

SUMMARY
Fibrotic interstitial lung diseases (fILDs) have poor survival rates and lack effective therapies. Despite evidence for immune mechanisms in lung fibrosis, immunotherapies have been unsuccessful for major types of fILD. Here, we review immunological mechanisms in lung fibrosis that have the potential to impact clinical practice. We first examine innate immunity, which is broadly involved across fILD subtypes. We illustrate how innate immunity in fILD involves a complex interplay of multiple cell subpopulations and molecular pathways. We then review the growing evidence for adaptive immunity in lung fibrosis, to provoke a re-examination of its role in clinical fILD. We close with future directions to address key knowledge gaps in fILD pathobiology: 1) longitudinal studies emphasizing early-stage clinical disease, 2) immune mechanisms of acute exacerbations, and 3) next-generation immunophenotyping integrating spatial, genetic, and single-cell approaches. Advances in these areas are essential for the future of precision medicine and immunotherapy in fILD.
Keywords (MeSH): Pulmonary fibrosis, Interstitial Lung Diseases, Idiopathic Pulmonary Fibrosis, Immune System, Fibroblasts
eTOC blurb
Fibrotic Interstitial lung diseases have poor survival rates, with current immunotherapies proving ineffective. This review examines underlying innate and adaptive immune mechanisms and potential clinical impacts. It lays the groundwork for future studies on early disease stages, acute exacerbation mechanisms, and advanced immunophenotyping to enhance therapeutic outcomes.
INTRODUCTION
Fibrosing interstitial lung diseases (fILDs) encompass several distinct clinical entities that share pathologic hallmarks of lung injury, inflammation, and fibrosis (Figure 1). The global prevalence of ILD is estimated to be 4.7 million people in 20191, the majority of whom had fILD. Clinically, fILD carries a poor prognosis, with 5-year survival rates below 50% depending on the underlying diagnosis2,3. These survival rates remain low, owing, in part, to a lack of effective, targeted therapies.
Figure 1. Fibrosing interstitial lung disease (fILD).
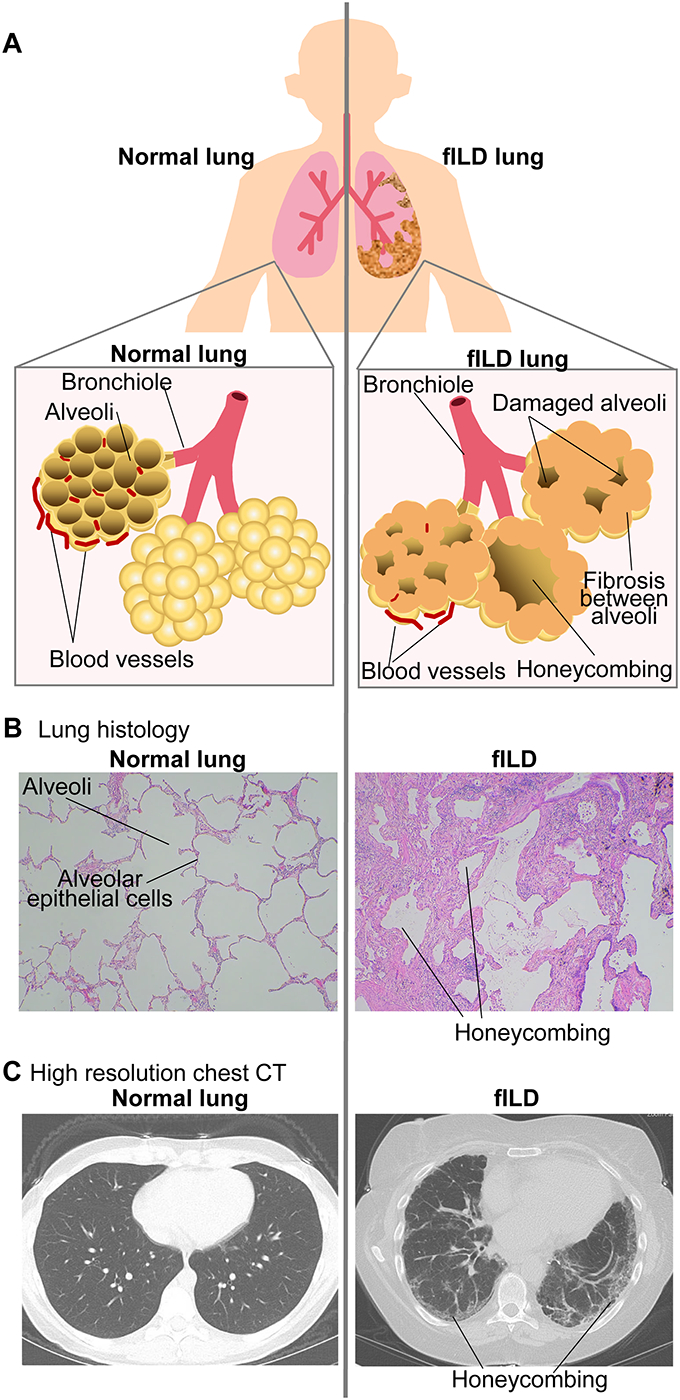
A. In fILD alveolar injury and abnormal repair leads to fibroblast accumulation, extracellular matrix deposition, and fibrosis of the interstitial spaces. The resulting end-stage “honeycombing” is seen in: B. lung histology; and C. high resolution chest CT imaging. B, C: images are obtained by authors.
Tremendous progress has been made toward understanding the pathobiology of fILD. Contemporary research efforts have focused on the contributions of non-immune cell populations, primarily epithelial and fibroblast cells. However, immune-mediated mechanisms have long been recognized. Here, we review recent advances in the immunobiology of fILD. After introducing fILD, we examine the innate immunology of fILD. Next, we discuss adaptive immune mechanisms in fILD. We review evidence that clinical subtypes of fILD defy simple classification regarding the involvement of adaptive immunity. We close with a discussion of how the future research agenda in fILD can address the gaps in knowledge preventing a more personalized approach to therapy.
CLASSIFICATION OF FIBROTIC ILD (fILD)
Over 200 distinct clinical disorders comprise the family of interstitial lung diseases. Of these, fILD are the most common and severe, with a median survival of 3–7 years3. Patients with fILD present with breathlessness, exercise intolerance, and cough. Clinically, these patients are subclassified based on associated medical comorbidities, radiographic findings, or tissue histology (Table 1)4. Regardless of classification, progressive disease leads to end-stage organ dysfunction due to excessive extracellular matrix (ECM) production and lung fibrosis. In the following subsections, we discuss specific morphological and clinical classifications of fILD that have particular relevance to our review.
Table 1.
Fibrosing interstitial lung diseases.
| Disease (year of diagnostic guideline) | Prevalence | Most common age of onset | Epidemiology, potential risk factors | Prognosis | HRCT findings | Histopathology | Clinical Characteristics | Auto-antibodies | Associated immune-related genetic variants |
|---|---|---|---|---|---|---|---|---|---|
| IPF (idiopathic UIP pattern) (2022)4 | 5 per 100,000 people, 3 million (worldwide) |
50–70 years old | Male dominant (male:female = 2:1), associated with cigarette smoking, environmental exposures, chronic viral infections | Estimated five-year survival: 45–70%, Median survival; 5 years |
UIP pattern; subpleural and basal predominance, honeycombing, reticular abnormality, absence of features inconsistent with UIP pattern (e.g. cysts, marked mosaic attenuation, predominant GGO) | Fibrosis and honeycombing predominantly in subpleural and paraseptal area, absence of certain features (e.g. hyaline membranes, organizing pneumonia, granulomas, marked interstitial inflammatory cell infiltrates, airway predominant) | No environmental exposure known to cause HP No SARD |
Anti-periplakin169, anti-HSPA2168 reported. | TOLLIP112, TLR3113, IL1RN191, IL8192, IL4193, TGFB1194, |
| NSIP202 | 1 to 9 per 100,000 people Further classified as cellular- (20%) and fibrosing- (80%) NSIP |
50–60 years old | Female dominant (60–70%), 60–70% were never smokers |
Five-year survival: 82.3%, Fibrosing has worse prognosis than cellular NSIP | NSIP pattern (basilar predominant GGO with or without subpleural sparing, reticular abnormality) | Temporally homogeneous inflammation and/or fibrosis, diffuse alveolar wall thickening by uniform fibrosis, Fibrotic foci and peripheral attenuation are typically absent |
No history of exposure associated with HP No SARD |
Not present. | Not reported. |
| SARD-ILD7,203 (distinct criteria for each subtype) | Varies by SARD (RA, 11%; SSc, 47%; MCTD, 56%; IIM, 41%; SLE, 6%) | Varies Smoking, male gender, presence of RF and anti-CCP antibody in RA-ILD | Varies | Varies | Varies (UIP predominant in RA-ILD, NSIP more common in other SARDs) | Varies | Concomitant with SARDs, such as rheumatoid arthritis, SSc, MCTD, SS, IIM, SLE | Auto-antibodies common* | RA-ILD; TOLLIP204, HLA-DRB1, SSc-ILD; STAT4205, IRF5205 |
| HP11 (2020) | 0.3–0.9 per 100,000 population | 50–60 years old | No gender preference Less common in current and past smokers Antigen exposure (e.g., fungal, bacterial, animal, protozoal, and insect) |
Varies among the subtypes. 58% of patients with fibrotic HP stayed alive 7 years after diagnosis | Acute HP (< 6 week) and subacute HP (6–23 week): upper lobe predominant GGO, mosaic attenuation, air trapping Chronic HP (>= 24 wks): upper lobe predominant reticular abnormality, peribronchovascular interstitial thickening, honeycombing, mosaic attenuation, air trapping, centrilobular nodules, relative sparing of the basis |
Nonfibrotic HP: cellular interstitial pneumonia, cellular bronchiolitis, poorly formed non-necrotizing granulomas Fibrotic HP: In addition to the above findings, chronic fibrosing interstitial pneumonia and/or airway-centered fibrosis are foci of organizing pneumonia |
History of environmental exposure known to cause HP No SARD |
Not present | HLA-DRB1*04206, TOLLIP207, TNF206 |
| uILD208,209 (2013) | 10–25% in ILD cohorts | 50–60 years old | Varies | Estimated five-year survival rate is 46–70% | Varies | Varies | No history of exposure associated with HP No SARDs |
Not present | Not assessed |
Common autoantibodies in SARD include e.g., RF, anti-CCP, anti-Scl70 (SSc), anti- centromere, anti-RNA polymerase III, anti-U1 RNP, anti-SS-A, anti-Jo-1 (myositis), anti-dsDNA. IPF, idiopathic pulmonary fibrosis; UIP, usual interstitial pneumonia; GGO, ground glass opacity; HP, hypersensitivity pneumonitis; SARD, systemic autoimmune rheumatic disease; iNSIP, idiopathic non-specific interstitial pneumonia; RA, rheumatoid arthritis; SSc, systemic sclerosis; MCTD, mixed connective tissue disorders; IIM, idiopathic inflammatory myopathy; SLE, systemic lupus erythematosus; RF, rheumatoid factor; CCP, cyclic citrullinated peptide; Scl-70, scleroderma-70; RA-ILD, rheumatoid arthritis associated interstitial lung disease; RNP, ribonucleoprotein; SS-A, Sjögren-syndrome-related antigen A; dsDNA, double stranded DNA; uILD, unclassifiable interstitial lung disease.
Morphological classifications
Usual interstitial pneumonia (UIP).
UIP is the archetypal histopathological lesion and chest imaging pattern found in fILD patients4. UIP is often idiopathic, that is, without a defined etiology. Histologically, UIP is defined by temporal and spatial heterogeneity of fibrotic lesions where fibroblastic infiltrates, mature fibrosis, “honeycomb” cysts, and scar tissue alternate with areas of normal lung parenchyma (Table 1)4. On chest high resolution computed tomography (HRCT) imaging, the UIP pattern consists of reticular opacities from thickened interstitium, honeycomb changes from cystic dilation and alveolar wall destruction, and enlarging airways caused by contraction of the fibrotic lung tissue (i.e., traction bronchiectasis). A key feature of UIP is that these findings arise predominantly in basilar and subpleural patterns. In the clinic, patients with UIP without known etiology carry the diagnosis idiopathic pulmonary fibrosis (IPF). IPF is the most common subtype of fILD and is the major focus of translational and experimental studies. UIP morphology can also manifest in a subpopulation of other fILD, such as systemic autoimmune rheumatic disease-related ILD (SARD-ILD).
Non-specific interstitial pneumonia (NSIP).
Radiographically, NSIP is defined by a pattern of inflammation and fibrosis without the architectural distortion found in UIP. Histologically, NSIP is more inflammatory than UIP, consisting of varying amounts of lymphocytes, plasma cells, and fibrosis characterized by collagen bundles with few fibroblasts. Unlike UIP, the lesions in NSIP exhibit temporal uniformity5. NSIP can be subcategorized into cellular (primarily inflammatory) and fibrotic (primarily fibrotic or mixed cellular and fibrotic) phenotypes. Cellular NSIP is more responsive to immunosuppressive treatment and has a better prognosis. Fibrotic NSIP is less reversible and has a poorer prognosis6. Like UIP, the morphology of NSIP can be idiopathic or can be found in a subpopulation of other clinical entities.
Clinical classifications
Systemic autoimmune rheumatic disease-related ILD (SARD-ILD).
While most cases of fILD are idiopathic, pulmonary fibrosis is also a serious complication of many SARDs. A recent meta-analysis determined the prevalence of clinically-apparent ILD in patients with SARDs ranges from 6–60%, with highest prevalence in mixed connective tissue disease (MCTD) and systemic sclerosis (SSc)7,8. SARD-ILD patients generally have better prognosis than patients with IPF. Lung pathology among SARD-ILD patients is heterogeneous, with fibrotic phenotypes contributing to excess morbidity and mortality9. Conversely, ~34% of patients with ILD can have features of SARDs, such as characteristic physical exam findings or the presence of autoantibodies10, without meeting full diagnostic criteria for a specific SARD. These patients with “interstitial pneumonia with autoimmune features” (IPAF) have a mildly better prognosis than IPF patients, but worse than SARD-ILD patients. The prevalence of histological subtypes, such as UIP or NSIP morphologies, differs among SARDs (Table 1)9.
Hypersensitivity pneumonitis (HP).
Like SARD-ILD, HP is an immune-mediated inflammatory and/or fibrotic lung disease. HP primarily affects the lung parenchyma and small airways arising from an immune reaction to inhaled antigens11. Histologically, HP can share fibrotic subfeatures of UIP, but also demonstrates peribronchiolar metaplasia and poorly formed, non-necrotizing granulomas. HRCT classifies HP as either non-fibrotic or fibrotic, and the extent of fibrosis correlates with symptom severity, disease progression, and patient survival11.
Despite differences in morphological and clinical classification schemes for fILD, progression of fibrosis remains the prime determinant of lung function and mortality. For this reason, clustering patients by shared lung morphology is gaining traction in the field. For example, some patients with rheumatoid arthritis-associated ILD (RA-ILD) resemble IPF patients with radiologic and histologic UIP patterns. These RA-UIP patients have increased mortality that more resembles the natural history of IPF patients than that of RA-ILD patients without UIP9. Hence, rather than classifying IPF as its own separate category, clinicians and researchers may benefit from considering IPF, RA-UIP, and other fILD with UIP features as one UIP group, distinct from RA patients without UIP. In the future, the current histopathological and radiological definitions of morphological subtypes should be refined by more granular immunophenotyping.
RESEARCH TOOLS
Working knowledge of clinical and experimental methods used to study immunological pathways in fILD is essential to understanding current efforts to refine our understanding of morphological subtypes. Over the past ten years, advances in biospecimen procurement, bioinformatic analysis, and experimental models have transformed our ability to study the immunology of fILD (Figure 2). Among these, human cohorts and animal models of fibrotic lung disease are indispensable. A growing number of established human cohorts and biorepositories helps define the natural history of disease and provides opportunities for molecular immunophenotyping. These translational findings can then be investigated in relevant animal models to test for novel mechanisms and potential treatment targets. The pairing of human cohort data with mechanistic studies in animal models has vastly improved our ability to translate laboratory findings to clinical practice.
Figure 2. Research methods in lung fibrosis.
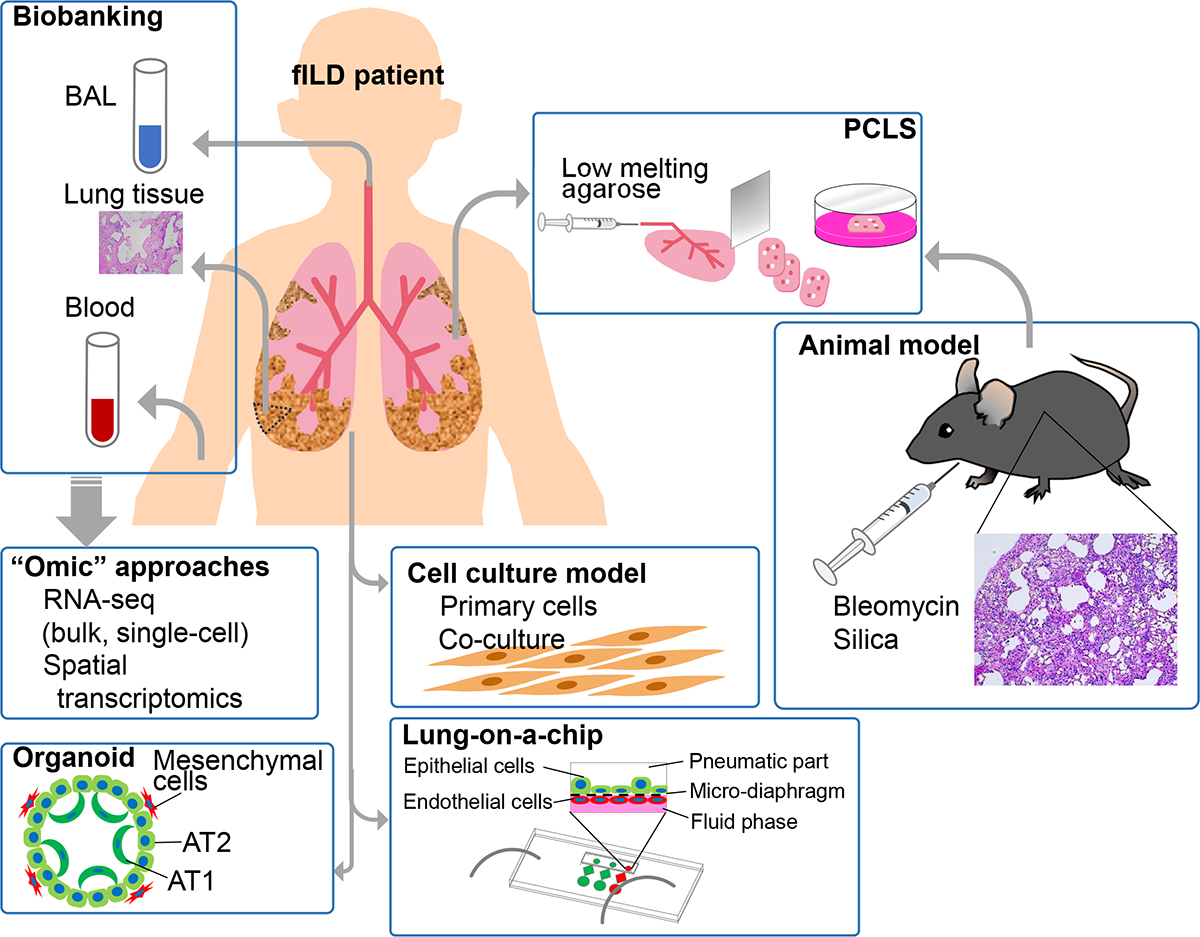
Human samples for translational studies of patients with fibrotic lung interstitial lung disease include bronchoalveolar lavage fluid (BAL), lung tissue obtained during biopsy or explant, and blood. Biobanked specimens can be analyzed by “omic” approaches, while primary cells may be isolated from fresh tissue samples for conventional two-dimensional tissue culture studies or three-dimensional organoids or complex in vitro model systems like “lung on a chip”. Lung tissue can also be used to generate precision cut lung slices (PCLS). Animal models induce lung fibrosis via bleomycin or silica exposure.
