

Abstract
S = 2 FeIV=O centers generated in the active sites of nonheme iron oxygenases cleave substrate C–H bonds at rates significantly faster than most known synthetic FeIV=O complexes. Unlike the majority of the latter, which are S = 1 complexes, [FeIV(O)(tris(2-quinolylmethyl)amine)(MeCN)]2+ (3) is a rare example of a synthetic S = 2 FeIV=O complex that cleaves C–H bonds 1000-fold faster than the related [FeIV(O)(tris(pyridyl-2-methyl)amine)(MeCN)]2+ complex (0). To rationalize this significant difference, a systematic comparison of properties has been carried out on 0 and 3 as well as related complexes 1 and 2 with mixed pyridine (Py)/quinoline (Q) ligation. Interestingly, 2 with a 2-Q-1-Py donor combination cleaves C–H bonds at 233 K with rates approaching those of 3, even though Mössbauer analysis reveals 2 to be S = 1 at 4 K. At 233 K however, 2 becomes S = 2, as shown by its 1H-NMR spectrum. These results demonstrate a unique temperature-dependent spin-state transition from triplet to quintet in oxoiron(IV) chemistry that gives rise to the high C–H bond cleaving reactivity observed for 2.
Keywords: Spin-State change, NMR, Oxoiron(IV), C–H bond cleavage rates
Since 2003, oxoiron(IV) intermediates in the quintet (S = 2) spin state have been identified as key oxidants in the mechanisms of nonheme iron oxygenases.1–3 A related S = 2 oxoiron(IV) species has also been characterized in an iron-containing zeolite that converts methane to methanol,4 mimicking the reaction catalyzed by soluble methane monooxygenase.5 However, in contrast to the enzymatic and zeolitic intermediates described thus far, an overwhelming majority of the >100 synthetic oxoiron(IV) complexes described to date have been found to have a triplet (S = 1) ground spin state (see Scheme 1 for a few examples).6 The latter complexes have typically been characterized by a combination of spectroscopic approaches, such as UV-vis, Mössbauer, resonance Raman or IR, and X-ray absorption spectroscopies.7–12 Mössbauer spectroscopy has established the spin state of the oxoiron(IV) unit at 4 K, while X-ray absorption spectroscopy has revealed metal-ligand distances that provide further insights into their geometry. In select cases, X-ray crystallography provides structures of these complexes when diffraction-quality crystals can be obtained.
Scheme 1.
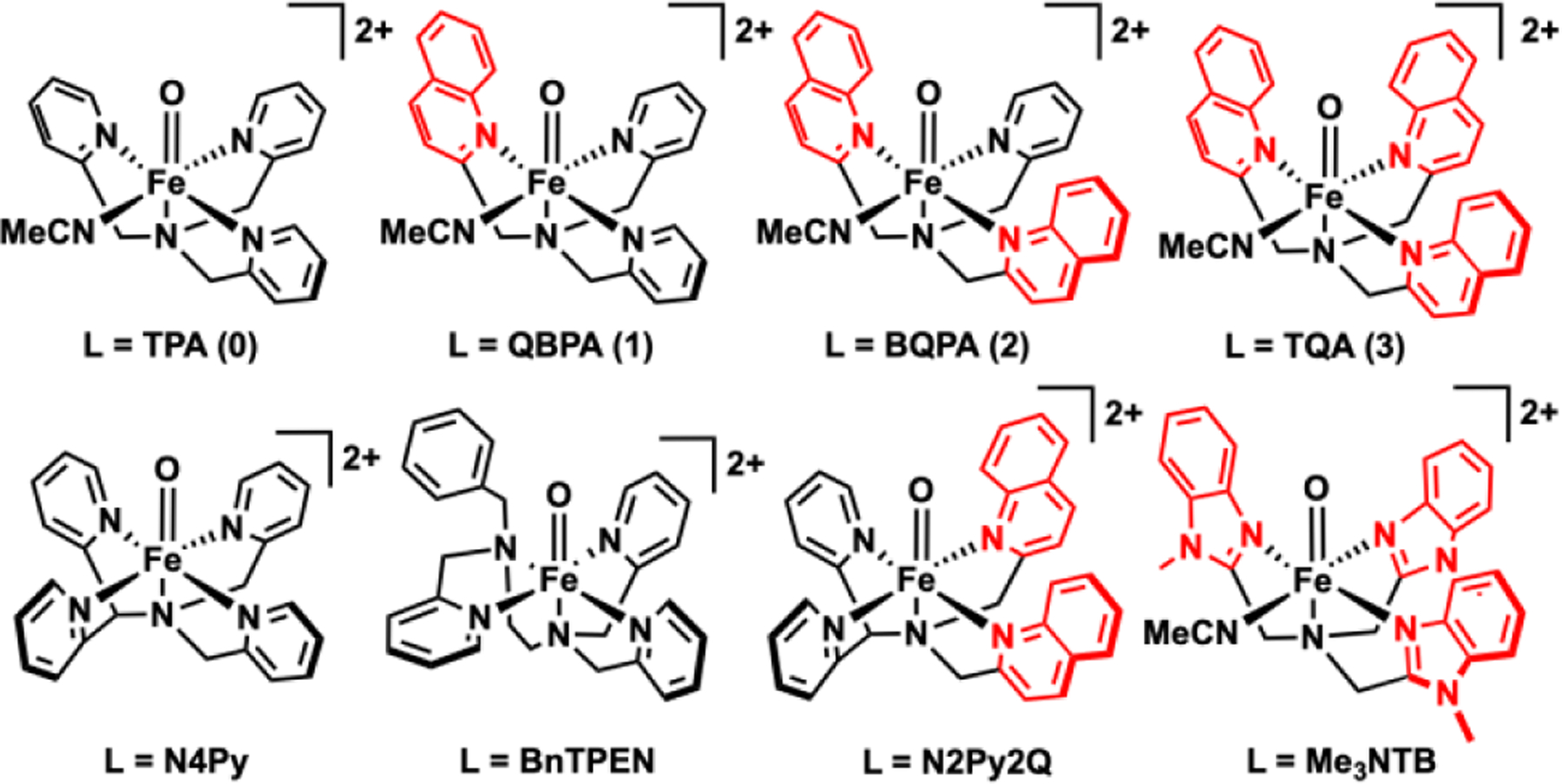
FeIV(O)L complexes discussed in this work.
1H-NMR spectroscopy has also proven to be quite useful for characterizing the Fe=O spin states of synthetic nonheme oxoiron(IV) complexes in liquid solution near ambient temperatures.13 The S = 1 [FeIV(O)(N4Py)]2+ complex (Scheme 1) shows a 1H-NMR spectrum with two sets of relatively sharp pyridine resonances, congruent with the two types of pyridines found in its crystal structure.14 Prominent peaks at 30 to 50 ppm and -10 to -20 ppm can be assigned to the distinct Py-β-H protons based on atom substitution and COSY experiments. Similarly, for the lower symmetry S = 1 [FeIV(O)(BnTPEN)]2+ complex (Scheme 1), the β-H resonances of the three distinct pyridines in this complex can also be easily distinguished and assigned. The two pyridines with planes oriented approximately parallel to the Fe=O axis exhibit shift patterns similar to those for the corresponding pyridines in [FeIV(O)(N4Py)]2+, but protons on the pyridine perpendicular to the Fe=O unit exhibit much smaller shifts, revealing an angle dependence for the delocalization of unpaired spin density from the Fe=O unit to the pyridine ring.13 DFT calculations of Borgogno et al. correctly predict these shift patterns,15 which match the resonances found for S = 1 [FeIV(O)(TPA)]2+ (0) (Scheme 1) and related complexes.
Much fewer synthetic examples of S = 2 complexes are described to date, representing only about 10% of the >100 synthetic nonheme oxoiron(IV) complexes characterized thus far.6 Most of the latter complexes are supported by tripodal ligands, such as TMG3tren, H3buae3-, (tpaPh)3-, and tBu3TACN (Scheme S1),16–19 that favor a trigonal arrangement of N-donors in the plane perpendicular to the Fe=O unit, resulting in a trigonal bipyramidal geometry. These complexes exhibit isomer shifts of 0.02–0.11 mm/s, which are lower in value than those typically found for oxoiron(IV) intermediates in enzyme active sites (0.22–0.31 mm/s),1 suggesting that the trigonal bipyramidal synthetic complexes do not resemble the enzymatic Fe=O units. In contrast, the isomer shifts of 0.21–0.24 mm/s for the S = 2 FeIV(O)TQA(X) complexes20–22 (X = MeCN, Cl−, and Br−) fall within the range of values defined by the enzymatic oxoiron(IV) intermediates, suggesting that a 6-coordinate geometry is favored for the oxoiron(IV) units in these enzyme active sites (Table 1). Other recent additions to the family of 6-coordinate S = 2 FeIV(O) a include Goldberg’s complex23 featuring a tridentate Me3TACN ligand and a bidentate disiloxide with a Mössbauer isomer shift of 0.22 mm/s and Long’s complex24 in a metal-organic framework with a Mössbauer isomer shift of 0.26 mm/s. However, unlike for the S = 1 complexes discussed above, 1H NMR spectra for S = 2 FeIV(O) complexes have not to date been reported.
Table 1.
