteins can affect age-related risk to liver diseases, other regulators must be considered.
The OMM and IMM include hardwired translocase complexes with a very intricately specialized protein import mechanism that drives mitochondrial dynamics. These translocases operate along five distinct protein import pathways, one of which is through the SAM50 channel (49, 50). Sam50 is one of the crucial proteins of the outer membrane of the mitochondria. SAM50 is a SAM complex component with a β-barrel domain (50). Although the role of Sam50 in mitochondria is still unclear, studies showed that Sam50 contributes to the preservation of cristae’s structural integrity. Sam50 maintains the formation of the cristae via interaction with mitofilin and CHCHD6, which are parts of the mitochondrial contact site and cristae organizing system (MICOS) (51). Furthermore, Sam50 aids in the maintenance of the mitochondrial network (50), establishing and maintaining mitochondrial integrity and mitochondrial metabolism, with its loss conferring volumetric losses in mitochondria structure (52). Recently, Sam50 has been linked to NAFLD pathogenesis (49), but the interconnection with aging liver mitochondrial structure remains poorly elucidated.
In our study, we use human models to explore the impairments of the liver due to aging, investigating changes in the structure and physical appearance of the liver in young and old groups. To establish the tandem roles of MICOS and SAM50, we used two-year-old mice, which represented an elderly model, and three-month-old mice, which represented a juvenile phenotype. We then studied mitochondrial morphological changes in both two-dimensional (2D) and three-dimensional (3D) which showed age-related losses in volume and complexity. We also find that metabolomic and lipogenic changes in aging may mediate mitochondrial structural alterations in the aging liver. We further learned how the MICOS complex is dysregulated in the aging liver. Notably, deletion of the MICOS genes resulted in impairments in mitochondrial calcium regulation and ROS production in the liver. Due to the changes observed in mitochondria function and structure, we also investigated the role of diet-dependent Sam50 depletion in the liver. Lastly, we modeled genetically-regulated gene expression (GReX) of SAMM50 in a biobank of 85,615 participants, and found that SAMM50 GReX was significantly associated with multiple liver diseases.
RESULTS:
Aging Causes Alterations in Liver Lipid Accumulation and Altered Disease Markers
Damage to the liver’s tissue and alterations in hepatic metabolism might result from abnormal lipid levels in the liver (31). Thus, we utilized magnetic resonance imaging to determine how lipid content is regulated during aging in human samples. By enrolling female and male participants (Figures 1A–D), we created a “young” cohort consisting of individuals from 18 to 55 years old and an “old” cohort of individuals older than 60 years old. For both sexes, the liver’s lipid percentage increased through aging (Figures 1E–F). This age-related influx in liver validated the progression towards liver diseases in humans. Specifically, when combining the male and female cohorts together (Figure 1G), the mean fat percent increased (from a mean of 1.41% ± 1.44% SD to 5.93% ± 6.11% SD). Additionally, the 75% percentile of fat percentage was higher, at 10.5% in the aged cohort, as compared to 2.10% in the young cohort. Notably, Grade 1 of fat fraction classification, representing mild hepatic steatosis has a fat percentile cutoff of 6.5% or higher (53). To further validate these results, we switch to our mouse model and used Oil Red O staining to visualize lipids in 3-month and 2-year murine liver tissue (Figures 1H–K). Specifically, we saw an increased amount of Oil Red O amount in the 2-year cohort signifying an increase in lipid amount across the aging process in the liver, with a more than 6-fold increase in lipid droplets in 2-year samples (Figure 1L; 12.2 ± 6.55 SD, 3months; 77.5 ± 20.4 SD, 2-years). An uptick of lipid droplets in hepatocytes is indicative of NAFLD (54). To further explore markers of potential disease progression and dystrophy outside of lipid accumulation, we also looked at liver mass, which was decreased in aged samples when normalized to body weight (Figure 1M; 4.56% ± 0.329% SD, 3-months; 3.93% + 0.376% SD, 2-years). Since mtDNA content reduction is a hallmark of NAFLD (55), we also showed an age-dependent loss in mtDNA content (Figure 1N; mean: 1, 3-months; 0.805 ± 0.119 SD, 2-years). Additionally, bile acid, which is metabolized in the liver, has increased concentrations with abnormal liver function and disease states (56). We also looked at the concentration of bile acid, but no significant difference was noted (Figure 1O; 0.871 μMol/μg ± 0.138 μMol/μg SD, 3-months; 0.89 μMol/μg ± 0.15 μMol/μg SD, 2-years). Finally, central to the pathology and a key marker of NAFLD is high triglycerides (57, 58). We observed that aging murine samples had significantly elevated triglycerides in the liver (Figure 1P; 0.30 mmol/L ± 0.10 mmol/L SD, 3-months; 0.748 mmol/L ± 0.15 mmol/L SD, 2-years), which was more drastic than the general increase in serum triglycerides (Figure 1Q; 0.211 mmol/L ± 0.100 mmol/L SD, 3-months; 0.379 mmol/L ± 0.118 mmol/L SD, 2-years). Thus, while we could not confirm aging human and murine samples had liver disease, these results support the observation of an age-related progression toward liver disease.
Figure 1: Comparative analyses of liver lipid content and morphology in young and old patients.

Cross-sectional imaging of in-phase (left) and out-of-phase (right) liver anatomy data from (A) males under 55 years old (aged 14–53 years old; n = 10), (B) females under 55 years old (aged 17–41 years old; n = 10), (C) males over 60 years old (aged 60–85 years old; n = 10), and (D) females over 60 years old (aged 64–96 years old; n = 10). (E) Computed fat percentage in males, (F) females, and (G) combined young and old cohorts. (H) Oil Red O staining at 20X and (I) 40X magnification in young and (J-K) old samples. (L) Quantification of Oil Red O staining per area in 3-month and 2-year murine liver samples (n=10). (M) Quantifications of liver weight relative to total body weight as a percent (n=10), (N) relative reverse transcription-quantitative polymerase chain reaction mtDNA content (n=8), (O) bile acids concentrations (n=10), (P) liver triglycerides concentration (n=8), (Q) and serum triglycerides concentration (n=8). For all panels, error bars indicate SEM, Mann–Whitney tests were used for statistical analysis, and significance value indicate *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, and ns indicates non-significant.
Ultrastructural Changes in Murine Liver Reveals Aging is Associated with Lower Volume and Complexity
Next, we sought to explain whether these age-related changes in liver mass and disease markers are correlative with atypical mitochondria structures in the liver. For these studies, we utilized aged C57BL/6J mice at 2 age points, 3-months and 2-years, which are generally understood to be good representations of “young” and “old” aged points akin to aging displayed in humans (59).
To begin with, we used TEM to measure changes in mitochondrial morphology in aged mice (Figures 2A–D). Qualitatively, our findings of lipid accumulation were confirmed, with aging also showing lipid droplets that showed less circularity. TEM analysis showed that mitochondria count increased while the average mitochondrion area decreased with aging (Figures 2E–J). To compare sex-dependent differences, mitochondrial counts, as normalized to μm cell area, in 3-month males and females were similar (0.15 ± 0.02 SD, males; 0.174 ± 0.03 SD, females), there was a slight divergence with aging, although both followed a similar trend (Figures 2E, 2H; 0.56 ± 0.130 0.15 SD, males; 0.738 ± 0.14 SD, females). This sex-difference which is more pronounced with age was similarly seen in average mitochondrion area when comparing 3-month samples (1.27 μm2 ± 1.34 μm2 SD, males; 1.21 μm2 ± 1.19 μm2 SD, females) with their 2-year counterparts (Figures 2F, 2I; 0.233 μm2 ± 0.442 μm2 SD, males; 0.558 μm2 ± 0.386 μm2 SD, females). However, in many ways, TEM is inadequate to quantify mitochondria, which exist in diverse 3D shapes (2). Yet, given the best in-class resolution of TEM (9), it is useful for measuring subcellular structures of cristae. In male and female murine samples, we found a decrease in cristae score both (Figures 2G, 2J), which is a semi-qualitative measure (9), that evaluates the relative amount and architectural integrity of cristae. Neither 3-month (3.72 ± 0.44 SD, males; 3.47 ± 0.62 SD, females) nor 2-year (2.29 ± 0.71 SD, males; 1.99 ± 0.87 SD, females) samples showed a significant sex-dependent difference in cristae score. Together, these results highlight that cristae integrity is lower in aged liver tissue samples. Additionally, we performed these studies in both male and female samples, which showed slight sex-dependent differences but were generally minimal with similar trends. Given the time-consuming nature of manual segmentation associated with 3D reconstruction, we proceeded with the study utilizing a male cohort.
Figure 2: Transmission Electron Microscopy and Serial Block Face-Scanning Electron Microscopy Shows Changes in Mitochondria Murine Liver Across Aging.

(A) Representative transmission electron micrographs in males and (B) females from 3-month and (C-D) 2-year murine liver tissue. (E) Mitochondrial quantifications of male mitochondria number (n=21, 3-months; n=24, 2-years) (F) mitochondrial area (n=1309, 3-months; n=333, 2-years) (G) and cristae score (n=555, 3-months; n=555, 2-years). (H) Mitochondrial quantifications of female mitochondria number (n=21, 3-months; n=25, 2-years) (I) mitochondrial area (n=1253, 3-months; n=1018, 2-years) (J) and cristae score (n=684, 3-months; n=684, 2-years). (K) Schematic depicting removal of the liver. (L) Following embedded fixation, Serial Block Face-Scanning Electron Microscopy (SBF-SEM) allows for ortho-slice alignment. (M) Manual segmentation of ortho slices was performed to yield (N) 3-dimensional (3-D) reconstructions of individually colored mitochondria. (O) Representative ortho slice images from 3-month murine liver tissue and (P) 2-year murine liver tissue. (O’) Representative ortho slice images with 3D reconstructions of mitochondria overlaid from 3-month murine liver tissue and (P’) 2-year murine liver tissue. (O”) Isolated 3D reconstructions of mitochondria from 3-month murine liver tissue and (P”) 2-year murine liver tissue (Q) Mitochondrial quantifications mitochondrial perimeter, (R) area, (S) and volume. For SBF-SEM, in total, 3-month samples both included 750 (n=250, per mouse) for a total of 1500 mitochondria, which were used for statistical analysis. For all panels, error bars indicate SEM, Mann–Whitney tests were used for statistical analysis, and significance values indicate **P ≤ 0.01, ***P ≤ 0.001, and ns, not significant.
With the 2D mitochondrial differences observed, we utilized 3D reconstruction of SBF-SEM to further investigate changes in mitochondrial structure. For each age cohort, we analyzed approximately 250 mitochondria from the three mice surveyed (Figure 2K), for a total of around 750 mitochondria for each age point. SBF-SEM allowed for 50 μM intervals in the z-axis to be imaged for a total of 300 slices, of which 50 slices were used for analysis. The x- and y-resolution was 10 μM, which allowed for about 50 ortho slices to be reconstructed and quantified for each stack (Figure 2L), which then undergo manual contour segmentation (Figure 2M), for the 3D reconstruction of mitochondria (Figure 2N). Displayed first in both age cohorts is a representative of each orthoslice, which allows for the identification of mitochondria (Figures 2O–P). From there, manual segmentation across each ortho slice was performed in the software Amira, for mitochondria to be reconstructed (Figures 2O’–P’). Additionally, for better visualization of each individually colored mitochondria, the ortho slice may be removed (Figures 2O”–P”). Once modeled, 3D reconstructions (Videos 1–2) can be quantified. We found that in comparing the young and aged mice, size significantly decreased in all metrics (Figures 2Q–S). Specifically, the perimeter, which represents the total of all external distances in the mitochondria was approximately 20% lower in aged samples (Figure 2Q; 10761 μm ± 1560 μm SD, 3-months; 8054 μm ± 450 μm SD, 2 year). Three-dimensional area, which represents a metric of OMM area, was decreased by nearly 50% in aged samples (Figure 2R; 7.66 μm2 ± 0.808 μm2 SD, 3-months; 4.76 μm2 ± 0.511 μm2 SD, 2-years). Finally, mitochondrial volume, which represents the total of all internal pixels within the 3D mitochondrial reconstruction, was approximately 30% lower in aged male murine liver samples (Figure 2S; 1.01 μm3 ± 0.182 μm3 SD, 3-months; 0.696 μm3 ± 0.107 μm3 SD, 2-years). Losses in mitochondrial volume may indicate decreased internal volume for ATP synthesis (60), but mitochondrial roles extend beyond their energetics, including through interfacing with endoplasmic reticulum (ER) to modulate calcium homeostasis (61). Thus, it is equally important to look at their morphology and capacity to form compact sites.
