

Abstract
INTRODUCTION
Amyloid beta (Aβ) impairs the cerebral blood flow (CBF) increase induced by neural activity (functional hyperemia). Tissue plasminogen activator (tPA) is required for functional hyperemia, and in mouse models of Aβ accumulation tPA deficiency contributes to neurovascular and cognitive impairment. However, it remains unknown if tPA supplementation can rescue Aβ‐induced neurovascular and cognitive dysfunction.
METHODS
Tg2576 mice and wild‐type littermates received intranasal tPA (0.8 mg/kg/day) or vehicle 5 days a week starting at 11 to 12 months of age and were assessed 3 months later.
RESULTS
Treatment of Tg2576 mice with tPA restored resting CBF, prevented the attenuation in functional hyperemia, and improved nesting behavior. These effects were associated with reduced cerebral atrophy and cerebral amyloid angiopathy, but not parenchymal amyloid.
DISCUSSION
These findings highlight the key role of tPA deficiency in the neurovascular and cognitive dysfunction associated with amyloid pathology, and suggest potential therapeutic strategies involving tPA reconstitution.
Highlights
Amyloid beta (Aβ) induces neurovascular dysfunction and impairs the increase of cerebral blood flow induced by neural activity (functional hyperemia).
Tissue plasminogen activator (tPA) deficiency contributes to the neurovascular and cognitive dysfunction caused by Aβ.
In mice with florid amyloid pathology intranasal administration of tPA rescues the neurovascular and cognitive dysfunction and reduces brain atrophy and cerebral amyloid angiopathy.
tPA deficiency plays a crucial role in neurovascular and cognitive dysfunction induced by Aβ and tPA reconstitution may be of therapeutic value.
Keywords: amyloid beta, atrophy, cerebral amyloid angiopathy, dementia, functional hyperemia, neuroinflammation
1. BACKGROUND
Alzheimer's disease (AD) and related dementias have emerged as a major challenge in contemporary health care. 1 , 2 , 3 The neuropathology of AD is characterized by the accumulation of amyloid beta (Aβ) in the brain parenchyma (amyloid plaques) and around blood vessels (cerebral amyloid angiopathy [CAA]), and of neurofibrillary tangles constituted by intracellular hyperphosphorylated tau. 4 , 5 , 6 , 7 Recent evidence underscores neurovascular dysfunction as an early manifestation of AD, exerting a detrimental impact on the disease's expression and progression, 4 , 5 , 8 , 9 and suggests that counteracting neurovascular dysfunction may be beneficial in AD patients. 1 , 5
Aβ attenuates the increase of cerebral blood flow (CBF) evoked by neural activity (functional hyperemia), or by vasoactive mediators acting on endothelial cells. 6 , 10 , 11 , 12 , 13 , 14 , 15 The mechanisms of the neurovascular dysfunction induced by Aβ remain to be fully elucidated, but we recently reported that the attenuation in functional hyperemia is accountable in part by a reduction in the activity of the tissue plasminogen activator (tPA). 14 tPA, a serine‐protease renowned for its role in fibrinolysis, 16 , 17 , 18 plays a key role in the full expression of functional hyperemia. 13 , 14 , 19 , 20 tPA, released by synaptic activity, 21 , 22 , 23 , 24 is required for the production of the vasodilator nitric oxide (NO) from neuronal NO synthase (nNOS) upon NMDA receptor (NMDAR) activation. 25 , 26 , 27 , 28 Thus, tPA is required for the full expression of the NMDAR‐nNOS dependent component of functional hyperemia. 13 , 14
In AD brains, as in mice overexpressing the amyloid precursor protein (APP) and Aβ, 14 , 29 , 30 , 31 tPA levels are reduced due to increased activity of the endogenous tPA inhibitor PAI‐1. 29 , 32 , 33 Thus, PAI‐1 upregulation and the resulting tPA deficiency underlie the reduction in functional hyperemia in Tg2576 mice overexpressing the Swedish mutation of APP. 14 , 31 Furthermore, prolonged rescue of tPA activity by intracerebral PAI‐1 inhibition reinstates neurovascular coupling, reduces CAA, and improves cognitive function in aged Tg2576 mice without affecting amyloid plaques. 14 These observations raise the possibility that tPA supplementation could be an effective therapeutic approach to counteract the deleterious effect of tPA deficiency on Aβ‐induced neurovascular pathology as well as cognitive function.
To more directly demonstrate that the beneficial effects of PAI‐I inhibition are related to rescuing tPA deficiency, we used administration of tPA in symptomatic Tg2576 mice to establish its impact on neurovascular regulation, cognitive function, and CAA. We found that intranasal administration of tPA, a well‐established route of tPA supplementation, 34 , 35 , 36 , 37 , 38 , 39 ameliorates CAA and rescues neurovascular and cognitive dysfunction in aged Tg2576 mice. The findings provide direct evidence that tPA deficiency is a critical contributor to the deleterious neurovascular and cognitive effects of amyloid pathology and raise the possibility that rescuing tPA activity may have translational potential.
2. METHODS
2.1. Mice
The Institutional Animal Care and Use Committee of Weill Cornell Medicine approved all experimental procedures. Experiments were performed in 11‐ to 15‐month‐old Tg2576 mice overexpressing the Swedish mutation of APP (K670/671L) 40 or age‐matched wild‐type (WT) littermates. All mice were males and derived from in‐house colonies. 41 , 42 , 43
2.2. Intranasal tPA administration
Tg2576 and WT littermates (age 11–12 months) were familiarized with the handling procedure and then randomly assigned to receive intranasal instillment of tPA (a generous gift from Genentech) as previously described 34 , 35 , 36 , 38 , 39 (Figure 1). Awake mice were instilled with tPA (0.8 mg/kg dissolved in dH2O/day; 10 μL/nostril) for 5 days/week over 12 weeks. tPA dosage was based on studies showing effectiveness in other disease models. 34 , 35 , 36 , 37 , 38 , 39 Control groups received vehicle (dH2O) with the same protocol as above.
FIGURE 1.
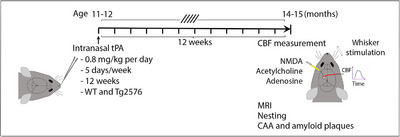
Experimental plan. tPA was intranasally administered at 11 to 12 months of age in WT and Tg2576 mice. Three months later, resting CBF (ASL‐MRI), CBF responses (laser‐Doppler flowmetry), brain atrophy (T2‐MRI), cognition (nesting), and CAA and amyloid plaques (histopathology) were assessed. ASL, arterial spin labeling; CAA, cerebral amyloid angiopathy; CBF, cerebral blood flow; MRI, magnetic resonance imaging; tPA, tissue plasminogen activator; WT, wild type.
