

Abstract
INTRODUCTION
We examined whether hypertension (HTN) was associated with Alzheimer's disease‐related biomarkers in cerebrospinal fluid (CSF) and how changes in blood pressure (BP) related to changes in CSF biomarkers over time.
METHODS
A longitudinal observation of cognitively healthy normotensive subjects (n = 134, BP < 140/90, with no antihypertensive medication), controlled HTN (n = 36, BP < 140/90, taking antihypertensive medication), and 35 subjects with uncontrolled HTN (BP ≥ 140/90). The follow‐up range was 0.5to15.6 years.
RESULTS
Total tau (T‐tau) and phospho‐tau181 (P‐tau 181) increased in all but controlled HTN subjects (group×time interaction: p < 0.05 for both), but no significant Aβ42 changes were seen. Significant BP reduction was observed in uncontrolled HTN, and it was related to increase in T‐tau (p = 0.001) and P‐tau 181 (p < 0.001).
DISCUSSION
Longitudinal increases in T‐tau and P‐tau 181 were observed in most subjects; however, only uncontrolled HTN had both markers increase alongside BP reductions. We speculate cumulative vascular injury renders the brain susceptible to relative hypoperfusion with BP reduction.
Highlights
Over the course of the study, participants with uncontrolled HTN at baseline showed greater accumulation of CSF total tau and phospho‐tau181 (P‐tau 181) than subjects with normal BP or with controlled HTN.
In the group with uncontrolled HTN, increases in total tau and P‐tau 181 coincided with reduction in BP.
We believe this highlights the role of HTN in vascular injury and suggests decline in cerebral perfusion resulting in increased biomarker concentrations in CSF.
Medication use was the main factor differentiating controlled from uncontrolled HTN, indicating that earlier treatment was beneficial for preventing accumulations of pathology.
Keywords: Alzheimer's disease, CSF biomarkers, neurodegeneration
1. INTRODUCTION
Approximately 1.2 billion people worldwide are affected by hypertension (HTN), with an estimated two‐thirds of Americans over the age of 60 exhibiting elevated blood pressure (BP) readings. 1 , 2 HTN negatively impacts on cerebral vasculature promoting atherosclerotic changes in larger brain arteries and arteriolosclerosis in smaller vessels. 3 Changes in vessel walls ultimately lead to reduced cerebral blood flow (CBF) and impaired cerebral autoregulation. 4 , 5 , 6 HTN is a risk factor for Alzheimer's disease (AD) and cognitive impairment. 7 , 8 , 9 A neuropathology report of community‐based subjects showed that higher late‐life systolic blood pressure (SBP) was associated with increased neurofibrillary tangles. 10 Similarly, a cerebrospinal fluid (CSF) study found that hypertensive patients had steeper longitudinal increases in total tau and phosphorylated tau. 11 Others observed positive associations between amyloid beta (Aβ) deposition measured by Pittsburgh compound B positron emission tomography (PET) and both SBP 12 and diastolic blood pressure (DBP), 13 while another study failed to replicate this finding. 14 Thus, it remains unclear whether HTN exacerbates AD pathology within the human brain. The mechanisms of this presumed association are also not fully understood, but reduced CBF and hypoxia is the leading explanation. Supporting evidence comes from animal studies showing that oligemia and hypoxia increase phosphorylated tau and Aβ. 15 , 16 It would follow that lowering BP should stop these changes. However, we have shown that in subjects with HTN there is an optimal BP range, outside of which CBF decreases 17 and white matter lesion (WML) burden increases. 18 This effect was not observed in normotensive subjects, suggesting that those with presumably impaired BP regulatory mechanisms may actually demonstrate hypoperfusion with low BP.
This study examined longitudinal trajectories of AD biomarkers and BP in cognitively normal (NL) subjects with normal baseline BP, as well as with controlled and uncontrolled HTN. We hypothesized that the group with uncontrolled HTN would (1) have more pathology at baseline than two other groups (we tested that in a larger cohort with at least one CSF assessment) and (2) would also show greater accumulation of AD pathology longitudinally (tested in a smaller subset with at least two lumbar punctures). Finally, we examined whether changes in BP were related to changes in CSF biomarkers in all three groups. Based on our prior work, we predicted (3) that subjects with uncontrolled HTN might show an increase in AD biomarkers even with BP reduction, consistent with impaired CBF regulation and inadequate CBF.
2. METHODS
2.1. Subjects
Our report is a retrospective analysis of longitudinally studied NL elderly subjects. Subjects were recruited from the community, and all signed Institutional Review Board (IRB)‐approved consent forms. This work was conducted between 1997 and 2017 and supported by National Institutes of Health grants awarded to Mony J. de Leon, with the goal of assessing the contributions of AD biomarkers to structural imaging in the early diagnosis of AD.
Participants diagnosed with probable AD dementia, non‐AD dementias, mild cognitive impairment, stroke, normal pressure hydrocephalus, and active substance abuse were excluded. All subjects underwent physical, and neurological exams, routine laboratory testing, and magnetic resonance imaging (MRI) to obtain a clinical diagnosis. The staging of cognitive functioning was based on a physician‐administered interview, including the Brief Cognitive Rating Scale, 19 the Global Deterioration Scale (GDS), 20 and the Clinical Dementia Rating (CDR). 21
NL subjects were defined as GDS = 1 (no complaint) or 2 (subjective cognitive complaint) at baseline. A total of 428 NL subjects who had undergone lumbar puncture (LP) were initially considered for this report. A total of 414 had documentation of HTN status (normotension, controlled, or uncontrolled HTN) at their first visit and are included in cross‐sectional analyses. Of the 414 subjects, 205 had two or more LP, and they constitute the main (longitudinal) group for this work. Seventy participants had three LPs, 35 had four, and 18 had five. Since only nine out of 205 subjects had more than five visits, we restricted our analysis at the fifth visit to prevent marked unbalance in the dataset. Figure 1 is a flow chart depicting participant inclusion.
FIGURE 1.
Flow chart describing final study sample.
2.2. Clinical assessments
At each time point participants underwent physical, and neurological exams, routine laboratory testing, MRI, and LP. MRI was performed to exclude the brain pathologies described earlier and is not included in the present analyses. Fasting blood samples were tested for complete blood count, liver function, metabolic, and lipid panels.
Body mass index (BMI) was calculated as weight/height2 [kilograms]/[meters]2. The quantitative insulin sensitivity check index (QUICKI) 22 was calculated as
| (1) |
Insulin resistance (IR) was defined as QUICKI ≥ 0.035.
Diabetes mellitus was established based on medical history, usage of glucose lowering medication, or fasting glucose plasma level ≥126 mg/dL. 23
Hypercholesterolemia was defined as low‐density lipoprotein (LDL) plasma level > 130 mg/dL or taking statins. 24
Smoking status was defined as positive if subject was a current smoker or smoked within last 10 years.