To aid in visualizing mitochondria, we present mitochondria 3D reconstructions from transverse (Figures 3A–B) and longitudinal (Figures 3A’–B’) viewpoints (Videos 3–4). To further verify this change across aging we looked at sphericity and mitochondrial complexity index (MCI) (62), which are analogous measures of mitochondrial morphology. Sphericity (calculated as ), as exhibited an approximate 20% higher mean value (Figure 3C; 0.638 ± 0.0211 SD, 3-months; 0.785 ± 0.0257 SD, 2-years). MCI (calculated as Surface Area3 /16 π 2volume2) expectedly showed an inverse decrease in the aged cohort (Figure 3D; 3.22 ± 0.240 SD, 3-months; 1.64 ± 0.236 SD, 2-years). Together, these validate that complexity is decreased in aged murine liver samples. To better visualize these changes, we organized mitochondria by their volume, in a method known as mito-otyping, to compare morphology across size (Figure 3E). In the three-month age cohort (Figure 3E), we observed an expected phenotype of normally sized mitochondria. However, we also observed some diversity in mitochondrial structure, as we noted elongation, branching, and other structures that prioritize surface area over volume. In contrast, the 2-year sample showed much less heterogeneity and mitochondrial structures mostly presented as tubular or compact (Figure 3F). For these metrics, while intra-sample heterogeneity is plentiful, there is minimal inter-sample heterogeneity or outliers (sFigures 1A–E).
Figure 3: Serial Block Face-Scanning Electron Microscopy Shows Changes in Mitochondrial Morphology in Liver Across Aging.

(A) 3D reconstructions of mitochondria displayed from the transverse viewpoint in 3-month and (B) 2-year murine liver tissue. (A’) Representative images of 3D reconstructions of mitochondria displayed from the longitudinal viewpoint in 3-month and (B’) 2-year murine liver tissue. (C) Based on these measurements, the mitochondrial sphericity and (D) complexity index was determined. (E) Mito-otyping displays diversity in mitochondrial morphology across each relative volume of mitochondria in 3-month and (F) 2-year murine liver tissue. (G-H”) Qualitative identification of wrappER. (G) Orthogonal (ortho) slice from 3-month and (H) 2-year samples, with (G’-H’) 3D reconstruction of mitochondria and endoplasmic reticulum overlaid. (G”-H”) Isolated 3D reconstruction of mitochondria, in purple, and endoplasmic reticulum, in blue. For SBF-SEM, in total, 3-month samples both included 750 (n=250, per mouse) for a total of 1500 mitochondria, which were used for statistical analysis. For all panels, error bars indicate SEM, Mann–Whitney tests were used for statistical analysis, and significance value indicate ****P ≤ 0.0001.
These changes in sphericity and morphology may impact the ability of mitochondria to form contact sites. Particularly, liver mitochondria are known to exhibit phenotypes in which ER wraps around them, known as wrappER, to modulate lipid flux (63–65). To investigate this paradigm, we qualitatively identified wrappER within liver tissue (Figures 3G–H). Qualitatively, we found that wrappER exists in both 3-month and 2-year murine liver tissue (Figures 3G’–H’), but in 3-month tissue wrappER contact is much more extensive (Figures 3G”–H”). This suggests that liver lipid homeostasis may be negatively impacted (63–65). However, to confirm these changes and further explore the implications of these mitochondrial morphological alterations, we analyzed metabolism and performed lipidomics in mice.
Global metabolic and lipidomic profiling reveals altered energy metabolism and storage in the aged liver.
Due to observed changes in mitochondria in the aged liver, we proceeded to analyze alterations in small biomolecules regulating energy metabolism, cellular homeostasis, and storage. Our investigation involved global metabolic and lipidomic profiling of young and aged liver tissues, revealing dynamic features and significant changes in aged mouse livers (Figures 4A–O; sFigures 2A–D). We noted an accumulation of metabolites related to Vitamin A metabolism, specifically retinoic acid and retinal (Figures 4B–C). While Vitamin A metabolism must be finely balanced, within breast cancers retinoid changes can be associated with controlling the redox status of cytochrome c (66). This finding supports the presence of metabolic shifts, altered cellular signaling, and impaired clearance mechanisms in the aging liver, all attributed to changes in mitochondrial function. Our metabolomics analysis also uncovered disruptions in tricarboxylic acid (TCA) cycle intermediates—succinate and malate, indicating mitochondrial dysfunction and impaired activity (Figures 4D–E). These changes can impact the production and regulation of these intermediates. Furthermore, we observed decreases in nucleotide monophosphates involved in purine and pyrimidine metabolism—GMP, CMP, UMP, AMP (Figures 4F–I). The synthesis of these nucleotides, which involves mitochondrial processes, is influenced by age-related mitochondrial dysfunction, affecting the overall nucleotide biosynthesis pathway supporting our observations. Our results additionally confirmed existing literature on dysregulated nicotinamide adenine dinucleotide (NAD+) metabolism in the aging liver (67). Significant depletions in tissue NAD+, NADP, NMN pools, and tissue ADP were detected (Figure 8J-M). Historical liver studies suggest that mitochondrial NAD(H) pools tend to be oxidized, playing a crucial role in energy homeostasis. Conversely, cytosolic NADP(H) pools tend to be highly reduced for reductive biosynthesis (68). Our data reveals disruptions to mitochondrial activity reverses these classical mechanisms. In summary, these findings collectively support the presence of altered energy metabolism and cellular homeostasis in the metabolically active liver due to mitochondrial dysfunction and impairment.
Figure 4: Global metabolomic and lipidomic profiling revealed metabolic dysregulation and disruptions in lipid classes with age in liver tissues.
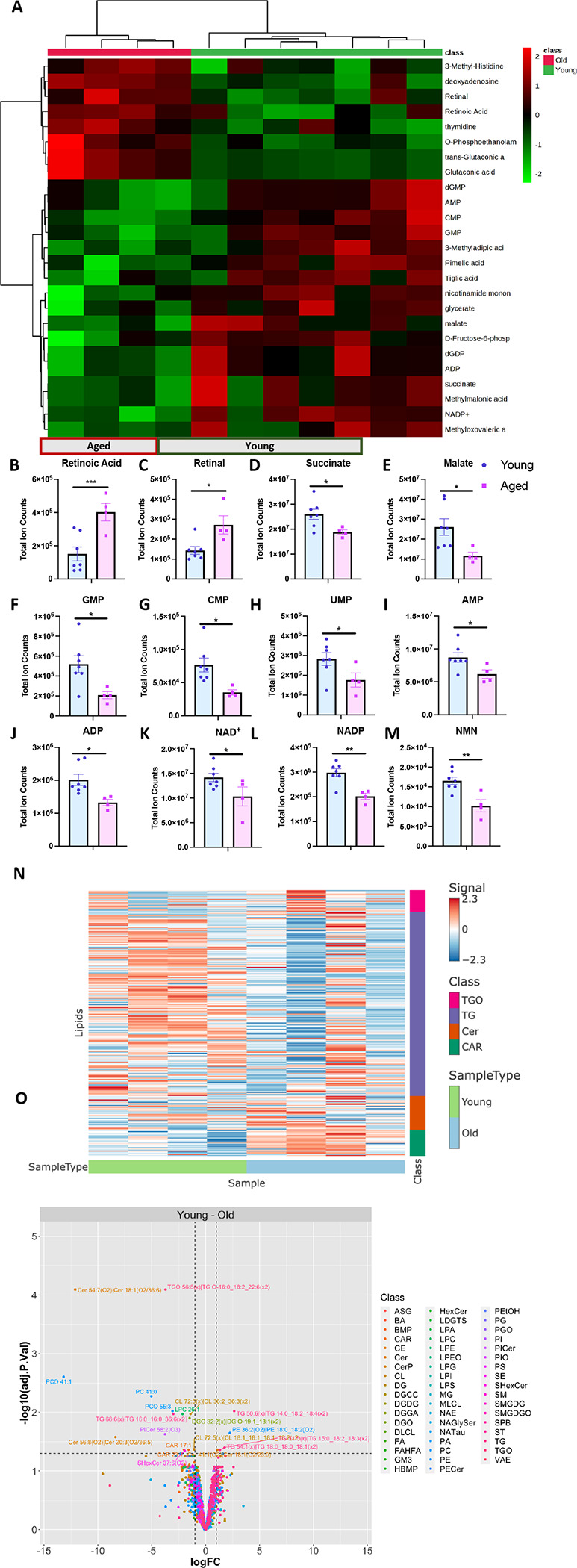
(A) Metabolomics heatmap showing the relative abundance of metabolites. (B-M) Metabolite pools illustrating the metabolic pathways that are altered with age in the liver– Vitamin A Metabolism, TCA Cycle, Nucleotide Metabolism, and NAD+ metabolism. (N) Heatmap showing enriched lipid classes based on comparison between young and old liver tissues. (O) Volcano plot labeling significant hits, which have adjusted p-value <0.05 and fold change (+ or −) greater than 1. For each tissue and metabolite in the heatmaps, the aged samples were normalized to the median of the young samples and then log2 transformed. Significantly different lipid classes represented in the figures are those with adjusted p-values < 0.05 (note: p-values were adjusted to correct for multiple comparisons using an FDR procedure) and log fold changes greater than 1 or less than −1. Young, n= 4; aged, n= 4. For all panels, error bars indicate SEM, ** indicates p< 0.01; and *p< 0.05, calculated with Student’s t-test.
Lipidomic profiling of both young and aged liver tissues unveiled age-related changes in lipid classes and chain lengths (Figure 9N-O; sFigures 2C–D). In the aging liver, significant alterations were observed in the triglycerides oligomers (TGO), triglycerides (TG), ceramide (Cer), and acylcarnitine (CAR) lipid classes compared to other lipid groups (Figures 4N–O, sFigure 2C). These lipid classes, each with distinct roles in liver mitochondria, contribute to various aspects of cellular metabolism and homeostasis. Disruptions in TGOs and TGs suggest imbalances in energy storage and release in the aging liver, possibly due to mitochondrial dysfunction. TGs, being the primary form of stored energy, can participate in mitochondrial beta-oxidation, providing acetyl-CoA for energy production. Conversely, Cer regulates apoptosis and modulates mitochondrial membrane permeability (69). Changes in Cer levels can influence mitochondrial integrity and function (70, 71). CARs play a crucial role in transporting fatty acids into the mitochondria for beta-oxidation, thus maintaining mitochondrial function (72). Additionally, significant differences in lipid chain lengths were noted with age in the liver, impacting membrane integrity, fluidity, and functionality (Figure 4D). These findings align with altered metabolomic and lipogenic changes attributed to the modified shape and function of mitochondria with age.
Murine Aging and Diet Exhibit Distinct Changes in the MICOS and SAM Complex
It is well established that the MICOS complex is critical for mitochondrial dynamics (73), and our group has previously investigated the effect of aging on the MICOS complex in kidney tubular cells (24), however, it is still unclear how aging affects the MICOS in aging liver. Studies have shown that Opa1, which is epistatic to the MICOS complex and physically interacts with components of the MICOS complex (43), decreases with age (74). With Opa1 as a positive control, we sought to determine if the MICOS complex mRNA expression is also decreased in murine liver with age. Consistent with previous studies, Opa1 mRNA decreased by over 50% between 3 months and 2 years (Figure 5A). Mitofilin also decreased significantly (Figure 5B). Similarly, Chchd3 and Chchd6 also progressively decreased with age but not as much as the decline of other transcripts (Figures 5C–D). While Opa1 interacts with the MICOS complex, it is not required for the formation of cristae junctions at which the MICOS complex forms, nor does Opa1 loss negatively affect MICOS components (54). This suggests that the loss of the MICOS complex across aging occurs in an Opa1-independent manner. This parallels our previous findings in skeletal muscle, cardiac, and kidney (24, 75, 76).
Figure 5. Aging Causes MICOS Loss, Diet Changes Affect SAM50 Expression, and SAM50 has Genetic Associations with Liver Diseases.

(A-D) Quantitative PCR shows changes in mRNA transcripts in (A) Opa1, (B) Mitofilin/Mic60, (C) Chchd3/Mic19, and (D) Chchd6/Mic25 between 3-month and 2-year murine liver samples. (E) Western blotting showing SAM50 protein levels, relative to tubulin, in 3-month and 2-year murine liver samples. (F) Western blotting showing MIC60 and SAM50 protein levels, relative to ß-actin, in low-fat diet and high-fat diet murine liver samples. (G-G’) Representative transmission electron micrographs from low-fat diet and (H-H’) high-fat diet murine liver tissue. Quantifications of (I) mitochondria number, as normalized to cell area (n=7, low fat diet; n=5, high fat diet), (J) individual mitochondrial area (n=592, low fat diet; n=266, high fat diet), (K) the sum of all mitochondrial area normalized to total cell area (n=7, low fat diet; n=5, high fat diet), (L) mitochondrial circularity index, (n=598, low fat diet; n=271, high fat diet), and (M) cristae score (n=425, low fat diet; n=425, high fat diet). (N-N’) Representative confocal fluorescence (using mCherry-Mito-7) from wildtype and (O-O’) Sam50-knockout fibroblasts. (P) Quantifications of mitochondrial length. (Q) Relative proportion of mitochondria that are either fragmented or tubular in wildtype and Sam50 KO fibroblasts, with the white area representing percentage of fragmentation and colored areas representing percentage of typical tubular. (R) Phenome-wide association study (PheWAS) results for SAMM50 GReX in individuals of European ancestry (n=70,440). The GReX for SAMM50 was tested for association across 1,704 clinical phenotypes extracted from the EHR. Association tests were run using logistic regression models, accounting for genetic ancestry (principle components/PC 1–10), sex, age, median age of medical record, and genotyping batch. Associations that met the Bonferroni-corrected threshold (red line, p < 2.934272 × 10−5) are labeled with phenotype name [see SFile 1 for all PheWAS results]. The blue line represents nominal significance (p = 0.05) For all panels, error bars indicate SEM, Mann–Whitney tests were used for statistical analysis, and significance value indicate ****P ≤ 0.0001, ns indicates non-significant.