Human cohorts
Human research cohorts provide a rich resource of clinical data and biospecimens for the study of fILD (Table 2). Retrospective studies are useful to define incidence, prevalence, risk factors, and natural history of ILD in population-based studies. In smaller-scale retrospective studies, a detailed review of clinical data, including imaging and pathology results, is feasible for careful phenotyping of ILD. Prospective studies can standardize clinical data collection, including additional research questionnaires, physical exams, as well as provide research biospecimens, such as peripheral blood, bronchoalveolar lavage fluid (BALF), and lung tissue. Molecular studies rely on a range of biobanks, from private institutions, like the Mass General Brigham Biobank, to national institutes, like the UK Biobank, and the Lung Tissue Research Consortium (LTRC) at the U.S. National Institutes of Health (available via the NIH BioLINCC, biolincc.nhlbi.nih.gov/studies/ltrc/).
Table 2.
Selected human cohorts investigating fILD prevalence, incidence, and progression.
| Study name | Study design | Location and sample size estimate | ILD pheno typing | Bio-specimen |
|---|---|---|---|---|
| Rochester Epidemiology Project210 | Population-based retrospective study | Olmsted County, MN, USA (n=500,000) | UIP | None |
| Taiwanese National Health Insurance Research Database211 | Population-based retrospective study | Taiwan (n=23,000,000) | ILD | None |
| Viral RNA in UIP212 | Retrospective study with retrieval of lung biopsy specimens | Birmingham, AL, USA (n=17) | UIP | Lung |
| Bronchoalveolar lavage in ILD213 | Prospective study of bronchoalveolar lavage | Fukuoka, Japan (n=31) | ILD | BAL, blood |
| LTRC214 | Prospective study of lung tissue | Multi-site, USA (n=4,200) | ILD, quantitative CT | Lung |
| UK Biobank215 | Prospective biobank study | Multi-site, United Kingdom (n=500,000) | UIP | Blood |
| MGB Biobank216 | Prospective biobank study | Boston, MA, USA (n=140,000) | ILD | Blood |
| Framingham Heart Study217 | Prospective general population study | Framingham, MA, USA (n=5,209) | ILA, quantitative CT | Blood |
| MESA218 | Prospective general population study | Multi-site, USA (n=6,814) | Quantitative CT | Blood |
| AGES176 | Prospective general population study | Multi-site, Iceland (n=5,320) | ILA | Blood |
| COPDGene176 | Prospective study of smokers | Multi-site, USA (10,371) | ILA, quantitative CT | Blood |
| Unaffected first-degree relatives of patients with pulmonary fibrosis175 | Prospective study among relatives of fILD | Boston, MA, USA (n=105) | ILA | Blood |
| UKILD-Long COVID177 | Prospective study of suspected ILD after COVID-19 | Multi-site, United Kingdom (n=12,000) | ILA, quantitative chest imaging | BAL, blood |
| VARA219 | Prospective registry of veterans with RA | Multi-site, USA (n=2,328) | ILD | Blood |
| Subclinical ILD in RA220 | Prospective study of patients with RA | Denver, CO, USA (n=194) | ILA, quantitative CT | Blood |
| BRASS-Lung178 | Prospective study of patients with RA | Boston, MA, USA (n=120) | ILA | Blood |
| SAIL-RA | Prospective study of patients with early RA | Multi-site, USA (n=160) | ILA | Blood |
| ANCHOR-RA | Prospective study of patients with RA | Multi-national (n=1,200) | ILA | Blood |
BAL, bronchoalveolar lavage fluid; CT, computed tomography; ILA, interstitial lung abnormalities; ILD, interstitial lung disease; UIP, usual interstitial pneumonia; RA, rheumatoid arthritis.
Experimental models of fibrotic ILD
Nearly all experimental studies of fILD employ rodent models. These models include administration of silica, asbestos, fluorescein isothiocyanate, thoracic radiation, and, most commonly, bleomycin12,13. In 2017, an American Thoracic Society workshop report described the bleomycin model as the best characterized model of lung fibrosis with highly relevant pathobiology12. Bleomycin is a chemotherapeutic glycopeptide that cleaves DNA thereby precipitating cellular injury. The most common model is a single bleomycin dose delivered to the lung. Epithelial cell injury is followed by inflammatory cell recruitment during the first 7 days. Ultimately, acute inflammation subsides leaving a chronic inflammatory milieu with fibrosis evident by day 14. Most studies end on day 21 (peak fibrogenic response), and fibrosis partially resolves over the subsequent month14. Bleomycin can also be delivered systemically by injection or osmotic pump. In these systemic models, initial injury is to the vasculature. After a single-dose of intrapulmonary bleomycin, the mouse lung shows patchy alveolar damage, alveolar hyperplasia, deposition of provisional matrix, inflammation, fibroproliferation, induction of senescence, and even areas of traction bronchiectasis15. In support of bleomycin’s clinical relevance, transitional epithelial cells noted in human UIP pathology have also been identified following bleomycin exposure in mouse lung16,17. However, a key feature of UIP pathology is fibroblastic foci, which are not reliably reproduced by this model. The partial resolution of fibrosis over time is also poorly representative of progressive fILD. Hence, a growing number of investigators use repeated dosing of bleomycin18 to model more persistent lung fibrosis.
While advances in gene sequencing and bench immunology have revolutionized our ability to investigate human disease, human cohorts and animal models have played an essential part in investigating the immunology of fILD. Continued advances in both their development and utilization is crucial for future discoveries of pathogenic immune axes and therapeutic targets.
NON-IMMUNE AXES IN LUNG FIBROSIS
Our review seeks to motivate further work in the immunology of fILD in part because the field has made the most progress on non-immune mechanisms of lung fibrosis. In normal lung, type 1 alveolar epithelial cells (AT1) are responsible for gas exchange and are renewed by type 2 alveolar epithelial cells (AT2), which possess stem cell-like properties. Recurrent injury or host-susceptibility factors, like age or early senescence, can exhaust AT2 renewal capacity and impair alveolar integrity19 (Figure 3). Consequently, dysfunctional lung epithelium produces pro-fibrotic cytokines and chemokines, such as transforming growth factor-β (TGF-β), platelet derived growth factor (PDGF), and matrix metalloproteinases (e.g., MMP-1 and MMP-7). Additionally, lung epithelial cells acquire mesenchymal phenotypes. Functionally, epithelial-to-mesenchymal transition is the direct transformation of epithelial cells into mesenchymal cells, contributing to the formation of myofibroblasts in pulmonary fibrosis19. Myofibroblasts express contractile proteins and secrete excessive ECM, thereby causing tissue fibrosis19. Compared to the substantial work investigating epithelial biology in fILD, understanding of the immune pathways in translational and experimental fILD represents a relative gap in knowledge in the pathobiology of fILD fibrosis.
Figure 3. Immune and stromal cell interactions drive fibrosis in fILD.
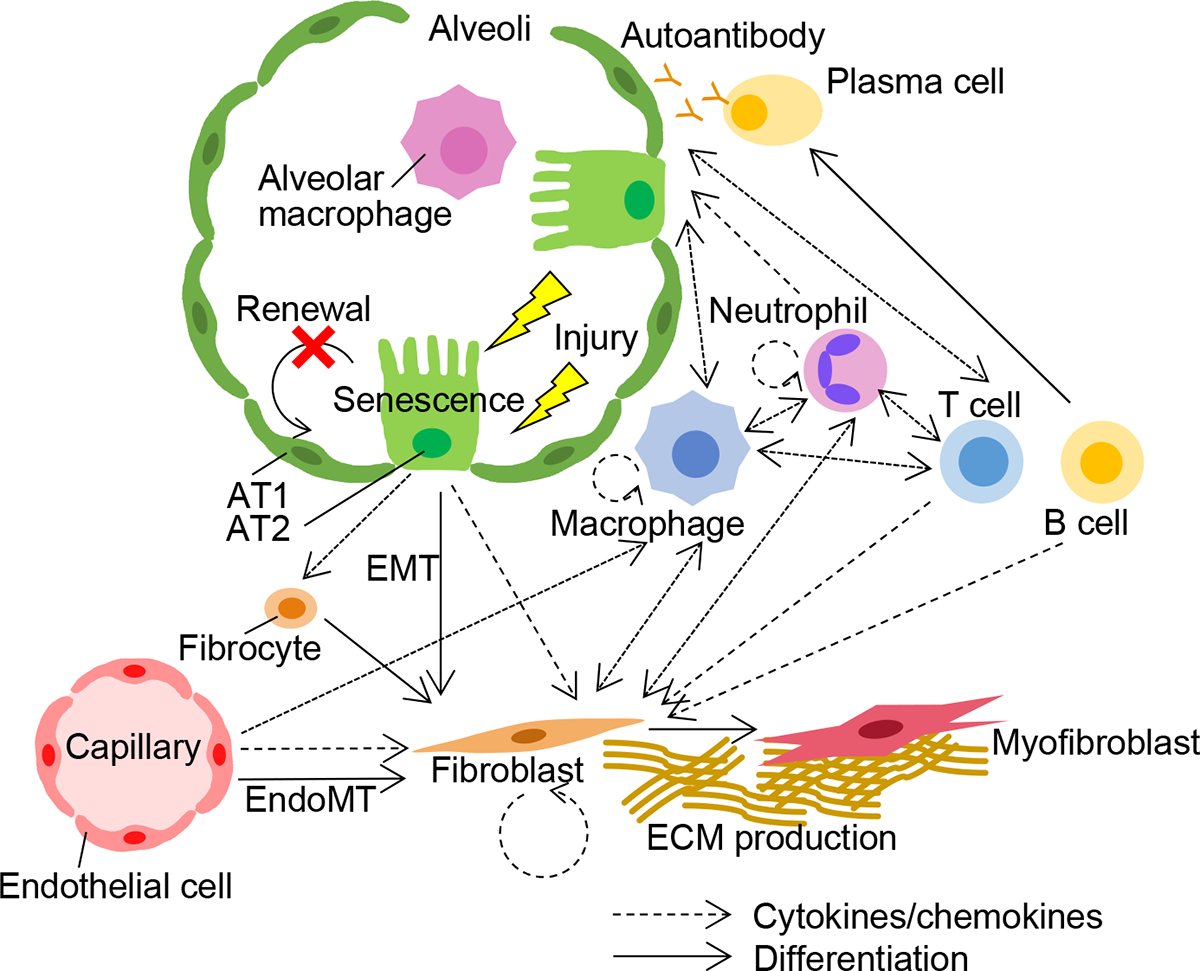
Epithelial cells exhibit aberrant repair responses after recurrent injury in the setting of risk factors like age, smoke exposure, or increased cell senescence. The dysfunctional epithelial cells profibrotic mediators, leading to a cascade of cell-cell interactions among the epithelium, endothelium, mesenchyme, and immune system drives lung fibrosis (i.e., deposition of extracellular matrix [ECM] in the lung interstitium by myofibroblasts). In addition to the proliferation of fibroblasts, the transition of type 2 alveolar epithelial cells (AT2) and endothelial cells (EMT and Endo-MT, respectively) to mesenchymal cells contribute to the accumulation of fibroblasts.
INNATE IMMUNE AXES IN LUNG FIBROSIS
Macrophage populations in lung fibrosis
Several lines of evidence implicate macrophages in a range of clinical fILD. Increased alveolar macrophages are observed in fibrotic lung tissue from both IPF and SARD-ILD patients15. Elevated monocyte counts in BALF are associated with poor prognosis in IPF patients20. In SSc-ILD patients, circulating classical CD14+ monocytes with dysregulated pro-fibrotic programs accumulate in fibrotic foci in their lungs21. Lung macrophages can be categorized by both cell lineage history and their anatomical location (i.e., airway, alveolar, and interstitial). For example, transcriptional and lineage analyses show that resident alveolar macrophages are self-renewing and distinct from monocyte-derived macrophages recruited from extra-pulmonary locations. These different myeloid cell populations likely have distinct contributions to the pathophysiology of lung fibrosis. For example, experimental models indicate monocyte-derived macrophages are more important drivers of disease than resident macrophages. Monocyte-derived alveolar macrophages reside near areas of fibrosis and express pro-fibrotic and extracellular matrix remodeling genes22–24. In line with these findings, deletion of monocyte-derived lung macrophages attenuates lung injury22–24, while deletion of resident alveolar macrophages did not affect fibrosis in an asbestosis-induced model of pulmonary fibrosis24. Monocyte-derived macrophages undergo apoptosis and decline in number during the later, fibrotic stages of murine lung fibrosis25.
M1 and M2 macrophages
Macrophages have been dichotomized into M1 and M2 phenotypes (Figure 4). M1 macrophages were originally defined as nitric oxide producing cells and are considered pro-inflammatory, a phenotype resembling “classically activated” by IFNγ, TNFα, GM-CSF, or lipopolysaccharide stimulation. M2 macrophages were originally defined as arginase-1 expressing cells and are anti-inflammatory, and include macrophages “alternatively activated” by IL-4 or IL-1326. Th1 cells produce IFNγ to stimulate M1 macrophages to produce pro-inflammatory cytokines, like IL-1β and TNFα. These stimuli induce characteristic M1 functions, such as upregulation of inducible nitric oxide synthase 2 (iNOS) and production of reactive oxygen species (ROS), inflammatory cytokines (e.g., IL-1β, IL-6, IL-12, TNFα, and CXCL10), and surface expression of MHC II, CD80, and CD86. M1 macrophages with their pro-inflammatory and ROS generating capabilities are critical to clearing infection, particularly of intracellular organisms and viruses. M1 macrophages also are implicated in numerous non-infectious, inflammatory contexts, ranging from atherosclerosis to autoimmune demyelinating disease. Conversely, M2 macrophages help resolve inflammation and augment tissue repair through production of TGF-β and various growth factors, such as fibroblast derived growth factor, PDGF, and vascular endothelial growth factor.
Figure 4. Th1/2, M1/M2, and Th17 axes in fILD.
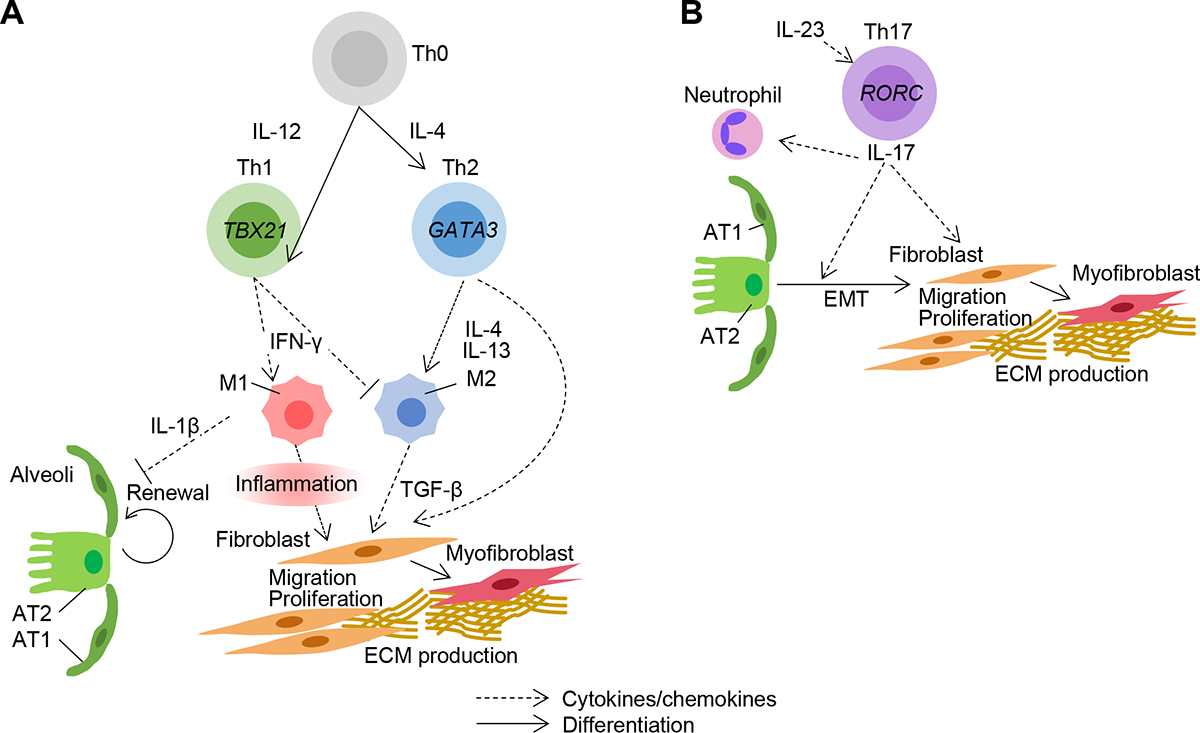
A. Naive T cells, also known as T-helper type 0 (Th0) cells, can differentiate into Th1 (via stimulation by IL-12) or Th2 cells (via IL-4). Th1 cells are on balance protective against lung fibrosis. Th1 cells promote M1 and inhibit M2 phenotypes in macrophages. M1 macrophages contribute to pathogenesis pathogenic by prolonged production of IL-1β that inhibits healthy epithelial cell renewal and inflammatory stimulation of fibroblast migration and proliferation. Th2 cells are pathogenic and drive M2 phenotypes. M2 macrophages secrete TGF-β that promotes the differentiation of fibroblasts into myofibroblasts producing ECM. B. Th17 cells are pathogenic and produce IL-17 that activates neutrophils and augments EMT. These mesenchymal cells migrate, proliferate, and differentiate into myofibroblasts producing ECM.
The M1/M2 classification has been an important framework for macrophage studies in fibrosis for over 20 years. M2 macrophages promote extracellular matrix deposition, angiogenesis, and tissue remodeling as part of wound healing. However, these homeostatic functions by M2 macrophages can go awry, leading to pathogenic fibrosis in a range of organs, including skin, kidney, liver and lung. Consistent with the disease paradigm of aberrant lung repair leading to fibrosis, M2 macrophages and monocytes are increased in fILD patients. Markers of M2 macrophages, like CD163, are elevated on lung macrophages in IPF patients27, and SSc-ILD patients have increased CD163+ M2 monocytes in circulation28. M2 macrophages promote myofibroblast differentiation via Wnt/β-catenin signaling29 or secretion of profibrotic mediators like TGF-β, galectin-330, and S100A431. Investigation of M2 macrophages continues to reveal important pathologic mechanisms, including the recent discovery of the AP-1 transcription factor Fra-2 driving type VI collagen expression and lung fibrosis32.
Considering fILD as a strictly “M2” disease is too simplistic, however. The failure of IPF clinical trials targeting the canonical M2 cytokine IL-1333,34 underscores this perspective. Beyond the general redundancy of pathological mechanisms in lung fibrosis, at least two factors likely contribute to the failure of targeting M2-related pathways. First, M2 macrophages can have different effects depending on the time course of injury. These cells can reduce injury and promote healing without scarring, as in experimental myocardial infarction35 and skeletal muscle injury36. In experimental liver injury, macrophage depletion during the early healing phase worsened fibrosis, while macrophage depletion during the later fibrotic phase reduced myofibroblast accumulation and scarring37. Second, the stimuli driving M2 macrophages are more complex than simply cytokines like IL-13. Additional upstream factors, including M1 macrophages, modulate the M2 response. For example, in the murine bleomycin model, adoptive transfer of Ly6chi (“inflammatory”) monocytes increases the abundance of M2 macrophages in the lung and promotes fibrosis27.