The comparisons of BMI, measures of glucose control, and lipids are given in Table 1 and Table S1 for descriptive purposes.
TABLE 1.
Characteristics of subjects studied longitudinally (n = 205, with 2 or more LPs).
| Normotension (n = 134) | Controlled hypertension (n = 36) | Uncontrolled hypertension (n = 35) | P | |
|---|---|---|---|---|
| Baseline | ||||
| Age (years) | 62.7 ± 9.5 | 67.4 ± 7.7 * | 68.0 ± 8.9 * | 0.001 |
| Education (years) | 16.8 ± 2.0 | 17.3 ± 2.1 | 17.1 ± 2.1 | 0.50 |
| Sex n (%female) | 89 (66%) | 18 (50%) | 19 (54%) | 0.13 |
| Median follow‐up time (years, range) | 2.7 (0.6 to 15.6) | 2.2 (0.5 to 7.2) | 2.7 (0.8 to 9.4) | 0.14 |
| APOE ε4 carriers n (%) | 43 (32%) | 9 (25%) | 12 (34%) | 0.65 |
| SBP (mmHg) | 116.4 ± 10.8 | 122.3 ± 9.8 | 147.2 ± 13.9 | N/A |
| DBP (mmHg) | 70.0 ± 8.9 | 72.6 ± 9.3 | 86.0 ± 10.7 | N/A |
| BMI a | 24.8 ± 3.6 | 28.1 ± 6.9 * | 27.5 ± 4.9 * | 0.002 |
| Glucose b | 79.8 ± 17.0 | 86.0 ± 15.1 * | 84.9 ± 12.7 | <0.001 |
| QUICKI c | 0.386 ± 0.041 | 0.351 ± 0.033 * | 0.368 ± 0.040 | <0.001 |
| Insulin resistance c n (%) | 22 (21%) | 12 (48%) | 11 (41%) | 0.009 |
| Diabetes mellitus a n (%) | 6 (5%) | 4 (11%) | 1 (3%) | 0.27 |
| Total cholesterol d (mg/dL) | 195.8 ± 36.0 | 179.7 ± 33.2 | 201.2 ± 38.3 † | 0.03 |
| HDL cholesterol e (mg/dL) | 63.9 ± 17.8 | 56.4 ± 15.5 | 60.2 ± 17.5 | 0.09 |
| LDL cholesterol f (mg/dL) | 113.5 ± 31.7 | 100.3 ± 27.4 | 121.8 ± 31.7 † | 0.01 |
| Triglycerides e (mg/dL) | 94.6 ± 51.5 | 114.8 ± 52.6 | 106.5 ± 53.7 | 0.06 |
| Statins g n (%) | 30 (23%) | 18 (50%) | 11 (32%) | 0.006 |
| Hypercholesterolemia h n (%) | 48 (38%) | 22 (61%) | 21 (62%) | 0.005 |
| Antihypertensive medication n (%) | NA | 36 (100%) | 10 (32%) | N/A |
| Smoking i n (%) | 9 (7%) | 1 (3%) | 4 (12%) | 0.31 |
| T‐tau (pg/mL) | 286.4 ± 12.3 | 308.7 ± 23.8 | 235.1 ± 24.2 | 0.27 |
| P‐tau 181 (pg/mL) | 44.0 ± 1.6 | 47.1 ± 3.0 | 38.9 ± 3.1 | 0.54 |
| Aβ42 (pg/mL) | 672.3 ± 20.0 | 669.5 ± 38.4 | 716.2 ± 39.6 | 0.29 |
| Follow‐up | ||||
| T‐tau (pg/mL) | 302.6 ± 12.6 | 337.7 ± 24.6 | 275.3 ± 24.8 | 0.66 |
| P‐tau 181 (pg/mL) | 47.0 ± 1.4 | 48.3 ± 2.8 | 44.5 ± 2.9 | 0.90 |
| Aβ42 (pg/mL) | 683.5 ± 19.6 | 702.1 ± 37.7 | 706.4 ± 39.3 | 0.83 |
| Dementia diagnosis at last visit NL/impaired/ missing n (%) | 125/6/3 (93/5/2%) | 34/0/2 (94/0/6%) | 30/2/3 (86/6/9%) | 0.27 |
| Hypertension diagnosis at last visit normotension/controlled/uncontrolled/missing n (%) | 106/5/12/11 (79/4/9/8%) | 4/25/5/2 (11/69/14/6%) | 10/12/10/3 (29/34/29/8%) | <0.001 |
Note: Kruskal‐Wallis ANOVA was used to compare groups for continuous variables (except for age, which was analyzed with ANOVA). Data are presented as mean ± standard deviation, unless otherwise specified. For CSF biomarkers we used ANCOVA adjusting for age and presented data as mean ± standard error. p values for T‐tau and P‐tau 181 come from ranked ANCOVA.
Median time was assessed with data restricted at fifth visit.
Abbreviation: APOE, Apolipoprotein E; BMI, Body Mass Index; DBP, Diastolic Blood Pressure; HDL, High Density Lipoprotein; LDL, Low Density Lipoprotein; NL, cognitively normal; NTN, normotensive; SBP, Systolic Blood Pressure; QUICKI, Quantitative insulin sensitivity check index.
Significant p values are in bold.
Data available for 196 subjects (129 normotensive individuals, 36 subjects with controlled hypertension [HTN], and 31 with uncontrolled HTN).
Data available for 200 subjects (131 normotensive individuals, 35 subjects with controlled HTN, and 34 with uncontrolled HTN).
Data available for 156 subjects (104 normotensive individuals, 25 subjects with controlled HTN, and 27 with uncontrolled HTN).
Data available for 195 subjects (127 normotensive individuals, 34 subjects with controlled HTN, and 34 with uncontrolled HTN).
Data available for 190 subjects (122 normotensive individuals, 34 subjects with controlled HTN, and 34 with uncontrolled HTN).
Data available for 186 subjects (120 normotensive individuals, 34 subjects with controlled HTN, and 32 with uncontrolled HTN).
Data available for 201 subjects (131 normotensive individuals, 36 subjects with controlled HTN, and 34 with uncontrolled HTN).
Data available for 203 subjects (134 normotensive individuals, 36 subjects with controlled HTN, and 33 with uncontrolled HTN).
Data available for 201 subjects (131 normotensive individuals, 36 subjects with controlled HTN, and 34 with uncontrolled HTN).
Different from normotensive group at < 0.05 corrected (Bonferroni).
Different from controlled hypertension at < 0.05 corrected.
BP was taken on the left upper arm using a sphygmomanometer in a sitting position after 5 min of rest. 25
Medication: The use and type of antihypertensive medications was recorded. The categories include angiotensin receptor blockers (ARBs), angiotensin converting enzyme (ACE) inhibitors, beta blockers, diuretics, and calcium channel blockers. We also recorded the use of statins and glucose‐lowering drugs.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the scientific literature on the relationships between CSF biomarkers of AD and vascular risk factors. The volume of scientific literature on CSF biomarkers in AD and vascular disease as a risk factor for AD has increased dramatically in recent years.
