

Abstract
Purpose:
Treatment paradigms for Isocitrate dehydrogenase (IDH) mutant gliomas are rapidly evolving. While typically indolent and responsive to initial treatment, these tumors invariably recur at higher grade and require salvage treatment. Homozygous deletion of the tumor suppressor gene CDKN2A/B frequently emerges at recurrence in these tumors, driving poor patient outcome. We investigated the effect of CDK-Rb pathway blockade on IDH-mutant glioma growth in vitro and in vivo using CDK4/6 inhibitors (CDKi).
Experimental Design:
Cell viability, proliferation assays and flow cytometry were used to examine the pharmacologic effect of two distinct CDKis, palbociclib and abemaciclib, in multiple patient-derived IDH-mutant glioma lines. Isogenic models were used to directly investigate the influence of CDKN2A/B status on CDKi sensitivity. Orthotopic xenograft tumor models were used to examine efficacy and tolerability of CDKi in vivo.
Results:
CDKi treatment leads to decreased cell viability and proliferative capacity in patient-derived IDH-mutant glioma lines, coupled with enrichment of cells in G1 phase. CDKN2A inactivation sensitizes IDH-mutant glioma to CDKi in both endogenous and isogenic models with engineered CDKN2A deletion. CDK4/6 inhibitor administration improves survival in orthotopically implanted IDH-mutant glioma models.
Conclusions:
IDH-mutant gliomas with deletion of CDKN2A/B are sensitized to CDK4/6 inhibitors. These results support investigation of the use of these agents in a clinical setting.
Introduction
IDH-mutant gliomas are primary brain tumors characterized by a mutation in the metabolic genes Isocitrate dehydrogenase 1 or 2. There are two major subtypes of IDH-mutant glioma: astrocytoma, with concomitant mutations in TP53 and ATRX, and oligodendroglioma, which harbors TERT promoter mutations and loss of chromosomes 1p and 19q. Compared to IDH wild-type glioblastoma, IDH-mutant diffuse gliomas exhibit more-indolent growth patterns and are generally responsive to upfront treatment with radiation therapy and alkylating chemotherapy (1–6). Recently, inhibition of mutant IDH enzyme activity with the small molecule inhibitor vorasidenib demonstrated clinical effectiveness, with prolonged median progression-free survival observed in patients with indolent phase disease, when compared to placebo (7). Despite the initial responsiveness to therapy, however, these tumors inevitably recur. Progressive IDH-mutant tumors frequently regrow at a higher grade and acquire new genomic alterations that drive more aggressive behavior (8).
Deletion of the tumor suppressor gene CDKN2A/B is the most commonly observed acquired genomic alteration in IDH-mutant tumors at recurrence, detected in 20 – 60% of tumors across different case series (9–12). Loss events are enriched in astrocytoma lineage compared to oligodendroglioma, as well as in those that have been previously treated with radiation therapy (9,12). Homozygous loss of CDKN2A/B is also associated with dramatically worsened prognosis when detected at initial diagnosis, with a median survival of 3 – 5 years (compared to 7 – 12 years in CDKN2A/B wild-type tumors) (14–16). This stark prognostic association is reflected in the updated WHO diagnostic criteria for IDH-mutant tumors, in which homozygous CDKN2A/B loss is sufficient for an IDH-mutant astrocytoma to be designated grade 4 (17). In the recurrent setting, acquisition of CDKN2A/B loss is also associated with shorter survival times (12).
Mechanistically, the CDKN2A locus encodes the p16(INK4A) and p14(ARF) tumor suppressors. As a kinase inhibitor protein, p16 plays a central role in restraining Cyclin Dependent Kinase (CDK) 4/6 activity to maintain cells in the G1 phase of the cell cycle. In the absence of p16, elevated CDK4/6 activity manifests as hyperphosphorylation of the downstream target the retinoblastoma protein (Rb), which in turn promotes cell replication via advancement through the G1/S transition (18,19). As evidence of the stepwise progression that occurs in IDH-mutant glioma progression, recent studies have reported intermediate outcomes for patients with IDH-mutant astrocytoma with hemizygous deletion of CDKN2A/B (20,21).
The observation that genetic disruption of the CDK-Rb pathway occurs in many cancer types led to the development of the specific CDK4/6 inhibitors (CDK4/6i) abemaciclib, ribociclib and palbociclib. Many tumor types have been reported to exhibit sensitivity to CDK4/6i in vitro but no predictive biomarkers for CDK4/6i responsiveness have yet been established (22). Despite the central role of p16 in restraining CDK4/6 activity, a study that surveyed a panel of >500 cancer cell lines for CDK4/6i responsiveness did not find a strong relationship between CDKN2A loss and abemaciclib sensitivity (22). In the clinical setting, CDK4/6 inhibitors have demonstrated a well-appreciated clinical benefit for the treatment of hormone receptor-positive, HER2-negative metastatic breast cancer in combination with endocrine therapy, yet this effect also has not been found to be dependent upon loss of p16 (23,24). In IDH wild-type glioblastoma, which is etiologically distinct from IDH-mutant glioma, no benefit to CDK4/6i has been observed (25,26).
These data notwithstanding, the strong correlation between prognosis and CDKN2A/B status implies that dysregulation of the CDK-Rb pathway is biologically relevant in IDH-mutant glioma. We therefore hypothesized that inhibition of CDK-Rb activity using CDK4/6 inhibitors will slow growth of IDH-mutant gliomas. Here we show inhibition of CDKN2A/B deleted IDH-mutant glioma growth by CDK4/6i in multiple patient-derived models of this disease. These results illustrate a unique scenario in which CDKN2A/B-deletion serves as a predictor of CDK4/6i responsiveness.
Materials and Methods
Cell culturing, reagents, and viral transduction
The patient-derived glioma lines MGG152 and MGG119 have been previously described and were maintained following the published protocol (8). TS603 and BT142-Luciferase tagged cells were kind gifts from T. Chan at the Memorial Sloan-Kettering Cancer Center and G. Riggins at the Sidney Kimmel Comprehensive Cancer Center, respectively, and maintained using a similar protocol. Cell lines were authenticated by short tandem repeat fingerprinting upon initial receipt and every 12–18 months thereafter (last performed April 2023). Testing for mycoplasma was performed every 6 months using Mycoplasma PCR Detection Kit (ABM, G238). All cell lines were used for no more than 10 passages. MGG152 CDKN2A −/− single guide (sg) RNA 1 & 3 cell lines were developed using CRISPR-CAS9 gene editing. sgRNAs targeting CDKN2A were obtained from GenScript’s GenCRISPR Plasmid Collection containing sgRNAs pre-validated by the Broad Institute and inserted into pLentiCRISPRv2 (GenScript, RRID:SCR_002891) all-in-one plasmid containing CRISPR associated protein 9 (Cas9 (Similar to RRID:Addgene_127644 without Neo resistance gene). Lentiviral particles were then generated by co-transfection of HEK293T cells (RRID:CVCL_0063) with pLentiCRISPRv2 plasmid, a packaging plasmid psPAX2 (Addgene, RRID:Addgene_12260), and an envelope plasmid VSV.G (Addgene, RRID:Addgene_14888) in a ratio of 2:1:1 with FuGENE HD (Promega). These lentiviral particles were then transduced into MGG152 cells using previously published protocols(27–29) and knock out was confirmed by western blot.
CDKN2A sgRNA sequences
sgRNA1: GGCCTCCGACCGTAACTATT
sgRNA3: GACCCGTGCACGACGCTGCC
Compounds and Chemicals
The following compounds were added to cell culture media where indicated: abemaciclib (Selleckchem, S5716), palbociclib (Selleckchem, S1116), temozolomide (Cayman Chemical, 14163), lomustine (Selleckchem, S1840), AGI-5198 (MedChemExpress, HY-18082), and dimethyl sulfoxide (Sigma-Aldrich).
Cell viability and cell proliferation analysis
Cell viability was assessed by measuring luminescence following 120 hours of indicated compound treatment using the Cell Titer-Glo Assay Kit (Promega) according to the manufacturer’s protocol with a microplate reader (Molecular Devices SpectraMax iD3, RRID:SCR_023920). Cell proliferation was assessed by measuring fluorescence following 120 hours of indicated compound treatment using the CYQUANT Cell Proliferation Assay (Invitrogen) according to the manufacturer’s protocol with a microplate reader (Molecular Devices SpectraMax iD3, RRID:SCR_023920; BioTek Synergy HTX multi-mode reader, RRID:SCR_019749).
