

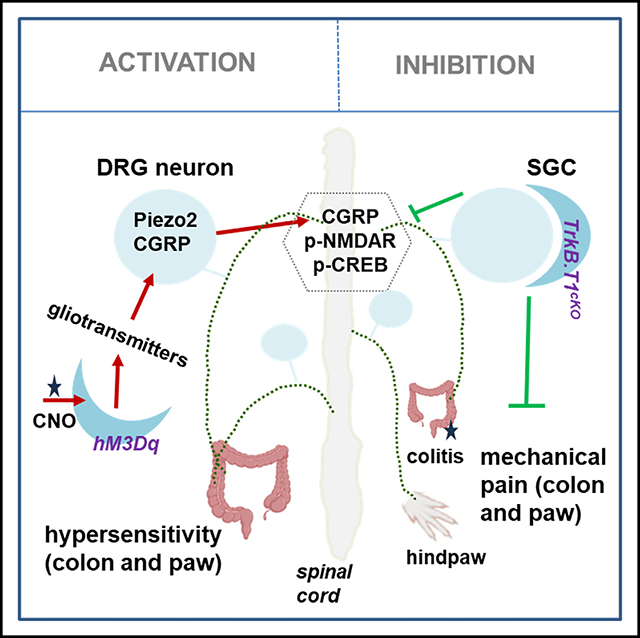
SUMMARY
Satellite glial cells (SGCs) of dorsal root ganglia (DRGs) are activated in a variety of chronic pain conditions; however, their mediation roles in pain remain elusive. Here, we take advantage of proteolipid protein (PLP)/creERT-driven recombination in the periphery mainly occurring in SGCs of DRGs to assess the role of SGCs in the regulation of chronic mechanical hypersensitivity and pain-like responses in two organs, the distal colon and hindpaw, to test generality. We show that PLP/creERT-driven hM3Dq activation increases, and PLP/creERT-driven TrkB.T1 deletion attenuates, colon and hindpaw chronic mechanical hypersensitivity, positively associating with calcitonin gene-related peptide (CGRP) expression in DRGs and phospho-cAMP response element-binding protein (CREB) expression in the dorsal horn of the spinal cord. Activation of Plp1+ DRG cells also increases the number of small DRG neurons expressing Piezo2 and acquiring mechanosensitivity and leads to peripheral organ neurogenic inflammation. These findings unravel a role and mechanism of Plp1+ cells, mainly SGCs, in the facilitation of chronic mechanical pain and suggest therapeutic targets for pain mitigation.
Graphical Abstract
In brief
Tiwari et al. reveal that Plp1+ satellite glial cells (SGCs) of dorsal root ganglia facilitate chronic colonic and somatic mechanical pain, in which activated SGCs release mediators to activate neurons that sense pain.
INTRODUCTION
Since the discovery of satellite glial cells (SGCs) of the spinal ganglia more than half a century ago,1,2 their roles in pain have recently drawn immense attention due to their proximity to pain-sensing neurons in dorsal root ganglia (DRGs).3 SGCs represent the largest glial subtype in DRGs and ensheath sensory neuron somata with a small gap (about 20 nm) between the glial and neuronal membrane, similar to the manner at synapses (a 20–40 nm gap), endowing efficient crosstalk between SGCs and sensory neurons.3–8 The modulatory role of SGCs on sensory neuron activity and thereby pain regulation is governed by mechanisms including but not limited to gap-junction-mediated intercellular calcium (Ca2+) waves and paracrine action of gliotransmitter release.9,10 For example, purinergic ligand 3′-O-(4-Benzoyl)benzoyl-ATP (BzATP) that acts on P2X7R in SGCs can transactivate small-size, presumably nociceptive, DRG neurons through Ca2+-gated pannexin1-mediated ATP paracrine action.10 In several pain modalities resulting from nerve injury or inflammation, the activity of SGCs is increased, manifested by the upregulation of the astrocytic marker glial fibrillary acidic protein (GFAP), connexin 43 (Cx43), and intracellular signaling molecules in DRGs.11–17 The reduction of Cx43 expression by RNA interference or carbenoxolone or genetic deletion attenuates nerve injury or inflammation-induced somatic pain.16,18,19
Proteolipid protein 1 (Plp1), which resides on the X chromosome, is also expressed in SGCs as well as in Schwann cells and enteric glia.20–22 However, Plp1 is not expressed by astrocytes, sympathetic glia, or neurons of DRGs of rodents and/or human.6,20,21,23 Recent studies using Plp1/creERT-driven genetic manipulation of Plp1-expressing (Plp1+) glia demonstrate that optogenetic activation of Plp1+ glia residing in the hindpaw of mice evokes sensory nerve electrical activity and animal painful behavior,24 and Plp1/creERT-driven conditional deletion of Cx43 from SGCs blocks sensory neuron intercellular crosstalk and reduces complete Freund’s adjuvant (CFA)-induced mechanical pain in the paw.16 These studies suggest a role of Plp1+ glia in facilitating somatic hypersensitivity. In terms of gastrointestinal (GI) physiology, PLP/creERT-guided enteric glia ablation increases total GI transit times in female mice but not male mice.25 Along the GI tract, Plp1 expression is the highest in the duodenum and successively decreases anally toward the colon, with a marked reduction of Plp1 expression levels in the colon during development post-natally from post-natal day (P)2 to P88.26 PLP/creERT-driven tdTomato expression in the colon is evident but very scarce.22 The present work reveals consistent results that PLP/creERT-driven mCherry expression or PLP/creERT-targeted TrkB.T1 conditional deletion mainly occurs in SGCs of DRGs with considerable low levels in the distal colon. With these unique features, we used PLP/creERT-guided genetic manipulation to examine the role of SGCs in the regulation of colonic function.
Acute pain has a protective role, while chronic pain is devastating, with up to 25% of the population having visceral pain at any one time. Previous studies show acute and transient analgesic effects, which peak at 30 min–1 h and diminish at 2 h post-activation of GFAP+ cells and spinal astrocytes,27,28 while optogenetic activation of spinal astrocytes evokes long-lasting (more than 1 day) somatic hypersensitivity and pain.29 The present study was undertaken to utilize PLP/creERT to examine whether SGCs also have roles in chronic pain in the colon and hindpaw in mice.
RESULTS
Chemogenetic activation of Plp1+ cells evoked chronic somatic and colonic mechanical hypersensitivity
We generated Plp1;hM3Dq;mCherry mice by breeding PLP/creERT mice with RC::L-hM3Dq mice (Figure S1A), which were further subjected to tamoxifen treatment to induce PLP/creERT-based recombination (Figure S1B). We divided these mice into three groups: (1) Plp1+;mCherry;hM3Dq mice (designated as Plp1;hM3Dq) that received clozapine N-oxide (CNO) treatment and were assigned as the “CNO” group, (2) Plp1;hM3Dq mice that received vehicle treatment and were assigned as the “vehicle” group, and (3) Plp1−;mCherry;hM3Dq mice (designated as neg;hM3Dq) that received CNO treatment and were assigned as the “CNO-off” group (Figure S1C). During the same time block, we treated these mice with either CNO (2 mg/kg body weight) or vehicle intraperitoneally (i.p.). A previous study showed that optogenetic activation of Plp1+ hindpaw cells rapidly evoked painful behavior in mice.24 We were curious whether painful behavior persisted after systemic activation of Plp1+ cells. We found that hindpaw mechanical hypersensitivity was evident 1 day post-CNO treatment of the CNO male group (left shift of responsive curves), but not the female group, when compared to vehicle or CNO-off target controls (Figures 1A and 1B, day 1). The hindpaw thermal sensitivity stayed unchanged among all groups (Figure 1C, day 1). In the subsequent 2 days following drug treatment (CNO, 1 mg/kg body weight, i.p.), the results were the same: only mechanical hypersensitivity was detected in the CNO male group (Figures 1A–1C). Of note, the mechanical pain threshold (3 responses out of 5 von Frey filament stimulations) was decreased from a 0.4 g average on day 1 to a 0.16 g average on day 3, suggesting that chronic activation of Plp1+ cells exacerbated mechanical painful sensation.
Figure 1. Sex-differential changes in somatic pain following CNO treatment of Plp1;hM3Dq mice.
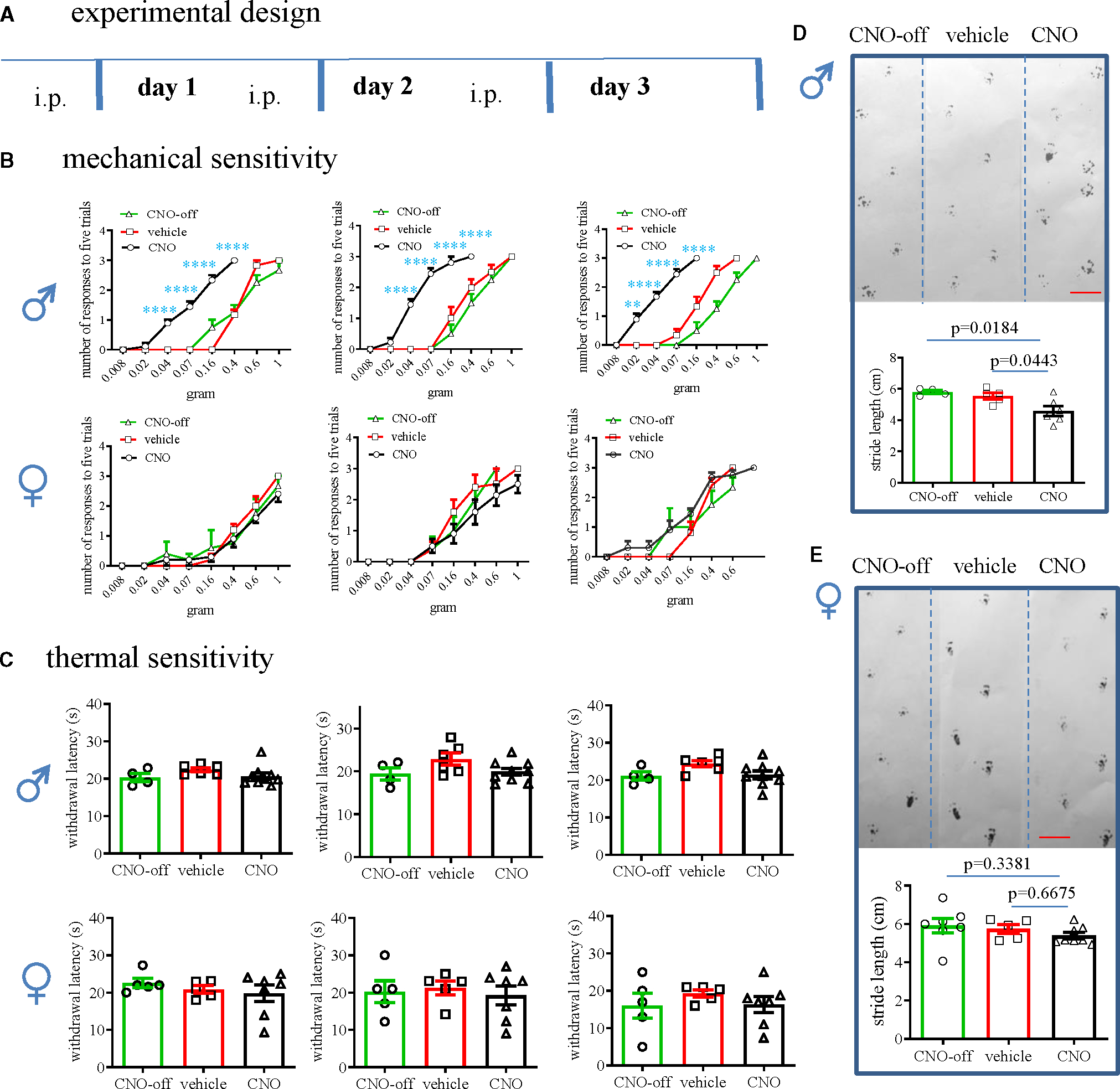
(A) Experimental design of chemogenetic activation of tamoxifen-induced Plp1-Cre-expressing glia.
(B) Assays of von Frey filament stimulation of the plantar surface of hindpaw for examination of somatic mechanical responses in male and female mice. Two-way ANOVA with Tukey’s multiple comparison test. CNO-treated neg;hM3Dq mice: n = 4 male and n = 5 female; vehicle-treated Plp1;hM3Dq mice (vehicle) n = 6 male and n = 5 female; CNO-treated Plp1;hM3Dq mice (CNO): n = 9 male and n = 10 female. **p = 0.0013 and ****p < 0.0001.
(C) Thermal sensitivity is examined by hot plate assay (CNO-off target: male n = 4, female n = 5; vehicle treated: male n = 6, female n = 5; CNO treated: male n = 9, female n = 7). One-way ANOVA with Tukey’s multiple comparison test.
(D) Gait assay of male mice (CNO-off target: n = 4; vehicle treated: n = 5; CNO treated: n = 6). One-way ANOVA with Tukey’s multiple comparison test.
(E) Gait assay of female mice (CNO-off target: n = 8; vehicle treated: n = 5; CNO treated: n = 7). One-way ANOVA with Tukey’s multiple comparison test. Bar: 2 cm.
All data are presented as mean ± SEM.
See also Figure S1.
On the last day (day 3), we performed a gait assay, which showed that the CNO group of male mice demonstrated shorter stride length and more erratic steps when compared to control mice (Figure 1D). CNO treatment did not affect the stride patterns in female Plp1;hM3Dq mice (Figure 1E). We also measured colonic mechanical sensitivity using colonometry that we developed,30,31 which showed that CNO treatment of Plp1;hM3Dq male mice induced colonic mechanical hypersensitivity manifested as increases in the amplitudes of intracolonic pressures (Figure 2A) and decreases in the lengths of fecal pellets (Figure 2B) when compared to controls. Colonic mechanical sensitivity in female mice stayed the same in all groups (Figure 2C). Calcitonin gene-related peptide (CGRP) was involved in a variety of chronic pain developments including visceral hypersensitivity, chronic somatic pain, and migraine and was increased in DRG neurons in pain.32,33 Following CNO treatment of Plp1;hM3Dq mice, CGRP expression was increased in DRG neurons of male mice, but not female mice, when compared to respective vehicle or off-target controls (Figures 2D and S2A), consistent with the sex-differential behavioral outcomes.
Figure 2. Assessments of colonic mechanical hypersensitivity and molecular changes in DRGs in both sexes following CNO treatment of Plp1;hM3Dq mice.
