

Abstract
Intermolecular functionalization of tertiary C–H bonds to construct fully substituted stereogenic carbon centers represents a formidable challenge: without the assistance of directing groups, state-of-the-art catalysts struggle to introduce chirality to racemic tertiary sp3-carbon centers. Direct asymmetric functionalization of such centers is a worthy reactivity and selectivity goal for modern biocatalysis. Here we present an engineered nitrene transferase (P411-TEA-5274), derived from a bacterial cytochrome P450, that is capable of aminating tertiary C–H bonds to provide chiral α-tertiary primary amines with high efficiency (up to 2300 total turnovers) and selectivity (up to >99% enantiomeric excess (e.e.)). The construction of fully substituted stereocenters with methyl and ethyl groups underscores the enzyme’s remarkable selectivity. A comprehensive substrate scope study demonstrates the biocatalyst’s compatibility with diverse functional groups and tertiary C–H bonds. Mechanistic studies elucidate how active-site residues distinguish between the enantiomers and enable the enzyme to perform this transformation with excellent enantioselectivity.
Introduction
Direct enantioselective functionalization of C(sp3)–H bonds is an ideal approach to synthesize high value-added chiral molecules from the viewpoint of atom- and step-economy.1–5 In recent years, many powerful catalytic methods have been developed toward this goal, with significant advances made for enantioselective functionalization of secondary C–H bonds (Figure 1a).1–10 In contrast, intermolecular asymmetric functionalization of racemic tertiary C–H bonds, particularly those with a pKa > 25 and in the absence of directing groups, toward the formation of fully substituted sp3-carbon stereocenters remains unexplored in synthetic chemistry (Figure 1b). The challenges of this transformation include the difficulty of precisely recognizing three different substituents attached to a tertiary carbon center (Figure 1b), especially when those substituents have only subtle steric and/or electronic differences; the lack of an intermolecular counterpart to recent breakthroughs in intramolecular enantioselective tertiary C–H functionalizations;11–15 and, finally, potential steric clash between the catalyst and the tertiary carbon. Chemists often achieve precise facial recognition by strategically incorporating bulky substituents or functional groups into a chiral catalyst. However, when it comes to the functionalization of a tertiary C–H bond, the steric clash between the bulky catalyst and the crowded surrounding of a tertiary carbon inhibits the approach of the catalyst to the tertiary C–H bond. Consequently, a tradeoff arises between increasing the catalyst’s steric bulk to better control stereoselectivity and decreasing it to avoid a steric clash with the tertiary sp3-carbon surroundings (Figure 1b).
Figure 1. Direct enantioselective functionalization of C(sp3)–H bonds.
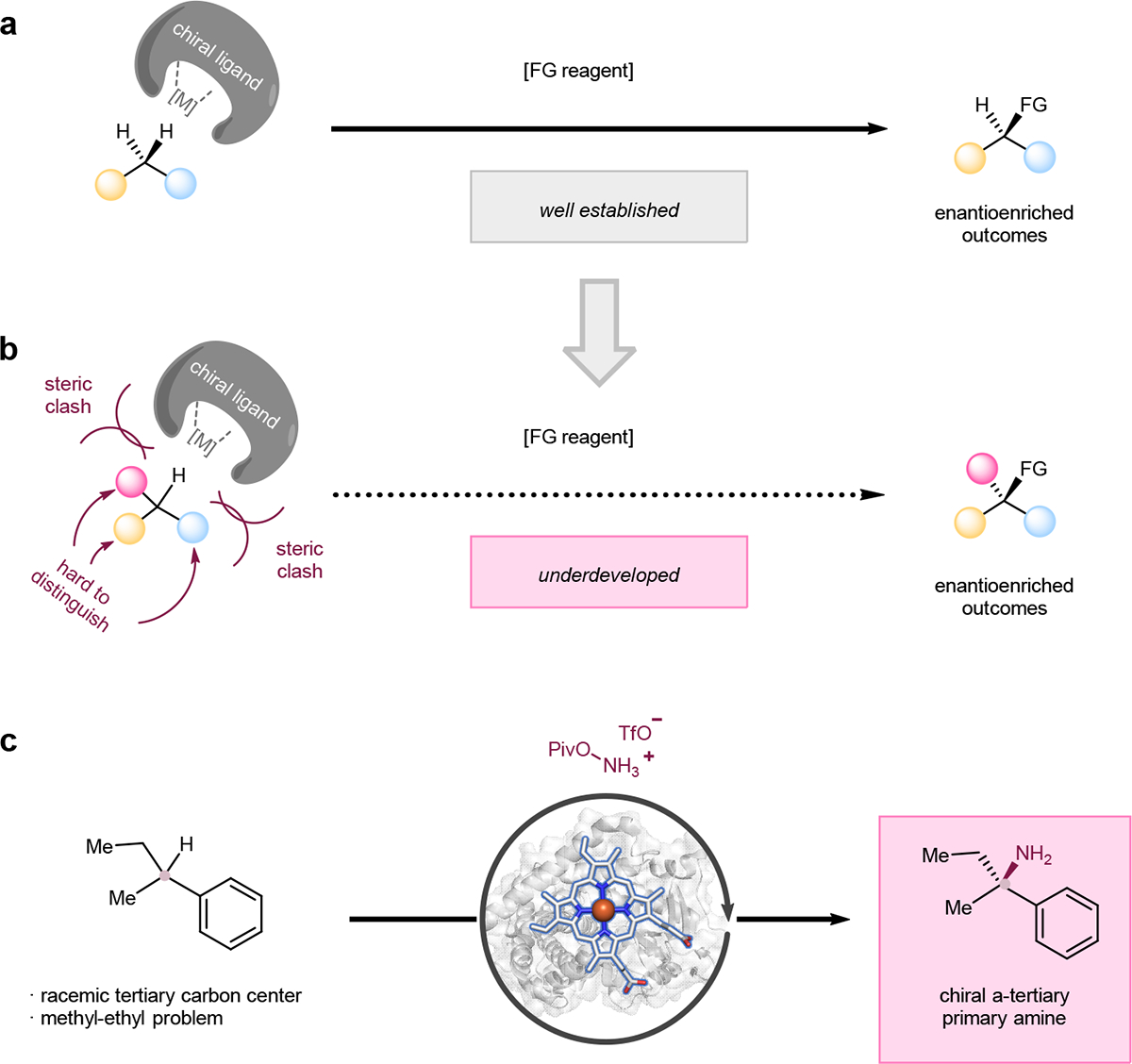
