

Abstract
BACKGROUND:
An imbalance of antiproliferative BMP (bone morphogenetic protein) signaling and proliferative TGF-β (transforming growth factor-β) signaling is implicated in the development of pulmonary arterial hypertension (PAH). The posttranslational modification (eg, phosphorylation and ubiquitination) of TGF-β family receptors, including BMPR2 (bone morphogenetic protein type 2 receptor)/ALK2 (activin receptor-like kinase-2) and TGF-βR2/R1, and receptor-regulated (R) Smads significantly affects their activity and thus regulates the target cell fate. BRCC3 modifies the activity and stability of its substrate proteins through K63-dependent deubiquitination. By modulating the posttranslational modifications of the BMP/TGF-β–PPARγ pathway, BRCC3 may play a role in pulmonary vascular remodeling, hence the pathogenesis of PAH.
METHODS:
Bioinformatic analyses were used to explore the mechanism of BRCC3 deubiquitinates ALK2. Cultured pulmonary artery smooth muscle cells (PASMCs), mouse models, and specimens from patients with idiopathic PAH were used to investigate the rebalance between BMP and TGF-β signaling in regulating ALK2 phosphorylation and ubiquitination in the context of pulmonary hypertension.
RESULTS:
BRCC3 was significantly downregulated in PASMCs from patients with PAH and animals with experimental pulmonary hypertension. BRCC3, by de-ubiquitinating ALK2 at Lys-472 and Lys-475, activated receptor-regulated Smad1/5/9 (Smad1/5/9), which resulted in transcriptional activation of BMP-regulated PPARγ, p53, and Id1. Overexpression of BRCC3 also attenuated TGF-β signaling by downregulating TGF-β expression and inhibiting phosphorylation of Smad3. Experiments in vitro indicated that overexpression of BRCC3 or the de-ubiquitin–mimetic ALK2-K472/475R attenuated PASMC proliferation and migration and enhanced PASMC apoptosis. In SM22α-BRCC3-Tg mice, pulmonary hypertension was ameliorated because of activation of the ALK2-Smad1/5-PPARγ axis in PASMCs. In contrast, Brcc3−/− mice showed increased susceptibility of experimental pulmonary hypertension because of inhibition of the ALK2-Smad1/5 signaling.
CONCLUSIONS:
These results suggest a pivotal role of BRCC3 in sustaining pulmonary vascular homeostasis by maintaining the integrity of the BMP signaling (ie, the ALK2-Smad1/5-PPARγ axis) while suppressing TGF-β signaling in PASMCs. Such rebalance of BMP/TGF-β pathways is translationally important for PAH alleviation.
Keywords: ALK2, BMP, BRCC3, pulmonary arterial hypertension
Mutation and dysfunction of BMPR2 (bone morphogenetic protein type 2 receptor) are associated with the development of idiopathic and heritable pulmonary arterial hypertension (PAH).1–4 BMPR2 tends to be downregulated in patients with PAH lacking genetic causes, possibly because of inflammatory or environmental hits.5–7 Upon BMP (bone morphogenetic protein) binding, BMPR2 binds to and phosphorylates its cognate type I receptor ALK2 (activin receptor-like kinase-2; also known as activin A receptor, type I) to activate the BMP-BMPR2-Smad1/5/9 signaling cascade. Phosphorylated (p) Smad1/5/9 translocates into the nucleus, which leads to activation of canonical transcription factors such as Id1 and noncanonical transcription factors including PPARγ and p53,8–10 which exert an antiproliferative and proapoptotic effect on pulmonary arterial smooth muscle cells (PASMCs). Upon TGF-β (transforming growth factor-β) or activin-A binding, TGF-β receptors signal through phosphorylated Smad2/3 to cause a proproliferative effect on pulmonary vascular cells.11 However, the BMP and TGF-β arms have some distinct effects on cell proliferation, hypertrophy, differentiation, and survival in vascular cells, depending on lineage and biological context.12,13
The posttranslational modification (eg, phosphorylation and ubiquitination) of TGF-β family receptors, including BMPR2/ALK2 and TGF-βR2/R1, and receptor-regulated (R) Smads (Smad1/5/9 or Smad2/3) significantly affects their activity and thus regulates the target cell fate.14,15 K48-linked ubiquitination of proteins is commonly associated with their proteasomal degradation, whereas K63-linked ubiquitination is involved in a variety of cellular events, including DNA repair, intracellular protein trafficking, and kinase signaling.16,17 As a member of the HECT (homologous to E6AP C terminus)–type E3 ubiquitin ligases, SMURF1 (Smad ubiquitination regulatory factor 1) ubiquitinates R-Smads (Smad1/5) for proteasomal degradation, thus blocking the BMP signaling. Additionally, SMURF1 can downregulate the BMP signaling through its degradation of BMPRI and Smad1/5 by I-Smads.14,18 In rodents with experimental pulmonary hypertension (PH), SMURF1 is significantly elevated.19 However, whether SMURF1 mediates the ubiquitination of BMP component proteins in pulmonary vasculature involved in PAH and any rebalancing mechanisms are currently unknown.
Protein ubiquitination is intricately regulated by E3 ligases and DUBs (deubiquitinating enzymes). Several DUBs, such as UCH37, USP15, CYLD, and AMSH, regulate TGF-β signaling pathways.20–23 Encoded by the BRCC3 gene, BRCC3 (BRCA1/BRCA2-containing complex subunit 3) is a member of the JAMM/MPN+ family of zinc metalloproteases that proteolytically cleaves the K63-linked polyubiquitin chains.24,25 BRCC3 was initially identified as an essential component of the BRCA1 complex that mediates homologous recombination during the repair of DNA double-strand breaks.26,27 Emerging evidence indicates that BRCC3 participates in regulating cell autophagy and NLRP3 inflammasome activation, in part because of its DUB activity.28
Phenotypic changes of PASMCs to a highly proliferative state are implicated in the development of pulmonary vascular remodeling and arteriole muscularization.29 In PASMCs from patients with PAH and animals with experimental PH, antiproliferative BMP signaling is downregulated, whereas the proproliferative signaling elicited by TGF-β, activins, and GDFs (growth differentiation factors) is enhanced. Indeed, inhibition of proproliferative TGF-β signaling by sotatercept, a fusion protein ligand trap of ActRIIA (activin receptor IIA),30,31 reduced pulmonary vascular resistance and increased the survival rate in patients with PAH (PulSAR and STELLAR trials).32–34
Given that BRCC3 is involved in (or required for) the antiproliferative BMP signaling proteins, we postulated that BRCC3 downregulation may have a causal role in PAH/PH. Results from this study indicate that BRCC3 level is lower in human PAH disease and rodent PH models. BRCC3 activates the ALK2-Smad1/5/9-PPARγ axis by deubiquitinating ALK2 in PASMCs. Thus, BRCC3 appears to antagonize SMURF1 in the disease pathogenesis, and upregulation and activation of BRCC3 may be a novel therapeutic strategy for PAH/PH.
METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Cell Cultures and Virus Infection
PASMCs were obtained from the Pulmonary Hypertension Breakthrough Initiative (PHBI). These cells, isolated from lung tissues of patients with idiopathic PAH (IPAH) and healthy controls, were cultured in smooth muscle cell (SMC) growth medium (SmGM-2; Lonza). PASMCs were also isolated from pulmonary arteries of adult Sprague-Dawley rats or various mouse models under a stereomicroscope and cultured in DMEM. Mouse lung tissues were isolated and placed in petri dishes containing precooled PBS. The lung tissues were repeatedly rinsed with PBS. The pulmonary arterioles were separated under a microscope, and the fibrous connective tissue of the outer membrane was removed. The arterial endothelium was gently scraped off with surgical forceps and cut into 1-mm³ tissue blocks, and DMEM containing 20% FBS was added. After 7 to 10 days, adhesive and proliferative PASMCs were harvested. Human embryonic kidney 293 (HEK293) cells were maintained in DMEM. PASMCs (passages 4–7) and HEK293 cells were maintained in an incubator under 95% air or 1% O2 (hypoxia treatment for 48–72 hours) and 5% CO2 at 37 °C. For siRNA transfection experiments, ALK2 siRNA or scramble control RNA (Genepharma) was transfected into PASMCs at 40 nM with Lipofectamine 2000 (Invitrogen) for 48 hours. Adenovirus-driven ectopic-expressing BRCC3 and the BRCC3-shRNA in cells was achieved by infecting cultured cells at 100 multiplicity of infection for 48 to 72 hours. The reagents used and their commercial sources were as follows: BMP2 (MCE), TGF-β1 (Sigma), LDN-214117 (Selleck), SU5416 (Cayman), monocrotaline (Sigma-Aldrich), MG132 (MCE), Microfil MV-122, MV-Diluent, MV curing agent (Flow Tech), and pioglitazone (MCE).
Human Lung Samples
Human lung specimens were from normal donor lungs or explanted lungs from patients with IPAH undergoing lung transplantation at the University of California, San Diego Medical Center. The study was approved by the University of California, San Diego institutional review board, and written informed consent was obtained from all participants. Demographic and clinical data for controls and patients with IPAH are in Table S1.
RNA Sequencing and In Silico Analyses
For RNA sequencing (RNA-seq) and data analysis, PASMCs were transfected with Ad-BRCC3 or Ad-Null (100 multiplicity of infection) for 72 hours, and total RNA was extracted with Trizol. The RNA integrity number was measured with the Agilent 2200 TapeStation-RNA R6K Bioanalyzer (Agilent Technologies), and high-quality RNA (RNA integrity number ≥8.0) was used. RNA libraries were prepared by using the NEBNex Ultra RNA Library Pre Kit (Illumina) following manufacturer protocols. The clustering of the index-code samples was performed on a cBot Cluster Generation System with the TruSeq PE Cluster Kit v3-cBot-HS (Illumina) and sequenced on an Illumina Hiseq platform. The RNA-seq reads were preprocessed by using FASTX-Toolkit 0.0.13.2 (http://hannonlab.cshl.edu/fastx_toolkit/) to obtain high-quality reads (Phred quality score ≥20 and reads >35 nt were retained). Reads were mapped and splice junctions were annotated to the UCSC human hg38 reference genome by using TopHat2 2.1.0. Then the annotated transcripts were assembled, and abundances were estimated and normalized by using Cufflinks 2.2.1. The gene counts obtained from RNA-seq were screened by using the R package DESseq2 for differentially expressed genes between Ad-Null and Ad-BRCC3 groups (log2-fold change >1 or <−1; FDR<0.1). The RNA-seq data set of lung tissue from patients with IPAH was from Gene Expression Omnibus (GEO) (GSE48149, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE48149), the human PASMCs data set was from GEO (GSE168905, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE168905), and the hypoxic rat PASMCs data set was from GEO (GSE72181, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE72181). Enrichment analysis of differentially expressed genes was performed using the online tool DAVID (https://david.ncifcrf.gov/). The program was run with default settings, with significant Gene Ontology terms and pathways identified ranking by –Log10(FDR) (obtained by hypergeometric test) shown in Figures 1 and 2.
Figure 1. BRCC3 is decreased in human and rodent PH.