Interpretation: Our results illustrate that in all subjects, CSF T‐tau and P‐tau 181 accumulated over time. However, in subjects with uncontrolled HTN, CSF biomarker increases also coincided with reductions in BP. This was not observed in individuals with normal baseline BP or controlled HTN.
Future directions: Prospective, longitudinal studies looking at vascular risk factors and other markers for preclinical AD should be performed to confirm this relationship.
2.3. Study groups
At baseline, all subjects were classified into one of the following groups:
Normotension was defined as BP < 140/90 mmHg and no antihypertensive treatment.
HTN was defined as current antihypertensive treatment and/or BP ≥ 140/90 mmHg 26 and divided into subjects with controlled HTN (current antihypertensive treatment) and BP < 140/90 mmHg and uncontrolled hypertension (BP ≥140/90 irrespective of treatment status).
Participants were again re‐assessed based on the foregoing criteria at their last visit; however, all the primary analyses are presented by groups established at baseline.
Since it is also important to consider groups based on their final outcome, we performed supplementary analyses dividing subjects into normotensive who stayed normotensive (n = 122), normotensive who became hypertensive (n = 12), controlled HTN (n = 36, as in original analysis), uncontrolled HTN who stayed uncontrolled (n = 13), and uncontrolled HTN whose BP normalized at the last visit (HTN– normotension, n = 22, 10/22 without medication). Subjects whose information was incomplete at the last visit (missing in Table 1) were categorized as if their status did not change over the study duration.
2.4. Lumbar puncture, CSF collection, and assays
As we previously described, clear CSF (15cc) was collected into polypropylene tubes using a 20‐gauge LP needle guided by fluoroscopy after a 2‐h fast. 27 The procedures occurred in the late morning and early afternoon. All CSF samples were kept on wet ice for a maximum of 1 h until centrifuged for 10 min at 2000 rpm at 4C. CSF aliquots of 0.25 mL were stored in coded polypropylene tubes at −80°C. Aβ42, total tau (T‐tau), and phospho‐tau181 (P‐tau 181) assays were conducted blind to all clinical data in batch mode. CSF Aβ42, T‐tau, and P‐tau 181 concentrations were measured using standard enzyme‐linked immunosorbent assays (ELISA) (INNOTEST, Fujirebio, Ghent, Belgium). Intra‐ and interassay coefficients of variation were <10% for each analyte. Our work was a retrospective analysis of already collected samples and existing information about biomarker concentrations.
CSF data were acquired over multiple batches. Since we detected the interbatch variability in Aβ42 levels, but not the tau measurements, Aβ values were z‐scored, recentered, and rescaled as previously described with a reference batch of 236 NL subjects. 27
AD profile (A+/T+) was defined as “biomarker evidence of Aβ (low Aβ42) and pathologic tau (high P‐tau 181)” as per National Institute on Aging‐Alzheimer's Association (NIA‐AA) research framework. 29 Since the lack of PET imaging or AD subjects for this cohort precluded us from establishing our own cutoff values, we used cutoffs proposed by Mulder at al., 28 namely, subjects were considered to have AD CSF profiles if their Aβ42 was below 550 pg/mL and P‐tau 181 was above 52 pg/mL.
2.5. Apolipoprotein E (APOE) genotyping
Genotyping was conducted using methods previously published by Main et al. 30 or using Kompetitive Allele Specific PCR (KASP) that enables biallelic scoring of single‐nucleotide polymorphisms (SNPs), as well as insertions and deletions at specific loci. The presence of rs429358 and rs7412 for the APOE alleles was assessed. 31 Study subjects were classified as APOE ε4‐positive, with the detection of either one or two ε4 alleles, and APOE ε4‐negative in the absence of ε4 alleles. ,
TABLE 2.
Change (per year) in T‐tau, P‐tau 181, Aβ42, SBP (systolic blood pressure), and DBP (diastolic blood pressure) in three groups (normotensive, controlled hypertension, and uncontrolled hypertension).
| Normotension (n = 134) | Controlled hypertension (n = 36) | Uncontrolled hypertension (n = 35) | |
|---|---|---|---|
| Log T‐tau | 0.02 (0.02 to 0.03), p < 0.001 | 0.02 (−0.00 to 0.05), p = 0.06 | 0.05 (0.03 to 0.07), p < 0.001 |
| T‐tau | 5.7 (3.1 to 8.2), p < 0.001 | 6.1 (−0.6 to 12.9), p = 0.076 | 12.2 (7.2 to 17.3), p < 0.001 |
| Log P‐tau 181 | 0.02 (0.01 to 0.02), p < 0.001 | 0.02 (−0.00 to 0.03), p = 0.06 | 0.04 (0.03 to 0.03), p < 0.001 |
| P‐tau 181 | 0.71 (0.39 to 1.03), p < 0.001 | 0.42 (−0.39 to 1.24), p = 0.31 | 1.67 (1.04 to 2.29), p < 0.001 |
| Aβ42 | 1.57 (−6.57 to 9.69), p = 0.70 | 9.97 (−9.18 to 29.14), p = 0.31 | −9.76 (−25.65 to 6.13), p = 0.23 |
| SBP a | 0.44 (−0.19 to 1.07), p = 0.17 | −0.73 (−2.32 to 0.87), p = 0.37 | −3.42 (−4.63 to −2.21), p < 0.001 |
| DBP a | −0.15 (−0.79 to 0.50), p = 0.66 | −0.28 (−1.76 to 1.19), p = 0.71 | −3.26 (−4.49 to −2.03), p < 0.001 |
Note: Respective p values for slopes (change with time) are derived from mixed models for repeated measures. CSF biomarker or BP were dependent variables, and time, group, and group × time interaction were predictors. Age at baseline was added as covariate in all the models. Parentheses indicate 95% CI for slopes. Due to adjustment for multiple comparisons, slopes were considered significantly different from 0 at p < 0.016 (bolded in table). The group × time interaction term for log‐transformed T‐tau was p = 0.045 and for P‐tau 181 p = 0.019. The group × time interaction term for untransformed T‐tau was p = 0.075 and for P‐tau 181 p = 0.017. Both for SBP and DBP the group × time interaction was significant at p < 0.001. No significant interaction was found for Aβ42.Abbreviations: DBP, diastolic blood pressure; SBP, systolic blood pressure.
Significant p values are in bold.
aData for 191 subjects.
2.6. Statistical analyses
Categorical variables were examined with χ2 tests. Analysis of variance (ANOVA) or Kruskal‐Wallis ANOVA were used to compare group means for continuous variables. Analysis of covariance (ANCOVA) or ranked ANCOVA were used when there was a need to adjust for age and sex. Normality was tested with the Shapiro‐Wilk test. The percentage of subjects meeting criteria for AD profile at baseline and last visit within each subgroup was compared with the McNemar test for paired proportions.