Properties of FeIV(O) complexes evaluated in this work, generated by treating iron(II) precursors with 1–2 equiv s-ArIO.
| Properties | 0 | 1 | 2 | 3 |
|---|---|---|---|---|
| λmax, nm (νmax, cm−1) ⌈ε, M−1 cm−1⌉ |
720 (13900) [300]25 |
775 (12900) [300]33 |
850 (11900) [200] |
650 [300]20 (15400) 900 [75] (11100) |
| δ (ΔEQ), mm/s | 0.01 (0.92)25 | 0.05 (0.70) |
0.10 (0.66) |
0.24 (-1.05) 20 |
| ν(Fe=O), cm−1 | 83025 | – | – | 838 (∆18O -35)20 |
|
S at 4 K (based on Mossbauer) |
1 | 1 | 1 | 2 |
|
S at 233 K (based on NMR analysis) |
1 | 1 | 2 | 2 |
| t½
(T in K) |
460 min (273) | 40 min (273) | 2 min (233) |
~15 min 20(233) |
|
k2', M−1s−1 (273 K, 233 K) cumene cyclohexane ethylbenzene |
0.026 ±0.001 0.00023 ±0.00002 0.01 ± 0.0005 0.0014 ±0.0003 |
0.10 ±0.005 0.0042 ±0.0004 0.043 ± 0.003 0.005 ±0.0003 |
1.15 ±0.014 0.011 ±0.001 0.98 ±0.02 |
1.0 0.031 1.05 |
This work presents a systematic comparison of the 1H-NMR spectra of a series of complexes with tetradentate ligands from TPA to TQA (Scheme 1), in which a sterically bulkier quinoline systematically replaces each pyridine donor of TPA. Two members of this series have been well characterized, with the TPA complex exhibiting an S = 1 spin state25 and the TQA complex having an S = 2 spin state based on Mössbauer studies at 4 K.20 The high-spin state of the latter derives from the presence of α-substituents on all three pyridine rings that lead to longer Fe–N bonds that weaken the ligand field about the oxoiron(IV) center and favor the S = 2 iron spin state. Intermediate members of the TPA-to-TQA series, namely 1 and 2, are less well characterized, if at all,26 and more detailed characterization is reported herein.
NMR spectroscopy turns out to be an excellent tool to gain insight into the magnetic properties of the FeIV=O centers in this family of complexes in liquid solution, as the chemical shifts of the pyridine ligand ring protons are sensitive to both the spin state of the oxoiron(IV) unit and the orientation of the pyridine rings relative to the oxoiron(IV) unit.13–15 This approach has allowed the oxoiron(IV) spin state to be monitored in solution at the same temperature as the reactivity studies, leading to the identification of 2 as the first example of a nonheme FeIV(O) complex that undergoes a change of spin state from S = 1 to S = 2 as a function of temperature.
Results and Discussion
The four numbered complexes shown in Scheme 1 represent a series of four nonheme FeIV=O complexes supported by a tripodal NN’3 ligand in which each of the pyridine N' donors sequentially replaced with sterically bulkier quinolines to form the following series of complexes, namely [FeIV(O)(TPA)(MeCN)]2+ (0), [FeIV(O)(QBPA)(MeCN)]2+ (1), [FeIV(O)(BQPA)(MeCN)]2+ (2), and [FeIV(O)(TQA)(MeCN)]2+ (3). Of these complexes,20, 25, 27–28 only 2 has not been previously reported. Complexes supported by related pentadentate N4Py29 and N2Py2Q30 are also useful for comparison. For this study, we have used FeII(OTf)2 instead of FeII(ClO4)2 to mitigate the risks associated with the explosive nature of perchlorates. All four complexes in this series can be generated from their ferrous precursors in acetonitrile at appropriate temperatures upon addition of 2-(tBuSO2)-C6H4IO (s-ArIO) in either 2,2,2-trifluoroethanol or dichloromethane-d2 solution. To gain insight into the oxidative reactivity patterns exhibited by this series of complexes, their physical properties are compared in Table 1, Complex 0 is the longest lived of the series, with a half-life of ~8 h at 273 K, followed by 1 upon substitution of one pyridine by the sterically bulkier quinoline with a shorter half-life of 40 min at 273 K. Sequential replacement of the remaining pyridines with quinolines gives to 2 and 3 with even shorter half-lives of 2 min and 15 min at 233 K, respectively.
Physical Properties
All four complexes exhibit absorption bands associated with nonheme FeIV=O complexes (Figure 1). For the 0 to 2 subset, λmax values decrease systematically from 720 nm (νmax = 13900 cm−1, ε = 300 M−1cm−1) for 0 to 775 nm (νmax = 12900 cm−1, ε = 300 M−1cm−1) for 1 and 840 nm (νmax = 11900 cm−1, ε = 200 M−1cm−1) for 2 in approximately 1000 cm−1 steps per substitution of a pyridine donor by quinoline, consistent with the weaker basicity of the quinoline,. A similar pattern is reported for the S = 1 Fe=O complexes of related pentadentate ligands N4Py29 (λmax 695 nm, νmax = 14400 cm−1, ε = 400 M−1cm−1) and N2Py2Q (λmax 770 nm, νmax = 13000 cm−1, ε = 380 M−1cm−1) (see Scheme 1 for ligand structuires).30 However the spectral pattern for 3 shows two features in the 600–900 nm region, having λmax values at 650 nm (νmax = 15400 cm−1, ε = 300 M−1cm−1) and 900 nm (νmax = 11100 cm−1, ε = 75 M−1cm−1). This difference likely reflects the switch in spin state to S = 2 in the case of 3.
Figure 1.
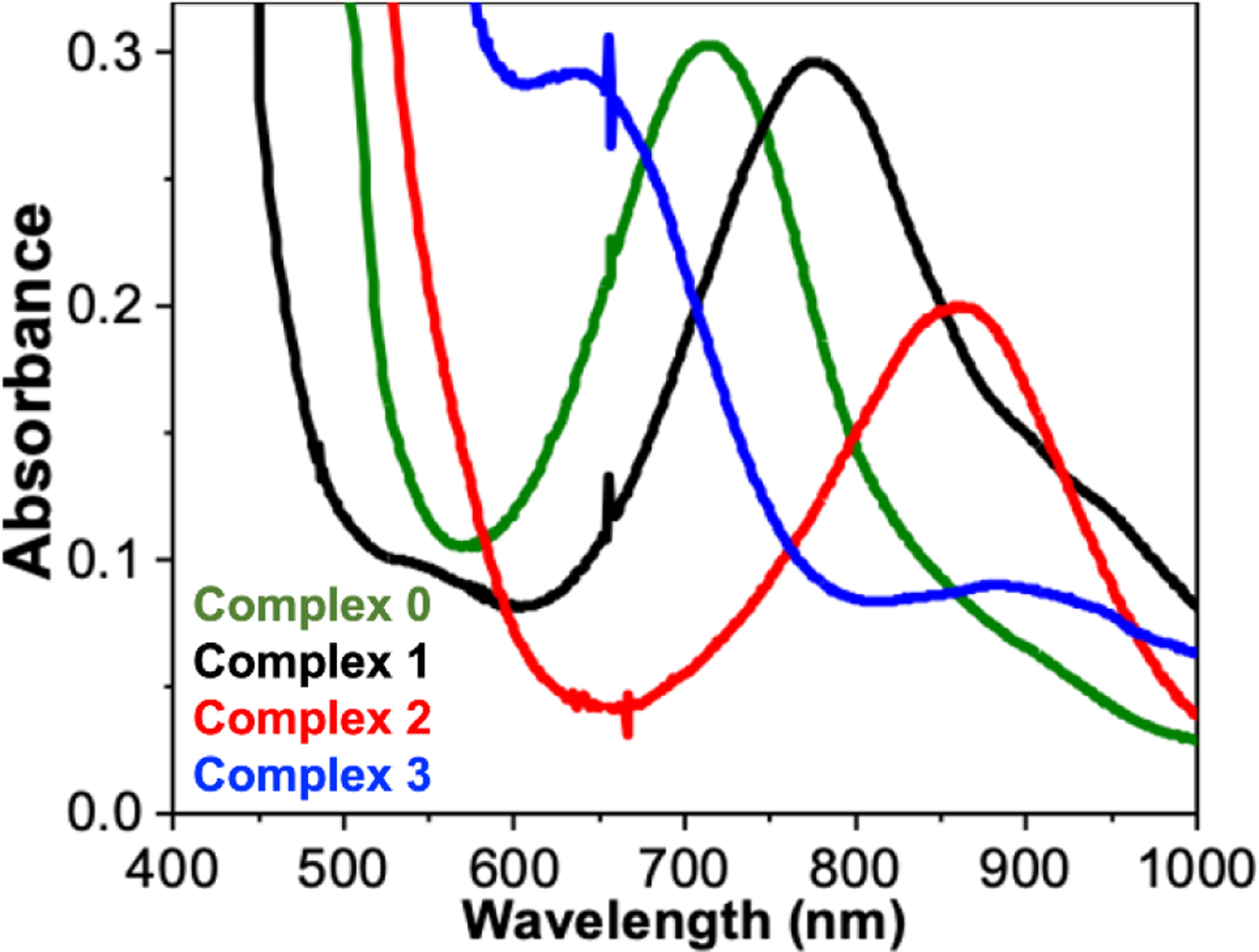
Electronic spectral features observed at 233 K of 1-mM solutions of FeIV=O iron(IV) complexes 0 (green), 1 (black), 2 (red) and 3 (blue) in MeCN obtained upon treatment of iron(II) precursors with s-ArIO.
Complex 0 exhibits a Mössbauer quadrupole doublet at 4.2 K with δ = 0.01 mm/s and ΔEQ = 0.92 mm/s,25 similar to values observed for other S = 1 oxoiron(IV) complexes.7 The replacement of one or two pyridines with quinolines on 1 respectively and 2 results in systematic increases in δ to 0.05 mm/s for 1 and 0.10 mm/s for 2 (Table 1).
These isomer shift differences nicely correlate with the lengthening of iron-ligand bonds based on the DFT optimized structures (Table S9) and follow a pattern originally pointed out by Neese in his earlier studies that correlate changes in metal-ligand distances with isomer shift.31 Nevertheless, all three complexes retain their S = 1 spin state at 4 K (see the SI for a detailed Mössbauer analysis in Figures S19-S20 and Table S8).