2.3. Magnetic resonance imaging
Magnetic resonance imaging (MRI) was performed on a 7.0 tesla 70/30 Bruker Biospec small‐animal MRI system with 450 mT/m gradient amplitude and a 4500 T/m/s slew rate, as previously reported. 44 Briefly, the animals were anesthetized with isoflurane (1%–2%) and placed in the MRI scanner. A volume coil was used for transmission and a surface coil for reception. The magnet coordinates of each animal varied a bit between mice, but the anatomical slice package was always aligned in the same way on each mouse, with the final slice in the forebrain immediately before the olfactory bulb. This allowed the ability to consistently landmark the anatomical images and then center the arterial spin labeling (ASL) slice package a set distance from the anatomical slice package. Anatomical localizer images were acquired to find the transversal slice corresponding to the somatosensory cortex. 44 This position was used for subsequent ASL imaging, which was based on a flow‐sensitive alternating inversion recovery rapid acquisition with relaxation enhancement (FAIR‐RARE) pulse sequence labeling the inflowing blood via the global inversion of the equilibrium magnetization. Three averages of one axial slice were acquired with a field of view of 15 × 15 mm, a spatial resolution of 0.234 × 0.234 × 2 mm3, an echo time (TE) of 5.368 ms, an effective TE of 26.84 ms, a repeat time (TR) of 10 seconds, and a rapid imaging with refocused echoes (RARE) factor of 36. For the computation of CBF, the Bruker ASL perfusion processing macro was used. 44 Turbo‐RARE anatomical images were acquired with the same field of view and orientation as the ASL images (resolution of 0.078 × 0.078 × 1 mm3, TE of 48 ms, TR of 2000 ms, and a RARE factor of 10). The total scan time (ASL plus anatomical images) was 39 minutes. The ASL–MRI and T2‐weighted MRI images were evaluated for resting CBF and for cortical and hippocampal anatomy, respectively, at the bregma level of −1.70 mm 45 using ImageJ 1.54 h (NIH). The average CBF value is reported as mL/100 g of tissue per minute (mL/100 g/minute). To measure the cortical thickness, two to three measurements were taken bilaterally in a coronal section at different levels from the midline and averaged to produce the mean thickness in mm. 44 To measure the hippocampal volume, the regions of interest (ROIs) representing the right and left hippocampi were drawn on T2‐weighted coronal sections and measured. The hippocampal volume (mm3) was then calculated using the following formula: the hippocampal volume = sum of ROI × 0.5 mm (a thickness of a T2‐weighted MRI section).
RESEARCH IN CONTEXT
Systematic review: Amyloid beta (Aβ) is a major contributor to Alzheimer's disease (AD) and attenuates functional hyperemia. Tissue plasminogen activator (tPA) is required for the full expression of the increase in cerebral blood flow (CBF) induced by neural activity and its deficiency contributes to the neurovascular and cognitive function impairments induced by Aβ, but it is unclear if tPA supplementation can prevent the Aβ‐induced impairments.
Interpretation: We found that intranasal administration of recombinant tPA in aged Tg2576 mice reverses the reduction in resting CBF and prevents brain atrophy. Additionally, tPA rescued the deficit in functional hyperemia, improved nesting behavior and reduced cerebral amyloid angiopathy.
Future directions: We will test the effectiveness and safety of tPA variants that do not affect blood clotting, as a potential therapeutic option for mitigating the cerebrovascular and cognitive effects of amyloid pathology.
2.4. CBF measurement
As detailed previously, 41 , 42 , 43 , 44 mice were anesthetized with isoflurane (induction, 5%; surgery, 1.5%) and maintained with urethane (750 mg/kg; ip) and α‐chloralose (50 mg/kg; ip). A femoral artery was cannulated to record arterial pressure and collect blood samples for blood gas analysis. The trachea was intubated, and mice were artificially ventilated with a mixture of N2 and O2. Arterial blood pressure (80–90 mmHg), blood gases (pO2, 120–140 mmHg; pCO2, 30–40 mmHg; pH, 7.3–7.4), and rectal temperature (37°C) were monitored and controlled. Throughout the experiment, the level of anesthesia was monitored by testing motor responses to tail pinch. The somatosensory cortex was exposed through a small craniotomy (2–2 mm). The dura was removed, and the exposed cortex was continuously bathed with a modified Ringer's solution (36–37°C; pH: 7.3–7.4) (see Iadecola 46 for composition). CBF was continuously monitored at the site of superfusion with a laser‐Doppler probe (Perimed Inc.) positioned stereotaxically on the neocortical surface and connected to a computerized data acquisition system. CBF values were expressed as a percent increase relative to the resting level. CBF was recorded after arterial pressure and blood gases were in a steady state. For functional hyperemia, the whiskers were mechanically stimulated for 30 seconds and the associated increase in CBF recorded over the somatosensory cortex. To test the NMDAR‐dependent component of functional hyperemia, NMDA (40 μM; Sigma‐Aldrich) was topically superfused on the cranial window. The concentration was previously established not to induce cortical spreading depression, which also causes CBF increases. 13 , 14 Acetylcholine (100 μM, Sigma) or adenosine (400 μM; Sigma) was topically superfused for 3 to 5 minutes to test endothelium‐dependent or smooth muscle vasoactivity, respectively, and the evoked CBF increases recorded. 13 , 14
2.5. Immunohistochemistry
Mice were anesthetized with sodium pentobarbital (120 kg/kg, ip) and perfused transcardially with phosphate‐buffered saline (PBS) followed by 4% paraformaldehyde (PFA) in PBS. Brains were removed, postfixed overnight, and sectioned coronally in a vibratome (section thickness: 40 μm). Free‐floating sections were permeabilized with 0.5% Triton X‐100 and non‐specific binding was blocked with 1% of normal donkey serum. Sections were randomly selected and incubated with the primary antibodies Iba‐1 (rabbit polyclonal, 1:500, Catalog # 019‐19741, Wako Chemicals) or glial fibrillary acidic protein (GFAP; mouse monoclonal, 1:1000, Catalog # G3893, Sigma) overnight at 4°C. After washing, sections were incubated with a fluorescein isothiocyanate–conjugated secondary antibody (1:200; Jackson ImmunoResearch Laboratories), mounted on slides and imaged with a confocal microscope (Leica SP8). In some experiments, sections were stained with thioflavin‐S (0.5%) to assess amyloid plaques and CAA with the blood vessels marker CD31 (see Section 2.6.2). The specificity of the immunofluorescence was verified by the omission of the primary and/or secondary antibody. All quantifications were performed by investigators blinded to the treatment on randomly selected fields within the neocortex.