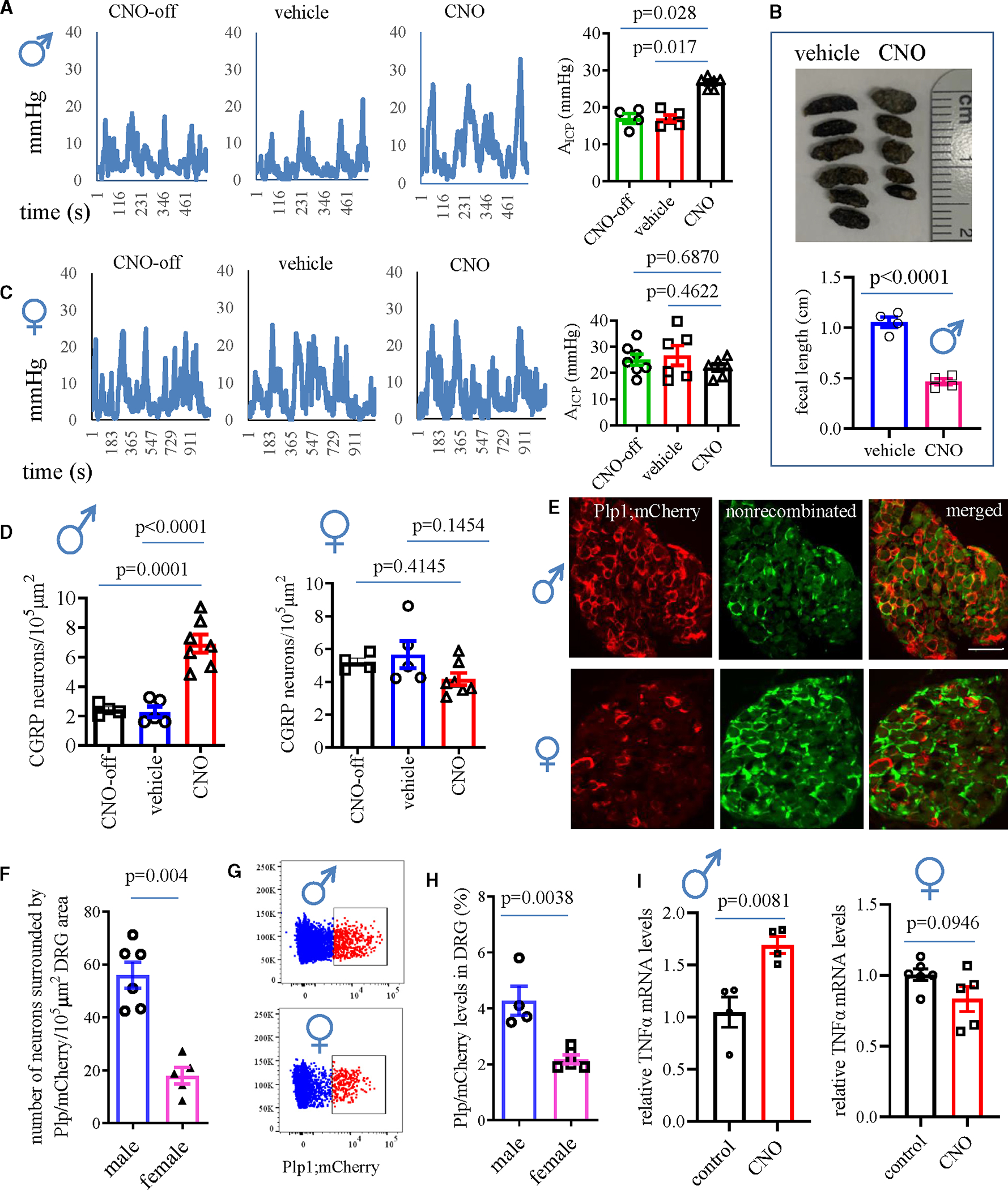
(A) Colonometrical recording of male mice (n = 6 CNO-treated Plp1;hM3Dq mice; n = 4 off-target control; n = 5 vehicle control). One-way ANOVA with Dunn’s multiple comparisons test.
(B) Fecal length from male mice (CNO-treated Plp1;hM3Dq mice: n = 4; vehicle-treated Plp1;hM3Dq mice: n = 4; unpaired two-tailed t test).
(C) Colonometrical recording of female Plp1;hM3Dq mice (n = 6 CNO; n = 7 off-target control; n = 6 vehicle control). One-way ANOVA with Dunn’s multiple comparisons test.
(D) CGRP expression in DRGs of female mice from different treatment groups: n = 4 CNO-treated neg;hM3Dq mice (CNO-off); n = 5 vehicle-treated Plp1;hM3Dq mice (vehicle); n = 7 CNO-treated Plp1;hM3Dq mice (CNO). One-way ANOVA with Tukey’s multiple comparison test.
(E) PLP/creERT-driven mCherry expression in DRGs showing mCherry expression in SGCs. Bar: 80 μm.
(F) Number of DRG neurons surrounded by SGCs that contain PLP/creERT-driven mCherry expression (male: n = 6; female n = 5). Unpaired two-tailed t test.
(G and H) Flow cytometry measurement of PLP/creERT-driven mCherry expression in DRGs (male n = 4, female n = 5). Unpaired two-tailed t test.
(I) Real-time PCR assessment of the mRNA levels of TNF-α in DRGs of male Plp1;hM3Dq mice (n = 4 control, n = 4 CNO treatment, unpaired two-tailed t test) and female Plp1;hM3Dq mice (n = 6 control, n = 5 CNO treatment; unpaired two-tailed t test).
All data are presented as mean ± SEM.
PLP/creERT-driven mCherry expression was highly present in SGCs of DRG
We examined the efficiency of tamoxifen-induced PLP/creERT-driven mCherry expression, which showed distinct levels of mCherry in DRGs of male and female mice. Specifically, the number of DRG neurons wrapped by mCherry-expressing SGCs was much higher in male mice than in female mice (Figures 2E and 2F). A separate and independent analysis (starting from tamoxifen treatment by a different experimenter) using flow cytometry to sort mCherry-expressing SGCs also demonstrated a higher number of mCherry-expressing SGCs in male than female Plp1;hM3Dq mice (Figures 2G and 2H). Tumor necrosis factor alpha (TNF-α), one of the SGC gliotransmitters34 that was released into Plp1;hM3Dq SGC culture medium after CNO (10 μM) treatment overnight (Figure S2B) thus serving as an indicator of SGC activation, also expressed at a higher level in DRGs after CNO treatment of male Plp1;hM3Dq mice but not female mice (Figure 2I). These findings suggested that Plp1+ SGCs in male mice had higher numbers and activity than in female mice, consistent with Plp1+ cell-facilitated sex-differential mechanical hypersensitivity.
PLP/creERT-driven hM3Dq activation in the paw skin of male mice (Figure 3A) can also contribute to evoked somatic mechanical pain.24 Consistent with previous findings,22,26 the distal colon of male mice did not contain much PLP/creERT-driven mCherry expression that was visible in the region of enteric plexuses (Figures 3A and S2C). A previous study reported that PLP/creERT-driven enteric glia ablation did not change GI transit times or the propagation frequency of colonic migrating motor complexes (CMMCs) in male mice,25 likely due to the low levels of Plp1 expression in the distal colon of male mice.22,26 Thus, the PLP/creERT line provided a unique model to study the role of SGCs in the regulation of colonic function due to its robust expression in SGCs compared to the colon (Figure 3B). Although several studies suggested that Plp1 was highly expressed in peripheral glia of mature mice,6,20,21,23 some cells in the spinal cord, but not as many as the SGCs of DRGs, also expressed PLP/creERT-driven mCherry (Figure S2D). The urinary bladder demonstrated strong mCherry expression (Figure S2E), suggesting efficient PLP/creERT-driven recombination in peripheral organs after tamoxifen treatment.
Figure 3. Effects of genetic inhibition of SGCs on colonic mechanical hypersensitivity.
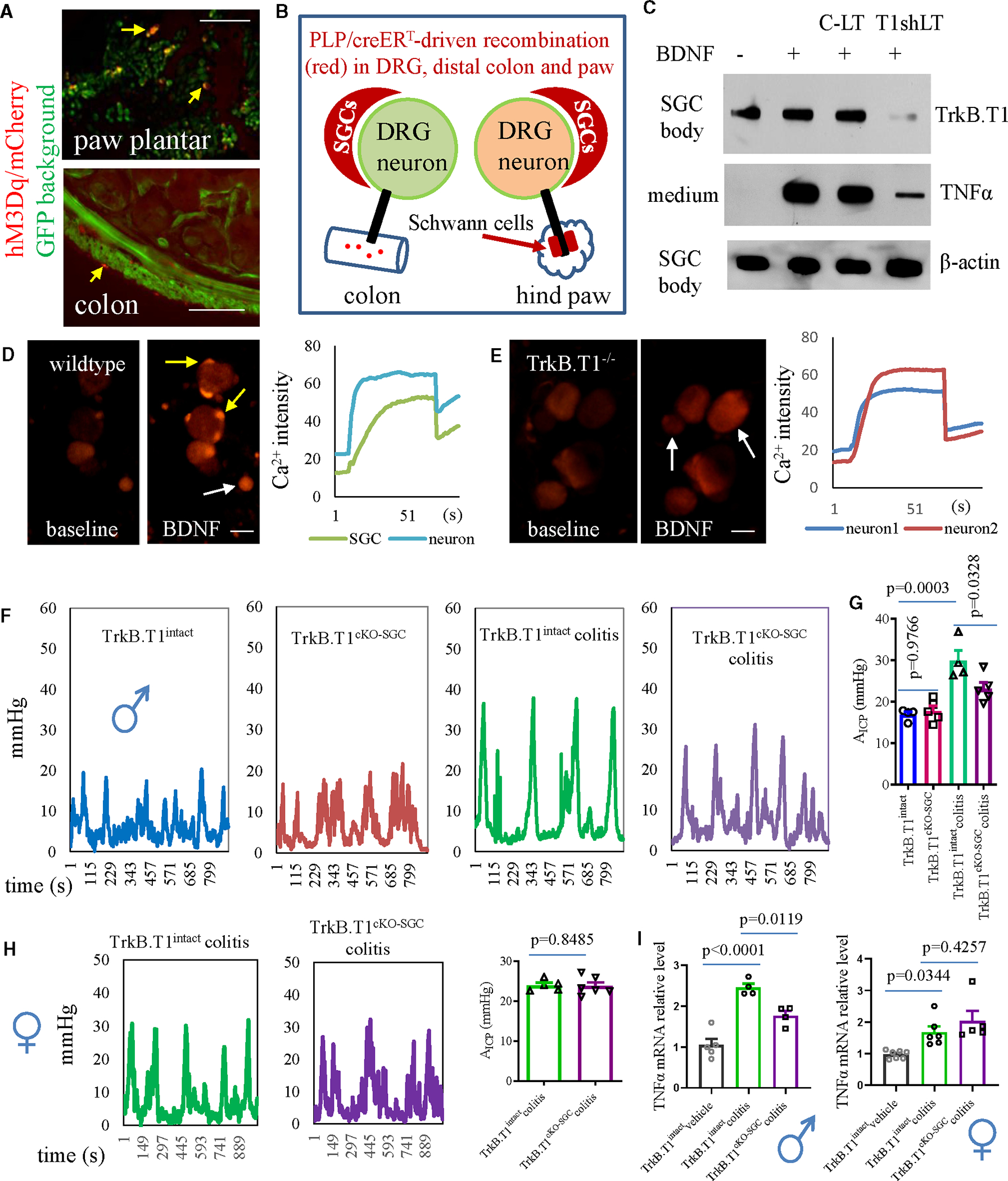
(A) PLP/creERT-driven mCherry expression in the plantar of hindpaw and distal colon (representative sections are from 7 mice). Bar: 200 μm.
(B) Diagram demonstrates the role of Plp1+ SGCs as a major driving force in colonic hypersensitivity.
(C) BDNF-induced TNF-α release from cultured SGCs is blocked by lentiviral particles carrying TrkB.T1 short hairpin RNA (shRNA; T1shLT) but not control lentiviral particles (C-LTs), noting that T1shLT pre-treatment reduces TrkB.T1, but not β-actin, expression levels in SGC cell bodies. Results are from slot blot assays.
(D) BDNF stimulation of cultured DRG neuron-SGC units from wildtype mice to elicit Ca2+ transient in SGCs (indicated by yellow arrows) and neurons (indicated by white arrow). Bar: 50 μm.
(E) TrkB.T1 deletion from SGCs blocks BDNF-induced Ca2+ activity in SGCs but not neurons (indicated by white arrows). Bar: 50 μm.
(F and G) Colonometrical recording of TrkB.T1intact and TrkB.T1cKO-SGC male mice with or without colitis (baseline: n = 4 TrkB.T1intact, n = 5 TrkB.T1cKO-SGC; colitic pain: n = 4 TrkB.T1intact, n = 5 TrkB.T1cKO-SGC; one-way ANOVA with Tukey’s multiple comparison test).
(H) Colonometrical recording of TrkB.T1intact (n = 5) and TrkB.T1cKO-SGC (n = 6) female mice with colitis (day 7, unpaired two-tailed t test).
(I) Inhibition of SGCs reduces colitis-evoked TNF-α upregulation in DRGs of male mice but not female mice (male: n = 5 TrkB.T1intact control, n = 4 TrkB.T1intact colitis, n = 4 TrkB.T1cKO-SGC colitis; female: n = 7 TrkB.T1intact control, n = 6 TrkB.T1intact colitis, n = 5 TrkB.T1cKO-SGC colitis; one-way ANOVA with Tukey’s multiple comparison test).
All data are presented as mean ± SEM.
See also Figures S2 and S3.
Genetic inhibition of SGCs attenuated chronic colonic mechanical hypersensitivity
In a variety of chronic pain states, brain-derived neurotrophic factor (BDNF) was upregulated in DRGs,35–38 and TrkB.T1, as a BDNF high-affinity receptor, was highly expressed in SGCs of DRGs that were characterized by in situ hybridization (Figure S3A). Moreover, TrkB.T1 mediated BDNF-induced TNF-α release from cultured SGCs (Figure 3C) as well as BDNF-evoked Ca2+ activity in SGCs of DRGs (Figure 3D), two biological parameters that were also regulated in SGCs by Plp1;hM3Dq-driven Gq-mediated action after CNO treatment (Figures 2I, S2B, and S4A. Therefore, we sought to delete TrkB.T1 from SGCs to achieve an inhibition of SGC activity. To do so, we crossed PLP/creERT mice with floxed TrkB.T1 mice, which generated heterozygotes in the first generation (Figure S3B). We continued backcrossing with homozygous floxed TrkB.T1 mice to generate conditional TrkB.T1 homozygous deletion (Plp1+/−;TrkB.T1fl/fl) and genetic background control mice (Plp1−/−;TrkB.T1fl/fl) for experiments (Figure S3C). To ensure that we had achieved effective TrkB.T1 deletion from SGCs after tamoxifen treatment, we performed tissue-specific genotyping, which showed that Plp1+/−;TrkB.T1fl/fl mice had TrkB.T1 deleted robustly from DRGs, with much less deletion in the distal colon (designated as TrkB.T1cKO-SGC mice [cKO, conditional knockout]), while Plp1−/−;TrkB.T1fl/fl mice did not show TrkB.T1 deletion from any of the tissues examined (designated as TrkB.T1intact mice) (Figure S3D). These results were consistent with the PLP/creERT-driven mCherry expression levels in these tissues/organs showing the strongest recombination in SGCs of DRGs and a lesser extent of recombination in the distal colon. Taking advantage of these features, we utilized TrkB.T1cKO-SGC mice to examine whether SGCs had a role in colonic mechanosensing and hypersensitivity, although it did not completely preclude the role of enteric glia.