To examine longitudinal changes in CSF biomarkers and BP, we used mixed models for repeated measures (MMRM), where biomarkers, SPB, and DBP were dependent variables, and time, group (normotension, controlled HTN, and uncontrolled HTN), and group × time interaction were independent variables. Initially, age at baseline and sex were added as covariates. Since sex did not contribute to any model, this variable was removed. Subject‐specific random effects for intercept and slope were used to assess individual variability in outcomes. Group‐specific slopes were estimated and deemed significant at p values adjusted using Holm‐Bonferroni formula.
To test whether changes in BP were related to changes in CSF biomarkers, we also used MMRM. Biomarkers were dependent variables and BP (SBP or DBP), group, and group × BP interaction were independent variables. SBP and DBP were tested in separate models. Subject‐specific random effects for intercept and slope were included unless otherwise noted.
We checked the linear models for violations of the models’ assumptions. If necessary, data were square root‐transformed or logarithmically transformed. If data normalization was not achieved, analyses were repeated using generalized estimating equations (GEEs) and results reported. Statistical significance was defined as a p value < 0.05. SPSS (version 25, SPSS, Inc., Chicago, IL, USA) software was used for all analyses.
3. RESULTS
3.1. General characteristics
The main (longitudinal) study group (n = 205) consisted of 126 (61%) females (age 63.4 ± 10.1 years; education 16.9 ± 2.0 years) and 79 (39%) males (age 66.2 ± 7.9 years; education 17.0 ± 2.2 years). Approximately 35% (n = 71 out of 205) had HTN. Median time between follow‐up visits was 2.0 years, and the follow‐up range was 0.5 to 15.6 years. The larger cross‐sectional group of 414 subjects consisted of 260 (63%) females (age 63.0 ± 10.7 years; education 16.9 ± 2.0 years) and 154 (37%) males (age 66.3 ± 9.7 years; education 17.1 ± 2.3 years), of whom 146/414 (35%) had HTN. Subjects studied more than once did not differ from those with only one LP in age, education, sex, APOE ε4, or HTN status, suggesting that these major variables did not affect the likelihood of longitudinal observation. Table 1 presents the baseline characteristics of subjects with longitudinal data separated by HTN status (normotensive, controlled HTN, and uncontrolled HTN). Similar comparisons for study participants with only one assessment (n = 414) are given in Table S1.
At the last study visit, 5% of normotensive, 0% of subjects with controlled HTN, and 6% of subjects with uncontrolled HTN received a diagnosis of cognitively impaired, and the proportions were not significantly different between groups (Table 1).
In the entire group DBP decreased significantly from first to last visit (GEE β = −0.55 mmHg per year, p < 0.007), but SBP remained unchanged (GEE β = −0.15 mmHg per year, p = 0.55).
The time × group interaction was significant at p < 0.001 for both SBP and DBP, indicating that trajectories of SBP and DBP differed between three groups (Table 2). In the uncontrolled HTN group, both SBP (slope −3.42 mmHg/year, p < 0.001) and DBP (slope −3.26 mmHg/year, p < 0.001) decreased significantly over time, but there were no significant changes for either SBP or DBP in the two other groups (Table 2, Figure 2).
FIGURE 2.
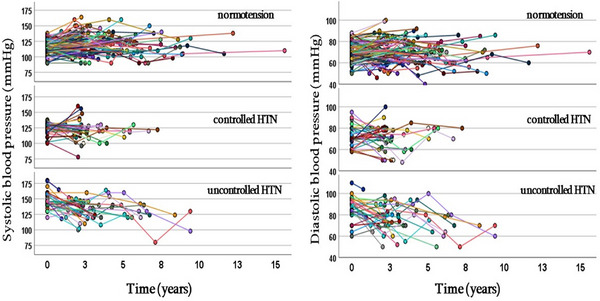
Individual trajectories of systolic and diastolic blood pressure over course of study in each of three study subgroups.
3.2. CSF biomarkers
CSF T‐tau, P‐tau 181, or Aβ42 did not differ at baseline between normotensive, controlled HTN, and uncontrolled HTN groups. This was true both for subjects studied longitudinally (n = 205) (Table 1 and Table 3) and in the larger sample with at least one LP (n = 414) (Table S1).
TABLE 3.
Frequency of AD CSF profile at baseline and follow‐up in each study subgroup.
|
Normotension (n = 130) |
Controlled hypertension (n = 34) |
Uncontrolled hypertension (n = 32) |
P (χ2 test) |
|
|---|---|---|---|---|
|
A+/T+ n (%) (baseline) |
10 (8%) | 5 (15%) | 1 (3%) | 0.22 |
|
A+/T+ n (%) (follow‐up) |
11 (9%) | 6 (18%) | 4 (13%) | 0.29 |
|
P value (McNemar test) |
1.0 | 1.0 | 0.25 |
Note: P values in bottom row are given for comparison between baseline and last assessment within subgroups (McNemar test for paired proportions) and in last column to compare frequencies of AD profiles between subgroups at baseline and follow‐up (χ2 test).
Results remained unchanged after exclusion of subjects with very high total tau (>1000 pg/mL).
3.2.1. Aβ42
Aβ42 remained unchanged in the entire group from first to last visit: GEE β = −0.12 pg/mL per year, p = 0.97. We did not find significant time × group interaction, indicating that Aβ42 dynamics were similar in all three groups. Moreover, changes in SBP or DBP were not associated with Aβ42 change.
3.2.2. P‐tau 181
In the entire group, P‐tau 181 increased significantly from first to last visit; GEE β = 1.1 pg/mL per year, p < 0.001. There was also a significant time × group interaction (MMRM, p = 0.019, log‐transformed data). P‐tau 181 increased significantly in the normotensive (slope 0.71 pg/mL/year, p < 0.001) and uncontrolled HTN groups (slope 1.67 pg/mL/year, p < 0.001), but not in subjects with controlled HTN. The slopes for the normotensive and uncontrolled HTN groups differed significantly from each other (Table 2, Figure 3).
FIGURE 3.
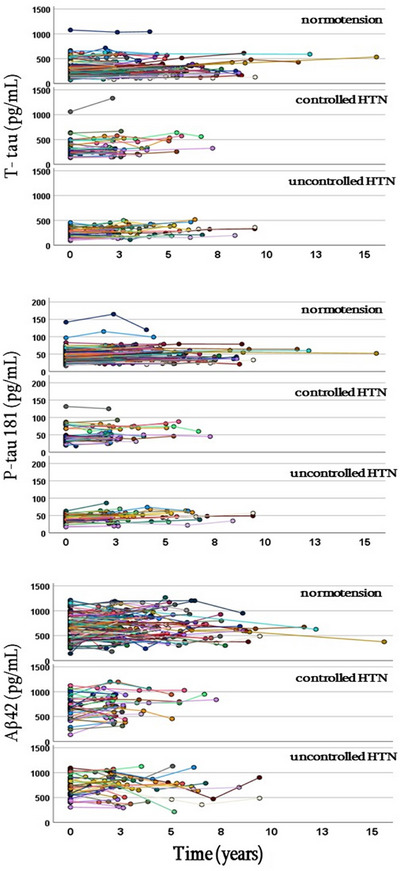
Individual trajectories of T‐tau (total tau), P‐tau 181 (phosphorylated tau), and Aβ42 (amyloid beta 42) over course of study in each of three study subgroups.