2.6. CAA and amyloid plaque
2.6.1. In vivo CAA imaging
Pial vessel CAA was imaged using 2‐photon microscopy. Optical access to the brain was achieved through a polished and reinforced thinned skull preparation sealed with cyanoacrylate glue and a cover glass. 43 , 44 , 47 Ten weeks after tPA instillation as above, mice were equipped with a cranial window and allowed 2 weeks to recover from the surgery. To label Aβ deposits, methoxy‐X04 (Tocris, dissolved in dimethyl sulfoxide at 100 mM) was intraperitoneally injected 1 day before imaging at a dose of 1 mg/100 g. 43 To fluorescently label the blood vessel, Texas Red dextran (40 μL, 2.5%, molecular weight [MW] = 70,000 kDa, Thermo Fisher Scientific) in saline was injected retro‐orbitally immediately before imaging. Imaging was performed on a commercial 2‐photon microscope (FVMPE; Olympus) with XLPlan N 25 Å ∼ 1.05 NA objective. Excitation pulses came from a solid‐state laser (InSight DS+; Spectraphysics) set at a wavelength of 830 nm. Image stacks were acquired through Fluoview software. During imaging, anesthesia was maintained with ≈ 1.5% isoflurane in an oxygen/nitrogen mix (21% oxygen), with slight adjustments made to the isoflurane to maintain the respiratory rate at ≈ 1 Hz. Animals were kept at 37°C with a feedback‐controlled heating pad. Two photon images are an average projection of three‐dimensional stacks using ImageJ (NIH).
2.6.2. CAA and amyloid plaque burden using immunohistochemistry
To quantify CAA and amyloid plaque burden, 14 , 41 , 43 coronal sections were stained with the blood vessels marker CD31 (goat polyclonal, 1:150, Catalog # AF3628, R&D Systems) followed by thioflavin‐S (0.5%), mounted on slides, and imaged with a confocal microscope (Leica SP8). CAA and plaque burden were quantified using ImageJ (NIH, v1.54 h) and expressed as a percentage (%) of the total area of the section.
2.7. Microhemorrhage detection
Microhemorrhages were assessed by performing Perl's Prussian blue staining and hepcidin immunolabeling. For Perl's Prussian blue staining, after 12 weeks of intranasal tPA treatment mice were anesthetized with intraperitoneal pentobarbital (200 mg/kg) and perfused transcardially with PBS followed by 4% PFA. The brains were removed and immersed first in 4% PFA overnight, and then the brains were embedded in paraffin and sectioned coronally in 6 μm using a microtome. Coronal sections were collected from three different planes of the brain (approximately 2.5 mm apart) and mounted on superfrost plus slides. The slides were incubated in a Prussian blue solution containing freshly prepared 5% potassium hexacyanoferrate trihydrate and 5% hydrochloric acid (Sigma). Sections were counterstained in 0.1% Nuclear fast red. Microhemorrhages were quantified in the somatosensory cortices using 20× images, and ImageJ (NIH) software was used for analysis. The count of Prussian blue‐positive deposits, indicative of hemorrhages, was averaged per mouse, with each mouse being treated as an individual data point. For hepcidin immunolabeling, coronal sections were co‐stained with hepcidin (rabbit polyclonal, 1:200, Catalog# ab30760, abcam) and the endothelial marker CD31 (goat polyclonal, 1:150, Catalog # AF3628, R&D Systems) followed by thioflavin‐S (0.5%), mounted on slides, and imaged with a confocal microscope (Leica SP8).
2.8. Nesting test
In the evening mice were placed in individual cages with preweighed nestlets (3 g/cage) and the next morning the remaining nestlets not assembled into a nest were weighed. 43 , 48 The nests were scored on a 5‐point rating scale based on the remaining nestlet ratio and shredded conditions: (1) nestlet not noticeably torn (>90% nestlet untorn), (2) nestlet partially torn (50%–90% nestlet untorn), (3) nestlet mostly shredded but no recognizable nest built (<50% nestlet untorn), (4) nest built recognizable but flat (<10% nestlet untorn), (5) nest near perfectly built like a crater (<10% nestlet untorn) with walls higher than mouse body height for more than 50% of its circumference. Shredded nestlets were expressed as %.
FIGURE 2.
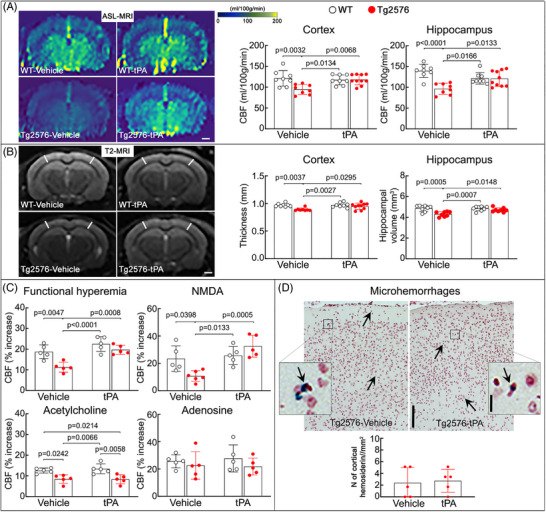
Intranasal tPA administration prevents neurovascular dysfunction and the associated brain atrophy in Tg2576 mice. A, Compared to vehicle‐treated WT littermates, vehicle‐treated Tg2576 mice (age 14–15 months) exhibit reduced neocortical and hippocampal CBF (ASL‐MRI; representative images on the left and quantification on the right), which is completely restored in intranasal tPA‐treated Tg2576 mice. N = 8–10 mice/group; two‐way ANOVA with Tukey test; data presented as mean ± SEM; scale bar, 1 mm. B, Neocortical thickness and hippocampal volume (T2‐MRI) at the level of the somatosensory cortex (−1.22 to −1.70 mm from bregma), are reduced in vehicle‐treated Tg2576 mice, but not in tPA‐treated Tg2576 mice. N = 8–10 mice/group; scale bar, 1 mm. C, In vehicle‐treated Tg2576 mice, CBF responses to whisker stimulation (functional hyperemia) and to the neocortical application of NMDA (40 μM) or acetylcholine (100 μM) are attenuated. In contrast, in intranasal tPA‐treated Tg2576 mice, functional hyperemia and CBF response to NMDA are rescued, but, as anticipated, 13 , 14 the CBF response to acetylcholine is still attenuated. The CBF response to adenosine (400 μM) is not affected. N = 5/group; two‐way ANOVA with Tukey test; data presented as mean ± SEM. D, Intranasal tPA does not affect microhemorrhages assessed by Prussian blue staining in Tg2576 mice. N = 5/group; data presented as mean ± SEM; scale bars, 200 and 10 μm. ANOVA, analysis of variance; ASL, arterial spin labeling; CBF, cerebral blood flow; MRI, magnetic resonance imaging; SEM, standard error of the mean; tPA, tissue plasminogen activator; WT, wild type.