The SAM complex, particularly its β-barrel channel Sam50, interacts with the MICOS complex to regulate cristae morphology (77–80). Particularly, overexpression of SAMM50 enhances fatty acid oxidation and reduces intracellular lipid accumulation, while polymorphisms in SAMM50 are associated with NAFLD (49), and loss of Sam50 in hepatocytes leads to cardiolipin-dependent mitochondrial membrane remodeling, mtDNA release, and liver injury (81). So, we also looked at SAM50 protein levels across aging. Interestingly, unlike the MICOS complex and Opa1, we saw no significant changes in SAM50 levels (Figure 5E). Notably, beyond aging, a key risk factor for NAFLD is a high-fat diet (HFD) which can cause lipid accumulation and dyslipidemia (82). Since past studies have shown that overexpression of SAMM50 can mitigate lipid accumulation (49), we focused on how an HFD also affects these regulators. To do this, we subjected male mice to an HFD, with fat constituting 60% of their diet, and a littermate cohort to a low-fat diet (LFD), with fat constituting only 5% of their calories, for a total of 12 weeks, with mice sampled at 20 weeks. Contrary to our age-related findings, we showed that while MIC60, a key component of the MICOS complex, remains unchanged, SAM50 shows a decrease with an HFD. Given the role of SAMM50 in mitigating lipid accumulation (49), this loss may increase susceptibility to lipid accumulation and dyslipidemia that is a hallmark of HFD (82).
To further investigate the impact of an HFD on mitochondrial ultrastructure, we performed TEM analysis in a 20-week-old murine cohort subjected to an LFD and HFD (Figures 5G–H”). Qualitatively, we observed more lipid droplets with less circularity in HFD samples. Looking at mitochondria, although mitochondrial count, when normalized to the μm cell area, was fewer in HFD samples (Figure 5I; 0.210 ± 0.114 SD, LFD; 0.128 ± 0.0972 SD, HFD), this change was not significant. Similar to aging, HFD samples exhibit a significantly higher average mitochondrion area (Figure 5J; 0.570 μm2 ± 0.401 μm2 SD, LFD; 0.923 μm2 ± 1.12 μm2 SD, HFD). When considering these two quantifications in tandem, we also calculated the percentage of mitochondrial area related to the total cell area, which showed no significant difference (Figure 5K; 11.8 μm2 ± 5.59 μm2 SD, LFD; 11.7 μm2 ± 5.10 μm2 SD, HFD). When looking at how mitochondrial shape changes with a HFD, we observed that mitochondrial had a higher circularity, indicative of potentially less complexity, with a HFD (Figure 5L; 0.781 ± 0.149 SD, LFD; 0.897 ± 0.0748 SD, HFD). Finally, looking at cristae score, similar to aging samples, HFD cohorts had a significantly lower cristae score (Figure 5M; 3.74 ± 0.441 SD, LFD; 2.04 ± 0.770 SD, HFD). Together, these findings show that a HFD can parallel age-related changes, and that HFD exhibit smaller mitochondria with aberrant cristae due, in part, to a loss of SAM50.
Previously, we have performed a rigorous 3D analysis of mitochondrial structure and networking in Sam50-deficient human and murine myotubes, showing that the deletion of Sam50 results in smaller and less complex mitochondria (52). To further investigate SAM50’s role in mitochondrial structure, we knocked out Sam50 in fibroblasts and used mito-mCherry confocal to investigate structure (Figures 5N–O”). Our results further validate our previous findings, showing that Sam50 deletion causes impairments in mitochondrial length (Figure 5P; 4.14 μm ± 1.57 μm SD, WT; 2.59 μm + 0.539 μm SD, Sam50 KO). Additionally, when comparing the percentage of fragmented mitochondria to tubular mitochondria, there was a significantly higher rate of fragmented mitochondria in Sam50 KO than in WT. (Figure 5Q; 3.33% ± 2.08% SD, WT; 57.3% + 18.0% SD, Sam50 KO). Notably, these findings parallel our findings in HFD of reduced mitochondrial size, indicating that loss of Sam50 may dictate the negative bioenergetics caused by an HFD (83, 84).
Clinical relevance of Sam50 in liver pathologies
Previously, we found that Sam50 is responsible for establishing and maintaining mitochondrial integrity, cristae structure, and mitochondrial metabolism (52). To identify the clinical implications of dysregulated SAMM50 gene expression in humans, we modeled genetically regulated gene expression (GReX) of SAMM50 across 85,615 individuals in the Vanderbilt biobank, BioVU (Table 1). Cross-tissue SAMM50 GReX was calculated using the genetic information for each individual, combined with SAMM50 gene expression models developed from the GTEX dataset (see Methods, sFigure3). The relationship between SAMM50 GReX and clinical phenotypes (n=1,704) and laboratory values (n=326) were then evaluated by logistic and linear regression models, respectively. In 70,440 BioVU individuals of European ancestry, we found that SAMM50 GReX is significantly associated with multiple liver disease diagnoses, including: chronic liver disease and cirrhosis, alcoholic liver damage, liver transplant, acute gastritis, and esophageal bleeding (p < 2.93427E-05, Figure 5R, SFile1). Within the 15,175 BioVU individuals of African ancestry, we did not find any significant associations between SAMM50 GReX and the clinical phenotypes tested; however, we did see a nominally significant association with acute gastritis (p=0.00142, SFile3). We did not find any significant associations between SAMM50 GReX and clinical lab values within the BioVU individuals of European or African ancestry (SFile2, SFile4).
Table 1: Demographics for BioVU Cohort.
Genetic Ancestry Abbreviations: (EA): European ancestry, (AA): African ancestry.
| Genetic Ancestry | European | African |
|---|---|---|
| N | 70,404 | 15,175 |
| Sex (%M) | 44.10% | 38.40% |
| Current Age | 57.04 ± 22.42 | 46.48 ± 21.39 |
| Median Age of Medical Record | 48.43 ± 22.24 | 38.32 ± 21.41 |
| Visits | 61.18 ± 75.14 | 55.02 ± 80.83 |
Knockdown of MIC60 and CHCHD6 reduces Ca2+ uptake and retention capacity in HepG2 cells
While this underscores how diet changes may contribute to liver diseases, we also wanted to establish how the MICOS complex contributes to age-related changes. To unravel the involvement of MIC60 and CHCHD6 in mitochondrial calcium (mCa2+) homeostasis, we monitored mCa2+ uptake and mCa2+ retention capacity in MIC60 and CHCHD6 knockdown human-derived HepG2 cells. Both MIC60 and CHCHD6 knockdown cells show reduced mCa2+ uptake (Figure 6A). Furthermore, to determine if the altered MICOS and cristae structure contributes to impaired mCa2+ retention capacity and involved in mitochondrial permeability transition pore opening, we measured the mCa2+ retention capacity in MIC60 and CHCHD6 knockdown cells. MIC60 as well as CHCHD6 knockdown cells underwent rapid permeability transition opening as compared to control cells (Figure 6B). We also found that mCa2+ retention capacity is significantly reduced in both MIC60 and CHCHD6 knockdown HepG2 cells (Figure 6C). These data suggest that altered MICOS complex and cristae disorganization increase the susceptibility of HepG2 cells to mCa2+ dysregulation and Ca2+-induced cell death.
Figure 6: Loss of MIC60 and CHCHD6 in HepG2 cells results in reduced mCa2+ uptake and calcium retention capacity and oxidative stress.

(A) Raw traces showing mitochondrial calcium uptake in permeabilized MIC60 and CHCHD6 knockdown HepG2 cells along with scr-siRNA transected controls. (B) Percentage change in mCa2+ uptake rate quantified from raw traces. (C) Recordings of mitochondrial calcium retention capacity in scr-siRNA, MIC60 siRNA, and CHCHD6 siRNA HepG2 cells. The circles indicate the number of calcium boluses taken up by specific cells. (D) Percentage change in mitochondrial calcium retention capacity quantified from recordings of mitochondrial calcium retention capacity. (E) Immunoblot confirming siRNA-mediated knockdown of CHCHD6 in HepG2 cells. (F) Immunoblot confirming siRNA-mediated knockdown of MIC60 in HepG2 cells. (G) 4′,6-diamidino-2-phenylindole (DAPI) staining, MitoPY1 (5 μM, 45 min at 370 c magnification of 60x), and merge channels in scramble-siRNA (control), MIC60-siRNA, and CHCHD6-siRNA transfected permeabilized HepG2 cells. (H) 4′,6-diamidino-2-phenylindole (DAPI) staining, MitoBright Deep Red (10 μM, 30 min at 37° c), DCFDA (10 μM, 30 min at 37° c, magnification of 60x), and merge channels in scramble-siRNA (control), MIC60-siRNA, and CHCHD6-siRNA transfected permeabilized HEK293 cells. (I) Plate reader-based reactive oxygen species (ROS) quantification. (J) Microscopy-based ROS quantification of MitoPY1 orange, (K) MitoSox Deep Red, and (L) DCFDA. For all statistical tests, one-way ANOVA statistical test was performed with Dunnett’s multiple comparisons test. N=5–10 for all calcium experiments, each indicated by dots, as run in triplicates. N=9–13 for all oxidative stress experiments, each indicated by dots, as run in triplicates. Significance values indicate **P ≤ 0.01 and ****P ≤ 0.0001.
Knockdown of MIC60 and CHCHD6 induces oxidative stress
Ca2+ directly affects oxidative stress signaling and reactive oxygen species (ROS) generation (85). To determine if MICOS affects ROS production, we evaluated total ROS, mitochondrial superoxide, and H2O2 production in MIC60 and CHCHD6 knockdown cells (Figures 6G–H). Mitochondrial H2O2 content, as measured by Mitochondria peroxy yellow 1, increased following MIC60 and CHCHD6 knockdown when quantified by both plate-reader based (Figure 6I) and microscopy-based (Figure 6J) ROS quantification. Silencing MIC60 and CHCHD6 in HepG2 cells significantly increased mitochondrial superoxide production and more general intracellular ROS, detected by MitoBright Deep Red (Figure 6K) and DCFDA (Figure 6L), respectively. These findings indicate that suppression of MIC60 and CHCHD6 disrupts mitochondrial ROS homeostasis, demonstrating that the MICOS complex is associated with oxidative stress.
Discussion:
Age-Related Changes in Liver Mitochondria Structure
Liver mitochondria have been imaged using 3D in rodent models since the early 1990s (86). Previously, in rat liver, 3D reconstruction revealed that cristae showed high variability in structure (87). 3D reconstruction also showed that there are extensive mitochondria endoplasmic reticulum contact sites (MERCs) in liver mitochondria [35]. Additionally, MERCs are sites for various purposes, such as calcium homeostasis and lipid homeostasis (88). In mice with high alcohol consumption, alcohol increased mitochondrial volume while conversely reducing the networking of mitochondria (89). Notably, the formation of mega mitochondria, a novel mitochondria 3D structure, arose in human non-alcoholic fatty liver disease (90). Beyond these 3D studies, previous studies using cryo-electron tomography (cryo-ET) have found that mitochondria fragmented and lamellar cristae lost integrity across the aging process (91). While these studies have aided in revealing previously unknown specific mitochondrial structures in the liver, the same has not yet been done for different mitochondrial structures that may arise across aging.
Here, we use SBF-SEM to attempt to answer how mitochondria change in an aged liver and the functional impact of such changes. We found that in aged states, the size and morphology of liver mitochondria are altered in both 2D and 3D. Using SBF-SEM and Amira for 3D morphology of mitochondria and comparing 2-year against 3-month, we observed fragmentation of mitochondria and decreased complexity. Importantly, we found that an aged murine liver has a decrease in area, perimeter, volume, and complexity in mitochondria. Mitochondria structure remains relevant in aging as structural decline may decrease mitochondrial function, thus reducing certain liver roles. For example, mitochondria dysfunction has been linked to an increased risk of liver damage upon drug treatment (92). Beyond this, in humans and mice with alcoholic liver disease, it has been suggested that megamitochondria form due to the lack of Drp1 and serve a protective role (93, 94). This may be due to the downregulation of the NR4A1/DNA-PKcs/p53 pathway which has been shown to activate Drp1 in other liver diseases (95). Aging and disease states affect each other; therefore, future experiments must look at the mitochondria 3D reconstruction in different disease states across aging to see if the confluence of these factors leads to the unique mitochondria 3D structures. Interestingly, while megamitochondria are a hallmark of hepatotoxicity and steatosis (94, 96, 97), and aging exhibited numerous hallmarks of progression towards liver diseases (Figure 1), we did not observe these megamitochondria in our sample, rather showing mostly smaller mitochondria. Since the formation of megamitochondria in Alcoholic Liver Disease follows a significant increase in Drp1 expression and mitochondrial fragmentation (98), it may be that our aged sample represents a pre-diseased state that can subsequently shift to megamitochondria as a compensatory response to reduce alcohol-induced toxicity (94, 98, 99).