a, General scenario for intermolecular enantioselective functionalization of secondary carbon centers. Secondary carbons offer less steric hindrance, which facilitates the construction of chirality. This type of transformation has been well documented. b, General scenario for intermolecular enantioselective functionalization of tertiary carbon centers. Tertiary carbons that possess chiral centers are difficult for catalysts to approach and recognize. This type of transformation is rarely reported. c, This work: biocatalytic, enantioselective primary amination of tertiary C–H bonds. M, metal catalysts; FG, functional groups; Piv, pivaloyl.
Unlike small-molecule catalysts, enzymes have large, three-dimensional (3D) structures determined by the folding of their polypeptide chain(s). With precisely sculpted 3D structures, enzymes can differentiate steric and electronic nuances as subtle as those between methane and ethane molecules.16 With precise adjustments of their folded structures, enzymes can also achieve excellent stereoselectivity while maintaining high activity by avoiding severe steric clashes with bulky substrates.17 We thus consider enzyme catalysis to be a promising approach to tackle existing challenges in the enantioselective functionalization of tertiary C–H bonds.
Cytochromes P450 represent nature’s most prevalent catalysts for C–H functionalization,10 with enzymes from this vast family directly activating C–H bonds for a wide range of oxidative transformations.18 Furthermore, cytochromes P450 are excellent candidates for directed evolution and discovery of non-natural activities due to their structural flexibility and remarkable promiscuity.19 Several reports have demonstrated abiological C–H functionalizations achieved by expanding the catalytic repertoire of the iron-haem-containing cytochrome P450 family.8,9,20 The activity and selectivity of these biocatalytic C–H functionalizations frequently complement state-of-the-art methodologies based on small-molecule catalysts, making them valuable additions to the synthetic chemist’s toolbox.8,21 Motivated by these precedents, we sought to leverage these enzymes for enantioselective intermolecular functionalization of tertiary C–H bonds.
Given the widespread presence of α-tertiary amines in bioactive compounds,22 the initial focus was on the primary amination of tertiary C–H bonds. Despite the existence of many methods for synthesizing chiral α-tertiary amines with stoichiometric amounts of auxiliary reagents, such as Ellman’s sulfinamides,23,24 catalytic strategies for enantioselective conversion of substrates into chiral α-tertiary amines are rare,11–13,22,25 because classical methods for synthesizing chiral α-secondary amines, including asymmetric reduction of imines,26 biocatalytic transamination,27 and reductive amination,28 are not applicable to the synthesis of chiral α-tertiary amines due to the absence of protons in the fully substituted carbon center. From a retrosynthetic perspective, intermolecular amination of tertiary C–H bonds provides an ideal and straightforward method for the assembly of chiral α-tertiary amines (Figure 1c). The Arnold group recently reported primary amination of these tertiary C–H bonds, but all the products were non-chiral.29,30 Here we present a biocatalytic system that exhibits remarkable ability to aminate tertiary C–H bonds, providing high-value chiral α-tertiary primary amines with exceptional efficiency and selectivity.
Results
Initial Discovery and Evolution of Tertiary C–H Primary Aminase P411-TEA-5274.