2.9. Statistics
Sample sizes were determined by power analysis using G*Power (v.3.1.9.2) based on previous works published by our lab on CBF regulation cognitive testing. 14 , 43 , 44 The experiments were randomized based on the random number generator (https:// www. random.com) and were performed and analyzed in a blinded fashion whenever possible. Data and image analyses were done using ImageJ 1.54f (NIH) or Prism 10 for MacOS (GraphPad Software). Data were tested for normal distribution by the D'Agostino–Person test and for outliers by the Grubbs test (extreme studentized deviate). Two‐group comparisons were analyzed using an unpaired two‐tailed t test, as indicated. Multiple comparisons were evaluated by one‐way or two‐way analysis of variance (ANOVA) and Tukey test. Differences were considered statistically significant for probability values less than 0.05. Data are expressed as the mean ± standard error of the mean.
3. RESULTS
3.1. Intranasal administration of tPA rescues resting CBF, brain atrophy, and neurovascular dysfunction in 14‐ to 15‐month‐old Tg2576 mice
To investigate whether tPA supplement rescues the neurovascular dysfunction induced by amyloid accumulation, we administered tPA (0.8 mg/kg/day) intranasally to Tg2576 mice. 34 , 36 , 37 , 38 , 39 tPA was instilled intranasally at an age (11–12 months) when Aβ pathology is accumulating and examined neurovascular and cognitive function once after 3 months of treatment 14 , 41 , 42 , 43 (Figure 1). We first assessed resting CBF (mL/100 g/min; ASL‐MRI) and found that in vehicle (dH2O)‐treated Tg2576 mice, CBF was attenuated in the neocortex and hippocampus compared to vehicle‐treated WT littermates (Figure 2A). In addition, in vehicle‐treated Tg2576 mice, the neocortex was thinner and hippocampal volume reduced compared to WT mice, assessed by T2‐MRI (Figure 2B). tPA administration prevented the attenuation in the neocortical and hippocampal CBF and the reduction in neocortical thickness and hippocampal volume (Figure 2A,B). Intranasal tPA did not affect these parameters in WT mice (Figure 2A,B), ruling out that the rescues in resting CBF, cortical thickness, and hippocampal volume resulted from non‐specific effects.
Then, we examined the effect of intranasal tPA on neurovascular regulation in anesthetized mice equipped with a cranial window. In vehicle‐treated Tg2576 mice, the increase in somatosensory cortex CBF evoked by whisker stimulation or by neocortical application of NMDA and the CBF response to acetylcholine were attenuated compared to vehicle‐treated WT littermates, while CBF response to adenosine was not affected (Figure 2C). Intranasal tPA administration prevented the attenuation in CBF induced by whisker stimulation or by neocortical superfusion of NMDA, but as anticipated, 13 , 14 the response to acetylcholine was not rescued (Figure 2C). Tg2576 mice exhibited rare microhemorrhages in neocortex, assessed by Prussian blue staining or immunolabeling for hepcidin, a regulator of iron levels, 49 which were not affected by intranasal tPA (Figure 2D; Figure S1 in supporting information). Thus, intranasal tPA prevents the attenuations in resting CBF, brain atrophy, and NMDA receptor–dependent functional hyperemia in aged Tg2576 mice.
3.2. Intranasal tPA improves nesting behavior and reduces CAA burden but not amyloid plaques in aged Tg2576 mice
Next, we investigated whether intranasal tPA administration improves cognitive impairment and Aβ pathology in aged Tg2576 mice. First, we examined the impact of intranasal tPA on nesting behavior, a sensitive indicator for cognitive alteration in Tg2576 mice at this age. 43 In vehicle‐treated Tg2576 mice, nesting behavior, assessed by the nesting score and shredded nestlet, was impaired compared to vehicle‐treated WT mice (Figure 3). Intranasal tPA administration did not affect nesting behavior in WT mice, but it markedly improved the nest building in Tg2576 mice (Figure 3). Considering the link between CAA burden and cognitive function, 14 , 42 , 43 , 50 we further investigated whether the improvement in the nest building behavior is associated with a reduction in CAA. As anticipated, 14 tPA administration attenuated CAA burden, but did not affect amyloid plaques (Figure 4A,B). Notably, intranasal tPA did not influence GFAP+ astrocytes and Iba1+ myeloid cells (Figure 5). In conclusion, our findings suggest that intranasal tPA improves nesting behavior and reduces CAA without affecting amyloid plaques in aged Tg2576 mice.
FIGURE 3.
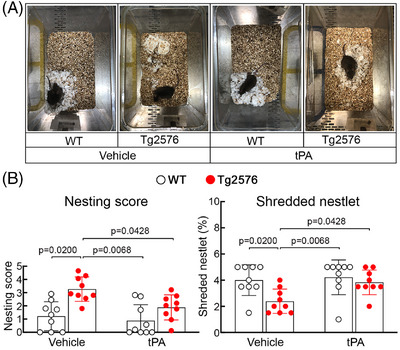
Intranasal tPA administration prevents cognitive dysfunction in aged Tg2576 mice. Nest building abilities (A, representative nest building images; B, quantification) are improved in intranasal tPA‐treated Tg2576 mice, compared to vehicle‐treated Tg2576 mice, as shown by a significantly higher nesting score and % shredded nestlets. N = 9/group; two‐way analysis of variance and Tukey test. Data presented as mean ± standard error of the mean. tPA, tissue plasminogen activator; WT, wild type.
FIGURE 4.