The baseline colonic mechanical sensitivity between TrkB.T1cKO-SGC and TrkB.T1intact male mice stayed the same (Figures 3F and 3G), suggesting that the absence of TrkB.T1 from SGCs did not disturb colonic sensory homeostasis. Similarly, PLP/creERT-driven enteric glia ablation did not change GI transit times or CMMC frequency in male mice.25 We and others have established that colitis induced by 2,4,6-trinitrobenzene sulfonic acid resulted in colonic mechanical pain in male mice measured on day 7.15,30,39 This phenomenon was also seen in TrkB.T1intact male mice (Figures 3F and 3G). However, in TrkB.T1cKO-SGC male mice, colitis-induced colonic hypersensitivity was significantly attenuated, although not completely suppressed, when compared to colitic TrkB.T1intact male mice (Figures 3F and 3G). Colonic mechanical sensitivity in female TrkB.T1cKO-SGC and TrkB.T1intact mice had no difference (Figures 3H and 3I). Moreover, colitis-induced TNF-α upregulation in DRGs was suppressed, in part, by TrkB.T1cKO-SGC in male mice but not in female mice (Figure 3I). Of note, colitis-induced TNF-α upregulation in DRGs could also emanate from other cells such as macrophages, which would not be reduced by SGC inhibition. This aligned with the partial inhibition of TNF-α levels in DRGs when SGCs were inhibited by TrkB.T1cKO-SGC.
Activation of SGCs upregulated Piezo2 in DRG neurons, explaining SGC-facilitated mechanical hypersensitivity
Piezo2 is a mechanotransducer that was implicated to participate in colonic and somatic mechanical pain.30,40–44 We therefore examined whether activation of SGCs can regulate Piezo2 expression and/or activity in DRG neurons. Using DRG explants from Plp1;ChR2-YFP (channelrhodopsin2-yellow fluorescent protein) mice, we treated one DRG with a fiber-coupled light-emitting diode (LED) at 470 nm blue light (Thorlabs) (Figure 4A) according to our published protocol,30 and the contralateral DRG served as control (Figures 4B and 4C). On the second day, we found that photostimulation increased the number of DRG neurons expressing Piezo2 (Figures 4B–4D), with a higher number of DRG neurons that simultaneously contained Piezo2 and were surrounded by Plp1+ SGCs (cells indicated by yellow arrows in Figure 4C′′ and summarized in Figure 4E). We measured the diameter of Piezo2+ DRG neurons from the same number of DRG sections of untreated (1,362 neurons were measured) and LED-treated (2,361 neurons were measured) DRGs, showing that the upregulation of Piezo2 by PLP/creERT-driven optogenetics mainly occurred in small DRG neurons (Figure 4F). Activation of Plp1+ SGCs also increased Piezo2 mRNA levels in DRGs (Figure 4G). Interestingly, activation of Plp1+ SGCs did not change either the mRNA or protein levels of the transient receptor potential vanilloid 1 (TRPV1) (Figures S5A–S5C). This could explain why Plp1+ SGCs participated in the regulation of mechanical but not thermal hypersensitivity, as found in our experiments (Figure 1) and others showing that PLP/creERT-driven Cx43 conditional deletion from SGCs reduced mechanical but not thermal hyperalgesia after CFA treatment of the hindpaw.16
Figure 4. Piezo2 expression in and mechanosensitivity of DRG neurons following optogenetic or chemogenetic activation of SGCs.
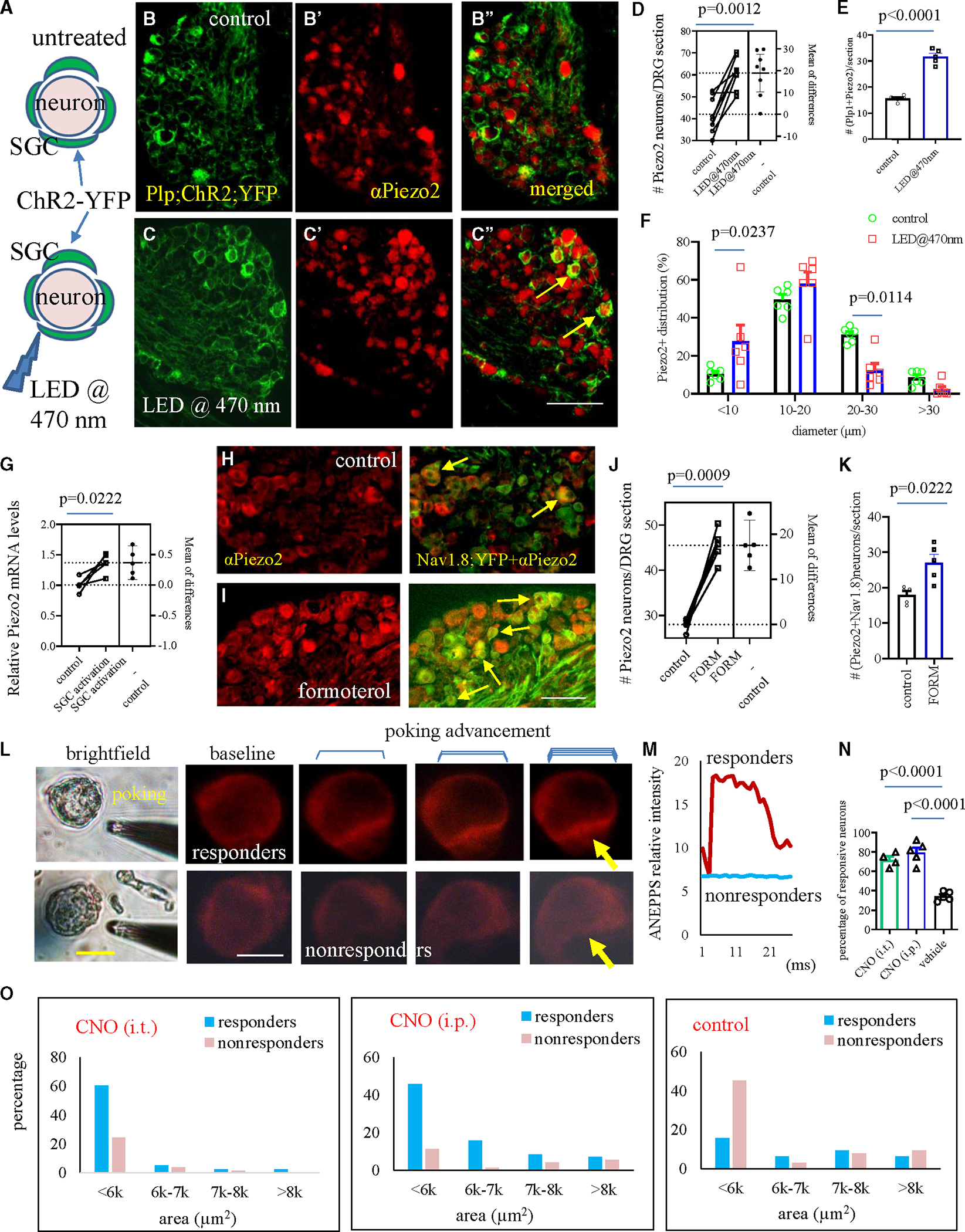
(A) Ex vivo experiments to activate SGCs by optogenetics.
(B and B′′) DRG sections show PLP/creERT-driven ChR2-YFP expression in SGCs (B) and Piezo2 immunoreactivity in DRG neurons (B′) that have few co-expressions of Piezo2 and YFP (B′′) in untreated DRG explants.
(C and C′′) Stimulation with LED@470 nm results in more neurons expressing Piezo2 with YFP+ SGC wrapping (C′′, indicated by yellow arrows). Bar: 80 μm.
(D) The number of DRG neurons expressing Piezo2 comparing between control and following SGC activation (n = 8, paired two-tailed t test).
(E) The number of DRG neurons that have Piezo2 and are wrapped by YFP+ SGCs comparing between control and following SGC activation (n = 5, paired two-tailed t test).
(F) Diameter-based distribution of Piezo2+ DRG neurons comparing between control and following SGC activation (n = 6, two-way ANOVA with Šídák’s multiple comparisons test).
(G) The relative Piezo2 mRNA levels in DRG explants with or without activation of SGCs. n = 5, paired two-tailed t test.
(H and I) Co-expression of Nav1.8;YFP and Piezo2 in DRG neurons before and after formoterol treatment. Bar: 100 μm.
(J) The number of DRG neurons expressing Piezo2 (n = 5, paired two-tailed t test).
(K) The number of DRG neurons co-expressing Piezo2 and Nav1.8;YFP (n = 5, paired two-tailed t test).
(L) DRG neurons that are positively responding to glass pipette poking (top, responders) or have no responses to poking (bottom, nonresponders). Bar: 40 μm.
(M) Di-8-ANEPPS fluorescent intensity curve comparing between responders and nonresponders.
(N) The percentage of responders from DRGs of Plp1;hM3Dq mice that receive CNO (i.t.) or CNO (i.p.) treatment compared to control mice (n = 4–5, one-way ANOVA with Tukey’s multiple comparison test).
(O) Activation of SGCs by either i.t. CNO or i.p. CNO increases the mechanosensitivity of small-diameter DRG neurons when compared to control.
See also Figures S4 and S5.
We next chemically activated SGCs by formoterol, the specific agonist of beta (β)2 adrenergic receptor that was expressed exclusively in SGCs measured in DRG.15 We treated DRG explants from Nav1.8;YFP mice with formoterol (10 μM) overnight and performed Piezo2 immunohistochemistry of DRG sections. Nav1.8;YFP mice were used to mark the nociceptors by Nav1.8;YFP. We found that activation of SGCs by formoterol increased the number of DRG neurons expressing Piezo2 (Figures 4H–4J), some of which co-expressed Nav1.8 (Figure 4K), suggesting an upregulation of Piezo2 in Nav1.8+ nociceptive neurons by SGC activation.
To measure the mechanosensitivity of DRG neurons, we dissected DRGs freshly for primary culture of enzymatically disassociated DRG neurons and examined DRG neuron responsiveness to mechanical stimulation using our recently established poking-voltage imaging coupling technique.30 The DRG neuron culture was obtained from Plp1;hM3Dq mice that received intrathecal (i.t.) injection of CNO (3 uL of 26 uM solution) and were sacrificed the second day after CNO treatment. As a comparison, DRG neuron cultures from Plp1;hM3Dq mice that received CNO i.p. injection and underwent behavioral tests as described above as well as control mice were also assessed. We incubated DRG neurons with a voltage-sensitive dye, Di-8-ANEPPS (Figure 4L), and randomly chose dissociated small to medium DRG single neurons from the bright-field view. The selected DRG neuron was poked by a tip-polished glass micropipette in a manner that the micropipette tip was pushed forward in a micrometer increment; simultaneously, the changes in the intensity of Di-8-ANEPPS fluorescence were recorded in a time-lapse rate of 1 kHz (Figure 4M). We found that CNO (i.t.) treatment of Plp1;hM3Dq mice resulted in 55 out of 78 successfully poked DRG neurons (an average of 70.5% from 4 mice) being mechanically responsive (designated as responders), CNO (i.p.) treatment of Plp1;hM3Dq mice resulted in 54 out of 69 successfully poked neurons (an average of 78.2% from 5 mice) being responders, and vehicle (i.p.)-treated Plp1;hM3Dq mice had 22 out of 64 successfully poked neurons (an average of 34.4% from 5 mice) being responders (Figure 4N). We measured the area of the soma of successfully poked DRG neurons and clustered them into a different range to check size-based distribution corresponding to their mechanical responsiveness. We found that the number of mechanosensitive DRG neurons (responders) was much higher in small-size neurons (e.g., smaller than 6,000 mm2 of freshly cultured neurons) from CNO-treated Plp1;hM3Dq mice when compared to control (Figure 4O), consistent with Piezo2 protein upregulation in nociceptive neurons.
Experiments from the above three distinct approaches, i.e., optogenetically, chemically, or chemogenetically activating SGCs, showed that SGC activation increased the number and activity of small-size and/or Nav1.8+ nociceptive DRG neurons expressing Piezo2, explaining the role of SGCs in the regulation of mechanical hypersensitivity.
SGC-to-DRG neuron crosstalk
A previous study reported that BzATP activated SGCs rapidly, followed by a delayed (about 2 min) activation of small-size DRG neurons, which was mediated by a paracrine action of ATP.10 Gliotransmitters are crucial in mediating SGC-to-sensory neuron paracrine crosstalk. We therefore used SGC-conditional medium (CM) (Figure S2B) to stimulate cultured DRG neurons to examine whether gliotransmitters from activated SGCs had roles in activating Piezo2+ DRG neurons and/or regulated Piezo2 expression to provide additional evidence on SGC regulation of DRG mechanosensory neurons (Figures 5A and 5B). We visualized Piezo2+ DRG neurons in culture by using DRGs from Piezo2;YFP mice and applied SGC-CM, off-target CM, or untreated CM for single-neuron Ca2+ imaging (Figures 5C–5E), in which SGC-CM elicited Ca2+ signals in a higher percentage of Piezo2;YFP+ DRG neurons when compared to untreated CM or off-target CM (Figure 5D). SGC-CM also increased the activity of some Piezo2− DRG neurons (Figure 5E, indicated by arrows). SGC-CM treatment of wild-type DRG explants increased the expression levels of Piezo2 in DRG neurons when compared to control (Figure 5F).
Figure 5. Assessments of paracrine interaction from SGCs to DRG neurons.