Beyond mitochondrial 3D structure, past studies have shown that fatty acids promote endoplasmic reticulum stress and liver injury in rodent models (100). Due to the increase of lipid droplets in both aging and HFD in TEM images, there may be an increase in mitochondria-lipid droplet contact sites (MLDCs) (61, 101). Beyond this, lipid droplet-endoplasmic reticulum contacts are also understood to help form lipid droplets and perform metabolism (54). Especially relevant is that MLDCs serve as a place for fatty acid homeostasis (101). Additionally, MERCs importantly play a role in lipid homeostasis and synthesis (102). MERCs are understood to regulate ER stress (103), so increases in fatty acids, triggered by lipid droplet formation, can increase MERC formation in the aged model. Conversely, we qualitatively showed a decrease in wrappER, which is known to maintain lipid flux through a 3-organelle contact site that involves peroxisomes (63). These wrappER sites, which contain sites of adhesion, regulate very-low-density lipoproteins (104). The age-related loss of mitochondrial complexity may impair the ability and relative surface area mitochondria in murine liver samples must form contact sites, including MERCs, thus interfering with functions including lipid homeostasis. Indeed, past reviews have suggested targeting MERCs in NAFLD due to the role of contact sites in glucose and lipid metabolism (105). However, in the future, a more rigorous analysis of age-related changes in contact sites within liver tissue is necessary.
Sam50 as a Regulator of Liver Disease
Due to the observed dysfunctions in mitochondrial function and structure, we investigated the role of SAM50, part of the SAM required for β-barrel protein assembly, in the liver (80). There is evidence that the IMM proteins MIC60 (Mitofilin) and CHCHD3 were part of a protein complex that includes the SAM components, known as the mitochondrial intermembrane space bridging (MIB) complex (80, 106). The preservation of cristae depends on interactions between the MIB complex’s OMM and IMM components. The destabilization of the MIB complex led to a decrease in the respiratory chain complex assembly. SAM50 depletion over time affects the protein contents of all large respiratory complexes containing subunits encoded by the mitochondria, suggesting a link between SAM50 and cristae structural integrity, respiratory complex assembly, and mitochondrial DNA (mtDNA) maintenance (107). Our previous study have shown that Sam50 plays a significant role in the maintenance of both the mitochondrial network and the structure of cristae and MICOS, especially in the context of liver injury (52). Mice with Sam50 deletion or liver-specific deletion produced by acetaminophen, an efficient antipyretic and analgesic, experience mtDNA release, which activates the cGAS-STING pathway and causes liver inflammation (81). Here, using phenome-wide and clinical lab-wide scans for SAMM50 GReX in a medical biobank, we saw a significant association between SAMM50 and multiple liver diseases including nonalcoholic or alcoholic liver disease and cirrhosis. This suggested that the expression of SAMM50 as a transcription factor may directly or indirectly contribute to mitochondrial defects of the liver.
To corroborate the clinical consequences of SAMM50 GReX in BioVU participants, we queried publicly available databases including: PheWeb (108), Biobank Japan PheWeb (109), and the FinnGen web browser (110). PheWeb reports genome-wide association results from the UK Biobank population (n > 400,000). The top phenotype associations for the SAMM50 locus within PheWeb included: Chronic liver disease and cirrhosis (p=2.8 X 10−24, cases=2,954), Other chronic nonalcoholic liver disease (p=1.2e-18, cases =1,687), Alcoholic liver damage (p=1.4 X 10−11, cases=836), Injury to other and unspecified nerves (p=2.2 X 10−10, cases=269), Portal hypertension (p=1.7 X 10−9, cases=527), and Liver abscess and sequelae of chronic liver disease (p=4.1 X 10−9, cases=974). BioBank Japan PheWeb similarly reports genome-wide association results from the BioBank Japan Project (n > 260,000). The top phenotype associations for the SAMM50 locus in BioBank Japan PheWeb included Cirrhosis (p=2.8 X 10−14, cases=2,551), as well as Aspartate transaminase (p=3.3e-87, cases=150,068), Alanine aminotransferase (p=3.0 X 10−71, cases=150,545), and platelet count (p=6.5 X 10−38, cases=148,623). The FinnGen web browser reports genome-wide association studies from the FinnGen data (n=342,499). Top results from FinnGen included several phenotypes related to liver diseases including cirrhosis (p=9.6 X 10−54, cases=3,548), diseases of liver (p=4.9e-49, cases=9,548), and nonalcoholic fatty liver disease (p=5.7 X 10−40, cases=1,908). This underscores the therapeutic potential of targeting SAMM50.
The MICOS Complex as a Regulator of Aging:
In this study, we demonstrated the disruptions in many aspects of mitochondria structure in both 2D and 3D analysis of mitochondria in aging liver. We also showed MICOS impairment in the aging liver. Therefore, we investigated whether MICOS regulates mitochondrial calcium influx and oxidative stress. Calcium influx in mitochondria can reflect cell viability (111, 112). Mitochondria mediation of calcium can impact many cell processes, such as apoptosis, signaling, and ATP production (113). In some cases, elevated mitochondria calcium uptake can occur antecedent to mitochondrial swelling, which results in a pathway often leading to apoptosis (114). However, there was a decline in calcium uptake as CHCHD6 and MIC60 were disrupted (Figure 6), suggestive of impaired cellular calcium signaling, which has previously been linked with ER stress and dysregulation of MERCs (113, 115). This provides a plausible disease link as ER calcium release promotes mitochondrial dysfunction, inducing oxidative stress and hepatotoxicity (116), with broader implications in NAFLD (117, 118). Since Miro clusters both interact with the MICOS complex, as well as regulate MERCs (119), Miro represents a potential future mechanistic avenue through which MICOS complex contributes to MERC tethering.
Mitochondria redox state has been proposed as a principal moderator of mitochondrial function in liver disease (120). Recent studies have continued to highlight a link between ROS and mitochondrial dynamics (38). Lipids can aid in stimulating the production of ROS (121). Additionally, many theories regarding aging have been formulated around the basis that mitochondria lose function due to ROS byproducts which accumulate across aging (122). There remains controversy about whether ROS are generated in the liver, while some studies have found they arise during aging in the liver (123), other studies conversely found that in the aging human liver, no superoxides are produced (124). Future studies should explore how ROS contributes to the observed 3D mitochondrial phenotypes, as oxidative stress may be a regulator of the phenotypes we observed. It is noteworthy that the loss of CHCHD6 and MIC60 leads to oxidative stress (Figure 6). This suggests that during aging, the liver undergo a vicious cycle wherein abnormal mitochondrial structures generate more harmful byproducts. These byproducts, in turn, worsen mitochondrial structural dysfunction, contributing to age-related oxidative stress.
The Interaction Effect of the SAM Complex and MICOS Complex in Aging and Diet:
In aged murine liver, decreases in the mtDNA numbers, urea cycle, mitochondrial function, locomotor activity, loss of enzymatic activity, and anti-oxidative stress are well established (123, 125, 126). Mitochondria also show diminished membrane potential across aging in murine liver (127) and declined mitophagy responses (128). Within our study, we looked at Sam50 in the context of aging. Although we did not observe a change in the protein level of SAM50 in the aging liver, there were defects in SAM50, but not MIC60, protein production during the HFD. Notably, our study contrasts previous quantitative proteomic analyses of liver mitochondria from HFD Diabetic mice, which found that Mitofilin, Sam50, and Chchd3 are all upregulated (129). While more research is warranted to resolve this controversy, our findings suggest that Sam50 may not regulate mitochondrial machinery during aging directly, but future studies performing an RNA sequencing scan of aged liver tissue, compared to young tissue, will provide some candidates that interact with Sam50.
We know that HFDs are linked with many liver complications. For example, oxidative stress, inflammation, and lipogenesis are some of the factors that might be linked to HFD and exacerbate the onset of non-alcoholic fatty liver disease (83). Additionally, HFD is associated with metabolic dysregulation of the liver (130). In this study, we found an increase in retinoic acid and retinal in the aged liver (Figure 4). Notably, there is evidence linking retinol-binding proteins and HFD. The enhanced supply of vitamin A and high-fat consumption in diet-induced obese mice is linked to the production of bisretinoid (131). Interestingly, obesity within hepatocytes has also been shown to increase MAMs (i.e., MERC-isolated biochemical fractions), which confers mitochondrial dysfunction (132, 133). High fat within a diet may further have an additive effect with aging to exacerbate age-related hepatic pathologies. For example, an HFD can alter mitochondrial structure, leading to decreased fatty acid oxidation (134). Within age-related hepatic steatosis, impaired fatty acid oxidation, and other factors that HFD can contribute to such as insulin resistance, are key risk factors (30). Notably, SAMM50 deficiency causes lipid accumulation and impaired fatty acid oxidation (49). Thus, HFDs can play a role in metabolism defects in the liver tissue, although this must be further investigated in the future.
The changes in SAM50 during HFD may be due to several factors. Our lipidomic studies confirm suggested dysregulations of lipids in the aged liver (Figure 4). Our observation of SAM50 decreased in HFD contrasts in vitro models of hepatic cellular steatosis, revealing that mitochondrial dysfunction is determined by oxidative stress instead of lipid buildup (135). This finding is novel since Sam50-deletion or overexpression, and the broader SAM complex, during HFD in the liver, have not been studied. However, past studies of HFD offer some insight. HFD can lead to pathological alterations and damage to the ultrastructure of the mitochondria and downregulation of MERCs regulators, MFN2 and OPA1 (84), which parallels the TEM ultrastructural changes we observed. Additionally, Mic19/Chchd3 was shown to be necessary in the liver (136). Sam50 is closely related to Mic19. Sam50 mediates mitochondrial outer and inner membrane interactions via the Sam50-Mic19-Mic60 axis (79). In the liver, Mic19/Chchd3 deletion reduces ER-mitochondrial contacts, disrupts mitochondrial lipid metabolism, disorganizes mitochondrial cristae, and causes unfolded protein stress response in mouse hepatocytes, leading to impairments of liver mitochondrial fatty acid β-oxidation and lipid metabolism (136). This may explain our results of declined Sam50 synthesis during HFD, but it remains unclear why Mic60 is not affected. Alternatively, linking MICOS and SAM complex changes, in hepatocytes both SAM50 and MIC60 cooperate to bind to cardiolipin, a phospholipid exclusively located in mitochondria (81). ROS also targets cardiolipin, which is associated with mitochondrial dysfunction in NAFLD (137). Cardiolipin inhibition has mitigated non-alcoholic steatohepatitis through NLRP3 inflammasome activation, suggesting a therapeutic role of cardiolipin (138). Sam50 depletion leads to cardiolipin externalization and subsequent liver inflammation (81). Interestingly, paralleling our findings of increased lipogenesis with aging (Figure 1), our lipidomic analysis showed altered lipogenesis across multiple lipid classes, which has implications extending beyond cardiolipin, since the liver is a principal site for lipid homeostasis (139). Thus, while aging exhibits a protective mechanism to maintain Sam50, the additional challenge of diet changes may cause loss of integrity in both the SAM complex and MICOS complex, which results in the buildup of lipids in the liver, raising the risk of cirrhosis, steatohepatitis, and NAFLD, among other hepatic conditions.
CONCLUSION
To broaden our understanding of age-related changes in male murine hepatic tissue, we have established how 2D and 3D ultrastructure undergo similar reductions in mitochondrial volume and connectivity, potentially due to an age-related loss of the MICOS complex. This is an important age-related regulator of mitochondrial structure and could be linked to functional changes across aging in the liver. Deletion of the MICOS complex in HepG2 cells results in impaired calcium uptake and increased oxidative stress, highlighting its significance. We have also established that Sam50, which is genetically associated with liver diseases, is maintained across aging but lost with a high-fat diet. Since a high-fat diet can exacerbate changes in lipid synthesis, catabolism, and mitochondrial alterations that already occur in hepatic aging (30, 83), this suggests the confluence of aging and a high-fat diet in increasing susceptibility to liver disease, in part due to the loss of both the MIB complex (i.e., MICOS and SAM complex). Together, this study demonstrates dysfunctions of mitochondrial structure in both 2D and 3D, mitochondrial calcium influx and ROS production, changes in metabolism and lipid content, and MICOS impairments in the aging liver, with alterations in Sam50 expression contributing to liver diseases.
EXPERIMENTAL PROCEDURES
Sex As a Biological Variable:
In this study, the consideration of sex as a biological variable was integral to our experimental design. All experimental procedures and analyses were conducted with careful consideration of sex differences. Since minimal sex-dependent differences were observed in TEM studies, we proceeded with a male murine model for all other studies. Sex differences in aging-related changes and longevity can affect relative mitochondrial morphology which is a limitation of our male model.
Human Cohort
All human samples were obtained from Brazilian cohorts according to the CAEE (Ethics Appreciation Presentation Certificate) guidelines. Samples from young and old individuals were collected and analyzed under CAEE number 77570224.2.0000.5281. Fat percentage was calculated as a function of in-phase and out-phase quantifications via the following calculator: https://www.ricardoromano.com/esteatoseRM (53).
Animal Care and Maintenance:
Per protocols previously described (140), the care and maintenance of the male C57BL/6J mice conformed to the National Institute of Healt’s guidelines for the use of laboratory animals. The University of Iowa’s Institutional Animal Care and Use Committee (IACUC) or University of Washington IACUC approved the housing and feeding of these mice. Anesthesia was achieved using a mixture of 5% isoflurane and 95% oxygen.