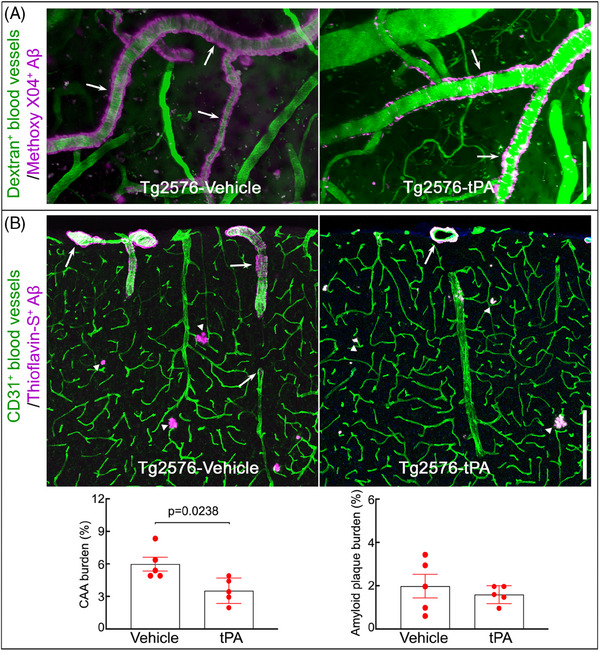
Intranasal tPA administration attenuates pial and neocortical CAA in aged Tg2576 mice. A, In vivo two‐photon excited fluorescent images illustrating methoxy‐XO4+ amyloid deposits (magenta) around somatosensory cortex blood vessels (green) identified by retroorbital injection of 70 kDa Texas Red dextran. Methoxy‐XO4+ Aβ deposits are reduced in intranasal tPA‐treated Tg2576 compared to vehicle‐treated Tg2576 mice. B, CAA burden (% thioflavin‐S+ area) in cortical sections labeled with thioflavin‐S and the blood vessel marker CD31 is reduced in intranasal tPA‐treated Tg2576 compared to vehicle‐treated Tg2576 mice, but amyloid plaque burden (% thioflavin‐S+ plaque area) was comparable between the two groups. In (A) and (B), arrows indicate CAA and arrowheads plaques. N = 5/group; unpaired t test; scale bars, 100 μm in (A) and 200 μm in (B); data presented as mean ± standard error of the mean. Aβ, amyloid beta; CAA, cerebral amyloid angiopathy; tPA, tissue plasminogen activator; WT, wild type.
FIGURE 5.
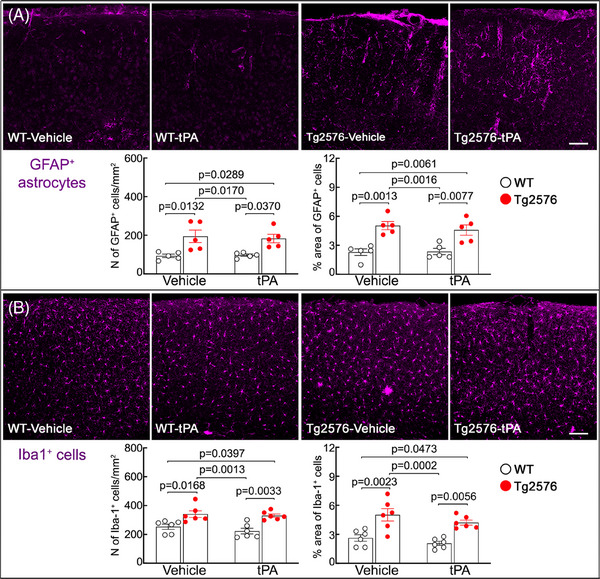
Intranasal tPA administration does not affect astrocytes and microglia/macrophages in the neocortex of the aged Tg2576 mice. The number and % areas of GFAP+ astrocytes (A) and Iba1+ microglia/macrophages (B) are higher in vehicle‐treated Tg2576 mice compared to vehicle‐treated WT littermates, which is not affected by intranasal tPA administration. N = 5/group; two‐way analysis of variance with Tukey's test; scale bar in (A) and (B), 100 μm; data presented as mean ± standard error of the mean. GFAP, glial fibrillary acidic protein; tPA, tissue plasminogen activator; WT, wild type.
4. DISCUSSION
The present observations demonstrated that tPA supplement counteracts neurovascular and cognitive dysfunction induced by Aβ accumulation in 14‐ to 15‐month‐old Tg2576 mice. Intranasal tPA not only prevents the CBF attenuation in the neocortex and hippocampus, but also improves the attenuation in CBF produced by neural activity and by neocortical superfusion of NMDA. These beneficial effects are associated with reduced atrophy, improved nesting behavior, and reduced CAA but not amyloid plaques. The findings suggest that tPA administration counteracts deleterious neurovascular and cognitive effects associated with Aβ pathology.
Functional hyperemia, the localized CBF increase evoked by neural activity, relies on various vasoactive factors that act on different parts of the brain vasculature. 5 , 9 , 51 tPA plays a key role in postsynaptic events coupling NMDA receptor activation to the nNOS‐NO–dependent component of the resulting hyperemic response. 13 , 14 , 20 tPA is released from active neurons 23 , 24 , 52 , 53 , 54 and is required for the full expression of the CBF increase triggered by neural activity. 13 , 14 , 19 These neurovascular effects are mediated by promoting the phosphorylation of nNOS and resulting in NO production during NMDA receptor activation. 13 We have previously demonstrated that tPA deficiency is implicated in the neurovascular and cognitive dysfunction induced by Aβ. 14 Aβ upregulates the tPA inhibitor PAI‐1 and causes tPA deficiency. 14 , 29 , 30 , 31 Thus, we found that suppressing such PAI‐1 upregulation rescues tPA activity and prevents Aβ from impairing neurovascular and cognitive function in aged Tg2576 mice. 14 In the current study, we sought to link the improvement produced by PAI‐1 inhibition directly to tPA. To this end, we instilled recombinant tPA intranasally for 12 weeks in aged Tg2576 mice. We found that intranasal tPA prevents cortical and hippocampal atrophy, resting CBF decline, and attenuation in functional hyperemia and cognitive function in Tg2676 mice. However, cognitive function was assessed only by nest building, and verification with other cognitive tests would be desirable.
The mechanisms by which tPA counteracts the deleterious consequences of amyloid pathology remain to be fully elucidated. In presymptomatic AD, resting CBF is attenuated and hemodynamic responses to neural activation are decreased, 55 , 56 , 57 , 58 , 59 indicating early contribution to disease progression. Therefore, one possibility is that tPA, by ameliorating functional hyperemia, provides more nutritional flow to the brain of Tg2576 mice, resulting in improved cerebral perfusion and attenuation of cerebral atrophy and cognitive dysfunction.