A, Bioinformatics analysis of GEO RNA-seq data (GES48149) for BRCC3 levels in human lung tissues from patients with idiopathic pulmonary artery hypertension (IPAH; n=8) and controls without IPAH (n=9). Western blot analysis of BRCC3 levels in lung tissues from patients with IPAH and nondiseased tissues (B); mouse models with experimental PH induced by 10% O2 with subcutaneous injection of SU5416 (SuHx) and control mice under normoxia (C); and rat models with SuHx-induced PH vs controls (D). E, Analysis of GEO RNA-seq data (GES168905) for BRCC3 expression level in human pulmonary arterial smooth muscle cells (PASMCs) from patients with or without IPAH. Western blot analysis of BRCC3 levels in lung PASMCs isolated from IPAH and control individuals (F); SuHx-induced PH mice and control mice (G); and SuHx-induced PH rat models and control rats (H). I, Gene Ontology (GO) enrichment analysis of GSE72181 using DAVID for the top upregulated and downregulated pathways (fold change in expression >1.5 or < −1.5; P<0.05) in PASMCs under normoxia vs hypoxia and plotted as –log(P value) [–Log10(P)]. J, Heatmap generated from GSE72181 revealing differentially expressed genes involved in the BMP pathway (18 differentially expressed genes), TGF-β pathway (16 differentially expressed genes), proliferation (25 differentially expressed genes), and apoptosis (16 differentially expressed genes) in PASMCs under normoxia or hypoxia. K, PASMCs cultured under hypoxia for 6, 12, 24, 48, or 72 hours. Western blot analysis of protein levels of BRCC3, BMPR2, ALK2, PPARγ, p53, and Id1 with those of β-actin as loading controls. Data in A through H and K are mean±SEM from 3 to 9 independent experiments. For data with normal distribution (A, D, and H), statistical significance was determined by 2-tailed Student t test with Welch correction between 2 indicated groups. Nonnormally distributed data in B, C, and E through G were analyzed by Mann-Whitney U test between 2 indicated groups. Nonnormally distributed data in K were analyzed by the Kruskal-Wallis test between multiple groups. *P<0.05 vs control patients (Ctrl) or normoxia (Nor) or vehicle (Veh) or 0 hour. ALK2 indicates activin receptor-like kinase-2; BMP, bone morphogenetic protein; BMPR2, bone morphogenetic protein type 2 receptor; BRCC3, BRCA1/BRCA2-containing complex subunit 3; GEO, Gene Expression Omnibus; PH, pulmonary hypertension; RNA-seq, RNA sequencing; and TGF-β, transforming growth factor-β.
Figure 2. BRCC3 overexpression ameliorates the BMP pathway in PASMCs.

A and B, RNAs extracted from PASMCs transfected with Ad-Null or Ad-BRCC3 (n=3, each condition) were subjected to RNA-seq analyses. A, GO enrichment analysis using DAVID for the top upregulated and downregulated genes (fold change in expression >1.5 or < –1.5; P<0.05) in PASMCs transfected with Ad-Null or Ad-BRCC3 and plotted as –log (P value) [–Log10(P)]. B, Heatmaps showing differentially expressed genes involved in BMP (9 differentially expressed genes) and TGF-β signaling pathways (9 differentially expressed genes), positive regulation of cell migration (34 differentially expressed genes), and SMC proliferation and apoptosis (57 differentially expressed genes) in PASMCs transfected with Ad-Null vs Ad-BRCC3. C, PASMCs were transfected with Ad-Null or Ad-BRCC3 for 24 hours, then exposed to hypoxia (1% O2) or normoxia for an additional 48 hours. Western blot analysis of protein levels of BMP2, BMPR2, ALK2, pSmad1/5, Smad1, PPARγ, p53, Id1, BRCC3, and β-actin. D, PASMCs were transfected with Ad-Null, Ad-BRCC3, or Ad-BRCC3 shRNA for 24 hours, then cultured under hypoxia for 48 hours and treated with vehicle or BMP2 (25 ng/mL) for 2 hours before harvesting. Western blot analysis of protein levels of pSmad1/5, Smad1, PPARγ, Id1, BRCC3, and β-actin. Data in C and D are mean±SEM from 4 to 6 independent experiments. Normally or nonnormally distributed data were analyzed by 1-way ANOVA (C) and 2-way ANOVA (D) or the Kruskal-Wallis test between multiple groups. Data in C, *P<0.05 vs Nor+Ad-Null, #P<0.05 vs Hyp+Ad-Null. Data in D, *P<0.05 vs Ad-Null in vehicle (Veh) group; #P<0.05 vs Ad-Null in BMP2 group. ALK2 indicates activin receptor-like kinase-2; BMP, bone morphogenetic protein; BMPR2, bone morphogenetic protein type 2 receptor; BRCC3, BRCA1/BRCA2-containing complex subunit 3; GO, Gene Ontology; PASMC, pulmonary arterial smooth muscle cell; RNA-seq, RNA sequencing; SMC, smooth muscle cell; and TGF-β, transforming growth factor-β.
To analyze the BRCC3/ALK2 protein-–protein interaction, ALK2-BRCC3 docking was performed by using HDOCK, a server used to automatically predict protein–protein interaction by a hybrid algorithm of template-based and -free docking 35 The chains A and B of the crystal structure of ALK2 (PDB ID: 6z36) and the chains B and G of the Cryo-EM structure of BRCC3 (PDB ID: 6h3c) were obtained from The Protein Data Bank.36 The top 10 interacting structures were predicted according to the corresponding docking and confidence scores. With a confidence score of the structure of 0.96, ALK2 likely interacts with BRCC3 based on the predicted structure.
PASMCs Migration, Proliferation Assays, and Flow Cytometry
Trans-well assay was used to evaluate the invasion and migration capacities of PASMCs. An amount of 105 cells in 500 μL serum-free medium was inoculated in the upper chamber. Medium containing 10% FBS (Gibco) was added to the lower chamber. After incubation for 48 hours, cells on the upper surface of the membrane were removed by wiping with a Q-tip, and the cells migrating into the other surface were fixed with paraformaldehyde and stained using 0.5% crystal violet (Sigma). The number of migrated cells was counted in 5 randomly selected fields under a microscope. Proliferation of PASMCs was assessed by the cell counting Kit-8 method (Dojindo Laboratories, Kumamoto, Japan), as we previously described.37 A Bio-Rad 680 microplate reader was used to detect absorbance at 450 nm to quantify PASMC proliferation. Flow cytometry was used to detect cell apoptosis. PASMCs were infected with Ad-BRCC3 virus and transfected with ALK2 K472R/K475R plasmid. After 24 hours, PASMCs were subjected to hypoxia conditions for 48 hours, then trypsinized, collected, and washed twice with PBS. After staining with annexin V–fluorescein isothiocyanate and propidium iodide for 10 minutes, cells were analyzed by flow cytometry to measure cell apoptosis rate.
Adenoviral Vectors
To generate adenovirus overexpressing BRCC3 (Ad-BRCC3), the mouse Brcc3 gene was amplified by polymerase chain reaction with pDEST-LTR-N-Flag-HA-BRCC3 (Cat.22540, Addgene, Cambridge, MA) as a template, then cloned into the adenovirus shuttle vector pDown-MCS-IRES/EGFP to obtain the recombinant plasmid pDown-BRCC3-GST-IRES-EGFP, from which the expression cassette of BRCC3-GST-IRES-EGFP was transferred to pAV.Desld with LR recombinant reaction to obtain the recombinant adenovirus plasmid pAV. Ex1d-CMV-BRCC3-GST-IRES-EGFP by Cyagen Bioscience (Guangzhou, China). The correct clone was linearized and transfected into HEK293A cells for packing, and the amplified adenovirus Ad-BRCC3 was obtained and identified. An adenovirus containing empty vector with an eGFP tag was a control (Ad-Null). To knockdown endogenous BRCC3, the specific shRNA targeting the mouse Brcc3 gene (Ad-GFP-U6-mBRCC-shRNA, Cat.shADV-254071) and scramble control shRNA (Ad-GFP-U6-shRNA, Cat. 1122) were purchased from Vector Biolabs (Malvern, PA). Experiments were conducted at 100 multiplicity of infection for both viruses. After 4 hours of incubation, the medium was removed, and cells were incubated in appropriate medium for 72 hours. The transfection efficiency was examined under a fluorescence microscope.
Animal Studies and Hemodynamics Measurements
Animal experiments in this study were approved by the Institutional Animal Ethics Committee of Yangzhou University. SMC-specific BRCC3-overexpressing transgenic mice (SM22α-BRCC3-Tg) were generated by using the SMC-specific promoter SM22α. Mouse Brcc3 cDNA and SM22α promotor were subcloned to obtain the expression clone. The construct was microinjected into the pronuclei of single-cell fertilized C57BL/6J embryos to generate BRCC3-Tg transgenic mice. To generate SMC-specific BRCC3 knockout mice (SMC-Brcc3−/−), the floxed BRCC3 allele homozygous mice (Brcc3flox/flox) were crossed initially with the Tagln-Cre line on the C57BL/6J background. The Tagln-Cre line was generated by placing the Cre recombinase expression cassette under the control of the Tagln gene promoter. Given Tagln encoding smooth muscle protein 22α, when crossed with mouse strains containing the flanking sequences of the loxP site, the Cre-mediated recombination led to the deletion of the flanking sequences in SMCs. Thus, the SMC-Brcc3−/− line was established by crossing Tagln-Cre line with Brcc3flox/flox mice. BRCC3 deletion was verified by detecting the BRCC3 level in lung tissue by Western blot analysis. Because of inconstant results often found in female rodents with experimental PH,38 male rats or mice were used in this study. For the monocrotaline-induced PH model, male rats (250–300 g) were administered a single dose of monocrotaline (60 mg/kg) by intraperitoneal injection and maintained for 5 weeks before euthanizing.
Mice at 8 to 12 weeks of age were exposed to hypoxia (10% O2) and subcutaneously injected with Su5416 (20 mg/kg) once a week for 5 weeks to induce PH. Animals under normoxia for 5 weeks were controls. Eight-week-old Sprague-Dawley rats were under 10% O2 with subcutaneous injection of SU5416 once at 20 mg/kg for 3 weeks followed by 2-week normoxia to induce PH. The right ventricular systolic pressure (RVSP) was measured by pulmonary artery catheterization connected to a pressure transducer with standardized protocols. Fulton index (right ventricular hypertrophy) was determined as the ratio of the right ventricular weight to that of the left ventricle + septum weight.
Histology and Immunofluorescence Staining
For investigating pulmonary vessel wall remodeling, formaldehyde-fixed, paraffin-embedded lung tissue sections were stained with hematoxylin and eosin (Beyotime) and elastic tissue stain and observed under a NanoZoomer Slide Scanner. For immunohistochemical staining, lung tissues were formalin-fixed, then embedded in paraffin and sectioned into 5-μm slices. After dewaxing, specimens were boiled and then incubated with 3% H2O2 at room temperature for 10 minutes to eliminate the activity of endogenous peroxidase. After washing with PBS, 10% normal goat serum was incubated at room temperature for 1 hour to block nonspecific bindings. Anti-SMAα antibody was added and incubated overnight at 4 °C. The slices were washed 3 times in PBS and incubated with a peroxidase-conjugated secondary antibody for 1 hour. Expression of the target protein was measured using the DAB kit (Vector Lab, USA). The sections were viewed by the use of a Nikon Ti-U inverted epifluorescence microscope equipped with appropriate filters (Nikon) at a 200× magnification. Five randomly selected fields from each slide were measured by a blinded observer, and the mean values were used for statistical analysis. For immunofluorescence staining, the slides with PASMCs were formalin-fixed, and then endogenous peroxidase activity was blocked, followed by incubation with anti-pSmad1/5, Smad3, and PPARγ antibody overnight at 4 °C. After PBS wash 3 times, specimens were incubated with horseradish peroxidase–conjugated goat antirabbit IgG secondary antibody for 50 minutes, followed by iF488-Tyramide incubation for 10 minutes in the dark. All slides were counterstained with DAPI, then mounted with the fluorescent mounting medium. The isotype-matched IgGs were used to validate antibody specificity. The secondary antibody alone was used as a negative control to distinguish nonspecific background signals.