Oil Red O
OCT blocks were cut into 7 μm thick sections, affixed to glass slides, brought to room temperature for 10 minutes, then stained with Oil Red O (Sigma-Aldrich), as previously described (141).
mtDNA Content
As previously described (142), mitochondrial DNA content was quantified using real-time PCR (RT-PCR) from DNA extracted and purified from gastrocnemius muscle tissue. Five nanograms of DNA were used for the quantification of mitochondrial (Cox1) and nuclear (β-actin) DNA markers. Mitochondrial DNA content was normalized to the genomic Rpl13a gene using specific primers for Cox1 and Rpl13a.
Bile Acid
From frozen liver tissue, 100 mg of tissue homogenized in 75% ethanol was incubated for 2 hours and then centrifuged at 6000g for 10 minutes. Once prepared, bile was measured with Mouse Total Bile Acids Assay Kit (Crystal Chem), per manufacturer’s instruction.
Triglyceride Levels
As previously described (142, 143), triglyceride levels were measured in the liver and in serum collected after a 6 h fast using the EnzyChrom™ Triglyceride Assay Kit (BioAssay Systems), with triglycerides extraction using a solution of isopropanol and Triton X 100.
Quantification of TEM Micrographs and Parameters Using ImageJ
Samples were fixed in a manner to avoid any bias, per established protocols (144). Following preparation, tissue was embedded in 100% Embed 812/Araldite resin with polymerization at 60 °C overnight. After ultrathin sections (90–100 nm) were collected, they were post-stained with lead citrate and imaged (JEOL 1400+ at 80 kV, equipped with a GatanOrius 832 camera). The National Institutes of Health (NIH) ImageJ software was used for quantification of TEM images, as described previously (9, 145).
Serial Block-Face Scanning Electron Microscope (SBF-SEM) Processing of Mouse Muscle Fibers
SBF-SEM was performed according to previously defined protocols (19, 144, 145). Anesthesia was induced in male mice using 5% isoflurane. Post skin and hair removal, the liver was treated with 2% glutaraldehyde in 100 mM phosphate buffer for 30 minutes, dissected into 1-mm³ cubes, and further fixed in a solution containing 2.5% glutaraldehyde, 1% paraformaldehyde, and 120 mM sodium cacodylate for 1 hour.
Fixation and subsequent steps collected onto formvar-coated slot grids (Pella, Redding CA), stained and imaged as previously described (19, 144, 145). This includes tissue washing with 100 mM cacodylate buffer, incubation in a mixture of 3% potassium ferrocyanide and 2% osmium tetroxide, followed by dehydration in an ascending series of acetone concentrations. The tissues were then embedded in Epoxy Taab 812 hard resin. Sectioning and imaging of sample was performed using a VolumeScope 2 SEM (Thermo Fisher Scientific, Waltham, MA). Conventional TEM analysis was performed on 300–400 serial sections from each sample, following staining and imaging protocols. Subsequently, analyzed, via imaging was performed under low vacuum/water vapor conditions with a starting energy of 3.0 keV and beam current of 0.10 nA. Sections of 50 nm thickness were cut allowing for imaging at 10 nm × 10 nm × 50 nm spatial resolution.
Segmentation and Quantification of 3D SBF-SEM Images Using Amira
SBF-SEM images were manually segmented in Amira to perform 3D reconstruction, as described previously (19). 300–400 slices were used and analyzed by a blind individual. 250 total mitochondria across from 3 mice were collected for each quantification. For 3D reconstruction of cardiomyocytes, 10 cells and a total of about 200 mitochondria. Quantification of 3D structures was performed using the Amira software with built-in parameters or previously described measurements (19).
LCMS Methods for Metabolomics:
Frozen tissues were weighed, ground with a liquid nitrogen in a cryomill (Retsch) at 25 Hz for 45 seconds, before extracting tissues 40:40:20 acetonitrile: methanol: water +0.5% FA +15% NH4HCO3 (146) with a volume of 40mL solvent per 1mg of tissue, vortexed for 15 seconds, and incubated on dry ice for 10 minutes. Tissue samples were then centrifuged at 16,000 g for 30 minutes. The supernatants were transferred to new Eppendorf tubes and then centrifuged again at 16,000 g for 25 minutes to remove any residual debris before analysis.
Extracts were analyzed within 24 hours by liquid chromatography coupled to a mass spectrometer (LC-MS). The LC–MS method was based on hydrophilic interaction chromatography (HILIC) coupled to the Orbitrap Exploris 240 mass spectrometer (Thermo Scientific) (147). The LC separation was performed on a xBridge BEH Amide column (2.1 × 150 mm, 3.5 μm particle size, Waters, Milford, MA). Solvent A is 95%: 5% H2O: acetonitrile with 20 mM ammonium acetate and 20mM ammonium hydroxide, and solvent B is 90%: 10% acetonitrile: H2O with 20 mM ammonium acetate and 20mM ammonium hydroxide. The gradient was 0 min, 90% B; 2 min, 90% B; 3 min, 75% B; 5 min, 75% B; 6 min, 75% B; 7 min, 75% B; 8 min, 70% B; 9 min, 70% B; 10 min, 50% B; 12 min, 50% B; 13 min, 25% B; 14min, 25% B; 16 min, 0% B; 18 min, 0% B; 20 min, 0% B; 21 min, 90% B; 25 min, 90% B. The following parameters were maintained during the LC analysis: flow rate 150 mL/min, column temperature 25 °C, injection volume 5 μL and autosampler temperature was 5 °C. For the detection of metabolites, the mass spectrometer was operated in both negative and positive ion mode. The following parameters were maintained during the MS analysis: resolution of 180,000 at m/z 200, automatic gain control (AGC) target at 3e6, maximum injection time of 30 ms and scan range of m/z 70–1000. Raw LC/MS data were converted to mzXML format using the command line “msconvert” utility (148). Data were analyzed via the EL-MAVEN software version 12.
LCMS Methods for Lipidomic Profiling
Tissue homogenization and extraction for lipids: Tissues were homogenized using a Retsch CryoMill. The homogenate was mixed with 1 mL of Extraction Buffer containing IPA/H2O/Ethyl Acetate (30:10:60, v/v/v) and Avanti Lipidomix Internal Standard (diluted 1:1000) (Avanti Polar Lipids, Inc. Alabaster, AL). Samples were vortexed and transferred to bead mill tubes for homogenization using a VWR Bead Mill at 6000 g for 30 seconds, repeated twice. The samples were then sonicated for 5 minutes and centrifuged at 15,000 g for 5 minutes at 4°C. The upper phase was transferred to a new tube and kept at 4°C. To re-extract the tissues, another 1 mL of Extraction Buffer (30:10:60, v/v/v) was added to the tissue pellet-containing tube. The samples were vortexed, homogenized, sonicated, and centrifuged as described earlier. The supernatants from both extractions were combined, and the organic phase was dried under liquid nitrogen gas.
Sample reconstitution for lipids: The dried samples were reconstituted in 300 μL of Solvent A (IPA/ACN/H2O, 45:35:20, v/v/v). After brief vortexing, the samples were sonicated for 7 minutes and centrifuged at 15,000 g for 10 minutes at 4°C. The supernatants were transferred to clean tubes and centrifuged again for 5 minutes at 15,000 g at 4°C to remove any remaining particulates. For LC-MS lipidomic analysis, 60 μL of the sample extracts were transferred to mass spectrometry vials.
LC-MS analysis for lipids: Sample analysis was performed within 36 hours after extraction using a Vanquish UHPLC system coupled with an Orbitrap Exploris 240™ mass spectrometer equipped with a H-ESI™ ion source (all Thermo Fisher Scientific). A Waters (Milford, MA) CSH C18 column (1.0 × 150 mm × 1.7 μm particle size) was used. Solvent A consisted of ACN:H2O (60:40; v/v) with 10 mM Ammonium formate and 0.1% formic acid, while solvent B contained IPA:ACN (95:5; v/v) with 10 mM Ammonium formate and 0.1% formic acid. The mobile phase flow rate was set at 0.11 mL/min, and the column temperature was maintained at 65 °C. The gradient for solvent B was as follows: 0 min 15% (B), 0–2 min 30% (B), 2–2.5 min 48% (B), 2.5–11 min 82% (B), 11–11.01 min 99% (B), 11.01–12.95 min 99% (B), 12.95–13 min 15% (B), and 13–15 min 15% (B). Ion source spray voltages were set at 4,000 V and 3,000 V in positive and negative mode, respectively. Full scan mass spectrometry was conducted with a scan range from 200 to 1000 m/z, and AcquireX mode was utilized with a stepped collision energy of 30% with a 5% spread for fragment ion MS/MS scan.
RNA Extraction and RT-qPCR
Using TRIzol reagent (Invitrogen), total RNA was isolated from tissues and further purified with the rNeasy kit (Qiagen Inc). RNA concentration was determined by measuring absorbance at 260 nm and 280 nm using a NanoDrop 1000 spectrophotometer (NanoDrop products, Wilmington, DE, USA). Approximately 1 μg of RNA was reverse-transcribed using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosciences, Carlsbad CA). Quantitative PCR (qPCR) was then performed using SYBR Green (Life Technologies, Carlsbad, CA) (149). For qPCR, 50 ng of cDNA was loaded into each well of a 384-well plate, with the reaction carried out on an ABI Prism 7900HT system (Applied Biosystems) with the following cycle: 1 cycle at 95°C for 10 min; 40 cycles of 95°C for 15 s; 59°C for 15 s, 72°C for 30 s, and 78°C for 10 s; 1 cycle of 95°C for 15 s; 1 cycle of 60°C for 15 s; and one cycle of 95°C for 15 s. GAPDH normalization was used to present the data as fold changes. qPCR primers used were from previously published sequences (142), as detailed in Table 2.
Table 2:
qPCR Primers Used
| Gene | Primers | |
|---|---|---|
| Opa1 | Forward | 5’-ACCAGGAGACTGTGTCAA-3’ |
| Reverse | 5’-TCTTCAAATAAACGCAGAGGTG-3’ | |
| Chchd3 | Forward | 5’-GAAAAGAATCCAGGCCCTTCCACGCGC-3’ |
| Reverse | 5’-CAGTGCCTAGCACTTGGCACAACCAGGAA-3’ | |
| Chchd6 | Forward | 5’-CTCAGCATGGACCTGGTAGGCACTGGGC-3’ |
| Reverse | 5’-GCCTCAATTCCCACATGGAGAAAGTGGC-3’ | |
| Mitofilin | Forward | 5’-CCTCCGGCAGTGTTCACCTAGTAACCCCTT-3’ |
| Reverse | 5’-TCGCCCGTCGACCTTCAGCACTGAAAACCTAT-3’ |
Western Blotting
Western blotting was performed as previously described (76). Briefly, following RIPA lysis buffer (1% NP40, 150 mM NaCl, 25 mM Tris base, 0.5% sodium deoxycholate, 0.1% SDS, 1% phosphatase inhibitor cocktails #2 (Sigma P5726–1ML) and #3 (Sigma P0044–1ML), and one cOmplete protease inhibitor tablet (Sigma 04693159001)), 3-month and 2-year tissue samples were quantified using a BCA Assay (Thermo Scientific VLBL00GD2). Equal amounts of proteins were run on 4%–20% Tris-glycine gels (Invitrogen WXP42012BOX). Protein was then transferred to a nitrocellulose membrane (Li-Cor 926–31092) that was incubated with primary antibodies overnight at 4°C: Mic60/mitofilin (Abcam ab110329), SAM50 (Proteintech 20824–1-AP), or tubulin (Novus NB100–690). Secondary antibodies [1:10,000; donkey anti-mouse IgG (H + L) (Invitrogen A32789) and donkey anti-rabbit IgG (H + L) (Invitrogen A32802)] were incubated with the membrane at room temperature for 1 h. Using the Li-Cor Odyssey CLx infrared imaging system, blots were imaged.
Confocal mCherry-Mito-7 Labeling
To label the mitochondria of cardiac fibroblasts, the mCherry-Mito-7 plasmid was transfected into the cells using a transfection reagent according to the manufacturer’s instructions (150) and as previously described (75). Briefly, following plasmid and transfection reagent dilution in Opti-MEM medium and incubation at room temperature for 20 minutes, the dilution was added to the culture medium of the cells, which were incubated for 24–48 hours to allow expression of the mCherry-Mito-7 protein. Localization in fibroblasts was visualized using a Leica SP8 Confocal Microscope. Cells were washed with PBS, fixed with 4% paraformaldehyde for 10 minutes, and mounted with DAPI-containing mounting medium. Fluorescent signals were observed using appropriate filters and recorded with a digital camera.