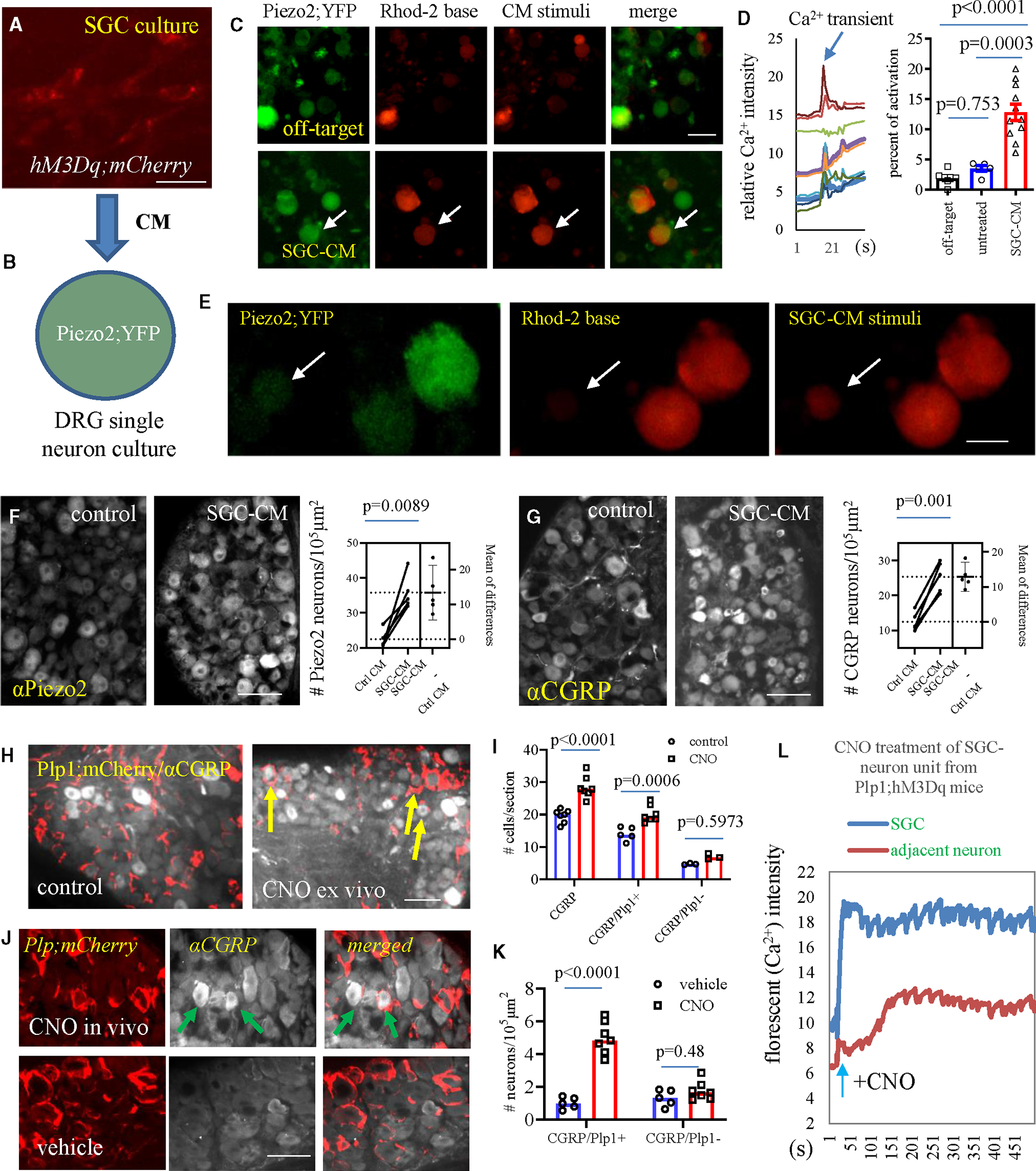
(A) Cultured SGCs from Plp1;hM3Dq mice treated with CNO or cultured SGCs from neg;hM3Dq mice treated with CNO (CNO-off target) to produce conditionalmedium (CM). Bar: 10 μm.
(B) Diagram to show application of SGC-CM to cultured DRG neurons.
(C and D) CNO-CM elicits Ca2+ transients in Piezo2+ DRG neurons (n = 5 CNO-off-treated wells, n = 5 basal CM-treated wells, and n = 11 CNO-CM-treated wells from 3 independent experiments). One-way ANOVA with Tukey’s multiple comparison test. Bar: 200 μm.
(E) CNO-CM stimulates activation of Piezo2− DRG neurons. Bar: 100 μm.
(F) Piezo2 expression after CNO-CM treatment of DRG explants (n = 5, paired two-tailed t test). Bar: 80 μm.
(G) CGRP expression after CNO-CM treatment of DRG explants (n = 5, paired two-tailed t test). Bar: 80 μm.
(H and I) CNO treatment of DRG explants from Plp1;hM3Dq mice increases CGRP upregulation in DRG neurons adjacent to Plp1+ SGCs (indicated by arrows, n = 7, two-way ANOVA with Šídák’s multiple comparisons test). Bar: 80 mm.
(J and K) CNO (i.p.) treatment of Plp1;hM3Dq mice (n = 7) increases CGRP upregulation in DRG neurons adjacent to Plp1+ SGCs (indicated by arrows) when compared to those from vehicle-treated Plp1;hM3Dq mice (n = 5). Two-way ANOVA with Šídák’s multiple comparisons test. Bar: 40 μm. All data are presented as mean ± SEM.
(L) Representative curves of SGC-to-DRG neuron crosstalk examined by Ca2+ imaging, noting a delayed activation of DRG neurons after CNO activation of SGCs in SGC-neuron unit culture from Plp1;hM3Dq mice.
See also Figure S4.
Following three paradigms of treatment, i.e., SGC-CM treatment of wild-type DRG explants (Figure 5G), CNO treatment of DRG explants from Plp1;hM3Dq mice (Figures 5H and 5I), or optogenetic activation of SGCs in DRG explants of Plp1;ChR2 mice (Figures S5D–S5F), CGRP expression was increased in DRG neurons. The number of CGRP+ neurons that were wrapped by Plp+ mCherry was increased after CNO treatment of Plp1;hM3Dq DRG explants ex vivo (Figures 5H and 5I) or intact Plp1;hM3Dq mice in vivo (Figures 5J and 5K). Using “SGC-DRG neuron unit” culture from Plp1;hM3Dq;mCherry mice, we found that CNO treatment rapidly evoked Ca2+ transients only in SGCs expressing hM3Dq;mCherry (Figures S4A–S4C and 5L) within the first minute of recording, suggesting that CNO specifically acted on SGCs in DRGs from Plp1;hM3Dq mice. Extending the recording time of Ca2+ imaging after CNO stimulation of SGC-neuron unit culture, we found that the activation of SGCs led to intracellular Ca2+ elevation in adjacent DRG neurons (Figures S4C and 5L) in an average time lapse of 86.18 ± 11.64 s (Figure S4D), suggesting a crosstalk from SGCs to adjacent DRG neurons that may explain the upregulation of Piezo2 and CGRP in DRG neurons adjacent to Plp1+ SGCs after SGC activation. Since TNF-α levels were elevated and released into the SGC culture medium after SGC activation (Figures 2I, S2B, 3C, and 3I), we stimulated cultured DRG neurons from Piezo2;GCaMP mice with TNF-α (1 ng/mL), which increased GCaMP intensity in a subpopulation of Piezo2+ DRG neurons (Figures S4E and S4F). Of note, a smaller percentage of Piezo2+ DRG neurons were activated by TNF-α (8.1% ± 1.1%) when compared to SGC-CM (12.8% ± 1.4%) treatment, suggesting additional gliotransmitters, e.g., ATP,10 in SGC-to-DRG neuron crosstalk.
Previous studies showed that astrocytes/SGCs demonstrated acute and transient antinociceptive action that peaked 30–60 min and diminished at 2 h post-glial activation.27,28 However, at 1 h post-activation of SGCs, the expression level of Piezo2, CGRP, or TRPV1 was not affected in DRGs (Figure S4G), suggesting that acute action of SGCs on DRG neurons did not involve the upregulation of pain-related neuronal markers (e.g., CGRP and Piezo2). Using either PLP/creERT;hM3Dq to drive SGC activation in the present study or GFAP-hM3Dq to drive SGC activation27 showed consistent results that at 2 h after CNO treatment, mechanical sensitivity/hypersensitivity (colonic sensitivity in Plp1;hM3Dq mice vs. somatic sensitivity in GFAP-hM3Dq mice) was not affected (Figure 6A). A study using optogenetic activation of spinal GFAP+ astrocytes showed long-lasting somatic hypersensitivity that peaked on day 1 and was back to normal on day 7.29 Combined with the present study, it appeared that the activation of these astrocytic glia (SGCs and astrocytes) had biphasic action that posed an antinociceptive effect rapidly and transiently,27,28 which diminished at about 2 h and then kicked on long-lasting pro-nociceptive action.29 Consistent with CNO (i.p.) treatment of Plp1;hM3Dq mice, the second day after i.t. injection of CNO to Plp1;hM3Dq mice, not only was the mechanosensitivity of DRG neurons upregulated (Figures 4N and 4O) but also colonic and hindpaw mechanical sensitivity was elevated when compared to sham control (Figures 6B and 6C).
Figure 6. Assessments of peripheral organ neurogenic inflammation after CNO treatment of Plp1;hM3Dq mice.
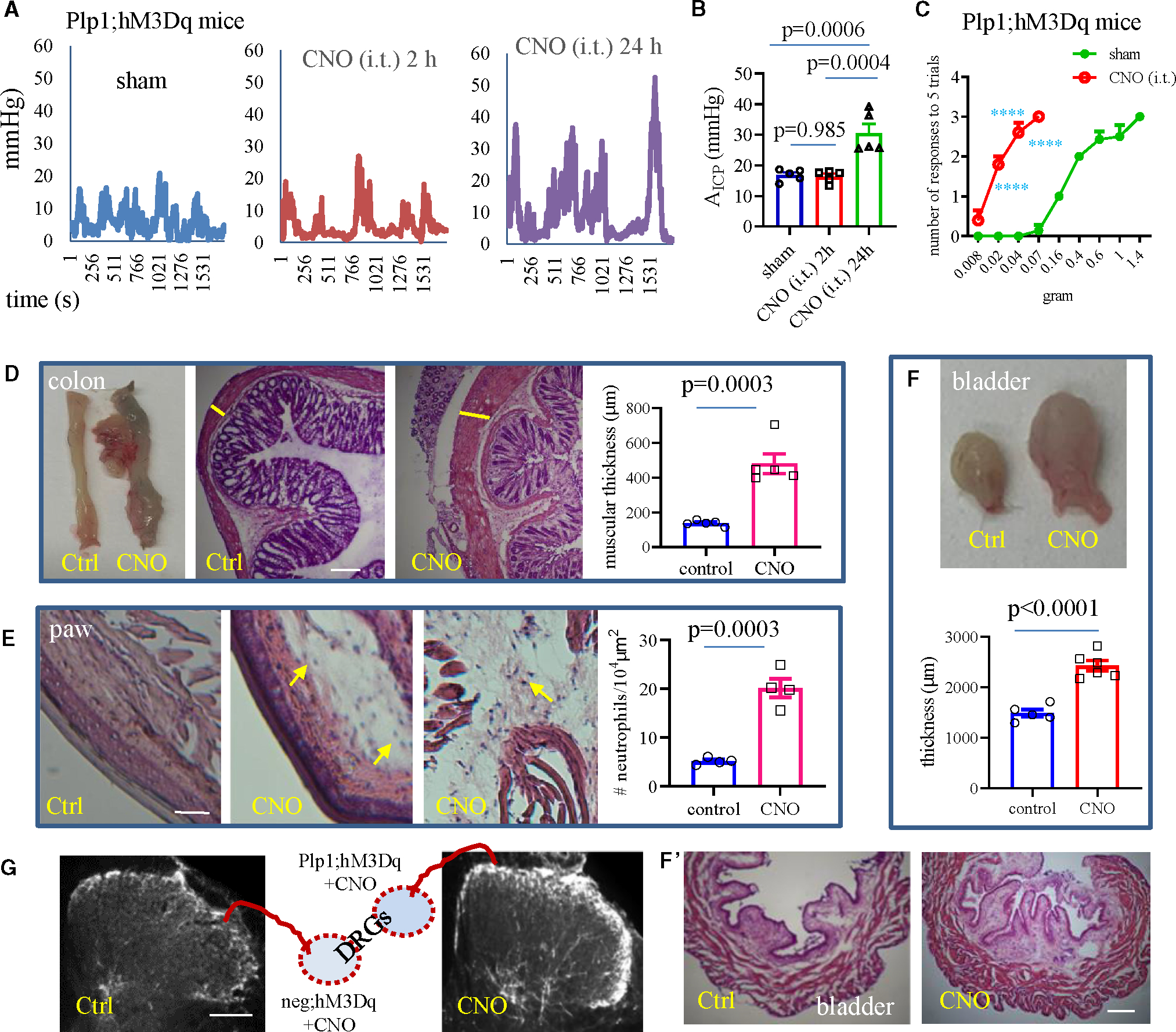
(A) Colonometrical recording of Plp1;hM3Dq mice following intrathecal (i.t.) injection of CNO or sham.
(B) A time-dependent change in colonic mechanical sensitivity after CNO (i.t.) treatment of Plp1;hM3Dq male mice when compared to sham control (n = 5 sham, n = 5 for 2 h, and n = 5 for 1 day post-CNO treatment). One-way ANOVA with Tukey’s multiple comparison test.
(C) CNO (i.t.) treatment of Plp1;hM3Dq male mice (n = 5) increases hindpaw mechanical sensitivity when compared to sham control (n = 7). Two-way ANOVA with Šídák’s multiple comparisons test. ****p < 0.0001.
(D) CNO (i.p.) treatment of Plp1;hM3Dq mice (n = 5) causes inflammation in the distal colon when compared to vehicle-treated control (n = 5). Unpaired two-tailed t test. Microscopic examination of H&E stain shows thickening of the muscular wall (indicated by yellow bars) after CNO treatment of Plp1;hM3Dq mice. Bar: 500 μm.
(E) CNO (i.p.) treatment of Plp1;hM3Dq mice (n = 4) causes inflammation in the hindpaw when compared to vehicle-treated control (n = 4). Unpaired two-tailed t test. Neutrophils are indicated by yellow arrows. Bar: 80 μm.
(F) CNO (i.p.) treatment of Plp1;hM3Dq mice (n = 6) causes inflammation in the urinary bladder when compared to vehicle-treated control (n = 5). Bar: 500 μm.
Unpaired two-tailed t test.
(G) CNO (i.p.) treatment of Plp1;hM3Dq mice evokes CGRP fiber intensity in the dorsal horn of the spinal cord extending to deep laminae when compared tovehicle-treated control (representative graphs are from n = 3 mice for both treatment). Bar: 100 μm.
All data are presented as mean ± SEM.
See also Figure S6.
PLP/creERT-regulated neurogenic inflammation and spinal central sensitization
Both CNO (i.p.) and CNO (i.t.) treatment of Plp1;hM3Dq mice induced neurogenic inflammation of the peripheral organs (Figures 6D–6F and S6A–S6C) and CGRP central release to the spinal cord (Figures 6G and S6H). Specifically, CNO (i.p. 1 dose for 1 day) treatment of Plp1;hM3Dq male mice achieved a similar degree of colonic inflammation compared to CNO (i.t., 1 dose for 1 day) treatment of Plp1;hM3Dq mice, manifested as ~ 2-fold increases in the thickness of the muscular wall of the distal colon (Figures S6A–S6C). CNO (i.p., 3 doses for 3 days) treatment of Plp1;hM3Dq mice achieved a greater degree of colonic inflammation when compared to a single dose of CNO injection, measured as about 3.5-fold increases in the thickness of the muscular wall of the distal colon (Figure 6D). Of note, CNO injected systemically can be rapidly diffused into plasma and maintain substrate activity in cerebrospinal fluid within 30 min for up to 24 h.45 Therefore, we did not preclude the effects of CNO on Plp1+ enteric glia after either route of injection. CNO treatment of Plp1;hM3Dq mice (i.p., 3 doses) also caused severe inflammatory infiltration in the skin of the hindpaw with increased numbers of neutrophils (Figure 6E) and inflammation of the urinary bladder showing as severe hypertrophy with increased suburothelial edema (Figure 6F), which could be combined effects from the activation of SGCs and Plp1+ cells in these organs.
