

Abstract
HIV-1 reverse transcriptase (RT) has received great attention as an attractive therapeutic target for acquired immune deficiency syndrome (AIDS), but the inevitable drug resistance and side effects have always been major challenges faced by non-nucleoside reverse transcriptase inhibitors (NNRTIs). This work aimed to identify novel chemotypes of anti-HIV-1 agents with improved drug-resistance profiles, reduced toxicity, and excellent druggability. A series of diarylpyrimidine (DAPY) derivatives were prepared via structural modifications of the leads K-5a2 and 25a. Among them, 15a with dimethylphosphine oxide moiety showed the most prominent antiviral potency against all of the tested viral panel, being 1.6-fold (WT, EC50 = 1.75 nmol/L), 3.0-fold (L100I, EC50 = 2.84 nmol/L), 2.4-fold (K103N, EC50 = 1.27 nmol/L), 3.3-fold (Y181C, EC50 = 5.38 nmol/L), 2.9-fold (Y188L, EC50 = 7.96 nmol/L), 2.5-fold (E138K, EC50 = 4.28 nmol/L), 4.8-fold (F227L/V106A, EC50 = 3.76 nmol/L) and 5.3-fold (RES056, EC50 = 15.8 nmol/L) more effective than that of the marketed drug ETR. Molecular docking results illustrated the detailed interactions formed by compound 15a and WT, F227L/V106A, and RES056 RT. Moreover, 15a·HCl carried outstanding pharmacokinetic (t1/2 = 1.32 h, F = 40.8%) and safety profiles (LD50 > 2000 mg/kg), which demonstrated that 15a HCl is a potential anti-HIV-1 drug candidate.
Key words: HIV-1, NNRTIs, NNIBP, Structural alert, Anti-HIV-1 drug candidate
Graphical abstract
15a·HCl exhibited enhanced drug resistance profiles against HIV-1 mutations and possessed improved solubility, favorable pharmacokinetic and safety profiles, which suggested that 15a·HCl could be a potential anti-HIV-1 drug candidate.

1. Introduction
In 2022, 39.0 million people were living with human immunodeficiency virus (HIV) infections according to the World Health Organization (WHO). Moreover, the number of new cases (1.3 million) and the deaths from HIV-related causes (630 thousand) showed the importance and urgency of the discovery of effective therapies to control HIV infection and transmission1.
Highly active antiretroviral therapy (HAART) is the treatment standard for the management of HIV infections in clinical practice at present, which composed of several classes of drugs targeting different stages of the virus life cycle, and has transformed acquired immune deficiency syndrome (AIDS) from a fatal disease to a controllable chronic disorder2, 3, 4. And the non-nucleoside reverse transcriptase inhibitor is the key backbone of HAART for its advantages of efficient antiviral activity and modest toxicity5,6. From 1996 to 2018, six NNRTIs were approved to treat HIV infections by the U.S. Food and Drug Administration (FDA), including the first-generation NNRTIs delavirdine (DLV), nevirapine (NVP), efavirenz (EFV) and the second-generation NNRTIs etravirine (ETR, 1), rilpivirine (RPV, 2), and doravirine (DOR)7. However, these drugs are challenged by the emergence of drug resistance during their widespread clinical application8,9. For example, the first-generation NNRTIs almost completely lost activity against mutant strains L100I, K103N, and Y181C, and reduced potency against the double mutant strains F227L/V106A and K103N/Y181C was found in the clinical treatment of HIV with the second-generation NNRTIs10, 11, 12, 13, 14. Moreover, both of them were always accompanied by severe rash and relatively high rates of neuropsychiatric central nervous system side effects15. Therefore, it is necessary to explore novel anti-HIV-1 drugs with highly effective anti-drug resistance and lower toxicity16.
In our previous efforts, we have designed and identified several potent NNRTIs as anti-HIV-1 drug candidates with ETR and RPV as leads. The representative piperidine-substituted thiophene[3,2-d]pyrimidine compounds 3 (K-5a2) and 4 (25a) (Fig. 1) exhibited higher anti-HIV-1 potency and favorable resistance profiles compared with ETR and RPV17,18. However, they all encountered the problem of poor solubility (SK-5a2≪ 1 μg/mL, S25a < 1 μg/mL, pH = 7.4; SK-5a2 = 0.05 μg/mL, S25a = 5.26 μg/mL, pH = 7.0), which lead to their lower bioavailability (FK-5a2 = 22.9%, F25a = 16.2%). The co-crystal structure (Fig. 2) revealed that the larger piperidine–CH2–benzenesulfonamide motif in the right-wing can fully occupy the tolerant region I of NNRTI-binding pocket (NNIBP), and the residues Pro225 and Pro236 located at the groove channel opening are pushed away to accommodate the larger benzenesulfonamide group, which protrudes to the solvent-exposed surface of the enzyme and helped improve the drug resistance and solubility of compounds. In addition, the double-hydrogen bonds between the surface-positioned sulfamide group with the carbonyl oxygen of Lys104 and the backbone nitrogen of Val106 is considered one of the key forces that maintain the binding of K-5a2 and 25a to the NNIBP19. However, the toxicity predictions displayed that the sulfamide group of K-5a2 and 25a is a structural alert for ocular toxicity, urolithiasis and phototoxicity by Derek Nexus Prediction. In addition, antibacterial drugs such as sulfaguanidine, succinylsulfathiazole, and sulfacyl, containing sulfonamide groups, have been reported to be phototoxic and nephrotoxic20.
Figure 1.

Chemical structures of ETR, RPV, and the piperidine-substituted thiophene[3,2-d]pyrimidine NNRTIs K-5a2 and 25a.
Figure 2.

Crystal structure of HIV-1 RT in complex with K-5a2 (PDB ID: 6c0j). (A) Top views of K-5a2 in the RT NNIBP. The inhibitor-binding pocket is in surface presentation and key residues are depicted as sticks. (B) Hydrogen bonds between K-5a2 and the main chains of NNIBP residues.
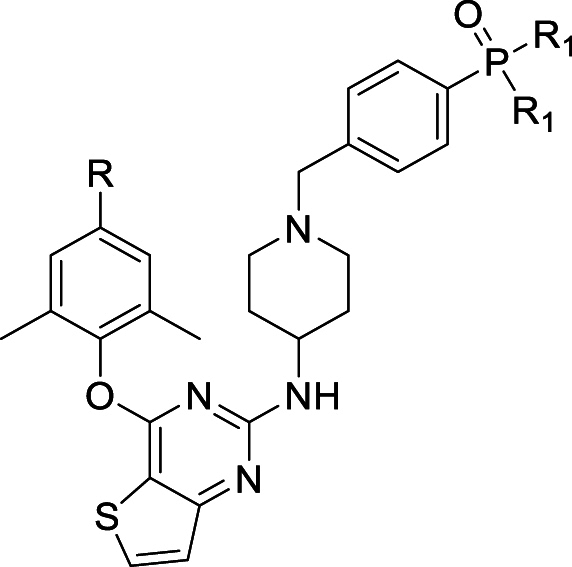
In this work, the privileged left wing and thiophene[3,2-d]pyrimidine central core of K-5a2 and 25a was kept, the potentially toxic sulfonamide group was replaced by dimethylphosphine oxide, substituted phosphonate ester and phosphoric acid utilizing the bioisosterism strategy. These three classes of privileged moieties are considered to be able to improve solubility and decrease toxicity of compounds, which were widely used in the design of anti-tumor and antiviral drugs21, 22, 23. Moreover, they can develop novel hydrogen bonds with NNIBP as hydrogen bond donors or acceptors. As shown in Fig. 3, the newly designed compounds 9a, 9b, and 10 could not only keep the interactions like K-5a2 in NNIBP but also bring hydrogen bonds formed by the newly introduced moieties with the backbone of Val106. Through the synthesis, activity evaluation, and mechanism confirmation, 15a was identified as the optimal compound, and the pharmacokinetics and safety profiles of its hydrochloride form 15a·HCl was further evaluated.
Figure 3.

The newly designed representative compounds 9a (A), 9b (B) and 10 (C), and their predicted binding modes with HIV-1 WT RT (PDB: 6c0j).
2. Results and discussion
2.1. Chemistry
Scheme 1, Scheme 2 showed the synthetic routes of target compounds 9a‒h, 10, 15a‒h and 16. Firstly, the starting material 2,4-dichlorothieno[3,2-d]pyrimidine (5) was reacted with 4-hydroxy-3,5-dimethylbenzonitrile afforded intermediate 6, which generated compound 7 via a nucleophilic reaction with tert-butyl 4-aminopiperidine-1-carboxylate and subsequent removal of t-butyloxy carbonyl (Boc) under the acidic conditions provided by trifluoroacetic acid (TFA). Nucleophilic addition of 7 with 1-(bromomethyl)-4-iodobenzene afforded intermediate 8, which was treated with dimethylphosphine oxide and substituted phosphite ester via Michaelis–Arbuzov reaction yielded compounds 9a‒h. Among them, compound 9c was finally treated with Me3SiBr and acetonitrile to give compound 10. In an analogous way, target compounds 15a‒h and 16 were synthesized, only with the difference that the starting material 5 was treated with 4-hydroxy-3,5-dimethylbenzaldehyde, and then through witting reaction to yield intermediate 12. And compound 15c was also reacted with Me3SiBr to give the target compound 16. In Scheme 3, the 15a·HCl is obtained by treating 15a with hydrogen chloride ethanol solution.
Scheme 1.

Synthesis of the target compounds 9a‒h and 10. Reagents and conditions: (i) K2CO3, DMF, r.t., 92% yield; (ii) K2CO3, DMF, 120 °C; TFA, DCM, r.t., 67% yield; (iii) K2CO3, DMF, r.t., 76% yield; (iv) Pd2(dba)3, Xantphos, Cs2CO3, 1,4-dioxane, 90 °C, 58%–69% yield; (v) Me3SiBr, acetonitrile, 0–80 °C, 67% yield.
Scheme 2.

Synthesis of the target compounds 15a‒h and 16. Reagents and conditions: (i) K2CO3, DMF, r.t., 90% yield; (ii) (EtO)2P(O)CH2CN, t-BuOK, THF, 0 °C to rt, 84% yield; (iii) K2CO3, DMF, 120 °C; TFA, DCM, r.t., 75% yield; (iv) K2CO3, DMF, r.t., 88% yield; (v) Pd2(dba)3, Xantphos, Cs2CO3, 1,4-dioxane, 90 °C, 55%–68% yield; (vi) Me3SiBr, acetonitrile, 0–80 °C, 63% yield.
Scheme 3.

Synthesis of compound 15a·HCl. Reagents and conditions: (i) Hydrogen chloride ethanol solution, ethyl acetate, DCM, 0 °C, 96% yield.
2.2. Biological activities
All the target compounds were screened for their antiviral activity against WT HIV-1 (IIIB) and a panel of NNRTIs-resistant strains, including L100I, K103N, Y181C, Y188L, E138K, F227L/V106A and RES056 (K103N/Y181C) in MT-4 cells by the MTT method24. ETR, RPV and DOR were acted as positive drugs. The values of EC50 (anti-HIV-1 potency), CC50 (cytotoxicity), and SI (selectivity index, CC50/EC50 ratio) values were determined and shown in Table 1, Table 2.
Table 1.