SAMM50 Analyses in Vanderbilt University Medical Center’s biobank, BioVU
To examine the clinical correlates of SAMM50 gene expression in a human population, we leveraged the BioVU biobank at Vanderbilt University Medical Center (151, 152). Genotype data linked to deidentified electronic health records (EHR) for 85,615 individuals were assessed. Genotype data were collected on Illumina’s Multi-Ethnic Genotyping Array (MEGA) and underwent quality control procedures as previously described (153). SAMM50 genetically-regulated gene expression (GReX) was calculated across all genotyped individuals incorporating training data from GTEX version 8 and utilizing PrediXcan, UTMOST, or JTI models based on the best performance (highest performance r2 for each gene-tissue pair) (154–157). MultiXcan was then performed to collapse individual tissue results into a single cross-tissue SAMM50 GReX model (158). The BioVU population was then stratified by genetic ancestry as previously described and within each group, SAMM50 GReX was tested for association with clinical phenotypes mapped from ICD9/10 codes and clinical lab values extracted from the EHR using the previously described QualityLabs pipeline (153, 159, 160). In total, we tested 1,704 phenotypes and 326 labs across 70,440 individuals of European ancestry and 1,361 phenotypes and 265 labs across 15,175 individuals of African ancestry. PheWAS analyses included logistic regressions for any mapped phenotype with at least 50 cases, requiring at least 2 instances of an ICD9/10 code on unique dates to be labeled as a case. LabWAS analyses included linear regressions for any lab values that met the QualityLabs QC criteria and had median measurements from at least 50 individuals. Covariates for both PheWAS and LabWAS included: principal components (PCs) 1–10 for genetic ancestry, sex, age, median age of medical record, and genotyping batch.
Knockdown of MIC60 and CHCHD6 in HepG2 cells.
The transfection of MIC60 and CHCHD6 siRNAs into HepG2 cells was carried out using Lipofectamine RNAiMax (Invitrogen) in accordance with the manufacturer’s instructions and as previously described (24). Following a 48-hour incubation period, the cells were utilized for mitochondrial calcium (mCa2+) and ROS measurements.
Measurement of mitochondrial calcium uptake and retention capacity in HepG2 cells.
mCa2+ uptake retention capacity in HepG2 cells were assessed using a ratiometric Ca2+ sensor Fura-FF, as detailed earlier (161), with slight modifications. In brief, cells (2.5×106) were washed with Ca2+/Mg2+-free DPBS (GIBCO), permeabilized in intracellular medium (ICM: 120 mM KCl, 10 mM NaCl, 1 mM KH2PO4, 20 mM HEPES-Tris, pH 7.2), and supplemented with thapsigargin and succinate. Fura-FF (1 μM) was added at the 0 s time point, and fluorescence emissions at 340- and 380-nm ex/510-nm em were monitored using a multi-wavelength excitation dual-wavelength emission fluorimeter (Delta RAM, PTI). To assess the mCa2+ uptake, a bolus of 5 μM Ca2+ and the mitochondrial uncoupler FCCP (10 μM) were added at specified time points with continuous stirring at 37°C. To assess mCa2+ retention capacity, following baseline recordings, a series of Ca2+ boluses (5 μM) were introduced at specified time points. Upon reaching a steady state, 10μM FCCP was added to collapse the Δψm and release matrix free-Ca2+. The number of Ca2+ boluses taken up by cells was counted to calculate mitochondrial CRC.
Evaluation of ROS levels
~0.2 million HepG2 cells were plated in 35 mm dishes. The next day, MIC60 and CHCHD6 siRNAs were transfected using Lipofectamine RNAiMax (Invitrogen) according to the manufacturing instructions. After incubation for 30 hrs, cells were co-stained for 30 min at 37°C with two different dyes for ROS detection: MitoBright ROS Deep Red (10 μM, Dojindo Laboratories) for mitochondrial superoxide, and DCFDA (10 μM, Invitrogen) for intracellular total ROS. Following the incubation with staining dyes, cells were washed three times with 1X HBSS and ROS analysis was done using a confocal microscope (FV4000, Olympus Life Science).
For mitochondrial H2O2 assessment, cells were stained with MitoPY1 (5 μM, Bio-Techne) for 45 min at 37°C. Cells were then washed with 1x HBSS and imaged using a confocal microscope (FV4000, Olympus Life Science). ImageJ was used for the quantification of fluorescence intensities.
Data Analysis
GraphPad Prism 10.2.3 (La Jolla, CA, USA), was used for all statistical analysis. All experiments involving SBF-SEM and TEM data had at least three independent experiments. Statistics were not handled by those conducting the experiments. The black bars represent the standard error of the mean. For all analysis, one-way ANOVA was performed with tests against each independent group and significance was assessed using Fisher’s protected least significant difference (LSD) test. *, **, ***, **** were set to show significant difference, denoting p < 0.05, p < 0.01, p < 0.001, and p < 0.0001, respectively.
Supplementary Material
Supplemental Figure 1: Heterogeneity in Mitochondrial Quantification Across Samples (A) Distribution of mitochondria for mouse heterogeneity (3 mice, each 250 mitochondria surveyed) in mitochondrial volume, (B) surface area, (C) perimeter, (D) sphericity, and (E) complexity index in young and old liver tissue.
Supplemental Figure 2: Global metabolomic and lipidomic profiling revealed metabolic dysregulation and disruptions in lipid classes with age in liver tissues. (A) Metabolic pathway analysis and (B) PCA plot analysis for metabolomics. (C) Lipid class enrichment (D) and lipid chain length enrichment based on comparison between young and old livers.
Supplemental Figure 3: Phenome-wide and clinical lab-wide scan for SAMM50 genetically-regulated gene expression (GReX) in a medical biobank. Clinical phenotypes and laboratory values from BioVU participants were extracted from Vanderbilt’s de-identified electronic health record database (n= 85,615, top left panel). Genetically-regulated gene expression for SAMM50 was calculated in BioVU participants using models built from the GTEx version 8 data (top right panel), which contains genotype data matched to RNA-Seq data from 838 donors across 49 tissues. Imputed gene expression was calculated and tested for association across up to 1,704 phenotypes and 329 clinical lab tests using logistic and linear regression models (bottom panel), accounting for genetic ancestry (principle components/PC 1–10), sex, age, median age of medical record and genotyping batch.
SFile 1: Phenome-wide association MultiXCan SAMM50 genetically-regulated gene expression (GReX) in individuals of European ancestry.
SFile 2: Laboratory-wide association MultiXCan SAMM50 genetically-regulated gene expression (GReX) in individuals of European ancestry.
SFile 3: Phenome-wide association MultiXCan SAMM50 genetically-regulated gene expression (GReX) in individuals of African ancestry.
SFile 4: Laboratory-wide association MultiXCan SAMM50 genetically-regulated gene expression (GReX) in individuals of African ancestry.
Video 1: 3D structure of 3-month murine liver tissue visualized to show volumetric changes.
Video 2: 3D structure of 2-year murine liver tissue visualized to show volumetric changes.
Video 3: 3D structure of 3-month murine liver tissue visualized to show complexity changes.
Video 4: 3D structure of 2-year murine liver tissue visualized to show complexity changes.
ACKNOWLEDGEMENTS
This project was funded by the National Institute of Health (NIH) NIDDK T-32, number DK007563 entitled Multidisciplinary Training in Molecular Endocrinology to Z.V.; National Institute of Health (NIH) NIDDK T-32, number DK007563 entitled Multidisciplinary Training in Molecular Endocrinology to A.C.; NSF MCB #2011577I to S.A.M.; The UNCF/Bristol-Myers Squibb E.E. Just Faculty Fund, Career Award at the Scientific Interface (CASI Award) from Burroughs Welcome Fund (BWF) ID # 1021868.01, BWF Ad-hoc Award, NIH Small Research Pilot Subaward to 5R25HL106365–12 from the National Institutes of Health PRIDE Program, DK020593, Vanderbilt Diabetes and Research Training Center for DRTC Alzheimer’s Disease Pilot & Feasibility Program. CZI Science Diversity Leadership grant number 2022– 253529 from the Chan Zuckerberg Initiative DAF, an advised fund of Silicon Valley Community Foundation to A.H.J. and O.A.A.; and National Institutes of Health grant HD090061 and the Department of Veterans Affairs Office of Research Award I01 BX005352 to J.G. Howard Hughes Medical Institute Hanna H. Gray Fellows Program Faculty Phase (Grant# GT15655 awarded to M.R.M); and Burroughs Wellcome Fund PDEP Transition to Faculty (Grant# 1022604 awarded to M.R.M). National Institutes of Health Grants: R21DK119879 (to C.R.W.) and R01DK-133698 (to C.R.W.), American Heart Association Grant 16SDG27080009 (to C.R.W.) and by an American Society of Nephrology KidneyCure Transition to Independence Grant (to C.R.W.). NIH Grants R01HL147818, R03HL155041, and R01HL144941 (A. Kirabo). NIH Grant R00DK120876 (D.T.), Harold S. Geneen Charitable Trust Awards Program (D.T.), Alzheimer’s Association AARG-NTF-23–1144888 (D.T.). NIH Grant R00AG065445 (P.J.), Alzheimer’s Association 24AARG-D-1191292 (P.J.), Wake ADRC REC and Development grant P30AG072947 (P.J.). American Heart Association Grant 23POST1020344 (A.K.). American Heart Association Grant 23CDA1053072 (M. S.). NIH K01AG062757 to (M.T.S.) The BioVU project at VUMC is supported by numerous sources: including the NIH funded Shared Instrumentation Grant S10OD017985 and S10RR025141; CTSA grants UL1TR002243, UL1TR000445, and UL1RR024975 from the National Center for Advancing Translational Sciences. Its contents are solely the responsibility of the authors and do not necessarily represent official views of the National Center for Advancing Translational Sciences. Genomic data are also supported by investigator-led projects that include U01HG004798, R01NS032830, RC2GM092618, P50GM115305, U01HG006378, U19HL065962, R01HD074711; and additional funding sources listed at https://victr.vumc.org/biovu-funding/.23CDA1053072 (M.S.). Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the NIH. The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript. We would also like to acknowledge the Huck Institutes’ Metabolomics Core Facility (RRID:SCR_023864) for use of the OE 240 LCMS and Drs. Imhoi Koo, Ashley Shay, and Sergei Koshkin for helpful discussions on sample preparation and analysis. We would also like to thank UCLA investigators for gifting us old and young human liver samples.
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
All authors have no competing interests.