The sample containing complex 2 exhibits two quadrupole doublets with a ~1:1 ratio. The doublet having δ = 0.10 mm/s corresponds to 2 with an S = 1 spin ground state, while the doublet with δ = 0.45 mm/s derives from a di-ferric decay product of 2. The spin state of 2 at low temperature has been confirmed by variable temperature/variable field (VT/VF) Mössbauer experiments (Figure 3). The spectral simulation using an S = 1 spin Hamiltonian for the spectral features from complex 2 together with that of a diamagnetic species that is assigned to the di-ferric decay product satisfactorily reproduces the experimental data (Figure 3), with 2 representing about 40% of the sample. Complex 2 persists up at 150 K, suggesting that 2 retains the S = 1 ground spin state even at this temperature (Figure 2). Over the entire temperature range (4.2 – 150 K) we used for the Mössbauer measurements, no signal assignable to an S = 2 FeIV=O species could be detected. Above 150 K, no useful additional information could be obtained as a result of a drastic drop in the Mössbauer signal intensity due to reduction of the Lamb-Mössbauer factor and accelerated sample decay over the period required to acquire an acceptable spectrum. In stark contrast, 3 exhibits a much higher isomer shift of δ = 0.24 mm/s at 4.2 K, which approaches the values found for TauD-J and related enzyme intermediates.20 This significant increase in δ signals a change of spin state to S = 2 for 3, an assignment that has previously confirmed further confirmed by VT/VF Mössbauer analysis.21
Figure 3.
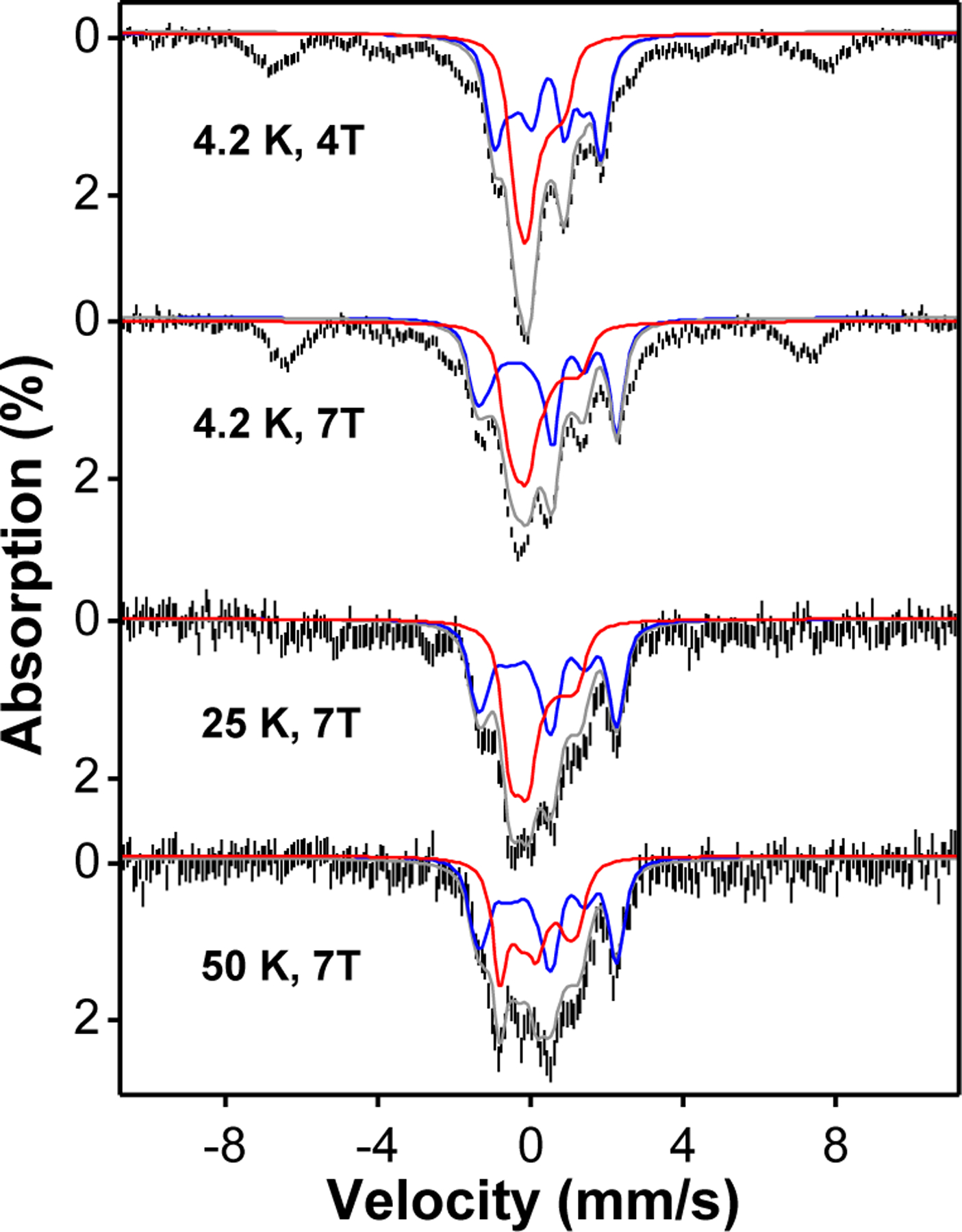
Variable-field-variable-temperature Mössbauer spectra of a sample containing 2 (black vertical bars) and the overall spectral simulations (grey solid lines) by including the simulations of 2 in an S = 1 spin state and the diamagnetic diferric decay product. The simulations of 2 are indicated by the red solid lines while the simulations of the diferric decay product are indicated by the blue lines. The simulation parameters for 2 are D ~ 35 cm−1, E/D = 0, gx = gy = gz = 2, Ax = Ay = -22.6 T, Az is not determined, δ = 0.10 mm/s, ΔEQ = 0.66 mm/s, η = 0. The absorption features observed at ± 8 mm/s are originated from high-spin mononuclear ferric species, representing 20 % of the iron in the sample.
Figure 2.
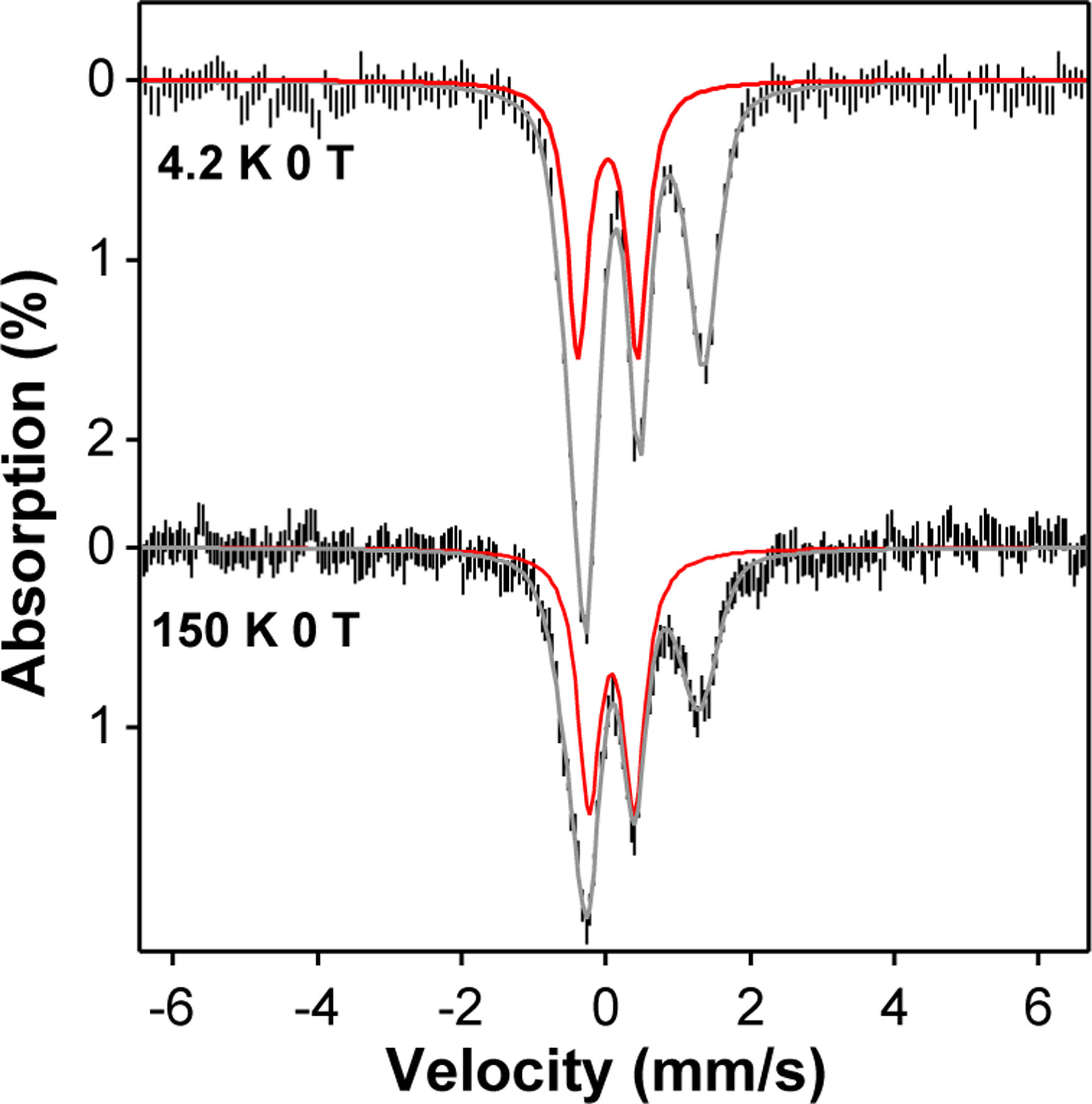
Zero field Mössbauer spectra of the sample containing complex 2 measured at 4.2 K and 150 K. The experimental data are shown as the vertical black bars, the overall simulations of the spectra are shown as the grey solid lines, and the spectral simulations of 2 are indicated as the red solid lines, which represents ~ 40 % of the total iron in the sample The simulation parameters are listed in Table S8.