Anti-HIV-1 (IIIB and K103N/Y181C) activity, cytotoxicity and SI values of 9a‒h, 10, 15a‒h and 16.
| Compd. | R | R1 | EC50 (nmol/L)a |
CC50 (μmol/L)b | SIc |
||
|---|---|---|---|---|---|---|---|
| IIIB | RES056 | IIIB | RES056 | ||||
| 9a | 2.20 ± 0.33 | 257 ± 95.2 | 215 ± 1.17 | 97,727 | 837 | ||
| 9b | CN | OCH3 | 3.63 ± 1.15 | 193 ± 40.4 | 54.5 ± 1.29 | 15,014 | 282 |
| 9c | CN | OCH2CH3 | 6.90 ± 1.92 | 445 ± 109 | 117 ± 7.75 | 16,957 | 263 |
| 9d | CN | O(CH2)3CH3 | 125 ± 39.0 | 6504 ± 483 | 7.80 ± 1.53 | 62 | 1 |
| 9e | CN | OCH2CF3 | 33.0 ± 7.63 | 1295 ± 120 | 17.4 ± 2.85 | 527 | 13 |
| 9f | CN | OCH(CH3)2 | 24.8 ± 14.8 | 514 ± 57.1 | 111 ± 33.6 | 4476 | 216 |
| 9g | CN | OC(CH3)3 | 99.3 ± 37.0 | >16,264 | 16.3 ± 4.28 | 164 | <1 |
| 9h | CN | OCH2CH(CH3)2 | 90.9 ± 50.8 | 13,201 ± 10,152 | 8.78 ± 9.12 | 97 | 1 |
| 10 | CN | OH | 16,495 ± 4081 | >227,442 | >227 | >14 | <1 |
| 15a | CV | CH3 | 1.75 ± 0.64 | 15.8 ± 2.62 | 117 ± 23.9 | 66,857 | 7405 |
| 15b | CV | OCH3 | 19.1 ± 13.9 | 603 ± 139 | 34.9 ± 2.86 | 1827 | 58 |
| 15c | CV | OCH2CH3 | 26.4 ± 15.1 | 110 ± 27.2 | 8.15 ± 0.64 | 309 | 74 |
| 15d | CV | O(CH2)3CH3 | 173 ± 54.0 | 1427 ± 173 | 12.2 ± 3.91 | 71 | 9 |
| 15e | CV | OCH2CF3 | 138 ± 55.8 | 747 ± 242 | 15.6 ± 2.67 | 113 | 21 |
| 15f | CV | OCH(CH3)2 | 29.2 ± 9.12 | 401 ± 321 | 4.45 ± 0.57 | 152 | 11 |
| 15g | CV | OC(CH3)3 | 20.6 ± 8.31 | 197 ± 30.5 | 7.40 ± 2.25 | 359 | 38 |
| 15h | CV | OCH2CH(CH3)2 | 151 ± 48.4 | 2198 ± 360 | 16.5 ± 2.05 | 109 | 8 |
| 16 | CV | OH | 3809 ± 1524 | 169,839 ± 16,344 | >217 | >57 | >1 |
| ETR | – | – | 2.81 ± 1.08 | 83.3 ± 21.1 | >4.59 | >1633 | >55 |
| RPVd | – | – | 1.00 ± 0.27 | 10.7 ± 7.96 | 3.98 | 3989 | 371 |
| DORd | – | – | 8.46 ± 3.64 | 128 ± 60.6 | >294 | >36,750 | >2296 |
EC50: concentration of compound required to achieve 50% protection of MT-4 cell cultures against HIV-1-induced cytotoxicity.
CC50: concentration required to reduce the viability of mock-infected cell cultures by 50%.
SI: the ratio of CC50/EC50.
The data were obtained from the same laboratory with the same method.
Table 2.
Activity against mutant HIV-1 strains of selected 9a‒h, 10, 15a‒h and 16.
| Compd. | EC50 (nmol/L)a |
|||||
|---|---|---|---|---|---|---|
| L100I | K103N | Y181C | Y188L | E138K | F227L/V106A | |
| 9a | 7.01 ± 1.76 | 2.66 ± 0.85 | 12.8 ± 2.62 | 11.3 ± 0.87 | 7.33 ± 1.36 | 21.5 ± 13.5 |
| 9b | 10.8 ± 3.42 | 3.72 ± 1.27 | 15.3 ± 10.1 | 9.95 ± 5.61 | 14.9 ± 8.60 | 8.14 ± 1.85 |
| 9c | 11.5 ± 3.27 | ≤4.62 | 13.2 ± 5.60 | 17.0 ± 11.0 | 12.3 ± 1.98 | 14.3 ± 4.55 |
| 9d | 230 ± 54.0 | 196 ± 13.6 | 311 ± 50.4 | 240 ± 58.3 | 241 ± 34.9 | 248 ± 37.3 |
| 9e | 165 ± 111 | 45.8 ± 4.61 | 248 ± 60.4 | 142 ± 63.3 | 137 ± 83.0 | 65.1 ± 19.5 |
| 9f | 29.5 ± 14.2 | 22.6 ± 7.83 | 73.5 ± 69.4 | 42.0 ± 11.7 | 41.3 ± 15.3 | 28.4 ± 13.4 |
| 9g | 310 ± 57.8 | 124 ± 26.2 | 719 ± 263 | 445 ± 157 | 179 ± 19.8 | 637 ± 492 |
| 9h | 135 ± 28.4 | 53.3 ± 19.0 | 182 ± 40.5 | 174 ± 94.9 | 124 ± 31.4 | 104 ± 53.1 |
| 10 | >227,442 | 146,756 ± 61,155 | >227,442 | >227,442 | 160,076 ± 33,319 | >227,442 |
| 15a | 2.84 ± 0.09 | 1.27 ± 0.68 | 5.38 ± 1.61 | 7.96 ± 3.89 | 4.28 ± 1.04 | 3.76 ± 0.46 |
| 15b | 85.7 ± 28.0 | 13.0 ± 3.80 | 193 ± 74.8 | 478 ± 158 | 64.5 ± 31.7 | 129 ± 83.3 |
| 15c | 20.1 ± 10.8 | 15.5 ± 1.21 | 44.1 ± 25.6 | 62.4 ± 22.7 | 56.6 ± 19.1 | 27.1 ± 10.7 |
| 15d | 265 ± 84.4 | 188 ± 17.0 | 779 ± 316 | 714 ± 157 | 678 ± 196 | 495 ± 168 |
| 15e | 140 ± 50.5 | 139 ± 65.7 | 584 ± 272 | 497 ± 49.2 | 423 ± 149 | 338 ± 37.1 |
| 15f | 56.1 ± 13.2 | 41.0 ± 7.63 | 156 ± 59.0 | 195 ± 32.3 | 162 ± 35.8 | 79.3 ± 13.0 |
| 15g | 25.8 ± 15.8 | 16.9 ± 7.68 | 46.4 ± 7.56 | 55.3 ± 22.2 | 59.7 ± 13.1 | 43.7 ± 7.52 |
| 15h | 712 ± 158 | 239 ± 27.0 | 1121 ± 106 | 860 ± 218 | 758 ± 249 | 886 ± 167 |
| 16 | 13,571 ± 7688 | 6269 ± 1904 | 30,390 ± 5042 | 133,369 ± 55,673 | 19,148 ± 5993 | 43,539 ± 6040 |
| ETR | 8.39 ± 3.40 | 3.04 ± 0.90 | 17.8 ± 5.74 | 22.1 ± 6.33 | 10.5 ± 2.49 | 18.2 ± 12.4 |
| RPVb | 1.51 ± 0.00 | 1.31 ± 0.36 | 4.73 ± 0.48 | 79.4 ± 0.77 | 5.75 ± 0.11 | 81.6 ± 21.2 |
| DORb | 13.3 ± 99.84 | 13.9 ± 4.83 | 26.8 ± 7.10 | 2085 ± 1697 | 37.4 ± 15.2 | 67,850 ± 25,160 |
EC50: concentration of compound required to achieve 50% protection of MT-4 cell cultures against HIV-1-induced cytotoxicity.
The data were obtained from the same laboratory with the same method.
As the results showed, the target compounds 9a‒h and 15a‒h exhibited moderate to excellent potency against HIV-1 IIIB with EC50 values ranging from 1.75 nmol/L to 173 nmol/L. Among them, 9a (EC50 = 2.20 nmol/L) and 15a (EC50 = 1.75 nmol/L) were proved to be the two most potent inhibitors, being comparable to that of ETR (EC50 = 2.81 nmol/L) and RPV (EC50 = 1.00 nmol/L), and being superior to that of DOR (EC50 = 8.46 nmol/L). Moreover, 9a (CC50 = 215 μmol/L, SI = 97,727) and 15a (CC50 = 117 μmol/L, SI = 66,857) also exhibited much lower cytotoxicity compared to that of RPV (CC50 = 3.98 μmol/L, SI = 3989), which contribute to its higher SI value. However, the activity of compounds 10 (EC50 = 16,495 nmol/L) and 16 (EC50 = 3809 nmol/L) with the phosphate group sharply decreased, which may be due to their polarity leading to poor cellular penetration and the sharply reduced activity. In addition, most compounds showed reduced potency against double mutant strain RES056. But, 15a exhibited excellent activity against RES056 with EC50 values of 15.8 nmol/L, being superior to that of ETR (EC50 RES056 = 83.3 nmol/L) and DOR (EC50 RES056 = 128 nmol/L), and equipotent to that of RPV (EC50 RES056 = 10.7 nmol/L).
The structure‒activity relationships (SARs) suggest that the substituents on the phenyl ring of right wing were essential for the anti-HIV-1 activity. The dimethylphosphine oxide group was more favorable for enhancement of activity than the substituted phosphonate ester group, and the activity of all of them is much superior to that of compounds with phosphoric acid group (9a vs 9b‒9h vs 10, and 15a vs 15b‒15h vs 16). Significantly, compounds 10 and 16 with the phosphoric acid substituent showed greatly reduced activity, which may be a representative example of the activity cliff concept. In addition, as the volume of the substituents increases, the anti-HIV-1 activity gradually decreases (9b vs 9c vs 9d vs 9e vs 9f vs 9g vs 9h and 15b vs 15c vs 15d vs 15e vs 15f vs 15g vs 15h). There is a benefit-risk relationship between the improved interactions with the tolerant region I of RT and the increased volume of compounds. Although an increase in compound volume may increase the interactions with RT, it also reduces the flexibility of the compound, leading to a sharp decrease in activity. Obviously, dimethylphosphine oxide substituent is the optimal solution for the tolerant region I. To sum up, there is an exploratory chemical space at the solvent-exposed surface to accommodate diverse groups, but there are certain requirements for the volume and polarity of the groups. The activity of dimethylphosphine oxide substituent is the best, but the introduction of groups with large volumes or high polarity can lead to a decrease in activity.
Furthermore, we further tested for the potency of the target compounds against HIV-1 single mutant strains L100I, K103N, Y181C, Y188L, E138K, and double mutant strain F227L/V106A. As shown in Table 2, the most promising RES056 inhibitor 15a also turned out to exhibit the most effective potency against other tested mutant strains, with EC50 values of 2.84 nmol/L (L100I), 1.27 nmol/L (K103N), 5.38 nmol/L (Y181C), 7.96 nmol/L (Y188L), 4.28 nmol/L (E138K) and 3.76 nmol/L (F227L/V106A), respectively. They were about 2.95-, 2.39-, 3.31-, 2.78-, 2.45- and 4.84-fold higher than that of ETR and 4.7-, 10.9-, 4.98-, 262-, 8.74- and 18,045 -fold higher than that of DOR, respectively. And they were equipotent or superior to that of RPV, especially for mutant strains Y188L and F227L/V106A, the activity of 15a was about 9.97- and 21.7-fold higher than that of RPV. Besides, 9a, 9b, and 9c also showed promising antiviral activity against tested mutant HIV-1 strains with EC50 values range 2.66–21.5 nmol/L, being equipotent or superior to that of ETR and RPV. These results contribute to the SARs that the substituents on the phenyl ring of the right wing sitting at the solvent-exposed surface made a marked impact on the activity, and the dimethylphosphine oxide group was proved to be the best choice for the right wing.
2.2.1. HIV-1 RT inhibitory activities
To verify the binding target and mechanism of these novel derivatives, the most potent compounds 9a‒c and 15a were selected to evaluate their inhibitory ability to recombinant WT HIV-1 RT enzyme25. As depicted in Table 3, 9a‒c and 15a exhibited modest inhibitory activity (IC50 = 0.31–0.76 μmol/L) to WT HIV-1 RT, being equipotent to that of NVP (IC50 = 0.43 μmol/L), but inferior to that of EFV (IC50 = 0.01 μmol/L), ETR (IC50 = 0.01 μmol/L) and RPV (IC50 = 0.02 μmol/L). However, the RT inhibitory activity is usually inconsistent with its activity in HIV cell culture. These differences may be attributed to the template-specific variations of polymerase progression and HIV1-RT-RNA binding affinity. Anyway, the HIV-1 RT inhibitory activity results could validate that the binding target of these target compounds is HIV-1 RT.