Pro-inflammatory cytokines produced by M1 macrophages, such as IL-1β38,39 and TNFα40, are also implicated in the pathogenesis of fILD. M1 macrophages are likely major sources of the increased IL-1β found in BALF from IPF patients38. In the bleomycin model, knocking out the IL-1β receptor, IL1R1, attenuates both lung inflammation and fibrosis39. While some amount of interstitial macrophage-derived IL-1β is necessary for physiologic type 1 alveolar epithelial cells differentiation and tissue repair, sustained IL-1β production inhibits regeneration and promotes the dysfunctional repair that is a hallmark of lung fibrosis41. Production of mature IL-1β requires the inflammasome, and both NLRP3 and AIM2 inflammasomes are activated in alveolar macrophages in human and murine lung fibrosis42,43. In line with a pathogenic role for M1 macrophages generally and the inflammasome specifically, mice deficient in NLRP3 were resistant to lung inflammation and fibrosis44
As an alternative to the M1/M2 scheme, macrophages can be classified as CCR2+ (“inflammatory”) or CX3CR1+ (“reparative”) phenotypes. This scheme may provide a more robust understanding of macrophage contributions to fILD. CCR2 is a cell-surface receptor expressed by macrophages and T cells that binds CCL2, CCL7, CCL8, CCL13, and other ligands. CCR2+ macrophages drive inflammation in a range of contexts, including the early phase of wound healing, myocardial infarction, and infection26. CX3CR1 is a cell-surface receptor that binds CX3CL1 (fractalkine). CX3CR1+ macrophages promote tissue repair in both normal and pathological fibrosis across several organs. Both CCR2+ and CX3CR1+ subpopulations are pathogenic in lung fibrosis. Expression of CCR2 and CX3CR1 ligands (CCL2 and CX3CL1, respectively) is increased in the lungs of IPF patients45,46. In experimental models, Ccr2-deficient mice had less macrophage infiltration into the lung and were protected from bleomycin-induced fibrosis47, with the important caveats that Ccr2-deficient mice have broad deficits in the recruitment of myeloid cells and cytokine production from T cells48. In the bleomycin model, mice with global deletion of CX3CR1 had attenuated pulmonary fibrosis with reduced infiltration of bone marrow-derived fibrocytes and CD206+ macrophages in the lung49. Mice with targeted deletion of CX3CR1+SiglecF+ macrophages were protected from lung fibrosis, with a reduction in Pdgfra+ and Pdgfrb+ fibroblasts50. Hence, pathogenic macrophages in fILD come from both traditional “inflammatory” and “reparative” subsets, whether classified by the M1/M2 or CCR2/CX3CR1 schema.
Advanced macrophage phenotyping
Enabled by high-throughput methods in RNA sequencing at single-cell resolution (scRNA-seq), there is a growing appreciation for macrophage populations distinguished by markers other than traditional subsets. Single-cell and other analyses have highlighted the expression of two monocyte-derived macrophage subpopulations – heparin-binding EGF-like growth factor (HBEGF+) macrophages51 and platelet-derived growth factor A (PDGFA+)50 macrophages – in both IPF lung and experimental lung fibrosis. HBEGF51,52 and PDGFA50 are expressed in myeloid cells from multiple organs and are known to promote fibrogenesis, stromal chemotaxis, and wound healing.
Single-cell transcriptomic studies performed on IPF lung also identified an expanded population of SPP1+ alveolar macrophages adjacent to areas of fibrosis53–55. Secreted phosphoprotein 1 (SPP1, osteopontin), which is found both in the ECM and intracellularly, is expressed in a wide range of cell types, including bone, smooth muscle, endothelium and myeloid cells. SPP1+ macrophages are implicated in the pathogenesis of fibrosis in multiple organs other than lung, such as heart, kidney, liver, and skin56. Inhibition of SPP1 with intranasal administration of small interfering RNA (siRNA) decreases pulmonary fibrosis in vivo. The profibrotic functions of SPP1 include the promotion of TGF-β-mediated epithelial-to-mesenchymal differentiation57, and migration of lung fibroblasts in vitro58. Beyond SPP1 itself, SPP1+ macrophages express other pro-fibrotic genes, such as IL1RN (IL-1 receptor antagonist), MMP9, and CHI3L153,55. The biology of SPP1+ lung macrophages may be informed by studies in other organs. For example, in a murine model of myocardial infarction, platelet-derived CXCL4 was critical for SPP1+ macrophage differentiation, and Cxcl4-deficient mice were protected against fibrosis after myocardial infarction and kidney ischemiareperfusion injury59. Platelet-bound antigen-antibody immune complexes induced SPP1 expression in monocytes in vitro through CSF1 (macrophage colony-stimulating factor)59. In lung macrophages, IL-6 cooperated with CSF1 to induce SPP1 expression58. When SSc patients with and without ILD were treated with tocilizumab (anti-IL-6 receptor blocking monoclonal antibody [mAb]), the circulating levels of SPP1 were reduced58. A role for CSF1 may not be limited to SARD-ILD, as CSF1 expression by macrophages increased at the terminus of the “cell trajectory” analysis of scRNA-seq of IPF lung, which raises the possibility of an autocrine loop amplifying macrophage recruitment, activation, and expression of SPP155.
Macrophage axes in clinical fILD
The subtypes of fILD vary in their relative amounts of inflammation and fibrosis (Table 1), and SARD-ILD and IPF clearly have distinct underlying immune drivers. However, these fILD share several innate immune axes centered on myeloid cells. First, SARD-ILD and IPF patients can share the same M2-like axes. Serum expression of CHI3L1 (a marker of M2 macrophages) predicts worsening pulmonary function in IPF patients60 and the development of ILD in patients with RA61. Experimentally, CHI3L1 promotes alternative activation of macrophages62, fibroblast proliferation, and expression of profibrotic transcriptional programs60. As another example, in patients with SSc-ILD, peripheral monocytes are primed for increased expression of MMP9 (a prominent biomarker in IPF patients63) when stimulated by profibrotic cytokines TGF-β, IL-4, IL-10, and IL-1321.
Second, both IPF patients64 and SSc-ILD65 patients have increased numbers of M1-like monocytes in circulation. IPF patients have circulating monocytes with an inflammatory phenotype characterized by increased expression of type I interferon (IFN)-responses and of CD64 (FcγR1), an IFNγ-response gene64. In addition, a subpopulation of CD64+ monocytes with increased expression of CCL2 and type I IFN genes were expanded in IPF lungs. The abundance of peripheral blood monocytes, or their expression of CD64, have also been associated with worse clinical outcomes, including decreased forced vital capacity (lung function), 6-minute-walk distance20, and radiographic fibrosis64. This macrophage/CCL2 axis identified in IPF has also been described in SSc patients, where increased levels of CCL2 in serum66 and BALF67 were associated with clinical ILD and reduced pulmonary function.
Third, increases in SPP1+ macrophages have been identified in lungs from SSc-ILD and HP patients, as well as IPF patients53–55,68,69. Elevated serum levels of SPP1 correlate with lung function decline in SSc-ILD58,60. The continued expansion of single-cell studies to include SARD-ILD and HP patients, along with the application of new technologies like spatial transcriptomics and proteomics, will likely reveal shared macrophage populations and functional pathways across fILD in increasingly finer detail. For example, HBEGF+ macrophages are a population in IPF lung, but HBEGF+ macrophages also mediate fibroblast invasiveness in the synovia of rheumatoid arthritis patients52, therefore meriting examination in future studies of RA-ILD.
In sum, fILD is driven by a broad spectrum of traditional inflammatory and reparative macrophage populations, along with new subpopulations, such as SPP1+ or HBEGF+ macrophages that have been uncovered using single-cell analysis. While IPF and SARD-ILD are distinct clinical entities with different treatment approaches, these diseases share macrophage axes and candidate therapeutic targets. A key challenge is translating insights obtained from studying a brief and self-resolving injury in mice to understanding the progression of clinical fILD over years. We hypothesize that many patients with earlier-stages of fILD, including both IPF and SARD-ILD, have both the inflammatory and reparative myeloid populations identified in experimental lung fibrosis. The spatial distribution of these functionally distinct myeloid populations may influence the differentiation of these disease subtypes. A critical question is how the mixture of myeloid populations evolve in patients with rapid progression of lung fibrosis. Some patients may have local exacerbations of their inflammatory myeloid populations, while other patients may be dominated by a ‘runaway’ expansion of their “reparative” monocytes and macrophages. We hypothesize that patients with both types of progression are found across disease subtypes, such as both IPF and SARD-ILD, but these speculations require further translational study across larger patient cohorts.
Neutrophils
Along with macrophages, neutrophils are key effector cells in acute lung inflammation and the early response to lung infection. Neutrophils respond to infection with anti-microbial granules, extracellular traps (NETs) and phagocytosis of microbes. Although IPF patients have increased infiltration of neutrophils in the lung70, neutrophils are far less studied in fILD than macrophages. Neutrophils may drive lung fibrosis by release of neutrophil elastase71,72, MMPs63, leukotrienes, and NETs73–75. These mechanisms are also implicated in lung fibrosis. For example, small molecule inhibition or genetic deletion of neutrophil elastase reduced lung fibrosis in murine models of lung fibrosis71,72. Neutrophil elastase acts on multiple cell types in the lung, promoting proliferation of fibroblasts and their differentiation into myofibroblasts72. In acute lung injury and cystic fibrosis, the protease activity of neutrophil elastase acts on lung epithelial cells to potentiate the expression of inflammatory cytokines and chemokines, epithelial cell senescence, and cell death. These mechanisms may also apply to lung fibrosis. Neutrophils are also a major source of MMPs, which regulate ECM degradation and tissue remodeling in fibrosis. The levels of multiple MMPs, such as MMP3, MMP8, MMP9, are elevated in both the BALF and lung tissue of IPF patients, and the MMP levels correlate with disease activity63. In vivo experiments support a profibrotic role for MMPs. For example, MMP3- and MMP8-deficient mice were protected from bleomycin-induced pulmonary fibrosis76,77. MMP8-deficient mice had greater lung inflammation, with increased expression of Cxcl10 and Ccl3 (MIP-1α). Deletion of either chemokine in MMP8-deficient mice reversed their phenotype, with reduced inflammation and increased fibrosis in the bleomycin model77.
To fight infection, neutrophils release NETs, an extracellular network of chromatin fibers studded with antimicrobial peptides and enzymes. In the murine bleomycin model, neutrophil activation and NETs correlated with disease severity78. NETs directly augment lung fibroblast proliferation and myofibroblast transdifferentiation in vitro. NETs contain several components, such myeloperoxidase, histone 3, and IL-17, that have profibrotic effects on fibroblasts73,79. In vitro, NETs promoted the secretion of inflammatory cytokines, such as IL-1β, TNFα, and IFNγ from macrophages, contributing to both tissue injury and downstream recruitment of neutrophils and macrophages80,81.
We lack a definitive demonstration of the role of neutrophils in experimental pulmonary fibrosis, as it is unclear whether prior studies82 achieved complete neutrophil depletion78. Further functional studies in vivo are critical to interpret the growing descriptive findings for neutrophils in clinical fILD. IPF patients have an increased number of neutrophils in their lungs83, driven by elevated chemoattractants (e.g., CXCL1, CXCL8, and G-CSF)84,85. Elevation of these neutrophil-attracting chemokines is associated with increased mortality in IPF86. In IPF lung, neutrophils co-localize with SPP1+ profibrotic macrophages. These SPP1+ macrophages had increased expression of TREM2 and CD9, which are markers for profibrotic, “scar-associated macrophages” in other organs68. Neutrophils adjacent to SPP1+ macrophages secreted MMP9, IL-17A, and GM-CSF. IL-17A can have profibrotic interactions with epithelial cells and fibroblasts in vitro87 and in vivo38,88. GM-CSF inhibited collagen deposition in the bleomycin model89 but may be profibrotic in other contexts. The sum effect of neutrophil-derived cytokines is yet to be determined. The limited translational studies to date have focused on neutrophils in IPF, but neutrophils likely influence other types of fILD. For example, in lungs with the NSIP pattern of lung fibrosis, neutrophil elastase and NETs are located near myofibroblasts and fibrotic lesions73. As short-lived cells, neutrophil populations can rapidly rise and fall and change phenotype. Hence, there is a particular need to understand the role of neutrophils in acute exacerbations of fILD.
Other myeloid cells
Beyond monocytes, macrophages, and neutrophils, less numerous granulocyte populations have been shown to make a demonstrable contribution to the pathobiology of pulmonary fibrosis. Plasmacytoid dendritic cells (pDC) distinguish SARD-ILD. pDCs are highly inflammatory cells in SARD, a finding not established in IPF. Increased number of pDC infiltrate the lung and skin tissue of SSc patients90,91; and pDC are found in other inflamed tissue from SARD patients, such as the synovium in RA and skin lesions in systemic lupus erythematosus. pDC are the major source of CXCL4, a cytokine/chemokine that directly induces myofibroblast differentiation and collagen synthesis in stromal cells and endothelial cells in vitro92. CXCL4 can also promote differentiation of monocytes into pro-inflammatory and pro-fibrotic dendritic cells93. Patients with SSc-ILD were found to have elevated serum levels of CXCL4 that correlated with the extent of both skin and lung fibrosis94. This finding may account for the observation that macrophage populations in the SARD-ILD, like SSc-ILD, had increased type I IFN responses compared to IPF even though both clinical phenotypes share SPP1+ (more abundant in more fibrotic areas of lung) and FABP4+ (more abundant in the areas of lung with normal histology) macrophages69. In experimental lung fibrosis, gene knockout or antibody blockade of CXCL4 reduced lung fibrosis92, suggesting a therapeutic avenue to break a pro-fibrotic positive feedback loop from pDCs to macrophages and fibroblasts.
Myeloid-derived suppressor cells (MDSCs) are a heterogenous population of myeloid cells characterized by their immunosuppressive activity, which plays a crucial role in the pathophysiology of various conditions such as infection, cancer, and autoimmune diseases. The two main subtypes of MDSCs are monocytic MDSCs and granulocytic MDSCs. Compared with control subjects and chronic obstructive pulmonary disease (COPD) patients, MDSCs are more abundant among peripheral blood mononuclear cells (PBMCs) of ILD patients, including IPF, HP, NSIP, and SARD-ILD. In particular, monocytic MDSCs, rather than granulocytic MDSCs, were more abundant in PBMC of IPF patients95. MDSCs are protective in other organ fibrosis models, such as kidney and liver96,97. As in these other models, MDSCs derived from IPF patients suppress T cell proliferation in vitro, a finding that suggests MDSCs modulate the expression of co-stimulatory molecules on peripheral T cells from IPF patients95. Despite these observations, increased circulating MDSCs negatively correlate with lung function in IPF patients95. This finding of more MDSCs in patients with worse fibrotic disease might seem to refute the in vitro data and findings in other organs; however, one can hypothesize that the MDSC mobilization is an insufficient homeostatic attempt to attenuate pulmonary fibrosis. Alternatively, T cell subsets can be either pathogenic or protective in lung fibrosis, as we describe further below, and MDSCs may have different effects on these T cell subpopulations.
Complement
Cell-specific chemokines and cytokines are not the only molecules that mediate cross-talk among innate immunity, epithelium and fibroblasts. Other innate immune mediators, such as complement and ligands of toll-like receptors (TLRs), are expressed across multiple cell types and contribute to the pathophysiology of lung fibrosis. IPF patients have elevated levels of the complement component 3a (C3a) and 5a (C5a) in the lung, and increased C1q deposition in the lung is associated with poor prognosis in IPF patients98. Further, IPF lung epithelial cells express less of complement inhibitory proteins (e.g., CD46, CD55)99. Like other innate immune pathways, complement is also implicated in other clinical fILD beyond IPF. In HP, immune complexes deposited in the lung can activate complement100, which may then amplify the activation of innate immunity. Autoantibody production in SARD-ILD and IPAF patients also raises the possibility of increased complement activation in these diseases when compared to IPF; however, this hypothesis needs to be tested experimentally.
AT2, bronchiolar epithelial cells, alveolar macrophages, and polymorphonuclear leukocytes can synthesize complement proteins following inflammatory cytokine stimulation by IL-6, IL-1, TNFα, and IFNγ. Serine proteases from alveolar macrophages cleave and activate C5, which initiates a range of inflammatory signaling cascades. In the murine bleomycin model, C5-deficient mice101 and C1q deficient mice102 had reduced lung fibrosis, compared to control mice. Complement factors can drive disease via several cellular targets, such as the induction of neutrophil proteolytic enzymes and reprogramming of macrophages towards a M2 phenotype103. Complement also acts directly on lung epithelium and stromal cells. C3a induced expression of TGFB by lung epithelial cells, and C3a and C5a amplified that effect by downregulating SMAD7, a negative regulator of TGF-β99. C5a induces lung fibroblasts to express MMPs101, and C1q stimulates fibroblast proliferation104. In other end-organs105 complement can promote epithelial-to-mesenchymal transition, but this function is yet to be determined in lung fibrosis.
The effect of complement in different stages of lung fibrosis needs delineation. For example, despite an eventual reduction in lung fibrosis, C5-deficient mice had exacerbated acute lung inflammation, which suggests the role of complement varies between the early inflammatory and later fibrotic phrases. In a recurrent theme, understanding how the shorter time course of murine models correlates to human disease is a challenge. Like neutrophils, complement may be most relevant to acute exacerbations or acute injury responses to injury.
Toll-like receptors
TLRs are evolutionarily conserved pattern recognition receptors in innate immunity. Like complement, TLRs can mediate the earliest responses to infection, inflammation, and tissue injury. Among TLRs, work in fILD has focused on TLR4, which both activates inflammatory responses from immune cells and upregulates profibrotic molecules in lung fibroblasts. Inhibiting MD2, an accessory receptor for TLR4, reduced myofibroblast differentiation and ECM remodeling. Consistent with these results, TLR4 deletion in fibroblasts in vivo resulted in less lung and skin fibrosis following the systemic administration of bleomycin, a model of SSc106. HMGB1 is a ligand for TLR4 that is released in broad inflammatory responses. HMGB1 promoted fibroblast proliferation in vitro, and anti-HMGB1 mAb reduced lung inflammation and fibrosis in the murine model107. ECM proteins tenascin-c and fibronectin extra type III domain A (FN-EDA, a fibronectin splice variant) bind TLR4. Both tenascin-c108 and FN-EDA109 stimulate myofibroblast differentiation and promote lung fibrosis in vivo. Tenascin-c, in particular, was upregulated in the skin and serum samples obtained from SSc patients compared with those in healthy controls. Tenascin-c deficient mice showed attenuated collagen accumulation and fibrosis severity in both lung and skin, as well as spontaneous resolution of fibrosis in these organs in the repetitive subcutaneous bleomycin-induced systemic sclerosis model, suggesting the role of Tenascin-c in maintaining the persistent fibrosis108. These data link TLR activation, inflammation and fibrogenesis. However, TLR signaling may also play a reparative, anti-fibrotic role following lung injury. For example, fibroblast-specific deletion of Tlr4 reduced bleomycin-induced lung fibrosis106. Interestingly, mice with a global deficiency of Tlr4 had more severe lung fibrosis, perhaps due to impaired epithelial repair and capacity for renewal110. Thus, while dysregulated TLR signaling is evident in the pathophysiology of fILD, cellular heterogeneity in expression and response profiles makes therapeutic targeting complex.
There is evidence implicating TLR and their ligands in clinical fILD. BALF levels of HMGB1 are increased in patients with IPF and HP, compared with control107. Tenascin-c and FN-EDA are increased in the lungs of patients with IPF109,111, HP111, and SSc-ILD108. A genome-wide association study on European-American patients identified common variants in the Toll-interacting protein (TOLLIP) gene encoding a regulatory molecule of TLR-NF-kB signaling pathways, and their association with IPF susceptibility112. Another study detected a loss-of-function variant in TLR3 associated with poor prognosis in IPF113. These associations are more striking in some SARD, where most genetic polymorphisms associated with increased risk of SSc are related to TLR or other aspects of innate immunity114. Polymorphism in TLR2 is associated with SSc and the development of pulmonary arterial hypertension, but an association between TLR polymorphisms and SSc-ILD has not been defined115. The extensive literature on the microbiome in pulmonary fibrosis suggests that the microbiome can induce inflammation via TLR signaling (as previously reviewed116). Multiple questions remain, such as the relative contribution of microbial and endogenous TLR ligands, the contribution of TLR over the course of clinical disease, and the mechanistic contribution, if any, of genetic associations. In a range of contexts, from infection to autoimmunity, innate immunity has a major role in activating and modulating the adaptive immune response, and vice versa. A direction for future studies is the cross-talk between innate immunity and adaptive immunity, which remains relatively undefined in clinical fILD.
ADAPTIVE IMMUNE AXES
The medical management of fILD depends on either immunosuppressive or antifibrotic therapies depending on the disease and the individual. These treatment decisions rely in part on clinical judgment, as the randomized controlled trial (RCT) data for immunosuppressive medications targeting adaptive immunity are limited and of lower certainty in fILD117. In certain scenarios, glucocorticoids and immunosuppressive medications are used for SARD-ILD patients118; hence, clinical practice suggests a possible role for adaptive immunity in some SARD-ILD patients. Supported by RCT studies, clinicians avoid glucocorticoid steroids and immunosuppressants in IPF patients119, regardless of disease stage or acute exacerbation. Thus, current clinical practice argues against a role for adaptive immunity in IPF. Furthermore, clinical trials targeting T cell-related pathways, such as the administration of IFNγ or anti-IL-13 blocking mAb, have failed in IPF34,120. Rather than immunosuppressive medications, two anti-fibrotic drugs, nintedanib and pirfenidone, are indicated for patients with IPF4. Nintedanib is a small molecule tyrosine kinase inhibitor that attenuates PDGF, fibroblast derived growth factor, and vascular endothelial growth factor receptor signaling. Pirfenidone is a small molecule with a myriad of anti-fibrotic, anti-inflammatory, and anti-oxidant properties; its precise mechanism of action is unclear. Both drugs modestly slow the rate of lung function decline in IPF121, and nintedanib has a similar effect in SSc122.