Only in the uncontrolled HTN group was reduction in DBP related to P‐tau 181 increases (p < 0.001). The group × DBP interaction was significant at p = 0.06 (Table 5). We did not find associations between change in SBP and change in P‐tau 181.
TABLE 5.
Relationships between change in DBP (diastolic blood pressure) and change in CSF biomarkers in three study subgroups (n = 191).
|
Normotension (n = 125) |
Controlled hypertension (n = 34) |
Uncontrolled hypertension (n = 32) |
|
|---|---|---|---|
| T‐tau | −0.72 (−1.72 to 0.27), p = 0.15 | −1.08 (−2.78 to 0.61), p = 0.21 | −1.34 (−2.55 to −0.11), p = 0.03 |
| P‐tau 181 | −0.14 (−0.25 to −0.03), p = 0.016 | −0.03 (−0.21 to 0.16), p = 0.80 | −0.30 (−0.44 to −0.16), p < 0.001 |
| Aβ42 | 0.12 (−2.06 to 2.30), p = 0.91 | 1.43 (−2.15 to 5.01), p = 0.43 | 0.89 (−1.71 to 3.48), p = 0.50 |
Note: Respective p values for slopes (change in biomarker with changes in DBP) are derived from mixed models for repeated measures. Biomarker was a dependent variable and DBP, group, and group × DBP interaction were predictors. Age at baseline was added as covariate in all models. Parentheses indicate 95% CI for slopes. Due to adjustment for multiple comparisons, slopes were considered significantly different from 0 at p < 0.016 (bolded in table). The group × DBP interaction term for P‐tau 181 was p = 0.06.
3.2.3. T‐tau
In the entire group, T‐tau increased significantly from first to last visit (GEE β = 9.3 pg/mL per year, p < 0.001). In MMRM we also observed a significant time × group interaction (p = 0.045, log‐transformed data). T‐tau increased in the normotensive (5.7/mL/year, p < 0.001) and uncontrolled HTN group (12.2 pg/mL/year, p < 0.001), but not in subjects with controlled HTN. These two slopes were significantly different from each other. Since logarithmic or square root transformations did not result in the normalization of the residuals, analysis was confirmed using GEE. Again, slopes differed between three groups (time × group interaction at p = 0.016). The steepest T‐tau increase was observed in the uncontrolled HTN (β = 0.044) group, and it was significantly greater than in the normotensive (β = 0.020) and controlled HTN (β = 0.015) groups (Table 2, Figure 3). Excluding subjects with very high tau (> 1000 pg/mL) did not change the results.
Only in the uncontrolled HTN group were changes in SBP associated with changes in T‐tau (SBP × group interaction p = 0.009, log‐transformed data); for example, the reduction in SBP was accompanied by an increase in T‐tau (p = 0.001) (Table 4). Since logarithmic or square root transformations of T‐tau did not result in the normalization of the residuals, analysis was confirmed with GEE: associations between changes in SBP and change in T‐tau in the HTN groups were significantly different (SBP × group interaction at p = 0.015). Among subjects with uncontrolled HTN, reduction in SBP was accompanied by increase in T‐tau, but no associations were found in the two other groups. We did not find associations between change in DBP and change in T‐tau.
TABLE 4.
Relationships between change in SBP (systolic blood pressure) and change in CSF biomarkers in three study subgroups (n = 191).
|
Normotension (n = 125) |
Controlled hypertension (n = 34) |
Uncontrolled hypertension (n = 32) |
|
|---|---|---|---|
| Log T‐tau | 0.001 (−0.001 to 0.003), p = 0.34 | −0.001 (−0.005 to 0.003), p = 0.76 | −0.004 (−0.007 to −0.002), p = 0.001 |
| T‐tau | 0.51 (−0.16 to 1.17), p = 0.14 | −0.50 (−1.71 to 0.71), p = 0.41 | −1.03 (−1.76 to (−0.30), p = 0.01 |
| Log P‐tau 181 | −0.001 (−0.003 to 0.001), p = 0.22 | 0.000 (−0.003 to 0.003), p = 0.99 | −0.002 (−0.005 to 0.000), p = 0.03 |
| P‐tau 181 | −0.07 (−0.16 to 0.02), p = 0.13 | 0.03 (−0.14 to 0.20), p = 0.71 | −0.11 (−0.22 to −0.01), p = 0.03 |
| Aβ42 | −0.35 (−1.96 to 1.25), p = 0.67 | −0.94 (−3.82 to 1.94), p = 0.52 | −0.39 (−2.21 to 1.44), p = 0.68 |
Note: Respective p values for slopes (change in biomarker with changes in SBP) are derived from mixed models for repeated measures. Biomarker was a dependent variable and SBP, group, and group × SBP interaction were predictors. Age at baseline was added as covariate in all models. Parentheses indicate 95% CI for slopes. Due to adjustment for multiple comparisons, slopes were considered significantly different from 0 at p < 0.016 (bolded in table). The group × SBP interaction term for both log transformed and untransformed T‐tau was p = 0.009.
All the results remained unchanged when BMI, smoking, baseline diabetes mellitus, and baseline hypercholesterolemia diagnoses were added to the models (supplemental material, p. 6).
3.3. Prevalence of AD CSF profile
For the subjects studied longitudinally, the number of individuals defined as having an AD CSF profile did not differ between three groups at baseline or at follow‐up. The number did not increase significantly from first to last visit within the groups (Table 3).
3.4. Additional analyses
Table S2 presents additional analyses of biomarkers and BP trajectories in the five groups: normotension–normotension, normotension‐HTN, controlled HTN, uncontrolled HTN–uncontrolled HTN, and uncontrolled HTN–normotension. The longitudinal dynamics of T‐tau or P‐tau 181 were similar for subjects with normal BP at baseline who remained normotensive at their last visit and those who developed HTN. Likewise, longitudinal dynamics of T‐tau or P‐tau 181 were similar for subjects with uncontrolled HTN at baseline whose BP remained uncontrolled and for those whose BP normalized. As for CSF Aβ42, the time × group interaction was at a trend level (p = 0.12): Subjects with uncontrolled baseline HTN whose status remained unchanged showed significant reduction in Aβ42 over time (p = 0.008).