Table 3.
Inhibitory activity against WT HIV-1 RT of 9a‒c and 15a.
| Compd. | IC50 (μmol/L)a | Compd. | IC50 (μmol/L)a |
|---|---|---|---|
| 9a | 0.31 ± 0.01 | 9b | 0.35 ± 0.05 |
| 9c | 0.76 ± 0.15 | 15a | 0.34 ± 0.09 |
| NVP | 0.43 ± 0.16 | EFV | 0.01 ± 0.00 |
| ETR | 0.01 ± 0.00 | RPVb | 0.02 ± 0.00 |
IC50: inhibitory concentration of test compound required to inhibit biotin deoxyuridine triphosphate (biotindUTP) incorporation into WT HIV-1 RT by 50%.
The data were obtained from the same laboratory with the same method.
2.2.2. Molecular docking analysis
To further explain the experimental activity results of the target compounds and completely understand the binding modes in RT, the molecular simulations for the selected compounds 9a, 9b, 9c, and 15a were worked out by the software Maestro (Maestro, Schrödinger, LLC, New York, NY, USA, 2019). Co-crystal structures of WT HIV-1 RT (PDB code: 6c0j), F227L/V106A HIV-1 RT (PDB code: 6duf), and RES056 HIV-1 RT (PDB code: 6c0r) were used as the input structures for docking calculations. Docking results were visualized with PyMOL2.6 and illustrated in Figure 4, Figure 519.
Figure 4.

(A) The predicted binding modes of 15a with WT HIV-1 RT (PDB: 6c0j), (B) F227L/V106A HIV-1 RT (PDB code: 6duf) and (C) RES056 HIV-1 RT (PDB: 6c0r). The hydrogen bonds are indicated with yellow dashed lines. Nonpolar hydrogen atoms are not shown for clarity.
Figure 5.

The predicted binding modes of 9a (A), 9b (B), and 9c (C) with RES056 HIV-1 RT (PDB: 6c0r). The hydrogen bonds are indicated with yellow dashed lines. Nonpolar hydrogen atoms are not shown for clarity.
To explain the excellent activity of 15a against highly resistant double mutant strains F227L/V106A and RES056 (K103N/Y181C), the molecular docking analysis of 15a with WT, F227L/V106A and RES056 RT were performed. The results were showed in Fig. 4, 15a exhibited the dominant U-shaped conformation in all three RT NNIBP and maintained the classical binding interactions: (i) the N atom of the pyrimidine core and the NH linking the central core and the right wing develops double hydrogen bonds with the backbone of Lys101; (ii) the left wing scaffold stretch into the hydrophobicity tunnel lined by Tyr181, Tyr188, Phe227 and Trp229, forming π‒π interactions with these residues; (iii) the S atom of the central core thiophene[3,2-d]pyrimidine could form water-mediated hydrogen bond with the main chains of Glu138; (iv) the right wing piperidine-linked aryl structure arches into the groove surrounded by Lys103, Lys104, Val106, Pro236 and Phe227, developing numerous van der Waals interactions with their lipophilic side chains, and guides the terminal dimethylphosphine oxide group to the solvent-exposed surface of RT, the N atom of piperidine forms hydrogen bonds with the backbones of Lys103 and Pro236 through a bridging water molecule, and the newly introduced dimethylphosphine oxide group on the phenyl ring generated newly hydrogen bond with the backbone of Val106. As for F227L/V106A RT, due to the less contribution of the interaction with Phe227 to maintaining the binding between 15a and RT, the binding force of the left wing is not affected by Phe227 to Leu227 substitution. Additionally, Val106 mutates to Ala106 contributing to the novel strong hydrogen bond between the dimethylphosphine oxide group and A106. When it comes to the RES056 RT, the Tyr181 to Cys181 substitution caused a loss of π‒π stacking interactions between the left wing and Tyr181, but it could be compensated by its enhanced hydrophobic interactions of the cyano vinyl group with Tyr188 and Trp229. However, the binding force of the right wing is not affected by the substitution of Lys103 to Asn103, with the reason that the flexibility of its right wing allows it to adjust freely to adapt to the mutation of amino acids to maintain the original hydrogen bond network.
Compounds 9a‒c maintain excellent antiviral activity against WT and other mutant strains, but their activity to double mutant strain RES056 was sharply decreased. As shown in Fig. 5, the compensation effect of the cyano vinyl group cannot be observed between the cyano group with Tyr 188 and Trp229, which greatly damaged the affinity between the compounds and RES056 RT. Additionally, the Lys103 to Asn103 substitution led to an increase in distance between the right wing of inhibitors and surrounding amino acids, the changes in the position of the right wing of compounds, and the disappearance of hydrogen bonding between the O atom and Val106 in WT RT. All these factors account for their reduced potency against RES056.
2.2.3. Molecular dynamics simulation
To further investigate the dynamic behavior and stability of 15a binding to HIV-1 WT and mutant RTs, 500 ns extensive molecular dynamics (MD) simulations were performed. The co-crystal structure of K-5a2 in complexes with HIV-1 RT (PDB code: 6c0j) was selected as the template for molecular dynamics simulation. The structures of the mutated strains were generated from this PDB-6c0j by site-specific mutation and minimized by Schrödinger Suites.
As shown in Fig. 6, the result of root-mean-square deviation (RMSD) illustrated that the binding of ligand‒RT complexes was stable throughout the 500 ns simulation. Moreover, the coordinates of 15a fluctuated less than 1 Å, which further verified that 15a existed in a few conformational forms in the HIV-1 WT RT and various mutant RTs and was truly stabilized in the allosteric site of HIV-1 RT.
Figure 6.

Time (ns) evolution of the rmsd (Å) profile determined for the complexes between 15a with HIV-1 WT and various mutant RTs.
2.2.4. Aqueous solubility of 15a and its salt form 15a·HCl
The introduction of multiple lipophilic aromatic rings to the DAPY NNRTIs resulted in their poor aqueous solubility (the solubility of ETR <1 μg/mL in pH 7.0) and lower bioavailability. Salification is confirmed as an effective way to enhance the solubility26. Therefore, the hydrochloride form of 15a with the most outstanding antiretroviral potency was prepared and the aqueous solubility of 15a and 15a·HCl were tested (Supporting Information Section 1). As shown in Table 4, the solubility of free 15a (S = 283 μg/mL, S = 36.4 μg/mL, and S = 27.6 μg/mL, respectively) at pH 2.0, 7.0 and 7.4 were moderately higher than that of ETR (S = 127 μg/mL, S ≪ 1 μg/mL, S < 1 μg/mL, respectively). As designed, 15a·HCl exhibited a significantly improved solubility at different pH values (S = 5965 μg/mL, S = 71.5 μg/mL, and S = 57.6 μg/mL, respectively) than that of 15a and ETR, which was selected for further in vivo pharmacokinetic and safety evaluation.
Table 4.
The aqueous solubility of compounds 15a and 15a·HCl.
| Compd. | Aqueous solubility (μg/mL) |
||
|---|---|---|---|
| pH = 2.0 | pH = 7.0 | pH = 7.4 | |
| 15a | 283 | 36.4 | 27.6 |
| 15a·HCl | 5965 | 71.5 | 57.9 |
| ETRa | 127 | <<1 | <1 |
Data were obtained from Ref. 27.
2.2.5. In vivo pharmacokinetics study of 15a·HCl
The authors declare that all experimental work complied with the institutional guidelines on animal studies (care and use of laboratory animals). The in vivo pharmacokinetic study of 15a·HCl was carried out by the Sprague Dawley (SD) rats PK models as previously reported (Supporting Information Section 2) 28. The PK parameters are provided in Table 5 and Fig. 7. The maximum concentration (Cmax) and AUC0‒t of 15a·HCl were 1785 ng/mL and 635 h·ng/mL, respectively, at an intravenous injection dose of 2 mg/kg. Additionally, 15a·HCl was characterized by lower clearance (CL = 52.1 mL/min/kg) and modest half-life (t1/2 = 0.60 h). After an oral dose of 10 mg/kg, 15a·HCl reached the maximum concentration (Cmax = 333 ng/mL) at 2.00 h, and the half-life of it was 1.32 h. Notably, 15a·HCl was demonstrated with an excellent oral bioavailability (F) of 40.8% and had great potential for development as an oral preparation.
Table 5.
Pharmacokinetic profiles of 15a·HCla.
| Subject | t1/2 (h) | Tmax (h) | Cmax (ng/mL) | AUC0‒t (h·ng/mL) | AUC0‒∞ (h·ng/mL) | CL (mL/min/kg) | F (%) |
|---|---|---|---|---|---|---|---|
| 15a·HCl (iv)b | 0.60 ± 0.03 | 0 ± 0.00 | 1785 ± 50.9 | 635 ± 9.85 | 640 ± 10.3 | 52.1 ± 0.82 | – |
| 15a·HCl (po)c | 1.32 ± 0.13 | 2.00 ± 0.00 | 333 ± 58.3 | 1270 ± 307 | 1303 ± 322 | – | 40.8 |
PK parameters (mean ± SD, n = 3).
Dosed intravenously at 2 mg/kg.
Dosed orally at 10 mg/kg.
Figure 7.

The plasma concentration–time profiles of 15a·HCl in rats following oral administration (10 mg/kg) and intravenous administration (2 mg/kg).
2.2.6. Safety assessment
Assessment of acute toxicity. The acute toxicity experiment of 15a·HCl was conducted on healthy Kunming mice (Supporting Information Section 3). 20 Kunming mice were divided into two groups; one group was given 15a·HCl with a dosage of 2000 mg/kg by oral administration, and the other was selected as a control, with given normal saline orally. Compared with the control group, the experimental mice did not exhibit any abnormal behaviors and showed no significant changes in body weight during the following 7 days (Fig. 8), which evidenced that 15a·HCl has no acute toxicity at a dosage of 2000 mg/kg in Kunming mice.
Figure 8.

Relative body weight changes of mice in two groups.
Assessment of subacute toxicity. To further evaluate the in vivo safety of 15a·HCl, we conducted the subacute toxicity experiment of it (Supporting Information Section 4). No apparent sign associated with animal toxicity and no behavioral abnormalities (lethargy, clonic convulsion, anorexia, or ruffled fur) of the treated animals were observed during the treatment period of orally administered mice treated with 50 mg/kg/2 days of 15a·HCl in 2 weeks. Additionally, the organ toxicity experiment was examined by hematoxylin-eosin (HE) staining. The results were depicted in Fig. 9, tissue section analysis displayed no apparent physiological abnormalities or lesions observed in the heart, liver, spleen, lungs, and kidneys, indicating the higher safety profiles of 15a·HCl.
Figure 9.

The results of subacute toxicity. HE stained images of major organs (heart, liver, spleen, lung, and kidney) collected from various groups (blank and 15a·HCl). The scale bar was 100 μmol/L.
3. Conclusions
In summary, we discovered a series of new thiophene[3,2-d]pyrimidine NNRTIs using K-5a2 and 25a as leads by replacing the sulfamide group with dimethylphosphine oxide, substituted phosphonate ester and phosphoric acid, to avoid the potentially toxic and get a clearer understanding of the SARs of the NNIBP tolerant region I. The anti-HIV-1 activity results indicated that most compounds exhibited outstanding antiretroviral activity against WT and a panel of mutant HIV-1 strains. Among them, 15a yielded the most potent activity (EC50 = 1.27–15.8 nmol/L), being superior to that of the marketed drugs ETR and RPV. In addition, it has significantly lower cytotoxicity (CC50 = 117 μmol/L) and higher SI values. SAR studies revealed that the introduction of groups with large volumes or high polarity to the solvent-exposed surface of NNIBP can lead to a decrease in activity. Molecular docking studies have been performed to show the binding modes of 15a, explaining why they are resilient to the highly resistant double mutant strains F227L/V106A and K103N/Y181C. Furthermore, 15a·HCl exhibited favorable pharmacokinetic profiles in SD rats (t1/2 = 1.32 h, F = 40.8%) and low-level toxicity in Kunming mice (LD50 > 2000 mg/kg). Therefore, 15a·HCl was considered to have promising potential for further development due to the effective potency and favorable profiles in vitro and in vivo.