References:
- 1.McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol CB. 2006;16(14):R551–560. [DOI] [PubMed] [Google Scholar]
- 2.Glancy B, et al. The Functional Impact of Mitochondrial Structure Across Subcellular Scales. Front Physiol. 2020;11. 10.3389/fphys.2020.541040. Accessed December 16, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Picard M, et al. Mitochondria: isolation, structure and function. J Physiol. 2011;589(18):4413–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–287. [DOI] [PubMed] [Google Scholar]
- 5.Li M, et al. Mitochondrial Fusion Via OPA1 and MFN1 Supports Liver Tumor Cell Metabolism and Growth. Cells. 2020;9(1):121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seo AY, et al. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J Cell Sci. 2010;123(15):2533–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bratic A, Larsson N-G. The role of mitochondria in aging. J Clin Invest. 2013;123(3):951–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haas RH. Mitochondrial Dysfunction in Aging and Diseases of Aging. Biology. 2019;8(2):48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lam J, et al. A Universal Approach to Analyzing Transmission Electron Microscopy with ImageJ. Cells. 2021;10(9):2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collins HE, et al. Mitochondrial Morphology and Mitophagy in Heart Diseases: Qualitative and Quantitative Analyses Using Transmission Electron Microscopy. Front Aging. 2021;2. 10.3389/fragi.2021.670267. Accessed September 8, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cogliati S, Enriquez JA, Scorrano L. Mitochondrial cristae: where beauty meets functionality. Trends Biochem Sci. 2016;41(3):261–273. [DOI] [PubMed] [Google Scholar]
- 12.Jenkins BC, et al. Mitochondria in disease: changes in shapes and dynamics. Trends Biochem Sci. 2024;49(4):346–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Long Q, et al. Modeling of Mitochondrial Donut Formation. Biophys J. 2015;109:892–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu X, Hajnóczky G. Altered fusion dynamics underlie unique morphological changes in mitochondria during hypoxia–reoxygenation stress. Cell Death Differ. 2011;18(10):1561–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vincent AE, et al. Mitochondrial Nanotunnels. Trends Cell Biol. 2017;27(11):787–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tyumentsev MA, et al. Mitochondria with Morphology Characteristic for Alzheimer’s Disease Patients Are Found in the Brain of OXYS Rats. Biochem Mosc. 2018;83(9):1083–1088. [DOI] [PubMed] [Google Scholar]
- 17.Wakabayashi T. Megamitochondria formation physiology and pathology. J Cell Mol Med. 2002;6(4):497–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gautier MK, Ginsberg SD. A method for quantification of vesicular compartments within cells using 3D reconstructed confocal z-stacks: Comparison of ImageJ and Imaris to count early endosomes within basal forebrain cholinergic neurons. J Neurosci Methods. 2021;350:109038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garza-Lopez E, et al. Protocols for Generating Surfaces and Measuring 3D Organelle Morphology Using Amira. Cells. 2022;11(1):65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Courson JA, et al. Serial Block-Face Scanning Electron Microscopy (SBF-SEM) of Biological Tissue Samples. J Vis Exp JoVE. 2021;(169): 10.3791/62045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marshall AG, et al. Serial Block Face-Scanning Electron Microscopy as a Burgeoning Technology. Adv Biol. 2023;e2300139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Russell MRG, et al. 3D correlative light and electron microscopy of cultured cells using serial blockface scanning electron microscopy. J Cell Sci. 2017;130(1):278–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vue Z, et al. 3D reconstruction of murine mitochondria reveals changes in structure during aging linked to the MICOS complex. Aging Cell. 2023;22(12):e14009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vue Z, et al. The MICOS Complex Regulates Mitochondrial Structure and Oxidative Stress During Age-Dependent Structural Deficits in the Kidney [preprint]. 2024;2024.06.09.598108. [DOI] [PubMed] [Google Scholar]
- 25.Gagliano N, Grizzi F, Annoni G. Mechanisms of Aging and Liver Functions. Dig Dis. 2007;25(2):118–123. [DOI] [PubMed] [Google Scholar]
- 26.Hunt NJ, et al. Hallmarks of Aging in the Liver. Comput Struct Biotechnol J. 2019;17:1151–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalra A, et al. Physiology, Liver. StatPearls. Treasure Island (FL): StatPearls Publishing; 2024. [Google Scholar]
- 28.Reddy SR, et al. Outcomes of Young Patients With Alcoholic Cirrhosis After First Hospitalization for Cirrhosis: A Carilion Clinic Experience. Cureus;13(7):e16695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim H, Kisseleva T, Brenner DA. Aging and liver disease. Curr Opin Gastroenterol. 2015;31(3):184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gong Z, et al. Hepatic lipid metabolism and non-alcoholic fatty liver disease in aging. Mol Cell Endocrinol. 2017;455:115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chung KW. Advances in Understanding of the Role of Lipid Metabolism in Aging. Cells. 2021;10(4):880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoffmann JJ, Becker T. Crosstalk between Mitochondrial Protein Import and Lipids. Int J Mol Sci. 2022;23(9):5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen G, Kroemer G, Kepp O. Mitophagy: An Emerging Role in Aging and Age-Associated Diseases. Front Cell Dev Biol. 2020;8. 10.3389/fcell.2020.00200. Accessed December 21, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishikawa T, et al. A switch in the source of ATP production and a loss in capacity to perform glycolysis are hallmarks of hepatocyte failure in advance liver disease. J Hepatol. 2014;60(6):1203–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Middleton P, Vergis N. Mitochondrial dysfunction and liver disease: role, relevance, and potential for therapeutic modulation. Ther Adv Gastroenterol. 2021;14:17562848211031394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang D, et al. Research progress on the mitochondrial mechanism of age-related non-alcoholic fatty liver. World J Gastroenterol. 2023;29(13):1982–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shutt TE, McBride HM. Staying cool in difficult times: Mitochondrial dynamics, quality control and the stress response. Biochim Biophys Acta BBA - Mol Cell Res. 2013;1833(2):417–424. [DOI] [PubMed] [Google Scholar]
- 38.Ježek J, Cooper KF, Strich R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants. 2018;7(1). 10.3390/antiox7010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abrisch RG, et al. Fission and fusion machineries converge at ER contact sites to regulate mitochondrial morphology. J Cell Biol. 2020;219(4):e201911122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Youle RJ, van der Bliek AM. Mitochondrial Fission, Fusion, and Stress. Science. 2012;337(6098):1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Belenguer P, Pellegrini L. The dynamin GTPase OPA1: More than mitochondria? Biochim Biophys Acta BBA - Mol Cell Res. 2013;1833(1):176–183. [DOI] [PubMed] [Google Scholar]
- 42.Chen H, et al. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160(2):189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu C, et al. OPA1 and MICOS Regulate mitochondrial crista dynamics and formation. Cell Death Dis. 2020;11(10):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Favaro G, et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat Commun. 2019;10(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anand R, et al. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol. 2014;204(6):919–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wai T, Langer T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol Metab. 2016;27(2):105–117. [DOI] [PubMed] [Google Scholar]
- 47.Takeichi Y, et al. Non-alcoholic fatty liver disease in mice with hepatocyte-specific deletion of mitochondrial fission factor. Diabetologia. 2021;64:2092–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galloway C, et al. Decreasing mitochondrial fission alleviates hepatic steatosis in a murine model of nonalcoholic fatty liver disease. Am J Physiol Gastrointest Liver Physiol. 2014;307 6:632–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Z, et al. The role of SAMM50 in non-alcoholic fatty liver disease: from genetics to mechanisms. FEBS Open Bio. 2021;11(7):1893–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lionello S, Marzaro G, Martinvalet D. SAM50, a side door to the mitochondria: The case of cytotoxic proteases. Pharmacol Res. 2020;160:105196. [DOI] [PubMed] [Google Scholar]
- 51.Shao B, et al. Ablation of Sam50 is associated with fragmentation and alterations in metabolism in murine and human myotubes. BioRxiv Prepr Serv Biol. 2023;2023.05.20.541602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shao B, et al. Ablation of Sam50 is associated with fragmentation and alterations in metabolism in murine and human myotubes. J Cell Physiol. 2024;n/a(n/a). 10.1002/jcp.31293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tang A, et al. Nonalcoholic Fatty Liver Disease: MR Imaging of Liver Proton Density Fat Fraction to Assess Hepatic Steatosis. Radiology. 2013;267(2):422–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma X, et al. Perspectives on Mitochondria–ER and Mitochondria–Lipid Droplet Contact in Hepatocytes and Hepatic Lipid Metabolism. Cells. 2021;10(9):2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Simões ICM, et al. Mitochondria in non-alcoholic fatty liver disease. Int J Biochem Cell Biol. 2018;95:93–99. [DOI] [PubMed] [Google Scholar]
- 56.Chiang JYL, Ferrell JM. Bile Acid Metabolism in Liver Pathobiology. Gene Expr. 2018;18(2):71–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.TOMIZAWA M, et al. Triglyceride is strongly associated with nonalcoholic fatty liver disease among markers of hyperlipidemia and diabetes. Biomed Rep. 2014;2(5):633–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Semova I, Biddinger SB. TRIGLYCERIDES IN NAFLD: GUILTY UNTIL PROVEN INNOCENT. Trends Pharmacol Sci. 2021;42(3):183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yanai S, Endo S. Functional Aging in Male C57BL/6J Mice Across the Life-Span: A Systematic Behavioral Analysis of Motor, Emotional, and Memory Function to Define an Aging Phenotype. Front Aging Neurosci. 2021;13. 10.3389/fnagi.2021.697621. Accessed February 8, 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Robertson GL, et al. DRP1-mediated mitochondrial fission is essential to maintain cristae morphology and bioenergetics [preprint]. 2022;2021.12.31.474637. [Google Scholar]
- 61.Giacomello M, Pellegrini L. The coming of age of the mitochondria–ER contact: a matter of thickness. Cell Death Differ. 2016;23(9):1417–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vincent AE, et al. Quantitative 3D Mapping of the Human Skeletal Muscle Mitochondrial Network. Cell Rep. 2019;26(4):996–1009.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ilacqua N, et al. A three-organelle complex made by wrappER contacts with peroxisomes and mitochondria responds to liver lipid flux changes. J Cell Sci. 2021;135(5):jcs259091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ilacqua N, Anastasia I, Pellegrini L. Isolation and analysis of fractions enriched in WrappER-associated mitochondria from mouse liver. STAR Protoc. 2021;2(3):100752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ilacqua N, et al. Expression of Synj2bp in mouse liver regulates the extent of wrappER-mitochondria contact to maintain hepatic lipid homeostasis. Biol Direct. 2022;17(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Surmacki JM, Abramczyk H. Confocal Raman imaging reveals the impact of retinoids on human breast cancer via monitoring the redox status of cytochrome c. Sci Rep. 2023;13(1):15049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McReynolds MR, et al. NAD+ flux is maintained in aged mice despite lower tissue concentrations. Cell Syst. 2021;12(12):1160–1172.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goodman RP, Calvo SE, Mootha VK. Spatiotemporal compartmentalization of hepatic NADH and NADPH metabolism. J Biol Chem. 2018;293(20):7508–7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ueda N. Ceramide-Induced Apoptosis in Renal Tubular Cells: A Role of Mitochondria and Sphingosine-1-Phoshate. Int J Mol Sci. 2015;16(3):5076–5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fugio LB, Coeli-Lacchini FB, Leopoldino AM. Sphingolipids and Mitochondrial Dynamic. Cells. 2020;9(3):581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Parra V, et al. Changes in mitochondrial dynamics during ceramide-induced cardiomyocyte early apoptosis. Cardiovasc Res. 2008;77(2):387–397. [DOI] [PubMed] [Google Scholar]
- 72.Longo N, Frigeni M, Pasquali M. CARNITINE TRANSPORT AND FATTY ACID OXIDATION. Biochim Biophys Acta. 2016;1863(10):2422–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li H, et al. Mic60/Mitofilin determines MICOS assembly essential for mitochondrial dynamics and mtDNA nucleoid organization. Cell Death Differ. 2016;23(3):380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tezze C, et al. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab. 2017;25(6):1374–1389.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vue Z, et al. Three-Dimensional Mitochondria Reconstructions of Murine Cardiac Muscle Changes in Size Across Aging. Am J Physiol-Heart Circ Physiol. [published online ahead of print: August 25, 2023]. 10.1152/ajpheart.00202.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vue Z, et al. 3D reconstruction of murine mitochondria reveals changes in structure during aging linked to the MICOS complex. Aging Cell. 2023;22(12):e14009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Körner C, et al. The C-terminal domain of Fcj1 is required for formation of crista junctions and interacts with the TOB/SAM complex in mitochondria. Mol Biol Cell. 2012;23(11):2143–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kozjak V, et al. An Essential Role of Sam50 in the Protein Sorting and Assembly Machinery of the Mitochondrial Outer Membrane *. J Biol Chem. 2003;278(49):48520–48523. [DOI] [PubMed] [Google Scholar]
- 79.Tang J, et al. Sam50–Mic19–Mic60 axis determines mitochondrial cristae architecture by mediating mitochondrial outer and inner membrane contact. Cell Death Differ. 2020;27(1):146–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ding C, et al. Mitofilin and CHCHD6 physically interact with Sam50 to sustain cristae structure. Sci Rep. 2015;5(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen L, et al. Loss of Sam50 in hepatocytes induces cardiolipin dependent mitochondrial membrane remodeling to trigger mtDNA release and liver injury. Hepatology. 2022;76(5):1389. [DOI] [PubMed] [Google Scholar]
- 82.Tsuru H, et al. HFD-induced hepatic lipid accumulation and inflammation are decreased in Factor D deficient mouse. Sci Rep. 2020;10:17593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lian C-Y, et al. High fat diet-triggered non-alcoholic fatty liver disease: A review of proposed mechanisms. Chem Biol Interact. 2020;330:109199. [DOI] [PubMed] [Google Scholar]
- 84.Zheng P, et al. High-fat diet causes mitochondrial damage and downregulation of mitofusin-2 and optic atrophy-1 in multiple organs. J Clin Biochem Nutr. 2023;73(1):61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.De Nicolo B, et al. Calcium and Reactive Oxygen Species Signaling Interplays in Cardiac Physiology and Pathologies. Antioxidants. 2023;12(2):353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Winslow JL, Hollenberg MJ, Lea PJ. Resolution limit of serial sections for 3D reconstruction of tubular cristae in rat liver mitochondria. J Electron Microsc Tech. 1991;18(3):241–248. [DOI] [PubMed] [Google Scholar]
- 87.Mannella CA. Structure and dynamics of the mitochondrial inner membrane cristae. Biochim Biophys Acta BBA - Mol Cell Res. 2006;1763(5):542–548. [DOI] [PubMed] [Google Scholar]
- 88.Mannella CA, et al. Electron microscopic tomography of rat-liver mitochondria and their interactions with the endoplasmic reticulum. BioFactors. 1998;8(3–4):225–228. [DOI] [PubMed] [Google Scholar]