We initiated the study by testing the coupling of sec-butylbenzene 1a and hydroxylamine ester 2a (Figure 2a) catalysed by an in-house collection of engineered cytochromes P411 (serine-ligated cytochrome P450 variants). These reagents were chosen for several reasons: First, compared to the elaborated starting materials used for many other tertiary C–H functionalizations, 1a is simple, inexpensive, and readily available.11–15 Second, the tertiary carbon center of 1a is racemic and attaches to a methyl and an ethyl substituent; distinguishing these is notoriously difficult in asymmetric catalysis.11,31,32 Last, hydroxylamine ester 2a has recently been shown to serve as a nitrene precursor for haemoprotein-catalysed secondary C–H primary amination;29 we wanted to expand that demonstration to primary amination of tertiary C–H bonds.
Figure 2. Directed evolution for enzymatic primary amination of tertiary C–H bonds.

a, Reaction conditions: 5 mM 1a, 10 mM 2a, E. coli whole cells harboring P411 variants (OD600 = 30) in M9-N aqueous buffer (pH 8.0), 5% (v/v) EtOH (co-solvent), 10 °C, anaerobic conditions, 20 h. b, Evolution trajectory of tertiary C–H primary aminase (P411-TEA) for the synthesis of α-tertiary primary amine 3a. c, The mutations (S72, M177, A327, M354, R395, A399, and Q403) that enhance activity or enantioselectivity are highlighted in the active site of closely related P411 variant E10 (PDB ID: 5UCW).35 d, Summary of beneficial mutations leading to P411-TEA-5274. TTN is defined as the (molar) amount of indicated product divided by the amount of haem protein in the reaction, as measured by the haemochrome assay (see Supplementary Method 7 for more details). TTN, total turnover number. Yields were calculated from HPLC calibration curves and the average of triplicate experiments (n = 3).
A panel of 48 cytochromes P411 previously engineered for nitrene transformations was screened in whole Escherichia coli cells against 1a and 2a under anaerobic conditions (Figure 2). The resulting reaction mixtures were monitored and analyzed after 20 h for the formation of α-tertiary primary amine 3a. Variant P411-TEA-5267 (TErtiary C–H Primary Aminase), which has three mutations (C324L, N395R, and G438V) with respect to P411BPA (previously engineered for benzylic C–H primary amination),29 produced the desired product 3a with a total turnover number (TTN) of 20 (Figure 2b; 1% yield, Supplementary Table 1). The primary amine 3a synthesized by P411-TEA-5267 was determined to have excellent stereopurity, with 90% e.e..
P411-TEA-5267 served as the starting point for directed evolution. Sequential rounds of site-saturation mutagenesis (SSM)33,34 and screening were performed to improve catalytic activity toward synthesis of 3a. We referred to the crystal structure of related P411 variant E1035 (haem domain only) and mainly targeted for mutagenesis amino acid residues proximal to the haem cofactor and/or residing on flexible loops, or those distal sites that have shown to play an important role in enhancing abiological carbene and nitrene transfer activities (Figure 2c).8 During each round of SSM, enzyme libraries were generated and screened for product formation in 96-well plates in the form of bacterial whole-cell catalysts. Three rounds of SSM and screening introduced mutations M354E, R395S, and A327V, leading to a three-fold improvement of the TTN from 20 to 64 (Figures 2b and 2d). Subsequent rounds of evolution introduced the M177Y and S72T mutations on the α-helices above the haem cofactor, as well as the A399G mutation on the loop directly below the haem cofactor. These mutations boosted the activity more than two-fold, reaching 140 TTN (Figure 2). The Q403A mutation located on the α-helix below the haem cofactor doubled the enzyme activity to 270 TTN (Figure 2). Interestingly, upon re-evaluation of position 395 in the loop beneath the haem cofactor, we found the S395V mutation increased the enzyme’s activity almost four-fold to 970 TTN (26% yield, Supplementary Table 2), culminating in the final variant, P411-TEA-5274 (Figure 2). In the standard conditions, an oxygen depletion system (catalase and glucose oxidase) was introduced to ensure a strict anaerobic environment. However, in the absence of this oxygen depletion system the yield and TTN only decrease slightly (entries 8 and 10, Supplementary Table 2), demonstrating the synthetic utility of this biotransformation. Notably, the enzyme’s enantioselectivity toward the formation of 3a also improved slightly during the evolution campaign, increasing from 90% e.e. (P411-TEA-5267, Figure 2d) to 92% e.e. (P411-TEA-5274, Figure 2d).
Substrate Scope Study of Tertiary C–H Primary Aminase P411-TEA-5274.