4. Experimental
4.1. Chemistry
All melting points were determined on a micro melting point apparatus (RY-1G, Tianjin TianGuang Optical Instruments). 1H NMR and 13C NMR spectra were recorded on a Bruker AV-400 spectrometer using solvents as indicated (DMSO-d6). Chemical shifts were reported in δ values (ppm) with TMS as the internal reference, and J values were reported in hertz (Hz); signals are abbreviated as s (singlet), d (doublet), t (triplet), and m (multiplet). Mass spectra were recorded on an LC Auto sampler device Standard G1313A instrument. TLC was performed on Silica Gel GF254 for TLC (Merck) and spots were visualized by irradiation with UV light (λ = 254 nm and 356 nm). Flash column chromatography was performed on columns packed with Silica Gel (200–300 mesh, Qingdao Haiyang Chemical Company). Solvents were of reagent grade and were purified and dried by standard methods when necessary. The purity of target compounds >95%, which was determined by analytical HPLC with HPLC conditions: methanol/water 80:20; flow rate 1.0 mL/min; UV detection from 210 to 400 nm; temperature, ambient; injection volume, 20 μL (Shimadzu SPD-20A/20AV (Supporting Information Section 5).
4-((2-Chlorothieno[3,2-d]pyrimidin-4-yl)oxy)-3,5-dimethylbenzonitrile (6). 2,4-Dichlorothieno[3,2-d]pyrimidine (5, 2.05 g, 10 mmol) was added to the mixed solution of 4-hydroxy-3,5-dimethylbenzonitrile (1.62 g, 11 mmol) and K2CO3 (1.70 g, 12 mmol) in 30 mL of N,N-dimethylformamide (DMF), and the mixture was stirred at room temperature for 5 h (monitored by TLC). Then pour it into ice water (50 mL) and left to stand for another 20 min. The obtained precipitated was filtrated and washed with cold water and recrystallized from DMF-H2O to provide intermediate 6 as a white solid in 92% yield. ESI-MS: m/z 316.2 [M+H]+. C15H10ClN3OS (315.02).
3,5-Dimethyl-4-((2-(piperidin-4-ylamino)thieno[3,2-d]pyrimidin-4-yl)oxy)benzonitrile (7). A solution of 6 (0.64 g, 2.0 mmol), N-Boc-4-aminopiperidine (0.48 g, 2.4 mmol), and anhydrous K2CO3 (0.42 g, 3.0 mmol) in DMF (10 mL) was heated at 120 °C for 8 h under magnetic stirring. Then, the mixture was cooled to room temperature and poured into ice water (40 mL). The resulting precipitate was collected by filtration and dried to give a crude product, which was recrystallized from ethyl acetate (EA)/petroleum ether (PE) to afford a white solid. Then dissolve the dry solid obtained in 10 ml dichloromethane (DCM) (4 mL) and add added trifluoroacetic acid (TFA) (0.74 mL, 10 mmol), and the mixture was stirred at room temperature for 6 h (monitored by TLC). Then, the reaction solution was alkalized to pH 9 with saturated sodium bicarbonate solution and extracted with DCM. The organic layer was washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give 7 as a white solid in 67% yield. ESI-MS: m/z 380.4 [M+H]+. C20H21N5OS (379.15).
4-((2-((1-(4-Iodobenzyl)piperidin-4-yl)amino)thieno[3,2-d]pyrimidin-4-yl)oxy)-3,5-dimethylbenzonitrile (8). Intermediate 7 (0.38 g, 1.0 mmol) was dissolved in anhydrous DMF (10 mL) in the presence of anhydrous K2CO3 (0.17 g, 1.2 mmol), followed by the addition of 1-(bromomethyl)-4-iodobenzene (0.33 g, 1.1 mmol). The reaction mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure, and then water (30 mL) was added, extracted with ethyl acetate (3 × 10 mL), and the organic phase was washed with saturated sodium chloride (10 mL), then dried over anhydrous Na2SO4 to give the corresponding crude product, which was purified by flash column chromatography and recrystallized from ethyl acetate (EA)/petroleum ether (PE) to afford the key intermediate 8 as a white solid in 76% yield. ESI-MS: m/z 596.2 [M+H]+. C27H26IN5OS (595.09).
General synthesis procedure for target compounds9a‒h. Pd2(dba)3 (0.01 g, 0.01 mmol), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (Xantphos) (0.01 g, 0.02 mmol) and Cs2CO3 (0.10 g, 0.30 mmol) were added to a mixture solution of intermediate 8 (0.12 g, 0.20 mmol) and dimethylphosphine oxide or phosphite ester (0.24 mmol) in dry 1,4-dioxane (5 mL). The mixture was stirred at 90 °C for 8–10 h (monitoring with TLC) under N2. Then, the mixture was diluted with 10 mL of water, and the aqueous layer was extracted with EA. The organic phase was dried over Na2SO4, filtered and evaporated under reduced pressure, purified by flash column chromatography, and recrystallized from EA/PE to afford the target compounds 9a‒h.
4-((2-((1-(4-(Dimethylphosphoryl)benzyl)piperidin-4-yl)amino)thieno[3,2-d]pyrimidin-4-yl)oxy)-3,5-dimethylbenzonitrile (9a). Recrystallized from EA/PE as a white solid, yield 64%, mp: 160–162 °C. 1H NMR (400 MHz, DMSO-d6) δ = 8.20 (d, J = 5.4, 1H, C6-thienopyrimidine-H), 7.72 (s, 2H, C2, C6-Ph′′-H), 7.69 (d, J = 8.1, 2H, C3, C5-Ph′-H), 7.46 (d, J = 5.4, 1H, C7-thienopyrimidine-H), 7.42 (d, J = 8.0, 2H, C2, C6-Ph′-H), 7.25 (s, 1H, NH), 3.47 (s, 2H, N–CH2), 2.84–2.67 (m, 4H, C2, C6-piperidine-H), 2.11 (s, 6H, 2 × CH3), 2.08 (s, 1H, C4-piperidine-H), 1.64 (s, 3H, P–CH3), 1.61 (s, 3H, P–CH3), 1.39–1.32 (m, 4H, C3, C5-piperidine-H). 13C NMR (100 MHz, DMSO-d6) δ 162.53, 160.54, 153.45, 133.24, 132.99, 131.83, 131.80, 131.71, 123.64, 119.00, 109.09, 99.99, 71.77, 71.71, 29.15, 29.09, 18.98, 16.24. HRMS: m/z C29H32N5O2PS: Calcd. 545.2014, Found 546.2120 [M+H]+, 568.1937 [M+Na]+. HPLC purity: 95.970%.
Dimethyl (4-((4-((4-(4-cyano-2,6-dimethylphenoxy)thieno[3,2-d]pyrimidin-2-yl)amino)piperidin-1-yl)methyl)phenyl)phosphonate (9b). Recrystallized from EA/PE as a white solid, yield 60%, mp: 244–246 °C. 1H NMR (400 MHz, DMSO-d6) δ = 8.13 (d, J = 5.4, 1H, C6-thienopyrimidine-H), 7.65 (s, 2H, C2, C6-Ph′′-H), 7.61 (d, J = 7.6, 2H, C3, C5-Ph′-H), 7.58 (d, J = 5.8, 1H, C7-thienopyrimidine-H), 7.40 (s, 2H, C2, C6-Ph′-H), 7.19 (s, 1H, NH), 3.59 (s, 3H, O–CH3), 3.57 (s, 3H, O–CH3), 3.43 (s, 2H, N–CH2), 3.13 (d, J = 7.8, 1H, C4-piperidine-H), 2.85–2.66 (m, 4H, C2, C6-piperidine-H), 2.04 (s, 6H, 2 × CH3), 1.32–1.16 (m, 4H, C3, C5-piperidine-H). 13C NMR (100 MHz, DMSO-d6) δ 168.05, 161.05, 156.13, 151.85, 150.64, 144.75, 144.46, 136.44, 133.36, 133.04, 131.74, 128.08, 127.96, 127.06, 119.71, 119.33, 118.86, 110.52, 103.26, 60.21, 21.22, 16.70, 14.55. HRMS: m/z C29H32N5O4PS: Calcd. 577.1913, Found 578.2099 [M+H]+. HPLC purity: 95.998%.
Diethyl (4-((4-((4-(4-cyano-2,6-dimethylphenoxy)thieno[3,2-d]pyrimidin-2-yl)amino)piperidin-1-yl)methyl)phenyl)phosphonate (9c). Recrystallized from EA/PE as a white solid, yield 65%, mp: 162–163 °C. 1H NMR (400 MHz, DMSO-d6) δ = 8.20 (d, J = 5.1, 1H, C6-thienopyrimidine-H), 7.72 (s, 2H, C2, C6-Ph′′-H), 7.68 (d, J = 8.1, 2H, C3, C5-Ph′-H), 7.65 (d, J = 5.6, 1H, C7-thienopyrimidine-H), 7.45 (dd, J = 7.8, 3.8, 2H, C2, C6-Ph′-H), 7.26 (s, 1H, NH), 4.06–3.95 (m, 4H, 2 × O–CH2), 3.49 (s, 2H, N–CH2), 2.74 (s, 2H, C2-piperidine-H), 2.11 (s, 6H, 2 × CH3), 2.06–1.83 (m, 2H, C6-piperidine-H), 1.78 (s, 2H, C3-piperidine-H), 1.44–1.39 (m, 2H, C5-piperidine-H), 1.33–1.27 (m, 1H, C4-piperidine-H), 1.23 (t, J = 7.0, 6H, 2 × CH2CH3). 13C NMR (100 MHz, DMSO-d6) δ 187.17, 144.02, 133.23, 133.00, 132.96, 132.90, 131.72, 131.66, 129.36, 129.25, 127.89, 126.64, 119.00, 109.06, 63.36, 63.32, 62.20, 62.08, 62.04, 52.76, 31.69, 29.07, 16.81, 16.78, 16.65, 16.61, 16.23. HRMS: m/z C31H36N5O4PS: Calcd. 605.2226, Found 606.2549 [M+H]+, 628.2379 [M+Na]+. HPLC purity: 96.334%.
Dibutyl (4-((4-((4-(4-cyano-2,6-dimethylphenoxy)thieno[3,2-d]pyrimidin-2-yl)amino)piperidin-1-yl)methyl)phenyl)phosphonate (9d). Recrystallized from EA/PE as a white solid, yield 62%, mp: 128–130 °C. 1H NMR (400 MHz, DMSO-d6) δ = 8.20 (d, J = 5.2, 1H, C6-thienopyrimidine-H), 7.72 (s, 2H, C2, C6-Ph′′-H), 7.68 (d, J = 8.0, 2H, C3, C5-Ph′-H), 7.65 (d, J = 5.1, 1H, C7-thienopyrimidine-H), 7.46 (dd, J = 7.9, 3.8, 2H, C2, C6-Ph′-H), 7.25 (s, 1H, NH), 3.78–3.70 (m, 4H, 2 × O–CH2), 3.49 (s, 2H, N–CH2), 2.74 (s, 2H, C2 -piperidine-H), 2.11 (s, 6H, 2 × CH3), 2.02 (s, 1H, C4-piperidine-H), 1.92–1.82 (m, 4H, 2 × O–CH2CH2), 1.77 (s, 2H, C6-piperidine-H), 1.44–1.37 (m, 4H, C3, C5-piperidine-H), 0.88 (s, 4H, 2 × CH2CH3), 0.86 (s, 6H, 2 × CH2CH3). 13C NMR (100 MHz, DMSO-d6) δ 180.00, 159.83, 153.80, 141.31, 133.25, 132.98, 131.81, 131.72, 124.97, 119.00, 109.10, 71.77, 71.71, 52.32, 29.15, 29.09, 18.97, 16.24. HRMS: m/z C35H44N5O4PS: Calcd. 661.2852, Found 662.3223 [M+H]+. HPLC purity: 96.298%.