Reactivity
Following earlier studies of other nonheme oxoiron(IV) complexes,7 we have compared the oxidative reactivities of complexes 0 – 3 with respect to a series of hydrocarbon substrates with C–H bonds that range in strength from 84 – 99.3 kcal mol−1. As observed in previous investigations, linear correlations are observed in the plots of the log(k2') values (where k2' = k2 divided by the number of equivalent substrate C−H bonds on the reactive substrate) versus C−H bond dissociation energies (BDE) (Figure 4). Measurements for 0 and 1 were carried out at 273 K, but those for 2 and 3 had to be obtained at 233 K due to their lower thermal stability. With the reaction rate adjusted by a Boltzmann factor reflecting the 40 K temperature difference, the reaction rates associated with 2 and 3 would be increased by at least an order of magnitude at 273 K, so that the oxidation rates for each substrate at 273 K would span over a range of 3 orders of magnitude for oxidants 0 – 3. We have also carried out reactivity comparisons of 0 to 3 at the same temperature (233 K) for the ethylbezene oxidation reaction (Table 1) and found almost one order magnitude lower k2 value at 233 K for complex 0 and 1 compared to the k2 at 273 K. As shown in Figure 4, the C–H bond cleavage rate for a particular substrate increases in the order of 0, 1, 2, and 3, even without correcting for the 40 K temperature decrease on going from the 0/1 pair to the 2/3 pair. Interestingly, the latter pair of complexes exhibits ethylbenzene oxidation rates that are essentially identical to each other within experimental error. However, with substrates having stronger C–H bonds, the oxidation rates of 2 and 3 diverge increasingly from each other with an increase in the strength of the scissile C–H bond, but the rate differences remain within an order of magnitude of each other. We attribute the increase in the C–H bond abstraction rates of complexes 0 – 3 to the lengthening of the average Fe–N bonds as the pyridine donors are replaced systematically with the sterically bulkier quinolines30 (Table 1), resulting in the progressive increase in the electrophilicity of the oxoiron(IV) center. The increasing reactivity also reflects the weakening of the ligand field about the oxoiron(IV) unit, providing greater access to the more reactive S = 2 spin state.
Figure 4.
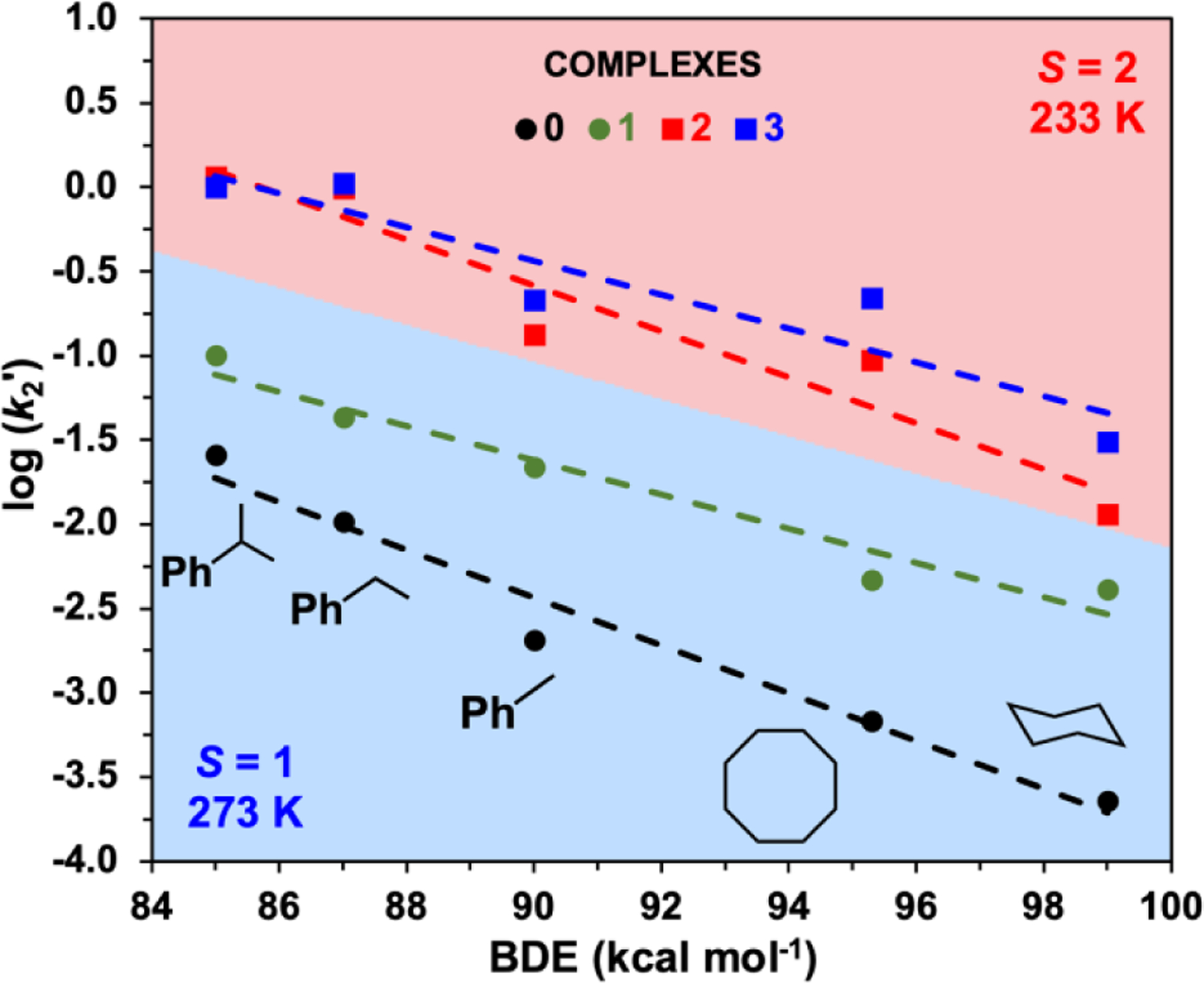
Evans-Polanyi plots for 0 and 1 (k2 at 273 K) and for 2 and 3 (k2 at 233 K) in MeCN. The 40 K drop in reaction temperature would be expected to decrease reaction rates by about an order of magnitude. The k2 values for the entire series of substrates from Figures S4-S6 are listed in Tables S3-S4 in the SI (error bars are instead listed in Tables S3-S4).
Unexpectedly, 2 is found to be significantly more reactive than 0 and 1 and resembles 3 much more closely in oxidation rates (Table 1). In fact, 2 and 3 exhibit essentially identical rates for cumene and ethylbenzene oxidation, which are at least 2 orders of magnitude faster than those of 0 and 1 after adjusting for the 40 K temperature difference (Figure 4). With substrates having stronger C–H bonds, 2 is not quite as powerful an oxidant as 3 but remains much more reactive than 1.
NMR Spectroscopy
Further insights have been obtained by comparing 1H-NMR data for 0 – 3 obtained at a similar temperature as the reactivity data. Unlike Mössbauer spectroscopy, 1H-NMR spectroscopy is a more effective probe of the electronic structure at the same temperature as the reactivity comparisons described earlier. Norlander and coworkers have recently reported the study of a series of stable oxoiron(IV) complexes with pentadentate ligand frameworks that exhibit values of 2.6–2.7 μB,11 exactly matching the theoretical value for S = 1 complexes with two unpaired electrons. Nam has also applied the Evans method to characterize the spin state of related FeIV=O complexes with tetradentate tripodal ligands, but was successful only for the least reactive and the thermally most stable complex 0, which was found to exhibit a magnetic moment of 3.1 μB.12 We have also attempted to carry out Evans susceptibility measurements on our FeIV=O complexes 2 and 3 but have found inconsistent results among the different sample preparations, likely due to their thermal lability that results in the formation of a mixture of compounds present in the samples. In contrast, NMR features of the iron(IV) complexes can be clearly distinguished from the decay product in our NMR experiments. We have thus taken advantage of the relatively favorable relaxation properties of the FeIV=O centers in 0 – 3 to assess the spin states of their respective paramagnetic centers at 233–273 K. These experiments provide some very valuable insights.
S = 1 Complexes 0 and 1 (233 K):
As observed for the well-characterized S = 1 [FeIV(O)(N4Py)]2+ complex (Figures S8-S9 and S11), the 1H-NMR spectrum of 0 at 233 K consists of resonances arising from two distinct sets of pyridine rings, with the latter showing a 2:1 ratio of signals reflecting the Cs symmetry expected for the 6-coordinate [FeIV(O)(TPA)]2+ complex with MeCN bound trans to one of the three pyridines (Figure 5A).32 The relatively sharp pyridine β, β', and γ proton signals can respectively be assigned to peaks at (55, 47), (-21, -25), and (8, 10) ppm, by analogy to the chemical shift pattern observed for [FeIV(O)(N4Py)]2+ (Figure 5B), the peaks of which have been assigned previously using atom-substitution and COSY experiments.14 Due to their proximity to the paramagnetic center, the α proton signals of 0 are much broader and less useful for this analysis. Similarly, the NMR spectrum of 1 (Figure 5D, Table 1) reveals two distinct pyridines with β-H signals at 58 and 45 ppm, β’-H signals at -26 and -17 ppm, and γ-H signals near 10 ppm, each with unit intensity. Thus, the two pyridines in 1 must be cis to each other. (The isomer with pyridines trans to each other would have a Cs-symmetric structure and give rise to only one set of pyridine peaks.) Furthermore, the paramagnetic shifts observed for 1 are comparable to those found for 0, indicative of a common S = 1 spin state for 0 and 1.