Nonetheless, accumulating clinical and translational evidence suggests an adaptive immune component of IPF pathogenesis may have been underappreciated. Here, we seek to provoke reconsideration of adaptive immunity across the range of fILD, including IPF. We first review the contribution of T and B lymphocytes to lung fibrosis and place these findings in clinical context.
T lymphocytes
T helper (Th) cells
T cells are broadly classified into CD8+ cytotoxic T-cells, which interact with MHC class I, and CD4+ Th cells, which bind MHC class II. CD8+ T cells are primarily responsible for injuring infected or cancerous cells, while CD4+ T cells play a crucial role in coordinating immune responses by secreted cytokines. Traditional CD4+ T cell subsets, including Th1 (T-bet+ IFNγ+), Th2 (GATA3+), and Th17 (RORγT+), have been implicated in ILD (Figure 4 and reviewed in123). Th1 cells, which are derived from Th0 cells upon stimulation with IL-12, play a central role in cell-mediated immunity. Th1 cells produce IFNγ and activate macrophages and neutrophils, contributing to anti-tumor immunity and host defense against intracellular pathogens. In contrast to Th1 cells, Th2 cells, which are induced upon stimulation with IL-4, potentiate humoral immunity. Through the production of cytokines such as IL-4 and IL-13, Th2 cells stimulate antibody production and activate eosinophils, as in allergic diseases123. Th17 cells contribute to host defense against bacterial and fungal infections and have been implicated in autoimmune diseases and tissue injury in lung disease123.
While inflammation is often viewed as a contributor to fibrosis progression, not all proinflammatory factors contribute to fibrosis. In contrast to pathogenic pro-inflammatory M1 or CCR2+ macrophages, pro-inflammatory Th1 pathways are protective in experimental lung fibrosis (Figure 3A). In the bleomycin model, administration of IL-12 attenuated lung fibrosis, while deficiency of the Th1 transcription factor Tbx21 (T-bet) in CD4+ cells exacerbated fibrosis124. After treatment with bleomycin, T-bet-deficient mice had increased levels of Th2 cytokines and TGF-β. Unsurprisingly, given the profibrotic role of M2-like macrophages, Th2 cells potentiate experimental lung fibrosis. Th2-biased mouse strains, such as DBA/2, have more severe and prolonged pulmonary fibrosis compared to Th1-biased mice, like C57BL/6J, in the chronic bleomycin model125 and the antigen-driven model of chronic HP126. Furthermore, mice lacking IL-13 or IL-4Rα have less severe lung fibrosis127. CRTH2 is a prominent receptor on Th2 cells and innate lymphoid type 2 cells. CRTH2 stimulation by chitinase 3 like 1 drives alternative activation of macrophages and experimental pulmonary fibrosis. This pathway likely occurs in human IPF62.
IL-17 producing T cells can promote lung fibrosis. IL-17A drove experimental lung and skin fibrosis following intratracheal and intradermal administration of bleomycin, respectively38,88,128. IL-17A stimulates lung epithelial-to-mesenchymal transition, fibroblast recruitment, proliferation, and ECM production87. IL-17A is also secreted by innate immune, epithelial, and endothelial cells. The relative contributions of IL-17A secreted by T cells, as opposed to these other cells, have not been well-defined. IL-23 stimulates IL-17A production by Th17 and γδ T cells in the early inflammatory phase of the murine bleomycin model, thereby augmenting lung fibrosis129. Although IL-23 also activates fibroblasts, its adverse effects in the bleomycin model are likely mediated primarily by lymphoid cells, as IL-23 did not induce myofibroblast proliferation, differentiation, or ECM production in human lung fibroblasts in vitro130.
As with macrophages, the pathogenic role of T cells and associated cytokines in fILD is more nuanced than classical phenotypic functions. T cell function changes over time in experimental lung fibrosis. For example IL-4 has an early anti-inflammatory role that changes to a profibrotic role later in the chronic stage131. Similarly, although IL-4 deficient mice have worse acute lung injury, they have less lung fibrosis over time131. This observation speaks to the importance of coordinated immunologic responses to lung injury where IL-4 modulates the early inflammatory response but drives an M2 macrophage-mediated pro-fibrotic response later in the disease course.
Th cells in clinical fILD
T cell populations clearly modulate the development and severity of lung fibrosis in experimental lung fibrosis. Correlating these insights with clinical disease has proven more difficult. In one example, IL-4 has a protective effect in the earlier, inflammatory phase of the murine model, the clinical correlation of this earlier inflammatory phase is uncertain131. Further, scRNA-seq analyses of end-stage IPF lung tissue showed comparatively little difference in the proportions of T cells and B cells compared to control, in contrast to the differences seen in macrophage populations53–55. T cells have a more obvious role in autoimmune disease (i.e., SARD-ILD) or in disease driven by environmental antigens (i.e., HP), than in IPF. In support of a role for T cells in SARD-ILD, the Th2 cytokines IL-4 and IL-13 are elevated in lungs from SSc-ILD patients132 and IL-17RA is increased in RA-ILD87. For clinical HP, the ratio of CD4+/CD8+ T cells in BALF is occasionally used to help distinguish this disease from other fILDs, like IPF or sarcoidosis. While Th1 profiles predominate in non-fibrotic types of HP, a transition to a Th2 phenotype is associated with fibrotic HP133.
Although the role of T cells in IPF patients is less conspicuous than in SARD-ILD patients, a growing body of translational data brings forward the question of Th cells in IPF patients. First, CD4+ and CD8+ T cells are increased in IPF, and increased CD8+ T cells in surgical lung biopsies were associated with worse lung function134. Experimental models have established the anti-fibrotic properties of Th1 cells and IFNγ and the pathogenic effect of Th2 cells. In concordance with this, IFNγ levels are decreased in the BALF or serum of IPF patients135,136. These translational and experimental findings may suggest a beneficial impact of Th1 and other IFNγ-producing cells. IPF patients have elevated levels of the Th2 cytokines IL-4 and IL-13136. Regarding Th17 cells, IPF patients have elevated expression of IL-17RA in their lungs87 and Th17 pathway genes (e.g., STAT3) in their PBMCs. Increased expression of this Th17 axis was associated with poor prognosis137. Hence, experimental and translational evidence has suggested a functional role for T cells in IPF. However, these findings must be reconciled with the lack of efficacy for glucocorticoids and other T cell-modulating medications in IPF. There are several possible explanations. Certainly, T cells may have little, if any, role in IPF. Alternatively, therapeutic intervention may require specific targeting of pathogenic T cells subsets rather than the nonspecific effects of glucocorticoid. Further, therapeutic intervention may need to be personalized to the phase of disease. For example, we can speculate that T cells have a role in the earliest stages of disease, prior to when most IPF patients first present to the clinic; but then, after non-immune axes are established, T cells then take a minor role in clinically apparent disease. In our discussion of “Future Directions,” we recommend longitudinal study of progressive IPF and its acute exacerbations, which may identify areas for more individualized intervention in adaptive immunity.
Regulatory T cells
Regulatory T cells (Treg) are CD4+ cells characterized by the expression of FOXP3 that play a crucial role in maintaining immune tolerance and immune homeostasis. Tregs manage other cell types by direct cytotoxicity, immune checkpoint activation, and secretion of cytokines like IL-10 and TGF-β1. The net role of Tregs in fibrosis is unclear. Adoptive transfer of Tregs attenuated pulmonary fibrosis after repeated doses of bleomycin in a chronic fibrosis model138; however, adoptive transfer of Tregs exacerbated lung fibrosis after a single dose of bleomycin139. Radiation-induced pulmonary fibrosis in mice was ameliorated by early depletion of Tregs with anti-CD25 mAb administered 2h after irradiation140. Depletion of Tregs increased Th1 and Th17 responses140. A protective role for Th17 cells in radiation-induced lung fibrosis that is distinct from their role in bleomycin models is just one of many possible explanations to reconcile the different results in the radiation, single dose bleomycin, and repeated dose bleomycin models.
Consistent with a protective role, the number of CD4+CD25+FOXP3+ Tregs in PBMC and BALF was decreased in ILD patients, including those with IPF and SARD-ILD. Tregs isolated from ILD patients had reduced proliferation and decreased ability to suppress Th1 and Th2 cells. More severely impaired Tregs were isolated from patients with more severe disease141. However, a different study found an increased proportion of activated Tregs in PBMC and BALF from IPF patients, compared with that of SSc patients and healthy controls142. These two studies reflect both a need for more fine immunophenotyping of Treg subpopulations in clinical fILD, but also a simple need for larger patient cohorts, as the latter study examined BALF from only 7 IPF patients. The latter study in particular examined patients with milder disease severity, so studies of Tregs in both earlier and later-stage clinical disease are needed. Finally, studies of the lung tissue itself are important. In contrast to the BALF findings, scRNA-seq demonstrated increased abundance of Treg in SSc-ILD lungs compared to that of healthy controls143.
Tissue-resident T cells
Just as resident lung macrophages are distinct from monocyte-derived infiltrating macrophages, tissue resident memory T cells (TRM) also have a distinct role in fILD. TRM cells persist long-term in peripheral tissues without ongoing antigen presentation. TRM cells populate mucosal organs such as the lung, skin, gut, and reproductive tract in both humans and mice. By being “on-site,” TRM can mount rapid defenses against respiratory infections. TRM cells promote proinflammatory responses in a variety of diseases, including psoriasis, asthma, and inflammatory bowel diseases. Similarly, increased CD8+ TRM cells have been found in the lungs of SSc-ILD patients143. Multiomic single-cell analysis with mass cytometry by time-of-flight (CyTOF) and single-cell RNA-seq identified increased relative abundance of CD103+CD4+ TRM, CD8+ TRM, and T effector memory cells expressing CD45RA (TEMRA) among T cells in IPF lung compared to control144. In IPF lung, these TRM cells demonstrated an activated IFNγ response phenotype and were associated with increased expression of TNF and perforin 1 (PRF1)144. Experimental models suggest a protective role for more inflammatory T cell axes. Consistent with this model, Th1 cell expansion with vaccinia vaccination after bleomycin improved fibrosis resolution, which was associated with increased lung TRM cells145. In another murine model, expansion of CD103lo CD4+ TRM by intranasal exposure to A. fumigatus drove fibrosis by enhanced production of IL-5 and IL-13146. These two studies demonstrate the need for further investigation of the heterogeneity of TRM contributions to the pathobiology of fILD.
Coactivating and immune checkpoint receptors
Coactivating receptors and immune checkpoints regulate the activation of immune responses and return to homeostasis. Recent studies raise the question whether these receptors have a role in fILD. Coactivating receptors may potentiate pathogenic T cells, as a study of 59 IPF patients found that increased expression of CD28 and ICOS in PBMCs was associated with worse lung transplant-free survival147. In lungs from IPF patients, another coactivator, CD40L, was expressed on activated T cells and B cells in tertiary lymphoid tissue148. Animal studies with anti-CD40L neutralizing mAb supported a pathogenic role for CD40L in autoimmune149 and radiation-induced pulmonary fibrosis150. CD40L+ T cells most likely interact with CD40 expressed on myeloid cells; however, CD40L+ T cells could also interact with fibroblasts, whose expression of CD40+ can be induced by IL-13148,151. Experimentally, engagement of CD40 stimulated fibroblast proliferation, adhesion, and IL-6 production152,153. The role of these and other coactivators on T cells merit further study in both IPF and SARD-ILD. For example, a polymorphism in the coactivator TNFSF4 (OX40L) is associated with susceptibility to SSc154, and OX40L was upregulated in the serum and fibrotic skin of SSc patients and correlated with worse prognosis in SSc-ILD155.
Recent studies have explored how immune checkpoints contribute to lung fibrosis. The immune checkpoint PD-1 is upregulated on T cells in the peripheral blood of IPF patients137,156. Several other subsets of PD-1hi T cells are increased in clinical fILD besides PD-1hi CD4+ T cells. PD-1hi Th17 subpopulations are increased in the lungs of sarcoidosis and IPF patients. Similarly, PD-1hiCD28null T cells are associated with poor prognosis in IPF patients147, and anti-PD-1 mAb accelerated fibrosis in humanized NSG mice given CD28null T cells157. Therapies targeting immune checkpoints may be beneficial in lung fibrosis. For example, PD-1hi CD4+ T cells from patients with sarcoidosis or IPF expressed TGF-β and stimulated collagen production in cocultured fibroblasts. This pathogenic effect was attenuated by PD-1 blockade137. In the bleomycin model, PD-1 knockout mice or mice treated with anti-PD-L1 mAb have reduced lung fibrosis137. Some studies of immune checkpoint blockade should be interpreted carefully, as these therapies may not act on T cell axes. A mass cytometric analysis of UIP lung tissue identified coexpression of CD47, FSP1, and PD-L1 in ACTA2+ fibroblasts in areas of lung fibrosis158, and fibroblasts with an invasive phenotype have especially high levels of PD-L1159. Treatment with anti-PD-L1 mAb had direct effects on fibroblast function159.
Like the role of T cells themselves, the role of their coactivators and immune checkpoints remains controversial in clinical fILD, particularly IPF. T cell coactivators and immune checkpoints merit further investigation via careful immunophenotyping with high-dimensional approaches, such as single-cell or spatial omics, that are coming to the fore. Certainly, any treatments modulating T cells would need to be approached cautiously, given the experience in oncology of treatment with immune checkpoint inhibitors causing ILD. Pre-existing interstitial lung abnormalities are known risk factors for ILD induced by immune checkpoint inhibitors160, but the mechanism of this association is not yet understood.
B lymphocytes
While it is not surprising that B cells contribute to SARD-ILD, they have also been implicated in UIP. IgA+ memory B cells are more abundant in the peripheral blood and lungs of IPF patients. In IPF lung, IgA+ memory B cells are colocalized with activated PD-1+ follicular T helper (Tfh)161. Before activated B cells terminally differentiate into plasma cells, plasmablasts are an intermediate cell type that express both B cell (CD19+) and plasma cell (CD38+CD27+) surface markers. Compared to healthy controls, IPF patients have an increased relative abundance of plasmablasts in circulation, which negatively correlates with pulmonary function162. Lung biopsies from IPF patients frequently contain CD20+ B cell aggregates and plasma cell infiltration163,164. In IPF lung, B cells can also organize into bronchus-associated lymphoid tissue (BALT). BALT is tertiary lymphoid organ with follicles containing germinal centers and follicular dendritic cells, and BALT is also prominent in many SSc and RA patients164. The presence of BALT correlates with increased expression of cytokines known to orchestrate interactions between T and B cells, such as CCL21, CXCL13, BAFF, and ICOSL164. Serum CXCL13 is associated with more severe disease and, together with MMP7, SPP1, CHI3L1, and CA-125, predicts transplant-free survival in IPF patients165,166. In addition to BALT, enlargement of lung-draining lymph nodes is common and predicts worse survival in fILD, including IPF167. In the future, mechanistic hypotheses for B cells can be tested in the murine bleomycin model, which develops germinal centers containing IgA+ and CD19+CD95+IgDlow B cells and plasma cells161.
The expansion of BALT and lymph nodes in clinical fILD raises the question of lung injury driven by antibodies. Serum and BALF IgG, including autoreactive antibodies, are increased in IPF patients168; and increased serum autoreactive IgA levels are associated with worse pulmonary function161. Furthermore, perivascular deposition of immune complexes is seen in UIP lung168. Several potential self-antigens for these antibodies have emerged. In one study, 40% of IPF patients had IgG auto-antibodies against periplakin, a component of desmosomes on lung epithelium; the level of this IgG auto-antibody correlated with decreased pulmonary function169. By contrast, IgG auto-antibodies against HSPA2 were associated with better survival in IPF patients, although the mechanism is unclear168. The potential pathobiological mechanisms for auto-antibodies require further study. In one hypothesis, anti-periplakin antibodies suppress alveolar epithelial cell migration in vitro and so may contribute to an aberrant lung injury repair169. Beyond their role in antibody production, peripheral B cells from IPF patients released more IL-6, IL-8, and MMP7 compared to healthy control after stimulation with microbial danger signals like CpG or β-glucan. These activated B cells also promoted the migration, proliferation, and activation of fibroblasts in vitro170. In support of a pathogenic role for B cells, CD19, a positive regulator of B cell activation, exacerbated lung injury in the murine bleomycin model171. In sum, while IPF patients are not considered to have an autoimmune disease, evidence suggests that B cells may be pathogenic in at least a subset of fILD without systemic autoimmune disease.
While T cells are relatively understudied in fILD, B cells are even less investigated and will benefit from inquiry with newer “omics” technologies. Bulk RNA-seq analysis of lung tissue from IPF patients identified increased expression of POU2AF1, a transcription factor mainly expressed in B cells. Mice lacking POU2AF1 were protected against bleomycin-induced pulmonary fibrosis172. More powerful discovery methods, like scRNA-seq, promise to finely define the role of B cells in IPF and other fILD. Single-cell approaches to B cell receptor and T cell receptor repertoire can detail the diverse range of lymphocyte receptors that enable recognition of antigens. Computational methods have been employed to elucidate shared specificity within B cell or T cell receptors, facilitating insights to immune responses. In addition, phage display techniques allow for the screening of target antigens173. Insights from other fibrotic organs may also drive future developments, particularly in how B cells interact with other cell types. For example, in a model of myocardial infarction, activated B cells secrete CCL7 and drive cardiac fibrosis by recruiting pro-fibrotic macrophages174. Such insights should inspire important investigations to examine similar B cell-macrophage interactions in fILD.
KNOWLEDGE GAPS AND FUTURE DIRECTIONS
A clear conclusion is the complexity of the immune response in fILD and the heterogeneity among patients and diseases. We recommend the fILD research agenda prioritize the following gaps in knowledge: First, immune mechanisms in earlier-stage fILD are poorly understood. Longitudinal studies of at-risk patients provide a valuable opportunity to study the initiation of disease and identify diagnostic and prognostic biomarkers. By screening patients before clinically-apparent ILD, one can detect subclinical or “preclinical” ILD. To investigate “preclinical” disease and its earliest-stages, prospective studies can enroll patients without a clinical diagnosis of ILD that are at elevated risk. Studies taking this approach target family members of patients with fILD175, smokers (COPDGene cohort176), post-COVID-19 patients177, and patients with SARDs without known ILD (BRASS-Lung, SAIL-RA, and ANCHOR-RA cohorts178). Collectively, observational studies inform the natural history of fibrotic lung disease, while associated biospecimens facilitate mechanistic investigations into its biologic underpinnings. Longitudinal studies also identify fILD patients that worsen at different rates. Identification of the regulators of disease progression, and their biomarkers, is important not only for the development of therapeutics but also for refinement of clinical trials. One study compared IPF patients with more stable disease to IPF patients with rapid progression of disease179. The lungs of IPF patients with rapid progression had increased expression of genes already implicated in disease, such as surfactant proteins and CCN1. Surprisingly, IPF patients with rapidly progressive disease showed the greatest differential transcriptional activity around the lung vasculature, rather than in fibroblastic foci179. This example highlights that longitudinal study of patients can illuminate new mechanisms not appreciated in end-stage patients that comprise the majority of single-cell analyses to date. However, opportunities for translational studies are highly limited, as fILD patients rarely undergo surgical lung biopsy and even bronchoscopy is becoming less common. One solution is multicenter efforts to pool biospecimens from patients undergoing lung surgery for ILD diagnosis or for removal of a lung cancer in the context of underlying fILD. Lung tissue from these clinically indicated surgeries should be complemented by research protocols that enroll patients for BAL and cryobiopsy or other endoscopic sampling of the lung tissue. While relatively few IPF patients have been immunophenotyped by high-throughput methods, there is an even more urgent need for immunophenotyping of non-IPF fILD.