Bis(2,2,2-trifluoroethyl) (4-((4-((4-(4-cyano-2,6-dimethylphenoxy)thieno[3,2-d]pyrimidin-2-yl)amino)piperidin-1-yl)methyl)phenyl)phosphonate (9e). Recrystallized from EA/PE as a white solid, yield 69%, mp: 147–148 °C. 1H NMR (400 MHz, DMSO-d6) δ = 8.19 (d, J = 5.4, 1H, C6-thienopyrimidine-H), 7.75 (d, J = 7.8, 2H, C3, C5-Ph′-H), 7.71 (s, 2H, C2, C6-Ph′′-H), 7.53 (d, J = 7.9, 2H, C2, C6-Ph′-H), 7.51 (d, J = 5.4, 1H, C7-thienopyrimidine-H), 7.26 (s, 1H, NH), 4.80–4.72 (m, 4H, 2 × O–CH2), 3.52 (s, 2H, N–CH2), 2.73 (s, 2H, C2-piperidine-H), 2.67 (s, 1H, C4-piperidine-H), 2.11 (s, 6H, 2 × CH3), 1.77 (s, 2H, C6-piperidine-H), 1.44 (s, 2H, C3-piperidine-H), 1.39 (s, 2H, C5-piperidine-H). 13C NMR (100 MHz, DMSO-d6) δ 165.47, 162.96, 158.36, 152.07, 150.55, 145.51, 137.35, 133.95, 132.12, 131.93, 128.77, 123.75, 119.97, 119.35, 116.85, 96.95, 49.39, 45.88, 34.27, 31.43, 22.53, 16.50, 14.43. HRMS: m/z C31H30F6N5O4PS: Calcd. 713.1660, Found 714.2020 [M+H]+. HPLC purity: 99.069%.
Diisopropyl (4-((4-((4-(4-cyano-2,6-dimethylphenoxy)thieno[3,2-d]pyrimidin-2-yl)amino)piperidin-1-yl)methyl)phenyl)phosphonate (9f). Recrystallized from EA/PE as a white solid, yield 58%, mp: 180–181 °C. 1H NMR (400 MHz, DMSO-d6) δ = 8.19 (d, J = 5.2, 1H, C6-thienopyrimidine-H), 7.71 (s, 2H, C2, C6-Ph′′-H), 7.66 (d, J = 7.9, 2H, C3, C5-Ph′-H), 7.63 (d, J = 5.3, 1H, C7-thienopyrimidine-H), 7.44 (dd, J = 8.7, 3.7, 2H, C2, C6-Ph′-H), 7.24 (s, 1H, NH), 4.54 (td, J = 12.8, 6.4, 2H, 2 × O–CH), 3.48 (s, 2H, N–CH2), 2.72–2.67 (m, 4H, C2, C6-piperidine-H), 2.11 (s, 6H, 2 × CH3), 1.99 (s, 1H, C4-piperidine-H), 1.44–1.39 (m, 4H, C3, C5-piperidine-H), 1.27 (d, J = 6.1, 6H, CH(CH3)2), 1.17 (d, J = 6.1, 6H, CH(CH3)2). 13C NMR (100 MHz, DMSO-d6) δ 162.69, 156.91, 152.41, 150.62, 146.32, 132.98, 131.95, 131.83, 128.77, 120.58, 120.04, 119.37, 118.73, 116.12, 96.77, 49.12, 45.11, 29.06, 16.55. HRMS: m/z C33H40N5O4PS: Calcd. 633.2539, Found 634.2683 [M+H]+, 656.2498 [M+Na]+. HPLC purity: 98.438%.
Di-tert-butyl (4-((4-((4-(4-cyano-2,6-dimethylphenoxy)thieno[3,2-d]pyrimidin-2-yl)amino)piperidin-1-yl)methyl)phenyl)phosphonate (9g). Recrystallized from EA/PE as a white solid, yield 64%, mp: 163–165 °C. 1H NMR (400 MHz, DMSO-d6) δ = 8.19 (d, J = 5.3, 1H, C6-thienopyrimidine-H), 7.71 (s, 2H, C2, C6-Ph′′-H), 7.66 (d, J = 8.1, 2H, C3, C5-Ph′-H), 7.64 (d, J = 8.1, 2H, C2, C6-Ph′-H), 7.47 (d, J = 5.4, 1H, C7-thienopyrimidine-H), 7.26 (s, 1H, NH), 3.50 (s, 2H, N–CH2), 2.80–2.62 (m, 4H, C2, C6-piperidine-H), 2.11 (s, 6H, 2 × CH3), 2.00–1.94 (m, 1H, C4-piperidine-H), 1.72–1.64 (m, 4H, C3, C5-piperidine-H), 1.25 (s, 18H, 2 × C(CH3)3). 13C NMR (100 MHz, DMSO-d6) δ 162.54, 160.54, 153.45, 133.24, 132.99, 131.80, 131.71, 123.64, 119.00, 109.09, 99.99, 71.77, 71.71, 52.58, 30.79, 29.15, 29.09, 23.58, 18.98, 16.24. HRMS: m/z C35H44N5O4PS: Calcd. 661.2852, Found 662.3101 [M+H]+, 684.2924 [M+Na]+. HPLC purity: 98.178%.
Diisobutyl (4-((4-((4-(4-cyano-2,6-dimethylphenoxy)thieno[3,2-d]pyrimidin-2-yl)amino)piperidin-1-yl)methyl)phenyl)phosphonate (9h). Recrystallized from EA/PE as a white solid, yield 60%, mp: 156–157 °C. 1H NMR (400 MHz, DMSO-d6) δ = 8.19 (d, J = 5.2, 1H, C6-thienopyrimidine-H), 7.71 (s, 2H, C2, C6-Ph′′-H), 7.68 (d, J = 8.0, 2H, C3, C5-Ph′-H), 7.65 (d, J = 5.8, 1H, C7-thienopyrimidine-H), 7.46 (d, J = 8.0, 2H, C2, C6-Ph′-H), 7.25 (s, 1H, NH), 3.78–3.60 (m, 4H, 2 × O–CH2), 3.49 (s, 2H, N–CH2), 2.72 (s, 2H, C2-piperidine-H), 2.11 (s, 6H, 2 × CH3), 2.08 (s, 1H, C4-piperidine-H),1.86 (tt, J = 13.3, 6.6, 2H, 2 × O–CH2CH), 1.81–1.73 (m, 2H, C6-piperidine-H), 1.66–1.61 (m, 2H, C3-piperidine-H), 1.39 (s, 2H, C5-piperidine-H), 0.87 (d, J = 6.7, 12H, 2 × CH(CH3)2). 13C NMR (100 MHz, DMSO-d6) δ 162.69, 156.77, 152.26, 150.56, 139.73, 132.00, 131.82, 129.50, 128.74, 121.31, 119.42, 118.77, 111.31, 96.88, 61.61, 50.67, 49.42, 43.53, 29.04, 16.58. HRMS: m/z C35H44N5O4PS: Calcd. 661.2852, Found 662.3036 [M+H]+, 684.2855 [M+Na]+. HPLC purity: 96.167%.
(4-((4-((4-(4-Cyano-2,6-dimethylphenoxy)thieno[3,2-d]pyrimidin-2-yl)amino)piperidin-1-yl)methyl)phenyl)phosphonic acid (10). 9c (0.12 g, 0.2 mmol) was dissolved in 5 mL of acetonitrile, and the acetonitrile solution of bromotrimethylsilane (Me3SiBr) (0.15 g, 1.0 mmol) was slowly dropped into it at 0 °C. Then, the mixture was stirred for 6 h at 80 °C (monitored by TLC). Then the solvent was cooled to room temperature and evaporated under reduced pressure, and the obtained residue was dissolved in 20 mL ethyl acetate. The organic phase was washed with saturated sodium chloride (35 mL) and then dried over anhydrous Na2SO4, filtered, and purified by flash column chromatography to give the target compounds 10. Recrystallized from EA/PE as a white solid, yield 67%, mp: 260‒261 °C. 1H NMR (400 MHz, DMSO-d6) δ = 9.60 (s, 1H, NH), 8.28 (d, J = 5.2, 1H, C6-thienopyrimidine-H), 7.78 (d, J = 7.6, 2H, C3, C5-Ph′-H), 7.74 (d, J = 7.6, 2H, C2, C6-Ph′-H), 7.62 (s, 2H, C2, C6-Ph′′-H), 7.28 (d, J = 5.3, 1H, C7-thienopyrimidine-H), 4.35 (s, 2H, 2 × P–OH), 3.35 (s, 2H, N–CH2), 3.14‒3.07 (m, 4H, C2, C6-piperidine-H), 2.12 (s, 6H, 2 × CH3), 1.98‒1.91 (m, 2H, C3-piperidine-H), 1.67 (s, 2H, C5-piperidine-H), 1.40 (s, 1H, C4-piperidine-H). 13C NMR (100 MHz, DMSO-d6) δ 180.91, 165.85, 161.41, 137.32, 133.14, 131.80, 131.67, 131.34, 131.24, 121.96, 118.95, 108.78, 51.19, 46.16, 29.08, 28.48, 18.92, 16.22. HRMS: m/z C27H28N5O4PS: Calcd. 549.1600, Found 550.1795 [M+H]+. HPLC purity: 98.56%.
4-((2-Chlorothieno[3,2-d]pyrimidin-4-yl)oxy)-3,5-dimethylbenzaldehyde (11). The synthetic method was similar to that described for 6, except that the starting material 5 (2.05 g, 10 mmol) was reacted with 4-hydroxy-3,5-dimethylbenzaldehyde (1.80 g, 12 mmol). White solid, 90% yield. ESI-MS: m/z 319.2 [M+H]+. C15H11ClN2O2S (318.02). HPLC purity: 99.564%.
(E)-3-(4-((2-Chlorothieno[3,2-d]pyrimidin-4-yl)oxy)-3,5-dimethylphenyl)acrylonitrile (12). A mixture of (EtO)2P(O)CH2CN (3.19 g, 18.0 mmol) and t-BuOK (2.20 g, 18.0 mmol) in THF (15 mL) was stirred for 0.5 h at 0 °C, and then a solution of 11 (2.86 g, 9.0 mmol) in THF (10 mL) and DCM (10 mL) was slowly added to it over 0.5 h. The mixture was stirred for another 4 h at room temperature (monitored by TLC) and then poured into ice water (50 mL). The precipitate was collected and washed with water to give intermediate 12 as a white solid with 84% yield. ESI-MS: m/z 342.2 [M+H]+. C17H12ClN3OS (341.04).
(E)-3-(3,5-Dimethyl-4-((2-(piperidin-4-ylamino)thieno[3,2-d]pyrimidin-4-yl)oxy)phenyl)acrylonitrile (13). The synthetic method was similar to that described for 7, except that N-Boc-4-aminopiperidine (0.48 g, 2.4 mmol) was reacted with 12 (0.68 g, 2.0 mmol). White solid, 75% yield. ESI-MS m/z: 406.3 [M+H]+. C22H23N5OS (405.16).
(E)-3-(4-((2-((1-(4-Iodobenzyl)piperidin-4-yl)amino)thieno[3,2-d]pyrimidin-4-yl)oxy)-3,5-dimethylphenyl)acrylonitrile (14). The synthetic method was similar to that described for 8, except that 1-(bromomethyl)-4-iodobenzene (0.33 g, 1.1 mmol) was reacted with 13 (0.41 g, 1.0 mmol l). White solid, 88% yield. ESI-MS m/z: 622.1 [M+H]+. C29H28IN5OS (621.11).
General synthesis procedure for target compounds15a‒h. The synthetic method was similar to that described for target compounds 9a‒h, except that the dimethylphosphine oxide or phosphite ester (0.24 mmol) was reacted with intermediate 14 (0.12 g, 0.20 mmol).