With C–H primary aminase P411-TEA-5274 in hand, we proceeded to explore its performance on various substrates (Figure 3). Representative substrates were designed with mixed kinds of C–H bonds (1a, Figure 3) and with diverse spatial hindrances (1a and 1c–1e, Figure 3), electronic effects (1f–1g, Figure 3), structures (1i–1j, Figure 3), and two different types of tertiary C–H bonds (1k–1l, Figure 3) to examine the impact of these factors on the activity and selectivity of the enzyme, as well as the enzyme’s regional selectivity. Importantly, we incorporated both methyl and ethyl groups at the tertiary carbon centers of most substrates (1a, 1c–1g and 1i–1l, Figure 3).11,31,32 These substrates allow us to assess the selectivity of the enzyme in conditions that present significant challenges for other systems.
Figure 3. Substrate scope study.

Reaction conditions: 5 mM 1a, 10 mM 2a, E. coli whole cells harboring P411-TEA-5274 (OD600 = 30) in M9-N aqueous buffer (pH 8.0), 5% (v/v) EtOH (co-solvent), 10 °C, anaerobic conditions, 20 h. a17% isolated yield at 1.0 mmol scale. b16% isolated yield at 0.5 mmol scale. c9% isolated yield at 0.5 mmol scale. d10% isolated yield at 0.5 mmol scale. TTN is defined as the (molar) amount of indicated product divided by the amount of haem protein in the reaction, as measured by the haemochrome assay (see Supplementary Method 7 for more details). TTN, total turnover number. e.e., enantiomeric excess. r.r., regioisomeric ratio.
To investigate enzyme regioselectivity, we tested substrate 1a (Figure 3). 1a has both an sp3 tertiary C–H bond and sp2 C–H bonds. Existing reports have demonstrated iron catalysts can efficiently catalyse primary amination of sp2 C–H bonds.36–38 In contrast, this enzyme exclusively catalysed the amination of the sp3 tertiary C–H bond of 1a, giving 3a with high efficiency (970 TTN, Figure 3) and exclusive regioselectivity (>99:1 regioselectivity ratio (r.r.), Figure 3) and enantioselectivity (92% e.e., Figure 3). A 1.0-mmol reaction was performed under the standard conditions, and primary amine 3a could be isolated in 17% isolated yield and with 92% e.e. by using simple acid−base extraction (Figure 3). Substrates (1b–1d) possess both an sp3 tertiary C–H bond and sp3 primary C–H bonds. Although the former has a smaller bond-dissociation energy (BDE) than the latter,39 the latter is kinetically more favorable for activation. This enzyme aminated the tertiary C–H bond of 1b–1d, delivering 3b–3d with high efficiency (up to 860 TTN). We next installed a methyl group (1c) and a methoxy group (1f) at the para-position of the phenyl ring. Interestingly, the enzyme’s activity toward the construction of 3c and 3f dropped to 310 TTN and 110 TTN, respectively. However, the enantioselectivities for both compounds were >99% e.e. (Figure 3). Conversely, when a methyl group was introduced at the meta-position of the phenyl ring, the enantioselectivity decreased to 60% e.e., but the enzyme’s activity was high (3d, 860 TTN, Figure 3). When a methyl group was introduced at the ortho-position, the enzyme struggled to convert 1e to 3e (Figure 3). When a fluoro-substituent was introduced at the para-position, the enzyme maintained high enantioselectivity, albeit with decreased activity (3g, 95% e.e., 73 TTN, Figure 3). When aminating an achiral tertiary carbon center, we found the efficiency was also good (3h, 300 TTN, Figure 3). In addition, when the aryl moiety was replaced with heteroaryl moieties, such as a pyridyl (1i) or a thienyl (1j) substituent, the enzyme retained satisfactory catalytic activity and good enantioselectivity (3i (52 TTN, 82% e.e.) and 3j (420 TTN, 92% e.e.)). Moreover, the biocatalytic system is not only applicable to benzylic tertiary C–H bonds but also to allylic (3k, 70 TTN, 90% e.e.) and propargylic tertiary C–H bonds (3l, 120 TTN, 67% e.e.). The absolute stereochemistry for enzymatic product 3f was assigned as S by comparing the elution order of the two enantiomers with a literature report40 and through X-ray crystallography (see Supplementary Table 8 and Supplementary Figure 21). The other α-tertiary primary amines 3 were assigned by analogy.
Experimental and Computational Study of P411-TEA-5274.
To better understand the mechanism of this biotransformation, we designed a series of substrate probes (Figure 4). First, enantiopure substrates (R)-1f and (S)-1f were synthesized (see SI for more details) and used to react with 2a under standard conditions catalysed by P411-TEA-5274 (Figures 4a and 4b). Experimental results showed that the efficiency of (R)-1f transforming into 3f (420 TTN, Figure 4a) was significantly higher (more than 16-fold difference) than that of (S)-1f transforming into 3f (26 TTN, Figure 4b), indicating the enzyme prefers the (R)-enantiomer. Moreover, we observed that (R)-1f was converted to (S)-3f (>99% e.e.), and (S)-1f was converted to (R)-3f (-85% e.e.), indicating that the enzyme achieves enantioselectivity via kinetic resolution and maintains the stereoconfiguration of the favored enantiomer.