Pain is accompanied by CGRP central release to the spinal cord, where CGRP acts on receptors to participate in spinal central sensitization, a hallmark of chronic pain.33 In all three paradigms (Figures S6D–S6F) that we used to activate different glial populations but all included SGCs, which were CNO (i.p.) treatment of Plp1;hM3Dq mice that activated SGCs and Schwann cells (Figure S6D), CNO (i.t.) treatment of Plp1;hM3Dq mice that focused on SGCs in DRGs (Figure S6E), and CNO (i.p.) treatment of GFAP;hM3Dq mice that activated SGCs and astrocytes (Figure S6F), we found increased intensity of CGRP fibers in the dorsal horn of the spinal cord (Figures 6G and S6H), suggesting a concept that SGC-mediated mechanical pain may involve spinal central sensitization. To further test this notion, we examined the levels of CGRP in the spinal cord of TrkB.T1cKO-SGC mice with colitis since TrkB.T1cKO-SGC attenuated colonic hypersensitivity (Figure 3). Consistent with our previous work,15 colitis increased CGRP levels in the dorsal horn of the spinal cord in TrkB.T1intact mice, with enhanced intensity of CGRP fibers extending to deep laminae (Figure 7A). TrkB.T1cKO-SGC reduced the intensity of CGRP nerve fibers in the spinal cord under the condition of colitis (Figure 7A). These results suggested that SGCs mediated colitis-induced CGRP release in the spinal cord, which was consistent with the result that direct activation of SGCs increased CGRP expression in DRG neurons and CGRP release to the spinal cord in CNO-treated Plp1;hM3Dq mice.
Figure 7. Assess the role of SGCs in the regulation of spinal cord activity.
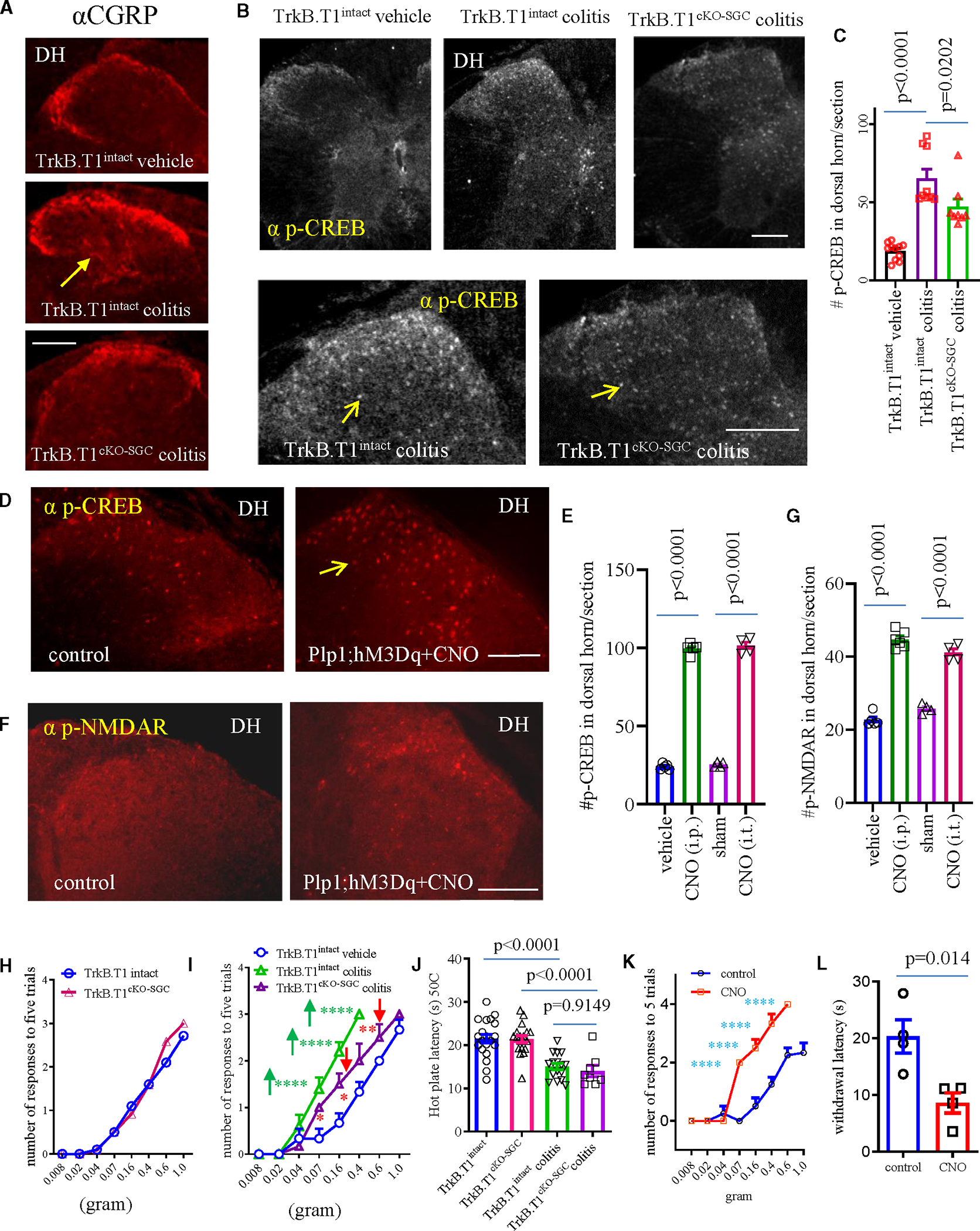
(A) CGRP fiber sprouting to the spinal cord and the expression of p-NMDAR in the dorsal horn. Bar: 50 μm.
(B and C) The expression of p-CREB in the spinal cord of TrkB.T1intact (n = 11 for control and n = 9 for colitis) and TrkB.T1cKO-SGC (n = 8) mice with or without colitis. One-way ANOVA with Tukey’s multiple comparisons test.
(D and E) The expression of p-CREB in the spinal dorsal horn after CNO treatment (i.t. or i.p.) of Plp1;hM3Dq mice. Bar: 50 μm (n = 4–6, one-way ANOVA with Tukey’s multiple comparisons test).
(F and G) The expression of p-NMDAR in the dorsal horn after CNO treatment (i.t. or i.p.) of Plp1;hM3Dq mice. Bar: 50 mm (n = 4–6, one-way ANOVA with Tukey’s multiple comparisons test).
(H) Effect of TrkB.T1cKO-SGC on baseline somatic mechanical sensitivity (n = 10 TrkB.T1intact; n = 12 TrkB.T1cKO-SGC). Two-way ANOVA with Šídák’s multiple comparisons test.
(I) TrkB.T1cKO-SGC attenuates colitis-induced mechanical pain (n = 6 TrkB.T1intact vehicle; n = 5 TrkB.T1intact colitis; n = 6 TrkB.T1cKO-SGC colitis). Two-way ANOVA with Šídák’s multiple comparisons test. ****p < 0.0001 and ***p < 0.001.
(J) Thermal sensitivity in TrkB.T1intact and TrkB.T1cKO-SGC mice with or without colitis (TrkB.T1intact vehicle: n = 20; TrkB.T1cKO-SGC vehicle: n = 19; TrkB.T1intact colitis: n = 16; TrkB.T1cKO-SGC colitis: n = 8). One-way ANOVA with Tukey’s multiple comparison test.
(K) CNO treatment of GFAP;hM3Dq mice evokes hindpaw mechanical pain (n = 4 control, n = 4 CNO treatment; two-way ANOVA with Šídák’s multiple comparisons test; ****p < 0.0001).
(L) CNO treatment of GFAP;hM3Dq mice evokes hindpaw thermal hypersensitivity (n = 4 control, n = 4 CNO treatment; unpaired two-tailed t test).
All data are presented as mean ± SEM.
See also Figures S6 and S7.
Spinal central sensitization is a hallmark of chronic pain, which can be manifested by the upregulation of the activated (phosphorylation) form of cAMP response element-binding protein (CREB) and N-methyl-D-aspartate receptor (NMDAR) and regulated by CGRP.33 In TrkB.T1intact and TrkB.T1cKO-SGC mice with or without colitis, we found that colitis induced phospho (p)-CREB upregulation in the dorsal horn, which was attenuated by TrkB.T1cKO-SGC (Figures 7B and 7C). Chemogenetic activation of SGCs (i.t. or i.p.) also promoted p-CREB expression in the dorsal horn (Figures 7D and 7E). These data aligned well to demonstrate a mediation role of SGCs in facilitating p-CREB expression in the spinal cord dorsal horn, where chronic pain signals are processed.
Chemogenetic activation of SGCs (i.t. or i.p.) increased p-NMDAR expression in the dorsal horn of the spinal cord (Figures 7F, 7G, and S7A), providing additional molecular readout in the chronic pain pathway that suggested a facilitation role of SGCs in driving spinal central sensitization. Post-colitis induction, the expression of p-NMDAR was upregulated in the spinal cord at both the dorsal horn and ventral horn, not only in the colon-innervating thoracolumbar spinal segments but also in the upper thoracic spinal cord that did not receive direct colonic sensory inputs (Figures S7B–S7D), suggesting a spread of neuronal activity along the spinal cord. Colitis-increased p-NMDAR expression in the spinal cord in TrkB.T1intact mice was partially attenuated by TrkB.T1cKO-SGC (Figures S7B–S7D).
Viscero-somatic cross-organ sensitization was manifested by spinal central sensitization. Therefore, changes in hindpaw sensitivity, in addition to spinal neural activity, in colitis can be additional functional assessments of the role of SGCs in mediating chronic pain. In TrkB.T1cKO-SGC and TrkB.T1inatct mice with or without colitis, we found that TrkB.T1cKO-SGC did not change the somatic basal mechanical sensitivity (Figure 7H) but reduced, at least in part, colitis-induced hindpaw referred mechanical pain (Figure 7I). However, TrkB.T1cKO-SGC did not have any effects on hindpaw thermal sensitivity at baseline and in inflammation (Figure 7J). In stark contrast, chemogenetic activation of GFAP+ glia (SGCs and astrocytes) increased hindpaw mechanical (Figure 7K) and thermal (Figure 7L) sensitivity measured on the second day post-CNO treatment (i.p. 2 mg/kg body weight) to assess the chronic effects on somatic sensitivity, consistent with a previous study showing that optogenetic activation of spinal astrocytes induced long-lasting (up to 7 days) mechanical hypersensitivity and thermal hyperalgesia peaked on day 1 post-photostimulation.29 Combined with the results from us and others,16,29 these data suggested that SGCs were prone to mediate chronic mechanical hypersensitivity but not thermal pain, while thermal hypersensitivity was likely mediated by astrocytes but not SGCs.
DISCUSSION
By combining a variety of in vivo and ex vivo approaches, we have identified the functional role of SGCs of DRGs in the mediation of chronic visceral and somatic mechanical pain. We show that the underlying mechanisms by which activated SGCs augment chronic mechanical pain are by increasing the number of DRG neurons expressing Piezo2 and mechanosensitivity that are small nociceptive neurons, as well as inducing CGRP upregulation in DRGs and subsequent CGRP release to induce neurogenic inflammation and spinal central sensitization. A paracrine crosstalk from SGCs to DRG neurons mediated by gliotransmitters plays a role in SGC-mediated Piezo2 and CGRP upregulation in DRG neurons. Our study also reveals that PLP/creERT-driven TrkB.T1 deletion mainly occurs in SGCs and is very scarce in the distal colon; thus, this unique mouse line, TrkB.T1cKO-SGC, can be used to study the role of SGCs in colonic pain circuits. Interestingly, TrkB.T1cKO-SGC reduces, at least in part, colitis-induced spinal neuron activation and hindpaw mechanical pain, suggesting an undescribed phenomenon of peripheral glia in the regulation of central activity. Finally, inhibition of SGCs does not affect baseline mechanical or thermal sensitivity, suggesting that SGCs are dispensable in maintaining basal sensory homeostasis, albeit crucial in chronic mechanical pain development. Therefore, SGCs could serve as targets for pain treatment with minimized adverse effects.
The interaction of SGCs and sensory neurons is well documented,46–48 and the importance of SGCs in pain processing has just begun to be recognized, such as that SGCs can respond to peripheral insults15 and their activation is associated with a variety of pain modalities.3 Here, we utilize three approaches to activate SGCs in intact mice (Figures S6D and S6E) for molecular and functional studies. Systemic activation of Plp1+ glia (SGCs and Schwann cells) leads to colonic and hindpaw mechanical pain but no changes in hindpaw thermal sensitivity. Systemic activation of GFAP+ glia (SGCs and astrocytes) results in mechanical and thermal hypersensitivity. The common cells activated in Plp1;hM3Dq mice and GFAP;hM3Dq mice are SGCs, and the common behavioral outcomes are mechanical hypersensitivity. We also use i.t. injection of CNO to Plp1;hM3Dq mice, which results in elevated colonic and hindpaw mechanical sensitivity. Using PLP/creERT-driven TrkB.T1 conditional deletion that inhibits the activity of SGCs, we find that colitis-induced visceral and somatic hypersensitivity are attenuated. TrkB.T1 in DRGs is expressed in SGCs and mediates BDNF-induced SGC activation. TrkB.T1 in GFAP+ SGCs and astrocytes also participates in chronic somatic pain.49 In resiniferatoxin (RTX)-induced mechanical allodynia, TrkB.T1 was increased in DRGs, and TrkB/T1 inhibitor ANA-12 blocked RTX-induced mechanical pain and sensory neuron activity.50 The present study shows that SGCs deficient in TrkB.T1 block BDNF-evoked Ca2+ activity in SGCs and reduce BDNF-induced TNF-α release from SGCs, suggesting the effectiveness of TrkB.T1 conditional deletion in the blockade of SGC activities and subsequent SGC-to-neuron crosstalk and mechanical hypersensitivity.
Piezo2 is newly identified as a mechanotransducer to mediate colonic and somatic mechanical pain.30,40,41,44 In DRGs, Piezo2 is expressed in various sizes of DRG neurons including the large neurons responsible for proprioception, low-threshold mechanoreceptors essential for touch sensation, and small-diameter neurons participating in pain.51–59 Here, we show that chemogenetic or optogenetic activation of SGCs increases Piezo2 expression in and mechanosensitivity of nociceptive neurons. In contrast, activation of SGCs fails to change the expression level of TRPV1, further proving that SGCs have a lesser role in the development of thermal hypersensitivity. CGRP is an excitatory neurotransmitter and a potent factor in the development of pain60 that is produced in nociceptive neurons to promote pain sensation by central release to the spinal cord for the generation of spinal central sensitization and by peripheral release to evoke neurogenic inflammation. CGRP expression in DRG neurons is also upregulated after the chemogenetic or optogenetic activation of SGCs. Concomitantly, enhanced CGRP intensity in the spinal dorsal horn and inflammatory infiltration of the distal colon, hindpaw, and urinary bladder are identified after chemogenetic activation of SGCs. Increased Ca2+ activity in small-size DRG neurons, likely including peptidergic neurons, after SGC activation10 can be a strong driving factor for the dense core vesicles to release CGRP. Pain-associated CGRP upregulation in the spinal cord is also attenuated by genetic inhibition of SGCs, subsequently inhibiting p-CREB in the dorsal horn. These findings further support a role of SGCs in the pain process, which involves SGC-neuron crosstalk leading to CGRP upregulation in and release from DRG neurons and subsequent CGRP central and peripheral action.