(E)-3-(4-((2-((1-(4-(Dimethylphosphoryl)benzyl)piperidin-4-yl)amino)thieno[3,2-d]pyrimidin-4-yl)oxy)-3,5-dimethylphenyl)acrylonitrile (15a). Recrystallized from EA/PE as a white solid, yield 67%, mp: 120‒122 °C. 1H NMR (400 MHz, DMSO-d6) δ = 8.17 (d, J = 5.3, 1H, C6-thienopyrimidine-H), 7.72 (dd, J = 11.1, 8.1, 2H, C3, C5-Ph′-H), 7.62 (d, J = 16.6, 1H, ArCH = ), 7.49 (s, 2H, C2, C6-Ph′′-H), 7.42 (d, J = 6.4, 2H, C2, C6-Ph′-H), 7.24 (d, J = 4.9, 1H, C7-thienopyrimidine-H), 6.88 (s, 1H, NH), 6.44 (d, J = 16.7, 1H, =CHCN), 3.89‒3.44 (m, 4H, C2, C6-piperidine-H), 2.74 (s, 2H, N–CH2), 2.08 (s, 6H, 2 × CH3), 1.82–1.77 (m, 1H, C4-piperidine-H), 1.65 (s, 3H, P–CH3), 1.62 (s, 3H, P–CH3), 1.51‒1.37 (m, 2H, C3-piperidine-H), 1.36‒1.24 (m, 2H, C5-piperidine-H). 13C NMR (100 MHz, DMSO-d6) δ 162.88, 160.67, 160.64, 150.47, 150.27, 142.76, 142.66, 131.90, 131.85, 130.18, 130.08, 129.25, 128.62, 119.40, 104.92, 96.86, 62.26, 52.79, 31.69, 18.57, 17.88, 16.54. HRMS: m/z C31H34N5O2PS: Calcd. 571.2171, Found 572.2488 [M+H]+. HPLC purity: 96.533%.
Dimethyl (E)-(4-((4-((4-(4-(2-cyanovinyl)-2,6-dimethylphenoxy)thieno[3,2-d]pyrimidin-2-yl)amino)piperidin-1-yl)methyl)phenyl)phosphonate (15b). Recrystallized from EA/PE as a white solid, yield 56%, mp: 142‒144 °C. 1H NMR (400 MHz, DMSO-d6) δ = 8.23 (d, J = 5.4, 1H, C6-thienopyrimidine-H), 7.69 (dd, J = 17.8, 9.7, 2H, C3, C5-Ph′-H), 7.61 (d, J = 16.9, 1H, ArCH = ), 7.49 (s, 2H, C2, C6-Ph′-H), 7.37 (d, J = 7.0, 2H, C2, C6-Ph′-H), 7.26 (d, J = 5.0, 1H, C7-thienopyrimidine-H), 6.91 (s, 1H, NH), 6.44 (d, J = 16.4, 1H, =CHCN), 3.71‒3.57 (m, 6H, 2 × O–CH3), 3.21 (d, J = 10.2, 4H, C2, C6-piperidine-H), 2.89 (s, 2H, N–CH2), 2.23‒2.09 (m, 4H, C3, C5-piperidine-H), 2.09 (s, 6H, 2 × CH3), 1.65 (s, 1H, C4-piperidine-H). 13C NMR (100 MHz, DMSO-d6) δ 150.51, 150.48, 150.46, 148.51, 147.20, 147.17, 131.90, 131.85, 130.16, 130.07, 129.24, 129.17, 129.13, 129.11, 119.38, 96.91, 96.89, 89.93, 62.27, 62.22, 61.87, 52.78, 31.70, 18.61, 17.91, 16.53. HRMS: m/z C31H34N5O4PS: Calcd. 603.2069, Found 604.2776 [M+H]+. HPLC purity: 98.421%.
Diethyl (E)-(4-((4-((4-(4-(2-cyanovinyl)-2,6-dimethylphenoxy)thieno[3,2-d]pyrimidin-2-yl)amino)piperidin-1-yl)methyl)phenyl)phosphonate (15c). Recrystallized from EA/PE as a white solid, yield 62%, mp: 189‒191 °C. 1H NMR (400 MHz, DMSO-d6) δ = 8.17 (d, J = 5.3, 1H, C6-thienopyrimidine-H), 7.68 (dd, J = 12.8, 7.8, 2H, C3, C5-Ph′-H), 7.62 (d, J = 16.5, 1H, ArCH = ), 7.49 (s, 2H, C2, C6-Ph′′-H), 7.37 (d, J = 7.4, 2H, C2, C6-Ph′-H), 7.24 (d, J = 5.4, 1H, C7-thienopyrimidine-H), 7.07 (s, 1H, NH), 6.43 (d, J = 16.7, 1H, =CHCN), 4.08–3.93 (m, 4H, 2 × O–CH2), 3.63–3.45 (m, 4H, C2, C6-piperidine-H), 2.78 (s, 2H, N–CH2), 2.08 (s, 6H, 2 × CH3), 1.81‒1.85 (m, 2H, C3-piperidine-H), 1.70‒1.53 (m, 2H, C5-piperidine-H), 1.42‒1.33 (m, 1H, C4-piperidine-H), 1.23 (t, J = 7.0, 6H, 2 × CH2CH3). 13C NMR (100 MHz, DMSO-d6) δ 162.68, 156.96, 152.41, 150.58, 146.79, 132.37, 131.94, 131.82, 128.74, 120.47, 120.13, 119.39, 118.73, 116.49, 96.79, 60.23, 59.27, 49.92, 48.10, 33.88, 29.03, 24.08, 21.23, 16.56, 14.56. HRMS: m/z C33H38N5O4PS: Calcd. 631.2382, Found 632.2730 [M+H]+. HPLC purity: 99.470%.
Dibutyl (E)-(4-((4-((4-(4-(2-cyanovinyl)-2,6-dimethylphenoxy)thieno[3,2-d]pyrimidin-2-yl)amino)piperidin-1-yl)methyl)phenyl)phosphonate (15d). Recrystallized from EA/PE as a white solid, yield 68%, mp: 136‒137 °C. 1H NMR (400 MHz, DMSO-d6) δ = 8.17 (d, J = 5.2, 1H, C6-thienopyrimidine-H), 7.67 (dd, J = 12.8, 7.8, 2H, C3, C5-Ph′-H), 7.61 (d, J = 16.2, 1H, ArCH = ), 7.48 (s, 2H, C2, C6-Ph′′-H), 7.45 (d, J = 12.0, 2H, C2, C6-Ph′-H), 7.32 (d, J = 5.1, 1H, C7-thienopyrimidine-H), 7.24 (s, 1H, NH), 6.43 (d, J = 16.7, 1H, =CHCN), 3.82‒3.63 (m, 4H, 2 × O–CH2), 3.48 (s, 2H, N–CH2), 3.08–2.81 (m, 2H, C2-piperidine-H), 2.72‒2.68 (m, 2H, C6-piperidine-H), 2.11 (s, 1H, C4-piperidine-H), 2.08 (s, 6H, 2 × CH3), 2.00–1.75 (m, 4H, 2 × O–CH2CH2), 1.33–1.32 (m, 4H, C3, C5-piperidine-H), 0.87–0.91 (m, 4H, 2 × CH2CH3), 0.85–0.81 (m, 6H, 2 × CH2CH3). 13C NMR (100 MHz, DMSO-d6) δ 162.69, 156.89, 152.38, 150.62, 145.68, 133.44, 131.96, 131.84, 129.70, 128.76, 122.61, 120.66, 120.07, 118.73, 116.75, 110.76, 99.99, 96.81, 49.28, 48.54, 45.85, 45.68, 43.31, 34.27, 16.68, 16.55. HRMS: m/z C37H46N5O4PS: Calcd. 687.3008, Found 688.3199 [M+H]+. HPLC purity: 96.745%.
Bis(2,2,2-trifluoroethyl) (E)-(4-((4-((4-(4-(2-cyanovinyl)-2,6-dimethylphenoxy)thieno[3,2-d]pyrimidin-2-yl)amino)piperidin-1-yl)methyl)phenyl)phosphonate (15e). Recrystallized from EA/PE as a white solid, yield 64%, mp: 162‒163 °C. 1H NMR (400 MHz, DMSO-d6) δ = 8.20 (d, J = 5.2, 1H, C6-thienopyrimidine-H), 7.77 (d, J = 7.6, 2H, C3, C5-Ph′-H), 7.62 (d, J = 16.3, 1H, ArCH = ), 7.49 (s, 2H, C2, C6-Ph′′-H), 7.46 (s, 2H, C2, C6-Ph′-H), 7.19 (d, J = 5.1, 1H, C7-thienopyrimidine-H), 6.80 (s, 1H, NH), 6.44 (d, J = 16.7, 1H, =CHCN), 4.12 (dt, J = 18.1, 9.1, 4H, 2 × O–CH2CF3), 3.07 (s, 2H, N–CH2), 2.08 (s, 6H, 2 × CH3), 1.64 (s, 4H, C3, C5-piperidine-H), 1.55‒1.35 (m, 1H, C4-piperidine-H), 1.28‒1.15 (m, 4H, C2, C6-piperidine-H). 13C NMR (100 MHz, DMSO-d6) δ 162.70, 156.78, 152.28, 150.57, 139.56, 132.01, 131.82, 130.91, 129.69, 129.25, 128.73, 121.24, 119.45, 118.76, 111.21, 96.86, 61.60, 52.14, 45.94, 34.13, 16.58. HRMS: m/z C33H32F6N5O4PS: Calcd. 739.1817, Found 740.2207 [M+H]+. HPLC purity: 96.201%.
Diisopropyl (E)-(4-((4-((4-(4-(2-cyanovinyl)-2,6-dimethylphenoxy)thieno[3,2-d]pyrimidin-2-yl)amino)piperidin-1-yl)methyl)phenyl)phosphonate (15f). Recrystallized from EA/PE as a white solid, yield 55%, mp: 142‒144 °C. 1H NMR (400 MHz, DMSO-d6) δ = 8.17 (d, J = 5.2, 1H, C6-thienopyrimidine-H), 7.67 (d, J = 7.9, 2H, C3, C5-Ph′-H), 7.61 (d, J = 16.5, 1H, ArCH = ), 7.48 (s, 2H, C2, C6-Ph′′-H), 7.43 (d, J = 5.1, 1H, C7-thienopyrimidine-H), 7.35 (d, J = 8.2, 2H, C2, C6-Ph′-H), 7.24 (s, 1H, NH), 6.43 (d, J = 16.7, 1H, =CHCN), 4.64–4.46 (m, 2H, 2 × O–CH), 3.48 (s, 2H, N–CH2), 2.73 (s, 2H, C2-piperidine-H), 2.11 (s, 1H, C4-piperidine-H), 2.08 (s, 6H, 2 × CH3), 1.77‒1.70 (m, 2H, C6-piperidine-H), 1.64 (s, 2H, C3-piperidine-H), 1.49‒1.31 (m, 2H, C5-piperidine-H), 1.28 (d, J = 6.2, 6H, CH(CH3)2), 1.17 (d, J = 6.2, 6H, CH(CH3)2). 13C NMR (100 MHz, DMSO-d6) δ 162.87, 160.63, 152.47, 150.42, 136.51, 134.24, 133.98, 133.88, 131.97, 131.88, 131.84, 131.69, 131.59, 129.41, 128.67, 128.55, 123.95, 123.62, 119.34, 113.91, 96.87, 70.55, 70.50, 52.65, 34.35, 32.94, 31.47, 24.21, 24.17, 24.07, 24.03, 16.51. HRMS: m/z C35H42N5O4PS: Calcd. 659.2695, Found 660.2829 [M+H]+. HPLC purity: 99.369%.