Western Blot Analysis
Cells were harvested and lysed in radioimmunoprecipitation assay buffer (Thermo Fisher Scientific) supplemented with protease inhibitor and phosphatase inhibitor cocktail (Roche). Membranes were probed with the following primary antibodies: Phospho-Rb (Ser807/811) (Cell Signaling Technology Cat# 8516, RRID:AB_11178658), Rb (Cell Signaling Technology Cat# 9313, RRID:AB_1904119), Cyclin B1 (Cell Signaling Technology Cat# 12231, RRID:AB_2783553), Cyclin A2 (Cell Signaling Technology Cat# 4656, RRID:AB_2071958), Cyclin E2 (Cell Signaling Technology Cat# 4132, RRID:AB_2071197), CDK2 (Cell Signaling Technology Cat# 2546, RRID:AB_2276129), CDK4 (Cell Signaling Technology Cat# 12790, RRID:AB_2631166), CDK6 (Cell Signaling Technology Cat# 3136, RRID:AB_2229289), phospho-Histone H3 (Ser10) (Cell Signaling Technology Cat# 3377 (also 3377S, 3377P), RRID:AB_1549592), p21 Waf1/Cip1 (Cell Signaling Technology Cat# 2947 (also 2947S, 2947P), RRID:AB_823586), p16 INK4A (Cell Signaling Technology Cat# 80772, RRID:AB_2799960), IDH R132H (Dianova Cat# DIA-H09, RRID:AB_2335716), Cyclophilin B (Cell Signaling Technology Cat# 43603, RRID:AB_2799247), and β-actin (Cell Signaling Technology Cat# 3700, RRID:AB_2242334). Secondary antibodies used based on the source animal (mouse or rabbit) of primary antibodies are as follows: ECL Anti-Rabbit IgG HRP-linked whole Ab from Donkey (Cytiva Cat# NA934, RRID:AB_772206) and ECL Anti-Mouse IgG HRP-linked whole Ab from sheep (Cytiva Cat# NA931, RRID:AB_772210). Membranes were exposed to ECL substrate (Bio-Rad) for 5 minutes before visualization on Gel Doc XRS+ (Bio-Rad, RRID:SCR_019690). Images were saved in Image Lab Software (Bio-Rad, RRID:SCR_014210), exported as TIFF files with 600 DPI resolution, and cropped as shown.
Cell Cycle Analysis
Cells were cultured at a density of 1×106 cells/mL and treated with 300nM Abemaciclib for 72 hours. On the day of data acquisition 10 μM of 5-ethynyl-2’-deoxyuridine (EdU) was added to the culturing media for 2 hours. DNA synthesis was determined using Click-IT® EdU Flow Cytometry Kit (Invitrogen) according to the manufacturer’s protocol. Propidium iodide/RNAse staining solution (Cell Signaling Technology) was added for 30 minutes following EdU staining to visualize the cell cycle profile for each sample. Samples were run on a BD LSRII flow cytometer (RRID:SCR_002159) and data was analyzed using FlowJo v10 (RRID:SCR_008520).
Xenograft Models
All animal experiments were approved by the Institutional Animal Care and Use Committee of Massachusetts General Hospital. For flank tumor experiments, 1.2 × 106 cells mixed 1:1 with Matrigel (Corning) were subcutaneously implanted in the right flank of 7- to 10-week-old female SCID mice (Charles River). Mice were monitored 2–3 times per week. At three weeks, mice were randomized to a vehicle group and abemaciclib (75 mg/kg) group. Abemaciclib (MedChemExpress, HY-16297) was formulated daily in 1% hydroxyethylcellulose in 25 mM phosphate buffer pH = 2. Treatment was administered orally 5 times per week. A digital caliper was used to measure tumor diameter 3 times a week. The volume (mm3) was calculated as length (mm) × width2 (mm2) × 0.5. To evaluate the status of phosphorylated Rb, tumor tissue was washed with PBS and homogenized in RIPA buffer and then western blot was performed probing for Rb, phospho-Rb and beta-actin. Quantitation was performed using densitometry within Image Lab (Bio-Rad).
For intracranial tumor experiments, 2 × 105 cells were orthotopically implanted into the right striatum of 8-week-old female SCID mice (Charles River) using a stereotactic frame. One week later, mice were randomized and assigned to vehicle or abemaciclib (90 mg/kg) group. Treatment was administered 5 times per week. Mice were monitored 2 – 3 times per week and euthanized when neurologic deficits or general body condition met criteria. For all experiments, mice were housed under a 12-hour light:dark cycle with standard bedding and ad libitum access to food.
Immunohistochemistry
Tumor-containing mouse brains were paraffin embedded to create formalin-fixed, paraffin-embedded (FFPE) blocks that were then sectioned for immunohistochemistry staining. The process of immunohistochemistry staining was performed using previously published protocols (30). The primary antibodies used to probe the tissue were as follows: Ki-67 (Dako), phospho-Rb (Cell Signaling Technology), and IDH R132H (Dianova).
Statistical Analysis
Statistical Analyses were performed with GraphPad Prism software v10 (RRID:SCR_002798). Nonlinear regression analysis was performed to create dose response curves. Differences were compared using unpaired Student’s t-test for 2 groups or ANOVA for multiple groups, with correction for multiple comparisons. Survival analyses were conducted in Prism following the method of Kaplan-Meier. All studies used a cutoff of significance of P < 0.05.
Data Availability
The data generated in this study are available upon request from the corresponding author.
Results
Sensitivity of IDH-mutant patient-derived glioma lines to CDK4/6 inhibitors
Alterations in cell cycle genes are frequently observed in recurrent IDH-mutant gliomas, primarily CDKN2A/B deletion, but also a low frequency of CDK4 and CDK6 amplification, CCND1 mutation and RB1 mutation (10,11,31). First, we screened our panel for loss of expression of p16, a protein product of CDKN2A, in four patient-derived, progressive IDH-mutant gliomas (astrocytic lines MGG152, MGG119, and BT142 and oligodendroglial line TS603). By this method, we confirmed frequent loss, with 3 of the 4 lines showing complete absence of protein product (Figure 1A). We then examined genetic alterations in the glioma lines based on a CLIA-approved sequencing panel employed at our institution, combined with published data (32). As expected, the astrocytic IDH-mutant glioma lines MGG152, MGG119 and BT142 have mutations in TP53 and mutation or alteration in ATRX. TS603, which was derived from a patient with an oligodendroglial tumor, exhibits a TERT promoter mutation (Figure 1B). Notably, we observed p16 loss in the MGG119 line, which did not have detectable genetic CDKN2A/B deletion by sequencing, either reflecting sensitivity limitations of this assay or alternative mechanisms of protein inactivation. We also note homozygous deletion of CDKN2A/B in BT142 (32), as well as the TS603 glioma line (Figure 1B), which though rare has been previously reported in oligodendroglioma.