Glial cells surround or have close proximity to neurons to supply nutrients to and modulate the activity of neuronal soma.46,61 At the basal physiological level, glial cells such as astrocytes are normally quiescent,62,63 but they can rapidly respond to neuronal activation during disease states62–64 and exaggerate pain.65 Recent studies targeting SGCs and astrocytes in the spinal cord in the acute phase of somatic hypersensitivity demonstrate analgesic effects, which completely diminished at 2 h post-treatment.27,28 In the chronic phase, the present study targeting SGCs and another study activating spinal astrocytes show the pronociceptive action of these glial cells.29 The transition from acute transient antinociception to chronic long-lasting pro-nociception governed by SGCs/astrocytes is interesting and important, with not much known about the underlying mechanisms. Signaling crosstalk between glial cells and sensory neurons, e.g., SGCs to neuronal soma and astrocytes to axon terminals, is documented to be rapid. Subsequent to Ca2+ elevation, sensory neuroplasticity such as Ca2+-mediated gene transcription and subsequent protein translation may play a role in chronic pain.
Limitations of the study
Among the popular markers that are selected for SGCs, Plp1 is identified to be strongly expressed in peripheral glia.6,20–23 Therefore, utilization of the PLP/creERT line minimizes the effects on central glia, which is beneficial in the clinical intervention of pain with less central adverse effects. Although the present study shows that PLP/creERT-driven recombination in the periphery mainly occurs in DRGs, the tested organs also express detectable levels of Plp1. Thus, our results do not preclude the role of Plp1+ cells in the peripheral organs in driving the observed changes. The term of SGCs used in the present study to represent Plp1+ cells in DRG is (1) due to strong Plp1 expression in SGCs and (2) for simplicity in description.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Communications regarding information presented in this paper can be directed to Liya Qiao (liya.qiao@vcuhealth.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Source data that were used to generate graphs in the paper are available upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Adult mice (2–3 months) with Black 6 background were used. Plp1; hM3Dq and neg; hM3Dq mice were generated by crossing PLP/creERT mice (JAX Stock # 005975) with RC::L-hM3Dq mice (JAX Stock # 026943) followed by genotypic screening and tamoxifen treatment. GFAP; hM3Dq mice were generated by crossing GFAP-Cre mice (JAX Stock # 012886) with RC::L-hM3Dq mice. Plp1; ChR2 mice were generated by crossing PLP/creERT mice with Ai32 mice (JAX Stock # 012569, B6) followed by genotypic screening and tamoxifen treatment. Piezo2; YFP mice were generated by crossing Piezo2-EGFP-IRES-Cre mice (Piezo2-Cre, JAX Stock # 027719) with Ai32 mice or R26-LSL-Gi-DREADD (JAX Stock # 026219) mice. Nav1.8; YFP mice were generated by crossing Nav1.8-Cre+/+ mice (a line that was created by Dr. John Wood, Wolfson Inst. UK)66 with Ai32 mice. Piezo2; tdTomato; GCaMP mice were generated by crossing Piezo2-Cre mice with PC-G5-tdT mice (JAX Stock # 024477). TrkB.T1 knockout mice were provided by Dr. Lino Tessarollo, NIH). Mice with inducible conditional TrkB.T1 deletion from Plp1-expressing glia (TrkB.T1cKO) were generated by using PLP/creERT mice and floxed TrkB.T1 (TrkB.T1fl/fl) mice which were then selected each step for needed genotypes followed by tamoxifen treatment and tissue-specific genotying to check for effectiveness of TrkB.T1 deletion from specific tissues. The genetic control for TrkB.T1cKO mice were their littermates (TrkB.T1intact) that were genotyped as Plp1 negative, floxed TrkB.T1 positive, and no tamoxifen-induced TrkB.T1 deletion in examined tissues. The sequences of primers for genotyping Nav1.8-Cre (13Salt and Cre 5a) and wildtype (13Salt and 12A) alleles were: 13Salt: GGAATGGGATGGAGCTTCTTAC; 12A: TTAC CCGGTGT GTGCTGTAGAAAG; CRE 5a: CAAATGTTGCTGGATAGTT TTTACTGCC. The sequences of primers for genotyping floxed TrkB.T1 were TrkBT1-5s: 5′-CCAGCTA TTGAGTAATGAATGAGTC-3’; TrkBT1-2: 5′-CTACCCATCCAGTGGGATCTT-3’; and TrkBT1-4a: 5′-CCACCGCGGTGGTACCATAACTTCG-3’. The rest of the genotyping utilized sequences of primers provided The Jackson Laboratory. All primers used for genotyping and other PCR were produced by Integrated DNA Technologies, Inc. (IDT: Coralville, Iowa).
Male and female mice were housed separately as 2–5 mice/per cage to ensure adequate social environment. Standard husbandry conditions with 12:12-h light cycles were provided. Animals had free access to regular food/water. For the part where sex was not specified, male mice were used. All experimental protocols involving animal use were approved by Virginia Commonwealth University Institutional Animal Care and Use Committee (IACUC). Animal care was in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) guidelines.
METHOD DETAILS
Drug administration
Tamoxifen was injected at a dose of 75 mg/kg bodyweight intraperitoneally (i.p.) once per day for consecutive 5 days according to a protocol provided by The Jackson Laboratory. Animals were used during the 2–3-week window post the last dose of tamoxifen injection. Tamoxifen-induced Cre recombination was examined postmortem to ensure Cre-based reporter (mCherry or YFP) gene expression or tissue-specific deletion of gene of target. Clozapine-N-oxide (CNO) was administered at 2 mg/kg bodyweight (i.p.) for the first day and 1 mg/kg bodyweight (i.p.) for subsequent days as one dose per day. CNO stock (prepared in DMSO) was diluted into saline for a total of 20 mL for injection (i.p.). Vehicle control contained the same amount of DMSO diluted into the same amount of saline and injected (i.p.) in the same manner as CNO injection. The CNO or vehicle treatment (i.p.) was given in the afternoon, and the behavioral recordings were taken the following morning. Intrathecal (i.t.) injection of CNO was performed at a single dose of 3 μL of 26 μM solution according to our previous experience.30 Sham control contained the same amount of DMSO and performed at the same manner as CNO (i.t.) injection. An insulin syringe was used to inject drug (i.t.) to a vertebra gap at the lower lumbar position in conscious or lightly anesthetized mice. Tail flick was observed to validate positive injections. Colonic inflammation was induced by intracolonic instillation of 2,4,6-trinitrobenzene sulfonic acid (TNBS) at a dose of 75 μL of 12.5 μg/μL solution in 30% EtOH. The same amount of 30% EtOH as vehicle was used in control animals. A PE-50 catheter was used to deliver TNBS or vehicle to the distal colon through the anus. The mouse tail was lifted for 1 min after drug installation to avoid drug leakage from the anus. After drug administration, all animals were observed for 20 min or until when their normal activity was resumed.
DRG ex vivo explant, in vitro single cell, and SGC-neuron unit culture
DRG explants were freshly dissected out and matched by spinal segments for experimental comparison. The freshly dissected DRG pairs were immediately cultured in Dulbecco’s Modified Eagle Medium (DMEM) for 2–4 h (fasting). One of the DRG explant of each pair was then randomly chosen for treatment by CNO (10 μM), SGC conditional medium (SGC-CM) or optogenetics using fiber coupled light-emitting diode (LED) @ 470 nm blue light (Thorlabs, Inc.). The contralateral DRG served as control. After overnight incubation, DRGs were subjected to immunohistochemistry or qPCR.
For single cell (neuron or SGCs) culture, freshly dissected DRGs were subject to enzymatic digestion in DMEM containing 2 mg/mL collagenase at 37°C and the duration of enzymatic process was 45 min. For SGC-neuron unit co-culture, the total duration of enzymatic process was 25 min. After centrifugation, cells were washed three times with DMEM and re-suspended into DMEM containing 5% fetal bovine serum (FBS) and seeded into culture plates. SGC culture was seeded into plastic culture plates and neurons in the culture were removed by splitting and washing. Agonists (10 μM CNO to Plp1; hM3Dq sample or 50 ng/mL BDNF to wildtype sample) were added to SGC culture for overnight and culture medium was collected as conditional medium (CM). For some of the BDNF-treated samples, lentiviral particles that carried either scramble sequence (C-LT: Forward: CGC GTA TAC CCC GTC CAA GCC TCG CAT TGA ATT CAA GAG ATT CAA TGC GAG GCT TGG ACT TTT TGG AAA T; Reserve: CGA TTT CCA AAA AGT CCA AGC CTC GCA TTG AAT CTC TTG AAT TCA ATG CGA GGC TTG GAC GGG GTA TA) or shRNA of TrkB.T1 (T1shLT: Forward: CGC GTA TAC CCC CAT AAG ATC CCC CTG GAT GTT CAA GAG ACA TCC AGG GGG ATC TTA TGT TTT TGG AAA T; Reverse: CGA TTT CCA AAA ACA TAA GAT CCC CCT GGA TGT CTC TTG AAC ATC CAG GGG GAT CTT ATG GGG GTA TA) were added to SGC culture one day prior to BDNF treatment. For slot blot, culture medium or cell homogenates were subject to slot blot apparatus (Bio-Rad) followed by standard immunoblotting procedures. DRG single neuron culture or SGC-neuron unit culture were seeded into Poly-L-Lysine (0.01% overnight) coated glass-bottom 96 wells plates, or onto coverslips for live cell optical imaging.
Immunohistochemistry
After fixation, DRGs and the spinal cord were incubated in 25% sucrose overnight at 4°C for cryoprotection. DRGs were sectioned at 12 μm thickness and the spinal cord at 20 μm thickness. The tissue sections were processed for on-slide immunostaining. Sections were incubated in primary antibodies diluted in PBST (0.3% Triton X-100 in 0.1 M PBS, pH 7.4) containing 5% normal donkey serum overnight at room temperature followed by incubation with fluorescence-conjugated species-specific secondary antibody for 2 h. The primary antibodies used were rabbit anti-Piezo2 (1:500, Novus Biologicals LLC, Cat# NBP1-78624), rabbit pAb to CGRP (1:1000, Abcam, Cat# AB47027), goat pAb to CGRP (1:2000, Abcam, Cat# AB36001), rabbit anti-p-CREB (1:500, Cell Signaling, Cat# 9198), rabbit anti-p-NMDA receptor (1:500, Cell Signaling, Cat#5355S), and rabbit anti-TRPV1 (1:1000, Millipore Sigma, Cat# SAB5700857). The secondary antibodies used were donkey anti rabbit (Cy3) (1:500, Jackson Immuno research, Cat# 711-165-152), donkey anti-goat 594 (1:500, Life Technologies, Cat# A11058), donkey anti-rabbit 594 (1:500, Life Technologies, A21207), and AMCA-conjugated AffiniPure donkey anti-goat (1:200, Jackson ImmunoResearch, Cat# 705-155-003). Immunostaining in the absence of primary or secondary antibody was assessed for background evaluation. The specificity of the primary antibodies used were validated in our previous studies30,33,67 or by manufactures. Slides were coverslipped with Citifluor (Citifluor Ltd., London). The processed sections were visualized under a Nikon or Zeiss fluorescent microscope.
Single cell calcium or voltage imaging
Cells were fasted for 2–4 h prior to imaging. For calcium imaging, cells were loaded with Fluo4-AM (2 μM) or Rhod-2 (2 μM) 30 min prior to stimulation with desired agonists including CNO (10 μM), C21 (1 mM), SGC conditional medium, or BDNF (10 ng/mL). Intracellular calcium levels were recorded by time-lapse capturing of dye intensity at a frame rate of 1 frame/per second (s). For voltage imaging, DRG neurons were pre-loaded with voltage sensor Di-8-ANEPPS (10 μM, Invitrogen) for 20 min and subjected to mechanical stimulation via glass micropipette poking driven by a piezo-driven micromanipulator with the polished glass microprobe tip toward the DRG neuron surface at an 80-degree angle. The voltage changes were detected by alteration in fluorescence intensity that was recorded at a frame rate of 1 kHz (1 frame/per ms).30
Total RNA extraction and quantitative real-time PCR
The RNAqueous Total RNA Isolation Kit (Thermo Fisher Scientific) was used to extract RNA from DRGs followed by reverse transcription using a cDNA synthesis kit High Capacity cDNA Reverse Transcription (Applied Biosystems). Quantitative real-time PCR (qPCR) was then performed using SYBR Green as indicator on StepOnePlus Systems (Applied Biosystems). Dissociation curve post-PCR reaction was monitored to verify the specificity of the qPCR reactions. The level of target mRNA was normalized against the expression of the internal control β-actin and was calculated with ΔCt method and expressed as fold changes (2−ΔΔCt fold). The primers used for qPCR was for Piezo2, CGRP, TRPV1, TNFα, and IL-6 as genes of interest. These primers are Piezo2: GTGGTATGCAACCCAGTACCC and GGCCATTCTCTATGGGCAGG; CGRP: GGACTTGGAGACAAACCACCA and GAGAGCAACCAGAGAGGAACTACA; TRPV1: CCGGCTTTTTGGGAAGGGT and GAGACAGGTAGGTCCATCCAC; TNFα: CAGGCGGTGCCTATGTCTC and CGATCACCCCG AAGTTCAGTAG; β-actin: GGCTGTATTCCCCTCCATCG and CCAGTTGGTAACAATGCCATGT.
Colonometry and colonometrogram analysis
Colonic mechanical sensitivity in free-moving mice was measured by colonometry.30,31 Specially, a three-way connector was used to couple a syringe infusion pump, a pressure transducer, and a polyethylene (PE)-50 intracolonic catheter that was inserted into the distal colon with the catheter tip 2.5 cm away from the anus. A rate of 1.2 mL/h saline infusion was used throughout for both male and female mice. The pressure transducer was connected to a bridge amplifier and a computer recording system (AD Instrument, Milford, MA) for data acquisition and analysis via LabChart 8 Pro and Reader. The colonic mechanosensitivity was presented by changes in the amplitude of intracolonic pressures (AICP) as results of colonic stretch-reflex contraction.