Di-tert-butyl (E)-(4-((4-((4-(4-(2-cyanovinyl)-2,6-dimethylphenoxy)thieno[3,2-d]pyrimidin-2-yl)amino)piperidin-1-yl)methyl)phenyl)phosphonate (15g). Recrystallized from EA/PE as a white solid, yield 62%, mp: 108‒110 °C. 1H NMR (400 MHz, DMSO-d6) δ = 8.17 (d, J = 5.2, 1H, C6-thienopyrimidine-H), 7.69 (d, J = 7.9, 2H, C3, C5-Ph′-H), 7.65 (d, J = 8.0, 2H, C2, C6-Ph′-H), 7.62 (d, J = 16.7, 1H, ArCH = ), 7.49 (s, 2H, C2, C6-Ph′′-H), 7.32 (d, J = 5.3, 1H, C7-thienopyrimidine-H), 6.89 (s, 1H, NH), 6.44 (d, J = 16.7, 1H, =CHCN), 3.54 (s, 2H, N–CH2), 2.83‒2.86 (m, 2H, C2-piperidine-H), 2.71‒2.77 (m, 2H, C6-piperidine-H), 2.08 (s, 6H, 2 × CH3), 1.96‒1.80 (m, 1H, C4-piperidine-H), 1.78‒1.57 (m, 4H, C3, C5-piperidine-H), 1.23 (s, 18H, 2 × C(CH3)3). 13C NMR (100 MHz, DMSO-d6) δ 162.89, 160.67, 160.65, 156.97, 156.59, 150.45, 136.47, 131.90, 131.86, 131.80, 131.78, 131.71, 131.68, 129.46, 128.61, 124.00, 123.66, 119.35, 96.88, 62.12, 62.06, 52.70, 52.62, 52.60, 31.48, 16.67, 16.61, 16.51. HRMS: m/z C37H46N5O4PS: Calcd. 687.3008, Found 688.3064 [M+H]+. HPLC purity: 99.582%.
Diisobutyl (E)-(4-((4-((4-(4-(2-cyanovinyl)-2,6-dimethylphenoxy)thieno[3,2-d]pyrimidin-2-yl)amino)piperidin-1-yl)methyl)phenyl)phosphonate (15h). Recrystallized from EA/PE as a white solid, yield 66%, mp: 157‒158 °C. 1H NMR (400 MHz, DMSO-d6) δ = 8.17 (d, J = 5.1, 1H, C6-thienopyrimidine-H), 7.69 (d, J = 7.9, 1H, C3-Ph′-H), 7.66 (d, J = 7.8, 1H, C5-Ph′-H), 7.61 (d, J = 16.1, 1H, ArCH = ), 7.48 (s, 2H, C2, C6-Ph′′-H), 7.47 (s, 2H, C2, C6-Ph′-H), 7.32 (d, J = 5.1, 1H, C7-thienopyrimidine-H), 7.24 (s, 1H, NH), 6.43 (d, J = 16.7, 1H, =CHCN), 3.77–3.65 (m, 4H, 2 × O–CH2), 3.51 (s, 2H, N–CH2), 2.76 (s, 1H, C4-piperidine-H), 2.08 (s, 6H, 2 × CH3), 1.91‒1.86 (m, 2H, 2 × CH(CH3)2), 1.85‒1.77 (m, 2H, C3-piperidine-H), 1.28 (d, J = 18.9, 2H, C5-piperidine-H), 0.87 (d, J = 6.6, 12H, 2 × CH(CH3)2), 0.85‒0.80 (m, 4H, C2, C6-piperidine-H). 13C NMR (100 MHz, DMSO-d6) δ 160.82, 160.63, 158.33, 150.43, 131.89, 131.84, 131.77, 131.69, 131.59, 128.64, 126.92, 126.77, 119.34, 96.90, 71.75, 71.69, 71.63, 62.87, 29.15, 29.08, 29.02, 18.97, 16.52. HRMS: m/z C37H46N5O4PS: Calcd. 687.3008, Found 688.3136 [M+H]+. HPLC purity: 98.160%.
(E)-(4-((4-((4-(4-(2-Cyanovinyl)-2,6-dimethylphenoxy)thieno[3,2-d]pyrimidin-2-yl)amino)piperidin-1-yl)methyl)phenyl)phosphonic acid (16). The synthetic method was similar to that described for 10, except that the Me3SiBr (0.15 g, 1.0 mmol) was reacted with 15c (0.13 g, 0.2 mmol). Recrystallized from EA/PE as a white solid, yield 63%, mp: 104‒105 °C. 1H NMR (400 MHz, DMSO-d6) δ = 8.12 (d, J = 5.1, 1H, C6-thienopyrimidine-H), 7.72 (s, 2H, C3, C5-Ph′-H), 7.52 (d, J = 16.4, 1H, ArCH = ), 7.41 (s, 2H, C2, C6-Ph′-H), 7.29 (s, 1H, C7-thienopyrimidine-H), 7.17 (s, 2H, C2, C6-Ph′′-H), 7.05 (s, 1H, NH), 6.35 (d, J = 16.0, 1H, =CHCN), 4.17 (s, 2H, 2 × P–OH), 3.40–3.31 (m, 4H, C3, C5-piperidine-H), 3.18 (s, 2H, N–CH2), 3.03 (d, J = 20.2, 2H, C2-piperidine-H), 2.00 (s, 6H, 2 × CH3), 1.80‒1.53 (m, 2H, C6-piperidine-H), 1.17–1.09 (m, 1H, C4-piperidine-H). 13C NMR (100 MHz, DMSO-d6) δ 165.46, 162.92, 158.43, 152.03, 150.48, 146.01, 137.29, 133.24, 132.09, 131.91, 128.75, 123.76, 120.13, 119.34, 116.81, 106.44, 96.96, 56.82, 49.81, 41.30, 31.48, 23.84, 16.52. HRMS: m/z C29H30N5O4PS: Calcd. 575.1756, Found 576.1878 [M+H]+. HPLC purity: 98.692%.
General synthesis procedure for compound15a·HCl. A solution of 15a (100 mg, 0.18 mmol) in EA/DCM (2 mL/2 mL) was stirred at 0 °C and added hydrogen chloride ethanol solution (1.6 mL, 4.8 mmol). After stirring for 2 h, white solid was formed. The residue was filtered and washed with ethyl acetate to obtain the 15a·HCl in 96% yield. 1H NMR (400 MHz, DMSO-d6) δ = 8.32 (d, J = 5.3, 1H, C6-thienopyrimidine-H), 7.86 (s, 2H, C3, C5-Ph′-H), 7.75 (s, 2H, C2, C6-Ph′′-H), 7.63 (s, 1H, C2-Ph′-H), 7.58 (d, J = 16.0, 1H, ArCH=), 7.50 (s, 1H, C6-Ph′-H), 7.38 (s, 1H, C7-thienopyrimidine-H), 7.25 (s, 1H, NH), 6.45 (d, J = 16.4, 1H, =CHCN), 4.32 (s, 2H, N–CH2), 3.14 (s, 4H, C3, C5-piperidine-H), 2.10 (s, 6H, 2×CH3), 1.82 (s, 2H, C2-piperidine-H), 1.69 (s, 3H, P–CH3), 1.66 (s, 3H, P–CH3), 1.47–1.36 (m, 1H, C4-piperidine-H), 1.36–1.21 (m, 2H, C6-piperidine-H). 13C NMR (100 MHz, DMSO-d6) δ 150.51, 150.46, 148.51, 147.84, 147.20, 147.17, 146.78, 131.90, 131.85, 130.16, 130.07, 129.24, 128.62, 119.38, 96.91, 96.87, 89.93, 87.64, 62.26, 52.78, 31.70, 18.61, 17.91, 16.53. HRMS: m/z C31H34N5O2PS (15a): Calcd. 571.2171, Found 572.2244 [M+H]+. HPLC purity: 98.583%.
4.2. In vitro anti-HIV-1 assay in MT-4 cells
The target compounds were evaluated for their activity and cytotoxicity against WT HIV-1 (IIIB), single mutant strains (L100I, K103N, Y181C, Y188L, and E138K), and double mutant strains F227L/V106A and RES056 utilizing the MTT method in MT-4 cells. Firstly, the stock solutions (10 × final concentrations) of test compounds were added in 25 μL volumes to two series of triplicate wells to allow simultaneous evaluation of their effects on mock and HIV-1 infected cell. Using a Biomek 3000 robot (Beckman Instruments, Fullerton, CA, USA), the compounds were diluted to five-fold serially in flat-bottomed 96-well microtiter trays, including untreated control HIV-1 and mock-infected cells for each sample. HIV-1 strain stocked (50 μL) at 100–300 CCID50 (50% cell culture infectious dose) or culture medium was added to either the infected or mock-infected wells of the microtiter tray. Mock-infected cells were used to evaluate the effect of test compounds on uninfected cells to assess its cytotoxicity. Exponentially growing MT-4 cells were centrifuged for 5 min at 1000 rpm and the supernatant was discarded. The MT-4 cells were resuspended at 6 × 105 cells/mL, and 50 μL volumes were transferred to the microtiter tray wells. Five days after infection, the viability of mock- and HIV-infected cells was examined spectrophotometrically by the MTT method. The 50% cytotoxic concentration (CC50) was defined as the concentration of the test compound that reduced the viability of the mock-infected MT-4 cells by 50%. The concentration achieving 50% protection from the cytopathic effect of the virus in infected cells was defined as the 50% effective concentration (EC50).
4.3. HIV-1 RT inhibition assays
The HIV-1 reverse transcriptase (RT) assay kit (Roche) was used to perform the RT inhibition assay. All the reagents for performing the RT reaction were from the kit and the particular ELISA procedures were carried out following the description in the kit protocol. Firstly, the reaction mixture containing the template/primer complex, viral nucleotides (dNTPs), and RT in the incubation buffer with or without inhibitors was incubated for 1 h at 37 °C. After that, the reaction mixture was transferred to a streptavidincoated microtiter plate and incubated for another 1 h at 37 °C to ensure that the retranscriptional cDNA chain that contained biotin-labeled dNTPs was bound to streptavidin. Then un-bound dNTPs were removed using washing buffer and anti-DIG-POD working solution was added. After incubation for another 1 h at 37 °C, the DIG-labeled dNTPs incorporated into cDNA were bound to the anti-DIG-POD antibody. The unbound anti-DIG-PODs were removed and the peroxide substrate (ABST) solution was added to the MTPs. A colored reaction proceeds during the cleavage of the substrate catalyzed by POD. The absorbance of the sample was determined at OD405 nm using a microtiter plate ELISA reader. The percentage inhibitory activity of RT inhibitors was calculated using Eq. (1) as given below:
| Inhibition (%) = (OD value with RT but without inhibitors‒OD value with RT and inhibitors)/(OD value with RT and inhibitors‒OD value without RT and inhibitors) × 100 | (1) |
4.4. Molecular docking experiment
Molecular docking was performed in the Maestro (Maestro, Schrödinger, LLC, New York, 2019) workspace using modules (Protein Preparation Wizard, LigPrep, and Receptor Glide Gener) in the Schrödinger software package (release 2019). The PDB (6c0j, 6duf, and 6c0r) were downloaded from the Protein Data Bank. The protein structures (PDB: 6c0j, 6duf, and 6c0r) were prepared using the Protein Preparation Wizard by assigning bond orders, using the CCD database, adding hydrogens, creating zero-order bonds to metals, creating disulfide bonds and generating het states using Epik at pH 7.0. And redundant molecules not suitable for this study were removed during system preparation. PROPKA was used to assign H-bonds at pH 7.0. Restrained minimization was then performed using the OPLS3 force field. The docking grid centered on the cocrystallized ligand was created using the Receptor Grid Gener using default settings without any constraints to define the binding site for the subsequent docking studies. The molecules 9a, 9b, 9c, and 15a were built using the 2D Sketcher and prepared using LigPrep by generating ionization states at pH 7.0 ± 2.0 and tautomers to generate suitable 3D conformations for docking. The prepared ligands were docked using Glide in standard precision (SP) mode with flexible ligand sampling. The docked results were visualized and analyzed in Maestro. The best binding poses were chosen according to the glide docking scores and shown by the software PyMOL.