Another possibility is that the beneficial effects of tPA are related to the reduction in CAA. The efficient clearance of Aβ from the brain requires intact neurovascular function. 6 , 9 , 60 Therefore, it is reasonable to assume that tPA by improving functional hyperemia in Tg2576 mice may enhance Aβ clearance from the brain through transvascular, paravascular (glymphatic), and perivascular pathways. 60 , 61 , 62 In support of this hypothesis, we recently found that rescuing neurovascular function in Tg2676 mice by deletion of the Aβ binding scavenging receptor CD36 is able to improve transvascular and paravascular clearance of Aβ from the brain. 43 In this regard, it is of interest that tPA reduced CAA but not amyloid plaques. The mechanisms of the selectivity of the amyloid clearing effects remains to be elucidated. Furthermore, the beneficial impact of additional protective actions of tPA in reducing APP processing, promoting Aβ enzymatic degradation, enhancing synaptic plasticity, and counteracting the plasma contact system cannot be ignored, 21 , 23 , 30 , 63 , 64 , 65 , 66 and their role needs to be addressed in future studies. Finally, sexual dimorphism, blood–brain barrier permeability, astrogliosis, and microglia in the beneficial effects of intranasal tPA warrant further investigation.
Our observation that tPA supplementation reduced vascular amyloid accumulation may have implications for counteracting the pathological effects of CAA, but with a critical caveat. CAA is a major cause of cortical hemorrhages in the elderly 2 , 50 and increases the risk of the amyloid‐related imaging abnormality (ARIA), a treatment‐limiting complication of Aβ immunotherapy. ARIA is observed with MRI in cortical areas in up to 47% of patients receiving the highest, most effective, dose and in some patients induces symptoms that prompt stopping therapy. 2 , 67 , 68 , 69 ARIA has been attributed to the overload of brain blood vessels by Aβ released from the breakdown of amyloid plaque resulting in vascular dysfunction and damage. 3 , 67 , 68 , 69 , 70 In this context, because rescuing tPA, either by PAI‐1 inhibition 14 or supplementation (present study), may improve vascular health and CAA, tPA treatment may be beneficial in protecting the brain from the deleterious effects of this condition. The important caveat is that tPA is an anticoagulant and may also reduce vascular stability by activating metalloproteases. 71 , 72 A recent case of a patient who had received Aβ immunotherapy and died of a massive hemorrhage after receiving tPA for ischemic stroke treatment 73 highlights the complexity of using this approach. Therefore, it would be imprudent to consider using tPA in cases of CAA or as an adjuvant to Aβ immunotherapy. Fortunately, tPA variants that do not interfere with the clotting system have been developed 38 , 74 , 75 and, after additional animal testing to verify their efficacy, could be contemplated as a possible therapeutic approach for CAA and related complications.
5. CONCLUSION
We found that tPA supplementation prevents the attenuation of CBF in the neocortex and hippocampus and rescues functional hyperemia. These vascular effects are associated with reducing brain atrophy, CAA burden, and cognitive dysfunction in Tg2576 mice. Taken together, these tPA rescue experiments provide more direct evidence for the pathogenic role of tPA deficiency in the detrimental neurovascular and cognitive effects of Aβ and strengthen the case for new therapeutic approaches based on counteracting tPA deficiency in conditions associated with amyloid pathology.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest. Author disclosures are available in the supporting information.
CONSENT STATEMENT
Consent was not necessary: no human subjects were included in the study.
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
Support from the Feil Family Foundation is gratefully acknowledged. Support also came from NIH grants R01‐NS097805 (LP), R01‐NS37853 (CI), Leon Levy Foundation (SJA), and the BrightFocus Foundation (AA, SJA).
Uekawa K, Anfray A, Ahn SJ, et al. tPA supplementation preserves neurovascular and cognitive function in Tg2576 mice. Alzheimer's Dement. 2024;20:4572–4582. 10.1002/alz.13878
Ken Uekawa and Antoine Anfray contributed equally to this study.
REFERENCES
- 1. Cummings J, Zhou Y, Lee G, Zhong K, Fonseca J, Cheng F. Alzheimer's disease drug development pipeline: 2023. Alzheimers Dement. 2023;9:e12385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jucker M, Walker LC. Alzheimer's disease: from immunotherapy to immunoprevention. Cell. 2023;186:4260‐4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Self WK, Holtzman DM. Emerging diagnostics and therapeutics for Alzheimer disease. Nat Med. 2023;29:2187‐2199. [DOI] [PubMed] [Google Scholar]
- 4. Apatiga‐Perez R, Soto‐Rojas LO, Campa‐Cordoba BB, et al. Neurovascular dysfunction and vascular amyloid accumulation as early events in Alzheimer's disease. Metab Brain Dis. 2022;37:39‐50. [DOI] [PubMed] [Google Scholar]
- 5. Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80:844‐866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Koizumi K, Wang G, Park L. Endothelial dysfunction and amyloid‐beta‐induced neurovascular alterations. Cell Mol Neurobiol. 2016;36:155‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scheltens P, De Strooper B, Kivipelto M, et al. Alzheimer's disease. Lancet. 2021;397:1577‐1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Iturria‐Medina Y, Sotero RC, Toussaint PJ, Mateos‐Perez JM, Evans AC, Alzheimer's Disease Neuroimaging I . Early role of vascular dysregulation on late‐onset Alzheimer's disease based on multifactorial data‐driven analysis. Nat Commun. 2016;7:11934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schaeffer S, Iadecola C. Revisiting the neurovascular unit. Nat Neurosci. 2021;24:1198‐1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Iadecola C, Zhang F, Niwa K, et al. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat Neurosci. 1999;2:157‐161. [DOI] [PubMed] [Google Scholar]