Figure 5.
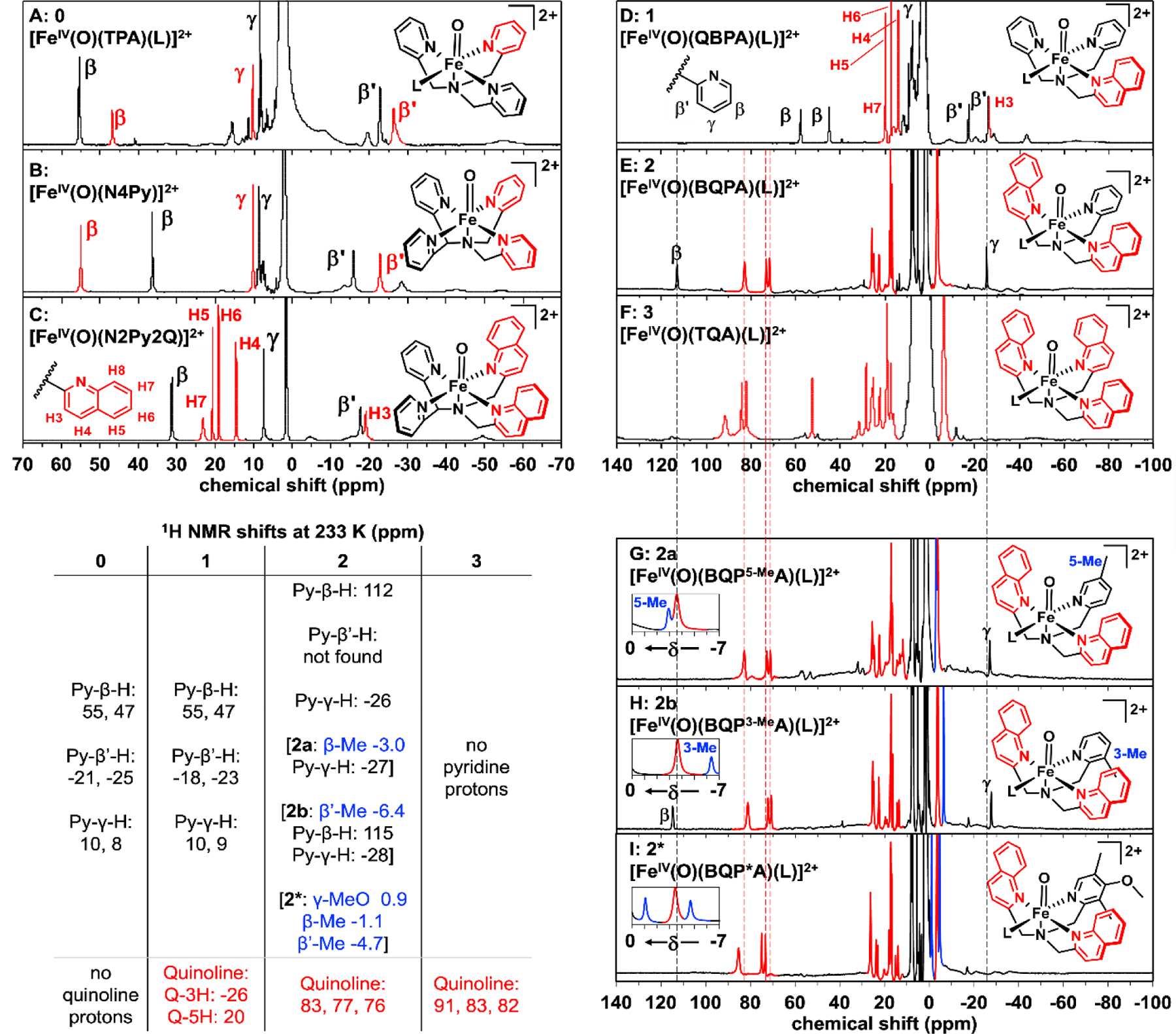
1H-NMR spectra of [FeIV(O)(L)]2+ complexes obtained at 233 K in CD3CN: (A) S = 1 0;32 (B) S = 1 [FeIV(O)(N4Py)]2+;13 (C) S = 1 [FeIV(O)(N2Py2Q)]2+;30 (D) S = 1 1; (E) S = 2 2; (F) S = 2 3; (G) S = 2 2a (Py 5-Me substituted); (H) S = 2 2b (Py 3-Me-substituted); (I) S = 2 2* (Py 3,5-Me2-4-OMe-substituted). The insets in panels G, H, and I show the regions between 0 and -7 ppm with signals from methyl groups that replace β-H and β’-H protons in the two variants highlighted in blue. Quinoline-derived signals are highlighted in red, except in panels A and B. Their assignments in panels E - I are based on predictions from DFT calculations using the Borgogno protocol.15 Dashed lines from panel E through panel I show slightly larger quinoline paramagnetic shifts in 3 when compared with those in 2, 2a, 2b and 2*. No peaks were discerned beyond 140 ppm (Figure S18)
Assignments for the quinoline peaks in 1 can be made by comparison with the 1H-NMR spectrum of the well-characterized S = 1 [FeIV(O)(N2Py2Q)]2+ complex (Scheme 1, Figure 5C).30 The latter shows eight sharp peaks with unit intensity, corresponding to ring protons of the pyridine (3 signals) and quinoline donors (5 signals), which are assigned in Figure 5C and Table S5 on the basis of their T1 values and linewidths that reflect their proximity to the metal center. Quinoline C3-H and C4-H signals can also be matched with the pyridine β’ and γ protons based on similarities in chemical shifts, linewidths and T1 values (see Table S5). The C5-H and C6-H assignments are further supported by the cross peak found between these two signals in the COSY spectrum of the complex (Figure S10). Due to their closer proximity to the FeIV=O unit, the quinoline C8-H and the pyridine α-H atoms give rise to much broader signals that make them more difficult to discern.
S = 2 Complex 3 (233 K):
In contrast, S = 2 complex 3 exhibits an NMR spectrum (Figure 5F) quite distinct from those of S = 1 complexes 0 and 1 (Figures 5A&D), due to the doubling in the number of unpaired electrons from 2 to 4. This difference results in a threefold increase in the magnetic susceptibility of 3 and the commensurate expansion of its observable chemical shift range. However, NMR spectra for such complexes are more challenging to obtain, due to their shorter lifetimes (Table 1). Thus, corresponding 1H-NMR data for the species formed upon decay have also been obtained to ensure correct identification of signals from the oxoiron(IV) center (Figures S14-S18). It is clear from one glance at the NMR spectrum of 3 (Figure 5F) that it exhibits more peaks than expected for a complex with a threefold-symmetric axis and that much larger paramagnetic shifts can be observed than found for S = 1, 1 (Figure 5D). The complexity of its spectrum favors the alternative six-coordinate configuration for 3 with a bound solvent, which should give rise to 12 peaks from aromatic ring protons, with 6 from the two quinolines trans to each other and 6 from the quinoline trans to the solvent with a 2:1 relative intensity ratio. However, closer scrutiny of the spectrum in Figure 5F shows that each of the three quinolines in 3 actually gives rise to distinct signals and is in fact in a unique environment.
The sharper peaks outside the diamagnetic region all show unit intensity, thereby excluding the possibility of twofold symmetry in the distortion of the six-coordinate oxoiron(IV) center in which the two trans quinoline donors twist slightly relative to each other to deviate from Cs symmetry. Such a distorted arrangement can be visualized in Figure S12 with the iron(II) precursor of 3 and is congruent with the DFT-optimized structure of 3. A similar distortion has also been found for the related S = 1 [FeIV(O)(Me3NTB)(solvent)]2+ complex by NMR (Scheme 1).34 Thus, the NMR data for 3 point to a six-coordinate C1-symmetric solution-state structure with one bound solvent as the dominant species at 233 K, with no detectable evidence for the 5-coordinate C3-symmetric isomer. These features are much more shifted than corresponding quinoline peaks found below 25 ppm in the spectra of the S = 1 complexes 1 and [FeIV(O)(N2Py2Q)]2+ (Figures 5C and 5D), as expected for an S = 2 complex with twice as many unpaired electrons. These results are corroborated by DFT calculations using the Borgogno protocol (Tables S10-S15).15
S = 2 Complex 2 (233 K):
In contrast to its Mössbauer spectrum at 4.2 K that identifies 2 as an S = 1 species, 2 exhibits a 1H-NMR spectrum at 233 K (Figure 5E) with signals that are significantly more shifted than would be expected for an S = 1 FeIV=O complex (Figure 5A-D) and more closely match the signals of the S = 2 complex 3 (Figure 5F). Complex 2 and 3 share a relatively sharp 3-peak pattern near 80 ppm that is absent in the spectrum of 1 and must arise from quinoline ligands. We prepared pyridine-ring-substituted variants of 2 to further distinguish the proton signals (Figures 5G, 5H and 5I). The quinoline peaks in 2, 2*, 2a and 2b are only slightly less paramagnetically shifted than those in complex 3, but these chemical shifts are significantly larger than those found for the quinoline peaks in 1. The 1H-NMR spectrum of 2 also shows peaks at 112 and –26 ppm that can be assigned to protons on the pyridine donor, based on comparisons with pyridine-ring-substituted derivatives of 2 (Figures 5G, 5H and 5I). The 112-ppm peak is absent in the spectrum of 2a (where a Me group replaces the Py-5-H atom) and in the spectrum of 2* (where Me groups replace Py-3-H and Py-5-H and an MeO group replaces Py-4-H atom), but it is present in the spectrum of 2b (where a Me group replaces the Py-3-H atom). Thus the 112-ppm peak in 2 can be assigned to the Py-5-H atom. On the other hand, the peak near –26-ppm observed in the spectra of 2, 2a and 2b disappears in the spectrum of 2* and can thus be assigned to the Py-γ-H atom. These pyridine protons in complexes 2 exhibit much larger shifts than found in 0 and 1, and the Py-γ-H shift is in fact opposite in sign, strongly supporting the assignment of an S = 2 spin state to complexes 2. Unfortunately, the pyridine β’-H proton predicted to be near 48 ppm by DFT cannot be identified in the spectra of 2 and its derivatives and may be hidden among the peaks between 0–30 ppm (Figure S12). However, the substitution of the β-H or β’-H proton with a methyl group gives rise to a β-Me (or 5-Me) peak at ~-3 ppm and a β’-Me (or 3-Me) peak near -6 ppm (Figures 3G-H, S11). Both β-Me & β’-Me peaks can be found in similar positions in the spectrum of 2* (Figure 5I). Based on the assignments of the Py-β-H and Py-γ-H signals, the significantly more shifted peaks observed for 2 at 233 K clearly point to a switch in spin state from S = 1 at 4 K to S = 2 at 233 K and align more closely with values predicted for the S = 2 isomers of 2 and [FeIV(O)(N2Py2Q)]2+ by DFT (Table 2 and illustrated in Figure 6).