4.5. Molecular dynamics simulation
Protein preparation: The HIV-1 RT crystal structures (PDB code: 6c0j) was prepared using the Protein Preparation Wizard module of Schrödinger suites (Release 2023-2). All original hydrogens were removed and re-added. The protein bond order was reassigned with the CCD database and H2O molecules beyond 8 Å of the bound ligand were deleted. The protonation state for the protein complex was set to pH 7.0. And the missing side chains were rebuilt using prime. The hydrogen bond order was reassigned with PROPKA and restrained minimization was carried with the OPLS4 force field. The generated protein complex structures were then used for docking. Ligands preparation: The ligand structures of 15a was generated with Chemdraw (version 20.0) and the structure was then transferred to 3D molecules via Chem3D (version 20.0), which were then prepared with the LigPrep module of Schrödinger suites. The ligands were prepared with OPLS4 force field and the ionization states were generated via Epik at pH 7.0. Maximum of 32 conformations were generated per ligand and the best-scoring conformation was used for docking. Ligands Docking: The protein binding pocket was identified by the co-crystalized ligands using the Receptor Grid Generation module. 15a was then docked into the HIV-1 RT prepared structure using Glide. The docking precision was set as XP and up to 10 docking poses were generated. All parameters were set as default unless mentioned. The binding conformations of each analogue with the best docking score were selected for analysis and next step MD simulations. The complex structures of 15a with RT were mutated to the corresponding mutation strains using the protein mutate module. The structures of the mutated strains were energy minimized using the minimize module with the default setting. Molecular Dynamics simulations: 500 ns MD simulations were performed for the binding poses of 15a with WT RT and various RT mutations. The protein‒ligand complex within the explicit solvent system with the OPLS4 force field was studied using the Desmond module of Schrödinger suite 2023-2. The atomic framework had been solvated with TIP3P water model with orthorhombic intermittent limit conditions for a 10 Å buffer region. The overlapping water molecules were eliminated and Na+ or Cl‒ were added as counter ions to neutralize the entire framework of atoms. An extra 0.15 mol/L NaCl was added to the system. The simulation was performed with an ensemble (NPT) of Nose–Hoover thermostat and barostat to maintain the constant temperature of 300 K and pressure of 1 bar in the system (Maestro, Schrödinger suits 2023-2). A hybrid energy minimization algorithm with 1000 steps of steepest descent followed by conjugate gradient algorithms was used. 500 ns molecular dynamics simulations (Maestro, Schrödinger suits 2023-2) were then performed and the post-dynamic simulation was analyzed using the simulation interactions diagram module. The binding free energy was calculated using the MM/GBSA module of Maestro based on the molecular dynamics simulations.
Author contributions
Xinyong Liu, Peng Zhan, and Dongwei Kang as the supervisors conceived the project and supplied the financial support. Yanying Sun conducted the synthesis of compounds. Zhenzhen Zhou and Zhongling Shi completed the structure confirmation. Yanying Sun and Zhenzhen Zhou completed the manuscript. Erik De Clercq and Christophe Pannecouque conducted and supervised the activity assay. The molecular docking was conducted by Fabao Zhao and Yanying Sun. Yanying Sun designed the pharmacokinetics, acute toxicity, and subacute toxicity experiment. Minghui Xie and Zongji Zhuo did the data analysis.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgments
We gratefully acknowledge financial support from the National Natural Science Foundation of China (NSFC Nos. 81973181, 82273773), Shandong Provincial Natural Science Foundation (ZR2020YQ61, ZR2020JQ31, China), Qilu Young Scholars Program of Shandong University and Taishan Scholar Program at Shandong Province. The technical assistance of Mr. Kris Uyttersprot and Mrs. Kristien Erven, for the HIV experiments is gratefully acknowledged.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting information to this article can be found online at https://doi.org/10.1016/j.apsb.2024.03.021.
Contributor Information
Dongwei Kang, Email: kangdongwei@sdu.edu.cn.
Peng Zhan, Email: zhanpeng1982@sdu.edu.cn.
Xinyong Liu, Email: xinyongl@sdu.edu.cn.
Appendix A. Supporting information
The following is the Supporting information to this article:
References
- 1.HIV data and statistics. https://www.who.int/teams/global-hiv-hepatitis-and-stis-programmes/hiv/strategic-information/hiv-dataand-statistics.
- 2.Fauci A.S., Lane H.C. Four decades of HIV/AIDS―much accomplished, much to do. N Engl J Med. 2020;383:1–4. doi: 10.1056/NEJMp1916753. [DOI] [PubMed] [Google Scholar]
- 3.Li G., Wang Y., De Clercq E. Approved HIV reverse transcriptase inhibitors in the past decade. Acta Pharm Sin B. 2022;12:1567–1590. doi: 10.1016/j.apsb.2021.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meng Y., Zhong J., Lv Y., Zou W. Research progress on HIV-1 immune escape mechanisms. AIDS Rev. 2022;24:133–138. doi: 10.24875/AIDSRev.21000068. [DOI] [PubMed] [Google Scholar]
- 5.Battini L., Bollini M. Challenges and approaches in the discovery of human immunodeficiency virus type-1 non-nucleoside reverse transcriptase inhibitors. Med Res Rev. 2019;39:1235–1273. doi: 10.1002/med.21544. [DOI] [PubMed] [Google Scholar]
- 6.Bec G., Meyer B., Gerard M.A., Steger J., Fauster K., Wolff P., et al. Thermodynamics of HIV-1 reverse transcriptase in action elucidates the mechanism of action of non-nucleoside inhibitors. J Am Chem Soc. 2013;135:9743–9752. doi: 10.1021/ja4018418. [DOI] [PubMed] [Google Scholar]
- 7.Namasivayam V., Vanangamudi M., Kramer V.G., Kurup S., Zhan P., Liu X., et al. The journey of HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs) from lab to clinic. J Med Chem. 2019;62:4851–4883. doi: 10.1021/acs.jmedchem.8b00843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Du S., Hu X., Menéndez-Arias L., Zhan P., Liu X. Target-based drug design strategies to overcome resistance to antiviral agents: opportunities and challenges. Drug Resist Updat. 2024;73 doi: 10.1016/j.drup.2024.101053. [DOI] [PubMed] [Google Scholar]
- 9.Du J., Guo J., Kang D., Li Z., Wang G., Wu J., et al. New techniques and strategies in drug discovery. Chin Chem Lett. 2020;31:1695–1708. [Google Scholar]
- 10.Lehman D.A., Wamalwa D.C., McCoy C.O., Matsen F.A., Langat A., Chohan B.H., et al. Low-frequency nevirapine resistance at multiple sites may predict treatment failure in infants on nevirapine-based treatment. J Acquir Immune Defic Syndr. 2012;60:225–233. doi: 10.1097/QAI.0b013e3182515730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cilento M.E., Kirby K.A., Sarafianos S.G. Avoiding drug resistance in HIV reverse transcriptase. Chem Rev. 2021;121:3271–3296. doi: 10.1021/acs.chemrev.0c00967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beyrer C., Pozniak A. HIV drug resistance―an emerging threat to epidemic control. N Engl J Med. 2017;377:1605–1607. doi: 10.1056/NEJMp1710608. [DOI] [PubMed] [Google Scholar]
- 13.Xu H.T., Asahchop E.L., Oliveira M., Quashie P.K., Quan Y., Brenner B.G., et al. Compensation by the E138K mutation in HIV-1 reverse transcriptase for deficits in viral replication capacity and enzyme processivity associated with the M184I/V mutations. J Virol. 2011;85:11300–11308. doi: 10.1128/JVI.05584-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng M., Wang D., Grobler J.A., Hazuda D.J., Miller M.D., Lai M.T. In vitro resistance selection with doravirine (MK-1439), a novel nonnucleoside reverse transcriptase inhibitor with distinct mutation development pathways. Antimicrob Agents Chemother. 2015;59:590–598. doi: 10.1128/AAC.04201-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y., De Clercq E., Li G. Current and emerging non-nucleoside reverse transcriptase inhibitors (NNRTIs) for HIV-1 treatment. Expert Opin Drug Met. 2019;15:813–829. doi: 10.1080/17425255.2019.1673367. [DOI] [PubMed] [Google Scholar]
- 16.Wang Z., Cherukupalli S., Xie M., Wang W., Jiang X., Jia R., et al. Contemporary medicinal chemistry strategies for the discovery and development of novel HIV-1 non-nucleoside reverse transcriptase inhibitors. J Med Chem. 2022;65:3729–3757. doi: 10.1021/acs.jmedchem.1c01758. [DOI] [PubMed] [Google Scholar]
- 17.Kang D., Fang Z., Li Z., Huang B., Zhang H., Lu X., et al. Design, synthesis, and evaluation of thiophene[3,2-d]pyrimidine derivatives as HIV-1 non-nucleoside reverse transcriptase inhibitors with significantly improved drug resistance profiles. J Med Chem. 2016;59:7991–8007. doi: 10.1021/acs.jmedchem.6b00738. [DOI] [PubMed] [Google Scholar]
- 18.Kang D., Fang Z., Huang B., Lu X., Zhang H., Xu H., et al. Structure-based optimization of thiophene[3,2-d]pyrimidine derivatives as potent HIV-1 non-nucleoside reverse transcriptase inhibitors with improved potency against resistance-associated variants. J Med Chem. 2017;60:4424–4443. doi: 10.1021/acs.jmedchem.7b00332. [DOI] [PubMed] [Google Scholar]
- 19.Yang Y., Kang D., Nguyen L.A., Smithline Z.B., Pannecouque C., Zhan P., et al. Structural basis for potent and broad inhibition of HIV-1 RT by thiophene[3,2-d]pyrimidine non-nucleoside inhibitors. Elife. 2018;7 doi: 10.7554/eLife.36340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khusro A., Aarti C., Buendía-Rodriguez G., Arasu M.V., Al-Dhabi N.A., Barbabosa-Pliego A. Adverse effect of antibiotics administration on horse health: an overview. J Equine Vet Sci. 2021;97 doi: 10.1016/j.jevs.2020.103339. [DOI] [PubMed] [Google Scholar]
- 21.Nam N.H., Kim Y., You Y.J., Hong D.H., Kim H.M., Ahn B.Z. Water soluble prodrugs of the antitumor agent 3-[(3-amino-4-methoxy)phenyl]-2-(3,4,5-trimethoxyphenyl)cyclopent-2-ene-1-one. Bioorgan Med Chem. 2003;11:1021–1029. doi: 10.1016/s0968-0896(02)00514-x. [DOI] [PubMed] [Google Scholar]
- 22.Juntunen J., Huuskonen J., Laine K., Niemi R., Taipale H., Nevalainen T., et al. Anandamide prodrugs. 1. water-soluble phosphate esters of arachidonylethanolamide and R-methanandamide. Eur J Pharm Sci. 2003;19:37–43. doi: 10.1016/s0928-0987(03)00044-7. [DOI] [PubMed] [Google Scholar]
- 23.Mäntylä A., Garnier T., Rautio J., Nevalainen T., Vepsälainen J., Koskinen A., et al. Synthesis, in vitro evaluation, and antileishmanial activity of water-soluble prodrugs of buparvaquone. J Med Chem. 2004;47:188–195. doi: 10.1021/jm030868a. [DOI] [PubMed] [Google Scholar]
- 24.Pannecouque C., Daelemans D., De Clercq E. Tetrazolium-based colorimetric assay for the detection of HIV replication inhibitors: revisited 20 years later. Nat Protoc. 2008;3:427–434. doi: 10.1038/nprot.2007.517. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki K., Craddock B.P., Okamoto N., Kano T., Steigbigel R.T. Poly A-linked colorimetric microtiter plate assay for HIV reverse transcriptase. J Virol Methods. 1993;44:189–198. doi: 10.1016/0166-0934(93)90054-u. [DOI] [PubMed] [Google Scholar]
- 26.Nair R., Lamare I., Tiwari N.K., Ravi P.R., Pillai R. In situ salification in polar solvents: a paradigm for enabling drug delivery of weakly ionic drugs as amorphous solid dispersion. AAPS PharmSciTech. 2018;19:326–337. doi: 10.1208/s12249-017-0808-z. [DOI] [PubMed] [Google Scholar]
- 27.Kang D., Feng D., Sun Y., Fang Z., Wei F., De Clercq E., et al. Structure-based bioisosterism yields HIV-1 NNRTIs with improved drug-resistance profiles and favorable pharmacokinetic properties. J Med Chem. 2020;63:4837–4848. doi: 10.1021/acs.jmedchem.0c00117. [DOI] [PubMed] [Google Scholar]
- 28.Gao S., Song L., Cheng Y., Zhao F., Kang D., Song S., et al. Discovery of novel sulfonamide substituted indolylarylsulfones as potent HIV-1 inhibitors with better safety profiles. Acta Pharm Sin B. 2023;13:2747–2764. doi: 10.1016/j.apsb.2023.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.