Figure 4. Mechanistic studies of enantioenriched enzymatic primary amination of tertiary C–H bonds.

a and b, Nitrene transfer reactions catalysed by P411-TEA-5274 using (R)- or (S)-1e. Reaction conditions: 5 mM (R)- or (S)-1f, 10 mM 2a, E. coli whole cells harboring P411-TEA-5274 (OD600 = 30) in M9-N aqueous buffer (pH 8.0), 5% (v/v) EtOH (co-solvent), 10 °C, anaerobic conditions, 20 h. c and d, Nitrene transfer reactions catalysed by P411-TEA-5274 using (E)- or (Z)-1m. Reaction conditions: 5 mM (E)- or (Z)-1m, 10 mM 2a, E. coli whole cells harboring P411-TEA-5274 (OD600 = 30) in M9-N aqueous buffer (pH 8.0), 5% (v/v) EtOH (co-solvent), 10 °C, anaerobic conditions, 20 h. E/Z ratios were determined by 1H NMR. e and f, Nitrene transfer reactions catalysed by P411-TEA-5274 using 1b or 1b’ (d7-1b). Reaction conditions: 5 mM 1b or 1b’, 10 mM 2a, E. coli whole cells harboring P411-TEA-5274 (OD600 = 30) in M9-N aqueous buffer (pH 8.0), 5% (v/v) EtOH (co-solvent), 10 °C, anaerobic conditions, 20 h. TTN is defined as the (molar) amount of indicated product divided by the amount of haem protein in the reaction, as measured by the haemochrome assay (see Supplementary Method 7 for more details). aTTNs were calculated based on all stereoisomers. b45% isolated yield at 0.6 mmol scale (see Supplementary Method 17).
Next, we synthesized substrates that incorporate a stereodefined olefin moiety ((E)-1m and (Z)-1m, Figures 4C and 4D), and subjected them to enzymatic reactions. Interestingly, scrambling of the olefin geometry was observed in the (Z)-substrate, with the (Z)-substrate yielding a significant proportion of the scrambled product that harbors a thermodynamically more stable product (E)-3m (E/Z > 99:1, Figure 4d). In line with established literature on related iron-based catalysts,11,41–43 this observed erosion of C=C stereochemistry supports the generation of a carbon-centered radical at the allylic position and a stepwise pathway, contradicting a concerted C–H insertion mechanism.
To understand the rate-determining step of this enzyme-catalysed reaction, the kinetic isotope effect (KIE) between cumene 1b and cumene-d7 1b’ was measured (Figures 4e and 4f). The non-competitive KIE was 9.5, while the competitive KIE was 22.9. These results contrast with previously measured KIEs for the benzylic C–H amidation of ethyl benzene,35 while aligning with those for the nitrogen insertion into unactivated C–H bonds,44 suggesting that the hydrogen atom transfer (HAT) is the rate-limiting step and a higher degree of tunneling than previously observed in these systems.44
To gain further insight into the reaction of 1a with the iron-nitrene intermediate in the active site of the enzyme, molecular dynamics (MD) simulations were performed on a mimic of the enantioselectivity-determining HAT transition state, employing the closely-related P411 enzyme E1035 as the starting point (beneficial mutations were introduced manually and a restraint was applied to the Nnitrene--H–Ctertiary distance; see Supplementary Figures 22–26). Simulations on the hydrogen atom transfer with substrate (R)-1a, which in the experiment delivers the major enantiomer (S)-3a, and the other enantiomer (S)-1a revealed that with both substrates the phenyl-substituent is preferentially oriented toward the left-hand side of the active site, corresponding to an N1-Fe-Nnitrene-Cphenyl dihedral angle of approximately 20° (Figure 5). In this orientation, the phenyl substituent is tightly positioned between a number of surrounding hydrophobic residues, with minimum C–C distances as close as 3.5 Å, 3.9 Å, and 3.9 Å between 1a and A87, A264, and M263, respectively, indicating the possibility of stabilizing C–H-π interactions (see Supplementary Figures 22 and 23 for details). This tight fit prevents significant movement of the carbon-centered radical species after hydrogen-atom transfer, precluding reorientation and rotation around the Cphenyl–Ctertiary bond, thus avoiding an interconversion of the radical intermediates formed from (R)-1a and (S)-1a and ablation of enantioselectivity. In addition, for substrate (R)-1a, the ethyl substituent is placed in a sterically accessible area between residues P268 and V328, while the methyl group is oriented toward the outward-facing side of the active site (see Supplementary Figures 22–26 for details). In contrast, for the other enantiomer, (S)-1a, the ethyl substituent mostly rotates upwards to avoid an otherwise close contact of the CH3 group of the ethyl substituent with the haem ring system. However, the rotation of the ethyl group causes a significant movement of F437 and the respective open-loop protein backbone, which results in exclusion of (S)-1a from the enzyme pocket or a destabilization of the HAT transition state, thereby disfavoring reaction with (S)-1a (see Supplementary Figures 22–26 for details). The computational results are consistent with the experimental findings based on the biotransformation of enantiomers (R)-1f (Figure 4a) and (S)-1f (Figure 4b).