Table 2.
Pyridine chemical shifts (in ppm), for [FeIV(O)]2+ complexes at 233 K in CD3CN. FWHM (in Hz) shown in parentheses.
| [FeIV(O)(N2Py2Q)]2+
(S = 1 from experiment) |
Complex 2 (S = 2 from experiment) |
||||||
|---|---|---|---|---|---|---|---|
| H | exp δ 233 K | calcd δ S = 1 |
calcd δ S = 2 |
H | exp δ 233 K | calcd δ S = 1 |
calcd δ S = 2 |
| α | broad | −44 | 141 | α | broad | −52 | 72 |
| β | 31 (100) | 35 | 156 | β | 112 (280) | 49 | 170 |
| β' | −18 (180) | −19 | 72 | β' | not found | −26 | 48 |
| γ | 7.4 (60) | 10 | −7.1 | γ | −26 (180) | 7.4 | −32 |
Figure 6.
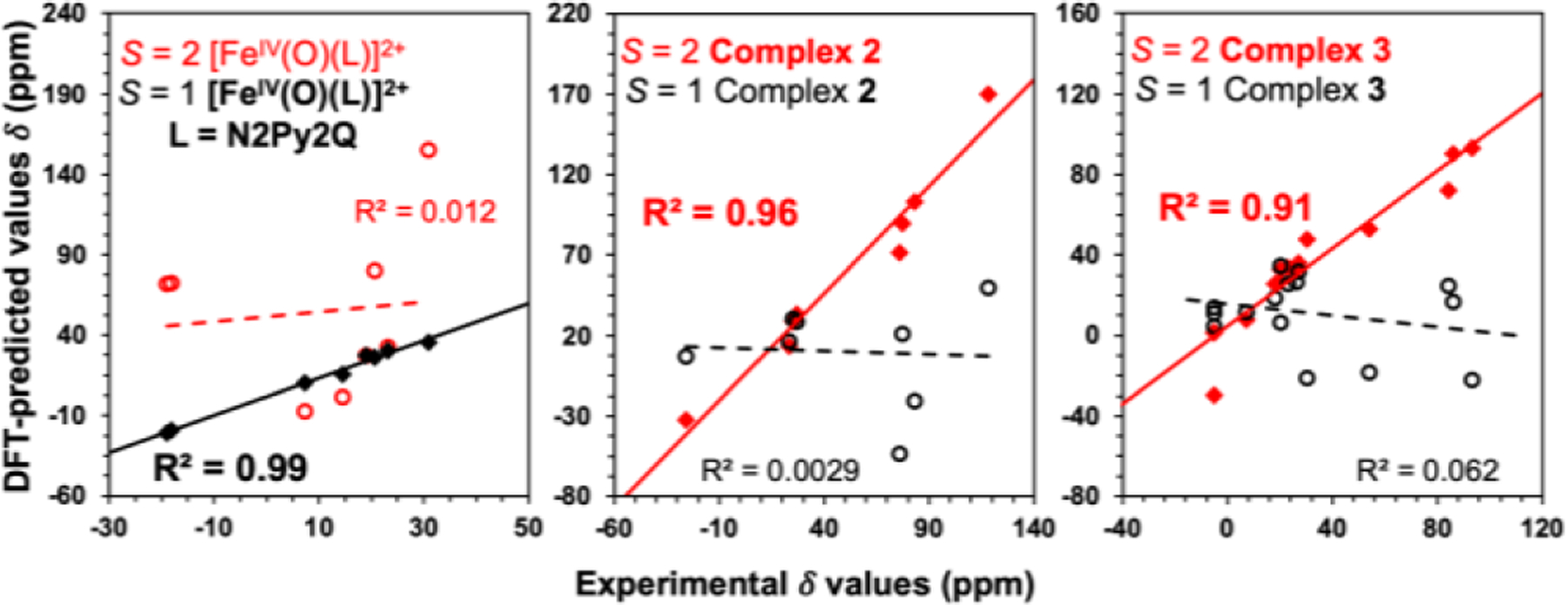
Linear correlation between the experimentally determined and DFT predicted 1H-NMR shifts of [FeIV(O)(N2Py2Q)]2+, 2 and 3. Solid lines show best fits for the calculated values with experimental data in S = 1 (for [FeIV(O)(N2Py2Q)]2+) or S = 2 (complexes 2 and 3) configurations, while dashed lines show poor fits for experimental data in the other spin state configuration.
Note that the shifts predicted for the S = 1 isomer of [FeIV(O)(N2Py2Q)]2+ align more closely with the experimentally obtained values at 233 K (R2 = 0.99), in contrast to the significantly greater scatter (R2 = 0.012) observed in Figure 6 for the predicted chemical shifts of its S = 2 isomer. On the other hand, the observed chemical shifts of 2 and 3 align more closely with the chemical shifts predicted in their S = 2 isomers, with respective R2 values of 0.96 and 0.91. Conversely a much greater scatter is found for observed chemical shifts in 2 and 3 with the calculated shifts in their S = 1 configuration, further supporting the intriguing conclusion that 2 must undergo a spin transition from from S = 1 to S = 2 upon warming to 233 K.
Conclusion
This work demonstrates the effects of systematically replacing pyridine donors with sterically bulkier quinolines on tetradentate ligand frameworks that support oxoiron(IV) complexes, leading to the identification of the first examples of oxoiron(IV) complexes that display two different spin states at different temparatures, specifically, namely 2 and its pyridine-ring-substituted variants. While the substitution of one pyridine donor with quinoline on the TPA framework to form 1 retains the S = 1 spin state at all temperatures studied, introducing the second quinoline elicits a change in the spin state at higher temperatures in 2, as indicated by its NMR peaks showing much larger paramagnetic shifts that approach those of the S = 2 complex 3 at 233 K. Congruent with this spectral change, the HAT reactivity found for 2 at 233 K approaches that of 3, (Figure 7) in contrast to the behavior of oxoiron(IV) complexes supported by pentadentate ligands for which replacing two pyridine donors in [FeIV(O)N4Py)]2+ with quinolines to form [FeIV(O)(N2Py2Q)]2+ retains the triplet spin state.30 Note however that S = 1 complex FeIV(O)(N2Py2Q)]2+ (k2 = 0.029 s−1 ) exhibits 500-fold enhanced reactivity compared to the S = 1 [FeIV(O)N4Py)]2+ complex (k2 = 0.000055 s−1 ) for cyclohexane oxidation6 at 298 K suggesting that the replacement of pyridine with quinoline still makes the Fe=O moiety more elctrophilic, even though there is no change in spin state. We have also found a similar increase in HAT reactivity from complexes 0 to 3 by the systematic replacement of pyridine with quinoline. We thus calculated the reduction potentials of this series of complexes by using DFT. Our results indeed reveal that higher reduction potentials, thus stronger electrophilicity, are found for complexes with more quinolines in the supporting ligand (Table S16). Nevertheless, this unique example of two spin states for the same ligand found in this family of complexes has broad implications in terms of access to the quintet state for an oxoiron(IV) complex. The hydrogen-atom abstraction reaction carried out by synthetic oxoiron(IV) complexes is often postulated to proceed via access to a low-lying excited quintet transition state during the reaction with the substrate. However, obtaining direct experimental evidence for such a transition has been very challenging. This particular hypothesis is the foundation for the two-state reactivity (TSR) paradigm of Shaik,35 which postulates that nonheme synthetic oxoiron(IV) complexes possess a low-lying excited quintet spin state that facilitates H-atom abstraction. This notion is often used to explain why S = 1 complexes are typically less reactive, so access to S = 2 spin state must occur in the course of hydrogen-atom abstraction to explain their reactivity. Unfortunately, transition states are not directly observable.
Figure 7.