- 11. Niwa K, Younkin L, Ebeling C, et al. Abeta 1‐40‐related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc Natl Acad Sci USA. 2000;97:9735‐9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nortley R, Korte N, Izquierdo P, et al. Amyloid beta oligomers constrict human capillaries in Alzheimer's disease via signaling to pericytes. Science. 2019;365:eaav9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Park L, Gallo EF, Anrather J, et al. Key role of tissue plasminogen activator in neurovascular coupling. Proc Natl Acad Sci USA. 2008;105:1073‐1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Park L, Zhou J, Koizumi K, et al. tPA deficiency underlies neurovascular coupling dysfunction by amyloid‐beta. J Neurosci. 2020;40:8160‐8173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tong XK, Lecrux C, Rosa‐Neto P, Hamel E. Age‐dependent rescue by simvastatin of Alzheimer's disease cerebrovascular and memory deficits. J Neurosci. 2012;32:4705‐4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gonias SL. Plasminogen activator receptor assemblies in cell signaling, innate immunity, and inflammation. Am J Physiol Cell Physiol. 2021;321:C721‐C734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Henderson SJ, Weitz JI, Kim PY. Fibrinolysis: strategies to enhance the treatment of acute ischemic stroke. J Thromb Haemost. 2018;16:1932‐1940. [DOI] [PubMed] [Google Scholar]
- 18. Thiebaut AM, Gauberti M, Ali C, et al. The role of plasminogen activators in stroke treatment: fibrinolysis and beyond. Lancet Neurol. 2018;17:1121‐1132. [DOI] [PubMed] [Google Scholar]
- 19. Anfray A, Drieu A, Hingot V, et al. Circulating tPA contributes to neurovascular coupling by a mechanism involving the endothelial NMDA receptors. J Cereb Blood Flow Metab. 2020;40(10):2038‐2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232‐243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baranes D, Lederfein D, Huang YY, Chen M, Bailey CH, Kandel ER. Tissue plasminogen activator contributes to the late phase of LTP and to synaptic growth in the hippocampal mossy fiber pathway. Neuron. 1998;21:813‐825. [DOI] [PubMed] [Google Scholar]
- 22. Diaz A, Jeanneret V, Merino P, McCann P, Yepes M. Tissue‐type plasminogen activator regulates p35‐mediated Cdk5 activation in the postsynaptic terminal. J Cell Sci. 2019;132:jcs224196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Qian Z, Gilbert ME, Colicos MA, Kandel ER, Kuhl D. Tissue‐plasminogen activator is induced as an immediate‐early gene during seizure, kindling and long‐term potentiation. Nature. 1993;361:453‐457. [DOI] [PubMed] [Google Scholar]
- 24. Samson AL, Medcalf RL. Tissue‐type plasminogen activator: a multifaceted modulator of neurotransmission and synaptic plasticity. Neuron. 2006;50:673‐678. [DOI] [PubMed] [Google Scholar]
- 25. Echagarruga CT, Gheres KW, Norwood JN. Drew PJ. nNOS‐expressing interneurons control basal and behaviorally evoked arterial dilation in somatosensory cortex of mice. eLife. 2020;9:e60533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. O'Gallagher K, Rosentreter RE, Elaine Soriano J, et al. The effect of a neuronal nitric oxide synthase inhibitor on neurovascular regulation in humans. Circ Res. 2022;131:952‐961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hosford PS, Gourine AV. What is the key mediator of the neurovascular coupling response? Neurosci Biobehav Rev. 2019;96:174‐181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cholet N, Seylaz J, Lacombe P, Bonvento G. Local uncoupling of the cerebrovascular and metabolic responses to somatosensory stimulation after neuronal nitric oxide synthase inhibition. J Cereb Blood Flow Metab. 1997;17:1191‐1201. [DOI] [PubMed] [Google Scholar]
- 29. Cacquevel M, Launay S, Castel H, et al. Ageing and amyloid‐beta peptide deposition contribute to an impaired brain tissue plasminogen activator activity by different mechanisms. Neurobiol Dis. 2007;27:164‐173. [DOI] [PubMed] [Google Scholar]
- 30. Jacobsen JS, Comery TA, Martone RL, Elokdah H, Crandall DL, Oganesian A, et al. Enhanced clearance of Abeta in brain by sustaining the plasmin proteolysis cascade. Proc Natl Acad Sci USA. 2008;105:8754‐8759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Melchor JP, Pawlak R, Strickland S. The tissue plasminogen activator‐plasminogen proteolytic cascade accelerates amyloid‐beta (Abeta) degradation and inhibits Abeta‐induced neurodegeneration. J Neurosci. 2003;23:8867‐8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dotti CG, Galvan C, Ledesma MD. Plasmin deficiency in Alzheimer's disease brains: causal or casual? Neurodegener Dis. 2004;1:205‐212. [DOI] [PubMed] [Google Scholar]
- 33. Sutton R, Keohane ME, VanderBerg SR, Gonias SL. Plasminogen activator inhibitor‐1 in the cerebrospinal fluid as an index of neurological disease. Blood Coagul Fibrinolysis. 1994;5:167‐171. [DOI] [PubMed] [Google Scholar]
- 34. Chen N, Chopp M, Xiong Y, et al. Subacute intranasal administration of tissue plasminogen activator improves stroke recovery by inducing axonal remodeling in mice. Exp Neurol. 2018;304:82‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dong Y, Hong W, Tang Z, Gao Y, Wu X, Liu H. Sevoflurane leads to learning and memory dysfunction via breaking the balance of tPA/PAI‐1. Neurochem Int. 2020;139:104789. [DOI] [PubMed] [Google Scholar]
- 36. Fonseca LC, Lopes JA, Vieira J, et al. Intranasal drug delivery for treatment of Alzheimer's disease. Drug Deliv Transl Res. 2021;11:411‐425. [DOI] [PubMed] [Google Scholar]
- 37. Liu Z, Li Y, Zhang L, et al. Subacute intranasal administration of tissue plasminogen activator increases functional recovery and axonal remodeling after stroke in rats. Neurobiol Dis. 2012;45:804‐809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pu H, Shi Y, Zhang L, Lu Z, Ye Q, Leak RK, et al. Protease‐independent action of tissue plasminogen activator in brain plasticity and neurological recovery after ischemic stroke. Proc Natl Acad Sci USA. 2019;116:9115‐9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xia Y, Pu H, Leak RK, et al. Tissue plasminogen activator promotes white matter integrity and functional recovery in a murine model of traumatic brain injury. Proc Natl Acad Sci USA. 2018;115:E9230‐E9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hsiao K, Chapman P, Nilsen S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99‐102. [DOI] [PubMed] [Google Scholar]
- 41. Park L, Zhou J, Zhou P, et al. Innate immunity receptor CD36 promotes cerebral amyloid angiopathy. Proc Natl Acad Sci USA. 2013;110:3089‐3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Park L, Zhou P, Pitstick R, et al. Nox2‐derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci USA. 2008;105:1347‐1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Uekawa K, Hattori Y, Ahn SJ, et al. Border‐associated macrophages promote cerebral amyloid angiopathy and cognitive impairment through vascular oxidative stress. Mol Neurodegener. 2023;18:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Park L, Hochrainer K, Hattori Y, et al. Tau induces PSD95‐neuronal NOS uncoupling and neurovascular dysfunction independent of neurodegeneration. Nat Neurosci. 2020;23:1079‐1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. Elsevier Academic Press; 2004. [Google Scholar]