Pulmonary Angiography
Mice were first anesthetized. Hearts and lungs were exposed by opening the chest cavity. Heparin (Sigma) was immediately injected into the pulmonary vasculature by the right ventricle (RV). The isolated lung was then perfused with warm PBS to wash out residual blood and then with Microfil polymers (Flow Tech Inc) (Microfil+diluent+curing agent). Lungs were washed with PBS, followed by gradient dehydration with ethanol. The dehydrated lungs were immersed in methyl salicylate (Merck) overnight to view the lung vasculature. ImageJ was used to quantify the total length of vascular branches, number of branches, and number of branch junctions in a given area of the peripheral regions of the lung.
Western Blot Analysis, Coimmunoprecipitation, Phos-Tag SDS-PAGE, and Quantitative Polymerase Chain Reaction
For Western blot analysis, total protein from lung tissues or cultured cells was extracted using RIPA lysis buffer with protease inhibitors (Thermo Scientific). Equal amounts of lysates were loaded onto 8% or 12% SDS-PAGE and immunoblotted with various antibodies. Bands were visualized by using the Pierce ECL Plus Western Blotting Substrate Kit (Thermo) and quantified with ImageJ. For immunoprecipitation, the respective antibody (2 μg for each immunoprecipitation sample) was incubated with Dynabeads Protein G (Invitrogen) for 2 hours at 4 °C, followed by 12-hour incubation with lysates. Immunoprecipitated products were resolved by SDS-PAGE and immunoblotted with the indicated antibodies. The antibodies used were anti-BRCC3 (Abcam, ab108411); anti-BMPR2 (Abcam, ab130206); anti-PPARγ (Santa Cruz, sc-7273); anti-p53 (Santa Cruz, sc-126); anti-Id1 (Abcam, ab283650); anti-BMP2 (Abcam, ab284387); anti-pSmad1/5 (Cell Signaling Technology, 13820); anti-Smad1 (Abcam, ab131371); anti-SMAα (Abcam, ab5694); anti–Ub K63 (Abcam, ab179434); anti–Ub K48 (Abcam, ab140601); anti–Bcl-XL (Cell Signaling Technology, 2764); anti-Bax (Abcam, abc2503); anti–cleaved caspase 3 (cle-Casp3) (Abcam, ab32042); anti-Bid (NOVUS, NB100–56106); anti-SMURF1 (Santa Cruz, sc-100616); anti–TGF-β (Cell Signaling Technology, 3709); anti-pSmad3 (Abcam, ab63403); anti-Smad3 (Cell Signaling Technology, 9523); anti-pSmad1/5/9 (Cell Signaling Technology, 13820); anti-Smad1/5/9 (Abcam, ab300164); anti–β-actin (Santa Cruz, sc-47778); anti–α-tubulin (Santa Cruz, sc-53646); and anti-ALK2 (Santa Cruz, sc-374523). Phos-tag reagent (Wako Chemicals) was used to determine the levels of phosphorylated ALK2 according to manufacturer instructions. Briefly, 7.5% polyacrylamide gels containing 75 μM Phos-tag and 100 μM MnCl2 were used to separate phosphorylated and unphosphorylated ALK2 by SDS-PAGE, followed by Western blot analysis. For quantitative polymerase chain reaction, total RNA was extracted from homogenized lung tissues or cells by using TRIzol reagent (Invitrogen). The cDNA was synthesized from 500 ng total RNA with the RevertAid First Strand cDNA Synthesis Kit (Thermo, USA) and quantitative polymerase chain reaction involved using an ABI PRISM 7500 sequence detection system (Applied Biosystems, CA). 18s were the internal controls. Primer sequences used for quantitative polymerase chain reaction and genotyping are in Table S2.
Statistics
Statistical analyses were performed with the use of SPSS 14.0 or GraphPad Prism 8. The numbers in figure legends indicate biological replicates performed in each experiment. Initially, the data sets were analyzed for normality with the Shapiro-Wilk test (P>0.05) and equal variance with the F test (P>0.05). For normally distributed data, the 2-tailed Student t test was used to compare 2 groups and 1-way ANOVA with Bonferroni post hoc test for multiple groups. Two-way ANOVA was used for comparing 2 independent variables. For data that were not normally distributed or experiments with small sample size (n≤5), the Mann-Whitney U test was used to compare 2 groups and Kruskal-Wallis with Dunn post hoc test for multiple groups. Data are expressed as mean±SEM, and P<0.05 was considered statistically significant.
RESULTS
BRCC3 Is Decreased in Human and Rodent PH
To decipher whether BRCC3 is involved in PAH, we first mined a GEO RNA-seq data set (GSE48149) generated from lung tissues of patients with IPAH and nondiseased controls. BRCC3 mRNA levels were lower in tissues from patients with IPAH than nondiseased controls (Figure 1A). Results from these mined data (ie, decreased BRCC3 expression) were validated in lung tissues from patients with IPAH, rodent models with Sugen5416/hypoxia (SuHx)-induced PH, and monocrotaline-induced PH (Figure 1B through 1D; Figure S1A). To explore the type of vascular tissue with decreased BRCC3, we mined the GSE168905 RNA-seq data set and found that the BRCC3 mRNA level was decreased in PASMCs from patients with IPAH (Figure 1E). The reduced BRCC3 expression was verified in PASMCs isolated from patients with IPAH and from rodent models with experimental PH (Figure 1F through 1H; Figure S1B). Next, to infer the signaling molecules and pathways associated with reduced level of BRCC3 in PASMCs, we studied the GSE72181 GEO RNA-seq data set generated from PASMCs under normoxia or hypoxia. Enrichment analysis showed that several PH-associated pathways, including “inflammatory response,” “positive regulation of cell proliferation,” “BMP signaling pathway,” “positive regulation of cell migration,” “TGF-β signaling pathway,” and “apoptosis,” were affected by hypoxia (Figure 1I). Of note, hypoxia-suppressed BRCC3 was concurrent with increased SMURF1 level, as revealed by a heatmap created from GSE72181 (Figure 1J). A total of 22 503 genes were detected in the RNA-seq, and the number of differential expressed genes was 1837 genes. Along with reduced BRCC3 level, genes involved in BMP signaling (eg, ALK2) and apoptosis (eg, BID), PPARγ, and p53 were downregulated (Figure 1J). However, there was little change of ALK1 and ALK3 mRNA in the GSE72181 data set (Figure S2A). By contrast, genes involved in TGF-β signaling (eg, TGFBR2) and proliferation (eg, PDGFB) in PASMCs were upregulated by hypoxia. To confirm these alterations in expression of genes involved in the BMP pathway, we subjected rat PASMCs to hypoxia ranging from 6 to 72 hours. Western blots in Figure 1K and Figure S2B show a time-dependent decrease in levels of BRCC3, BMPR2, ALK2, PPARγ, p53, and Id1 (a BMP pathway marker gene) but increase in that of SMURF1 in PASMCs by hypoxia. All together, these results by others and us suggest an inverse correlation between SMURF1 versus BRCC3 but a positive relation between BRCC3 and the BMP pathway in PASMCs during IPAH onset in humans and experimental PH in rodents.39
BRCC3 Overexpression Ameliorates the BMP Pathway in PASMCs
Because of the decreased BRCC3 level in PASMCs, we transfected PASMCs with adenovirus Ad-BRCC3 or Ad-Null to examine the BRCC3-regulated transcriptomes. DAVID analysis showed that genes involved in several PH-related pathways, eg, positive regulation of cell proliferation (GO:0008284), response to hypoxia (GO:0001666), TGF-β signaling pathway (rno04350, KEGG), positive regulation of restricted SMAD phosphorylation (GO:0010862), positive regulation of BMP signaling pathway (GO:0030513), positive regulation of SMC migration (GO:0014911), and were affected by BRCC3 overexpression (Figure 2A). A total of 17 724 genes were detected in the RNA-seq, and the number of differentially expressed genes was 1008. The corresponding heatmap in Figure 2B demonstrates the upregulation of genes in the BMP signaling pathway, such as Smad5, PPARγ, p53, and Id1. By contrast, genes in the TGF-β signaling pathway, such as TGFBR1 and Smad2/3, were downregulated by BRCC3 overexpression. Because of the noticeable changes in RNA-seq data, we examined the expression levels of molecules involved in the BMP pathway in PASMCs under normoxia or hypoxia and with or without BRCC3 overexpression. As anticipated, cells under hypoxia showed decreased levels of BMP2, BMPR2, ALK2, ALK1, ALK3, ALK6, Actr2a, pSmad1/5, PPARγ, p53, and Id1. However, BRCC3 overexpression partially rectified some of their expression (Figure 2C; Figure S3). We also treated PASMCs with BMP2 under hypoxia, then investigated the role of BRCC3 overexpression or knockdown in regulating the BMP pathway. Like BMP2 stimulation, BRCC3 overexpression increased the level of pSmad1/5, PPARγ, and Id1. However, BRCC3 knockdown abolished the BMP2-induced pSmad1/5, PPARγ, and Id1 expression (Figure 2D). We speculated that ALK2 underwent both K48- and K63-dependent ubiquitination under hypoxia because hypoxia decreased ALK2 expression (presumably by K48-dependent degradation), and BRCC3 supplementation increased ALK2 activity (presumably by K63-dependent modification). Indeed, hypoxia-increased ALK2 ubiquitination was both K48- and K63-dependent (Figure S4).
BRCC3 Overexpression in PASMCs Confers Protection Against PH
According to data from in vitro experiments in Figure 2, BRCC3 likely exerts a PH-protective effect by potentiating the BMP pathway. Thus, we established a BRCC3 gain-of-function mouse line with BRCC3 expression driven by the SM22α promoter (termed SM22α-BRCC3-Tg mice; Figure S5A). We compared the SuHx-induced PH in C57BL/6J mice and BRCC3-Tg mice (Figure S5B). As anticipated, BRCC3 was preferentially overexpressed in PASMCs in these mice (Figure S2C). Male SM22α-BRCC3-Tg mice and their age-matched wild-type (WT) littermates were subjected to SuHx stress to induce PH or kept under normoxia as controls. Compared with WT counterparts, SM22α-BRCC3-Tg mice receiving SuHx had reduced RVSP (Figure 2A). The less severe PH phenotype in SM22α-BRCC3-Tg mice was also supported by right ventricular (RV)±dP/dt (Figure 2B), RV index (Figure 2C), and Fulton index (RV/left ventricular+septum weight; Figure 2D). Also, in rats that underwent monocrotaline and SuHx protocol, Ad-BRCC3 administration alleviated the PH phenotype, as indicated by the drastically reduced RVSP, (RV)±dP/dt, and Fulton index (Figures S1C and S6). In agreement with hemodynamic measurements, the PH-associated vascular damage was mitigated in SM22α-BRCC3-Tg versus WT mice, as demonstrated by angiography. The PH-associated decreases in total length of branches and number of branches and junctions were significantly reverted in BRCC3-Tg mice (Figure 2E). Compared with WT mice under hypoxia, SM22α-BRCC3-Tg mice exhibited reduced vascular remodeling (ie, wall thickness, as revealed by hematoxylin and eosin staining; Figure 2F), medial wall thickness (demonstrated by elastic fiber staining; Figure 2G), and hypertrophy of the middle smooth muscle layer (shown by anti-SMAα staining; Figure 2H). With the deubiquitinase activity of BRCC3 (K63-dependent), lung tissues from SM22α-BRCC3-Tg mice exhibited a lower level of K63-linked ubiquitination (Figure 2I). Moreover, the expression of Bcl-XL (a proliferation marker) was decreased, whereas that of Bax, cle-Casp3, and Bid (apoptosis markers) was increased in lung tissues of SM22α-BRCC3-Tg mice (Figure 2J). Overall, results in Figure 2 suggest that BRCC3 overexpression in PASMCs from SM22α-BRCC3-Tg mice ameliorated experimental PH, possibly by BRCC3-mediated protein deubiquitylation.