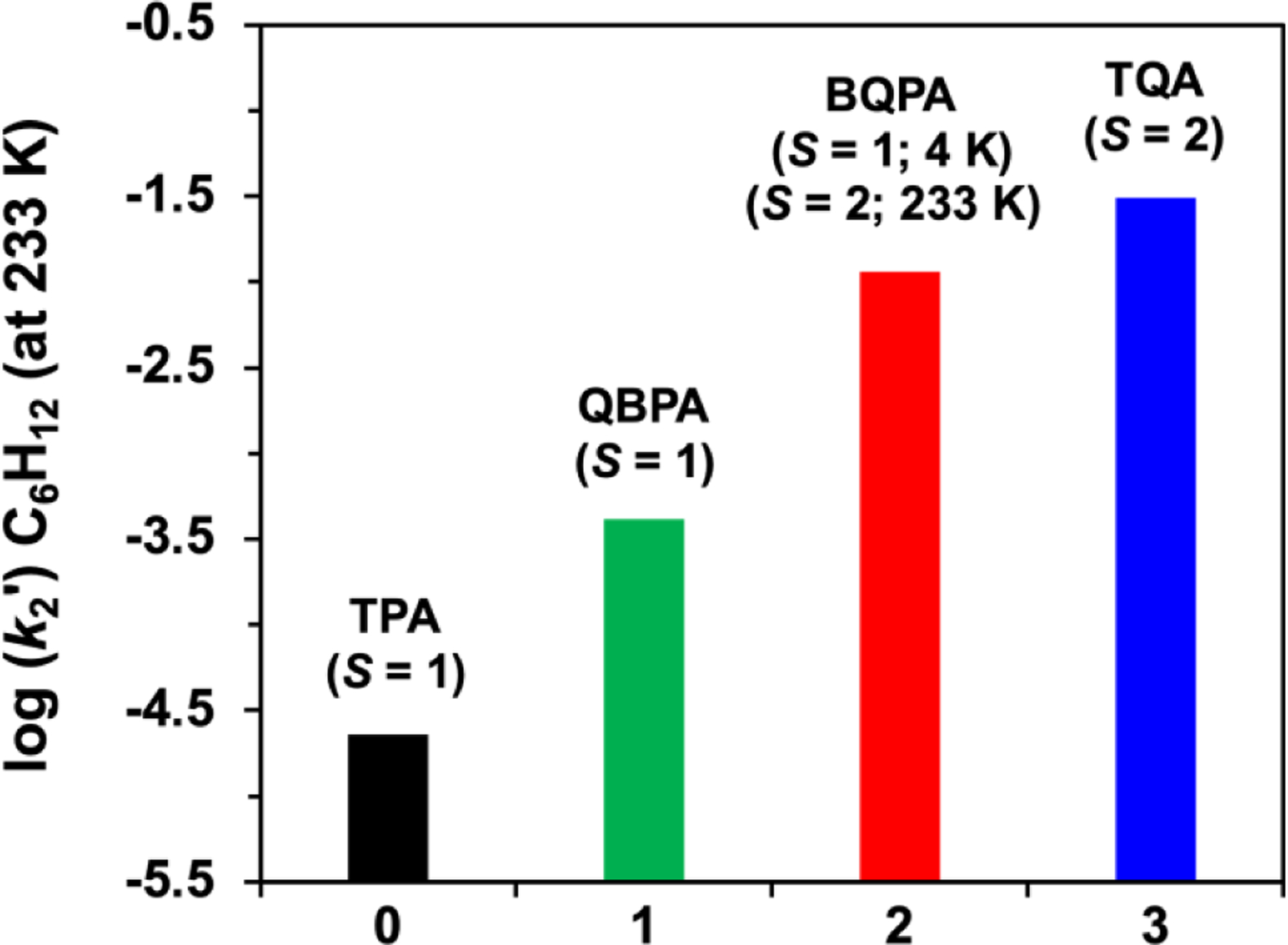
Comparison of log(k2') values of cyclohexane oxidation rates for complexes 0 – 3 at 233 K. (where k2' = k2/number of equivalent substrate C-H bonds that may be cleaved). The oxidation rates for 2 and 3 were obtained at 233 K, while those for 0 and 1 were measured at 273 K and then decreased by a factor of 10 to adjust for the difference in reaction temperatures.
Overall, a larger picture would also look at the thermodynamics of the HAT reaction whereby the energy difference between the reactants and products should also be taken into account.36–39 Nevertheless, our work has identified the first example of an oxoiron(IV) complex supported by a tetradentate tripodal ligand that gives rise to a triplet ground state at 4 K but converts into a complex with a quintet ground spin state at higher temperatures.
In the course of examining the NMR properties of these nonheme oxoiron(IV) complexes, we have obtained for the first time the NMR spectra of quintet oxoiron(IV) complexes, revealing a set of paramagnetically shifted peaks that span a range of about 150 ppm, which is much larger than the 60-ppm range found for corresponding S = 1 complexes (Figure 5). Complex 2 is the first oxoiron(IV) complex to exhibit a spin state change from S = 1 at 4 K to S = 2 at 233 K. Its S = 1 spin state is established by Mössbauer analysis at 4.2 K, while significant population of the S = 2 spin state at 233 K is demonstrated by the NMR studies. We observe not only a much broader chemical shift range for 2 relative to 1, but also chemical shift patterns for the pyridine and quinoline protons in 2 significantly different from those observed for the S = 1 complexes 0 and 1. These observations indicate that 2 and 3 share the same spin state at 233 K, resulting in C–H bond cleavage rates for 2 that are 2–3 orders of magnitude faster than those for 0 and 1 and approaching rates observed for 3. Our observation of an oxoiron(IV) complex that exhibits different ground spin states at different temperatures is unprecedented and highlights an unexpected aspect of the chemistry of these oxoiron(IV) complexes.
Supplementary Material
ACKNOWLEDGMENT
The authors acknowledge the support of National Institutes of Health grants R35 GM-131721 to L.Q. and R01 GM-125924 to Y.G. J.E.M.N.K. thanks the Alexander von Humboldt Foundation for a Feodor Lynen Research Fellowship.
Footnotes
Corresponding Authors
The supporting information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c10694.
Experimental details; synthesis and characterization for all compounds, including full spectroscopic analysis; and full computational details and optimized structures
Contributor Information
Waqas Rasheed, Department of Chemistry, University of Minnesota, Minneapolis, MN 55455 (USA).
Nabhendu Pal, Department of Chemistry, University of Minnesota, Minneapolis, MN 55455 (USA).
Ahmed M. Aboelenen, Department of Chemistry, University of Minnesota, Minneapolis, MN 55455 (USA)
Saikat Banerjee, Department of Chemistry, University of Minnesota, Minneapolis, MN 55455 (USA).
Ruixi Fan, Department of Chemistry, Carnegie Mellon University, Pittsburgh, PA 15213 (USA).
Jin Xiong, Department of Chemistry, Carnegie Mellon University, Pittsburgh, PA 15213 (USA).
Williamson N. Oloo, Department of Chemistry, University of Minnesota, Minneapolis, MN 55455 (USA)
Johannes E. M. N. Klein, Department of Chemistry, University of Minnesota, Minneapolis, MN 55455 (USA)
Yisong Guo, Department of Chemistry, Carnegie Mellon University, Pittsburgh, PA 15213 (USA).
Lawrence Que, Jr., Department of Chemistry, University of Minnesota, Minneapolis, MN 55455 (USA).