Figure 5. Molecular dynamics simulations of iron haem-catalysed tertiary C–H amination.

(A) and (B) Molecular dynamics simulations of HAT transition state mimics with (R)-1a (A) and (S)-1a (B). Plot of the N1-Fe-Nnitrene-Cphenyl dihedral angle over time and respective probability density plot. During the simulations, the Nnitrene--H–Ctertiary distance (dashed yellow line) was restrained. Structures correspond to representative structures of the most populated cluster. Non-relevant, non-polar hydrogens are omitted for clarity. The color scheme for atoms of amino acid residues: carbon = blue, oxygen = red, and sulphur = yellow; for the atoms of 1a and the iron-nitrene intermediate: carbon = grey, iron = orange, nitrogen = blue, and oxygen = red.
Conclusions
We have developed an engineered enzyme, P411-TEA-5274, that can directly aminate tertiary C–H bonds, efficiently and selectively to produce high-value chiral α-tertiary primary amines. This biocatalyst provides an alternative to small-molecule catalysts, which struggle to functionalise tertiary C–H bonds, and demonstrates remarkable enantioselectivity and activity. The substrate scope study demonstrated the compatibility of this biotransformation toward various substrates, the exclusive regioselectivity to tertiary C–H bonds, and the broad applicability against different tertiary C–H bonds. Experimental and computational investigations indicate that the enzyme’s excellent enantioselectivity arises from its enantiomeric substrate specificity. Given the prevalence of α-tertiary amines in bioactive molecules, this work provides a straightforward disconnection strategy to construct these fragments. Leveraging this work in the future, we hope to expand the limited repertoire of catalysts for asymmetric intermolecular functionalization of tertiary C–H bonds.
Methods
Expression of P411-TEA variants
E. coli (E. cloni BL21(DE3)) cells carrying plasmid encoding the appropriate P411-TEA variant were grown overnight in 5 mL Luria-Bertani medium with 0.1 mg/mL ampicillin (LBamp). Preculture (1 mL) was used to inoculate 50 mL of Hyper Broth medium with 0.1 mg/mL ampicillin (HBamp) in an Erlenmeyer flask (125 mL). This culture was incubated at 37 °C and 220 rpm for 2.5 hours, cooled on ice for 30 min, and induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside and 1.0 mM 5-aminolevulinic acid (final concentrations). Expression was conducted at 22 °C, 140 rpm for 20–22 h. E. coli cells were then transferred to a conical centrifuge tube (50 mL) and pelleted by centrifugation (4,000g, 3 min, and 4 °C). Supernatant was removed and the resulting cell pellet was resuspended in M9-N (pH = 8.0) buffer to OD600 = 38. An aliquot of this cell suspension (3 mL) was taken to determine protein concentration using the pyridine haemochromagen assay after lysis by sonication.
Primary amination of tertiary C–H bonds using whole E. coli cells harboring P411-TEA
All the biocatalytic reactions were set up in an anaerobic chamber (oxygen level: <40 ppm). M9-N medium (pH 8.0) and D-glucose solution (500 mM in M9-N, pH 8.0) were placed in the anaerobic chamber for at least 24 hours. Harvested cells were resuspended with M9-N buffer (pH 8.0) in to OD600 = 38, and 320 μL of resuspended cells were aliquoted to 2-mL screw cap vials. The screw-cap vials with cells were precooled to 0 °C on an ice bath. 30 μL of the precooled glucose solution, 10 μL of a stock solution containing glucose oxidase (from Aspergillus niger, 1,000 U/mL) and catalase (from bovine liver, 14,000 U/mL) in double-distilled water were added to make the total volume to 360 μL. The resulting mixtures stayed on the ice bath for another 2 minutes, and then the hydrocarbon substrate 1 (20 μL, 0.1 M stock in ethanol) and the nitrene precursor 2a (20 μL, 0.2 M stock in water) were added in a sequential manner. The reactions were then shaken at 10 °C for 20 hours at 250 rpm.
Supplementary Material
Acknowledgment
This work is supported by the National Institute of General Medical Science of the NIH (grant no. R01GM138740). Support by National Science Foundation Division of Chemistry (CHE-2153972 to K.N.H.) and the Alexander von Humboldt-Foundation (Feodor Lynen Fellowship, T.R.) is gratefully acknowledged. We thank Dr. Scott C. Virgil for the maintenance of the Caltech Center for Catalysis and Chemical Synthesis (3CS). We thank Dr. Mona Shahgoli for mass spectrometry assistance. We thank Dr. David Vander Velde for the maintenance of the Caltech NMR facility. We thank Dr. Michael K. Takase and Lawrence M. Henling for assistance with X-ray crystallographic data collection. We also thank Dr. Sabine Brinkmann-Chen for the helpful discussions and comments on the manuscript.