BRCC3 Enhances ALK2-Smad1/5 Signaling in PASMCs
Upon BMP binding, BMPR2 binds to the cognate receptor ALK2 with ensuing phosphorylation of ALK2. The activated ALK2 then phosphorylates Smad1/5 at its C terminus (pSmad1/5 Ser-463/465), which leads to the nuclear entry of Smad1/5 to transactivate downstream genes, including PPARγ, p53, and Id1.9,40,41 We explored whether BRCC3 regulates the ALK2-Smad1/5 axis in vivo. BRCC3 overexpression in lungs of SM22α-BRCC3-Tg mice partially restored the levels of pALK2, pSmad1/5, PPARγ, p53, and Id1. However, the expression of SMURF1 was decreased (Figure 3A). Consistent with these data from mouse lungs, cultured PASMCs infected with Ad-BRCC3 showed increased levels of pALK2 and pSmad1/5 (Figure 3B) and nuclear translocation of pSmad1/5 under both normoxia and hypoxia (Figure 3C). However, BRCC3 overexpression did not affect the expression of BMPR2 or level of pBMPR2 (Figure 3B), which indicates little effect of BRCC3 on BMPR2. In the reciprocal BRCC3 loss-of-function experiments, treatment with LDN (an ALK2 functional inhibitor) or ALK2 knockdown attenuated the BRCC3-enhanced pSmad1/5 (Figure 3D and 3E) and its nuclear translocation (Figure 3F and 3G).
Figure 3. BRCC3 overexpression in PASMCs attenuates experimental PH.
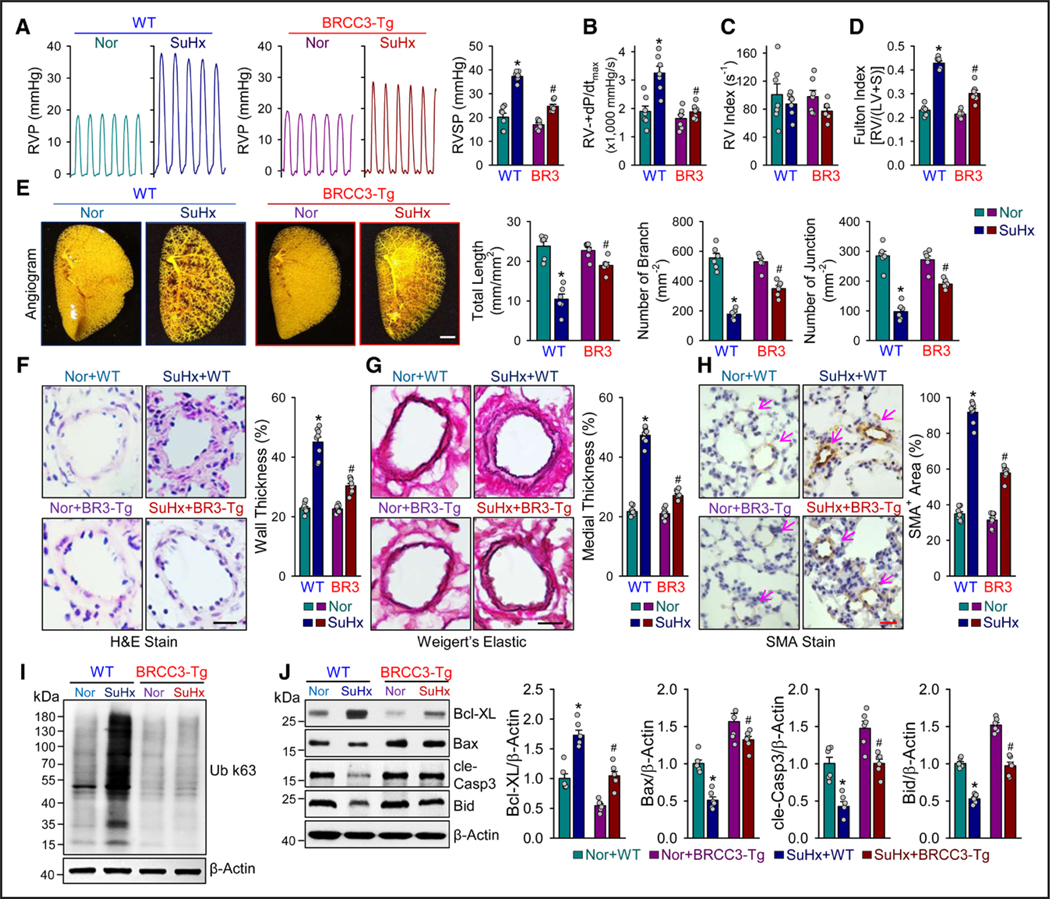
A through E, SM22α-BRCC3-Tg mice and wild-type (WT) littermates were subjected to normoxia, or 10% O2 with subcutaneous injection of SU5416 at 20 mg/kg/week (SuHx) to induce PH. Mice in the WT+Nor, WT+SuHx, BRCC3-Tg+Nor, and BRCC3-Tg+SuHx groups were euthanized. A, Representative records of right ventricular pressure (RVP) and summarized RV systolic pressure (RVSP), summarized values of RV±dp/dtmax (B), RV index (C), Fulton index (ratio of weight of right ventricle [RV] to that of left ventricle [LV] and septum [S] [RV/LV+S]; D), and angiography of pulmonary vasculature (E). Summarized data (mean±SEM; n=6 lungs) showing the total length of branches, number of branches, and number of junctions of the left lungs. Scale bar=50 mm. F, Hematoxylin and eosin (H&E) staining of pulmonary arteries. Scale bar=20 μm. G, Weigert elastic staining revealing the elastic fibers (dark blue) and collagen fibers (red). The area between the 2 layers of elastic fibers is the media layer, whereas the adventitial layer is indicated by the stained collagen fibers in red. Scale bar=20 μm. H, Immunohistochemical staining of SMAα. Summarized data (mean±SEM) showing the pulmonary arterial wall thickness and PASMC hypertrophy. Scale bar=20 μm. I, Western blot analysis of anti-Ub K63 to detect protein ubiquitination (n=3). J, Western blot analysis of Bcl-XL, Bax, cle-Casp3, Bid, and β-actin. Data in A through H and J are mean±SEM (6–8 mice per group). Normally distributed data in A through H and J were analyzed by 2-way ANOVA between multiple groups. *P<0.05 vs WT+Nor; #P<0.05 vs WT+SuHx. BRCC3 indicates BRCA1/BRCA2-containing complex subunit 3; cle-Casp3, cleaved caspase 3; PASMC, pulmonary arterial smooth muscle cell; and PH, pulmonary hypertension.
BRCC3 Regulation of ALK2 in PASMCs
Because BRCC3 regulation of Smad1/5 seemed to be dependent on ALK2, we determined whether BRCC3 affects ALK2 ubiquitination in vitro and in vivo. With hypoxia induction of the K63- and K48-dependent ubiquitination of ALK2 in cultured PASMCs, BRCC3 overexpression attenuated both types of ubiquitination (Figure 4A; Figure S7A). In vivo, levels of K63- and K48-dependent ubiquitination of ALK2 were elevated in lung tissues of WT mice subjected to SuHx stress, which was lower in SM22α-BRCC3-Tg mice (Figure 4B; Figure S7B). Anti-BRCC3 and anti-ALK2 pulldown assay revealed that BRCC3 and ALK2 were coimmunoprecipitated from cultured PASMCs (Figure 4C and 4D). Also, SMURF1 was immunoprecipitated by anti-BRCC3 or anti-ALK2 (Figure 4C and 4D), which suggests a protein complex containing ALK2, BRCC3, and SMURF1 in PASMCs. In line with the hypoxia downregulation of BRCC3 in vascular SMCs, the interaction between BRCC3 and ALK2 was decreased under hypoxia (Figure 4E). However, hypoxia increased the interaction between SMURF1 and ALK2, which was abolished by BRCC3 overexpression (Figure 4F). Decreased interaction between SMURF1 and ALK2 was also evident in lung tissue in SM22α-BRCC3-Tg mice compared with WT littermates (Figure 4G). Together, data in Figure 4A through 4G suggest an ALK2/BRCC3/SMURF1 ternary complex in PASMCs. PH onset or hypoxia in vitro increased the level of SMURF1 and decreased that of BRCC3, thus decreasing the interaction between BRCC3 and ALK2 and leading to increased ubiquitination of ALK2. Because SMURF1 mediates the K48-dependent ubiquitination,43 hypoxia in vitro and SuHx stress in vivo increased both types of ubiquitination of ALK2, which led to proteosome-regulated ALK2 degradation (K48-dependent) and hindered its kinase activity (K63-dependent). Putatively, BRCC3 overexpression decreased K63-dependent ubiquitination of ALK2 and also hindered SMURF1 binding to ALK2 to attenuate K48-dependent ubiquitination of ALK2, thus elevating ALK2 level and ALK2 phosphorylation of Smad1/5 during the onset of PH (Figure 4H).
Figure 4. BRCC3 enhances ALK2-Smad1/5 signaling in PASMCs.

A, SM22α-BRCC3-Tg mice and WT littermates were subjected to SuHx to induce PH. The lung tissues were isolated from euthanized mice, and Western blot analysis was performed to detect BMPR2, pALK2, ALK2, pSmad1/5, Smad1, SMURF1, PPARγ, p53, Id1, and β-actin. B through E, PASMCs were transfected with Ad-Null or Ad-BRCC3 for 48 hours. In B, cells were lysed, followed by Western blot analysis to detect pALK2, ALK2, pSmad1/5, Smad1, pBMPR2, BMPR2, BRCC3, and β-actin. In C, transfected PASMCs were exposed to hypoxia (1% O2) or normoxia for an additional 48 hours. The pSmad1/5 nuclear translocation was assessed by immunofluorescence microscopy with an antibody against pSmad1/5 (red); nuclei were counterstained with DAPI. Scale bar=50 μm. In D, cells were treated with or without LDN (10 mmol) for an additional 8 hours. In E, cells were transfected with control siRNA or ALK2 siRNA for 6 hours before Ad-Null or Ad-BRCC3 infection. Cells were lysed, followed by Western blot analysis to detect pSmad1/5, Smad1, ALK2, BRCC3, and β-actin. In F and G, PASMCs were treated with LDN or vehicle or transfected with ALK2 siRNA or control siRNA. Cells were infected with Ad-BRCC3 or Ad-Null, and then pSmad1/5 nuclear translocation was observed by immunofluorescence staining. Scale bar=50 μm. Data in A through G are mean±SEM from 5 or 6 independent experiments. Normally distributed data in A and C through G were analyzed by 2-way ANOVA between multiple groups. Normally and Nonnormally distributed data in B were analyzed by 2-tailed Student t test with Welch correction or the Mann-Whitney U test between 2 indicated groups. *P<0.05 vs WT+Nor (A), Ad-Null (B), Nor+Ad-Null (C), Veh+Ad-Null (D and F), and Ctrl SiR+Ad-Null (E and G). #P<0.05 vs WT+SuHx (A), Hyp+Ad-Null (C), Veh+Ad-BRCC3 (D and F), and Ctrl SiR+Ad-BRCC3 (E and G). ALK2 indicates activin receptor-like kinase-2; BMPR2, bone morphogenetic protein type 2 receptor; BRCC3, BRCA1/BRCA2-containing complex subunit 3; PASMC, pulmonary arterial smooth muscle cell; PH, pulmonary hypertension; SMURF1, Smad ubiquitination regulatory factor 1; SuHx, Sugen5416/hypoxia; and WT, wild-type.