REFERENCES
- 1.Bollinger J,M; Chiang W-C; Matthews ML; Martinie RJ; Boal A,K; Krebs C, 2-Oxoglutarate-Dependent Oxygenases (Eds.: Hausinger RP, Schofield CJ). The Royal Society of Chemistry, Cambridge: 2015; p pp. 95–122. [Google Scholar]
- 2.Kal S; Que L, Dioxygen activation by nonheme iron enzymes with the 2-His-1-carboxylate facial triad that generate high-valent oxoiron oxidants. JBIC, J. Biol. Inorg. Chem 2017, 22 (2), 339–365. [DOI] [PubMed] [Google Scholar]
- 3.Krebs C; Galonić Fujimori D; Walsh CT; Bollinger JM, Non-Heme Fe(IV)–Oxo Intermediates. Acc. Chem. Res 2007, 40 (7), 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Snyder BER; Böttger LH; Bols ML; Yan JJ; Rhoda HM; Jacobs AB; Hu MY; Zhao J; Alp EE; Hedman B; Hodgson KO; Schoonheydt RA; Sels BF; Solomon EI, Structural characterization of a non-heme iron active site in zeolites that hydroxylates methane. Proc. Natl. Acad. Sci. U. S. A 2018, 115 (18), 4565–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banerjee R; Jones JC; Lipscomb JD, Soluble Methane Monooxygenase. Annu. Rev. Biochem 2019, 88 (1), 409–431. [DOI] [PubMed] [Google Scholar]
- 6.Abelson CS; Aboelenen AM; Rasheed W; Que L, 8.17 - Synthetic Nonheme High-Valent Iron-Oxo Complexes Structures and Oxidative Function. In Comprehensive Coordination Chemistry III, Constable EC; Parkin G; Que L Jr, Eds. Elsevier: Oxford, 2021; pp 412–454. [Google Scholar]
- 7.McDonald AR; Que L, High-valent nonheme iron-oxo complexes: Synthesis, structure, and spectroscopy. Coord. Chem. Rev 2013, 257 (2), 414–428. [Google Scholar]
- 8.Comba P; Nunn G; Scherz F; Walton PH, Intermediate-spin iron(iv)-oxido species with record reactivity. Faraday Discuss. 2022, 234 (0), 232–244. [DOI] [PubMed] [Google Scholar]
- 9.Abu-Odeh M; Bleher K; Johnee Britto N; Comba P; Gast M; Jaccob M; Kerscher M; Krieg S; Kurth M, Pathways of the Extremely Reactive Iron(IV)-oxido complexes with Tetradentate Bispidine Ligands. Chem. - Eur. J 2021, 27 (44), 11377–11390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Munshi S; Sinha A; Yiga S; Banerjee S; Singh R; Hossain MK; Haukka M; Valiati AF; Huelsmann RD; Martendal E; Peralta R; Xavier F; Wendt OF; Paine TK; Nordlander E, Hydrogen-atom and oxygen-atom transfer reactivities of iron(iv)-oxo complexes of quinoline-substituted pentadentate ligands. Dalton Trans 2022, 51 (3), 870–884. [DOI] [PubMed] [Google Scholar]
- 11.Li Y; Singh R; Sinha A; Lisensky GC; Haukka M; Nilsson J; Yiga S; Demeshko S; Gross SJ; Dechert S; Gonzalez A; Farias G; Wendt OF; Meyer F; Nordlander E, Nonheme FeIV═O Complexes Supported by Four Pentadentate Ligands: Reactivity toward H- and O- Atom Transfer Processes. Inorganic Chemistry 2023, 62 (45), 18338–18356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee NY; Mandal D; Bae SH; Seo MS; Lee Y-M; Shaik S; Cho K-B; Nam W, Structure and spin state of nonheme FeIVO complexes depending on temperature: predictive insights from DFT calculations and experiments. Chem Sci 2017, 8 (8), 5460–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rasheed W; Fan R; Abelson CS; Peterson PO; Ching W-M; Guo Y; Que L, Structural implications of the paramagnetically shifted NMR signals from pyridine H atoms on synthetic nonheme FeIV=O complexes. JBIC, J. Biol. Inorg. Chem 2019, 24 (4), 533–545. [DOI] [PubMed] [Google Scholar]
- 14.Klinker EJ; Kaizer J; Brennessel WW; Woodrum NL; Cramer CJ; Que L Jr., Structures of Nonheme Oxoiron(IV) Complexes from X-ray Crystallography, NMR Spectroscopy, and DFT Calculations. Angew. Chem., Int. Ed 2005, 44 (24), 3690–3694. [DOI] [PubMed] [Google Scholar]
- 15.Borgogno A; Rastrelli F; Bagno A, Characterization of Paramagnetic Reactive Intermediates: Predicting the NMR Spectra of Iron(IV)–Oxo Complexes by DFT. Chem. - Eur. J 2015, 21 (37), 12960–12970. [DOI] [PubMed] [Google Scholar]
- 16.England J; Guo Y; Farquhar ER; Young VG Jr; Münck E; Que L Jr, The Crystal Structure of a High-Spin Oxoiron(IV) Complex and Characterization of Its Self-Decay Pathway. J. Am. Chem. Soc 2010, 132 (25), 8635–8644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bigi JP; Harman WH; Lassalle-Kaiser B; Robles DM; Stich TA; Yano J; Britt RD; Chang CJ, A High-Spin Iron(IV)–Oxo Complex Supported by a Trigonal Nonheme Pyrrolide Platform. J. Am. Chem. Soc 2012, 134 (3), 1536–1542. [DOI] [PubMed] [Google Scholar]
- 18.Warm K; Paskin A; Kuhlmann U; Bill E; Swart M; Haumann M; Dau H; Hildebrandt P; Ray K, A Pseudotetrahedral Terminal Oxoiron(IV) Complex: Mechanistic Promiscuity in C−H Bond Oxidation Reactions. Angew. Chem., Int. Ed 2021, 60 (12), 6752–6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lacy DC; Gupta R; Stone KL; Greaves J; Ziller JW; Hendrich MP; Borovik AS, Formation, Structure, and EPR Detection of a High Spin FeIV—Oxo Species Derived from Either an FeIII—Oxo or FeIII—OH Complex. J. Am. Chem. Soc 2010, 132 (35), 12188–12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biswas AN; Puri M; Meier KK; Oloo WN; Rohde GT; Bominaar EL; Münck E; Que L, Modeling TauD-J: A High-Spin Nonheme Oxoiron(IV) Complex with High Reactivity toward C–H Bonds. J. Am. Chem. Soc 2015, 137 (7), 2428–2431. [DOI] [PubMed] [Google Scholar]
- 21.Puri M; Biswas AN; Fan R; Guo Y; Que L Jr., Modeling Non-Heme Iron Halogenases: High-Spin Oxoiron(IV)–Halide Complexes That Halogenate C–H Bonds. J. Am. Chem. Soc 2016, 138 (8), 2484–2487. [DOI] [PubMed] [Google Scholar]
- 22.Puri M; Que L Jr., Toward the Synthesis of More Reactive S = 2 Non-Heme Oxoiron(IV) Complexes. Acc. Chem. Res 2015, 48 (8), 2443–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gordon JB; Albert T; Dey A; Sabuncu S; Siegler MA; Bill E; Moënne-Loccoz P; Goldberg DP, A Reactive, Photogenerated High-Spin (S = 2) FeIV(O) Complex via O2 Activation. J. Am. Chem. Soc 2021, 143 (51), 21637–21647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hou K; Börgel J; Jiang HZH; SantaLucia DJ; Kwon H; Zhuang H; Chakarawet K; Rohde RC; Taylor JW; Dun C; Paley MV; Turkiewicz AB; Park JG; Mao H; Zhu Z; Alp EE; Zhao J; Hu MY; Lavina B; Peredkov S; Lv X; Oktawiec J; Meihaus KR; Pantazis DA; Vandone M; Colombo V; Bill E; Urban JJ; Britt RD; Grandjean F; Long GJ; DeBeer S; Neese F; Reimer JA; Long JR, Reactive high-spin iron(IV)-oxo sites through dioxygen activation in a metal–organic framework. Science 2023, 382 (6670), 547–553. [DOI] [PubMed] [Google Scholar]
- 25.Lim MH; Rohde J-U; Stubna A; Bukowski MR; Costas M; Ho RYN; Münck E; Nam W; Que L Jr., An FeIV=O Complex of a Tetradentate Tripodal Nonheme Ligand. Proc. Natl. Acad. Sci. USA 2003, 100 (7), 3665–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paine TK; Costas M; Kaizer J; Que L Jr, Oxoiron(IV) complexes of the tris(2-pyridylmethyl)amine ligand family: effect of pyridine α-substituents. JBIC, J. Biol. Inorg. Chem 2006, 11 (3), 272–276. [DOI] [PubMed] [Google Scholar]
- 27.Zang Y; Kim J; Dong Y; Wilkinson EC; Appelman EH; Que L, Models for Nonheme Iron Intermediates: Structural Basis for Tuning the Spin States of Fe(TPA) Complexes. J. Am. Chem. Soc 1997, 119 (18), 4197–4205. [Google Scholar]
- 28.Chen K; Que L, Stereospecific Alkane Hydroxylation by Non-Heme Iron Catalysts: Mechanistic Evidence for an FeVO Active Species. Journal of the American Chemical Society 2001, 123 (26), 6327–6337. [DOI] [PubMed] [Google Scholar]
- 29.Lubben M; Meetsma A; Wilkinson EC; Feringa B; Que L Jr., Nonheme Iron Centers in Oxygen Activation: Characterization of an Iron(III) Hydroperoxide Intermediate. Angew. Chem., Int. Ed 1995, 34 (13–14), 1512–1514. [Google Scholar]
- 30.Rasheed W; Draksharapu A; Banerjee S; Young VG Jr.; Fan R; Guo Y; Ozerov M; Nehrkorn J; Krzystek J; Telser J; Que L Jr., Crystallographic Evidence for a Sterically Induced Ferryl Tilt in a Non-Heme Oxoiron(IV) Complex that Makes it a Better Oxidant. Angew. Chem., Int. Ed 2018, 57 (30), 9387–9391. [DOI] [PubMed] [Google Scholar]
- 31.Neese F, Prediction and interpretation of the 57Fe isomer shift in Mössbauer spectra by density functional theory. Inorg. Chim. Acta 2002, 337, 181–192. [Google Scholar]
- 32.Duban EA; Bryliakov KP; Talsi EP, The Active Intermediates of Non-Heme-Iron-Based Systems for Catalytic Alkene Epoxidation with H2O2/CH3COOH. Euro. JIC 2007, 2007 (6), 852–857. [Google Scholar]
- 33.Paine TK; Costas M; Kaizer J; Que L Jr., Oxoiron(IV) complexes of the tris(2-pyridylmethyl)amine ligand family: effect of pyridine α-substituents. J. Biol. Inorg. Chem 2006, 11 (11), 272–276. [DOI] [PubMed] [Google Scholar]
- 34.Banerjee S; Rasheed W; Fan R; Draksharapu A; Oloo WN; Guo Y; Que L Jr., NMR Reveals That a Highly Reactive Nonheme FeIV=O Complex Retains Its Six-Coordinate Geometry and S=1 State in Solution. Chem. - Eur. J 2019, 25 (41), 9608–9613. [DOI] [PubMed] [Google Scholar]
- 35.Shaik S; Hirao H; Kumar D, Reactivity of High-Valent Iron–Oxo Species in Enzymes and Synthetic Reagents: A Tale of Many States. Acc. Chem. Res 2007, 40 (7), 532–542. [DOI] [PubMed] [Google Scholar]
- 36.Mayer JM, Understanding Hydrogen Atom Transfer: From Bond Strengths to Marcus Theory. Acc. Chem. Res 2011, 44 (1), 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saouma CT; Mayer JM, Do spin state and spin density affect hydrogen atom transfer reactivity? Chem. Sci 2014, 5 (1), 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barman SK; Yang M-Y; Parsell TH; Green MT; Borovik AS, Semiempirical method for examining asynchronicity in metal–oxido-mediated C–H bond activation. Proc. Natl. Acad. Sci. U. S. A 2021, 118 (36), e2108648118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schneider JE; Goetz MK; Anderson JS, Statistical analysis of C–H activation by oxo complexes supports diverse thermodynamic control over reactivity. Chem. Sci 2021, 12 (11), 4173–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.