Somatic mechanical and thermal sensitivity assays
Mice were placed individually into plexiglass chambers placed above a mesh stand (IITC Life Science Inc., CA) to allow for acclimation to the environment for 30 min. When animal had all four paws resting on the floor, a von Frey filament was applied perpendicularly to the plantar surface of the hindpaw from underneath of the mesh floor. The mice were each consecutively tested for a filament size, and this was repeated five times before moving on to a filament size of higher force. The tests were blinded to minimize bias. A positive response was considered when a withdrawal behavior (paw licking, shaking, or withdrawal) was noted during or immediately after application. Three positive responses out of the five stimulations were considered as painful responses. Pain threshold was determined by the weight of von Frey filament that evoked three positive responses.
Thermal sensitivity was measured by a hot plate assay. Mice were placed in a clear plexiglass cylinder placed on a temperature-controlled metal plate heated to 52°C. Acute nociception was measured in terms of latency of the animal to show signs of and/or either lifting or licking of the hindpaw, at which point the animal was removed. Each test was not exceeded beyond 30 s to avoid tissue damage.
Gait assay and stride analysis
The hindpaws of mice were gently brushed with water-based dyes and animals were dropped into one end of a chamber with a dimension of 50 cm (L) × 5.5 cm (W) × 15.5 cm (H) for freely walking toward the other end. The paw prints were captured by inks onto regular printing papers placed underneath the chamber. The stride length was defined as distance between the adjacent same toe prints. The stride length from the same animal was averaged as one number to be included in the statistical analysis. All procedures and analysis styles stayed the same among all experimental animals.
In situ hybridization
DRG sections (10 mm thickness) were mounted onto poly-L-lysine-coated slides and postfixed in 4% paraformaldehyde for 10 min. After treatment with acetic anhydride (0.25% v/v) and proteinase K (10 μg/mL) and subsequent dehydration in a graded series of ethanol (70% for 1 min, 80% for 1 min, 95% for 1 min × 2, 100% for 1 min × 2), tissues were incubated in buffer containing 50% formamide, 5xSSC, 500 μg/mL yeast tRNA, 0.1% Tween 20 (pH = 6.0 adjusted by citric acid) and RNA probes that were generated by in vitro transcription. The primers for PCR of cDNA of interests as templates for generating RNA probes were that TrkB.T1 Forward GCCGAGCTCCGCCAGTCTGTTCCTTCTGT and Reverse CGGGGTACCACAGTG GGTCAACAAGCCAA. PCR was performed using a DIG Probe Synthesis Kit (Roche Diagnostics). PCR products were incorporated into cloning vector pSP73 (Promega Corporation) for cDNA cloning and amplification, and in vitro transcription. After hybridization, the slides were processed with anti-DIG antibody.
Flow cytometry
DRGs were digested in digestion media (DMEM, 10 mM HEPES, 5 mg/mL BSA, 100 μg/mL DNase 1) containing 1.6 mg/mL collagenase Type 4 for 30 min at 37°C. After digestion, cells were filtered through a 40 μm strainer, centrifuged, and washed with FACS buffer (10% fetal bovine serum (FBS), 1 mM EDTA in PBS). Following resuspension in FACS buffer, dead cells were excluded by using live/dead cells labeling Zombie aqua kit (Biolegend # 423101) as described previously.30 DRGs from Plp1; hM3Dq mice were directly applied for cell sorting and the amount of SGCs were detected by transgenetically expressed mCherry. Cell sorting was performed on a BD Fortessa cell analyzer followed by data analysis using FlowJo software.
Hematoxylin and Eosin (H&E) stain
The distal colon (10 mm thickness) and urinary bladder (10 μm thickness) were sectioned transversely and the plantar tissue of the hindpaw (7 μm) was sectioned sagittally to include all layers. Tissues were fixed with 4% paraformaldehyde at room temperature for 30 min. Slides were stained with an H&E staining kit according to the protocol provided by the manufacture (Richard-AllanScientific, Kalamazoo, MI). The sections were examined with a Nikon brightfield microscope.
QUANTIFICATION AND STATISTICAL ANALYSIS
Immunostaining
The positively stained neurons in DRG that showed visible nucleus were measured for diameters and counted with a built-in software in a blind fashion to minimize any potential bias. Data from multiple sections of a given DRG were pooled and averaged as one data point. The section areas that contained neuronal soma, avoiding the areas that had extensive nerve fibers, were measured for normalizing the expression levels of protein of interest, which was presented as number of positive neurons per unit area. To avoid double counting, we chose every third section in the serial cutting for each specific antibody analysis. The positively stained neurons in the spinal cord were counted and presented as number of neurons per section. Data from all spinal cord sections for a protein of interest from a single animal were averaged and presented as one data point.
Data and statistical analysis
We used Imaging J, LabChart 8, FlowJo, Zeiss ZEN pro, Nikon NIS-ELEMENTS-BR, and NIS-ELEMENTS-ADVANCED for data analysis. We used GraphPad Prism 9 for statistical analysis. The results from each study were presented as mean ± SEM. One-way ANOVA, two-way ANOVA, or t test were performed according to specific experimental design and the number of variables. Detailed statistical approaches were described in Figure legends. Differences between means at a level of p ≤ 0.05 were considered to be significant.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| PIEZO2 Antibody | Novus Biologicals | Cat# NBP1-78624; RRID:AB_11005294 |
| Anti-CGRP antibody | Abcam | Cat# AB47027; RRID:AB_1141573 |
| Anti-CGRP antibody | Abcam | Cat# AB36001; RRID:AB_725807 |
| Phospho-CREB (Ser133) (87G3) Rabbit mAb | Cell Signaling Technology | Cat# 9198; RRID:AB_2561044 |
| Phospho-NMDAR2B (Ser1284) Antibody | Cell Signaling Technology | Cat# 5355; RRID:AB_10922589 |
| Anti-TRPV1 (VR1) Antibody | Alomone Labs | Cat# ACC-030; RRID:AB_2313819 |
| Cy™3 AffiniPure™ Donkey Anti-Rabbit IgG (H + L) | Jackson ImmunoResearch Labs | Cat# 711-165-152; RRID:AB_2307443 |
| Donkey anti-Goat IgG, Alexa Fluor™ 594 | Thermo Fisher Scientific | Cat# A-11058; RRID:AB_2534105 |
| AMCA-AffiniPure Donkey Anti-Goat IgG (H + L) | Jackson ImmunoResearch Labs | Cat# 705-155-003; RRID:AB_2340408 |
| mCherry (E5D8F) Rabbit mAb | Cell Signaling Technology | Cat# 43590; RRID:AB_2799246 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Tamoxifen | Sigma-Aldrich | Cat#T5648 |
| Poly-L-Lysine | Millipore Sigma | Cat#P6282 |
| BDNF | Thermo Fisher Scientific | Cat#PHC7074 |
| Clozapine-N-oxide | Tocris Bioscience | Cat#4936 |
| Fluo-4, AM | Invitrogen | Cat#F14201 |
| Rhod-2, AM | Invitrogen | Cat#R1244 |
| Di-8-ANEPPS | Thermo Fisher Scientific | Cat#D3167 |
| TNBS | Sigma-Aldrich | Cat#92822 |
| Tumor Necrosis Factor-α human | Sigma-Aldrich | Cat#H8916 |
|
| ||
| Critical commercial assays | ||
|
| ||
| H&E staining kit | Epredia | Cat#999001 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6J mice | Jackson Lab | Stock # 000664 |
| Plp1-CreERT mice | Jackson Lab | Stock # 005975 |
| RC::L-hM3Dq mice | Jackson Lab | Stock # 026943 |
| GFAP-Cre mice | Jackson Lab | Stock #012886 |
| Ai32 mice | Jackson Lab | Stock #012569 |
| PC-G5-tdT mice | Jackson Lab | Stock # 024477 |
| Piezo2-EGFP-IRES-Cre mice | Jackson Lab | Stock# 027719 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Piezo2 qPCR | Integrated DNA Technologies, Inc. | GTGGTATGCAACCCAGTACCC and GGCCATTCTCTATGGGCAGG |
| CGRPqPCR | Integrated DNA Technologies, Inc. | GGACTTGGAGACAAACCACCA and GAGAGCAACCAGAGAGGAACTACA |
| TRPV1 qPCR | Integrated DNA Technologies, Inc. | CCGGCTTTTTGGGAAGGGT and GAGACAGGTAGGTCCATCCAC |
| TNFα qPCR | Integrated DNA Technologies, Inc. | CAGGCGGTGCCTATGTCTC and CGATCACCCCGAAGTTCAGTAG |
| Beta actin qPCR | Integrated DNA Technologies, Inc. | GGCTGTATTCCCCTCCATCG and CCAGTTGGTAACAATGCCATGT |
| Nav1.8-Cre genotyping | Integrated DNA Technologies, Inc. | 13Salt: GGAATGGGATGGAGCTTCTTAC; 12A: TTACCCGGTGTGTGCTGTAGAAAG; CRE 5a: CAAATGTTGCTGGATAGTT TTTACTGCC. |
| floxed TrkB.T1 | Integrated DNA Technologies, Inc. | TrkBT1-5s: 5’-CCAGCTATTGAGTAATGAATGAGTC-3’; TrkBT1-2: 5’-CTACCCATCCAGTGGGATCTT-3’; and TrkBT1-4a: 5’-CCACCGCGGTGGTACCATAACTTCG-3’. |
| TrkB.T1 shRNA | Integrated DNA Technologies, Inc. | Forward: CGC GTA TAC CCC CAT AAG ATC CCC CTG GAT GTT CAA GAG ACA TCC AGG GGG ATC TTA TGT TTT TGG AAA T; Reverse: CGA TTT CCA AAA ACA TAA GAT CCC CCT GGA TGT CTC TTG AAC ATC CAG GGG GAT CTT ATG GGG GTA TA |
| shRNA scramble sequence | Integrated DNA Technologies, Inc. | CGC GTA TAC CCC GTC CAA GCC TCG CAT TGA ATT CAA GAG ATT CAA TGC GAG GCT TGG ACT TTT TGG AAA T; Reserve: CGA TTT CCA AAA AGT CCA AGC CTC GCA TTG AAT CTC TTG AAT TCA ATG CGA GGC TTG GAC GGG GTA TA |
| TrkB.T1 RNA probe for In situ hybridization | Integrated DNA Technologies, Inc. | Forward GCCGAGCTCCGCCAGTCTGTTCCTTCTGT and Reverse CGGGGTACCACAGTGGGTCAACAAGCCAA. |
|
| ||
| Software and algorithms | ||
|
| ||
| GraphPad Prism 9 | GraphStats | https://www.graphpad.com/ |
| ImageJ/FIJI | Public software | https://imagej.nih.gov/ij/ |
| FlowJo v10.6.2 | BD Biosciences | https://www.flowjo.com/solutions/flowjo/downloads/previous-versions |
| LabChart 8 | ADInstruments | https://www.adinstruments.com/ |
| NIS Elements Advanced Version 5.30 | Nikon Instruments Inc. | https://www.microscope.healthcare.nikon.com/ |
| Imager.Z1 with ZEN pro software | Zeiss | https://www.zeiss.com/microscopy/en/products/software/zeiss-zen.html |
Highlights.
Activation of SGCs facilitates colonic and hindpaw mechanical hypersensitivity
SGC inhibition attenuates, in part, chronic colonic and somatic mechanical pain
A paracrine action from SGCs to sensory neurons upregulates Piezo2 and CGRP
SGC regulation involves neurogenic inflammation and spinal central sensitization
ACKNOWLEDGMENTS
This study was supported by grants NIH R01 DK118137 (L.Y.Q.) and R01 DK 121131 (L.Y.Q.). Cell sorting was performed at the Virginia Commonwealth University Flow Cytometry Shared Resource, supported, in part, with funding from NIH-NCI Cancer Center Support Grant P30 CA016059. We thank Dr. Lino Tessarollo for providing TrkB.T1 KO mice and floxed TrkB.T1 mice.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114230.