Figure 1:
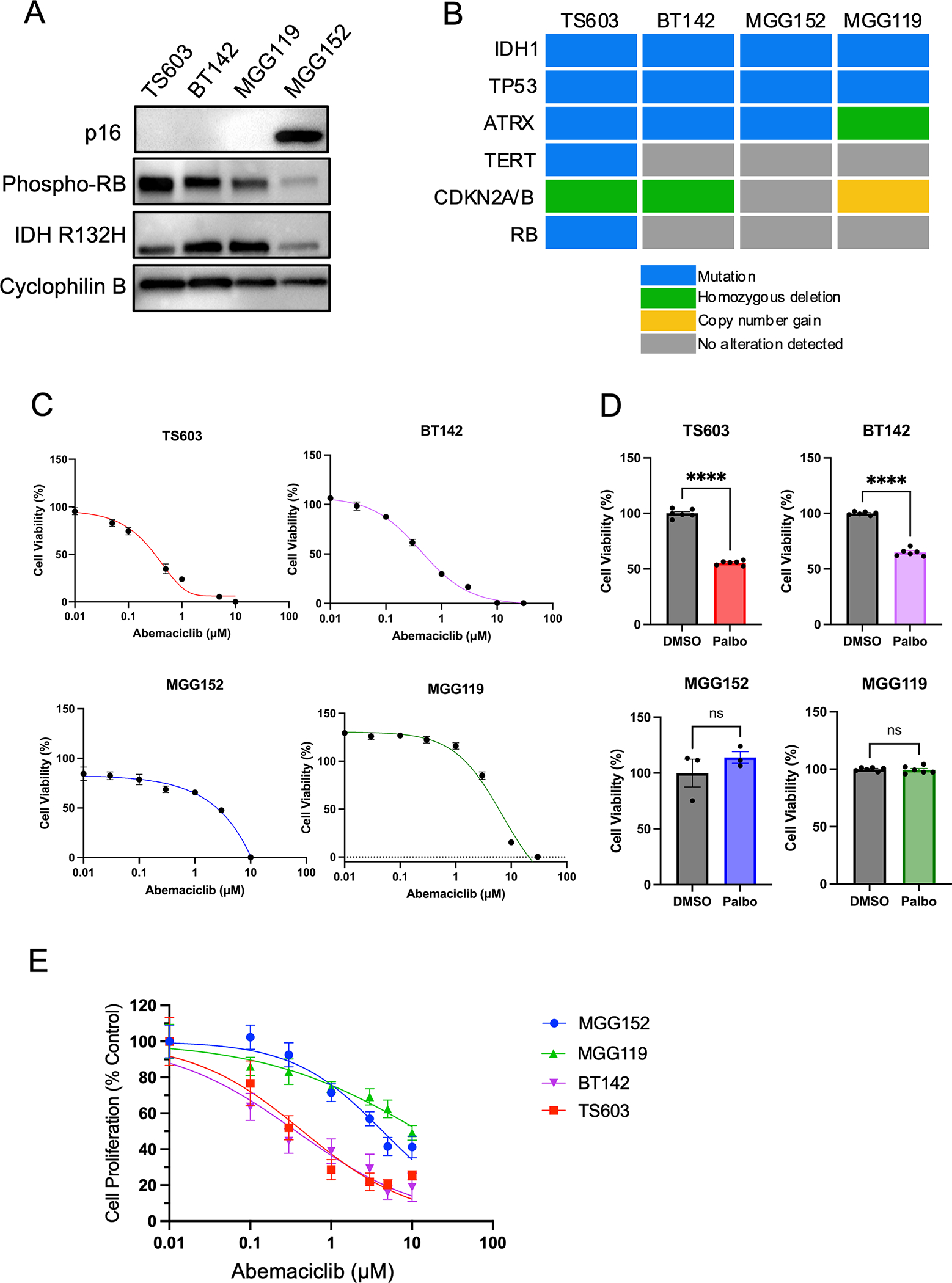
CDK4/6 inhibitors decrease cell viability in IDH-mutant patient-derived glioma cell lines. A. Western blot of IDH-mutant glioma lines for p16, phosphorylated-Rb, IDH1 R132H mutation. Cyclophilin B serves as a loading control. B. Mutation profiles of IDH-mutant glioma lines. C. Relative cell viability of IDH-mutant glioma lines after exposure to abemaciclib at the indicated concentrations for 5 days. n = 6 for each concentration, bars ± SEM. D. Relative cell viability of IDH-mutant glioma lines after exposure to palbociclib at a dose of 1 μM for 5 days. n = 3–5 for each concentration, bars ± SEM. Comparisons based on unpaired t-test, ns: non-significant, **** p < 0.0001. E. Relative cell proliferation of IDH-mutant glioma lines after exposure to abemaciclib at the indicated concentration for 5 days. n = 6 for each concentration, bars ± SEM. Data in C, D and E are representative of at least three biological replicates.
We then tested whether patient-derived glioma lines are sensitive to CDK4/6 inhibitors. We examined sensitivity to two such inhibitors, abemaciclib and palbociclib. Using a cell viability assay on a panel of patient-derived IDH-mutant glioma lines derived from recurrent tumors, we observed a significant decrease in cell viability in two IDH-mutant glioma lines following exposure to these drugs (Figure 1C, D). Importantly, the two sensitive models, TS603 and BT142, exhibited IC50 below 1 μM for abemaciclib, a level which has been shown to correlate with clinically-beneficial kinase inhibition in other cancer types (33,34). We observed a similar sensitivity to abemaciclib using the CyQUANT proliferation assay (Figure 1E). Because the four glioma lines exhibit different growth speeds, we quantitated the doubling time to examine if CDK4/6i sensitivity is associated with growth rate. TS603 exhibited the shortest doubling time (5.0 days), followed by MGG152 (6.3 days) and BT142 (6.7 days), while MGG119 had the longest (8.4 days). The relative insensitivity of MGG119 may be related to slower growth rate in addition to lack of homozygous deletion of CDKN2A/B. In subsequent studies, we chose abemaciclib as the CDKi because of its known ability to penetrate the blood brain barrier (26,35).
Responsiveness to CDK4/6 inhibitor is associated with loss of CDKN2A
Our data suggest that glioma lines with CDKN2A/B deletion were generally more sensitive to CDK4/6 inhibitor treatment in vitro. In the hypo-phosphorylated state, the retinoblastoma (Rb) protein binds to and represses E2F transcription factor family members, maintaining cells in the G1 phase of the cell cycle. Activation of CDK4/6 by coupling to cyclin D, typically triggered by mitogenic signals, leads to hyperphosphorylation of Rb, release of E2F, and transcriptional activation to promote transition into S phase (18,19). CDK4/6 inhibitor p16 blocks this signaling cascade. Consistent with this well-established biochemical mechanism, we observed that IDH-mutant glioma lines lacking p16 expression display higher levels of phospho-Rb at baseline (Figure 1A).
To directly test whether CDKN2A deletion could serve as a biomarker for CDK4/6 inhibitor sensitivity in the context of progressive IDH-mutant glioma, we used CRISPR-based gene editing to knockout CDKN2A in MGG152, a CDKN2A intact glioma line. Following successful engineered inactivation of p16 using two distinct sgRNAs, we observed a concomitant increase in phospho-Rb at serine 807/811 (Figure 2A), as would be expected in the setting of unrestrained CDK4/6 activity. This phosphorylation of Rb was reversed with abemaciclib treatment, further evidence of on-target CDK4/6 activity from p16 inactivation (Figure 2A). Most importantly, CDKN2A deletion in MGG152 glioma cells resulted in increased sensitivity to abemaciclib treatment (Figure 2B), consistent with our hypothesis.