To further investigate the molecular mechanism by which BRCC3 affects ALK2 ubiquitination, we performed protein–protein docking analysis to simulate the interaction between ALK2 and BRCC3. According to the published proteomic atlas,44,45 Lys-472 and Lys-475 are 2 putative ubiquitination sites (Figure 4I). Located at the kinase domain, these 2 Lys residues are highly conserved among mammalian species (Figure S8). The simulated 3-dimensional structure revealed that Lys-472 and Lys-475 are in an α-helix interacting with BRCC3 (Figure 4I). Thus, we constructed ALK2 K472R, K475R, and K472R/K475R mutants with Lys-472 or Lys-475 replaced by an Arg so that these mutants are “deubiquitin-mimetic” at Lys-472 or Lys-475. HEK293 cells overexpressing ALK2 K472R, K475R, or K472R/K475R exhibited lower levels of K63-dependent ubiquitination than cells expressing the ALK2 WT (Figure 4J). With respect to downstream BMP signaling, HEK293 cells overexpressing these deubiquitin-mimetics showed increased levels of pALK2, pSmad1/5, PPARγ, p53, and Id1 compared with cells overexpressing the WT (Figure 4K). Moreover, pSmad1/5 nuclear entry was enhanced by ALK2 K472R/K475R overexpression (Figure 4L). Results in Figure 4 suggest that BRCC3 regulates BMP signaling by BRCC3 deubiquitination of ALK2 Lys-472 and Lys-475.
BRCC3 Regulates the PASMC Phenotype
During PAH pathogenesis in vivo or under hypoxic conditions in vitro, PASMCs are converted from a contractile to a synthetic phenotype that is proliferative and migrating, which contributes to vascular remodeling.46 This phenotypic change is concurrently affected by the deactivated BMP pathway and activated TGF-β pathway.47,48 Because of the causality of BRCC3 regulation of ALK2 and the downstream BMP signaling, we investigated the corresponding changes in PASMC phenotype. BRCC3 or ALK2 K472R/K475R overexpression largely reversed the hypoxia induction of PASMC proliferation and migration (Figure 5A and 5B). In contrast, PASMC apoptosis was increased by BRCC3 or ALK2 K472R/K475R overexpression (Figure 5C). The BRCC3-decreased proliferation and -enhanced apoptosis of PASMCs were in line with the expression levels of Bcl-XL, Bax, cle-Casp3, and Bid (Figure 5D). Because of its significant inhibition of the PASMC phenotypic change, BRCC3 may synergistically hinder the TGF-β pathway. Indeed, TGF-β level was reduced in vitro in hypoxic PASMCs with overexpressed BRCC3 (Figure 5E) and lower in lung tissues from SuHx-exerted SMA22α-BRCC3-Tg mice than WT littermates (Figure 5F). Moreover, BRCC3 overexpression remedied the hypoxia or TGF-β1 induction of genes involved in PAH (eg, CTGF [connective tissue growth factor], OPN [osteopontin], PAI-1 [plasminogen activator inhibitor-1], and periostin [Figure 5G]). On R-Smads in TGF-β signaling (ie, Smad3), BRCC3 overexpression attenuated TGF-β1–induced Smad3 phosphorylation (pSmad3) and consequent nuclear entry (Figure 5H and 5I). PPARγ is a key nexus between the antiproliferative BMP pathway and pro-proliferative TGF-β pathway in vascular SMCs.49 With the concurrent downregulation of BRCC3 and PPARγ in PASMC (Figure 1I and 1J) and BRCC3 overexpression rectifying the hypoxia-downregulated PPARγ (Figure 2D), BRCC3 overexpression substantiated PPARγ nuclear entry (Figure 5J), like that of pSmad1/5 (Figure 3C). In all, data in Figure 5 accentuate the beneficial role of BRCC3 in maintaining the PASMC phenotype by activating the BMP pathway and downregulating the TGF-β pathway, merging at PPARγ activation.
Figure 5. BRCC3-ALK2 axis regulates BMP signaling by BRCC3 deubiquitination of ALK2 Lys-472 and Lys-475.

PASMCs transfected with or without Ad-BRCC3 were cultured under normoxia or hypoxia for 48 hours. Cell extracts were immunoprecipitated (IP) with anti-ALK2 and immunoblotted (IB) with anti-Ub K63 in A, anti-BRCC3 in E, and anti-SMURF1 in F. The input controls were immunoblotted with various antibodies as indicated. Cell lysates from cultured PASMCs were immunoprecipitated with anti-BRCC3 in C or anti-ALK2 in D and immunoblotted with anti-ALK2, anti-BRCC3, or anti-SMURF1. The input controls were lysates immunoblotted with anti-BRCC3, anti-ALK2, anti-SMURF1, or anti–β-actin. Lung tissues from WT/Nor, WT/SuHx, and BRCC3-Tg/SuHx groups of mice were immunoprecipitated with anti-ALK2 and immunoblotted with anti-Ub K63 in B and anti-SMURF1 in G. H, Schematic diagram of the competitive binding of BRCC3 and SMURF1 with ALK2. I, Schematic diagram of BRCC3 interaction with ALK2. ALK2 is depicted in blue and BRCC3 in green. The moiety highlighted in red in ALK2 are experimentally verified ubiquitination sites.42 ALK2 K472 and K475 were predicted to be in the interaction interface, which includes all residue pairs within 5.0 Å between the interacting molecules. J through L, HEK293 cells were transfected with expression plasmid encoding ALK2-WT, ALK2-K472R, ALK2-K475R, or ALK2-K472R/K475R (KR) together with Ub plasmids. Cell extracts were immunoprecipitated with anti-ALK2 and immunoblotted with anti-Ub K63 in J. The input protein was immunoblotted with various as indicated in K. L, PASMCs were transfected with ALK2-WT or ALK2-K472R/K475R plasmids. Nuclear translocation was quantified by immune fluorescence staining with pSmad1/5 (red), and nuclei were counterstained with DAPI (blue). Scale bar=50 μm. Data in A through L are mean±SEM from 3 to 7 independent experiments. Normally distributed data were analyzed by 1-way ANOVA in K and 2-way ANOVA in L between multiple groups. Data in K and L, *P<0.05 vs ALK2-WT or ALK2-WT in normoxia group. #P<0.05 vs ALK2-WT in hypoxia group. ALK2 indicates activin receptor-like kinase-2; BMP, bone morphogenetic protein; BRCC3, BRCA1/BRCA2-containing complex subunit 3; PASMC, pulmonary arterial smooth muscle cell; SMURF1, Smad ubiquitination regulatory factor 1; SuHx, Sugen5416/hypoxia; and WT, wild-type.
Pioglitazone and ALK2 K472R/K475R Rectify BRCC3 Loss
Given the decreased level of BRCC3 in PASMCs in patients with IPAH and rodent PH models (Figure 1E through 1H), we established an SMC-Brcc3−/− mouse line by crossing Brcc3flox/flox mice with Tagln-Cre mice to examine whether BRCC3 loss in PASMCs aggravates PH (Figure S9A and S9B). Because the expression of BRCC3 was decreased in lung tissue from these mice (Figure S9C), levels of pALK2 and pSmad1/5, PPARγ, p53, and Id1 were all substantially decreased, but the level of TGF-β was increased compared with WT littermates (Figure 6A). These results were in line with the increased K63-ubiquitination of ALK2 (Figure 6B). Then, male SMC-Brcc3−/− mice and their WT littermates at 12 weeks old were subjected to SuHx stress to induce PH or kept under normoxia as controls. Among the 4 groups, SMC-Brcc3−/− mice under SuHx exhibited the most severe phenotype of PH, as indicated by the most elevated RVSP, (RV)±dP/dt, and Fulton index and varying degrees of vascular pruning present in SMC-Brcc3−/− mice at baseline and worsened by SuHx treatment (Figure 6C through 6E). Of note, levels of RVSP, (RV)±dP/dt, Fulton index, PA medial thickness, and baseline vascular pruning in SMC-Brcc3−/− mice under normoxia were close to those of the WT under SuHx (Figure 6C), which suggests spontaneous PH and pulmonary vascular remodeling in SMC-Brcc3−/− mice. Because PPARγ is a common downstream transcription factor of the BMP/TGF-β pathway, we examined whether pharmacological activation of PPARγ by thiazolidinedione in SMC-Brcc3−/− mice could rescue the PH phenotype. Pioglitazone administration at 10 mg/kg/d for 4 weeks indeed rectified the PH and vascular remodeling caused by BRCC3 loss (Figure 6C through 6E). In human IPAH, the impaired BRCC3-ALK2 axis is evident because lung tissues from patients with IPAH showed lower levels of BRCC3, ALK2, and pSmad1/5 in connection with increased level of SMAα (Figure 6F; Figure S10). Given the decreased level of BRCC3 in PASMCs in patients with IPAH (Figure 1F), these PASMCs were infected with Ad-BRCC3 or transfected with ALK2 K472R/K475R expression plasmid. With an attempt to potentiate the BRCC3-ALK2 axis, BRCC3 or ALK2 K472R/K475R overexpression increased the levels of PPARγ, p53, and Id1 (Figure 6G). Overall, Figure 6 suggests that BRCC3 ablation impaired the BMP signaling in PASMCs, thus leading to aggravated PAH in humans and PH in mice. Intervention at BRCC3 or ALK2 (eg, K472R/K475R) could rectify the PAH phenotype.
Figure 6. BRCC3 regulates PASMC phenotype.