Second, the most abrupt changes in lung function occur during acute exacerbations of pulmonary fibrosis, and the underlying mechanisms of acute exacerbation are poorly understood. Acute exacerbation is a rapid clinical deterioration that can be either idiopathic or associated with possible triggers, like infection or lung surgery. The pathology of acute exacerbation is typically diffuse alveolar damage superimposed on the pre-existing lung fibrosis. Although acute exacerbation is a major cause of morbidity and mortality in fILD, few studies have investigated the underlying molecular mechanisms. In contrast, acute exacerbations of obstructive lung diseases, like asthma or COPD, have been better defined at the cellular and molecular level. The innate immune system has been implicated in the pathogenesis of acute exacerbations. Acute exacerbation of IPF involves an increase in BALF levels of IL-23180 and neutrophils181, along with serum levels of neutrophil elastase182. Further, the number of BALF neutrophils correlated with patient mortality86. In an experimental model of acute ILD exacerbation induced by lipopolysaccharide, treatment with anti-IL-23 mAb was given to interrupt the activation of neutrophils. This treatment led to decreases in IL-17A levels, airway inflammation, and fibrosis180. Macrophages are also implicated in acute exacerbations. In scRNA-seq analysis of IPF lung, CCL18+ and SPP1+ macrophages partially overlap58, and CCL18+ macrophages are also found in SSc-ILD lung183. BALF levels of CCL18 predicted acute exacerbation of IPF patients181, and elevated levels of serum CCL18 was associated with mortality184. As one pathogenic mechanism, CCL18 can directly induce the production of collagen from lung fibroblasts185. To date, only bulk RNA-seq analysis, rather than more cutting-edge methods such as single-cell analysis, have been used to study acute exacerbations. Bulk RNA-seq analysis of BAL from IPF patients demonstrated increased expression of innate immune chemokines and their signaling during acute exacerbation186. Owing to smaller numbers of affected patients and the acuity of exacerbations, a fuller understanding of acute exacerbations will require both the assembly of large, multi-center biorepositories for sample collection, and the application of newer “omic” approaches.
Third, as highlighted above, currently available immunophenotyping data in clinical fILD is often based on older technologies. Newer, high-dimensional methods, such as single-cell analysis or high-resolution spatial ‘omics’ methods will provide a more granular profile of immune cells. Spatial approaches are important given the lung’s complex architecture, the spatial heterogeneity of fibrosis, and cell-cell crosstalk within fibrotic niches187. For example, spatial transcriptomic analyses of UIP lung found that the expression of regulatory genes, such as NFKBIZ, were associated with histological features of fibrosis188, but even histologically uninvolved areas had dysregulated gene expression patterns189. The early focus of these spatial studies have been epithelial and stromal cells; however, spatial “omic” methods are also ideal for examination of immune cells. Spatial transcriptomic analysis of human fILD lung will help contrast the cell-to-cell interactions for B cells within BALT, lymph nodes and elsewhere in the lung. Multimodal approaches will be of particular importance for immune cells. Combining single-cell transcriptomics with proteomics is highly effective for examining lymphoid subpopulations. To complement RNA-seq, single-cell assay for transposase-accessible chromatin (ATAC)-seq can identify the regulatory elements for immune cells, which are less defined than their effector programs. A critical advance will be combining genetic and single-cell approaches to define expression quantitative trait loci (eQTLs) in immune cell subpopulations. An example in healthy individuals combining genomic variants with single-cell analysis found that the behavior of eQTL variants depended on the T cell subpopulation190. Genome-wide association studies in IPF identified the most common genetic risk factor, MUC5B, as well as many rare but highly pathogenic variants (e.g., TERT, TERC) that do not have obvious immune-related mechanisms19. However, there are also mutational variants within several immune-related genes that have clear associations with IPF, such as TOLLIP112, TLR3113, IL1RN191, IL8192, IL4193, TGFB1194. Single-cell and other approaches can identify the cell types expressing these and other genomic associations. Subsequently, murine models with these genetic variants can test their function, as has been done for non-immune axes. For example, mice expressing human polymorphisms associated with epithelial dysfunction develop spontaneous lung fibrosis and ILD195–197. These findings could have therapeutic implications, as industry considers genomic associations to significantly de-risk therapeutic development for a candidate target gene.
Research initiatives to address the preceding gaps in knowledge should be complemented by two underutilized research approaches. First, the overlap of pro-fibrotic immunological programs across organ systems should be explored. Advances in high-throughput immunophenotyping may reveal fibrotic programs shared by lung and other organs. For example, our cross-tissue human fibroblast atlas at single-cell resolution of fILD lung, RA synovium, inflammatory bowel disease bowel, and Sjogren’s lip biopsies identified two fibroblast cell states shared across these four diseases – CXCL10+CCL19+ inflammatory fibroblasts near T cells and SPARC+COL3A1+ perivascular fibroblasts198. These add to known cross-organ findings, such as the expansion of SPP1+ macrophages in rheumatoid arthritis synovium, chronic kidney disease and heart failure59, CD9+TREM2+ macrophages in hepatic fibrosis68, or HBEGF+ macrophages in both lung fibrosis and rheumatoid arthritis synovium52,199. Finding pathogenic genes affecting multiple organs is important for drug discovery, as a therapeutic target with multiple disease indications is a more attractive investment for drug development. Second, how we can simplify yet replicate immune interactions in experimentally tractable model systems is essential for therapeutic development. Next-generation immunophenotyping will lead to an increasing number of candidate genes whose mechanisms need validation at the bench. Despite extensive efforts, most promising drug targets for fILD have failed in human clinical trial phases II and III. These trial failures stem from the complex pathobiology of ILDs, personalized drug responses, and over-reliance on simple in vitro and animal models. Consequently, research attention has turned towards newer in vitro models, like three-dimensional (3D) approaches ranging from precision cut lung slices (PCLS) and organoids to “lung-on-a-chip”200 (Figure 2). PCLS treated with a profibrotic cocktail recapitulated some of the transcriptional profiles of IPF lungs at single-cell resolution, including an increase in aberrant basaloid cells, collagen expressing ectopic endothelial cells and the gene signature of SPP1+ macrophages201. To better understand the interaction of immune cells with the epithelium and stroma, we encourage the co-culture of PCLS with immune cells. Since PCLS has its own weaknesses, combining newer in vitro models with murine studies will help complement their respective limitations and may improve their clinical translation.
Our review of the immune response in clinical and experimental fILD underscores its mechanistic complexity. Although tremendous progress has been made in defining immune cell types driving pulmonary fibrosis, important knowledge gaps have limited the clinical translation of immunotherapies for these diseases. Future immunotherapies will need to be tailored to individual patients and their stage of disease. Such “personalized medicine” will require earlier disease recognition, advanced immunophenotyping, and more representative experimental model systems. Through these ongoing investigative efforts, we are optimistic the field will develop approaches to reprogram profibrotic immune responses for patients with fILD, for whom treatment options are limited.
Acknowledgments
We acknowledge that we have omitted many important contributions to the field and their references due to space restriction. We appreciate the extensive manuscript editing by L.T. Merriam. T.J.D. is supported by the National Institutes of Health/National Heart, Lung, and Blood Institute (R01HL155522). F.Z. is supported by the Pharmaceutical Research and Manufacturers of America and the Arthritis National Research Foundation. J.A.S. is supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01 AR080659, R01 AR077607, P30 AR070253, and P30 AR072577), the R. Bruce and Joan M. Mickey Research Scholar Fund, and the Llura Gund Award funded by the Gordon and Llura Gund Foundation. W.M.O. is supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health (R01HL167718). E.Y.K. is supported by American Lung Association (DA-827785) and the BWH Bell Family Award.
Declaration of interests
In disclosures unrelated to this work: M.K. received research funding from GlaxoSmithKline. T.J.D.received research support from Bayer and Bristol Myers Squibb, consulting fees from Boehringer Ingelheim and L.E.K. consulting, and has been part of a clinical trial funded by Genentech, all unrelated to this study. L.K.D. received research grants from the Brazilian Ministry of Health (PROADI-SUS), Boehringer Ingelheim, and Bristol-Myers-Squibb. J.S.L. received grants from the NIH and Boehringer Ingelheim, an unrestricted research gift from Pliant, and consulting fees from Blade, Boehringer Ingelheim, United Therapeutics, Astra Zenca, and Eleven P15, all outside the submitted work. J.S.L. serves on the DSMB for UT, Avalyn and is an advisor for the Pulmonary Fibrosis Foundation, all outside the submitted work. L.K.D. received consulting fees from Boehringer Ingelheim, Roche, and Bristol-Myers-Squibb. J.A.S. has received research support from Bristol Myers Squibb and performed consultancy for AbbVie, Amgen, Boehringer Ingelheim, Bristol Myers Squibb, Gilead, Inova Diagnostics, Janssen, Optum, Pfizer, and ReCor unrelated to this work. C.M.H. serves in a scientific advisory role for the following companies: Lung Therapeutics, Lassen Therapeutics, Rubedo Life Sciences, and Structure Therapeutics. C.M.H. also consults for the Three Lakes Foundation. C.M.H. receives research funding from Kyowa Kirin Co, Ltd. B.B.M. is a grant review consultant for Boehringer-Ingelheim, the Pulmonary Fibrosis Foundation and the National Scleroderma Foundation. W.M.O. has received consulting fees from Nikang Therapeutics on a topic unrelated to the present manuscript. E.Y.K. receives research funding in fILD from Bayer AG, Roche Pharma Research and Early Development, and 10X Genomics. E.Y.K. has a PCT patent application (US2022/075673) concerning a method to treat fibrosis that is not mentioned in this manuscript. E.Y.K. has a financial interest in Novartis AG unrelated to this work. Other authors have nothing to declare. The funders had no role in the decision to publish or preparation of this manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard University, its affiliated academic health care centers, or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Zeng Q, and Jiang D (2023). Global trends of interstitial lung diseases from 1990 to 2019: an age–period–cohort study based on the Global Burden of Disease study 2019, and projections until 2030. Front. Med 10, 1141372. 10.3389/fmed.2023.1141372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zheng Q, Cox IA, Campbell JA, Xia Q, Otahal P, de Graaff B, Corte TJ, Teoh AKY, Walters EH, and Palmer AJ (2022). Mortality and survival in idiopathic pulmonary fibrosis: a systematic review and meta-analysis. ERJ Open Res 8. 10.1183/23120541.00591-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, Swigris JJ, Taniguchi H, and Wells AU (2017). Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primer 3, 17074. 10.1038/nrdp.2017.74. [DOI] [PubMed] [Google Scholar]
- 4.Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, Kreuter M, Lynch DA, Maher TM, Martinez FJ, et al. (2022). Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med 205, e18–e47. 10.1164/rccm.202202-0399ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doyle TJ, and Dellaripa PF (2017). Lung manifestations in the rheumatic diseases. Chest 152, 1283–1295. 10.1016/j.chest.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Travis WD, Hunninghake G, King TE, Lynch DA, Colby TV, Galvin JR, Brown KK, Chung MP, Cordier J-F, du Bois RM, et al. (2008). Idiopathic nonspecific interstitial pneumonia: report of an American Thoracic Society project. Am. J. Respir. Crit. Care Med 177, 1338–1347. 10.1164/rccm.200611-1685OC. [DOI] [PubMed] [Google Scholar]
- 7.Joy GM, Arbiv OA, Wong CK, Lok SD, Adderley NA, Dobosz KM, Johannson KA, and Ryerson CJ (2023). Prevalence, imaging patterns and risk factors of interstitial lung disease in connective tissue disease: a systematic review and meta-analysis. Eur. Respir. Rev. Off. J. Eur. Respir. Soc 32, 220210. 10.1183/16000617.0210-2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tana C, Drent M, Nunes H, Kouranos V, Cinetto F, Jessurun NT, and Spagnolo P (2022). Comorbidities of sarcoidosis. Ann. Med 54, 1014–1035. 10.1080/07853890.2022.2063375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doyle TJ, and Dellaripa PF (2017). Lung manifestations in the rheumatic diseases. Chest 152, 1283–1295. 10.1016/j.chest.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oldham JM, Adegunsoye A, Valenzi E, Lee C, Witt L, Chen L, Husain AN, Montner S, Chung JH, Cottin V, et al. (2016). Characterisation of patients with interstitial pneumonia with autoimmune features. Eur. Respir. J 47, 1767–1775. 10.1183/13993003.01565-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raghu G, Remy-Jardin M, Ryerson CJ, Myers JL, Kreuter M, Vasakova M, Bargagli E, Chung JH, Collins BF, Bendstrup E, et al. (2020). Diagnosis of Hypersensitivity Pneumonitis in Adults. An Official ATS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med 202, e36–e69. 10.1164/rccm.202005-2032ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jenkins RG, Moore BB, Chambers RC, Eickelberg O, Königshoff M, Kolb M, Laurent GJ, Nanthakumar CB, Olman MA, Pardo A, et al. (2017). An Official American Thoracic Society Workshop Report: Use of Animal Models for the Preclinical Assessment of Potential Therapies for Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol 56, 667–679. 10.1165/rcmb.2017-0096ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore B, Lawson B, Oury WE, Sisson TD, H T, Raghavendran K, and Hogaboam CM (2013). Animal Models of Fibrotic Lung Disease. Am. J. Respir. Cell Mol. Biol 49, 167–179. 10/f5ctv3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T, Meldrum E, Sanders YY, and Thannickal VJ (2014). Reversal of Persistent Fibrosis in Aging by Targeting Nox4-Nrf2 Redox Imbalance. Sci. Transl. Med 6, 231ra47. 10.1126/scitranslmed.3008182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, Chen C-I, Anekalla KR, Joshi N, Williams KJN, et al. (2017). Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med 214, 2387–2404. 10.1084/jem.20162152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang P, Gil de Rubio R, Hrycaj SM, Gurczynski SJ, Riemondy KA, Moore BB, Omary MB, Ridge KM, and Zemans RL (2020). Ineffectual Type 2-to-Type 1 Alveolar Epithelial Cell Differentiation in Idiopathic Pulmonary Fibrosis: Persistence of the KRT8hi Transitional State. Am. J. Respir. Crit. Care Med 201, 1443–1447. 10.1164/rccm.201909-1726LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strunz M, Simon LM, Ansari M, Kathiriya JJ, Angelidis I, Mayr CH, Tsidiridis G, Lange M, Mattner LF, Yee M, et al. (2020). Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun 11, 3559. 10.1038/s41467-020-17358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Degryse AL, Tanjore H, Xu XC, Polosukhin VV, Jones BR, McMahon FB, Gleaves LA, Blackwell TS, and Lawson WE (2010). Repetitive intratracheal bleomycin models several features of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol 299, L442–452. 10.1152/ajplung.00026.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moss BJ, Ryter SW, and Rosas IO (2022). Pathogenic Mechanisms Underlying Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol 17, 515–546. 10.1146/annurev-pathol-042320-030240. [DOI] [PubMed] [Google Scholar]
- 20.Kreuter M, Lee JS, Tzouvelekis A, Oldham JM, Molyneaux PL, Weycker D, Atwood M, Kirchgaessler K-U, and Maher TM (2021). Monocyte Count as a Prognostic Biomarker in Patients with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med 204, 74–81. 10.1164/rccm.202003-0669OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rudnik M, Hukara A, Kocherova I, Jordan S, Schniering J, Milleret V, Ehrbar M, Klingel K, Feghali-Bostwick C, Distler O, et al. (2021). Elevated Fibronectin Levels in Profibrotic CD14+ Monocytes and CD14+ Macrophages in Systemic Sclerosis. Front. Immunol 12, 642891. 10.3389/fimmu.2021.642891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, Chen C-I, Anekalla KR, Joshi N, Williams KJN, et al. (2017). Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med 214, 2387–2404. 10.1084/jem.20162152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCubbrey AL, Barthel L, Mohning MP, Redente EF, Mould KJ, Thomas SM, Leach SM, Danhorn T, Gibbings SL, Jakubzick CV, et al. (2018). Deletion of c-FLIP from CD11bhi Macrophages Prevents Development of Bleomycin-induced Lung Fibrosis. Am. J. Respir. Cell Mol. Biol 58, 66–78. 10.1165/rcmb.2017-0154OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joshi N, Watanabe S, Verma R, Jablonski RP, Chen C-I, Cheresh P, Markov NS, Reyfman PA, McQuattie-Pimentel AC, Sichizya L, et al. (2020). A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M-CSF/M-CSFR signalling in monocyte-derived alveolar macrophages. Eur. Respir. J 55. 10.1183/13993003.00646-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janssen WJ, Barthel L, Muldrow A, Oberley-Deegan RE, Kearns MT, Jakubzick C, and Henson PM (2011). Fas Determines Differential Fates of Resident and Recruited Macrophages during Resolution of Acute Lung Injury. Am. J. Respir. Crit. Care Med 184, 547–560. 10.1164/rccm.201011-1891OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gordon S, and Plüddemann A (2017). Tissue macrophages: heterogeneity and functions. BMC Biol 15, 53. 10.1186/s12915-017-0392-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, Van Rooijen N, Haslett C, Howie SE, Simpson AJ, et al. (2011). Ly6Chi Monocytes Direct Alternatively Activated Profibrotic Macrophage Regulation of Lung Fibrosis. Am. J. Respir. Crit. Care Med 184, 569–581. 10.1164/rccm.201010-1719OC. [DOI] [PubMed] [Google Scholar]