Figure 2:
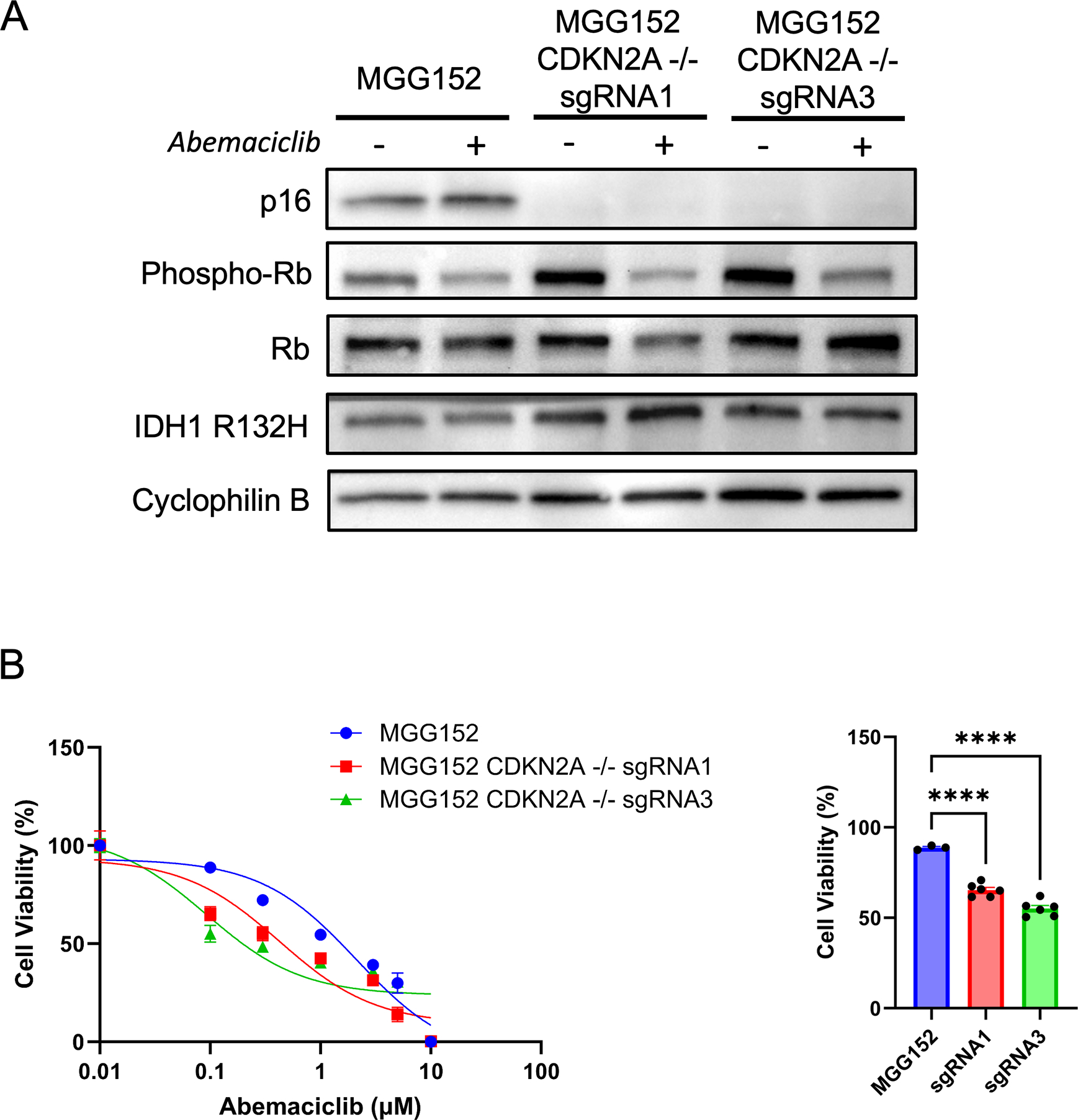
Loss of p16 enhances sensitivity of IDH-mutant glioma lines to CDK4/6 inhibition. A. Western blot of parental and CDKN2A knockout MGG152 lines showing loss of p16 expression and changes in Rb phosphorylation in the presence of abemaciclib treatment (300 nM). Cyclophilin B is used as a loading control. B. Cell viability assay of MGG152 lines after exposure to abemaciclib at the indicated concentrations for 5 days. C. Cell viability following abemaciclib treatment at 100 nM. n = 3 for each concentration, bars ± SEM. Data are representative of three biological replicates. **** p < 0.0001.
Abemaciclib treatment results in G1/S block
As detailed above, aberrant activation of CDK4/6 and subsequent Rb hyperphosphorylation promotes transition through the cell cycle. Consistent with this, in our glioma model, we observed a decrease in the fraction of cells in G1 phase when CDKN2A is deleted (Figure 3A). Additionally, we find that inhibition of CDK4/6 with abemaciclib resulted in an enrichment in G1 population and a decrease in G2/M phase (Figure 3A, Supplemental Figure 1). This effect is consistent with on-target activity of cyclin-dependent kinases at the G1/S transition and was observed in all IDH-mutant glioma lines, regardless of the effect that abemaciclib had on proliferation. The enrichment in the G1 population, however, was most apparent in CDKN2A-deleted lines. In addition, we observed a statistically significant enrichment in the proportion of cells in G2/M phase. This G2/M block was present only in the abemaciclib-sensitive glioma lines TS603 and BT142 (Figure 3A, 3B), suggesting that inhibition of CDK4/6 activity independent of the G1/S checkpoint may be driving the anti-tumor benefit. This observation raises the mechanistic implication that the CDKi sensitivity from CDKN2A knockout in IDH-mutant glioma reflects an induced oncogenic addiction to CDK4/6 in this context.
Figure 3:
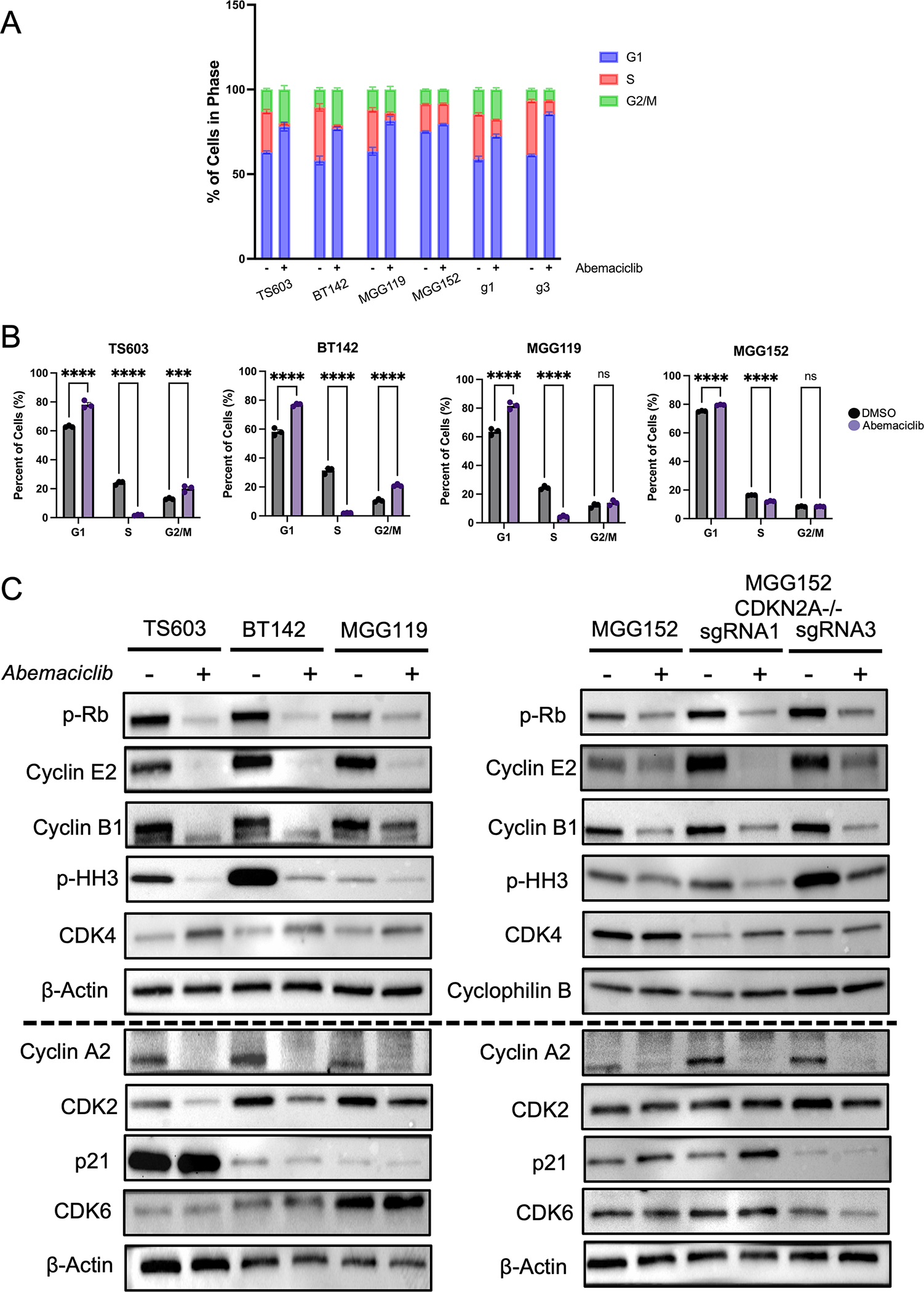
Abemaciclib leads to block at G1/S transition in IDH-mutant glioma. A. Cell cycle profile based on PI and EdU staining following exposure to DMSO (−) or abemaciclib (+) at a dose of 300 nM. The percentage of cell population in each cell cycle phase is shown. B. Cell cycle data showing changes in cell fraction following abemaciclib. n = 3, mean values ± SEM. Data are representative of at least two biological replicates. See Supplemental Figure 1 for raw data. C. Western blot analysis of cell cycle protein expression following exposure to DMSO (−) or 300 nM abemaciclib (+). Β-actin and cyclophilin B serve as loading control for each membrane. Two separate blots are used to present the data, with the horizontal dotted line delineating the separate membranes. G1, g3: single guide RNA 1 or 3 used for deletion of CDKN2A. ns, non-significant. ***, p < 0.001. ****, p < 0.0001.