4. DISCUSSION
There are several major findings from our study. First, among NL community‐dwelling middle‐aged and older adults, CSF AD biomarkers did not differ at baseline between normotensive individuals and subjects with controlled and uncontrolled HTN.
Second, we observed significant longitudinal increases in T‐tau and P‐tau 181 in subjects who were normotensive or had uncontrolled HTN at baseline. Remarkably, the increase in both markers was twice as high among subjects with uncontrolled HTN than in normotensive individuals. , ,
Third, the biomarker slopes were comparable for normotensive subjects who stayed normotensive and those who developed HTN. The same was true for subjects with baseline uncontrolled HTN: the slopes did not differ for those whose HTN stayed uncontrolled and those in whom BP normalized (Table S2). When Aβ42 changes were analyzed across three groups at baseline, no difference was found. However, splitting uncontrolled HTN groups based on their final outcome revealed that only subjects whose BP was uncontrolled throughout the study experienced significant reduction in Aβ42 (Table S2). Still, analyses concerning five groups must be considered with caution as groups were small and the models unbalanced. Overall, our longitudinal results are consistent with previous findings in which high BP has been linked to increased tau at post mortem assessment, 10 and measures of arterial stiffness were related to higher concentrations of T‐tau and P‐tau 181 in CSF. 32 They also add to older reports showing more atherosclerotic plaques in the circle of Willis in patients with sporadic AD. 33 , 34 , 35 We found some evidence for association between chronically uncontrolled HTN and changes in CSF Aβ42. These findings contribute to existing conflicting evidence. Some studies showed that vascular risk was associated with NFT development, but not with neuritic plaques. 10 , 36 , 37 Others observed a link between CSF Aβ42 reduction or positive Aβ PET with BP and vascular risk. 38 , 39 , 40 The lack of significant CSF Aβ42 decline in the HTN groups may be attributed to antihypertensive medication. Diuretic use was previously associated with reduced Aβ42 oligomerization 41 and angiotensin receptor blockers with reduced plaque deposition and dementia risk. 42 , 43 Consistently, in our analysis of the five groups, Aβ42 reduction was observed only in the subgroup that stayed unmedicated with high BP. The frequency of AD profile across groups and time did not differ and overall was rather low. Since supposedly a third of cognitively healthy older adults may harbor the AD CSF pattern, 44 the validity of using cutoff values not derived from our own populations may be disputed. However, consistent with this low frequency, a very small number of participants developed cognitive impairment at follow‐up. Taken together, the clinical importance of the continuous changes may seem questionable. Nonetheless, although not big enough to cross the thresholds of positivity, changes were significant and, remarkably, for T‐tau and P‐tau, two times greater for uncontrolled HTN than for normotensive and controlled HTN groups. We believe this indicates that biomarker dynamics are relevant but rather slow.
In subjects with controlled HTN, slopes for T‐tau and P‐tau were not significant, but they were numerically similar to the slopes for normotensive subjects. If the group had been bigger, they might have reached significance, although, the fact that the slopes for uncontrolled HTN were twice as steep still suggests protective effects of treatment. Minor increases in tau biomarkers seen in the normotensive group were anticipated, as these may be attributed to normal aging or to other unaccounted risk factors.
We assumed that subjects with uncontrolled HTN might represent a group with a higher vascular burden with corresponding impairments of CBF regulation. If true, then a reduction of BP should be paradoxically accompanied by increases in CSF markers of AD pathology due to relative hypoperfusion. In the uncontrolled HTN group, reduced SBP was indeed associated with increases in T‐tau, and reductions in DBP were associated with increases in P‐tau 181. This study extends our earlier observation that cognitively healthy HTN subjects with reductions in BP experienced increases in P‐tau 181 and memory decline. 45 Somewhat similarly, autopsy studies have demonstrated that subjects with low pre‐mortem SBP and high Aβ load also have more tau post mortem. 46 However, secondary analyses indicate that a paradoxical BP–tau relationship might not be true for all subjects in this subgroup: similar T‐tau and P‐tau 181 increases were seen in subjects whose BP stayed uncontrolled and in those whose BP normalized (Table S2), despite obviously divergent BP trajectories. In addition, Aβ42 significantly dropped only in participants with uncontrolled HTN that stayed uncontrolled. We posit that multiple mechanisms played a role. In some subjects, further vascular damage (uncontrolled BP) was associated with a rise in tau markers, but in those with BP reduction, tau increased due to presumed hypoperfusion. Both scenarios could elicit ischemia. An earlier PET study showed a decrement in CBF together with increased oxygen extraction fraction and tau deposition in the hemisphere ipsilateral to atherosclerotic carotid artery, suggesting that ischemia facilitates tau deposition in humans. 47 We previously observed a similar pattern when analyzing WML growth. In subjects with uncontrolled HTN at baseline, WML progressed irrespective of BP trajectories. 18 Impaired CSF/interstitial fluid clearance due to reduced vascular function and concomitant CBF reduction might have also contributed to our findings of increased tau accumulation and Aβ42 reduction; this requires further study.
Finally, medication use was the main factor differentiating controlled from uncontrolled HTN. This implies that earlier treatment was beneficial for preventing accumulations of pathology.
Our study has multiple limitations. Our group had more female than male participants. Participants were predominantly Caucasian, with higher‐than‐average educational attainment. Furthermore, there was a significant difference between the median age of our normo‐ and hypertensive subjects and higher rates of obesity and metabolic disorders within the HTN groups. Also, the median time of overall follow‐up (2 years) was rather short, and a longer period of observation could result in more pronounced changes between the groups. The classification of patients within HTN groups was based on a single measurement during an outpatient visit. Similarly, for follow‐up assessments, BP was taken only once at each visit. Additionally, in rare instances, antihypertensive drugs may have been prescribed for indications other than HTN. While this could have induced bias, it is noteworthy that 80% of subjects classified as normotensive at baseline remained normotensive at their last visit. Also, 70% of subjects with controlled HTN remained in the same category. In the uncontrolled HTN group, we expected more subjects to change status as they likely underwent interventions. Both uncontrolled and controlled HTN groups were much smaller than the normotensive group. This was a clear disadvantage and prevented us from assessing the effects of various classes of antihypertensive mediaction and statins and, most importantly, the effects of BP normalization in the uncontrolled group.
A major limitation of this study was the retrospective character of our analysis. We used already existing values of biomarker concentrations from assays performed earlier. ELISA was then a widely used method. It has much higher intra‐ and interassay (15%) variability compared to newer fully automated methods (< 5%). 48 Since the lack of PET imaging or AD subjects for this cohort precluded establishing our own cutoff values, we arbitrarily used cutoffs from other group, potentially adding bias to our analysis. We did not have Aβ40 values and could not calculate an Aβ42/ Aβ40 ratio, which is considered a more sensitive indicator of abnormality, as it better predicts PET amyloid status than Aβ42 alone. 49 Finally, some participants might have suffered a minor stroke, resulting in increased T‐tau or P‐tau, which we could have erroneously interpreted as related to HTN.