- 28.Mathai SK, Gulati M, Peng X, Russell TR, Shaw AC, Rubinowitz AN, Murray LA, Siner JM, Antin-Ozerkis DE, Montgomery RR, et al. (2010). Circulating monocytes from systemic sclerosis patients with interstitial lung disease show an enhanced profibrotic phenotype. Lab. Invest 90, 812–823. 10.1038/labinvest.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hou J, Shi J, Chen L, Lv Z, Chen X, Cao H, Xiang Z, and Han X (2018). M2 macrophages promote myofibroblast differentiation of LR-MSCs and are associated with pulmonary fibrogenesis. Cell Commun. Signal. CCS 16, 89. 10.1186/s12964-018-0300-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mackinnon AC, Gibbons MA, Farnworth SL, Leffler H, Nilsson UJ, Delaine T, Simpson AJ, Forbes SJ, Hirani N, Gauldie J, et al. (2012). Regulation of transforming growth factor-β1-driven lung fibrosis by galectin-3. Am. J. Respir. Crit. Care Med 185, 537–546. 10.1164/rccm.201106-0965OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang W, Ohno S, Steer B, Klee S, Staab-Weijnitz CA, Wagner D, Lehmann M, Stoeger T, Königshoff M, and Adler H (2018). S100a4 Is Secreted by Alternatively Activated Alveolar Macrophages and Promotes Activation of Lung Fibroblasts in Pulmonary Fibrosis. Front. Immunol 9, 1216. 10.3389/fimmu.2018.01216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ucero AC, Bakiri L, Roediger B, Suzuki M, Jimenez M, Mandal P, Braghetta P, Bonaldo P, Paz-Ares L, Fustero-Torre C, et al. (2019). Fra-2–expressing macrophages promote lung fibrosis. J. Clin. Invest 129, 3293–3309. 10.1172/JCI125366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maher TM, Costabel U, Glassberg MK, Kondoh Y, Ogura T, Scholand MB, Kardatzke D, Howard M, Olsson J, Neighbors M, et al. (2021). Phase 2 trial to assess lebrikizumab in patients with idiopathic pulmonary fibrosis. Eur. Respir. J 57, 1902442. 10.1183/13993003.02442-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parker JM, Glaspole IN, Lancaster LH, Haddad TJ, She D, Roseti SL, Fiening JP, Grant EP, Kell CM, and Flaherty KR (2018). A Phase 2 Randomized Controlled Study of Tralokinumab in Subjects with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med 197, 94–103. 10.1164/rccm.201704-0784OC. [DOI] [PubMed] [Google Scholar]
- 35.Morimoto H, Takahashi M, Shiba Y, Izawa A, Ise H, Hongo M, Hatake K, Motoyoshi K, and Ikeda U (2007). Bone Marrow-Derived CXCR4+ Cells Mobilized by Macrophage Colony-Stimulating Factor Participate in the Reduction of Infarct Area and Improvement of Cardiac Remodeling after Myocardial Infarction in Mice. Am. J. Pathol 171, 755–766. 10.2353/ajpath.2007.061276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arnold L, Henry A, Poron F, Baba-Amer Y, Van Rooijen N, Plonquet A, Gherardi RK, and Chazaud B (2007). Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med 204, 1057–1069. 10.1084/jem.20070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, and Iredale JP (2005). Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Invest 115, 56–65. 10.1172/JCI200522675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW, and Wynn TA (2010). Bleomycin and IL-1β–mediated pulmonary fibrosis is IL-17A dependent. J. Exp. Med 207, 535–552. 10.1084/jem.20092121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gasse P, Mary C, Guenon I, Noulin N, Charron S, Schnyder-Candrian S, Schnyder B, Akira S, Quesniaux VFJ, Lagente V, et al. (2007). IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J. Clin. Invest, JCI32285. 10.1172/JCI32285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, Hussell T, Feldmann M, and Udalova IA (2011). IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol 12, 231–238. 10.1038/ni.1990. [DOI] [PubMed] [Google Scholar]
- 41.Choi J, Park J-E, Tsagkogeorga G, Yanagita M, Koo B-K, Han N, and Lee J-H (2020). Inflammatory Signals Induce AT2 Cell-Derived Damage-Associated Transient Progenitors that Mediate Alveolar Regeneration. Cell Stem Cell 27, 366–382.e7. 10.1016/j.stem.2020.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lasithiotaki I, Giannarakis I, Tsitoura E, Samara KD, Margaritopoulos GA, Choulaki C, Vasarmidi E, Tzanakis N, Voloudaki A, Sidiropoulos P, et al. (2016). NLRP3 inflammasome expression in idiopathic pulmonary fibrosis and rheumatoid lung. Eur. Respir. J 47, 910–918. 10.1183/13993003.00564-2015. [DOI] [PubMed] [Google Scholar]
- 43.Cho SJ, Moon J-S, Nikahira K, Yun HS, Harris R, Hong KS, Huang H, Choi AMK, and Stout-Delgado H (2020). GLUT1-dependent glycolysis regulates exacerbation of fibrosis via AIM2 inflammasome activation. Thorax 75, 227–236. 10.1136/thoraxjnl-2019-213571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, and Tschopp J (2008). Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677. 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iyonaga K, Takeya M, Saita N, Sakamoto O, Yoshimura T, Ando M, and Takahashi K (1994). Monocyte chemoattractant protein-1 in idiopathic pulmonary fibrosis and other interstitial lung diseases. Hum. Pathol 25, 455–463. 10.1016/0046-8177(94)90117-1. [DOI] [PubMed] [Google Scholar]
- 46.Rivas-Fuentes S, Herrera I, Salgado-Aguayo A, Buendía-Roldán I, Becerril C, and Cisneros J (2020). CX3CL1 and CX3CR1 could be a relevant molecular axis in the pathophysiology of idiopathic pulmonary fibrosis. Int. J. Med. Sci 17, 2357–2361. 10.7150/ijms.43748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okuma T, Terasaki Y, Kaikita K, Kobayashi H, Kuziel WA, Kawasuji M, and Takeya M (2004). C-C chemokine receptor 2 (CCR2) deficiency improves bleomycin-induced pulmonary fibrosis by attenuation of both macrophage infiltration and production of macrophage-derived matrix metalloproteinases. J. Pathol 204, 594–604. 10.1002/path.1667. [DOI] [PubMed] [Google Scholar]
- 48.Boring L, Gosling J, Chensue SW, Kunkel SL, Farese RV, Broxmeyer HE, and Charo IF (1997). Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J. Clin. Invest 100, 2552–2561. 10.1172/JCI119798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ishida Y, Kimura A, Nosaka M, Kuninaka Y, Hemmi H, Sasaki I, Kaisho T, Mukaida N, and Kondo T (2017). Essential involvement of the CX3CL1-CX3CR1 axis in bleomycin-induced pulmonary fibrosis via regulation of fibrocyte and M2 macrophage migration. Sci. Rep 7, 16833. 10.1038/s41598-017-17007-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, et al. (2019). Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol 20, 163–172. 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hult EM, Gurczynski SJ, O’Dwyer DN, Zemans RL, Rasky A, Wang Y, Murray S, Crawford HC, and Moore BB (2022). Myeloid- and Epithelial-derived Heparin-Binding Epidermal Growth Factor-like Growth Factor Promotes Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol 67, 641–653. 10.1165/rcmb.2022-0174OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuo D, Ding J, Cohn IS, Zhang F, Wei K, Rao DA, Rozo C, Sokhi UK, Shanaj S, Oliver DJ, et al. (2019). HBEGF+ macrophages in rheumatoid arthritis induce fibroblast invasiveness. Sci. Transl. Med 11. 10.1126/scitranslmed.aau8587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, Fernandez R, Akbarpour M, Chen C-I, Ren Z, et al. (2019). Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med 199, 1517–1536. 10.1164/rccm.201712-2410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morse C, Tabib T, Sembrat J, Buschur KL, Bittar HT, Valenzi E, Jiang Y, Kass DJ, Gibson K, Chen W, et al. (2019). Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur. Respir. J 54. 10.1183/13993003.02441-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, Chu SG, Raby BA, DeIuliis G, Januszyk M, et al. (2020). Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv 6, eaba1983. 10.1126/sciadv.aba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ouyang JF, Mishra K, Xie Y, Park H, Huang KY, Petretto E, and Behmoaras J (2023). Systems level identification of a matrisome-associated macrophage polarisation state in multi-organ fibrosis. eLife 12, e85530. 10.7554/eLife.85530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hatipoglu OF, Uctepe E, Opoku G, Wake H, Ikemura K, Ohtsuki T, Inagaki J, Gunduz M, Gunduz E, Watanabe S, et al. (2021). Osteopontin silencing attenuates bleomycin-induced murine pulmonary fibrosis by regulating epithelial–mesenchymal transition. Biomed. Pharmacother 139, 111633. 10.1016/j.biopha.2021.111633. [DOI] [PubMed] [Google Scholar]
- 58.Gao X, Jia G, Guttman A, DePianto DJ, Morshead KB, Sun K-H, Ramamoorthi N, Vander Heiden JA, Modrusan Z, Wolters PJ, et al. (2020). Osteopontin Links Myeloid Activation and Disease Progression in Systemic Sclerosis. Cell Rep. Med 1, 100140. 10.1016/j.xcrm.2020.100140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoeft K, Schaefer GJL, Kim H, Schumacher D, Bleckwehl T, Long Q, Klinkhammer BM, Peisker F, Koch L, Nagai J, et al. (2023). Platelet-instructed SPP1+ macrophages drive myofibroblast activation in fibrosis in a CXCL4-dependent manner. Cell Rep 42, 112131. 10.1016/j.celrep.2023.112131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou Y, Peng H, Sun H, Peng X, Tang C, Gan Y, Chen X, Mathur A, Hu B, Slade MD, et al. (2014). Chitinase 3–Like 1 Suppresses Injury and Promotes Fibroproliferative Responses in Mammalian Lung Fibrosis. Sci. Transl. Med 6. 10.1126/scitranslmed.3007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu R, Liu X, Deng X, Li S, Wang Y, Zhang Y, Ke D, Yan R, Wang Q, Tian X, et al. (2023). Serum CHI3L1 as a Biomarker of Interstitial Lung Disease in Rheumatoid Arthritis. Front. Immunol 10.3389/fimmu.2023.1211790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cao Y, Rudrakshala J, Williams R, Rodriguez S, Sorkhdini P, Yang AX, Mundy M, Yang D, Palmisciano A, Walsh T, et al. (2022). CRTH2 Mediates Profibrotic Macrophage Differentiation and Promotes Lung Fibrosis. Am. J. Respir. Cell Mol. Biol 67, 201–214. 10.1165/rcmb.2021-0504OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McKeown S, Richter AG, O’Kane C, McAuley DF, and Thickett DR (2009). MMP expression and abnormal lung permeability are important determinants of outcome in IPF. Eur. Respir. J 33, 77–84. 10.1183/09031936.00060708. [DOI] [PubMed] [Google Scholar]
- 64.Fraser E, Denney L, Antanaviciute A, Blirando K, Vuppusetty C, Zheng Y, Repapi E, Iotchkova V, Taylor S, Ashley N, et al. (2021). Multi-Modal Characterization of Monocytes in Idiopathic Pulmonary Fibrosis Reveals a Primed Type I Interferon Immune Phenotype. Front. Immunol 12, 623430. 10.3389/fimmu.2021.623430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trombetta AC, Soldano S, Contini P, Tomatis V, Ruaro B, Paolino S, Brizzolara R, Montagna P, Sulli A, Pizzorni C, et al. (2018). A circulating cell population showing both M1 and M2 monocyte/macrophage surface markers characterizes systemic sclerosis patients with lung involvement. Respir. Res 19, 186. 10.1186/s12931-018-0891-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hasegawa M, Fujimoto M, Matsushita T, Hamaguchi Y, Takehara K, and Sato S (2011). Serum chemokine and cytokine levels as indicators of disease activity in patients with systemic sclerosis. Clin. Rheumatol 30, 231–237. 10.1007/s10067-010-1610-4. [DOI] [PubMed] [Google Scholar]
- 67.Schmidt K, Martinez-Gamboa L, Meier S, Witt C, Meisel C, Hanitsch LG, Becker MO, Huscher D, Burmester GR, and Riemekasten G (2009). Bronchoalveoloar lavage fluid cytokines and chemokines as markers and predictors for the outcome of interstitial lung disease in systemic sclerosis patients. Arthritis Res. Ther 11, R111. 10.1186/ar2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fabre T, Barron AMS, Christensen SM, Asano S, Bound K, Lech MP, Wadsworth MH, Chen X, Wang C, Wang J, et al. (2023). Identification of a broadly fibrogenic macrophage subset induced by type 3 inflammation. Sci. Immunol 8, eadd8945. 10.1126/sciimmunol.add8945. [DOI] [PubMed] [Google Scholar]
- 69.Valenzi E, Bulik M, Tabib T, Morse C, Sembrat J, Trejo Bittar H, Rojas M, and Lafyatis R (2019). Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Ann. Rheum. Dis 78, 1379–1387. 10.1136/annrheumdis-2018-214865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Richter AG, Perkins GD, Chavda A, Sapey E, Harper L, and Thickett DR (2011). Neutrophil chemotaxis in granulomatosis with polyangiitis (Wegener’s) and idiopathic pulmonary fibrosis. Eur. Respir. J 38, 1081–1088. 10.1183/09031936.00161910. [DOI] [PubMed] [Google Scholar]
- 71.Takemasa A, Ishii Y, and Fukuda T (2012). A neutrophil elastase inhibitor prevents bleomycin-induced pulmonary fibrosis in mice. Eur. Respir. J 40, 1475–1482. 10.1183/09031936.00127011. [DOI] [PubMed] [Google Scholar]
- 72.Gregory AD, Kliment CR, Metz HE, Kim K-H, Kargl J, Agostini BA, Crum LT, Oczypok EA, Oury TA, and Houghton AM (2015). Neutrophil elastase promotes myofibroblast differentiation in lung fibrosis. J. Leukoc. Biol 98, 143–152. 10.1189/jlb.3HI1014-493R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chrysanthopoulou A, Mitroulis I, Apostolidou E, Arelaki S, Mikroulis D, Konstantinidis T, Sivridis E, Koffa M, Giatromanolaki A, Boumpas DT, et al. (2014). Neutrophil extracellular traps promote differentiation and function of fibroblasts: NETs induce fibrosis via differentiation of fibroblasts. J. Pathol 233, 294–307. 10.1002/path.4359. [DOI] [PubMed] [Google Scholar]
- 74.Yan S, Li M, Liu B, Ma Z, and Yang Q (2023). Neutrophil extracellular traps and pulmonary fibrosis: an update. J. Inflamm 20, 2. 10.1186/s12950-023-00329-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Suzuki M, Ikari J, Anazawa R, Tanaka N, Katsumata Y, Shimada A, Suzuki E, and Tatsumi K (2020). PAD4 Deficiency Improves Bleomycin-induced Neutrophil Extracellular Traps and Fibrosis in Mouse Lung. Am. J. Respir. Cell Mol. Biol 63, 806–818. 10.1165/rcmb.2019-0433OC. [DOI] [PubMed] [Google Scholar]
- 76.Yamashita CM, Dolgonos L, Zemans RL, Young SK, Robertson J, Briones N, Suzuki T, Campbell MN, Gauldie J, Radisky DC, et al. (2011). Matrix Metalloproteinase 3 Is a Mediator of Pulmonary Fibrosis. Am. J. Pathol 179, 1733–1745. 10.1016/j.ajpath.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Craig VJ, Quintero PA, Fyfe SE, Patel AS, Knolle MD, Kobzik L, and Owen CA (2013). Pro-fibrotic Activities for Matrix Metalloproteinase-8 During Bleomycin-mediated Lung Injury. J. Immunol. Baltim. Md 1950 190, 4283–4296. 10.4049/jimmunol.1201043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Warheit-Niemi HI, Huizinga GP, Edwards SJ, Wang Y, Murray SK, O’Dwyer DN, and Moore BB (2022). Fibrotic Lung Disease Alters Neutrophil Trafficking and Promotes Neutrophil Elastase and Extracellular Trap Release. ImmunoHorizons 6, 817–834. 10.4049/immunohorizons.2200083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang S, Jia X, Zhang Q, Zhang L, Yang J, Hu C, Shi J, Jiang X, Lu J, and Shen H (2020). Neutrophil extracellular traps activate lung fibroblast to induce polymyositis-related interstitial lung diseases via TLR9-miR-7-Smad2 pathway. J. Cell. Mol. Med 24, 1658–1669. 10.1111/jcmm.14858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen X, Li Y, Qin L, He R, and Hu C (2021). Neutrophil Extracellular Trapping Network Promotes the Pathogenesis of Neutrophil-associated Asthma through Macrophages. Immunol. Invest 50, 544–561. 10.1080/08820139.2020.1778720. [DOI] [PubMed] [Google Scholar]
- 81.Soehnlein O, Kai-Larsen Y, Frithiof R, Sorensen OE, Kenne E, Scharffetter-Kochanek K, Eriksson EE, Herwald H, Agerberth B, and Lindbom L (2008). Neutrophil primary granule proteins HBP and HNP1–3 boost bacterial phagocytosis by human and murine macrophages. J. Clin. Invest 118, 3491–3502. 10.1172/JCI35740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Thrall RS, Phan SH, McCormick JR, and Ward PA (1981). The development of bleomycin-induced pulmonary fibrosis in neutrophil-depleted and complement-depleted rats. Am. J. Pathol 105, 76–81. [PMC free article] [PubMed] [Google Scholar]
- 83.Ham J, Kim J, Ko YG, and Kim HY (2022). The Dynamic Contribution of Neutrophils in the Chronic Respiratory Diseases. Allergy Asthma Immunol. Res 14, 361–378. 10.4168/aair.2022.14.4.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xaubet A, Agustí C, Luburich P, Barberá JA, Carrión M, Ayuso MC, Roca J, and Rodriguez-Roisin R (1998). Interleukin-8 expression in bronchoalveolar lavage cells in the evaluation of alveolitis in idiopathic pulmonary fibrosis. Respir. Med 92, 338–344. 10.1016/s0954-6111(98)90118-4. [DOI] [PubMed] [Google Scholar]
- 85.Ashitani J, Mukae H, Taniguchi H, Ihi T, Kadota J, Kohno S, and Matsukura S (1999). Granulocyte-colony stimulating factor levels in bronchoalveolar lavage fluid from patients with idiopathic pulmonary fibrosis. Thorax 54, 1015–1020. 10.1136/thx.54.11.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kinder BW, Brown KK, Schwarz MI, Ix JH, Kervitsky A, and King TE (2008). Baseline BAL Neutrophilia Predicts Early Mortality in Idiopathic Pulmonary Fibrosis. Chest 133, 226–232. 10.1378/chest.07-1948. [DOI] [PubMed] [Google Scholar]
- 87.Zhang J, Wang D, Wang L, Wang S, Roden AC, Zhao H, Li X, Prakash YS, Matteson EL, Tschumperlin DJ, et al. (2019). Profibrotic effect of IL-17A and elevated IL-17RA in idiopathic pulmonary fibrosis and rheumatoid arthritis-associated lung disease support a direct role for IL-17A/IL-17RA in human fibrotic interstitial lung disease. Am. J. Physiol.-Lung Cell. Mol. Physiol 316, L487–L497. 10.1152/ajplung.00301.2018. [DOI] [PubMed] [Google Scholar]
- 88.Mi S, Li Z, Yang H-Z, Liu H, Wang J-P, Ma Y-G, Wang X-X, Liu H-Z, Sun W, and Hu Z-W (2011). Blocking IL-17A Promotes the Resolution of Pulmonary Inflammation and Fibrosis Via TGF-β1–Dependent and –Independent Mechanisms. J. Immunol 187, 3003–3014. 10.4049/jimmunol.1004081. [DOI] [PubMed] [Google Scholar]
- 89.Piguet PF, Grau GE, and de Kossodo S (1993). Role of granulocyte-macrophage colony-stimulating factor in pulmonary fibrosis induced in mice by bleomycin. Exp. Lung Res 19, 579–587. 10.3109/01902149309031729. [DOI] [PubMed] [Google Scholar]
- 90.Ah Kioon MD, Tripodo C, Fernandez D, Kirou KA, Spiera RF, Crow MK, Gordon JK, and Barrat FJ (2018). Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci. Transl. Med 10, eaam8458. 10.1126/scitranslmed.aam8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kafaja S, Valera I, Divekar AA, Saggar R, Abtin F, Furst DE, Khanna D, and Singh RR (2018). pDCs in lung and skin fibrosis in a bleomycin-induced model and patients with systemic sclerosis. JCI Insight 3, e98380, 98380. 10.1172/jci.insight.98380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Affandi AJ, Carvalheiro T, Ottria A, de Haan JJ, Brans MAD, Brandt MM, Tieland RG, Lopes AP, Fernández BM, Bekker CPJ, et al. (2022). CXCL4 drives fibrosis by promoting several key cellular and molecular processes. Cell Rep 38, 110189. 10.1016/j.celrep.2021.110189. [DOI] [PubMed] [Google Scholar]
- 93.Silva-Cardoso SC, Tao W, Angiolilli C, Lopes AP, Bekker CPJ, Devaprasad A, Giovannone B, van Laar J, Cossu M, Marut W, et al. (2020). CXCL4 Links Inflammation and Fibrosis by Reprogramming Monocyte-Derived Dendritic Cells in vitro. Front. Immunol 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Van Bon L, Affandi AJ, Broen J, Christmann RB, Marijnissen RJ, Stawski L, Farina GA, Stifano G, Mathes AL, Cossu M, et al. (2014). Proteome-wide Analysis and CXCL4 as a Biomarker in Systemic Sclerosis. N. Engl. J. Med 370, 433–443. 10.1056/NEJMoa1114576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fernandez IE, Greiffo FR, Frankenberger M, Bandres J, Heinzelmann K, Neurohr C, Hatz R, Hartl D, Behr J, and Eickelberg O (2016). Peripheral blood myeloid-derived suppressor cells reflect disease status in idiopathic pulmonary fibrosis. Eur. Respir. J 48, 1171–1183. 10.1183/13993003.01826-2015. [DOI] [PubMed] [Google Scholar]