We therefore examined the broader impact of abemaciclib on the cell cycle progression machinery in IDH-mutant glioma by immunoblot for key cell cycle proteins. Abemaciclib treatment resulted in a strong decrease in levels of phospho-Rb. Consistent with the data from flow cytometry, CDK4/6 inhibition led to a decrease in proteins associated with G2 and M phase, such as cyclin E2, cyclin A2, cyclin B1 and phospho-HH3 (Figure 3C). We observed an increase in CDK4 levels following drug exposure. The increase in CDK4 levels is unexpected but has been reported following abemaciclib in lung cancer cells in vitro (36). While cyclin protein levels are well-known to vary in abundance in a cell-cycle dependent manner (37), the regulation of CDK levels is less well-studied. However, CDK4 protein levels have been reported to decrease during the mitosis in a ubiquitin-proteasome-dependent manner via the Anaphase Promoting Complex/Cyclosome (APC/C) (38,39). This raises the possibility that the increase in CDK4 is related to enrichment in G1 phase, a time when APC/C-driven degradation is low. CDK6 levels were unchanged. Interestingly, abemaciclib can lead to cellular senescence and increased p21 levels in some cancer cell models (40,41), however, we found baseline p21 levels variable across IDH-mutant glioma lines and unaffected by abemaciclib treatment (Figure 3C). The most consistently elevated protein across p16-deleted lines was cyclin E2, which was uniformly decreased upon CDKi treatment, potentially serving as an additional biomarker of induced CDKi sensitivity in the context of IDH-mutant glioma. In glioma lines lacking p16, CDK2 expression decreased with CDKi treatment, as has been previously described (42), while there was no change in MGG152 parental or engineered lines (Figure 3C). The mechanism for this difference is unclear.
Abemaciclib in combination with alkylating chemotherapy or IDH inhibitor confers no added benefit
Patients with IDH mutated gliomas are often treated with the alkylating chemotherapeutics temozolomide (TMZ) and lomustine (CCNU), prompting us to investigate the utility of combination therapy of these agents combined with CDKi. However, we did not observe an additive effect from co-treatment with abemaciclib and temozolomide (Figure 4A) or abemaciclib and lomustine (Supplemental Figure 2). This lack of effect was observed regardless of CDKN2A/B status.
Figure 4:
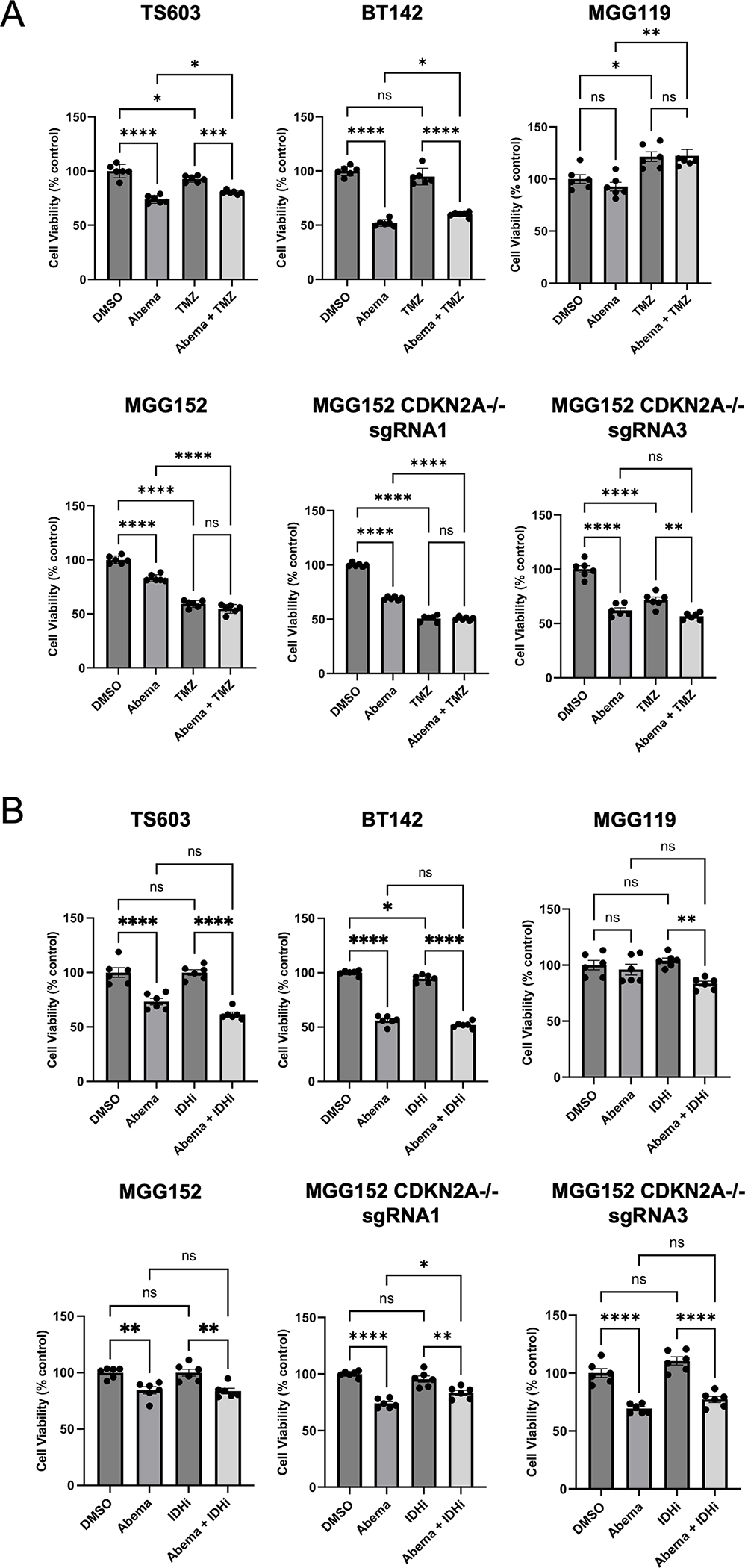
Combination treatment with standard therapies does not lead to an added benefit. A. Relative cell viability of IDH-mutant glioma lines treated with DMSO, abemaciclib 0.1 μM, temzolomide (TMZ) 100 μM or combination of abemaciclib plus TMZ for 5 days. n = 6 for each treatment, mean values ± SEM. (B) Relative cell viability of IDH-mutant glioma lines treated with DMSO, abemaciclib 0.1 μM, AGI-5198 5 μM (IDHi) or combination of abemaciclib plus AGI-5198 for 5 days. n = 6 for each treatment, mean values ± SEM. Comparisons were made using One-way ANOVA adjusted for multiple comparisons. ns, non-significant. *, p < 0.05. **, p < 0.01. ***, p < 0.001. ****, p < 0.0001.
Additionally, the IDH1/2 inhibitor vorasidenib has been recently demonstrated to improve progression-free survival in treatment-naïve patients with grade 2 IDH-mutant gliomas. Anticipating that use of IDH inhibitors will be considered for other populations of patients with IDH-mutant gliomas, we investigated whether there is a benefit from combination of abemaciclib and IDH inhibitor. In vitro administration of the IDH inhibitor AG-5198 resulted in marked decrease in D-2HG levels (27,30,43), however, IDHi did not influence cell proliferation in these models (Figure 4B). Further, we did not observe an additive benefit when administered concurrently with abemaciclib (Figure 4B).
Abemaciclib delays growth of orthotopically implanted IDH-mutant gliomas
Given the potential for differences between cell growth dependencies in vitro versus in vivo, we next tested the anti-tumor efficacy of abemaciclib monotherapy in in vivo. Abemaciclib treatment 5 days per week, at a clinically relevant dose level, led to significant slowing of tumor growth in a canonical TS603 flank xenograft model when compared to tumor growth in vehicle-treated animals (Figure 5A). We examined the level of phosphorylated Rb and total Rb in tumor samples and observed a significant decrease in the ratio of phospho-Rb to total Rb in the abemaciclib group, consistent with evidence of intratumoral target engagement (Figure 5B; Supplemental Figure 3A).