- 89.Das S, et al. Mitochondrial morphology and dynamics in hepatocytes from normal and ethanol-fed rats. Pflugers Arch. 2012;464(1):101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shami GJ, et al. Three-dimensional ultrastructure of giant mitochondria in human non-alcoholic fatty liver disease. Sci Rep. 2021;11:3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Daum B, et al. Age-dependent dissociation of ATP synthase dimers and loss of inner-membrane cristae in mitochondria. Proc Natl Acad Sci. 2013;110(38):15301–15306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Labbe G, Pessayre D, Fromenty B. Drug-induced liver injury through mitochondrial dysfunction: mechanisms and detection during preclinical safety studies. Fundam Clin Pharmacol. 2008;22(4):335–353. [DOI] [PubMed] [Google Scholar]
- 93.Altamirano J, et al. A histologic scoring system for prognosis of patients with alcoholic hepatitis. Gastroenterology. 2014;146(5):1231–1239.e1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Palma E, et al. Dynamin-1-Like Protein Inhibition Drives Megamitochondria Formation as an Adaptive Response in Alcohol-Induced Hepatotoxicity. Am J Pathol. 2019;189(3):580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhou H, et al. DNA-PKcs promotes alcohol-related liver disease by activating Drp1-related mitochondrial fission and repressing FUNDC1-required mitophagy. Signal Transduct Target Ther. 2019;4:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yamada T, et al. Prevention and regression of megamitochondria and steatosis by blocking mitochondrial fusion in the liver. iScience. 2022;25(4):103996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tandler B, Hoppel CL. Giant Mitochondria (Megamitochondria). In: Lennarz WJ, Lane MD, eds. Encyclopedia of Biological Chemistry (Second Edition). Waltham: Academic Press; 2013:375–376. [Google Scholar]
- 98.Palma E, et al. Perturbations in mitochondrial dynamics are closely involved in the progression of Alcoholic Liver Disease. Alcohol Clin Exp Res. [published online ahead of print: 2020]. 10.1111/acer.14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Palma E, et al. Perturbation of mitochondrial dynamics in Alcoholic Liver Disease. J World Mitochondria Soc. 2016;2. 10.18143/JWMS_V2I2_1948. [DOI] [Google Scholar]
- 100.Wang D, Wei Y, Pagliassotti MJ. Saturated Fatty Acids Promote Endoplasmic Reticulum Stress and Liver Injury in Rats with Hepatic Steatosis. Endocrinology. 2006;147(2):943–951. [DOI] [PubMed] [Google Scholar]
- 101.Renne MF, Hariri H. Lipid Droplet-Organelle Contact Sites as Hubs for Fatty Acid Metabolism, Trafficking, and Metabolic Channeling. Front Cell Dev Biol. 2021;9. 10.3389/fcell.2021.726261. Accessed February 12, 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kim H, et al. Structural basis for mitoguardin-2 mediated lipid transport at ER-mitochondrial membrane contact sites. Nat Commun. 2022;13(1):3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Marchi S, Patergnani S, Pinton P. The endoplasmic reticulum–mitochondria connection: One touch, multiple functions. Biochim Biophys Acta BBA - Bioenerg. 2014;1837(4):461–469. [DOI] [PubMed] [Google Scholar]
- 104.Anastasia I, et al. Mitochondria-rough-ER contacts in the liver regulate systemic lipid homeostasis. Cell Rep. 2021;34(11):108873. [DOI] [PubMed] [Google Scholar]
- 105.Wang J, et al. Mutual interaction between endoplasmic reticulum and mitochondria in nonalcoholic fatty liver disease. Lipids Health Dis. 2020;19(1):72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Huynen MA, et al. Evolution and structural organization of the mitochondrial contact site (MICOS) complex and the mitochondrial intermembrane space bridging (MIB) complex. Biochim Biophys Acta BBA-Mol Cell Res. 2016;1863(1):91–101. [DOI] [PubMed] [Google Scholar]
- 107.Ott C, et al. Sam50 Functions in Mitochondrial Intermembrane Space Bridging and Biogenesis of Respiratory Complexes. Mol Cell Biol. 2012;32(6):1173–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gagliano Taliun SA, et al. Exploring and visualizing large-scale genetic associations by using PheWeb. Nat Genet. 2020;52(6):550–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kanai M, et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat Genet. 2018;50(3):390–400. [DOI] [PubMed] [Google Scholar]
- 110.Kurki MI, et al. FinnGen: Unique genetic insights from combining isolated population and national health register data [preprint]. 2022;2022.03.03.22271360. [Google Scholar]
- 111.Duchen MR, Szabadkai G. Roles of mitochondria in human disease. Essays Biochem. 2010;47:115–137. [DOI] [PubMed] [Google Scholar]
- 112.Alonso MT, et al. Calcium microdomains in mitochondria and nucleus. Cell Calcium. 2006;40(5–6):513–525. [DOI] [PubMed] [Google Scholar]
- 113.Bertero E, Maack C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ Res. 2018;122(10):1460–1478. [DOI] [PubMed] [Google Scholar]
- 114.Hoek JB, et al. Calcium ion-dependent signalling and mitochondrial dysfunction: mitochondrial calcium uptake during hormonal stimulation in intact liver cells and its implication for the mitochondrial permeability transition. Biochim Biophys Acta BBA - Mol Basis Dis. 1995;1271(1):93–102. [DOI] [PubMed] [Google Scholar]
- 115.Csordás G, Weaver D, Hajnóczky G. Endoplasmic Reticulum–Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 2018;28(7):523–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Egnatchik RA, et al. ER calcium release promotes mitochondrial dysfunction and hepatic cell lipotoxicity in response to palmitate overload. Mol Metab. 2014;3(5):544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kim YS, Kim SG. Endoplasmic reticulum stress and autophagy dysregulation in alcoholic and non-alcoholic liver diseases. Clin Mol Hepatol. 2020;26(4):715–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Amaya MJ, Nathanson MH. Calcium Signaling in the Liver. Compr Physiol. 2013;3(1):515–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Modi S, et al. Miro clusters regulate ER-mitochondria contact sites and link cristae organization to the mitochondrial transport machinery. Nat Commun. 2019;10(1):4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cichoż-Lach H, Michalak A. Oxidative stress as a crucial factor in liver diseases. World J Gastroenterol WJG. 2014;20(25):8082–8091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li P-L, Gulbins E. Bioactive Lipids and Redox Signaling: Molecular Mechanism and Disease Pathogenesis. Antioxid Redox Signal. 2018;28(10):911–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Barja G. The mitochondrial free radical theory of aging. Prog Mol Biol Transl Sci. 2014;127:1–27. [DOI] [PubMed] [Google Scholar]
- 123.Navarro A, Boveris A. Rat brain and liver mitochondria develop oxidative stress and lose enzymatic activities on aging. Am J Physiol-Regul Integr Comp Physiol. 2004;287(5):R1244–R1249. [DOI] [PubMed] [Google Scholar]
- 124.Maeso-Díaz R, et al. Effects of aging on liver microcirculatory function and sinusoidal phenotype. Aging Cell. 2018;17(6):e12829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nahata M, et al. Decline in Liver Mitochondria Metabolic Function Is Restored by Hochuekkito Through Sirtuin 1 in Aged Mice With Malnutrition. Front Physiol. 2022;13. 10.3389/fphys.2022.848960. Accessed February 12, 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Barazzoni R, Short KR, Nair KS. Effects of Aging on Mitochondrial DNA Copy Number and Cytochromec Oxidase Gene Expression in Rat Skeletal Muscle, Liver, and Heart *. J Biol Chem. 2000;275(5):3343–3347. [DOI] [PubMed] [Google Scholar]
- 127.Serviddio G, et al. Bioenergetics in aging: mitochondrial proton leak in aging rat liver, kidney and heart. Redox Rep. 2007;12(1–2):91–95. [DOI] [PubMed] [Google Scholar]
- 128.Moreira OC, et al. Mitochondrial Function and Mitophagy in the Elderly: Effects of Exercise. Oxid Med Cell Longev. 2017;2017:2012798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Guo Y, et al. Quantitative Proteomic and Functional Analysis of Liver Mitochondria from High Fat Diet (HFD) Diabetic Mice. Mol Cell Proteomics MCP. 2013;12(12):3744–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Graham DS, et al. Ability of high fat diet to induce liver pathology correlates with the level of linoleic acid and Vitamin E in the diet. PloS One. 2023;18(6):e0286726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kim HJ, et al. A high fat diet fosters elevated bisretinoids. J Biol Chem. 2023;299(6):104784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Parlakgül G, et al. Spatial mapping of hepatic ER and mitochondria architecture reveals zonated remodeling in fasting and obesity. Nat Commun. 2024;15(1):3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Arruda AP, et al. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med. 2014;20(12):1427–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Softic S, et al. Dietary Sugars Alter Hepatic Fatty Acid Oxidation via Transcriptional and Post-translational Modifications of Mitochondrial Proteins. Cell Metab. 2019;30 4:735–7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lockman KA, et al. Oxidative stress rather than triglyceride accumulation is a determinant of mitochondrial dysfunction in in vitro models of hepatic cellular steatosis. Liver Int Off J Int Assoc Study Liver. 2012;32(7):1079–1092. [DOI] [PubMed] [Google Scholar]
- 136.Dong J, et al. Mic19 depletion impairs endoplasmic reticulum-mitochondrial contacts and mitochondrial lipid metabolism and triggers liver disease. Nat Commun. 2024;15(1):168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Paradies G, et al. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J Gastroenterol WJG. 2014;20(39):14205–14218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Liu J, et al. Cardiolipin inhibitor ameliorates the non-alcoholic steatohepatitis through suppressing NLRP3 inflammasome activation. Eur Rev Med Pharmacol Sci. 2019;23(18):8158–8167. [DOI] [PubMed] [Google Scholar]
- 139.Alves-Bezerra M, Cohen DE. Triglyceride metabolism in the liver. Compr Physiol. 2017;8(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hinton A, et al. ATF4 Dependent Increase in Mitochondrial-Endoplasmic Reticulum Tethering Following OPA1 Deletion in Skeletal Muscle. Journal of Cellular Physiology. [published online ahead of print: 2024]. 10.1101/2022.09.12.507669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Stahl EC, et al. Inflammation and Ectopic Fat Deposition in the Aging Murine Liver Is Influenced by CCR2. Am J Pathol. 2020;190(2):372–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Pereira RO, et al. OPA 1 deficiency promotes secretion of FGF 21 from muscle that prevents obesity and insulin resistance. EMBO J. 2017;36(14):2126–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Tam J, et al. Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J Clin Invest. 2010;120(8):2953–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hinton A, et al. A Comprehensive Approach to Sample Preparation for Electron Microscopy and the Assessment of Mitochondrial Morphology in Tissue and Cultured Cells. Adv Biol. 2023;e2200202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Neikirk K, et al. Systematic Transmission Electron Microscopy-Based Identification and 3D Reconstruction of Cellular Degradation Machinery. Adv Biol. 2023;7(6):2200221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lu W, et al. Extraction and Quantitation of Nicotinamide Adenine Dinucleotide Redox Cofactors. Antioxid Redox Signal. 2018;28(3):167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang L, et al. Peak Annotation and Verification Engine for Untargeted LC–MS Metabolomics. Anal Chem. 2019;91(3):1838–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Adusumilli R, Mallick P. Data Conversion with ProteoWizard msConvert. In: Comai L, Katz JE, Mallick P, eds. Proteomics: Methods and Protocols. New York, NY: Springer; 2017:339–368. [DOI] [PubMed] [Google Scholar]
- 149.Boudina S, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56(10):2457–2466. [DOI] [PubMed] [Google Scholar]
- 150.Olenych SG, et al. The fluorescent protein color palette. Curr Protoc Cell Biol. 2007;Chapter 21:Unit 21.5. [DOI] [PubMed] [Google Scholar]
- 151.Roden DM, et al. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin Pharmacol Ther. 2008;84(3):362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Danciu I, et al. Secondary use of clinical data: the Vanderbilt approach. J Biomed Inform. 2014;52:28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Dennis JK, et al. Clinical laboratory test-wide association scan of polygenic scores identifies biomarkers of complex disease. Genome Med. 2021;13(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Consortium GTEx. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369(6509):1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gamazon ER, et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet. 2015;47(9):1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hu Y, et al. A statistical framework for cross-tissue transcriptome-wide association analysis. Nat Genet. 2019;51(3):568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zhou D, et al. A unified framework for joint-tissue transcriptome-wide association and Mendelian randomization analysis. Nat Genet. 2020;52(11):1239–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Barbeira AN, et al. Integrating predicted transcriptome from multiple tissues improves association detection. PLoS Genet. 2019;15(1):e1007889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Denny JC, et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinforma Oxf Engl. 2010;26(9):1205–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Carroll RJ, Bastarache L, Denny JC. R PheWAS: data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinforma Oxf Engl. 2014;30(16):2375–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Tomar D, et al. MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell Rep. 2016;15(8):1673–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Heterogeneity in Mitochondrial Quantification Across Samples (A) Distribution of mitochondria for mouse heterogeneity (3 mice, each 250 mitochondria surveyed) in mitochondrial volume, (B) surface area, (C) perimeter, (D) sphericity, and (E) complexity index in young and old liver tissue.
Supplemental Figure 2: Global metabolomic and lipidomic profiling revealed metabolic dysregulation and disruptions in lipid classes with age in liver tissues. (A) Metabolic pathway analysis and (B) PCA plot analysis for metabolomics. (C) Lipid class enrichment (D) and lipid chain length enrichment based on comparison between young and old livers.
Supplemental Figure 3: Phenome-wide and clinical lab-wide scan for SAMM50 genetically-regulated gene expression (GReX) in a medical biobank. Clinical phenotypes and laboratory values from BioVU participants were extracted from Vanderbilt’s de-identified electronic health record database (n= 85,615, top left panel). Genetically-regulated gene expression for SAMM50 was calculated in BioVU participants using models built from the GTEx version 8 data (top right panel), which contains genotype data matched to RNA-Seq data from 838 donors across 49 tissues. Imputed gene expression was calculated and tested for association across up to 1,704 phenotypes and 329 clinical lab tests using logistic and linear regression models (bottom panel), accounting for genetic ancestry (principle components/PC 1–10), sex, age, median age of medical record and genotyping batch.
SFile 1: Phenome-wide association MultiXCan SAMM50 genetically-regulated gene expression (GReX) in individuals of European ancestry.
SFile 2: Laboratory-wide association MultiXCan SAMM50 genetically-regulated gene expression (GReX) in individuals of European ancestry.
SFile 3: Phenome-wide association MultiXCan SAMM50 genetically-regulated gene expression (GReX) in individuals of African ancestry.
SFile 4: Laboratory-wide association MultiXCan SAMM50 genetically-regulated gene expression (GReX) in individuals of African ancestry.
Video 1: 3D structure of 3-month murine liver tissue visualized to show volumetric changes.
Video 2: 3D structure of 2-year murine liver tissue visualized to show volumetric changes.
Video 3: 3D structure of 3-month murine liver tissue visualized to show complexity changes.
Video 4: 3D structure of 2-year murine liver tissue visualized to show complexity changes.