- 46. Iadecola C. Nitric oxide participates in the cerebrovasodilation elicited from cerebellar fastigial nucleus. Am J Physiol. 1992;263:R1156‐R1161. [DOI] [PubMed] [Google Scholar]
- 47. Santisteban MM, Ahn SJ, Lane D, et al. Endothelium‐macrophage crosstalk mediates blood‐brain barrier dysfunction in hypertension. Hypertension. 2020;76:795‐807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Deacon RM. Assessing nest building in mice. Nat Protoc. 2006;1:1117‐1119. [DOI] [PubMed] [Google Scholar]
- 49. Nemeth E, Ganz T. Hepcidin and iron in health and disease. Annu Rev Med. 2023;74:261‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Greenberg SM, Bacskai BJ, Hernandez‐Guillamon M, Pruzin J, Sperling R, van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease—one peptide, two pathways. Nat Rev Neurol. 2020;16:30‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Iadecola C. The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron. 2017;96:17‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wu F, Torre E, Cuellar‐Giraldo D, et al. Tissue‐type plasminogen activator triggers the synaptic vesicle cycle in cerebral cortical neurons. J Cereb Blood Flow Metab. 2015;35:1966‐1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yepes M, Wu F, Torre E, Cuellar‐Giraldo D, Jia D, Cheng L. Tissue‐type plasminogen activator induces synaptic vesicle endocytosis in cerebral cortical neurons. Neuroscience. 2016;319:69‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lochner JE, Honigman LS, Grant WF, et al. Activity‐dependent release of tissue plasminogen activator from the dendritic spines of hippocampal neurons revealed by live‐cell imaging. J Neurobiol. 2006;66:564‐577. [DOI] [PubMed] [Google Scholar]
- 55. Janik R, Thomason LA, Chaudhary S, et al. Attenuation of functional hyperemia to visual stimulation in mild Alzheimer's disease and its sensitivity to cholinesterase inhibition. Biochim Biophys Acta. 2016;1862:957‐965. [DOI] [PubMed] [Google Scholar]
- 56. McDade E, Kim A, James J, et al. Cerebral perfusion alterations and cerebral amyloid in autosomal dominant Alzheimer disease. Neurology. 2014;83:710‐717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ruitenberg A, den Heijer T, Bakker SL, et al. Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam Study. Ann Neurol. 2005;57:789‐794. [DOI] [PubMed] [Google Scholar]
- 58. Swinford CG, Risacher SL, Wu YC, et al. Altered cerebral blood flow in older adults with Alzheimer's disease: a systematic review. Brain Imaging Behav. 2023;17:223‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tomoto T, Tarumi T, Chen J, Pasha EP, Cullum CM, Zhang R. Cerebral vasomotor reactivity in amnestic mild cognitive impairment. J Alzheimers Dis. 2020;77:191‐202. [DOI] [PubMed] [Google Scholar]
- 60. Tarasoff‐Conway JM, Carare RO, Osorio RS, et al. Clearance systems in the brain‐implications for Alzheimer disease. Nat Rev Neurol. 2015;11:457‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Holstein‐Ronsbo S, Gan Y, Giannetto MJ, et al. Glymphatic influx and clearance are accelerated by neurovascular coupling. Nat Neurosci. 2023;26:1042‐1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Weller RO, Sharp MM, Christodoulides M, Carare RO, Mollgard K. The meninges as barriers and facilitators for the movement of fluid, cells and pathogens related to the rodent and human CNS. Acta Neuropathol. 2018;135:363‐385. [DOI] [PubMed] [Google Scholar]
- 63. Badimon A, Torrente D, Norris EH. Vascular dysfunction in Alzheimer's disease: alterations in the plasma contact and fibrinolytic systems. Int J Mol Sci. 2023;24:7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kingston IB, Castro MJ, Anderson S. In vitro stimulation of tissue‐type plasminogen activator by Alzheimer amyloid beta‐peptide analogues. Nat Med. 1995;1:138‐142. [DOI] [PubMed] [Google Scholar]
- 65. Rodriguez G, Eren M, Haupfear I, et al. Pharmacological inhibition of plasminogen activator inhibitor‐1 prevents memory deficits and reduces neuropathology in APP/PS1 mice. Psychopharmacology. 2023;240:2641‐2655. [DOI] [PubMed] [Google Scholar]
- 66. Tucker HM, Kihiko M, Caldwell JN, et al. The plasmin system is induced by and degrades amyloid‐beta aggregates. J Neurosci. 2000;20:3937‐3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Filippi M, Cecchetti G, Spinelli EG, Vezzulli P, Falini A, Agosta F. Amyloid‐Related imaging abnormalities and beta‐amyloid‐targeting antibodies: a systematic review. JAMA Neurol. 2022;79:291‐304. [DOI] [PubMed] [Google Scholar]
- 68. Hampel H, Elhage A, Cho M, Apostolova LG, Nicoll JAR, Atri A. Amyloid‐related imaging abnormalities (ARIA): radiological, biological and clinical characteristics. Brain. 2023;146:4414‐4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early Alzheimer's disease. N Engl J Med. 2023;388:9‐21. [DOI] [PubMed] [Google Scholar]
- 70. Sims JR, Zimmer JA, Evans CD, et al. Donanemab in early symptomatic Alzheimer disease: the TRAILBLAZER‐ALZ 2 randomized clinical trial. JAMA. 2023;330:512‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Adibhatla RM, Hatcher JF. Tissue plasminogen activator (tPA) and matrix metalloproteinases in the pathogenesis of stroke: therapeutic strategies. CNS Neurol Disord Drug Targets. 2008;7:243‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lapchak PA, Chapman DF, Zivin JA. Metalloproteinase inhibition reduces thrombolytic (tissue plasminogen activator)‐induced hemorrhage after thromboembolic stroke. Stroke. 2000;31:3034‐3040. [DOI] [PubMed] [Google Scholar]
- 73. Reish NJ, Jamshidi P, Stamm B, et al. Multiple cerebral hemorrhages in a patient receiving lecanemab and treated with t‐PA for stroke. N Engl J Med. 2023;388:478‐479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Olson ST, Swanson R, Day D, Verhamme I, Kvassman J, Shore JD. Resolution of Michaelis complex, acylation, and conformational change steps in the reactions of the serpin, plasminogen activator inhibitor‐1, with tissue plasminogen activator and trypsin. Biochemistry. 2001;40:11742‐11756. [DOI] [PubMed] [Google Scholar]
- 75. Yi JS, Kim YH, Koh JY. Infarct reduction in rats following intraventricular administration of either tissue plasminogen activator (tPA) or its non‐protease mutant S478A‐tPA. Exp Neurol. 2004;189:354‐360. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information