Figure 5:
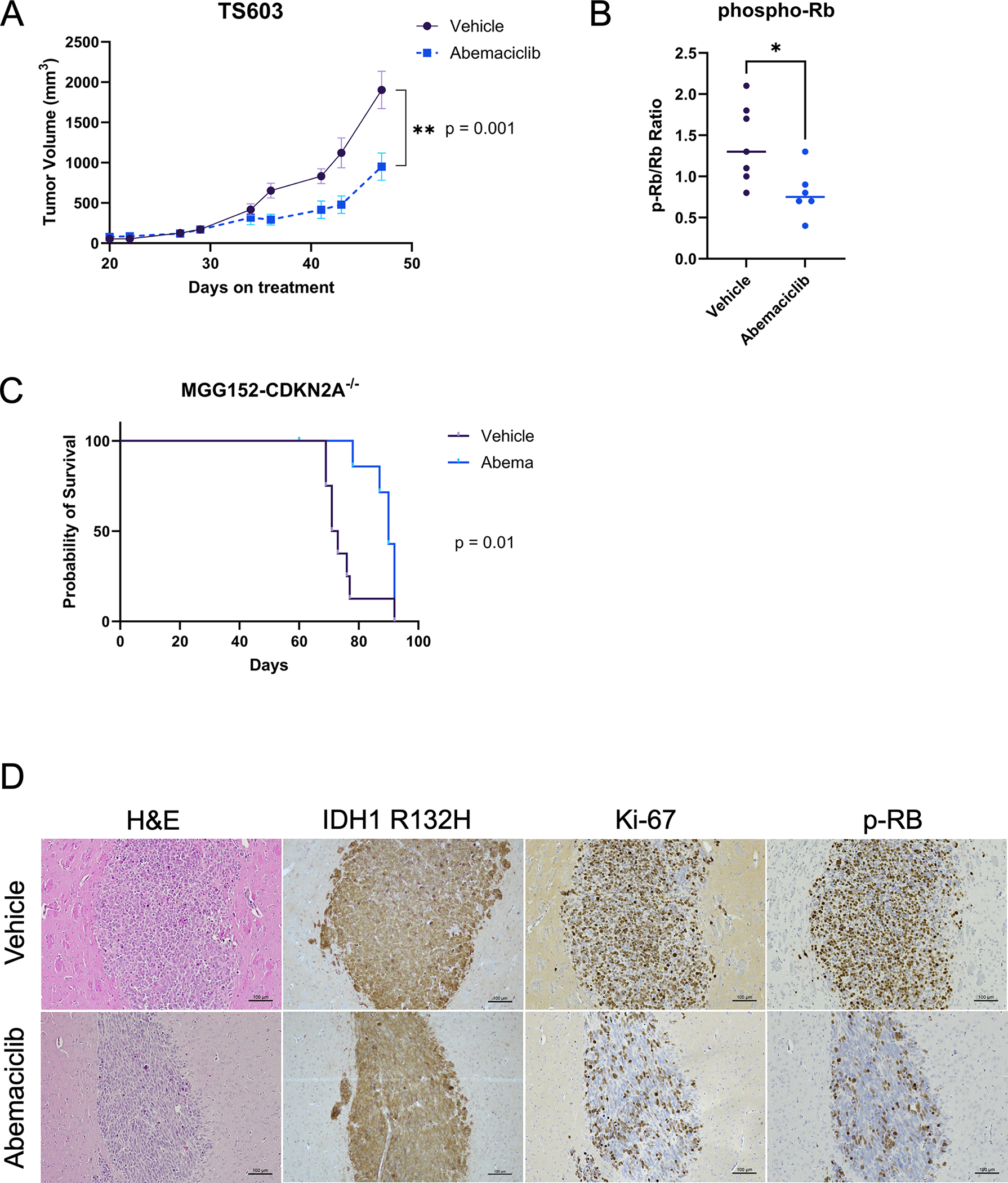
Abemaciclib slows tumor growth in IDH-mutant glioma models with CDKN2A deletion. (A) Tumor volumes of TS603 subcutaneous xenografts in animals treated with abemaciclib (N = 6) or vehicle control (N = 7) five times per week. Bars ± SEM. (B) Ratio of phospho-Rb to total Rb detected in each tumor taken at study endpoint show reduction to a mean of 0.8 in abemaciclib treated tumors compared to 1.4 for vehicle treated tumors. Comparison based on unpaired t-test, p = 0.02. (C) Survival curve of mice bearing intracranial xenografts of MGG152-CDKN2A−/− sgRNA3 treated five times per week with abemaciclib (N = 8) or vehicle (N = 8). One animal in Abemaciclib group censored due to unrelated dermatologic issue. (D) Representative images of immunohistochemistry of tumors extracted from vehicle or abemaciclib-treated animals, showing IDH1 R132H, Ki-67 and phosphorylated Rb (p-Rb).
We then proceeded to test the impact of abemaciclib on the survival of animals with orthotopically implanted IDH-mutant, CDKN2A-deleted gliomas using the MGG152-CDKN2A-knockout glioma model. Here, we observed a significant prolongation of median survival in the abemaciclib-treated group (Figure 5C), with abemaciclib-treated animals living a median of 90 days compared to 72 days for vehicle treated animals (p=0.01; HR 0.37; 95% CI 0.12 – 1.12). Treatment was tolerable, even when given for an extended period (Supplemental Figure 3B, C). Immunohistochemistry demonstrated a decrease in Ki-67 and phospho-Rb expression in abemaciclib-treated tumors, consistent with expected, on-target effects of CDK4/6i (Figure 5D).
Discussion
Here, we provide the first report of IDH1 mutant glioma sensitivity to CDK4/6 inhibitors, including abemaciclib. We tested 4 endogenous IDH-mutant glioma lines in vitro and noted 2 lines with increased responsiveness to either of two different CDK4/6 inhibitors. Since the most responsive glioma lines lacked expression of p16, a tumor suppressor protein encoded by CDKN2A, we performed further controlled, isogenic experiments demonstrating that genetic deletion of CDKN2A in a glioma line without endogenous loss enhances the anti-tumor efficacy of CDK4/6 inhibition. These data provide strong evidence that CDKN2A, and the encoded protein p16, play a critical role in determining sensitivity to this drug class in IDH-mutant gliomas.
To date, no relationship between CDKN2A deletion or p16 loss and CDK4/6i responsiveness has been established. Indeed, “intermediate” sensitivity to abemaciclib was observed in a large panel of adherent cancer cell lines. Eighteen glioma lines were included in this panel, 10 of which had homozygous deletion of CDKN2A. None of these glioma lines had an IDH mutation based on a repository of next generation sequencing data of cancer cell lines (22,44). In contrast, our findings in IDH-mutant gliomas are distinct from these findings. This difference likely reflects the different natural histories of these two glioma sub-types, and the emergence of CDKN2A/B deletion during stepwise, clonal progression in the evolution of IDH-mutant glioma. Thus, in contrast to glioblastoma or breast cancer, CDKN2A deletion has the potential to serve as a biomarker for patient selection of CDKi in the setting of progressive IDH-mutant glioma.
It is notable that, in responsive cell lines with CDKN2A loss, we observed a prominent decrease in CDK2 levels following CDK4/6 inhibitor treatment. Adaptive activation of CDK2 is a known resistance pathway in CDK4/6 inhibitor-treated breast cancers (45,46), raising the possibility that agents that target CDK2 in tandem with CDK4/6 may provide additional anti-tumor benefit.