Footnotes
Competing Interests Statement
The authors declare no competing interests.
Data Availability
Crystallographic data are available free of charge from the Cambridge Crystallographic Data Centre under no. CCDC 2287786 (3f derivative (S)-N-(2-(4-methoxyphenyl)butan-2-yl)benzamide). The original materials and data that support the findings of this study are available within the paper and its Supplementary Information or can be obtained from the corresponding author upon reasonable request.
References
- 1.Chu JCK & Rovis T Complementary Strategies for Directed C(sp3)–H Functionalization: A Comparison of Transition-Metal-Catalyzed Activation, Hydrogen Atom Transfer, and Carbene/Nitrene Transfer. Angew. Chem., Int. Ed 57, 62–101 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dalton T, Faber T & Glorius F C–H Activation: Toward Sustainability and Applications. ACS Cent. Sci 7, 245–261 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Newton CG, Wang SG, Oliveira CC & Cramer N Catalytic Enantioselective Transformations Involving C–H Bond Cleavage by Transition-Metal Complexes. Chem. Rev 117, 8908–8976 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Saint-Denis TG, Zhu RY, Chen G, Wu QF & Yu JQ Enantioselective C(sp3)–H Bond Activation by Chiral Transition Metal Catalysts. Science 359, eaao4798 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rogge T et al. C–H activation. Nat. Rev. Methods Primers 1 (2021). [Google Scholar]
- 6.Davies HML & Beckwith REJ Catalytic Enantioselective C–H Activation by Means of Metal-Carbenoid-Induced C–H Insertion. Chem. Rev 103, 2861–2904 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Zhang C, Li ZL, Gu QS & Liu XY Catalytic Enantioselective C(sp3)–H Functionalization Involving Radical Intermediates. Nat. Commun 12, 475 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Y & Arnold FH Navigating the Unnatural Reaction Space: Directed Evolution of Heme Proteins for Selective Carbene and Nitrene Transfer. Acc. Chem. Res 54, 1209–1225 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang RK, Huang X & Arnold FH Selective C–H Bond Functionalization with Engineered Heme Proteins: New Tools to Generate Complexity. Curr. Opin. Chem. Biol 49, 67–75 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lewis JC, Coelho PS & Arnold FH Enzymatic Functionalization of Carbon–Hydrogen Bonds. Chem. Soc. Rev 40, 2003–2021 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Y, Cho I, Qi X, Liu P & Arnold FH An Enzymatic Platform for the Asymmetric Amination of Primary, Secondary and Tertiary C(sp3)–H Bonds. Nat. Chem 11, 987–993 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang C-J et al. Cu-Catalysed Intramolecular Radical Enantioconvergent Tertiary β-C(sp3)–H Amination of Racemic Ketones. Nat. Catal 3, 539–546 (2020). [Google Scholar]
- 13.Lang K, Li C, Kim I & Zhang XP Enantioconvergent Amination of Racemic Tertiary C–H Bonds. J. Am. Chem. Soc 142, 20902–20911 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ye CX, Shen X, Chen S & Meggers E Stereocontrolled 1,3-Nitrogen Migration to Access Chiral α-Amino Acids. Nat. Chem 14, 566–573 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ye C-X, Dansby DR, Chen S & Meggers E Expedited Synthesis of α-Amino Acids by Single-Step Enantioselective α-Amination of Carboxylic Acids. Nat. Synth 2, 645–652 (2023). [Google Scholar]
- 16.Hahn CJ et al. Crystal Structure of a Key Enzyme for Anaerobic Ethane Activation. Science 373, 118–121 (2021). [DOI] [PubMed] [Google Scholar]
- 17.Zanger UM & Schwab M Cytochrome P450 Enzymes in Drug Metabolism: Regulation of Gene Expression, Enzyme Activities, and Impact of Genetic Variation. Pharmacol. Ther 138, 103–141 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Huang X & Groves JT Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev 118, 2491–2553 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poulos TL Cytochrome P450 Flexibility. Proc. Natl. Acad. Sci 100, 13121–13122 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brandenberg OF, Fasan R & Arnold FH Exploiting and Engineering Hemoproteins for Abiological Carbene and Nitrene Transfer Reactions. Curr. Opin. Biotechnol 47, 102–111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen K & Arnold FH Engineering New Catalytic Activities in Enzymes. Nat. Catal 3, 203–213 (2020). [Google Scholar]
- 22.Hager A, Vrielink N, Hager D, Lefranc J & Trauner D Synthetic Approaches Towards Alkaloids Bearing α-Tertiary Amines. Nat. Prod. Rep 33, 491–522 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Liu G, Cogan DA & Ellman JA Catalytic Asymmetric Synthesis oftert-Butanesulfinamide. Application to the Asymmetric Synthesis of Amines. . J. Am. Chem. Soc 119, 9913–9914 (1997). [Google Scholar]