REFERENCES
- 1.Pannese E (1964). Number and Structure of Perisomatic Satellite Cells of Spinal Ganglia under Normal Conditions or during Axon Regeneration and Neuronal Hypertrophy. Z. Zellforsch. Mikrosk. Anat. 63, 568–592. 10.1007/BF00339491. [DOI] [PubMed] [Google Scholar]
- 2.Pannese E (1956). [Research on the morphology of the perineuronal satellite cells in the spinal and sympathetic ganglia of mammals. I. Phasecontrast findings]. Boll. Soc. Ital. Biol. Sper. 32, 72–74. [PubMed] [Google Scholar]
- 3.Hanani M, and Spray DC (2020). Emerging importance of satellite glia in nervous system function and dysfunction. Nat. Rev. Neurosci. 21, 485–498. 10.1038/s41583-020-0333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pannese E (2010). The structure of the perineuronal sheath of satellite glial cells (SGCs) in sensory ganglia. Neuron Glia Biol. 6, 3–10. . [DOI] [PubMed] [Google Scholar]
- 5.Xia CM, Colomb DG Jr., Akbarali HI, and Qiao LY (2011). Prolonged sympathetic innervation of sensory neurons in rat thoracolumbar dorsal root ganglia during chronic colitis. Neuro Gastroenterol. Motil. 23, 801–e339. 10.1111/j.1365-2982.2011.01728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mapps AA, Thomsen MB, Boehm E, Zhao H, Hattar S, and Kuruvilla R (2022). Diversity of satellite glia in sympathetic and sensory ganglia. Cell Rep. 38, 110328. 10.1016/j.celrep.2022.110328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohara PT, Vit JP, Bhargava A, Romero M, Sundberg C, Charles AC, and Jasmin L (2009). Gliopathic Pain: When Satellite Glial Cells Go Bad. Neuroscientist 15, 450–463. 10.1177/1073858409336094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rozanski GM, Nath AR, Adams ME, and Stanley EF (2013). Low voltage-activated calcium channels gate transmitter release at the dorsal root ganglion sandwich synapse. J. Physiol. 591, 5575–5583. 10.1113/jphysiol.2013.260281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suadicani SO, Cherkas PS, Zuckerman J, Smith DN, Spray DC, and Hanani M (2010). Bidirectional calcium signaling between satellite glial cells and neurons in cultured mouse trigeminal ganglia. Neuron Glia Biol. 6, 43–51. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Z, Zhang C, Song X, Cui X, Liu J, Ford NC, He S, Zhu G, Dong X, Hanani M, and Guan Y (2022). BzATP Activates Satellite Glial Cells and Increases the Excitability of Dorsal Root Ganglia Neurons In Vivo. Cells-Basel 11. 10.3390/cells11152280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang TY, Belzer V, and Hanani M (2010). Gap junctions in dorsal root ganglia: Possible contribution to visceral pain. Eur. J. Pain 14, 49.e1–57. 10.1016/j.ejpain.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 12.Hanani M (2015). Role of satellite glial cells in gastrointestinal pain. Front. Cell. Neurosci. 9, 412. 10.3389/fncel.2015.00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Warwick RA, and Hanani M (2013). The contribution of satellite glial cells to chemotherapy-induced neuropathic pain. Eur. J. Pain 17, 571–580. 10.1002/j.1532-2149.2012.00219.x. [DOI] [PubMed] [Google Scholar]
- 14.Cherkas PS, Huang TY, Pannicke T, Tal M, Reichenbach A, and Hanani M (2004). The effects of axotomy on neurons and satellite glial cells in mouse trigeminal ganglion. Pain 110, 290–298. 10.1016/j.pain.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 15.Shen S, Tiwari N, Madar J, Mehta P, and Qiao LY (2022). Beta 2-adrenergic receptor mediates noradrenergic action to induce cyclic adenosine monophosphate response element-binding protein phosphorylation in satellite glial cells of dorsal root ganglia to regulate visceral hypersensitivity. Pain 163, 180–192. 10.1097/j.pain.0000000000002330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim YS, Anderson M, Park K, Zheng Q, Agarwal A, Gong C, Saijilafu, Young L., He S., LaVinka PC., et al. (2016). Coupled Activation of Primary Sensory Neurons Contributes to Chronic Pain. Neuron 91, 1085–1096. 10.1016/j.neuron.2016.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGinnis A, and Ji RR (2023). The Similar and Distinct Roles of Satellite Glial Cells and Spinal Astrocytes in Neuropathic Pain. Cells 12, 965. 10.3390/cells12060965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamakita S, Horii Y, Takemura H, Matsuoka Y, Yamashita A, Yamaguchi Y, Matsuda M, Sawa T, and Amaya F (2018). Synergistic activation of ERK1/2 between A-fiber neurons and glial cells in the DRG contributes to pain hypersensitivity after tissue injury. Mol. Pain 14, 1744806918767508. 10.1177/1744806918767508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohara PT, Vit JP, Bhargava A, and Jasmin L (2008). Evidence for a role of connexin 43 in trigeminal pain using RNA interference in vivo. J. Neurophysiol. 100, 3064–3073. 10.1152/jn.90722.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Avraham O, Deng PY, Jones S, Kuruvilla R, Semenkovich CF, Klyachko VA, and Cavalli V (2020). Satellite glial cells promote regenerative growth in sensory neurons. Nat. Commun. 11, 4891. 10.1038/s41467-020-18642-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hall AK, and Landis SC (1992). Division and migration of satellite glia in the embryonic rat superior cervical ganglion. J. Neurocytol. 21, 635–647. 10.1007/bf01191725. [DOI] [PubMed] [Google Scholar]
- 22.Rao M, Nelms BD, Dong L, Salinas-Rios V, Rutlin M, Gershon MD, and Corfas G (2015). Enteric glia express proteolipid protein 1 and are a transcriptionally unique population of glia in the mammalian nervous system. Glia 63, 2040–2057. 10.1002/glia.22876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nguyen MQ, von Buchholtz LJ, Reker AN, Ryba NJ, and Davidson S (2021). Single-nucleus transcriptomic analysis of human dorsal root ganglion neurons. Elife 10, e71752. 10.7554/eLife.71752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abdo H, Calvo-Enrique L, Lopez JM, Song J, Zhang MD, Usoskin D, El Manira A, Adameyko I, Hjerling-Leffler J, and Ernfors P (2019). Specialized cutaneous Schwann cells initiate pain sensation. Science 365, 695–699. 10.1126/science.aax6452. [DOI] [PubMed] [Google Scholar]
- 25.Rao M, Rastelli D, Dong L, Chiu S, Setlik W, Gershon MD, and Corfas G (2017). Enteric Glia Regulate Gastrointestinal Motility but Are Not Required for Maintenance of the Epithelium in Mice. Gastroenterology 153, 1068–1081.e7. 10.1053/j.gastro.2017.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patyal P, Fil D, and Wight PA (2023). in the enteric nervous system is preferentially expressed during early postnatal development in mouse as DM20, whose expression appears reliant on an intronic enhancer. Front. Cell. Neurosci. 17, 1175614. 10.3389/fncel.2023.1175614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie AX, Madayag A, Minton SK, McCarthy KD, and Malykhina AP (2020). Sensory satellite glial Gq-GPCR activation alleviates inflammatory pain via peripheral adenosine 1 receptor activation. Sci. Rep. 10, 14181. 10.1038/s41598-020-71073-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu Q, Ford NC, He S, Huang Q, Anderson M, Chen Z, Yang F, Crawford LK, Caterina MJ, Guan Y, and Dong X (2021). Astrocytes contribute to pain gating in the spinal cord. Sci. Adv. 7, eabi6287. 10.1126/sciadv.abi6287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nam Y, Kim JH, Kim JH, Jha MK, Jung JY, Lee MG, Choi IS, Jang IS, Lim DG, Hwang SH, et al. (2016). Reversible Induction of Pain Hypersensitivity following Optogenetic Stimulation of Spinal Astrocytes. Cell Rep. 17, 3049–3061. 10.1016/j.celrep.2016. 11.043. [DOI] [PubMed] [Google Scholar]
- 30.Madar J, Tiwari N, Smith C, Sharma D, Shen S, Elmahdi A, and Qiao LY (2023). Piezo2 regulates colonic mechanical sensitivity in a sex specific manner in mice. Nat. Commun. 14, 2158. 10.1038/s41467-023-37683-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qiao LY, and Madar J (2021). An objective approach to assess colonic pain in mice using colonometry. PLoS One 16, e0245410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iyengar S, Ossipov MH, and Johnson KW (2017). The role of calcitonin gene-related peptide in peripheral and central pain mechanisms including migraine. Pain 158, 543–559. 10.1097/j.pain.0000000000000831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kay JC, Xia CM, Liu M, Shen S, Yu SJ, Chung C, and Qiao LY (2013). Endogenous PI3K/Akt and NMDAR act independently in the regulation of CREB activity in lumbosacral spinal cord in cystitis. Exp. Neurol. 250, 366–375. 10.1016/j.expneurol.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang X, Chen Y, Wang C, and Huang LYM (2007). Neuronal somatic ATP release triggers neuron-satellite glial cell communication in dorsal root ganglia. Proc. Natl. Acad. Sci. USA 104, 9864–9869. 10.1073/pnas.0611048104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sikandar S, Minett MS, Millet Q, Santana-Varela S, Lau J, Wood JN, and Zhao J (2018). Brain-derived neurotrophic factor derived from sensory neurons plays a critical role in chronic pain. Brain 141, 1028–1039. 10.1093/brain/awy009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Obata K, and Noguchi K (2006). BDNF in sensory neurons and chronic pain. Neurosci. Res. 55, 1–10. 10.1016/j.neures.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 37.Cappoli N, Tabolacci E, Aceto P, and Dello Russo C (2020). The emerging role of the BDNF-TrkB signaling pathway in the modulation of pain perception. J. Neuroimmunol. 349, 577406. 10.1016/j.jneuroim.2020.577406. [DOI] [PubMed] [Google Scholar]
- 38.Yu SJ, Grider JR, Gulick MA, Xia CM, Shen S, and Qiao LY (2012). Up-regulation of brain-derived neurotrophic factor is regulated by extracellular signal-regulated protein kinase 5 and by nerve growth factor retrograde signaling in colonic afferent neurons in colitis. Exp. Neurol. 238, 209–217. 10.1016/j.expneurol.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lamb K, Zhong F, Gebhart GF, and Bielefeldt K (2006). Experimental colitis in mice and sensitization of converging visceral and somatic afferent pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G451–G457. 10.1152/ajpgi.00353.2005. [DOI] [PubMed] [Google Scholar]
- 40.Murthy SE, Loud MC, Daou I, Marshall KL, Schwaller F, Kühnemund J, Francisco AG, Keenan WT, Dubin AE, Lewin GR, and Patapoutian A (2018). The mechanosensitive ion channel Piezo2 mediates sensitivity to mechanical pain in mice. Sci. Transl. Med. 10, eaat9897. 10.1126/scitranslmed.aat9897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu J, McGinnis A, and Ji RR (2023). Piezo2 mediates visceral mechanosensation: A new therapeutic target for gut pain? Neuron 111, 450–452. 10.1016/j.neuron.2023.01.011. [DOI] [PubMed] [Google Scholar]
- 42.Xie Z, Feng J, Hibberd TJ, Chen BN, Zhao Y, Zang K, Hu X, Yang X, Chen L, Brookes SJ, et al. (2023). Piezo2 channels expressed by colon-innervating TRPV1-lineage neurons mediate visceral mechanical hypersensitivity. Neuron 111, 526–538.e4. 10.1016/j.neuron.2022.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eijkelkamp N, Linley JE, Torres JM, Bee L, Dickenson AH, Gringhuis M, Minett MS, Hong GS, Lee E, Oh U, et al. (2013). A role for Piezo2 in EPAC1-dependent mechanical allodynia. Nat. Commun. 4, 1682. 10.1038/ncomms2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Szczot M, Liljencrantz J, Ghitani N, Barik A, Lam R, Thompson JH, Bharucha-Goebel D, Saade D, Necaise A, Donkervoort S, et al. (2018). PIEZO2 mediates injury-induced tactile pain in mice and humans. Sci. Transl. Med. 10, eaat9892. 10.1126/scitranslmed.aat9892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raper J, Morrison RD, Daniels JS, Howell L, Bachevalier J, Wichmann T, and Galvan A (2017). Metabolism and Distribution of Clozapine-N-oxide: Implications for Nonhuman Primate Chemogenetics. ACS Chem. Neurosci. 8, 1570–1576. 10.1021/acschemneuro.7b00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hanani M (2005). Satellite glial cells in sensory ganglia: from form to function. Brain research. Brain Res. Rev. 48, 457–476. 10.1016/j.brainresrev.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 47.Pannese E (1981). The satellite cells of the sensory ganglia. Adv. Anat. Embryol. Cell Biol. 65, 1–111. [DOI] [PubMed] [Google Scholar]
- 48.Qiao LY, and Tiwari N (2020). Spinal neuron-glia-immune interaction in cross-organ sensitization. Am. J. Physiol. Gastrointest. Liver Physiol. 319, G748–G760. 10.1152/ajpgi.00323.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cao T, Matyas JJ, Renn CL, Faden AI, Dorsey SG, and Wu J (2020). Function and Mechanisms of Truncated BDNF Receptor TrkB.T1 in Neuropathic Pain. Cells 9, 1194. 10.3390/cells9051194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei X, Wang L, Hua J, Jin XH, Ji F, Peng K, Zhou B, Yang J, and Meng XW (2021). Inhibiting BDNF/TrkB.T1 receptor improves resiniferatoxin-induced postherpetic neuralgia through decreasing ASIC3 signaling in dorsal root ganglia. J. Neuroinflammation 18, 96. 10.1186/s12974-021-02148-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Woo SH, Lukacs V, de Nooij JC, Zaytseva D, Criddle CR, Francisco A, Jessell TM, Wilkinson KA, and Patapoutian A (2015). Piezo2 is the principal mechanotransduction channel for proprioception. Nat. Neurosci. 18, 1756–1762. 10.1038/nn.4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ranade SS, Woo SH, Dubin AE, Moshourab RA, Wetzel C, Petrus M, Mathur J, Bégay V, Coste B, Mainquist J, et al. (2014). Piezo2 is the major transducer of mechanical forces for touch sensation in mice. Nature 516, 121–125. 10.1038/nature13980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lewis AH, Cui AF, McDonald MF, and Grandl J (2017). Transduction of Repetitive Mechanical Stimuli by Piezo1 and Piezo2 Ion Channels. Cell Rep. 19, 2572–2585. 10.1016/j.celrep.2017.05.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu J, Lewis AH, and Grandl J (2017). Touch, Tension, and Transduction - The Function and Regulation of Piezo Ion Channels. Trends Biochem. Sci. 42, 57–71. 10.1016/j.tibs.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Woo SH, Ranade S, Weyer AD, Dubin AE, Baba Y, Qiu Z, Petrus M, Miyamoto T, Reddy K, Lumpkin EA, et al. (2014). Piezo2 is required for Merkel-cell mechanotransduction. Nature 509, 622–626. 10.1038/nature13251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shin SM, Moehring F, Itson-Zoske B, Fan F, Stucky CL, Hogan QH, and Yu H (2021). Piezo2 mechanosensitive ion channel is located to sensory neurons and nonneuronal cells in rat peripheral sensory pathway: implications in pain. Pain 162, 2750–2768. 10.1097/j.pain.0000000000002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Usoskin D, Furlan A, Islam S, Abdo H, Lönnerberg P, Lou D, Hjerling-Leffler J, Haeggström J, Kharchenko O, Kharchenko PV, et al. (2015). Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 18, 145–153. 10.1038/nn.3881. [DOI] [PubMed] [Google Scholar]
- 58.Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, Dubin AE, and Patapoutian A (2010). Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 330, 55–60. 10.1126/science.1193270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Szczot M, Pogorzala LA, Solinski HJ, Young L, Yee P, Le Pichon CE, Chesler AT, and Hoon MA (2017). Cell-Type-Specific Splicing of Piezo2 Regulates Mechanotransduction. Cell Rep. 21, 2760–2771. 10.1016/j.celrep.2017.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Benemei S, Nicoletti P, Capone JG, and Geppetti P (2009). CGRP receptors in the control of pain and inflammation. Curr. Opin. Pharmacol. 9, 9–14. 10.1016/j.coph.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 61.Hanani M (2010). Satellite glial cells in sympathetic and parasympathetic ganglia: in search of function. Brain Res. Rev. 64, 304–327. 10.1016/j.brainresrev.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 62.Stobart JL, Ferrari KD, Barrett MJP, Glück C, Stobart MJ, Zuend M, and Weber B (2018). Cortical Circuit Activity Evokes Rapid Astrocyte Calcium Signals on a Similar Timescale to Neurons. Neuron 98, 726–735.e4. 10.1016/j.neuron.2018.03.050. [DOI] [PubMed] [Google Scholar]
- 63.Fiacco TA, and McCarthy KD (2018). Multiple Lines of Evidence Indicate That Gliotransmission Does Not Occur under Physiological Conditions. J. Neurosci. 38, 3–13. 10.1523/Jneurosci.0016-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Savtchouk I, and Volterra A (2018). Gliotransmission: Beyond Blackand-White. J. Neurosci. 38, 14–25. 10.1523/Jneurosci.0017-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stern P (2016). Glial cells contribute to pain. Science 354, 1114–1115. 10.1126/science.354.6316.1114-e. [DOI] [PubMed] [Google Scholar]
- 66.Chen L, Huang J, Zhao P, Persson AK, Dib-Hajj FB, Cheng X, Tan A, Waxman SG, and Dib-Hajj SD (2018). Conditional knockout of NaV1.6 in adult mice ameliorates neuropathic pain. Sci. Rep. 8, 3845. 10.1038/s41598-018-22216-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qiao LY, and Grider JR (2007). Up-regulation of calcitonin gene-related peptide and receptor tyrosine kinase TrkB in rat bladder afferent neurons following TNBS colitis. Exp. Neurol. 204, 667–679. 10.1016/j.expneurol.2006.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data that were used to generate graphs in the paper are available upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.