Importantly, we show an in vivo anti-tumor benefit in two distinct patient-derived models of CDKN2A-deleted IDH-mutant glioma. While this is encouraging, we note that the durability of response could still be improved and, thus, there may be benefit from incorporation in a combination regimen in future studies. Nevertheless, given the very poor prognosis of patients with IDH-mutant glioma with CDKN2A deletion, the work presented here provides strong preclinical rationale for investigating CDK4/6 inhibitors in this clinical setting, where even a small change in tumor growth rate could provide substantial clinical benefit. We have focused our investigation here on recurrent disease due to the availability of IDH-mutant glioma models and the high frequency of CDKN2A/B alterations at this stage. However, IDH-mutant gliomas found to have CDKN2A/B deletion at initial diagnosis typically follow an aggressive course. These tumors are unlikely to respond to vorasidenib or other IDH inhibitors, which have shown the strongest benefit in indolent disease. For this reason, consideration should also be given to exploration of CDK4/6 inhibitors in this setting. Standard upfront treatment for patients with IDH-mutant glioma with CDKN2A/B deletion involves a combination of radiation and temozolomide based on an extrapolation from the CATNON trial (6), which shows a survival advantage for patients with higher grade IDH-mutant gliomas receiving adjuvant temozolomide. Abemaciclib could be integrated into this treatment paradigm. Since we do not observe increased efficacy when combining temozolomide with abemaciclib in our models, a potential starting point could be exploration of abemaciclib with concurrent radiation therapy, which displays a synergistic relationship in other tumor types (36,47,48). The wide availability of this class of drugs suggest that these results are readily translatable. Taken together, our preclinical data support further clinical investigation of CDK4/6 inhibition in IDH-mutant glioma with CDKN2A/B deletion.
Supplementary Material
Translational Relevance.
IDH-mutant gliomas (astrocytoma and oligodendroglioma) typically recur despite initial responsiveness to standard therapies like radiation treatment and chemotherapy. There is an urgent need for improved treatments at progression. Homozygous loss of CDKN2A/B is observed at high frequency in the recurrent tumor setting, as well as in a small subset of IDH-mutant gliomas at initial diagnosis. Deletion of this locus is associated with poor outcomes in both contexts. Our study shows that CDK-Rb pathway hyperactivity in CDKN2A/B-deleted IDH-mutant tumors can be blocked with CDK4/6 inhibitors. CDK4/6 inhibition prevents proliferation, slows tumor growth, and improves survival in preclinical patient-derived IDH-mutant glioma models with CDKN2A/B loss. Our study thus presents strong preclinical rationale for investigating CDK4/6 inhibitors in patients with IDH-mutant glioma harboring CDKN2A/B loss.
Acknowledgements
This work is supported by the American Cancer Society ACS-IRG-21-130-10 (J. Miller), NINDS K08NS119796 (J. Miller), MGH Transformative Scholar Award (J. Miller), Seeman Family MGH Scholar Funds (J. Miller), NCI Brain SPORE P50CA165962 (J. Miller, D. Cahill), NCI R01CA276765 (D. Cahill) and Tawingo Fund (D. Cahill). We acknowledge assistance from the MGH Flow Cytometry Core, MGH Center for Comparative Medicine, Gregory R. Wojtkiewicz of the MGH Center for Systems Biology Mouse Imaging Program. We thank our patients for their donation of critical research materials and members of the Translational Neuro-Oncology Laboratory for helpful discussions.
Footnotes
Author Disclosures
J.J.M. reports consulting fees from Servier. D.P.C reports financial compensation from Lilly, GlaxoSmithKline, Incephalo, Boston Pharmaceuticals, Servier, Boston Scientific and Pyramid Biosciences for advisory input, and has also received financial compensation and travel reimbursement from Merck for invited lectures, and from the US NIH and DOD for clinical trial and grant review.
References
- 1.Buckner JC, Shaw EG, Pugh SL, Chakravarti A, Gilbert MR, Barger GR, et al. Radiation plus Procarbazine, CCNU, and Vincristine in Low-Grade Glioma. N Engl J Med 2016;374(14):1344–55 doi 10.1056/NEJMoa1500925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bell EH, Zhang P, Shaw EG, Buckner JC, Barger GR, Bullard DE, et al. Comprehensive Genomic Analysis in NRG Oncology/RTOG 9802: A Phase III Trial of Radiation Versus Radiation Plus Procarbazine, Lomustine (CCNU), and Vincristine in High-Risk Low-Grade Glioma. J Clin Oncol 2020:JCO1902983 doi 10.1200/JCO.19.02983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van den Bent MJ, Brandes AA, Taphoorn MJ, Kros JM, Kouwenhoven MC, Delattre JY, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol 2013;31(3):344–50 doi 10.1200/JCO.2012.43.2229. [DOI] [PubMed] [Google Scholar]
- 4.Cairncross G, Wang M, Shaw E, Jenkins R, Brachman D, Buckner J, et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol 2013;31(3):337–43 doi 10.1200/JCO.2012.43.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cairncross JG, Wang M, Jenkins RB, Shaw EG, Giannini C, Brachman DG, et al. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J Clin Oncol 2014;32(8):783–90 doi 10.1200/JCO.2013.49.3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van den Bent MJ, Tesileanu CMS, Wick W, Sanson M, Brandes AA, Clement PM, et al. Adjuvant and concurrent temozolomide for 1p/19q non-co-deleted anaplastic glioma (CATNON; EORTC study 26053–22054): second interim analysis of a randomised, open-label, phase 3 study. Lancet Oncol 2021;22(6):813–23 doi 10.1016/S1470-2045(21)00090-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mellinghoff IK, van den Bent MJ, Blumenthal DT, Touat M, Peters KB, Clarke J, et al. Vorasidenib in IDH1- or IDH2-Mutant Low-Grade Glioma. N Engl J Med 2023;389(7):589–601 doi 10.1056/NEJMoa2304194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wakimoto H, Tanaka S, Curry WT, Loebel F, Zhao D, Tateishi K, et al. Targetable Signaling Pathway Mutations Are Associated with Malignant Phenotype in IDH-Mutant Gliomas. Clinical Cancer Research 2014;20(11):2898–909 doi 10.1158/1078-0432.ccr-13-3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bai H, Harmancı AS, Erson-Omay EZ, Li J, Coşkun S, Simon M, et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nature genetics 2016;48(1):59–66 doi 10.1038/ng.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barthel FP, Johnson KC, Varn FS, Moskalik AD, Tanner G, Kocakavuk E, et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature 2019;576(7785):112–20 doi 10.1038/s41586-019-1775-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jonsson P, Lin AL, Young RJ, DiStefano NM, Hyman DM, Li BT, et al. Genomic Correlates of Disease Progression and Treatment Response in Prospectively Characterized Gliomas. Clin Cancer Res 2019;25(18):5537–47 doi 10.1158/1078-0432.CCR-19-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kocakavuk E, Anderson KJ, Varn FS, Johnson KC, Amin SB, Sulman EP, et al. Radiotherapy is associated with a deletion signature that contributes to poor outcomes in patients with cancer. Nature genetics 2021;53(7):1088–96 doi 10.1038/s41588-021-00874-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fortin Ensign SP, Jenkins RB, Giannini C, Sarkaria JN, Galanis E, Kizilbash SH. Translational significance of CDKN2A/B homozygous deletion in isocitrate dehydrogenase-mutant astrocytoma. Neuro Oncol 2023;25(1):28–36 doi 10.1093/neuonc/noac205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu VM, O’Connor KP, Shah AH, Eichberg DG, Luther EM, Komotar RJ, et al. The prognostic significance of CDKN2A homozygous deletion in IDH-mutant lower-grade glioma and glioblastoma: a systematic review of the contemporary literature. J Neurooncol 2020;148(2):221–9 doi 10.1007/s11060-020-03528-2. [DOI] [PubMed] [Google Scholar]
- 15.Shirahata M, Ono T, Stichel D, Schrimpf D, Reuss DE, Sahm F, et al. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol 2018;136(1):153–66 doi 10.1007/s00401-018-1849-4. [DOI] [PubMed] [Google Scholar]