In conclusion, we found that participants with uncontrolled HTN showed greater accumulation of T‐tau and P‐tau than subjects with normal BP or with controlled HTN. Moreover, the increases occurred irrespective of BP trajectories in this subgroup. Aβ reduction seemed to be linked to pervasive high BP. We believe this highlights the role of HTN in vascular injury and probable decline in cerebral perfusion resulting in increased biomarker concentrations in CSF. This implies that early treatment of HTN may have protective effects. Considering conflicting findings from previous studies, we believe further research is required to elucidate relationship between HTN and preclinical AD outcomes.
CONFLICT OF INTEREST STATEMENT
AB, LG, YL: none. Author disclosures are available in the supporting information.
CONSENT STATEMENT
Subjects were recruited from the community, and all signed Institutional Review Board‐approved consent forms. This work was conducted between 1997 and 2017.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The authors are most grateful to Ms. Cynthia Fox for editing this paper. Study funding came from NIH grants AG022374, AG12101, AG05603, HL111724, NS104364, AG058913, and AG057570.
Biskaduros A, Glodzik L, Saint Louis LA, et al. Longitudinal trajectories of Alzheimer's disease CSF biomarkers and blood pressure in cognitively healthy subjects. Alzheimer's Dement. 2024;20:4389–4400. 10.1002/alz.13800
Adrienne Biskaduros and Lidia Glodzik contributed equally to this work.
REFERENCES
- 1. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the american college of cardiology/american heart association task force on clinical practice guidelines. Hypertension. 2018;71:e13‐e115. doi: 10.1161/HYP.0000000000000065 [DOI] [PubMed] [Google Scholar]
- 2. Collaboration NCDRF . Worldwide trends in hypertension prevalence and progress in treatment and control from 1990 to 2019: a pooled analysis of 1201 population‐representative studies with 104 million participants. Lancet. 2021;398:957‐980. doi: 10.1016/S0140-6736(21)01330-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Grinberg LT, Thal DR. Vascular pathology in the aged human brain. Acta Neuropathol. 2010;119:277‐290. doi: 10.1007/s00401-010-0652-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Veglio F, Paglieri C, Rabbia F, Bisbocci D, Bergui M, Cerrato P. Hypertension and cerebrovascular damage. Atherosclerosis. 2009;205:331‐341. doi: 10.1016/j.atherosclerosis.2008.10.028 [DOI] [PubMed] [Google Scholar]
- 5. Kennelly SP, Lawlor BA, Kenny RA. Blood pressure and the risk for dementia: a double edged sword. Ageing Res Rev. 2009;8:61‐70. doi: 10.1016/j.arr.2008.11.001 [DOI] [PubMed] [Google Scholar]
- 6. Iadecola C, Gottesman RF. Neurovascular and cognitive dysfunction in hypertension. Circ Res. 2019;124:1025‐1044. doi: 10.1161/circresaha.118.313260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Harrison SL, Ding J, Tang EY, et al. Cardiovascular disease risk models and longitudinal changes in cognition: a systematic review. PLoS One. 2014;9:e114431. doi: 10.1371/journal.pone.0114431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kivipelto M, Ngandu T, Fratiglioni L, et al. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Archives of Neurology. 2005;62:1556‐1560. doi: 10.1001/archneur.62.10.1556 [DOI] [PubMed] [Google Scholar]
- 9. McGrath ER, Beiser AS, DeCarli C, et al. Blood pressure from mid‐ to late life and risk of incident dementia. Neurology. 2017;89:2447‐2454. doi: 10.1212/wnl.0000000000004741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arvanitakis Z, Capuano AW, Lamar M, et al. Late‐life blood pressure association with cerebrovascular and Alzheimer disease pathology. Neurology. 2018;91:e517‐e525. doi: 10.1212/wnl.0000000000005951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bos I, Vos SJB, Schindler SE, et al. Vascular risk factors are associated with longitudinal changes in cerebrospinal fluid tau markers and cognition in preclinical Alzheimer's disease. Alzheimers Dement. 2019;15(9):1149‐1159. doi: 10.1016/j.jalz.2019.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Langbaum JB, Chen K, Launer LJ, et al. Blood pressure is associated with higher brain amyloid burden and lower glucose metabolism in healthy late middle‐age persons. Neurobiology of Aging. 2012;33:827. e811‐e829. doi: 10.1016/j.neurobiolaging.2011.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Toledo JB, Toledo E, Weiner MW, et al. Cardiovascular risk factors, cortisol, and amyloid‐beta deposition in Alzheimer's disease neuroimaging initiative. Alzheimers Dement. 2012;8:483‐489. doi: 10.1016/j.jalz.2011.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vemuri P, Knopman DS, Lesnick TG, et al. Evaluation of amyloid protective factors and Alzheimer disease neurodegeneration protective factors in elderly individuals. JAMA Neurol. 2017;74:718‐726. doi: 10.1001/jamaneurol.2017.0244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Koike MA, Green KN, Blurton‐Jones M, Laferla FM. Oligemic hypoperfusion differentially affects tau and amyloid‐{beta}. Am J Pathol. 2010;177:300‐310. doi: 10.2353/ajpath.2010.090750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Raz L, Bhaskar K, Weaver J, et al. Hypoxia promotes tau hyperphosphorylation with associated neuropathology in vascular dysfunction. Neurobiol Dis. 2019;126:124‐136. doi: 10.1016/j.nbd.2018.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Glodzik L, Rusinek H, Tsui W, et al. Different relationship between systolic blood pressure and cerebral perfusion in subjects with and without hypertension. Hypertension. 2019;73:197‐205. doi: 10.1161/HYPERTENSIONAHA.118.11233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Woldstad C, Rusinek H, Sweeney E, et al. Quadratic relationship between systolic blood pressure and white matter lesions in individuals with hypertension. J Hypertens. 2023;41:35‐43. doi: 10.1097/HJH.0000000000003292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reisberg B, Ferris SH. Brief cognitive rating scale (BCRS). Psychopharmacol Bull. 1988;24:629‐636. [PubMed] [Google Scholar]
- 20. Reisberg BSS, Franssen EH, de Leon MJ, et al. Clinical stages of normal aging and Alzheimer's disease: the GDS staging system. Neuroscience Research Communications. 1993;13(1):551‐554. Suppl.. [Google Scholar]
- 21. Morris JC. The clinical dementia rating (CDR): current version and scoring rules. Neurology. 1993;43:2412‐2414. doi: 10.1212/wnl.43.11.2412-a [DOI] [PubMed] [Google Scholar]
- 22. Katz A, Nambi SS, Mather K, et al. Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans. J Clin Endocrinol Metab. 2000;85:2402‐2410. doi: 10.1210/jcem.85.7.6661 [DOI] [PubMed] [Google Scholar]