A through C, Rat PASMCs were transfected with Ad-Null or Ad-BRCC3 for 48 hours and transfected with plasmid encoding ALK2-WT or ALK2-KR (1 μg/mL), then exposed to normoxia or hypoxia for an additional 48 hours. The harvested cells were subjected to cell counting Kit-8 (CCK-8) assay (A), Trans-well assay (B), and flow cytometry assays. Scale bar=100 μm. (C). D and E, PASMCs were transfected with Ad-Null, Ad-BRCC3, or Ad-BRCC3 shRNA for 24 hours, then cultured under normoxia or hypoxia for 48 hours. Western blot analysis of protein levels of Bcl-XL, Bax, cle-Casp3, Bid, and β-actin (D) and TGF-β (E). F, Lung tissues from WT or BRCC3-Tg mice subjected to normoxia or SuHx were analyzed by Western blot using anti–TGF-β. G through J, PASMCs were transfected with Ad-Null or Ad-BRCC3 for 24 hours, then cultured under normoxia or hypoxia for 48 hours and treated with vehicle or TGF-β (5 ng/mL) for 24 hours before harvesting. G, The CTGF, OPN, PAI-1, and Periostin mRNAs were quantified by quantitative polymerase chain reaction. H, Western blot analysis of pSmad3. I, Immunofluorescence staining analysis of Smad3 nuclear entry. Scale bar=50 μm. J, Immunofluorescence staining of PPARγ nuclear entry. Scale bar=50 μm. Data in A through J are mean±SEM from 6 to 8 independent experiments. Normally distributed data in were analyzed by 1-way ANOVA (A and B) and 2-way ANOVA (D through I) between multiple groups. Normally distributed data in C and J were analyzed by 2-tailed Student t test with Welch correction between 2 indicated groups. Data in A through C, *P<0.05 vs Nor+Ad-Null or Nor+ALK2-WT, #P<0.05 vs Hyp+Ad-Null or Hyp+ALK2-WT. Data in D and E, *P<0.05 vs Nor+Ad-Null, #P<0.05 vs Hyp+Ad-Null. Data in F, *P<0.05 vs WT+Nor, #P<0.05 vs WT+SuHx. Data in G through I, *P<0.05 vs Nor+Ad-Null, #P<0.05 vs Nor+Ad-Null+TGF-β1, ϕP<0.05 vs Hyp+Ad-Null, δP<0.05 vs Hyp+Ad-Null+TGF-β1. BRCC3 indicates BRCA1/BRCA2-containing complex subunit 3; cle-Casp3, cleaved caspase 3; CTGF, connective tissue growth factor; OPN, osteopontin; PAI-1, plasminogen activator inhibitor-1; PASMC, pulmonary arterial smooth muscle cell; SuHx, Sugen5416/hypoxia; TGF-β, transforming growth factor-β; and WT, wild-type.
DISCUSSION
In this study, we are first time to examine the role of BRCC3 in experimental PH in rodents and PAH in humans. Summarized in Figure 7, the major findings are that: (1) BRCC3 level was decreased in lung tissues and PASMCs from PH animal models and patients with IPAH; (2) SM22α-BRCC3-Tg mice exhibited a protective effect against PH, and SMC-BRCC3 knockout mice showed aggravated PH; (3) BRCC3 rectified BMP signaling by its deubiquitination of ALK2 and hence activation of the downstream Smad1/5; (4) BRCC3 and ALK2 K472R/K475R preserved the integrity of the ALK2-Smad1/5-PPARγ axis, which recapitulated the effect of pioglitazone; and (5) BRCC3 overexpression in PASMCs from patients with IPAH rebalanced the antiproliferative and antimigrating pathways.
Figure 7. Pioglitazone and ALK2-K472R/K475R mitigate PH.

A, Western blot analysis of pALK2, ALK2, pSmad1/5, Smad1, PPARγ, p53, Id1, and BRCC3 in lung tissue of WT mice and SMC-Brcc3−/− (KO) mice. B, Lung tissues of WT and KO mice were immunoprecipitated with anti-ALK2 or IgG, then immunoblotted with anti-Ub K63. The input protein was immunoblotting with antibodies against BRCC3, ALK2, or β-actin (n=3). C through E, KO mice and WT littermates were subjected to normoxia as controls or SuHx to induce PH. KO mice were treated with pioglitazone (PIO) for 4 weeks (10 mg/kg, daily). Mice in the WT+Nor, BRCC3 KO+Nor, BRCC3 KO+Nor+PIO, WT+SuHx, BRCC3 KO+SuHx, and BRCC3 KO+SuHx+PIO groups were euthanized. C, Representative records of RVP and summarized RVSP, summarized values of RV±dp/dtmax, Fulton index. D, Angiography of the pulmonary vasculature. Summarized data (mean±SEM, n=6 lungs) on the right showing the total length of branches, number of branches, and number of junctions of the left lungs. Scale bar=50 mm. E, Hematoxylin and eosin (H&E) staining of pulmonary arteries. Scale bar=20 μm. F, Representative immunofluorescent staining of of BRCC3 (red), SMAα (green), DAPI (blue), and merge images in distal pulmonary arteries from IPAH and nondiseased individuals. Scale bar=50 μm (n=6). G, Western blot analysis of pALK2, ALK2, pSmad1/5/9, Smad1/5/9, PPARγ, p53, Id1, BRCC3, and β-actin in PASMCs from patients with IPAH vs nondiseased individuals (n=4). Normally distributed data in A and C through F are mean±SEM from 6 independent experiments and analyzed by 2-tailed Student t test with Welch correction (A and F) and 2-way ANOVA between multiple groups (C through E). Nonnormally distributed data in G were analyzed by the Kruskal-Wallis test between multiple groups (n=4). Data in A, *P<0.05 vs WT. Data in C through E, *P<0.05 vs WT in normoxia (Nor) group; #P<0.05 vs BRCC3-KO in normoxia group; ϕP<0.05 vs WT in SuHx group; δP<0.05 vs BRCC3-KO in SuHx group. Data in F and G, *P<0.05 vs Nor; #P<0.05 vs IPAH control. ALK2 indicates activin receptor-like kinase-2; BRCC3, BRCA1/BRCA2-containing complex subunit 3; IPAH, idiopathic pulmonary arterial hypertension; PASMC, pulmonary arterial smooth muscle cell; PH, pulmonary hypertension; SMC, smooth muscle cell; SuHx, Sugen5416/hypoxia; and WT, wild-type.
The signaling and function of BMP/TGF-β family receptors and downstream R-Smads are tightly regulated by their ubiquitination status.14,50 Functioning as a deubiquitinase, BRCC3 can deubiquitinate the K63-linked polyubiquitinating chains.24,25 Our results showed that BRCC3 was downregulated in PASMCs by hypoxia in vitro and during PH onset in vivo. Concordant with the decreased level of BRCC3, K63-dependent ALK2 ubiquitination was increased. Although ALK2 can also undergo K48-linked ubiquitination,45 BRCC3-mediated ALK2 deubiquitination seemed to involve K63-dependent ubiquitination at Lys-472 and Lys-475. Functionally, K63-dependent ALK2 ubiquitination at the kinase domain decreased ALK2 kinase activity rather than ALK2 degradation, as revealed by attenuated pSmad1/5, a substrate of ALK2. Mechanistically, Lys-472 and Lys-475 seem to be positioned at the interface between ALK2 and BRCC3, which facilitates their deubiquitination by BRCC3. This argument is made considering that overexpression of ALK2 K472R, ALK2 K475R, and ALK2 K472/475R all decreased ALK2 ubiquitination with ensuing increased Smad1/5 phosphorylation and nuclear entry (Figure 4J through 4L). These “deubiquitin-mimetic” mutants likely function as active mutants to activate BMP signaling.
Coimmunoprecipitation experiments in Figure 4C and 4D demonstrated that SMURF1 binds to BRCC3 and ALK2 to form a ternary complex in unstimulated PASMCs. Upon pathophysiological stimulation such as hypoxia and SuHx, BRCC3 downregulation together with SMURF1 upregulation resulted in increased ALK2 ubiquitination (Figure 4E through 4G). Because SMURF1 can catalyze K48-linked ubiquitination,45 whereas BRCC3 attenuated SMURF1-mediated K48-ubiquitination of ALK2, BRCC3 likely prevented SMURF1 from binding to ALK2 to decrease K48-dependent ALK2 ubiquitination (Figure 4F and 4G). Like ALK2, BMPR2 was downregulated in vitro by hypoxia and in vivo in PH mice (Figure 2C; Figure 3A).51 However, BRCC3 overexpression, although rectifying the level of ALK2 and pSmad1/5, did not potentiate that of BMPR2 (Figure 3A). Thus, ALK2, but not BMPR2, may be indispensable for the activation of Smad1/5 upon BMP stimulation.52 The kinase activity of type I receptor is increased by the phosphorylation of Ser/Thr in the GS domain by type II receptors.53 Because pALK2 was enhanced in BRCC3 gain-of-function experiments (Figure 3B), BRCC3 deubiquitination of Lys-472 and Lys-475 is likely to change the conformation of the GS domain of ALK2. These results suggest that BRCC3 positively regulated the BMP signaling by deubiquitination of ALK2 and then promoted its activation by type II receptors (eg, BMPR2). Besides our newly uncovered mechanisms, BMP2 has been reported to induce BRCC3.54 We also found that BMP2 increased levels of BRCC3 and pSmad1/5 in PASMCs in a time-dependent manner (Figure S11A and S11B). Furthermore, BRCC3 was increased by BMP6 and BMP9 and decreased by TGF-β1 in PASMCs (Figure S11C). The induction of BRCC3 occurred after that of pSmad1/5. This suggests that BRCC3 positively regulates the BMP signaling pathway in a feedforward manner. Additionally, BRCC3 inhibits vascular calcification associated with chronic kidney disease by inhibiting β-catenin phosphorylation and inducing the contractile protein SMAα in vascular SMCs.55 Therefore, the beneficial role of BRCC3 may include its regulation of other PAH-related pathways such as β-catenin.
PPARγ plays a pivotal role in cardiovascular homeostasis.56 With respect to the BMP/TGF-β antagonism in vascular SMCs, PPARγ simultaneously regulates antiproliferative BMP and pro-proliferative TGF-β signaling in a yin-yang fashion.49 During the progression of PAH, PPARγ is downregulated, which causes PASMC proliferation and secretion of extracellular matrix.57 Pioglitazone has been previously shown to reverse PH in a SuHx rat model.58 PPARγ binds to the complex of Smad2/3 and Smad4 in PASMCs to inhibit the TGF-β–induced PH in mice, and the BMP2-stimulated PPARγ inhibits human PASMC proliferation and the expression of TGF-β target genes.9,49 Here, we showed that BRCC3 regulated PPARγ by ALK2, and pioglitazone rescued the PH phenotype in SMC-BRCC3 knockout mice (Figure 6C through 6G). As a positive control, the PPARγ agonists indeed rescued the phenotypic changes caused by BRCC3 deficiency.
The pathophysiology of PAH involves the slanted BMP/TGF-β pathways (ie, downregulated BMP signaling versus upregulated TGF-β signaling) that drive the proliferative phenotype of PASMCs.47,59 We found that BRCC3 upregulated pSmad1/5 and downregulated pSmad3 in parallel. The opposite effect of BRCC3 on BMP versus TGF-β signaling was at transcriptional and translational levels (Figures 2B and 5E through 5H). Significantly, BRCC3 overexpression attenuated SuHx-induced TGF-β in vivo and hypoxia-induced TGF-β as well as TGF-β1-induced pSmad3 in vitro (Figure 5E, 5F, and 5H). Thus, the profound effect of BRCC3 on ALK2-pSmad1/5 would complement that by ACTRIIA-Fc on Smad2/3 by restoring the balance between the anti- and pro-proliferative pathways. However, supraphysiological activation of ALK2 is contradictory, given the ALK2 (R206H) mutation involved in fibrodysplasia ossificans progressiva, a genetic autosomal dominant musculoskeletal disease featuring heterotopic ossification.60 With the underlying mechanism of this disease involving autophosphorylation of Smad1/5 by R206H mutation of ALK2 and enhanced BMP signaling in the absence of an exogenous ligand,61,62 the usefulness of BRCC3 agonists in alleviating PAH warrants further study. It is interesting that the expression level of BRCC3 in human umbilic vein endothelial cells and human pulmonary arterial endothelial cells was significantly downregulated by hypoxia (Figure S12). However, the expression level of MDM2 was lower in the hypoxic PASMCs, which is contradictory to that in endothelial cells,63 and the molecular insights underlying this discrepancy are currently unknown.
In conclusion, this study demonstrates the causality between the downregulated BRCC3 and compromised ALK2-Smad1/5-PPARγ signaling during the onset of PAH. BRCC3 gain-of-function would inhibit the hyperproliferation, phenotypic transformation, apoptosis resistance, and extracellular matrix production in PASMCs, thereby alleviating experimental PH in rodents and decreasing PAH susceptibility in humans.