- 16.Appay R, Dehais C, Maurage CA, Alentorn A, Carpentier C, Colin C, et al. CDKN2A homozygous deletion is a strong adverse prognosis factor in diffuse malignant IDH-mutant gliomas. Neuro Oncol 2019;21(12):1519–28 doi 10.1093/neuonc/noz124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Louis DN, Wesseling P, Aldape K, Brat DJ, Capper D, Cree IA, et al. cIMPACT-NOW update 6: new entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol 2020;30(4):844–56 doi 10.1111/bpa.12832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sherr CJ. A New Cell-Cycle Target in Cancer - Inhibiting Cyclin D-Dependent Kinases 4 and 6. N Engl J Med 2016;375(20):1920–3 doi 10.1056/NEJMp1612343. [DOI] [PubMed] [Google Scholar]
- 19.Rubin SM, Sage J, Skotheim JM. Integrating Old and New Paradigms of G1/S Control. Mol Cell 2020;80(2):183–92 doi 10.1016/j.molcel.2020.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kocakavuk E, Johnson KC, Sabedot TS, Reinhardt HC, Noushmehr H, Verhaak RGW. Hemizygous CDKN2A deletion confers worse survival outcomes in IDHmut-noncodel gliomas. Neuro Oncol 2023;25(9):1721–3 doi 10.1093/neuonc/noad095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hickman RA, Gedvilaite E, Ptashkin R, Reiner AS, Cimera R, Nandakumar S, et al. CDKN2A/B mutations and allele-specific alterations stratify survival outcomes in IDH-mutant astrocytomas. Acta Neuropathol 2023;146(6):845–7 doi 10.1007/s00401-023-02639-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gong X, Litchfield LM, Webster Y, Chio LC, Wong SS, Stewart TR, et al. Genomic Aberrations that Activate D-type Cyclins Are Associated with Enhanced Sensitivity to the CDK4 and CDK6 Inhibitor Abemaciclib. Cancer Cell 2017;32(6):761–76 e6 doi 10.1016/j.ccell.2017.11.006. [DOI] [PubMed] [Google Scholar]
- 23.Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol 2015;16(1):25–35 doi 10.1016/S1470-2045(14)71159-3. [DOI] [PubMed] [Google Scholar]
- 24.VanArsdale T, Boshoff C, Arndt KT, Abraham RT. Molecular Pathways: Targeting the Cyclin D-CDK4/6 Axis for Cancer Treatment. Clin Cancer Res 2015;21(13):2905–10 doi 10.1158/1078-0432.CCR-14-0816. [DOI] [PubMed] [Google Scholar]
- 25.Rahman R, Trippa L, Lee EQ, Arrillaga-Romany I, Fell G, Touat M, et al. Inaugural Results of the Individualized Screening Trial of Innovative Glioblastoma Therapy: A Phase II Platform Trial for Newly Diagnosed Glioblastoma Using Bayesian Adaptive Randomization. J Clin Oncol 2023;41(36):5524–35 doi 10.1200/JCO.23.00493. [DOI] [PubMed] [Google Scholar]
- 26.Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Gandhi L, et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non–Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discovery 2016(July):740–54 doi 10.1158/2159-8290.CD-16-0095. [DOI] [PubMed] [Google Scholar]
- 27.Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F, Lelic N, et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015;28(6):773–84 doi 10.1016/j.ccell.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tateishi K, Higuchi F, Miller JJ, Koerner MVA, Lelic N, Shankar GM, et al. The Alkylating Chemotherapeutic Temozolomide Induces Metabolic Stress in IDH1-Mutant Cancers and Potentiates NAD(+) Depletion-Mediated Cytotoxicity. Cancer Res 2017;77(15):4102–15 doi 10.1158/0008-5472.CAN-16-2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller JJ, Fink A, Banagis JA, Nagashima H, Subramanian M, Lee CK, et al. Sirtuin activation targets IDH-mutant tumors. Neuro Oncol 2021;23(1):53–62 doi 10.1093/neuonc/noaa180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagashima H, Lee CK, Tateishi K, Higuchi F, Subramanian M, Rafferty S, et al. Poly(ADP-ribose) Glycohydrolase Inhibition Sequesters NAD(+) to Potentiate the Metabolic Lethality of Alkylating Chemotherapy in IDH-Mutant Tumor Cells. Cancer Discov 2020;10(11):1672–89 doi 10.1158/2159-8290.CD-20-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med 2015;372(26):2481–98 doi 10.1056/NEJMoa1402121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen Y, Grisdale CJ, Islam SA, Bose P, Lever J, Zhao EY, et al. Comprehensive genomic profiling of glioblastoma tumors, BTICs, and xenografts reveals stability and adaptation to growth environments. Proc Natl Acad Sci U S A 2019;116(38):19098–108 doi 10.1073/pnas.1813495116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wander SA, Cohen O, Gong X, Johnson GN, Buendia-Buendia JE, Lloyd MR, et al. The Genomic Landscape of Intrinsic and Acquired Resistance to Cyclin-Dependent Kinase 4/6 Inhibitors in Patients with Hormone Receptor-Positive Metastatic Breast Cancer. Cancer Discov 2020;10(8):1174–93 doi 10.1158/2159-8290.CD-19-1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gelbert LM, Cai S, Lin X, Sanchez-Martinez C, Del Prado M, Lallena MJ, et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest New Drugs 2014;32(5):825–37 doi 10.1007/s10637-014-0120-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raub TJ, Wishart GN, Kulanthaivel P, Staton BA, Ajamie RT, Sawada GA, et al. Brain Exposure of Two Selective Dual CDK4 and CDK6 Inhibitors and the Antitumor Activity of CDK4 and CDK6 Inhibition in Combination with Temozolomide in an Intracranial Glioblastoma Xenograft. Drug Metab Dispos 2015;43(9):1360–71 doi 10.1124/dmd.114.062745. [DOI] [PubMed] [Google Scholar]
- 36.Naz S, Sowers A, Choudhuri R, Wissler M, Gamson J, Mathias A, et al. Abemaciclib, a Selective CDK4/6 Inhibitor, Enhances the Radiosensitivity of Non-Small Cell Lung Cancer In Vitro and In Vivo. Clin Cancer Res 2018;24(16):3994–4005 doi 10.1158/1078-0432.CCR-17-3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murray AW. Recycling the cell cycle: cyclins revisited. Cell 2004;116(2):221–34 doi 10.1016/s0092-8674(03)01080-8. [DOI] [PubMed] [Google Scholar]
- 38.Chen H, Xu X, Wang G, Zhang B, Wang G, Xin G, et al. CDK4 protein is degraded by anaphase-promoting complex/cyclosome in mitosis and reaccumulates in early G(1) phase to initiate a new cell cycle in HeLa cells. J Biol Chem 2017;292(24):10131–41 doi 10.1074/jbc.M116.773226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murphy JM, Jeong K, Ahn EE, Lim SS. Nuclear focal adhesion kinase induces APC/C activator protein CDH1-mediated cyclin-dependent kinase 4/6 degradation and inhibits melanoma proliferation. J Biol Chem 2022;298(6):102013 doi 10.1016/j.jbc.2022.102013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dowless M, Lowery CD, Shackleford T, Renschler M, Stephens J, Flack R, et al. Abemaciclib Is Active in Preclinical Models of Ewing Sarcoma via Multipronged Regulation of Cell Cycle, DNA Methylation, and Interferon Pathway Signaling. Clin Cancer Res 2018;24(23):6028–39 doi 10.1158/1078-0432.CCR-18-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kartika ID, Kotani H, Iida Y, Koyanagi A, Tanino R, Harada M. Protective role of cytoplasmic p21Cip1/Waf1 in apoptosis of CDK4/6 inhibitor-induced senescence in breast cancer cells. Cancer Med 2021;10(24):8988–99 doi 10.1002/cam4.4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cornwell JA, Crncec A, Afifi MM, Tang K, Amin R, Cappell SD. Loss of CDK4/6 activity in S/G2 phase leads to cell cycle reversal. Nature 2023;619(7969):363–70 doi 10.1038/s41586-023-06274-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013;340(6132):626–30 doi 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghandi M, Huang FW, Jane-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019;569(7757):503–8 doi 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Freeman-Cook K, Hoffman RL, Miller N, Almaden J, Chionis J, Zhang Q, et al. Expanding control of the tumor cell cycle with a CDK2/4/6 inhibitor. Cancer Cell 2021;39(10):1404–21.e11 doi 10.1016/j.ccell.2021.08.009. [DOI] [PubMed] [Google Scholar]
- 46.Asghar US, Kanani R, Roylance R, Mittnacht S. Systematic Review of Molecular Biomarkers Predictive of Resistance to CDK4/6 Inhibition in Metastatic Breast Cancer. JCO Precis Oncol 2022;6:e2100002 doi 10.1200/PO.21.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Michaud K, Solomon DA, Oermann E, Kim JS, Zhong WZ, Prados MD, et al. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res 2010;70(8):3228–38 doi 10.1158/0008-5472.CAN-09-4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Whittaker S, Madani D, Joshi S, Chung SA, Johns T, Day B, et al. Combination of palbociclib and radiotherapy for glioblastoma. Cell Death Discov 2017;3:17033 doi 10.1038/cddiscovery.2017.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available upon request from the corresponding author.