- 23. 2. Classification and diagnosis of diabetes: standards of medical care in diabetes‐2020. Diabetes care. 2020;43:S14‐S31. doi: 10.2337/dc20-S002 [DOI] [PubMed] [Google Scholar]
- 24. Ibrahim MAEJI . Hypercholesterolemia. 2023. Accessed 29 Jan 2024. https://www.ncbi.nlm.nih.gov/books/NBK459188/
- 25. Muntner P, Shimbo D, Carey RM, et al. Measurement of blood pressure in humans: a scientific statement from the american heart association. Hypertension. 2019;73:e35‐e66. doi: 10.1161/hyp.0000000000000087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chobonian A, Bakris GL, Black HR, et al. The seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure. JAMA. 2003;289:2560‐2572. [DOI] [PubMed] [Google Scholar]
- 27. de Leon MJ, Pirraglia E, Osorio RS, et al. The nonlinear relationship between cerebrospinal fluid Abeta42 and tau in preclinical Alzheimer's disease. PloS one. 2018;13:e0191240. doi: 10.1371/journal.pone.0191240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mulder C, Verwey NA, van der Flier WM, et al. Amyloid‐beta(1‐42), total tau, and phosphorylated tau as cerebrospinal fluid biomarkers for the diagnosis of Alzheimer disease. Clinical Chemistry. 2010;56:248‐253. doi: 10.1373/clinchem.2009.130518 [DOI] [PubMed] [Google Scholar]
- 29. Jack CR Jr, Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. doi: 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Main BF, Jones PJ, MacGillivray RT, Banfield DK. Apolipoprotein E genotyping using the polymerase chain reaction and allele‐specific oligonucleotide primers. J Lipid Res. 1991;32:183‐187. [PubMed] [Google Scholar]
- 31. Evans S, Dowell NG, Tabet N, Tofts PS, King SL, Rusted JM. Cognitive and neural signatures of the APOE E4 allele in mid‐aged adults. Neurobiology of Aging. 2014;35:1615‐1623. doi: 10.1016/j.neurobiolaging.2014.01.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moore EE, Liu D, Li J, et al. Association of aortic stiffness with biomarkers of neuroinflammation, synaptic dysfunction, and neurodegeneration. Neurology. 2021;97:e329‐e340. doi: 10.1212/WNL.0000000000012257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roher AE, Esh C, Kokjohn TA, et al. Circle of willis atherosclerosis is a risk factor for sporadic Alzheimer's disease. Arterioscler Thromb Vasc Biol. 2003;23:2055‐2062. doi: 10.1161/01.ATV.0000095973.42032.44 [DOI] [PubMed] [Google Scholar]
- 34. Beach TG, Wilson JR, Sue LI, et al. Circle of Willis atherosclerosis: association with Alzheimer's disease, neuritic plaques and neurofibrillary tangles. Acta Neuropathol. 2007;113:13‐21. doi: 10.1007/s00401-006-0136-y [DOI] [PubMed] [Google Scholar]
- 35. Honig LS, Kukull W, Mayeux R. Atherosclerosis and AD: analysis of data from the US National Alzheimer's Coordinating Center. Neurology. 2005;64:494‐500. doi: 10.1212/01.WNL.0000150886.50187.30 [DOI] [PubMed] [Google Scholar]
- 36. Bos I, Vos SJB, Schindler SE, et al. Vascular risk factors are associated with longitudinal changes in cerebrospinal fluid tau markers and cognition in preclinical Alzheimer's disease. Alzheimers Dement. 2019;15:1149‐1159. doi: 10.1016/j.jalz.2019.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zetterberg H, Mortberg E, Song L, et al. Hypoxia due to cardiac arrest induces a time‐dependent increase in serum amyloid beta levels in humans. PLoS One. 2011;6:e28263. doi: 10.1371/journal.pone.0028263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hughes TM, Wagenknecht LE, Craft S, et al. Arterial stiffness and dementia pathology: atherosclerosis Risk in Communities (ARIC)‐PET Study. Neurology. 2018;90:e1248‐e1256. doi: 10.1212/wnl.0000000000005259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gottesman RF, Schneider AL, Zhou Y, et al. Association between midlife vascular risk factors and estimated brain amyloid deposition. Jama. 2017;317:1443‐1450. doi: 10.1001/jama.2017.3090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sible IJ, Nation DA, Alzheimer's Disease Neuroimaging I . Visit‐to‐visit blood pressure variability and csf alzheimer disease biomarkers in cognitively unimpaired and mildly impaired older adults. Neurology. 2022;98:e2446‐e2453. doi: 10.1212/WNL.0000000000200302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhao W, Wang J, Ho L, Ono K, Teplow DB, Pasinetti GM. Identification of antihypertensive drugs which inhibit amyloid‐beta protein oligomerization. J Alzheimers Dis. 2009;16:49‐57. doi: 10.3233/JAD-2009-0925 [DOI] [PubMed] [Google Scholar]
- 42. Glodzik L, Rusinek H, Kamer A, et al. Effects of vascular risk factors, statins, and antihypertensive drugs on PiB deposition in cognitively normal subjects. Alzheimers Dement (Amst). 2016;2:95‐104. doi: 10.1016/j.dadm.2016.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li NC, Lee A, Whitmer RA, et al. Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: prospective cohort analysis. BMJ. 2010;340:b5465. doi: 10.1136/bmj.b5465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mattsson N, Rosén E, Hansson O, et al. Age and diagnostic performance of Alzheimer disease CSF biomarkers. Neurology. 2012;78:468‐476. doi: 10.1212/WNL.0b013e3182477eed [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Glodzik L, Rusinek H, Pirraglia E, et al. Blood pressure decrease correlates with tau pathology and memory decline in hypertensive elderly. Neurobiology of Aging. 2014;35:64‐71. doi: 10.1016/j.neurobiolaging.2013.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Oveisgharan S, Capuano AW, Kapasi A, et al. Association of low systolic blood pressure with postmortem amyloid‐beta and tau. J Alzheimers Dis. 2020;78:1755‐1764. doi: 10.3233/JAD-200412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yamauchi H, Kagawa S, Kusano K, Ito M, Okuyama C. Misery perfusion and tau deposition in atherosclerotic major cerebral artery disease: a (18)F‐Florzolotau positron emission tomography study. Stroke. 2022;53:e500‐e503. doi: 10.1161/strokeaha.122.040493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hansson O, Seibyl J, Stomrud E, et al. CSF biomarkers of Alzheimer's disease concord with amyloid‐β PET and predict clinical progression: a study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement. 2018;14:1470‐1481. doi: 10.1016/j.jalz.2018.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Alcolea D, Pegueroles J, Muñoz L, et al. Agreement of amyloid PET and CSF biomarkers for Alzheimer's disease on Lumipulse. Ann Clin Transl Neurol. 2019;6:1815‐1824. doi: 10.1002/acn3.50873 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information