Supplementary Material
Figure 8. Schematic illustration of the protective role of BRCC3 in PH alleviation.

Under normal conditions, BRCC3 preserves the deubiquitination of ALK2, thus sustaining the phosphorylation of Smad1/5 and its nuclear entry to maintain the integrity of BMP pathway, which contributes to the contractile phenotype of PASMCs. Upon the onset of PAH, reduction of BRCC3 and increase in SMURF1 mediates the K63-linked ubiqutination of ALK2, leading to decreased ALK2 activity and phosphorylation and nuclear entry of Smad1/5, which promotes the proliferative phenotype of PASMCs. Pharmacological or genetic rectification of the BRCC3-ALK2 axis to rebalance BMP/TGF-β pathways may ameliorate PH in rodents and humans. ALK2 indicates activin receptor-like kinase-2; BMP, bone morphogenetic protein; BRCC3, BRCA1/BRCA2-containing complex subunit 3; PAH, pulmonary arterial hypertension; PASMC, pulmonary arterial smooth muscle cell; PH, pulmonary hypertension; SMURF1, Smad ubiquitination regulatory factor 1; and TGF-β, transforming growth factor-β.
Clinical Perspective.
What Is New?
BRCC3 (BRCA1/BRCA2-containing complex subunit 3) expression is decreased in lung tissues from patients with idiopathic pulmonary arterial hypertension and animals with experimental pulmonary hypertension.
BRCC3 deubiquitylation of ALK2 (activin receptor-like kinase-2) activates the BMP (bone morphogenetic protein) arm of the TGF-β (transforming growth factor-β) signaling in lung vascular smooth muscle cells.
BRCC3 gain-of-function may decrease pulmonary arterial hypertension susceptibility in humans.
What Are the Clinical Implications?
Downregulated BRCC3 is involved in the pulmonary arterial hypertension pathogenesis, and BRCC3 upregulation and activation may be a novel therapeutic strategy for pulmonary arterial hypertension/pulmonary hypertension.
Restoration of the BMP signaling in pulmonary vasculature to rebalance the TGF-β/BMP system by BRCC3 elevation can mitigate pulmonary arterial hypertension–associated vascular remodeling.
Acknowledgments
H.S., P.A.T., A.M., J.X.-J.Y., J.Y.-J.S., and K.G. designed research; H.S., Y.G., D.G., M.T., Q. Yin, F.H., T.-Y.W.W., Y.C., Q. Yang, Z.L., X.L., and Y.Y. performed research; H.S., Y.G., Q. Yin, Z.C., J.X.-J.Y., and K.G. analyzed data; T.-Y.L., Z.L., T.-Y.W.W., and F.H. predicted biological information; H.S., J.Y.-J.S., K.G., J.X.-J.Y., J.W., and Z.Z. wrote and edited the article. All authors made substantial contributions to the subsequent version of the article and approved the final version for submission. The authors acknowledge Dr. Chen Wang, Dr. Liang Bai, and Yiman Zhang at Xi’an Jiaotong University for their consultation and useful discussion. The authors are grateful to the PHBI for providing human lung cells for this study. PHBI is funded by the Cardiovascular Medical Research and Education Fund.
Sources of Funding
This work was supported by the National Natural Science Foundation of China (81270197, 81470381, 81770262, and 81970225 to K.G.; 82100428 to S.H.; 82170317 to Y.Y.; 82170069 to J.W.); the Natural Science Foundation of Jiangsu Province (BK20210142 to S.H.; BK20201223 to Y.Y.); the High-End Talent Support Program of Yangzhou University (to S.H.); the Jiangsu Association for Science & Technology Youth Science & Technology Talents Lifting Project (to S.H.); and the Jiangsu Provincial Medical Key Discipline Cultivation Unit (JSDW202251 to K.G).
Nonstandard Abbreviations and Acronyms
- ALK2
activin receptor-like kinase-2
- ActRIIA
activin receptor IIA
- BRCC3
BRCA1/BRCA2-containing complex subunit 3
- BMP
bone morphogenetic protein
- BMPR2
bone morphogenetic protein type 2 receptor
- cle-Casp3
cleaved caspase 3
- CTGF
connective tissue growth factor
- DUB
deubiquitinating enzyme
- GEO
Gene Expression Omnibus
- GDF
growth differentiation factor
- HEK293
human embryonic kidney 293
- HECT
homologous to E6AP C terminus
- IPAH
idiopathic pulmonary arterial hypertension
- OPN
osteopontin
- p
phosphorylated
- PAH
pulmonary arterial hypertension
- PAI-1
plasminogen activator inhibitor-1
- PASMC
pulmonary arterial smooth muscle cell
- PH
pulmonary hypertension
- PHBI
Pulmonary Hypertension Breakthrough Initiative
- RNA-seq
RNA sequencing
- RV
right ventricle
- RVSP
right ventricular systolic pressure
- SMC
smooth muscle cell
- SuHx
Sugen5416/hypoxia
- SMURF1
Smad ubiquitination regulatory factor 1
- TGF-β
transforming growth factor-β
- WT
wild-type
Footnotes
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/CIRCULATIONAHA.123.066430.
Disclosures
None.
REFERENCES
- 1.Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger R, Brida M, Carlsen J, Coats A, Escribano-Subias P, Ferrari P, et al. 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2023;61:2200879. doi: 10.1183/13993003.00879-2022 [DOI] [PubMed] [Google Scholar]
- 2.Morrell NW, Aldred MA, Chung WK, Elliott CG, Nichols WC, Soubrier F, Trembath RC, Loyd JE. Genetics and genomics of pulmonary arterial hypertension. Eur Respir J. 2019;53:1801899. doi: 10.1183/13993003.01899-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2012;122:4306–4313. doi: 10.1172/JCI60658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA III, Loyd JE, Nichols WC, Trembath RC; International PPH Consortium. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–84. doi: 10.1038/79226 [DOI] [PubMed] [Google Scholar]
- 5.Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, Morrell NW. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105:1672–1678. doi: 10.1161/01.cir.0000012754.72951.3d [DOI] [PubMed] [Google Scholar]
- 6.Thenappan T, Ormiston ML, Ryan JJ, Archer SL. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ. 2018;360:j5492. doi: 10.1136/bmj.j5492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res. 2014;115:165–175. doi: 10.1161/CIRCRESAHA.113.301141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang J, Li X, Al-Lamki RS, Southwood M, Zhao J, Lever AM, Grimminger F, Schermuly RT, Morrell NW. Smad-dependent and smad-independent induction of id1 by prostacyclin analogues inhibits proliferation of pulmonary artery smooth muscle cells in vitro and in vivo. Circ Res. 2010;107:252–262. doi: 10.1161/CIRCRESAHA.109.209940 [DOI] [PubMed] [Google Scholar]
- 9.Hansmann G, de Jesus Perez VA, Alastalo TP, Alvira CM, Guignabert C, Bekker JM, Schellong S, Urashima T, Wang L, Morrell NW, et al. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest. 2008;118:1846–1857. doi: 10.1172/JCI32503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chau JF, Jia D, Wang Z, Liu Z, Hu Y, Zhang X, Jia H, Lai KP, Leong WF, Au BJ, et al. A crucial role for bone morphogenetic protein-Smad1 signalling in the DNA damage response. Nat Commun. 2012;3:836. doi: 10.1038/ncomms1832 [DOI] [PubMed] [Google Scholar]
- 11.Guignabert C, Humbert M. Targeting transforming growth factor-β receptors in pulmonary hypertension. Eur Respir J. 2021;57:2002341. doi: 10.1183/13993003.02341-2020 [DOI] [PubMed] [Google Scholar]
- 12.Morikawa M, Derynck R, Miyazono K. TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol. 2016;8:a021873. doi: 10.1101/cshperspect.a021873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Massagué J, Sheppard D. TGF-β signaling in health and disease. Cell. 2023;186:4007–4037. doi: 10.1016/j.cell.2023.07.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu H, Kavsak P, Abdollah S, Wrana JL, Thomsen GH. A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature. 1999;400:687–693. doi: 10.1038/23293 [DOI] [PubMed] [Google Scholar]
- 15.Heldin CH, Moustakas A. Signaling receptors for TGF-β family members. Cold Spring Harb Perspect Biol. 2016;8:a022053. doi: 10.1101/cshperspect.a022053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clague MJ, Urbé S. Ubiquitin: same molecule, different degradation pathways. Cell. 2010;143:682–685. doi: 10.1016/j.cell.2010.11.012 [DOI] [PubMed] [Google Scholar]
- 17.Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328 [DOI] [PubMed] [Google Scholar]
- 18.Murakami K, Mathew R, Huang J, Farahani R, Peng H, Olson SC, Etlinger JD. Smurf1 ubiquitin ligase causes downregulation of BMP receptors and is induced in monocrotaline and hypoxia models of pulmonary arterial hypertension. Exp Biol Med (Maywood). 2010;235:805–813. doi: 10.1258/ebm.2010.009383 [DOI] [PubMed] [Google Scholar]
- 19.Rothman AM, Arnold ND, Pickworth JA, Iremonger J, Ciuclan L, Allen RM, Guth-Gundel S, Southwood M, Morrell NW, Thomas M, et al. MicroRNA-140–5p and SMURF1 regulate pulmonary arterial hypertension. J Clin Invest. 2016;126:2495–2508. doi: 10.1172/JCI83361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wicks SJ, Haros K, Maillard M, Song L, Cohen RE, Dijke PT, Chantry A. The deubiquitinating enzyme UCH37 interacts with Smads and regulates TGF-beta signalling. Oncogene. 2005;24:8080–8084. doi: 10.1038/sj.onc.1208944 [DOI] [PubMed] [Google Scholar]
- 21.Eichhorn PJ, Rodón L, Gonzàlez-Juncà A, Dirac A, Gili M, Martínez-Sáez E, Aura C, Barba I, Peg V, Prat A, et al. USP15 stabilizes TGF-β receptor I and promotes oncogenesis through the activation of TGF-β signaling in glioblastoma. Nat Med. 2012;18:429–435. doi: 10.1038/nm.2619 [DOI] [PubMed] [Google Scholar]
- 22.Tang Y, Reissig S, Glasmacher E, Regen T, Wanke F, Nikolaev A, Gerlach K, Popp V, Karram K, Fantini MC, et al. Alternative splice forms of CYLD mediate ubiquitination of SMAD7 to prevent TGFB signaling and promote Colitis. Gastroenterology. 2019;156:692–707.e7. doi: 10.1053/j.gastro.2018.10.023 [DOI] [PubMed] [Google Scholar]