- 24.Ellman JA, Owens TD & Tang TP N-tert-Butanesulfinyl Imines: Versatile Intermediates for the Asymmetric Synthesis of Amines. Acc. Chem. Res 35, 984–995 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Gao X, Turek-Herman JR, Choi YJ, Cohen RD & Hyster TK Photoenzymatic Synthesis of α-Tertiary Amines by Engineered Flavin-Dependent “Ene”-Reductases. J. Am. Chem. Soc 143, 19643–19647 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barrios-Rivera J, Xu Y, Wills M & Vyas VK A Diversity of Recently Reported Methodology for Asymmetric Imine Reduction. Org. Chem. Front 7, 3312–3342 (2020). [Google Scholar]
- 27.Slabu I, Galman JL, Lloyd RC & Turner NJ Discovery, Engineering, and Synthetic Application of Transaminase Biocatalysts. ACS Catal 7, 8263–8284 (2017). [Google Scholar]
- 28.Afanasyev OI, Kuchuk E, Usanov DL & Chusov D Reductive Amination in the Synthesis of Pharmaceuticals. Chem. Rev 119, 11857–11911 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Jia ZJ, Gao S & Arnold FH Enzymatic Primary Amination of Benzylic and Allylic C(sp3)–H Bonds. J. Am. Chem. Soc 142, 10279–10283 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Liu Z et al. An Enzymatic Platform for Primary Amination of 1-Aryl-2-Alkyl Alkynes. J. Am. Chem. Soc 144, 80–85 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Y, Shi S-L, Niu D, Liu P & Buchwald SL Catalytic Asymmetric Hydroamination of Unactivated Internal Olefins to Aliphatic Amines. Science 349, 62–66 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fandrick KR et al. A General Copper-BINAP-Catalyzed Asymmetric Propargylation of Ketones with Propargyl Boronates. J. Am. Chem. Soc 133, 10332–10335 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Kille S et al. Reducing Codon Redundancy and Screening Effort of Combinatorial Protein Libraries Created by Saturation Mutagenesis. ACS Synth. Biol 2, 83–92 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Reetz MT & Carballeira JD Iterative Saturation Mutagenesis (ISM) For Rapid Directed Evolution of Functional Enzymes. Nat. Protoc 2, 891–903 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Prier CK, Zhang RK, Buller AR, Brinkmann-Chen S & Arnold FH Enantioselective, Intermolecular Benzylic C–H Amination Catalysed by an Engineered Iron-Haem Enzyme. Nat. Chem 9, 629–634 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D’Amato EM, Borgel J & Ritter T Aromatic C–H Amination in Hexafluoroisopropanol. Chem. Sci 10, 2424–2428 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Legnani L, Prina Cerai G & Morandi B Direct and Practical Synthesis of Primary Anilines through Iron-Catalyzed C–H Bond Amination. ACS Catal 6, 8162–8165 (2016). [Google Scholar]
- 38.Liu J et al. Fe-Catalyzed Amination of (Hetero)Arenes with a Redox-Active Aminating Reagent under Mild Conditions. Eur. J. Chem 23, 563–567 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Luo Y-R Comprehensive Handbook of Chemical Bond Energies 19–145 (Taylor & Francis, 2007). [Google Scholar]
- 40.Pan Y et al. Kinetic Resolution of α-Tertiary Propargylic Amines through Asymmetric Remote Aminations of Anilines. ACS Catal 11, 8443–8448 (2021). [Google Scholar]
- 41.Hennessy ET, Liu RY, Iovan DA, Duncan RA & Betley TA Iron-Mediated Intermolecular N-Group Transfer Chemistry with Olefinic Substrates. Chem. Sci 5, 1526–1532 (2014). [Google Scholar]
- 42.Jacobs BP, Wolczanski PT, Jiang Q, Cundari TR & MacMillan SN Rare Examples of Fe(IV) Alkyl-Imide Migratory Insertions: Impact of Fe–C Covalency in (Me2IPr)Fe(=NAd)R2 (R = neoPe, 1-nor). J. Am. Chem. Soc 139, 12145–12148 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Singh R, Kolev JN, Sutera PA & Fasan R Enzymatic C(sp3)–H Amination: P450-Catalyzed Conversion of Carbonazidates into Oxazolidinones. ACS Catal 5, 1685–1691 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Athavale SV et al. Enzymatic Nitrogen Insertion into Unactivated C–H Bonds. J. Am. Chem. Soc 144, 19097–19105 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Crystallographic data are available free of charge from the Cambridge Crystallographic Data Centre under no. CCDC 2287786 (3f derivative (S)-N-(2-(4-methoxyphenyl)butan-2-yl)benzamide). The original materials and data that support the findings of this study are available within the paper and its Supplementary Information or can be obtained from the corresponding author upon reasonable request.