- 96.Höchst B, Mikulec J, Baccega T, Metzger C, Welz M, Peusquens J, Tacke F, Knolle P, Kurts C, Diehl L, et al. (2015). Differential induction of Ly6G and Ly6C positive myeloid derived suppressor cells in chronic kidney and liver inflammation and fibrosis. PloS One 10, e0119662. 10.1371/journal.pone.0119662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hsieh C-C, Lin C-L, He J-T, Chiang M, Wang Y, Tsai Y-C, Hung C-H, and Chang P-J (2018). Administration of cytokine-induced myeloid-derived suppressor cells ameliorates renal fibrosis in diabetic mice. Stem Cell Res. Ther 9, 183. 10.1186/s13287-018-0915-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kou W, Li B, Shi Y, Zhao Y, Yu Q, Zhuang J, Xu Y, and Peng W (2022). High complement protein C1q levels in pulmonary fibrosis and non-small cell lung cancer associated with poor prognosis. BMC Cancer 22, 110. 10.1186/s12885-021-08912-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gu H, Mickler EA, Cummings OW, Sandusky GE, Weber DJ, Gracon A, Woodruff T, Wilkes DS, and Vittal R (2014). Crosstalk between TGF-β1 and complement activation augments epithelial injury in pulmonary fibrosis. FASEB J 28, 4223–4234. 10.1096/fj.13-247650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yoshizawa Y, Nomura A, Ohdama S, Tanaka M, Morinari H, and Hasegawa S (2009). The Significance of Complement Activation in the Pathogenesis of Hypersensitivity Pneumonitis: Sequential Changes of Complement Components and Chemotactic Activities in Bronchoalveolar Lavage Fluids. Int. Arch. Allergy Appl. Immunol 87, 417–423. 10.1159/000234712. [DOI] [PubMed] [Google Scholar]
- 101.Addis-Lieser E, Köhl J, and Chiaramonte MG (2005). Opposing Regulatory Roles of Complement Factor 5 in the Development of Bleomycin-Induced Pulmonary Fibrosis1. J. Immunol 175, 1894–1902. 10.4049/jimmunol.175.3.1894. [DOI] [PubMed] [Google Scholar]
- 102.Ogawa T, Shichino S, Ueha S, Ogawa S, and Matsushima K (2022). Complement protein C1q activates lung fibroblasts and exacerbates silica-induced pulmonary fibrosis in mice. Biochem. Biophys. Res. Commun 603, 88–93. 10.1016/j.bbrc.2022.02.090. [DOI] [PubMed] [Google Scholar]
- 103.Galvan MD, Foreman DB, Zeng E, Tan JC, and Bohlson SS (2012). Complement Component C1q Regulates Macrophage Expression of Mer Tyrosine Kinase To Promote Clearance of Apoptotic Cells. J. Immunol 188, 3716–3723. 10.4049/jimmunol.1102920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rennard SI, Chen YF, Robbins RA, Gadek JE, and Crystal RG (1983). Fibronectin mediates cell attachment to C1q: a mechanism for the localization of fibrosis in inflammatory disease. Clin. Exp. Immunol 54, 239–247. [PMC free article] [PubMed] [Google Scholar]
- 105.Llorián-Salvador M, Byrne EM, Szczepan M, Little K, Chen M, and Xu H (2022). Complement activation contributes to subretinal fibrosis through the induction of epithelial-to-mesenchymal transition (EMT) in retinal pigment epithelial cells. J. Neuroinflammation 19, 182. 10.1186/s12974-022-02546-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bhattacharyya S, Wang W, Qin W, Cheng K, Coulup S, Chavez S, Jiang S, Raparia K, De Almeida LMV, Stehlik C, et al. TLR4-dependent fibroblast activation drives persistent organ fibrosis in skin and lung. JCI Insight 3, e98850. 10.1172/jci.insight.98850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hamada N, Maeyama T, Kawaguchi T, Yoshimi M, Fukumoto J, Yamada M, Yamada S, Kuwano K, and Nakanishi Y (2008). The Role of High Mobility Group Box1 in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol 39, 440–447. 10.1165/rcmb.2007-0330OC. [DOI] [PubMed] [Google Scholar]
- 108.Bhattacharyya S, Wang W, Morales-Nebreda L, Feng G, Wu M, Zhou X, Lafyatis R, Lee J, Hinchcliff M, Feghali-Bostwick C, et al. (2016). Tenascin-C drives persistence of organ fibrosis. Nat. Commun 7, 11703. 10.1038/ncomms11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Muro AF, Moretti FA, Moore BB, Yan M, Atrasz RG, Wilke CA, Flaherty KR, Martinez FJ, Tsui JL, Sheppard D, et al. (2008). An essential role for fibronectin extra type III domain A in pulmonary fibrosis. Am. J. Respir. Crit. Care Med 177, 638–645. 10.1164/rccm.200708-1291OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liang J, Zhang Y, Xie T, Liu N, Chen H, Geng Y, Kurkciyan A, Mena JM, Stripp BR, Jiang D, et al. (2016). Hyaluronan and TLR4 promote surfactant protein C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nat. Med 22, 1285–1293. 10.1038/nm.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Estany S, Vicens-Zygmunt V, Llatjós R, Montes A, Penín R, Escobar I, Xaubet A, Santos S, Manresa F, Dorca J, et al. (2014). Lung fibrotic tenascin-C upregulation is associated with other extracellular matrix proteins and induced by TGFβ1. BMC Pulm. Med 14, 120. 10.1186/1471-2466-14-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Noth I, Zhang Y, Ma S-F, Flores C, Barber M, Huang Y, Broderick SM, Wade MS, Hysi P, Scuirba J, et al. (2013). Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir. Med 1, 309–317. 10.1016/S2213-2600(13)70045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.O’Dwyer DN, Armstrong ME, Trujillo G, Cooke G, Keane MP, Fallon PG, Simpson AJ, Millar AB, McGrath EE, Whyte MK, et al. (2013). The Toll-like receptor 3 L412F polymorphism and disease progression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med 188, 1442–1450. 10.1164/rccm.201304-0760OC. [DOI] [PubMed] [Google Scholar]
- 114.Bhattacharyya S, and Varga J (2018). Endogenous ligands of TLR4 promote unresolving tissue fibrosis: Implications for systemic sclerosis and its targeted therapy. Immunol. Lett 195, 9–17. 10.1016/j.imlet.2017.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.A rare polymorphism in the gene for Toll-like receptor 2 is associated with systemic sclerosis phenotype and increases the production of inflammatory mediators 10.1002/art.33325. [DOI] [PubMed] [Google Scholar]
- 116.Lipinski JH, Moore BB, and O’Dwyer DN (2020). The evolving role of the lung microbiome in pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol 319, L675–L682. 10.1152/ajplung.00258.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lombardi F, Stewart I, Fabbri L, Adams W, Kawano-Dourado L, Ryerson CJ, and Jenkins G (2024). Mycophenolate and azathioprine efficacy in interstitial lung disease: a systematic review and meta-analysis. BMJ Open Respir. Res 11, e002163. 10.1136/bmjresp-2023-002163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wijsenbeek M, and Cottin V (2020). Spectrum of Fibrotic Lung Diseases. N. Engl. J. Med 383, 958–968. 10.1056/NEJMra2005230. [DOI] [PubMed] [Google Scholar]
- 119.Idiopathic Pulmonary Fibrosis Clinical Research Network, Raghu G, Anstrom KJ, King TE, Lasky JA, and Martinez FJ (2012). Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med 366, 1968–1977. 10.1056/NEJMoa1113354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.King TE, Albera C, Bradford WZ, Costabel U, Hormel P, Lancaster L, Noble PW, Sahn SA, Szwarcberg J, Thomeer M, et al. (2009). Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicentre, randomised, placebo-controlled trial. Lancet Lond. Engl 374, 222–228. 10.1016/S0140-6736(09)60551-1. [DOI] [PubMed] [Google Scholar]
- 121.King TE, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, et al. (2014). A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med 370, 2083–2092. 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 122.Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD, Raghu G, Sauter W, Girard M, Alves M, et al. (2019). Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. N. Engl. J. Med 380, 2518–2528. 10.1056/NEJMoa1903076. [DOI] [PubMed] [Google Scholar]
- 123.Ruterbusch M, Pruner KB, Shehata L, and Pepper M (2020). In Vivo CD4+ T Cell Differentiation and Function: Revisiting the Th1/Th2 Paradigm. Annu. Rev. Immunol 38, 705–725. 10.1146/annurev-immunol-103019-085803. [DOI] [PubMed] [Google Scholar]
- 124.Xu J, Mora AL, LaVoy J, Brigham KL, and Rojas M (2006). Increased bleomycin-induced lung injury in mice deficient in the transcription factor T-bet. Am. J. Physiol.-Lung Cell. Mol. Physiol 291, L658–L667. 10.1152/ajplung.00006.2006. [DOI] [PubMed] [Google Scholar]
- 125.Chung MP, Monick MM, Hamzeh NY, Butler NS, Powers LS, and Hunninghake GW (2003). Role of repeated lung injury and genetic background in bleomycin-induced fibrosis. Am. J. Respir. Cell Mol. Biol 29, 375–380. 10.1165/rcmb.2003-0029OC. [DOI] [PubMed] [Google Scholar]
- 126.Mitaka K, Miyazaki Y, Yasui M, Furuie M, Miyake S, Inase N, and Yoshizawa Y (2010). Th2-biased immune responses are important in a murine model of chronic hypersensitivity pneumonitis. Int. Arch. Allergy Immunol 154, 264–274. 10.1159/000321114. [DOI] [PubMed] [Google Scholar]
- 127.Singh B, Kasam RK, Sontake V, Wynn TA, and Madala SK (2017). Repetitive intradermal bleomycin injections evoke T-helper cell 2 cytokine-driven pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol 313, L796–L806. 10.1152/ajplung.00184.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lei L, Zhao C, Qin F, He Z-Y, Wang X, and Zhong X-N (2016). Th17 cells and IL-17 promote the skin and lung inflammation and fibrosis process in a bleomycin-induced murine model of systemic sclerosis. Clin. Exp. Rheumatol 34 Suppl 100, 14–22. [PubMed] [Google Scholar]
- 129.Gasse P, Riteau N, Vacher R, Michel M-L, Fautrel A, di Padova F, Fick L, Charron S, Lagente V, Eberl G, et al. (2011). IL-1 and IL-23 mediate early IL-17A production in pulmonary inflammation leading to late fibrosis. PloS One 6, e23185. 10.1371/journal.pone.0023185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lau J, Wang S, Davis J, Matteson E, and Vassallo R (2019). Role of IL23 in the development of fibrotic interstitial lung disease. Eur. Respir. J 54. 10.1183/13993003.congress-2019.PA2432. [DOI] [Google Scholar]
- 131.Huaux F, Liu T, McGarry B, Ullenbruch M, and Phan SH (2003). Dual roles of IL-4 in lung injury and fibrosis. J. Immunol 170, 2083–2092. 10.4049/jimmunol.170.4.2083. [DOI] [PubMed] [Google Scholar]
- 132.Pellicano C, Vantaggio L, Colalillo A, Pocino K, Basile V, Marino M, Basile U, and Rosato E (2023). Type 2 cytokines and scleroderma interstitial lung disease. Clin. Exp. Med 10.1007/s10238-023-01125-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Barrera L, Mendoza F, Zuñiga J, Estrada A, Zamora AC, Melendro EI, Ramírez R, Pardo A, and Selman M (2008). Functional Diversity of T-Cell Subpopulations in Subacute and Chronic Hypersensitivity Pneumonitis. Am. J. Respir. Crit. Care Med 177, 44–55. 10.1164/rccm.200701-093OC. [DOI] [PubMed] [Google Scholar]
- 134.Daniil Z, Kitsanta P, Kapotsis G, Mathioudaki M, Kollintza A, Karatza M, Milic-Emili J, Roussos C, and Papiris SA (2005). CD8+ T lymphocytes in lung tissue from patients with idiopathic pulmonary fibrosis. Respir. Res 6, 81. 10.1186/1465-9921-6-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Prior C, and Haslam PL (1992). In vivo levels and in vitro production of interferon-gamma in fibrosing interstitial lung diseases. Clin. Exp. Immunol 88, 280–287. 10.1111/j.1365-2249.1992.tb03074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Emura M, Nagai S, Takeuchi M, Kitaichi M, and Izumi T (2008). In vitro production of B cell growth factor and B cell differentiation factor by peripheral blood mononuclear cells and bronchoalveolar lavage T lymphocytes from patients with idiopathic pulmonary fibrosis. Clin. Exp. Immunol 82, 133–139. 10.1111/j.1365-2249.1990.tb05416.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Celada LJ, Kropski JA, Herazo-Maya JD, Luo W, Creecy A, Abad AT, Chioma OS, Lee G, Hassell NE, Shaginurova GI, et al. (2018). PD-1 up-regulation on CD4+ T cells promotes pulmonary fibrosis through STAT3-mediated IL-17A and TGF-β1 production. Sci. Transl. Med 10, eaar8356. 10.1126/scitranslmed.aar8356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kamio K, Azuma A, Matsuda K, Usuki J, Inomata M, Morinaga A, Kashiwada T, Nishijima N, Itakura S, Kokuho N, et al. (2018). Resolution of bleomycin-induced murine pulmonary fibrosis via a splenic lymphocyte subpopulation. Respir. Res 19, 71. 10.1186/s12931-018-0783-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Birjandi SZ, Palchevskiy V, Xue YY, Nunez S, Kern R, Weigt SS, Lynch JP, Chatila TA, and Belperio JA (2016). CD4+CD25hiFoxp3+ Cells Exacerbate Bleomycin-Induced Pulmonary Fibrosis. Am. J. Pathol 186, 2008–2020. 10.1016/j.ajpath.2016.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Xiong S, Guo R, Yang Z, Xu L, Du L, Li R, Xiao F, Wang Q, Zhu M, and Pan X (2015). Treg depletion attenuates irradiation-induced pulmonary fibrosis by reducing fibrocyte accumulation, inducing Th17 response, and shifting IFN-γ, IL-12/IL-4, IL-5 balance. Immunobiology 220, 1284–1291. 10.1016/j.imbio.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 141.Kotsianidis I, Nakou E, Bouchliou I, Tzouvelekis A, Spanoudakis E, Steiropoulos P, Sotiriou I, Aidinis V, Margaritis D, Tsatalas C, et al. (2009). Global impairment of CD4+CD25+FOXP3+ regulatory T cells in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med 179, 1121–1130. 10.1164/rccm.200812-1936OC. [DOI] [PubMed] [Google Scholar]
- 142.Hou Z, Ye Q, Qiu M, Hao Y, Han J, and Zeng H (2017). Increased activated regulatory T cells proportion correlate with the severity of idiopathic pulmonary fibrosis. Respir. Res 18, 170. 10.1186/s12931-017-0653-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Padilla CM, Valenzi E, Tabib T, Nazari B, Sembrat J, Rojas M, Fuschiotti P, and Lafyatis R (2023). Increased CD8+ tissue resident memory T cells, regulatory T cells and activated natural killer cells in systemic sclerosis lungs. Rheumatology, kead273. 10.1093/rheumatology/kead273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Serezani APM, Pascoalino BD, Bazzano JMR, Vowell KN, Tanjore H, Taylor CJ, Calvi CL, McCall AS, Bacchetta MD, Shaver CM, et al. (2022). Multiplatform Single-Cell Analysis Identifies Immune Cell Types Enhanced in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol 67, 50–60. 10.1165/rcmb.2021-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Collins SL, Chan-Li Y, Oh M, Vigeland CL, Limjunyawong N, Mitzner W, Powell JD, and Horton MR Vaccinia vaccine–based immunotherapy arrests and reverses established pulmonary fibrosis. JCI Insight 1, e83116. 10.1172/jci.insight.83116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ichikawa T, Hirahara K, Kokubo K, Kiuchi M, Aoki A, Morimoto Y, Kumagai J, Onodera A, Mato N, Tumes DJ, et al. (2019). CD103hi Treg cells constrain lung fibrosis induced by CD103lo tissue-resident pathogenic CD4 T cells. Nat. Immunol 20, 1469–1480. 10.1038/s41590-019-0494-y. [DOI] [PubMed] [Google Scholar]
- 147.Herazo-Maya JD, Noth I, Duncan SR, Kim S, Ma S-F, Tseng GC, Feingold E, Juan-Guardela BM, Richards TJ, Lussier Y, et al. (2013). Peripheral blood mononuclear cell gene expression profiles predict poor outcome in idiopathic pulmonary fibrosis. Sci. Transl. Med 5, 205ra136. 10.1126/scitranslmed.3005964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Marchal-Sommé J, Uzunhan Y, Marchand-Adam S, Valeyre D, Soumelis V, Crestani B, and Soler P (2006). Cutting edge: nonproliferating mature immune cells form a novel type of organized lymphoid structure in idiopathic pulmonary fibrosis. J. Immunol. Baltim. Md 1950 176, 5735–5739. 10.4049/jimmunol.176.10.5735. [DOI] [PubMed] [Google Scholar]
- 149.Zhang-Hoover J, Sutton A, and Stein-Streilein J (2001). CD40/CD40 ligand interactions are critical for elicitation of autoimmune-mediated fibrosis in the lung. J. Immunol. Baltim. Md 1950 166, 3556–3563. 10.4049/jimmunol.166.5.3556. [DOI] [PubMed] [Google Scholar]
- 150.Adawi A, Zhang Y, Baggs R, Rubin P, Williams J, Finkelstein J, and Phipps RP (1998). Blockade of CD40-CD40 ligand interactions protects against radiation-induced pulmonary inflammation and fibrosis. Clin. Immunol. Immunopathol 89, 222–230. 10.1006/clin.1998.4606. [DOI] [PubMed] [Google Scholar]
- 151.Kaufman J, Sime PJ, and Phipps RP (2004). Expression of CD154 (CD40 ligand) by human lung fibroblasts: differential regulation by IFN-gamma and IL-13, and implications for fibrosis. J. Immunol. Baltim. Md 1950 172, 1862–1871. 10.4049/jimmunol.172.3.1862. [DOI] [PubMed] [Google Scholar]
- 152.Yellin MJ, Winikoff S, Fortune SM, Baum D, Crow MK, Lederman S, and Chess L (1995). Ligation of CD40 on fibroblasts induces CD54 (ICAM-1) and CD106 (VCAM-1) up-regulation and IL-6 production and proliferation. J. Leukoc. Biol 58, 209–216. 10.1002/jlb.58.2.209. [DOI] [PubMed] [Google Scholar]
- 153.Atamas SP, Luzina IG, Dai H, Wilt SG, and White B (2002). Synergy between CD40 ligation and IL-4 on fibroblast proliferation involves IL-4 receptor signaling. J. Immunol. Baltim. Md 1950 168, 1139–1145. 10.4049/jimmunol.168.3.1139. [DOI] [PubMed] [Google Scholar]
- 154.Gourh P, Arnett FC, Tan FK, Assassi S, Divecha D, Paz G, McNearney T, Draeger H, Reveille JD, Mayes MD, et al. (2010). Association of TNFSF4 (OX40L) polymorphisms with susceptibility to systemic sclerosis. Ann. Rheum. Dis 69, 550–555. 10.1136/ard.2009.116434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Elhai M, Avouac J, Hoffmann-Vold AM, Ruzehaji N, Amiar O, Ruiz B, Brahiti H, Ponsoye M, Fréchet M, Burgevin A, et al. (2016). OX40L blockade protects against inflammation-driven fibrosis. Proc. Natl. Acad. Sci 113, E3901–E3910. 10.1073/pnas.1523512113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ni K, Liu M, Zheng J, Wen L, Chen Q, Xiang Z, Lam K-T, Liu Y, Chan GC-F, Lau Y-L, et al. (2018). PD-1/PD-L1 Pathway Mediates the Alleviation of Pulmonary Fibrosis by Human Mesenchymal Stem Cells in Humanized Mice. Am. J. Respir. Cell Mol. Biol 58, 684–695. 10.1165/rcmb.2017-0326OC. [DOI] [PubMed] [Google Scholar]