- 23.Itoh F, Asao H, Sugamura K, Heldin CH, ten Dijke P, Itoh S. Promoting bone morphogenetic protein signaling through negative regulation of inhibitory Smads. EMBO J. 2001;20:4132–4142. doi: 10.1093/emboj/20.15.4132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cooper EM, Cutcliffe C, Kristiansen TZ, Pandey A, Pickart CM, Cohen RE. K63-specific deubiquitination by two JAMM/MPN+ complexes: BRISC-associated Brcc36 and proteasomal Poh1. EMBO J. 2009;28:621–631. doi: 10.1038/emboj.2009.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeqiraj E, Tian, Piggott CA, Pillon MC, Duffy NM, Ceccarelli DF, Keszei AF, Lorenzen K, Kurinov I, Orlicky S, et al. Higher-order assembly of BRCC36-KIAA0157 is required for DUB activity and biological function. Mol Cell. 2015;59:970–983. doi: 10.1016/j.molcel.2015.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen JJ, Silver D, Cantor S, Livingston DM, Scully R. BRCA1, BRCA2, and Rad51 operate in a common DNA damage response pathway. Cancer Res. 1999;59:1752s–1756s. [PubMed] [Google Scholar]
- 27.Rabl J, Bunker RD, Schenk AD, Cavadini S, Gill ME, Abdulrahman W, Andrés-Pons A, Luijsterburg MS, Ibrahim A, Branigan E, et al. Structural basis of BRCC36 function in DNA repair and immune regulation. Mol Cell. 2019;75:483–497.e9. doi: 10.1016/j.molcel.2019.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell. 2013;49:331–338. doi: 10.1016/j.molcel.2012.11.009 [DOI] [PubMed] [Google Scholar]
- 29.Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol. 2011;8:443–455. doi: 10.1038/nrcardio.2011.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dussiot M, Maciel TT, Fricot A, Chartier C, Negre O, Veiga J, Grapton D, Paubelle E, Payen E, Beuzard Y, et al. An activin receptor IIA ligand trap corrects ineffective erythropoiesis in β-thalassemia. Nat Med. 2014;20:398–407. doi: 10.1038/nm.3468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pearsall RS, Canalis E, Cornwall-Brady M, Underwood KW, Haigis B, Ucran J, Kumar R, Pobre E, Grinberg A, Werner ED, et al. A soluble activin type IIA receptor induces bone formation and improves skeletal integrity. Proc Natl Acad Sci U S A. 2008;105:7082–7087. doi: 10.1073/pnas.0711263105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Humbert M, McLaughlin V, Gibbs J, Gomberg-Maitland M, Hoeper MM, Preston IR, Souza R, Waxman AB, Ghofrani HA, Escribano Subias P, et al. Sotatercept for the treatment of pulmonary arterial hypertension: PULSAR open-label extension. Eur Respir J. 2023;61:2201347. doi: 10.1183/13993003.01347-2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoeper MM, Badesch DB, Ghofrani HA, Gibbs J, Gomberg-Maitland M, McLaughlin VV, Preston IR, Souza R, Waxman AB, Grünig E, et al. Phase 3 trial of sotatercept for treatment of pulmonary arterial hypertension. N Engl J Med. 2023;388:1478–1490. doi: 10.1056/NEJMoa2213558 [DOI] [PubMed] [Google Scholar]
- 34.Humbert M, McLaughlin V, Gibbs J, Gomberg-Maitland M, Hoeper MM, Preston IR, Souza R, Waxman A, Escribano Subias P, Feldman J, et al. Sotatercept for the treatment of pulmonary arterial hypertension. N Engl J Med. 2021;384:1204–1215. doi: 10.1056/NEJMoa2024277 [DOI] [PubMed] [Google Scholar]
- 35.Yan Y, Tao H, He J, Huang SY. The HDOCK server for integrated protein-protein docking. Nat Protoc. 2020;15:1829–1852. doi: 10.1038/s41596-020-0312-x [DOI] [PubMed] [Google Scholar]
- 36.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jain PP, Lai N, Xiong M, Chen J, Babicheva A, Zhao T, Parmisano S, Zhao M, Paquin C, Matti M, et al. TRPC6, a therapeutic target for pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2021;321:L1161–L1182. doi: 10.1152/ajplung.00159.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bal E, Ilgin S, Atli O, Ergun B, Sirmagul B. The effects of gender difference on monocrotaline-induced pulmonary hypertension in rats. Hum Exp Toxicol. 2013;32:766–774. doi: 10.1177/0960327113477874 [DOI] [PubMed] [Google Scholar]
- 39.He Y, Cao X, Guo P, Li X, Shang H, Liu J, Xie M, Xu Y, Liu X. Quercetin induces autophagy via FOXO1-dependent pathways and autophagy suppression enhances quercetin-induced apoptosis in PASMCs in hypoxia. Free Radic Biol Med. 2017;103:165–176. doi: 10.1016/j.freeradbiomed.2016.12.016 [DOI] [PubMed] [Google Scholar]
- 40.Daly AC, Randall RA, Hill CS. Transforming growth factor beta-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol Cell Biol. 2008;28:6889–6902. doi: 10.1128/MCB.01192-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salazar VS, Gamer LW, Rosen V. BMP signalling in skeletal development, disease and repair. Nat Rev Endocrinol. 2016;12:203–221. doi: 10.1038/nrendo.2016.12 [DOI] [PubMed] [Google Scholar]
- 42.Wang C, Tan X, Tang D, Gou Y, Han C, Ning W, Lin S, Zhang W, Chen M, Peng D, et al. GPS-Uber: a hybrid-learning framework for prediction of general and E3-specific lysine ubiquitination sites. Brief Bioinform. 2022;23:bbab574. doi: 10.1093/bib/bbab574 [DOI] [PubMed] [Google Scholar]
- 43.Seo D, Jung SM, Park JS, Lee J, Ha J, Kim M, Park SH. The deubiquitinating enzyme PSMD14 facilitates tumor growth and chemoresistance through stabilizing the ALK2 receptor in the initiation of BMP6 signaling pathway. EBioMedicine. 2019;49:55–71. doi: 10.1016/j.ebiom.2019.10.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elia AE, Boardman AP, Wang DC, Huttlin EL, Everley RA, Dephoure N, Zhou C, Koren I, Gygi SP, Elledge SJ. Quantitative proteomic atlas of ubiquitination and acetylation in the DNA damage response. Mol Cell. 2015;59:867–881. doi: 10.1016/j.molcel.2015.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Satpathy S, Krug K, Jean Beltran PM, Savage SR, Petralia F, Kumar-Sinha C, Dou Y, Reva B, Kane MH, Avanessian SC, et al. ; Clinical Proteomic Tumor Analysis Consortium. A proteogenomic portrait of lung squamous cell carcinoma. Cell. 2021;184:4348–4371.e40. doi: 10.1016/j.cell.2021.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res. 2006;99:675–691. doi: 10.1161/01.RES.0000243584.45145.3f [DOI] [PubMed] [Google Scholar]
- 47.Morrell NW, Yang X, Upton PD, Jourdan KB, Morgan N, Sheares KK, Trembath RC. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-beta(1) and bone morphogenetic proteins. Circulation. 2001;104:790–795. doi: 10.1161/hc3201.094152 [DOI] [PubMed] [Google Scholar]
- 48.Jeffery TK, Morrell NW. Molecular and cellular basis of pulmonary vascular remodeling in pulmonary hypertension. Prog Cardiovasc Dis. 2002;45:173–202. doi: 10.1053/pcad.2002.130041 [DOI] [PubMed] [Google Scholar]
- 49.Calvier L, Chouvarine P, Legchenko E, Hoffmann N, Geldner J, Borchert P, Jonigk D, Mozes MM, Hansmann G. PPARγ links BMP2 and TGFβ1 pathways in vascular smooth muscle cells, regulating cell proliferation and glucose metabolism. Cell Metab. 2017;25:1118–1134.e7. doi: 10.1016/j.cmet.2017.03.011 [DOI] [PubMed] [Google Scholar]
- 50.Liu J, Jin J, Liang T, Feng XH. To Ub or not to Ub: a regulatory question in TGF-β signaling. Trends Biochem Sci. 2022;47:1059–1072. doi: 10.1016/j.tibs.2022.06.001 [DOI] [PubMed] [Google Scholar]
- 51.Wang C, Xing Y, Zhang J, He M, Dong J, Chen S, Wu H, Huang HY, Chou CH, Bai L, et al. MED1 regulates BMP/TGF-β in endothelium: implication for pulmonary hypertension. Circ Res. 2022;131:828–841. doi: 10.1161/CIRCRESAHA.122.321532 [DOI] [PubMed] [Google Scholar]
- 52.Star GP, Giovinazzo M, Langleben D. ALK2 and BMPR2 knockdown and endothelin-1 production by pulmonary microvascular endothelial cells. Microvasc Res. 2013;85:46–53. doi: 10.1016/j.mvr.2012.10.012 [DOI] [PubMed] [Google Scholar]
- 53.Willis SA, Mathews LS. Regulation of activin type I receptor function by phosphorylation of residues outside the GS domain. FEBS Lett. 1997;420:117–120. doi: 10.1016/s0014-5793(97)01499-3 [DOI] [PubMed] [Google Scholar]
- 54.Zhang JM, Wang KN, Zhang Y, Zhang JZ, Yuan XP, Zou GJ, Cao Z, Zhang CJ. BRCC36 promotes intestinal mucosal barrier injury caused by BMP2 after ischemia reperfusion via inhibiting PPARγ signaling. Biosci Biotechnol Biochem. 2022;86:331–339. doi: 10.1093/bbb/zbab210 [DOI] [PubMed] [Google Scholar]
- 55.Xie C, Chen C, Wu L, Xiong Y, Xing C, Mao H. BRCC36 prevents vascular calcification in chronic kidney disease through the β-catenin signalling pathway. Exp Cell Res. 2022;413:113051. doi: 10.1016/j.yexcr.2022.113051 [DOI] [PubMed] [Google Scholar]
- 56.Montaigne D, Butruille L, Staels B. PPAR control of metabolism and cardiovascular functions. Nat Rev Cardiol. 2021;18:809–823. doi: 10.1038/s41569-021-00569-6 [DOI] [PubMed] [Google Scholar]
- 57.Calvier L, Boucher P, Herz J, Hansmann G. LRP1 deficiency in vascular SMC leads to pulmonary arterial hypertension that is reversed by PPARγ activation. Circ Res. 2019;124:1778–1785. doi: 10.1161/CIRCRESAHA.119.315088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Legchenko E, Chouvarine P, Borchert P, Fernandez-Gonzalez A, Snay E, Meier M, Maegel L, Mitsialis SA, Rog-Zielinska EA, Kourembanas S, et al. PPARγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci Transl Med. 2018;10:eaao0303. doi: 10.1126/scitranslmed.aao0303 [DOI] [PubMed] [Google Scholar]
- 59.Cai J, Pardali E, Sánchez-Duffhues G, ten Dijke P. BMP signaling in vascular diseases. FEBS Lett. 2012;586:1993–2002. doi: 10.1016/j.febslet.2012.04.030 [DOI] [PubMed] [Google Scholar]
- 60.Katagiri T, Tsukamoto S, Nakachi Y, Kuratani M. Recent topics in fibrodysplasia ossificans progressiva. Endocrinol Metab (Seoul). 2018;33:331–338. doi: 10.3803/EnM.2018.33.3.331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shen Q, Little SC, Xu M, Haupt J, Ast C, Katagiri T, Mundlos S, Seemann P, Kaplan FS, Mullins MC, et al. The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. J Clin Invest. 2009;119:3462–3472. doi: 10.1172/JCI37412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fukuda T, Kohda M, Kanomata K, Nojima J, Nakamura A, Kamizono J, Noguchi Y, Iwakiri K, Kondo T, Kurose J, et al. Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. J Biol Chem. 2009;284:7149–7156. doi: 10.1074/jbc.M801681200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shen H, Zhang J, Wang C, Jain PP, Xiong M, Shi X, Lei Y, Chen S, Yin Q, Thistlethwaite PA, et al. MDM2-mediated ubiquitination of angiotensin-converting enzyme 2 contributes to the development of pulmonary arterial hypertension. Circulation. 2020;142:1190–1204. doi: 10.1161/CIRCULATIONAHA.120.048191 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.