- 157.Habiel DM, Espindola MS, Kitson C, Azzara AV, Coelho AL, Stripp B, and Hogaboam CM (2019). Characterization of CD28null T cells in idiopathic pulmonary fibrosis. Mucosal Immunol 12, 212–222. 10.1038/s41385-018-0082-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Cui L, Chen S-Y, Lerbs T, Lee J-W, Domizi P, Gordon S, Kim Y, Nolan G, Betancur P, and Wernig G (2020). Activation of JUN in fibroblasts promotes pro-fibrotic programme and modulates protective immunity. Nat. Commun 11, 2795. 10.1038/s41467-020-16466-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Geng Y, Liu X, Liang J, Habiel DM, Kulur V, Coelho AL, Deng N, Xie T, Wang Y, Liu N, et al. (2019). PD-L1 on invasive fibroblasts drives fibrosis in a humanized model of idiopathic pulmonary fibrosis. JCI Insight 4. 10.1172/jci.insight.125326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Shimoji K, Masuda T, Yamaguchi K, Sakamoto S, Horimasu Y, Nakashima T, Miyamoto S, Iwamoto H, Fujitaka K, Hamada H, et al. (2020). Association of Preexisting Interstitial Lung Abnormalities With Immune Checkpoint Inhibitor–Induced Interstitial Lung Disease Among Patients With Nonlung Cancers. JAMA Netw. Open 3, e2022906. 10.1001/jamanetworkopen.2020.22906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Heukels P, van Hulst JAC, van Nimwegen M, Boorsma CE, Melgert BN, von der Thusen JH, van den Blink B, Hoek RAS, Miedema JR, Neys SFH, et al. (2019). Enhanced Bruton’s tyrosine kinase in B-cells and autoreactive IgA in patients with idiopathic pulmonary fibrosis. Respir. Res 20, 232. 10.1186/s12931-019-1195-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Xue J, Kass DJ, Bon J, Vuga L, Tan J, Csizmadia E, Otterbein L, Soejima M, Levesque MC, Gibson KF, et al. (2013). Plasma B lymphocyte stimulator and B cell differentiation in idiopathic pulmonary fibrosis patients. J. Immunol. Baltim. Md 1950 191, 2089–2095. 10.4049/jimmunol.1203476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kim HC, Song JS, Park S, Yoon H-Y, Lim SY, Chae EJ, Jang SJ, and Song JW (2020). Histologic features suggesting connective tissue disease in idiopathic pulmonary fibrosis. Sci. Rep 10, 21137. 10.1038/s41598-020-78140-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Rangel-Moreno J, Hartson L, Navarro C, Gaxiola M, Selman M, and Randall TD (2006). Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J. Clin. Invest 116, 3183–3194. 10.1172/JCI28756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Vuga LJ, Tedrow JR, Pandit KV, Tan J, Kass DJ, Xue J, Chandra D, Leader JK, Gibson KF, Kaminski N, et al. (2014). C-X-C Motif Chemokine 13 (CXCL13) Is a Prognostic Biomarker of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med 189, 966–974. 10.1164/rccm.201309-1592OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Adegunsoye A, Alqalyoobi S, Linderholm A, Bowman WS, Lee CT, Pugashetti JV, Sarma N, Ma S-F, Haczku A, Sperling A, et al. (2020). Circulating Plasma Biomarkers of Survival in Antifibrotic-Treated Patients With Idiopathic Pulmonary Fibrosis. Chest 158, 1526–1534. 10.1016/j.chest.2020.04.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Adegunsoye A, Oldham JM, Bonham C, Hrusch C, Nolan P, Klejch W, Bellam S, Mehta U, Thakrar K, Pugashetti JV, et al. (2019). Prognosticating Outcomes in Interstitial Lung Disease by Mediastinal Lymph Node Assessment. An Observational Cohort Study with Independent Validation. Am. J. Respir. Crit. Care Med 199, 747–759. 10.1164/rccm.201804-0761OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mills R, Mathur A, Nicol LM, Walker JJ, Przybylski AA, Mackinnon AC, Howie SEM, Wallace WAH, Dransfield I, and Hirani N (2019). Intrapulmonary Autoantibodies to HSP72 Are Associated with Improved Outcomes in IPF. J. Immunol. Res 2019, 1845128. 10.1155/2019/1845128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Taillé C, Grootenboer-Mignot S, Boursier C, Michel L, Debray M-P, Fagart J, Barrientos L, Mailleux A, Cigna N, Tubach F, et al. (2011). Identification of Periplakin as a New Target for Autoreactivity in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med 183, 759–766. 10.1164/rccm.201001-0076OC. [DOI] [PubMed] [Google Scholar]
- 170.Ali MF, Egan AM, Shaughnessy GF, Anderson DK, Kottom TJ, Dasari H, Van Keulen VP, Aubry M-C, Yi ES, Limper AH, et al. (2021). Antifibrotics Modify B-Cell-induced Fibroblast Migration and Activation in Patients with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol 64, 722–733. 10.1165/rcmb.2020-0387OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Komura K, Yanaba K, Horikawa M, Ogawa F, Fujimoto M, Tedder TF, and Sato S (2008). CD19 regulates the development of bleomycin-induced pulmonary fibrosis in a mouse model. Arthritis Rheum 58, 3574–3584. 10.1002/art.23995. [DOI] [PubMed] [Google Scholar]
- 172.McDonough JE, Ahangari F, Li Q, Jain S, Verleden SE, Herazo-Maya J, Vukmirovic M, DeIuliis G, Tzouvelekis A, Tanabe N, et al. (2019). Transcriptional regulatory model of fibrosis progression in the human lung. JCI Insight 4, e131597, 131597. 10.1172/jci.insight.131597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Dobson CS, Reich AN, Gaglione S, Smith BE, Kim EJ, Dong J, Ronsard L, Okonkwo V, Lingwood D, Dougan M, et al. (2022). Antigen identification and high-throughput interaction mapping by reprogramming viral entry. Nat. Methods 19, 449–460. 10.1038/s41592-022-01436-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zouggari Y, Ait-Oufella H, Bonnin P, Simon T, Sage AP, Guérin C, Vilar J, Caligiuri G, Tsiantoulas D, Laurans L, et al. (2013). B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat. Med 19, 1273–1280. 10.1038/nm.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hunninghake GM, Quesada-Arias LD, Carmichael NE, Martinez Manzano JM, Poli De Frías S, Baumgartner MA, DiGianni L, Gampala-Sagar SN, Leone DA, Gulati S, et al. (2020). Interstitial Lung Disease in Relatives of Patients with Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med 201, 1240–1248. 10.1164/rccm.201908-1571OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Putman RK, Gudmundsson G, Axelsson GT, Hida T, Honda O, Araki T, Yanagawa M, Nishino M, Miller ER, Eiriksdottir G, et al. (2019). Imaging Patterns Are Associated with Interstitial Lung Abnormality Progression and Mortality. Am. J. Respir. Crit. Care Med 200, 175–183. 10.1164/rccm.201809-1652OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wild JM, Porter JC, Molyneaux PL, George PM, Stewart I, Allen RJ, Aul R, Baillie JK, Barratt SL, Beirne P, et al. (2021). Understanding the burden of interstitial lung disease post-COVID-19: the UK Interstitial Lung Disease-Long COVID Study (UKILD-Long COVID). BMJ Open Respir. Res 8, e001049. 10.1136/bmjresp-2021-001049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Esposito AJ, Sparks JA, Gill RR, Hatabu H, Schmidlin EJ, Hota PV, Poli S, Fletcher EA, Xiong W, Frits ML, et al. (2022). Screening for preclinical parenchymal lung disease in rheumatoid arthritis. Rheumatol. Oxf. Engl 61, 3234–3245. 10.1093/rheumatology/keab891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sharma NS, Patel K, Sari E, Shankar S, Gastanadui MG, Moncada-Giraldo D, Soto-Vazquez Y, Stacks D, Hecker L, Dsouza K, et al. (2023). Active transcription in the vascular bed characterizes rapid progression in idiopathic pulmonary fibrosis. J. Clin. Invest 133, e165976. 10.1172/JCI165976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Senoo S, Taniguchi A, Itano J, Oda N, Morichika D, Fujii U, Guo L, Sunami R, Kanehiro A, Tokioka F, et al. (2021). Essential role of IL-23 in the development of acute exacerbation of pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol 321, L925–L940. 10.1152/ajplung.00582.2020. [DOI] [PubMed] [Google Scholar]
- 181.Schupp JC, Binder H, Jäger B, Cillis G, Zissel G, Müller-Quernheim J, and Prasse A (2015). Macrophage Activation in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. PLOS ONE 10, e0116775. 10.1371/journal.pone.0116775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Sugino K, Nakamura Y, Muramatsu Y, Hata Y, Shibuya K, and Homma S (2016). Analysis of blood neutrophil elastase, glutathione levels and pathological findings in postoperative acute exacerbation of idiopathic pulmonary fibrosis associated with lung cancer: Two case reports. Mol. Clin. Oncol 5, 402–406. 10.3892/mco.2016.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Christmann RB, Hayes E, Pendergrass S, Padilla C, Farina G, Affandi AJ, Whitfield ML, Farber HW, and Lafyatis R (2011). Interferon and alternative activation of monocyte/macrophages in systemic sclerosis-associated pulmonary arterial hypertension. Arthritis Rheum 63, 1718–1728. 10.1002/art.30318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Prasse A, Probst C, Bargagli E, Zissel G, Toews GB, Flaherty KR, Olschewski M, Rottoli P, and Müller-Quernheim J (2009). Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med 179, 717–723. 10.1164/rccm.200808-1201OC. [DOI] [PubMed] [Google Scholar]
- 185.Prasse A, Pechkovsky DV, Toews GB, Jungraithmayr W, Kollert F, Goldmann T, Vollmer E, Müller-Quernheim J, and Zissel G (2006). A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am. J. Respir. Crit. Care Med 173, 781–792. 10.1164/rccm.200509-1518OC. [DOI] [PubMed] [Google Scholar]
- 186.Carleo A, Deluca D, Bargagli E, Wuyts W, Kaminski N, and Prasse A (2019). The Transciptome of BAL Cells at Acute Exacerbation of IPF reveals New Insights. Eur. Respir. J 54. 10.1183/13993003.congress-2019.PA597. [DOI] [Google Scholar]
- 187.Madissoon E, Oliver AJ, Kleshchevnikov V, Wilbrey-Clark A, Polanski K, Richoz N, Ribeiro Orsi A, Mamanova L, Bolt L, Elmentaite R, et al. (2023). A spatially resolved atlas of the human lung characterizes a gland-associated immune niche. Nat. Genet 55, 66–77. 10.1038/s41588-022-01243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Eyres M, Bell JA, Davies ER, Fabre A, Alzetani A, Jogai S, Marshall BG, Johnston DA, Xu Z, Fletcher SV, et al. (2022). Spatially resolved deconvolution of the fibrotic niche in lung fibrosis. Cell Rep 40, 111230. 10.1016/j.celrep.2022.111230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Blumhagen RZ, Kurche JS, Cool CD, Walts AD, Heinz D, Fingerlin TE, Yang IV, and Schwartz DA (2023). Spatially distinct molecular patterns of gene expression in idiopathic pulmonary fibrosis. Respir. Res 24, 287. 10.1186/s12931-023-02572-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Nathan A, Asgari S, Ishigaki K, Valencia C, Amariuta T, Luo Y, Beynor JI, Baglaenko Y, Suliman S, Price AL, et al. (2022). Single-cell eQTL models reveal dynamic T cell state dependence of disease loci. Nature 606, 120–128. 10.1038/s41586-022-04713-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Korthagen NM, van Moorsel CHM, Kazemier KM, Ruven HJT, and Grutters JC (2012). IL1RN genetic variations and risk of IPF: a meta-analysis and mRNA expression study. Immunogenetics 64, 371–377. 10.1007/s00251-012-0604-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ahn M-H, Park B-L, Lee S-H, Park S-W, Park J-S, Kim D-J, Jang A-S, Park J-S, Shin H-K, Uh S-T, et al. (2011). A promoter SNP rs4073T>A in the common allele of the interleukin 8 gene is associated with the development of idiopathic pulmonary fibrosis via the IL-8 protein enhancing mode. Respir. Res 12, 73. 10.1186/1465-9921-12-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Vasakova M, Sterclova M, Matej R, Olejar T, Kolesar L, Skibova J, and Striz I (2013). IL-4 polymorphisms, HRCT score and lung tissue markers in idiopathic pulmonary fibrosis. Hum. Immunol 74, 1346–1351. 10.1016/j.humimm.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 194.Xin L, Jiang M, Su G, Xie M, Chen H, Liu X, Xu M, Zhang G, and Gong J (2018). The association between transforming growth factor beta1 polymorphism and susceptibility to pulmonary fibrosis: A meta-analysis (MOOSE compliant). Medicine (Baltimore) 97, e11876. 10.1097/MD.0000000000011876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Katzen J, Wagner BD, Venosa A, Kopp M, Tomer Y, Russo SJ, Headen AC, Basil MC, Stark JM, Mulugeta S, et al. (2019). An SFTPC BRICHOS mutant links epithelial ER stress and spontaneous lung fibrosis. JCI Insight 4, e126125, 126125. 10.1172/jci.insight.126125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Nureki S-I, Tomer Y, Venosa A, Katzen J, Russo SJ, Jamil S, Barrett M, Nguyen V, Kopp M, Mulugeta S, et al. (2018). Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J. Clin. Invest 128, 4008–4024. 10.1172/JCI99287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Mahavadi P, Korfei M, Henneke I, Liebisch G, Schmitz G, Gochuico BR, Markart P, Bellusci S, Seeger W, Ruppert C, et al. (2010). Epithelial stress and apoptosis underlie Hermansky-Pudlak syndrome-associated interstitial pneumonia. Am. J. Respir. Crit. Care Med 182, 207–219. 10.1164/rccm.200909-1414OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Korsunsky I, Wei K, Pohin M, Kim EY, Barone F, Major T, Taylor E, Ravindran R, Kemble S, Watts GFM, et al. (2022). Cross-tissue, single-cell stromal atlas identifies shared pathological fibroblast phenotypes in four chronic inflammatory diseases. Muhasebe Enstitüsü Derg. - J. Account. Inst 3, 481–518.e14. 10.1016/j.medj.2022.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hult E, Gurczynski S, and Moore B (2021). Protection of Lyz2Cre+ HBEGF−/− mice from bleomycin-induced lung fibrosis. FASEB J 35. 10.1096/fasebj.2021.35.S1.01999. [DOI] [Google Scholar]
- 200.Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY, and Ingber DE (2010). Reconstituting organ-level lung functions on a chip. Science 328, 1662–1668. 10/fg99sq. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lang NJ, Gote-Schniering J, Porras-Gonzalez D, Yang L, Sadeleer LJD, Jentzsch RC, Shitov VA, Zhou S, Ansari M, Agami A, et al. (2023). Ex vivo tissue perturbations coupled to single-cell RNA-seq reveal multilineage cell circuit dynamics in human lung fibrogenesis. Sci. Transl. Med [DOI] [PubMed] [Google Scholar]
- 202.Romagnoli M, Nannini C, Piciucchi S, Girelli F, Gurioli C, Casoni G, Ravaglia C, Tomassetti S, Gurioli C, Gavelli G, et al. (2011). Idiopathic nonspecific interstitial pneumonia: an interstitial lung disease associated with autoimmune disorders? Eur. Respir. J 38, 384–391. 10.1183/09031936.00094910. [DOI] [PubMed] [Google Scholar]
- 203.Park JH, Kim DS, Park I-N, Jang SJ, Kitaichi M, Nicholson AG, and Colby TV (2007). Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am. J. Respir. Crit. Care Med 175, 705–711. 10.1164/rccm.200607-912OC. [DOI] [PubMed] [Google Scholar]
- 204.Newton CA, Oldham JM, Ley B, Anand V, Adegunsoye A, Liu G, Batra K, Torrealba J, Kozlitina J, Glazer C, et al. (2019). Telomere length and genetic variant associations with interstitial lung disease progression and survival. Eur. Respir. J 53, 1801641. 10.1183/13993003.01641-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Dieudé P, Guedj M, Wipff J, Ruiz B, Hachulla E, Diot E, Granel B, Sibilia J, Tiev K, Mouthon L, et al. (2009). STAT4 is a genetic risk factor for systemic sclerosis having additive effects with IRF5 on disease susceptibility and related pulmonary fibrosis. Arthritis Rheum 60, 2472–2479. 10.1002/art.24688. [DOI] [PubMed] [Google Scholar]
- 206.Falfán-Valencia R, Camarena A, Pineda CL, Montaño M, Juárez A, Buendía-Roldán I, Pérez-Rubio G, Reséndiz-Hernández JM, Páramo I, Vega A, et al. (2014). Genetic susceptibility to multicase hypersensitivity pneumonitis is associated with the TNF-238 GG genotype of the promoter region and HLA-DRB1*04 bearing HLA haplotypes. Respir. Med 108, 211–217. 10.1016/j.rmed.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 207.Isshiki T, Koyama K, Homma S, Sakamoto S, Yamasaki A, Shimizu H, Miyoshi S, Nakamura Y, and Kishi K (2021). Association of rs3750920 polymorphism in TOLLIP with clinical characteristics of fibrosing interstitial lung diseases in Japanese. Sci. Rep 11, 16250. 10.1038/s41598-021-95869-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Ryerson CJ, Urbania TH, Richeldi L, Mooney JJ, Lee JS, Jones KD, Elicker BM, Koth LL, King TE, Wolters PJ, et al. (2013). Prevalence and prognosis of unclassifiable interstitial lung disease. Eur. Respir. J 42, 750–757. 10.1183/09031936.00131912. [DOI] [PubMed] [Google Scholar]
- 209.Guler SA, Ellison K, Algamdi M, Collard HR, and Ryerson CJ (2018). Heterogeneity in Unclassifiable Interstitial Lung Disease. A Systematic Review and Meta-Analysis. Ann. Am. Thorac. Soc 15, 854–863. 10.1513/AnnalsATS.201801-067OC. [DOI] [PubMed] [Google Scholar]
- 210.Baqir M, Vasirreddy A, Vu AN, Moua T, Chamberlain AM, Frank RD, and Ryu JH (2021). Idiopathic pulmonary fibrosis and gastroesophageal reflux disease: A population-based, case-control study. Respir. Med 178, 106309. 10.1016/j.rmed.2021.106309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Chen H-H, Yong Y-M, Lin C-H, Chen Y-H, Chen D-Y, Ying J-C, and Chao W-C (2020). Air pollutants and development of interstitial lung disease in patients with connective tissue disease: a population-based case-control study in Taiwan. BMJ Open 10, e041405. 10.1136/bmjopen-2020-041405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Yin Q, Strong MJ, Zhuang Y, Flemington EK, Kaminski N, de Andrade JA, and Lasky JA (2020). Assessment of viral RNA in idiopathic pulmonary fibrosis using RNA-seq. BMC Pulm. Med 20, 81. 10.1186/s12890-020-1114-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Hata K, Yanagihara T, Matsubara K, Kunimura K, Suzuki K, Tsubouchi K, Eto D, Ando H, Uehara M, Ikegame S, et al. (2023). Mass cytometry identifies characteristic immune cell subsets in bronchoalveolar lavage fluid from interstitial lung diseases. Front. Immunol 14, 1145814. 10.3389/fimmu.2023.1145814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Salisbury ML, Xia M, Murray S, Bartholmai BJ, Kazerooni EA, Meldrum CA, Martinez FJ, and Flaherty KR (2016). Predictors of idiopathic pulmonary fibrosis in absence of radiologic honeycombing: A cross sectional analysis in ILD patients undergoing lung tissue sampling. Respir. Med 118, 88–95. 10.1016/j.rmed.2016.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Ma Y, Cui F, Li D, Wang J, Tang L, Xie J, Hu Y, and Tian Y (2023). Lifestyle, Genetic Susceptibility, and the Risk of Idiopathic Pulmonary Fibrosis: A Large Prospective Cohort Study. Chest, S0012-3692(23)00504-4. 10.1016/j.chest.2023.04.008. [DOI] [PubMed] [Google Scholar]
- 216.McDermott G, Gill R, Gagne S, Byrne S, Huang W, Cui J, Prisco L, Zaccardelli A, Martin L, Kronzer VL, et al. (2022). Associations of the MUC5B promoter variant with timing of interstitial lung disease and rheumatoid arthritis onset. Rheumatol. Oxf. Engl 61, 4915–4923. 10.1093/rheumatology/keac152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kliment CR, Araki T, Doyle TJ, Gao W, Dupuis J, Latourelle JC, Zazueta OE, Fernandez IE, Nishino M, Okajima Y, et al. (2015). A comparison of visual and quantitative methods to identify interstitial lung abnormalities. BMC Pulm. Med 15, 134. 10.1186/s12890-015-0124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Podolanczuk AJ, Oelsner EC, Barr RG, Hoffman EA, Armstrong HF, Austin JHM, Basner RC, Bartels MN, Christie JD, Enright PL, et al. (2016). High attenuation areas on chest computed tomography in community-dwelling adults: the MESA study. Eur. Respir. J 48, 1442–1452. 10.1183/13993003.00129-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Natalini JG, Baker JF, Singh N, Mahajan TD, Roul P, Thiele GM, Sauer BC, Johnson CR, Kawut SM, Mikuls TR, et al. (2021). Autoantibody Seropositivity and Risk for Interstitial Lung Disease in a Prospective Male-Predominant Rheumatoid Arthritis Cohort of U.S. Veterans. Ann. Am. Thorac. Soc 18, 598–605. 10.1513/AnnalsATS.202006-590OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Matson SM, Deane KD, Peljto AL, Bang TJ, Sachs PB, Walts AD, Collora C, Ye S, Demoruelle MK, Humphries SM, et al. (2022). Prospective Identification of Subclinical Interstitial Lung Disease in a Rheumatoid Arthritis Cohort Is Associated with the MUC5B Promoter Variant. Am. J. Respir. Crit. Care Med 205, 473–476. 10.1164/rccm.202109-2087LE. [DOI] [PMC free article] [PubMed] [Google Scholar]